 Bilal Shaker
Bilal Shaker Sajjad Ahmad
Sajjad Ahmad Junhao Shen
Junhao Shen Hyung Wook Kim
Hyung Wook Kim Dokyun Na
Dokyun Na- 1Department of Biomedical Engineering, Chung-Ang University, Seoul, South Korea
- 2Department of Health and Biological Sciences, Abasyn University, Peshawar, Pakistan
- 3College of Life Sciences, Sejong University, Seoul, South Korea
Porphyromonas gingivalis is a Gram-negative pathogenic bacterium associated with chronic periodontitis. The development of a chimeric peptide-based vaccine targeting this pathogen could be highly beneficial in preventing oral bone loss as well as other severe gum diseases. We applied a computational framework to design a multi-epitope-based vaccine candidate against P. gingivalis. The vaccine comprises epitopes from subunit proteins prioritized from the P. gingivalis reference strain (P. gingivalis ATCC 33277) using several reported vaccine properties. Protein-based subunit vaccines were prioritized through genomics techniques. Epitope prediction was performed using immunoinformatic servers and tools. Molecular modeling approaches were used to build a putative three-dimensional structure of the vaccine to understand its interactions with host immune cells through biophysical techniques such as molecular docking simulation studies and binding free energy methods. Genome subtraction identified 18 vaccine targets: six outer-membrane, nine cytoplasmic membrane-, one periplasmic, and two extracellular proteins. These proteins passed different vaccine checks required for the successful development of a vaccine candidate. The shortlisted proteins were subjected to immunoinformatic analysis to map B-cell derived T-cell epitopes, and antigenic, water-soluble, non-toxic, and good binders of DRB1*0101 were selected. The epitopes were then modeled into a multi-epitope peptide vaccine construct (linked epitopes plus adjuvant) to enhance immunogenicity and effectively engage both innate and adaptive immunity. Further, the molecular docking approach was used to determine the binding conformation of the vaccine to TLR2 innate immune receptor. Molecular dynamics simulations and binding free energy calculations of the vaccine–TLR2 complex were performed to highlight key intermolecular binding energies. Findings of this study will be useful for vaccine developers to design an effective vaccine for chronic periodontitis pathogens, specifically P. gingivalis.
Introduction
Porphyromonas gingivalis is a rod-shaped, Gram-negative oral bacterium asserted as a pivotal pathogen in the growth of chronic periodontitis (1). Periodontitis is caused by the inflammation of periodontal tissues and may result in tooth loss (2). P. gingivalis is detected in 85% of periodontitis sites, is rarely detected in healthy patients (3), and can modulate periodontal protective mechanisms (4–6). P. gingivalis may exacerbate cognitive impairments in patients with Alzheimer’s disease; oral infection by P. gingivalis led to the activation of a complement pathway in the mouse brain tissue (7), and periodontitis-induced Alzheimer’s disease model mice showed impaired cognitive function compared to non-induced mice (8).
Vaccines are biological products that stimulate and induce acquired immunity against a particular pathogen and are the most efficient and cost-effective method to prevent infectious diseases (9, 10). There were several attempts to develop attenuated vaccines against P. gingivalis, but satisfactory levels of protection were not observed. For example, Polak et al. (11) observed humoral immune responses elicited against P. gingivalis in the immunized mice, but the vaccine failed to prevent periodontitis. Leone et al. (12) demonstrated that immunized mice against P. gingivalis showed increased inflammation and fibroblast apoptosis, and exacerbated bone loss through the up-regulation of innate immune response. Immunization with specific proteins such as fimbriae and RgpA from P. gingivalis, instead of whole bacterial materials, was found to be more protective against periodontitis and oral bone loss (13–22). These results emphasize the need for identifying proteins responsible for boosted vaccination. Since the key components inducing immune response are the epitopes included in proteins, there is a need to identify responsible antigenic sequences from proteins and optimize the sequences for enhanced vaccines (22).
Over the past decades, the vaccine field has been revolutionized owing to the advances in structural biology, genomics, computational biology, and biotechnology (23, 24). A new technology, named reverse vaccinology, allows for identifying virulence factors of pathogens responsible for unmet diseases from their genomic sequences, discovering antigenic sequences, and optimizing the sequences via computational methods (25). The reverse vaccinology approach was first applied to Neisseria meningitidis serogroup B for which no broad-spectrum vaccines were available due to the variations in their outer-membrane proteins and the potential cross-reactivity of their capsular polysaccharide with human tissues (26). After this successful attempt, reverse vaccinology has been accepted as an effective method for vaccine discovery.
In this study, we aimed to develop a vaccine to provide protective immunity against P. gingivalis that causes chronic periodontitis in humans, and is likely correlated with the severity of Alzheimer’s disease symptoms. We adopted a pan-genomic reverse vaccinology strategy to shed light on the core proteins shared across all strains of the pathogen and unique proteins present in selected strains, which allows for the development of both broad- and narrow-spectrum vaccines (27). Subtractive proteomic analysis further discarded the proteins of the pathogen homologous to human proteins. After the prioritization of potential vaccine target proteins, multiple epitopes that can interact with major histocompatibility complex (MHC) class I and II proteins were predicted from the target proteins to maximize both B- and T-cell immune responses (28). Subsequently, world human population coverage by predicted MHC class I and II was also analyzed (29). To boost the immune system, a multi-epitope chimeric protein was designed to ensure sufficient solubility and stability via molecular dynamics simulations (30). The designed vaccine protein contained the predicted epitopes tagged using a flexible linker, and Pam3CSK4 chain C (PDB ID: 2Z7X) was used as a vaccine adjuvant (31–33) to boost the antigenicity of predicted epitopes and guide epitope recognition and processing. The computational methodology developed in this study will facilitate the development of reliable and effective vaccines to fight against pathogens. In addition, the designed vaccine construct may provide a new method to treat chronic periodontitis and alleviate the symptoms of Alzheimer’s disease.
Material and Methods
Antigenic Protein Selection From Proteome of P. gingivalis
The complete proteome of P. gingivalis (ATCC 33277) was obtained from the National Center for Biotechnology Information database (34). To shortlist proteins for vaccine construction, the whole proteome of the pathogen was subjected to a subtractive pipeline (35). First, duplicate or very similar proteins were discarded using the Cluster Database at High Identity with Tolerance (CD-HIT) suite with a sequence identity cut-off of 0.9 (36). Second, proteins predicted to be located within the cytoplasm using the PSORTb subcellular localization prediction tool (37) were discarded because cytoplasmic proteins cannot be recognized by host immune systems; only proteins in the outer or periplasmic membranes play important roles in the attachment, infection, and survival of pathogens (38). Third, the proteins homologous to human proteins were discarded to avoid autoimmune responses using the protein Basic Local Alignment Search Tool (BLASTp) with a cutoff E-value of 10-4 (39, 40). The remaining proteins were subjected to the subsequent processes including virulency-, molecular weight-, and antigenicity prediction.
Virulent proteins are promising candidates for vaccine design because they play an important role in pathogenicity, interact with host pathways, and thus are conserved across the strains of pathogens (41–43). Therefore, they are major targets for vaccines against multiple strains. Of the selected proteins in previous processes, those similar to known virulence factors deposited in the Virulence Factor Database (44) were selected. The search was carried out by using BLASTp with a sequence identity >30% and a bit score >100.
The potential antigenicity of the selected virulent proteins that were non-homologous to human proteins and were not located within the cytoplasm was predicted by using VaxiJen (45). The proteins with a predicted antigenicity >0.4 (default cutoff of VaxiJen) and a molecular mass <110 kDa were selected (42, 43) and used as target proteins for epitope search.
Epitope Prediction Within Selected Antigenic Proteins
Potential epitopes to stimulate cell-mediated immune responses were predicted on the selected proteins from the previous processes by using NetCTL with a default setting (46). NetCTL predicted cytotoxic T lymphocyte (CTL) epitopes capable of interacting with class I MHC proteins and thereby stimulating the activation of CTLs based on the binding affinity between class I MHC proteins and epitopes, proteasomal C-terminal cleavage, and transporter associated with antigen processing (TAP) transport efficiency (46). The predicted CTL epitopes could elicit cell-mediated immunity to inhibit the development of the pathogen and induce the generation of memory T cells to prevent future infection (47).
B-cell epitopes are specific peptides within antigens recognized by B-cell receptors and antibodies produced from the activated B cells. Stimulation of humoral immune response is also important because it can clear pathogens by inducing an antibody-mediated immune response. Linear B-cell epitopes within the selected proteins were predicted using the ABCpred server (48).
Full activation of stimulated immune cells (T and B cells) requires additional stimulation from helper T lymphocytes (HTL) to avoid autoimmune responses. HTL recognizes epitopes loaded onto class II MHC proteins in B-cell membranes and other antigen-presenting cells. The HTL epitopes were predicted by using an online tool served by the Immune Epitope Database (IEDB) (49). This tool ranked predicted epitopes based on a percentile score calculated by comparing the scores generated using three methods (SMM-align, Combinatorial library, and Sturniolo) with the scores of five million random 15-mer peptides generated from the proteins in the UniProt database (50, 51).
Interferon-Gamma Epitope Prediction
The IFNepitope webserver was used to predict the MHC-II (HTL) epitopes that can induce interferon-gamma with an 82% accuracy (52). The server uses machine learning-based models including motif-based model, support vector machine-based model, and hybrid approach (motif and support vector machine) to predict the interferon-producing property of epitopes and assigns support vector machine scores to each input epitope.
CTL and HTL Epitope Screening
Predicted CTL and HTL epitopes were further shortlisted by applying multiple filters including toxicity, antigenicity, MHC binding affinity, and solubility. Online web servers VaxiJen (45), ToxinPred (53), MHCPred (54), and peptide solubility calculator (https://pepcalc.com/peptide-solubility-calculator.php) were used to predict the toxicity, antigenicity, MHC binding affinity, and water solubility of each epitope, respectively. Epitopes predicted as non-toxic, antigenic, and water-soluble positive binders were selected for vaccine design.
Population Coverage by CTL and HTL Epitopes
Human leukocyte antigen (HLA) pattern varies among ethnic groups and geographical areas. Thus, the estimation of population coverage by the predicted CTL and HTL epitopes is important to design an effective vaccine. The population coverage tool of IEDB (http://tools.iedb.org/population/) was used to estimate the world human population coverage by the predicted CTL and HTL epitopes (49).
Multi-Epitope Peptide Vaccine Construction
In this step, shortlisted epitopes were linked using two linkers, AAY and GPGPG (55). The AAY linker was used to fuse CTL epitopes, whereas the GPGPG linker was used to fuse HTL epitopes. These linkers help separate the epitopes to avoid junctional epitope (neo-epitopes) formation and improve epitope presentation (56–59). Furthermore, Pam3CSK4 chain C (PDB ID: 2Z7X) was used as a vaccine adjuvant to increase the immunogenic property of the vaccine construct (33). Adjuvant was linked at the N-terminus of the vaccine construct by using the EAAAK linker (60).
Antigenicity and Allergenicity Prediction of Vaccine Construct
VaxiJen v2.0 (45) was used to predict the antigenicity of the vaccine construct. This server generates the antigenic score of the query sequence with the precision of 70–89% by using an alignment-independent algorithm. The non-allergenic behavior of the vaccine was predicted using the AllerCatPro server (61), which predicts the allergenic potential of peptides based on the three-dimensional (3D) structural similarity and compares their amino acid sequence with a dataset of known allergen proteins. The dataset used in this server was derived from the union of the major databases such as Food Allergy Research and Resources Program, Comprehensive Protein Allergen Resource, WHO/International Union of Immunological Societies, UniProtKB, and Allergome (62).
Physicochemical Properties, Structure Prediction, Refinement, and Validation
ProtParam (63) was employed to determine the physicochemical properties of the vaccine construct, namely molecular weight, amino acid composition, theoretical isoelectric point value (PI), stability index, in vitro and in vivo half-life estimation, and GRAVY (grand average of hydropathicity). Further, the vaccine construct sequence was subjected to the PDBSUM server (64) for secondary structure prediction. The Robetta server (65) was used to generate the 3D structure of the vaccine construct. Robetta performs comparative modeling if there is a suitable template in a database; if there is no template, then the ab initio structure prediction method is employed. The fragment-guided molecular dynamics (66) algorithm was used for structure refinement at the atomic level. Subsequently, the refined predicted model was validated by employing VARIFY 3D (67), ProSA-web server (68), and the Ramachandran plot (69).
Protein Disulfide Engineering
Protein model stability can be enhanced by adopting a disulfide bond formation approach. Disulfide bonds strengthen the geometric conformation of the protein structure and stabilize the structure. The protein structure was subjected to an online server DbD2 (70) for disulfide engineering. This web server can detect residue pairs with the ability to form disulfide bonds if the individual amino acid mutated to cysteine.
Molecular Docking With TLR2
Molecular docking is an in silico technique to evaluate the binding affinity between ligand and receptor molecules (71). Human toll-like receptor 2 (TLR2) was used in this study to determine the binding affinity between the vaccine construct and human TLRs. The 3D structure of TLR2 was retrieved from the Protein Data Bank (PDB) (72) (PDB ID: 5D3I). The PatchDock server (73) was used to perform molecular docking, and the 3D structure of human TLR2 was uploaded as a receptor and vaccine structure as a ligand. Subsequently, the FireDock server (74) was used to refine the docked complex obtained from the PatchDock server.
Molecular Dynamics Simulation
Molecular dynamics simulation (75) was performed to understand the stability and protein-protein interactions at the atomic level to support the predictions. Amber v.20 (76) software was used to run a 100 ns simulation of the selected complex. The LeaP module was utilized to add hydrogen atoms to the docked complex of the vaccine and the receptor (77). Then counter ions (Na+) were introduced to neutralize the simulated system. Subsequently, a truncated octahedral box of the TIP3P water model with buffer was submerged with the neutralized system (78). The solvated system was minimized first for the 1500 steps of the steepest descent method and then for the 1000 steps of the conjugated gradient method by using the ff14SB force field (79). In the next step, the system was heated up to 100 ps, reaching an equilibrium after 100 ps. The SHAKE algorithm (80) was applied to constrain bonds involving hydrogen atoms. A molecular dynamics simulation run was accomplished for 100 ns to determine the dynamics of the complex and evaluate the docked conformation and stability of the ligand. The CPPTRAJ module in Amber was used to perform trajectory analysis (81).
MMPBSA/MMGBSA Binding Free Energy of the Complex
Two efficient energy calculation methods were used to estimate the binding free energy, namely MMPBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) and MMGBSA (Molecular Mechanics-Generalized Born Surface Area), embedded in the MMPBSA.py module of Amber (82). The net energy of the system was calculated using equation (1).
The terms in equation (1) include several energy calculations including van der Waals energy, internal energy from molecular mechanics, electrostatic energy, and polar and non-polar contribution towards solvation energy.
Results and Discussion
Several advanced sequencing techniques are being used for microorganism genome sequencing and thus hundreds of thousands of complete genomes are now available in sequence databases allowing the prediction of vaccine and drug targets (83, 84). In this study, immunoinformatics and subtractive proteomics techniques were applied to identify suitable proteins and design multiple epitope-based vaccines against P. gingivalis infection (85). Schematic diagram of the complete methodology to design a multi-epitope peptide vaccine against P. gingivalis is displayed in Figure 1. The complete proteome of P. gingivalis strain ATCC33277, comprising 1835 proteins, was collected from the National Center for Biotechnology Information (86). In the first step, the complete protein sequences of the pathogen were compared with each other to remove those that share the sequence identity of the set cut-off as stated in the methodology. For this purpose, the CD-HIT tool (87) was employed to identify 1788 non-paralogous and non-redundant proteins at a 90% identity threshold. These unique sequences are considered excellent starting materials in the vaccine and drug discovery process (88). These non-paralogous proteins were subjected to the PSORTb server (37) to predict the subcellular localization of proteins. The server categorized the proteins based on their location within the cell as 333 cytoplasmic-membrane, 8 extracellular, 39 outer-membrane, 16 periplasmic, and 1392 cytoplasmic proteins (Figure 2). Subsequently, cytoplasmic proteins were discarded because they are inaccessible to the host immune system and least suitable for vaccine design (38). Additionally, these proteins are enzymatic in nature to catalyze cellular processes and possess hydrophobic pockets for substrate binding (89). On the contrary, secretome/surfome/periplasmic proteins are surface-exposed, and host immune response can recognize antigenic epitopes of such proteins more efficiently for producing prompt and targeted immune reactions (90).
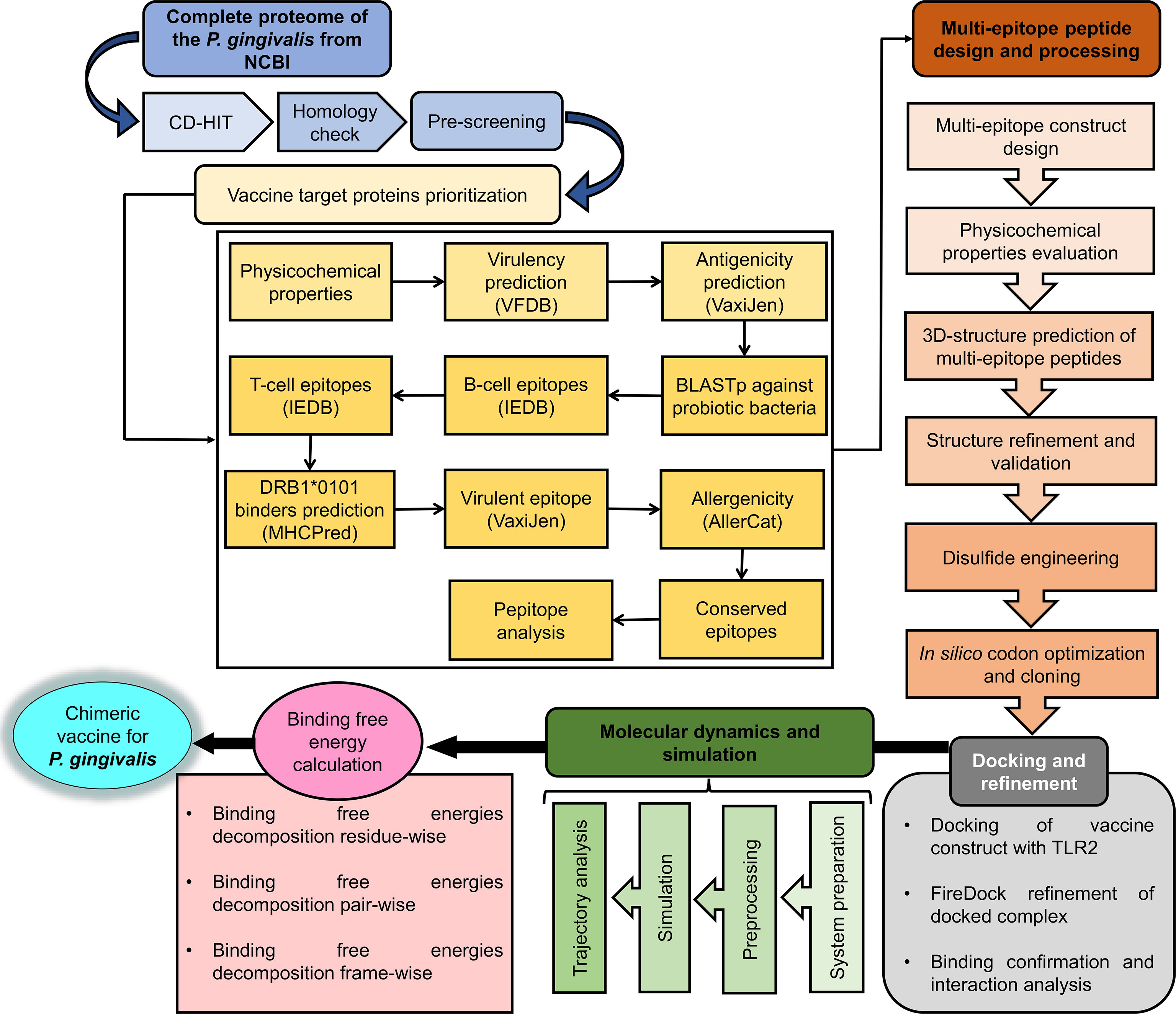
Figure 1 Stepwise workflow designed for in silico vaccine design against P. gingivalis.
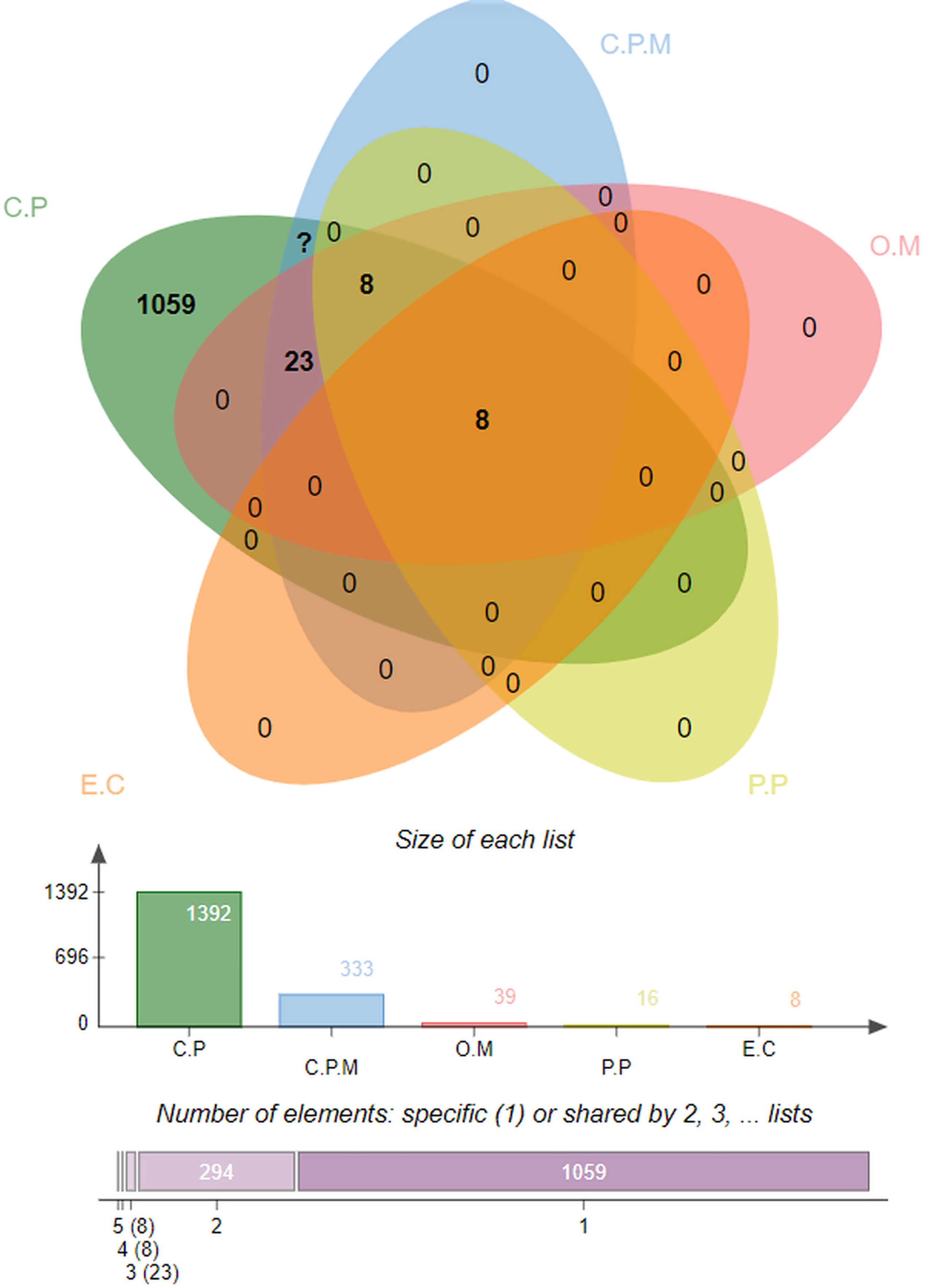
Figure 2 Percent-wise distribution of proteins within the cell, where C.P denotes Cytoplasmic, C.P.M is for Cytoplasmic-membrane, O.M represents Outer-membrane, P.P used for Periplasmic, and E.C is for Extracellular.
Virulent Protein Analysis
Virulent proteins are crucial for pathogen survival and cause infection; they help the pathogen in bypassing the host immunity, invading host cells, dissemination, intracellular survival, and proliferation, which make them suitable for vaccine design (91). The BLASTp tool of the Virulence Factor Database was used to identify virulent proteins. Virulent proteins are explored often as potent vaccine targets and are responsible for infection establishment and pathological conditions (43). Proteins with identity scores >30% and a bit score >100% were considered virulent and selected for further evaluation. As a result, 30 cytoplasmic-membrane, 2 extracellular, 6 outer-membrane, and 4 periplasmic proteins were determined as virulent.
Antigenic and Small-Size Protein Prediction
Different physicochemical properties (antigenicity and size) of the pooled virulent proteins were further evaluated to guide the selection of suitable vaccine candidates for experimental evaluation that can be readily used in vaccine development (40, 42). VaxiJen server (45) predicted the antigenicity of the virulent proteins. Proteins predicted as antigenic were selected, and subsequently, the ProtParam tool (63) was used to calculate the molecular weight of each antigenic protein. Since proteins with smaller molecular weight are easier to isolate and purify for structural and functional studies (41), proteins with molecular weight lower than 110 kDa were selected. Further, selected proteins were submitted to the BLASTp (92) tool of the National Center for Biotechnology Information to search for P. gingivalis for human homologs at the 10-4 cut-off value. Homolog proteins (sequence identity >30%) were discarded subsequently. Selecting human homologs could initiate the autoimmune response, negatively impacting the host’s healthy cells and tissues (93). Nine cytoplasmic-membrane, two extracellular, one periplasmic, and six outer-membrane proteins were antigenic with molecular weights lower than 110 kDa, and they were not homologous to the human proteome. A list of selected proteins is shown in Table 1.
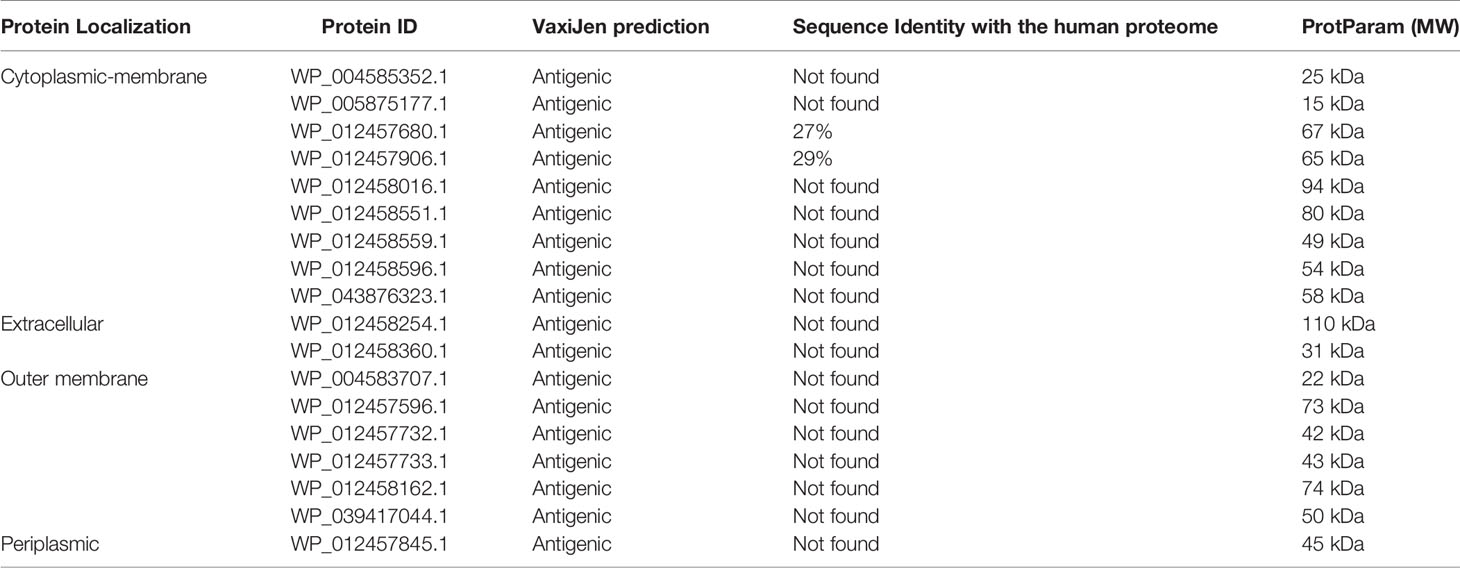
Table 1 Selected proteins from P. gingivalis proteome for multiple epitope prediction.
MHC-I and MHC-II Epitope Prediction
MHC-1 molecules are expressed on the cell surface of all nucleated cells. They present peptide fragments derived from intracellular proteins and play an important role in alerting the immune system against virally infected cells (94). NetCTL 1.2 server (46) was used to predict the MHC-I binding epitopes (9-mer) of each protein; 150 MHC-1 epitopes for cytoplasmic membrane, 41 epitopes for extracellular, 93 epitopes for outer-membrane, and 12 epitopes for periplasmic proteins were collected based on their high binding affinity. The IEDB MHC-II server (49) was used to predict MHC-II binding epitopes (15-mer) against a reference set of seven HLAs. MHC-II molecules are central to effective adaptive immune response and expressed on specialized antigen-presenting cells like B cells, dendritic cells, thymic epithelial cells, and monocytes (95). The top ten MHC-II binding epitopes with low percentile rank were retrieved from each protein. Both MHC-I and MHC-II epitopes were further subjected to a screening pipeline to determine suitable epitope candidates for a potent vaccine construct.
Screening of MHC-I Epitopes
Antigenic, toxic, and water-soluble MHC-I epitopes were predicted using the VaxiJen server (45), ToxinPred (53), and peptide solubility calculator (https://pepcalc.com/peptide-solubility-calculator.php), respectively. Subsequently, half-maximal inhibitory concentration (IC50) values of MHC-I epitopes binding to HLA DRB1*0101 were predicted using the MHCPred server (54), and epitopes with IC50 <100 nm were selected.
Collectively, 16 MHC-I epitopes were selected for an effective vaccine construct based on their antigenicity, non-toxicity, water solubility, and IC50 binding with the receptor (Table 2). The IFNepitope server (52) was also used to search for interferon-gamma inducing epitopes. Positive binding epitopes were predicted and shortlisted for constructing the vaccine.
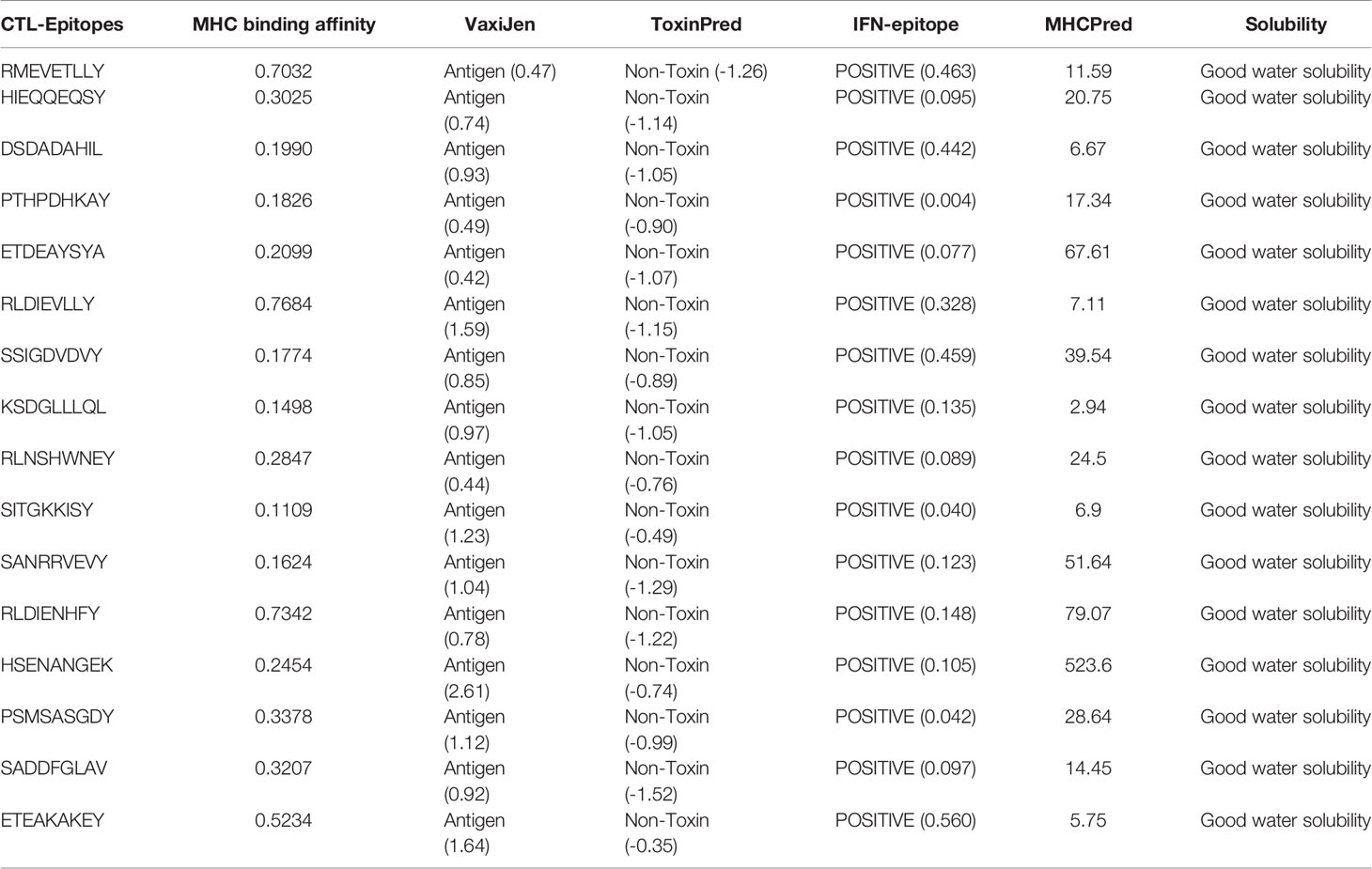
Table 2 Selected MHC-I epitopes for vaccine construct.
Screening of MHC-II Epitopes
Predicted MHC-II epitopes were also introduced to the screening pipeline, and antigenic, non-toxic, positive interferon inducer, water-soluble, and low percentile-ranked epitopes were selected for vaccine design. Eleven selected 15-mer MHC-II epitopes are listed in Table 3.
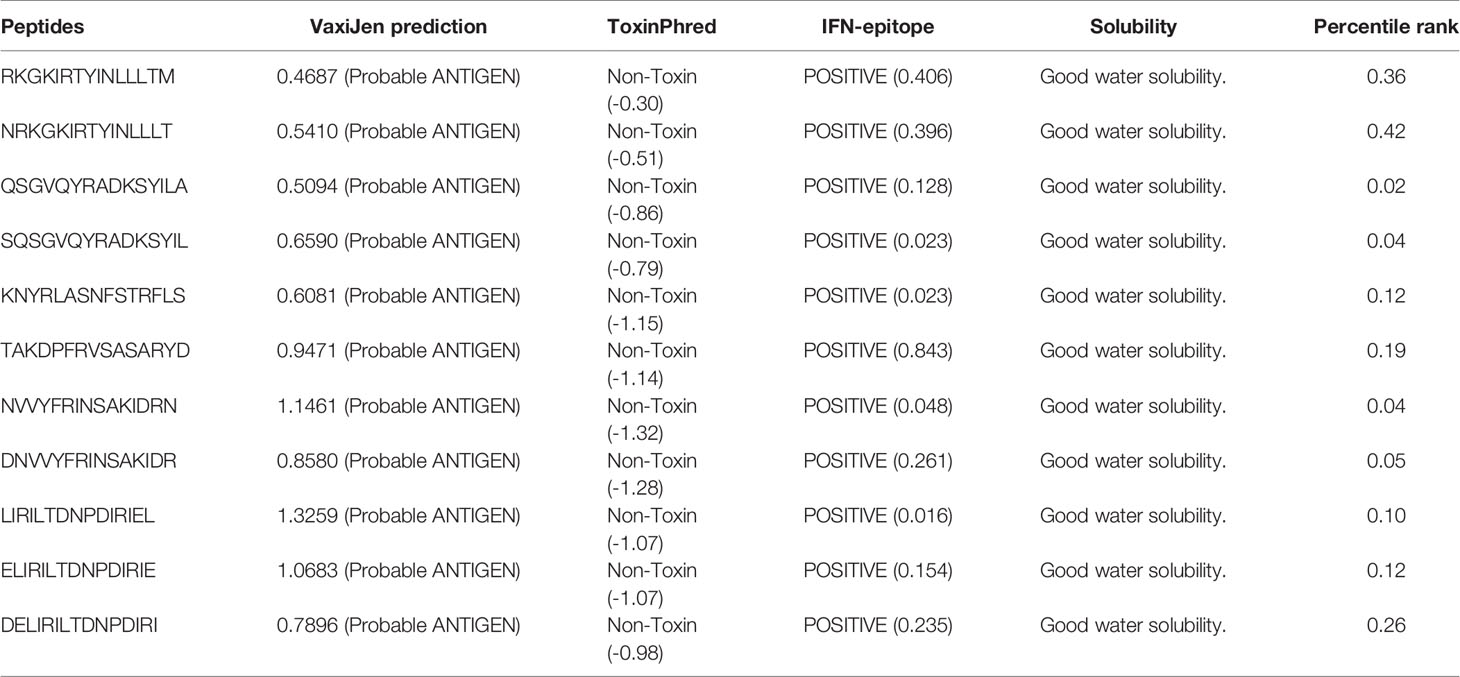
Table 3 Selected potent MHC-II epitope candidates.
Population Coverage Analysis
The cumulative percentage of population coverage was estimated using the predicted epitopes in the vaccine construct in highly populated countries of South Asia, East Asia, Europe, and North America (Supplementary Table 1). The result indicates that the multi-epitope strategy could cover 99.93% of the human population: specifically, around 99% for European and North American, and 40-60% for Asian and African. The high coverage in European and American population and low coverage in Asian and African population represent that our vaccine construct may work better in European and American population. This might be due to biased HLA information in IEDB: when IEDB HLA dataset was analyzed, about half of the HLA dataset in IEBD was from European and American.
Vaccine Construction
Organism- and peptide-based vaccines can successfully reduce the mortality and morbidity caused by infectious diseases (96). Peptide-based vaccines offer an attractive alternative to organism-based vaccines as they induce a specific immune response, are easy to synthesize and use in clinical settings, are cost-effective, minimize the risk of antigen-induced anaphylaxis, and are flexible in changing antigens (97). Nevertheless, during isolation, peptides are weakly immunogenic and require proper adjuvating (96). In this study, 16 CTL epitopes and 11 HTL epitopes were used to construct a vaccine sequence. The AAY and GPGPG linkers were used to fuse MHC-I and MHC-II epitopes, respectively. As TLR2 expression increases during P. gingivalis infection, TLR2 agonist Pam3CSK4 chain c (PDB ID; 2Z7X) was linked to the N-terminal of the vaccine construct using the EAAAK linker. The final vaccine construct composed of 419 residues containing both T-cell and B-cell epitopes were designed (Figure 3A). The vaccine construct is approximately 45.5 kDa. The construct can be easily purified by fusing with a short 6×histidine peptide or a fusion agent such as glutathione-S-transferase or maltose-binding protein. Even the fusion with the fusion agent (approximately 20-30 kDa), the recombinant protein would be easily produced and purified from E. coli via conventional protein expression techniques (98–100).
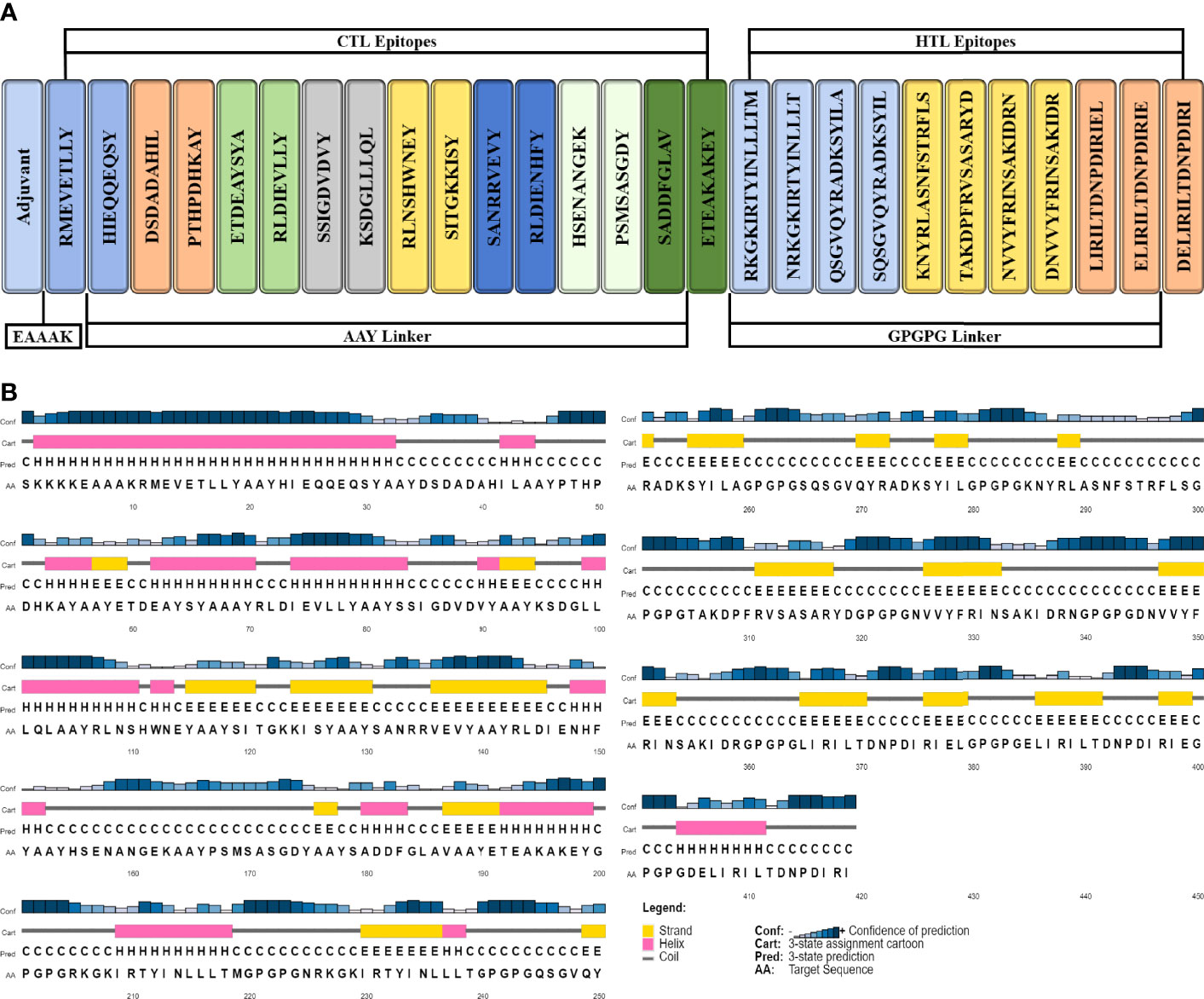
Figure 3 Schematic illustration of 419-amino-acid-long vaccine construct sequence. (A) Representation of adjuvant and epitope arrangement in vaccine construct and (B) 2D structure prediction of vaccine construct.
B-Cell Epitope Prediction
The acquired/adaptive/specific immunity responses of the immune system are highly specialized and systemic in eradicating pathogens from the body or halting their growth (101). B-cells are major lymphocytes of adaptive immunity and are involved in generating humoral and cell-mediated immunity against specific and unwanted invader pathogens (101). In this study, the ABCpred server (48) predicted 14 protein sequence-based linear B-cell epitopes (20-mer) with a 0.8+ score (Table 4 and Supplementary Figure 1). The DiscoTop2.0 server (102) predicted 202 discontinuous B-cell epitopes from the protein structure (Supplementary Table 2).
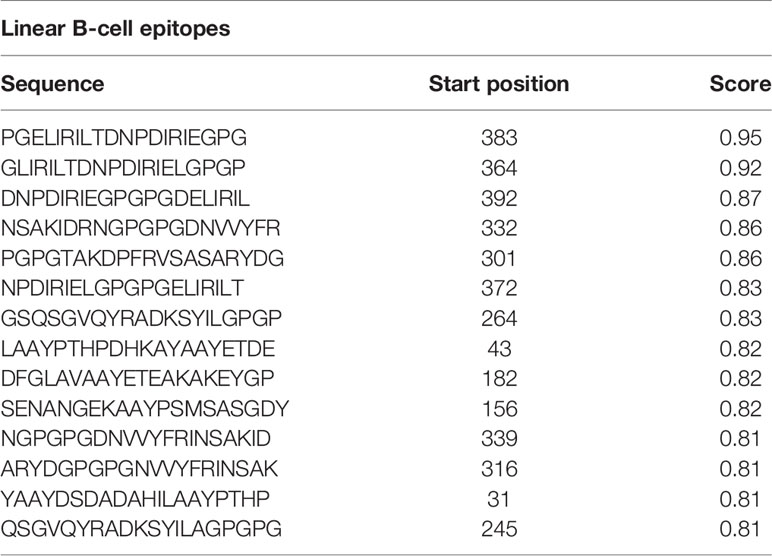
Table 4 Predicted linear B-cell epitopes.
Antigenicity and Allergenicity Prediction of Vaccine Construct
VaxiJen (45) and AllerCatPro (61) servers were used to predict antigenicity and allergenicity of the vaccine construct, respectively. The VaxiJen server calculated an antigenic score of 0.89 for the vaccine construct at 0.4 thresholds designating the vaccine potential to trigger host immune response. AllerCatPro (61) showed no evidence (no sequence and structural similarity with known allergens) for vaccine allergenicity prediction.
Prediction of Physicochemical Properties
The ProtParam tool (63) was employed to compute the physicochemical properties of the vaccine construct. The molecular weight was 45.5 kDa and the theoretical PI was 7.69, whereas the vaccine construct was classified as stable with an instability index of 29.95. Furthermore, the estimated half-life (in vivo) of the vaccine construct in yeast and Escherichia coli was >20 and >10 h, respectively. The aliphatic index indicates the thermostability of the vaccine and was calculated as 77.49, whereas the GRAVY was calculated as -0.488.
Secondary Structure Prediction
The secondary structure was generated by using the PSIPRED workbench (103). The vaccine structure was composed of 26.2% alpha helices, 23.6% beta strands, and 50.1% coils (Figure 3B).
3D Structure Prediction and Validation
The Robetta server (65) was employed to model the 3D structure of the vaccine (Figure 4). Robetta performed ab initio modeling to model the target sequence and generated five structures. Subsequently, model-1 was selected for additional analysis after a brief structural evaluation. Several computational tools were employed to validate the 3D structure of the vaccine construct. The ERRAT server (104) was used to predict the overall quality of the 3D structure with a quality score of 86.91, and ProSA-web server (68) was used to calculate the Z-score of the structure as -5.93.
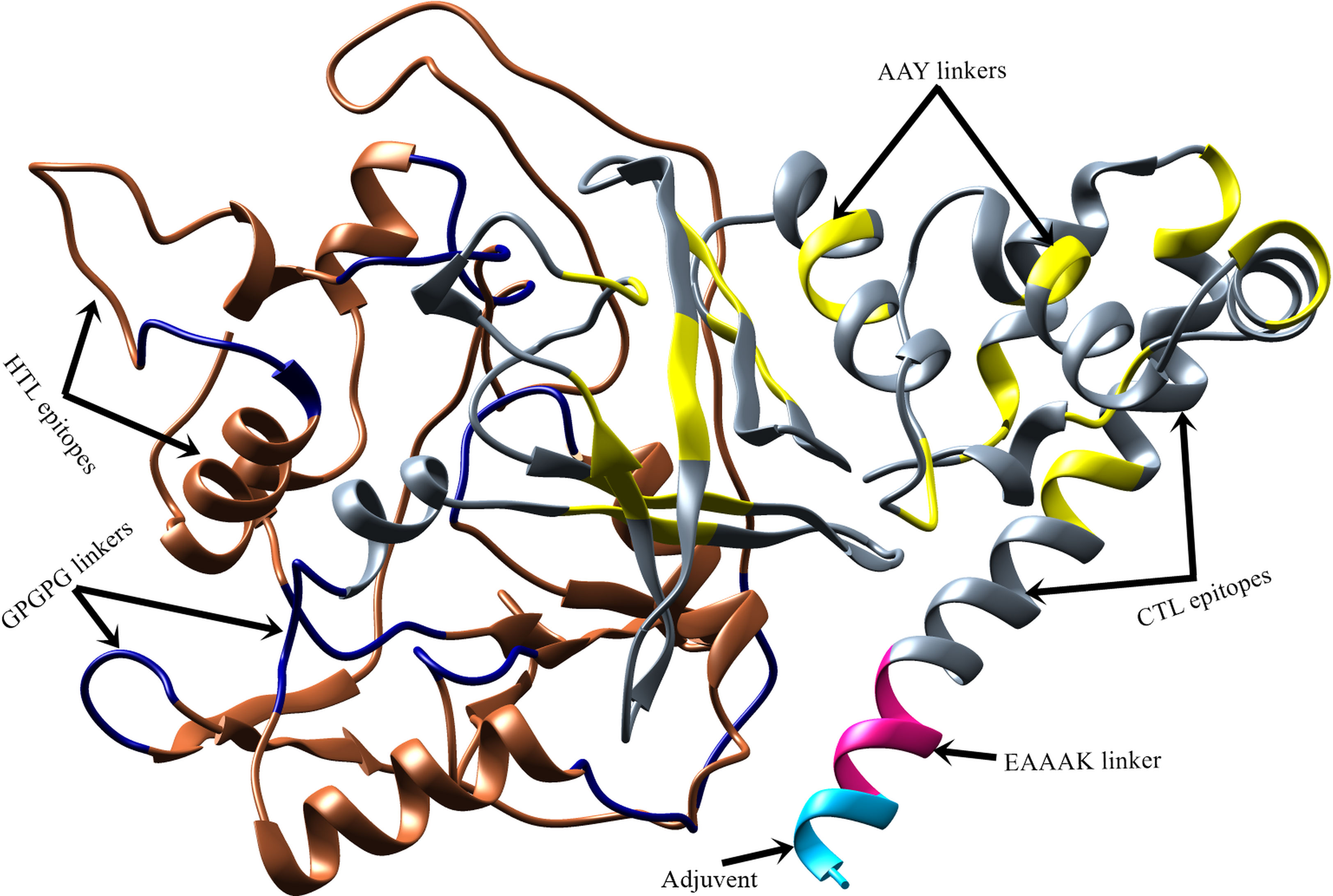
Figure 4 Predicted 3D structure of vaccine construct. Vaccine adjuvant is shown in cyan color, EAAAK linker in pink, CTL epitopes are in grey, AAY linkers and GPGPG linkers are in yellow and navy-blue color, respectively, while HTL epitopes are in sienna color.
Structure Refinement
The accuracy of the predicted structure was improved using structure refinement tools (e.g., fragment-guided molecular dynamics algorithm). The evaluation of the refined vaccine structure on the Ramachandran plot showed that 88.4% of the residues were in most favored regions, 10.1% in additional allowed regions, 0.9% in generously allowed regions, and -0.6% in disallowed regions (Supplementary Figure 2C). In contrast, the unrefined structure had 42.6%, 40.7%, 11.9%, and 4.8% residues in most favored, additionally allowed, generously allowed, and disallowed regions, respectively.
Disulfide Engineering of Vaccine Construct
Residues in the high-mobility region of the protein sequence were mutated with cysteine to perform disulfide engineering. Both inter- and intra-chain disulfide bonds were evaluated. A total of 49 pairs of amino acid residues with the capability to form disulfide bonds were predicted using the DbD2 server. After the evaluation of the pair residues in terms of chi3 and energy values, only two pairs were found potent for disulfide bond formation. Those residues were Ala92–Asp97 and Ala176 and Tyr190 (Supplementary Figure 2B). These residues were replaced with cysteine residues. The values of chi3 between -87 and +97 were considered for residue screening while the energy value was <2.5. The main objective of disulfide engineering was to make the vaccine construct less susceptible to host proteases by replacing susceptible residues with another one, e.g., cysteine, and to increase overall stability of the construct by introducing a disulfide bond (105, 106). The addition of the disulfide bond may alter the thermodynamic stability and disrupt the rate of folding and unfolding of the construct (106, 107). According to the Ramachandran plot (Supplementary Figure 2) of the construct, the introduction of the disulfide bond improved the stability of the construct by introducing conformational constraints to the backbone (108). The improved stability and removal of protease-susceptible residues may represent an improved immunogenicity by increasing the half-life of the construct in human body (108).
Molecular Docking
The protein-peptide molecular docking technique was employed to predict the best binding mode of the vaccine construct to TLR2. As a membrane surface receptor, TLR2 is the most promiscuous TLR with respect to the pathogen-associated molecular pattern recognition derived from bacteria, virus, parasites, and fungi and its activation can result in the functioning of the intracellular signaling pathway of nuclear factor-kappa B and cytokine production leading to innate immunity activation (109). The PatchDock server (73) was used to generate the docked pose and electrostatic interactions between the vaccine construct and receptor (TLR2). Subsequently, the docked complex was further refined by using the FireDock tool (74). FireDock generated 10 docked solutions for the docked complexes with respective global energy values, and solution 9 of the docked complex with the lowest global energy was selected (Figure 5) (-29.77 kJ/mol global energy with TLR2, Table 5). The lowest global energy indicates the highest binding affinity between vaccines and receptors. Within 3Å of the vaccine construct Leu555, Trp558, Gln553, Leu575, Pro549, His573, Ala552, Asp557, Arg574, Pro559, Asp560, Ala32, Asp31, Ser30, Arg39, Asp37, Gly34, Ser29, Glu178, Glu103, Ser42, Ala53, Val35, Cys30, Arg155, His104, Val80, Phe43, Gln152, Lys127, Ile82, Tyr128, Ser101, Leu151, and Asn150 residues from TLR2 were involved in both hydrophilic and hydrophobic interactions. The selected docked complex was subjected to the molecular dynamics simulation for further stability analysis.
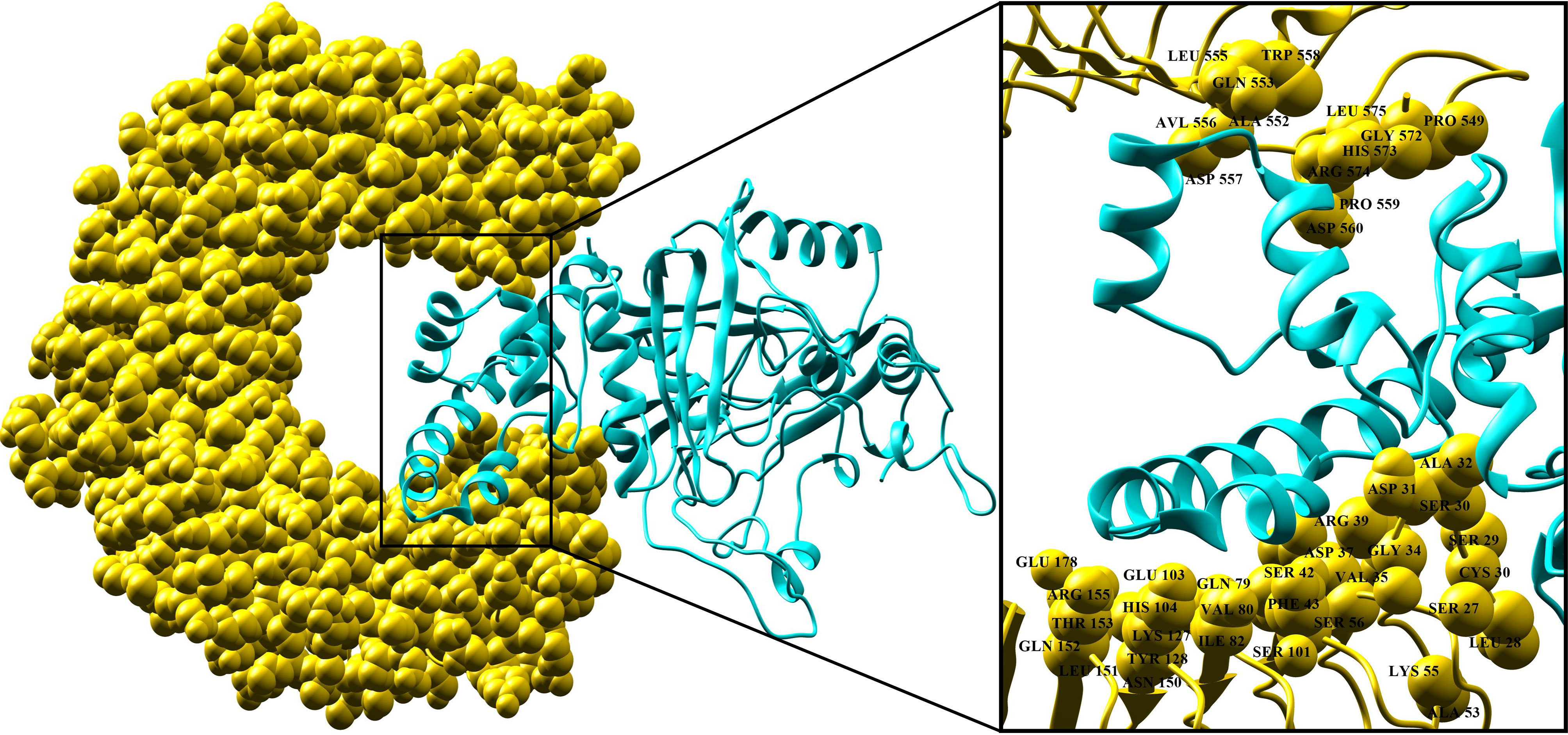
Figure 5 Docking pose of vaccine and TLR-2 receptor. TLR-2 receptor is in yellow and vaccine construct is in cyan. Interaction residues of the receptor are shown on the right-hand side.
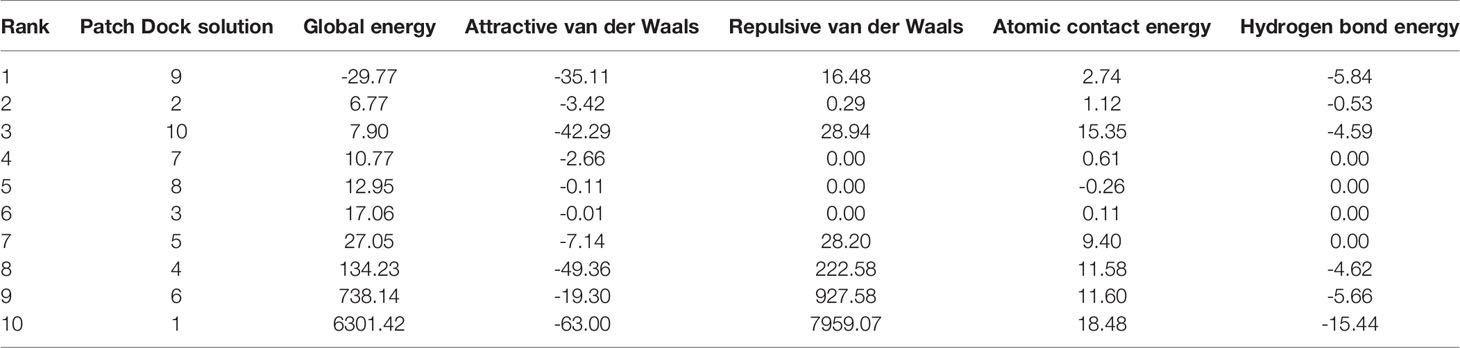
Table 5 Top ten best refined docked solutions generated using FireDock. Energy is presented in kJ/mol.
Molecular Dynamics Simulation
The molecular dynamics simulation was performed to explore the physical movements at the atomic level and to confirm the stability of the docked complex. The root mean square deviation and root mean square fluctuation was determined to estimate the dynamic behavior and stability of the complex for the run of 100 ns. The root mean square deviation is the distance measure between backbone carbon alpha of superimposed proteins (110), and the graph showed an initial fluctuation of 7.6Å at 2 ns, increasing to 14Å (Figure 6A). The system reached equilibrium at 10–30 ns; some major fluctuations were observed at 30–64 ns; afterward, the system was re-equilibrated and remained stable until the end of the simulation run with an average root mean square deviation of 10.6 Å. Residual flexibility of the complex was analyzed via root mean square fluctuation (111) to understand the fluctuations of the residues and whether these variations affect the complex. Figure 6B revealed that residues of the vaccine construct (1–419) have mild fluctuations with a mean root mean square fluctuation of 3.08Å, which indicates the stability and uninterrupted interactions between the receptor and the construct. Major fluctuations with the highest peak of 12.5 Å were observed at C-terminal residues (700–976) (Figure 6B).
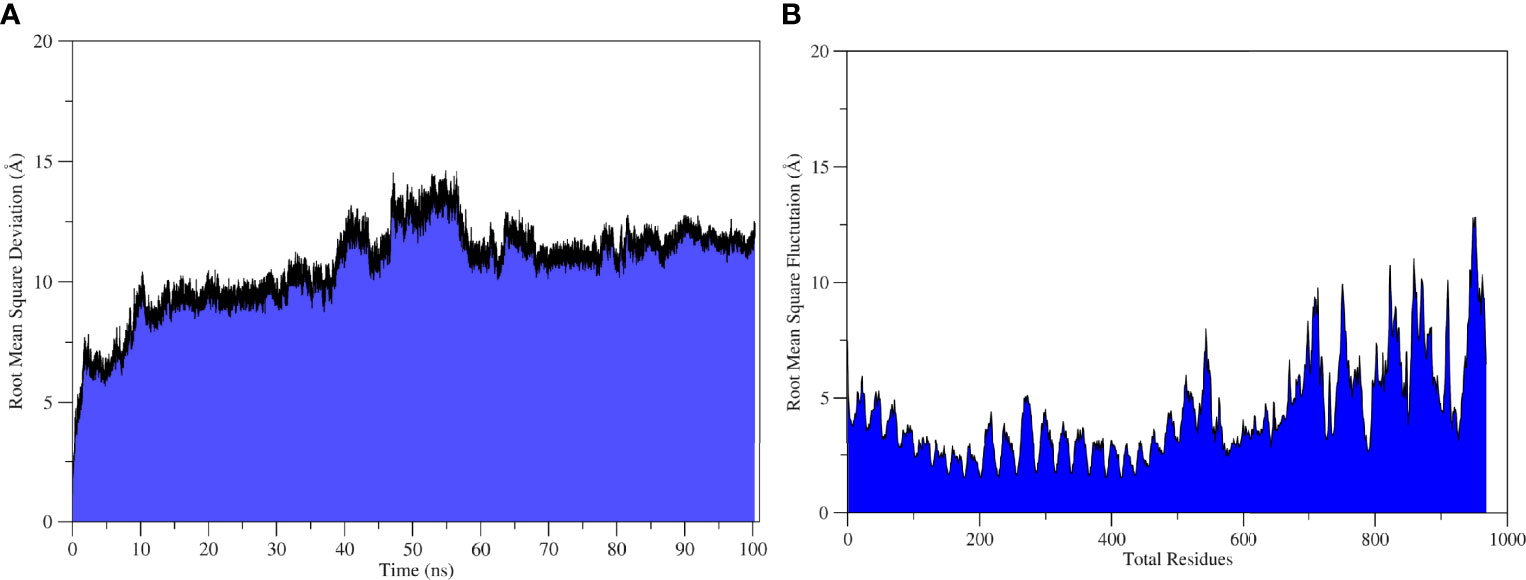
Figure6 Statistical analysis of TLR2-Vaccine simulation trajectories. (A) Root Mean Square Deviation analysis, (B) root mean square fluctuation analysis.
MMPB/GB-SA Binding Free Energy
The MMPB/GB-SA methods are the most popular and preferred methods used to calculate the binding free energy in biomolecular studies such as protein-protein interactions. These methods are user-friendly, more accurate than docking/scoring methods, and considerably less expensive than the free energy perturbation method (112). The binding energies of the system understudies in both methods (MMPBSA/GBSA) are tabulated in Table 6. The system exhibited highly stable and very robust binding with a net binding energy of -43.27 kcal/mol and -68.99 kcal/mol in MMGBSA and MMPBSA, respectively. This energy was dominated by the gas-phase energy of -291.06 kcal/mol in both systems. Solvation was unfavorable as 247.79 kcal/mol in MMGBSA and 222.06 kcal/mol in MMPBSA. The gas-phase energy was mostly contributed by electrostatic energy (-162.74 kcal/mol) and less by van der Waals energy (-128.32 kcal/mol) in both methods.
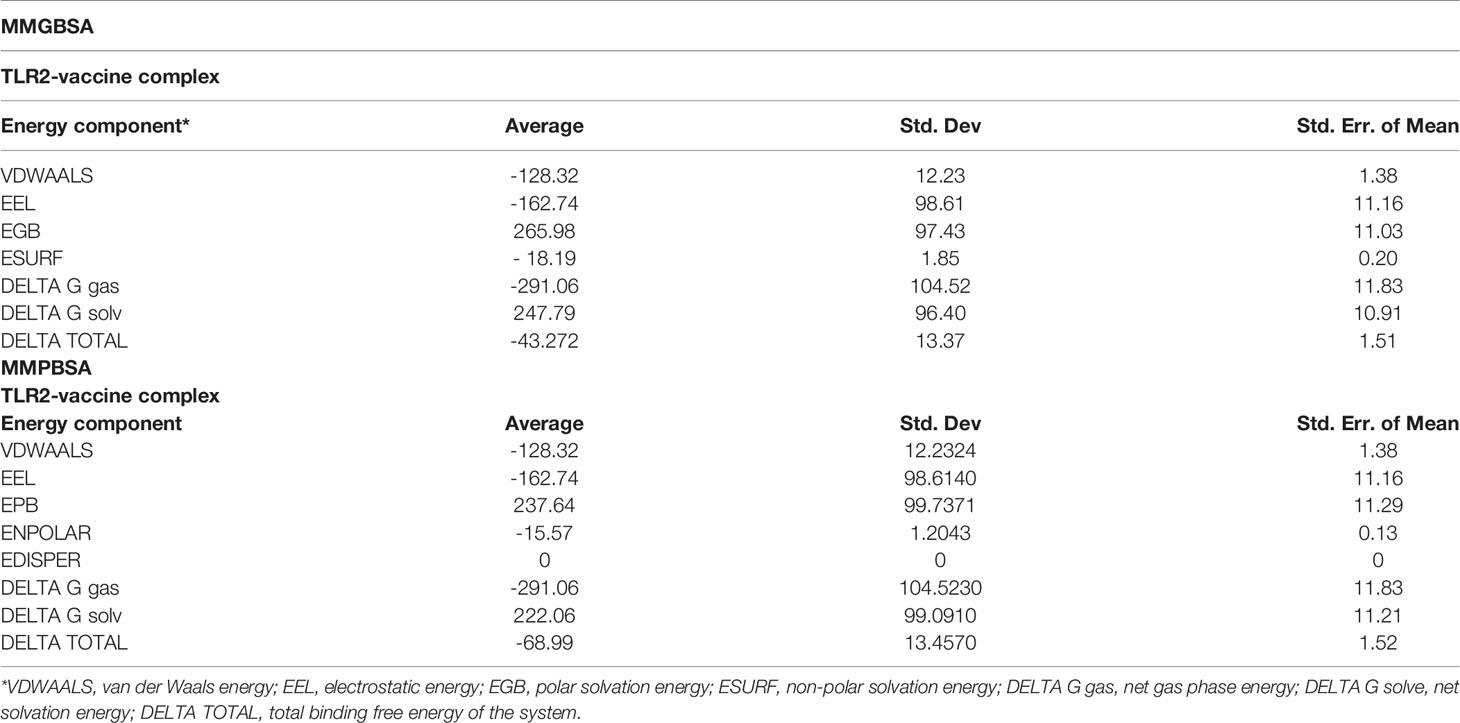
Table 6 Free energy of binding, estimated by MMGBSA and MMPBSA for the complex.
Conclusions
We used immunoinformatic techniques in combination with subtractive proteomics to design a potent and safe multi-epitope vaccine that can be used against P. gingivalis infections. This study begins with the retrieval of the complete proteome of the pathogen and removal of irrelevant proteins to determine the most suitable protein targets composed of non-allergic, antigenic, virulent, and high-affinity binders with DRB*0101 multi-epitope peptides. Predicted epitopes showed higher worldwide human population coverage of 99.93%. Immunoinformatic tools and online web servers were used to predict B and T cells from the selected proteins. CTL and HTL epitopes were fused using appropriate linkers. Molecular docking showed the strong binding affinity of the vaccine construct to the innate immune receptor (TLR2), thus allowing adaptive immunity and prompt response against the pathogen. The molecular dynamics simulations illustrated the highly stable molecular interactions and predicted the binding mode of the construct. Further, binding free energy calculations support the docking and simulation findings as a highly stable nature of the complex. The designed vaccine construct was immunogenic in our in silico evaluation; nevertheless, the extent to which it could prevent P. gingivalis infection is unknown. Immunoinformatics approaches have been successfully applied to a number of pathogens to screen for protective antigens, such as the Meningococcus B (MenB) vaccine developed recently. Similarly, Chlamydia, Staphylococcus aureus, and group A Streptococcus are successfully addressed using such in silico methods. The vaccine construct developed in this study is ready to be evaluated in experimental studies to disclose its immunogenicity against the P. gingivalis pathogen.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author Contributions
BS: project design, investigation, data curation, original draft preparation, figure preparation, and referencing. SA: Figure preparation, reference management, reviewing, and editing. JS: Figure preparation, reference management, reviewing, and editing. HWK: Supervision, conceptualization, methodology, and writing—reviewing and editing. DN: Supervision, conceptualization, methodology, patient’s sampling acquisition, writing—reviewing and editing, and project management. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI21C1659). This research was also supported by the Chung-Ang University Research Grants, 2020.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.806825/full#supplementary-material
Abbreviations
MHC, Major histocompatibility complex; TLR, toll-like receptor; HTL, helper T lymphocytes; CTL, cytotoxic T lymphocyte.
References
1. Bostanci N, Belibasakis GN. Porphyromonas Gingivalis: An Invasive and Evasive Opportunistic Oral Pathogen. FEMS Microbiol Lett (2012) 333:1–9. doi: 10.1111/j.1574-6968.2012.02579.x
2. How KY, Song KP, Chan KG. Porphyromonas Gingivalis: An Overview of Periodontopathic Pathogen Below the Gum Line. Front Microbiol (2016) 7:53. doi: 10.3389/fmicb.2016.00053
3. Yang H-W, Huang Y-F, Chou M-Y. Occurrence of Porphyromonas Gingivalis and Tannerella Forsythensis in Periodontally Diseased and Healthy Subjects. J Periodontol (2004) 75:1077–83. doi: 10.1902/jop.2004.75.8.1077
4. Curtis MA, Zenobia C, Darveau RP. The Relationship of the Oral Microbiotia to Periodontal Health and Disease. Cell Host Microbe (2011) 10:302–6. doi: 10.1016/j.chom.2011.09.008
5. Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas Gingivalis as a Potential Community Activist for Disease. J Dent Res (2012) 91:816–20. doi: 10.1177/0022034512453589
6. Lamont RJ, Jenkinson HF. Life Below the Gum Line: Pathogenic Mechanisms of Porphyromonas Gingivalis. Microbiol Mol Biol Rev (1998) 62:1244–63. doi: 10.1128/mmbr.62.4.1244-1263.1998
7. Poole S, Singhrao SK, Chukkapalli S, Rivera M, Velsko I, Kesavalu L, et al. Active Invasion of Porphyromonas Gingivalis and Infection-Induced Complement Activation in ApoE-/- Mice Brains. J Alzheimer’s Dis (2014) 43:67–80. doi: 10.3233/JAD-140315
8. Ishida N, Ishihara Y, Ishida K, Tada H, Funaki-Kato Y, Hagiwara M, et al. Periodontitis Induced by Bacterial Infection Exacerbates Features of Alzheimer’s Disease in Transgenic Mice. NPJ Aging Mech Dis (2017) 3:1–7. doi: 10.1038/s41514-017-0015-x
9. Levine, Myron M, Rosanna L. Vaccines and Vaccination in Historical Perspective. New Generation Vaccines (1990) 2:1–11. doi: 10.3109/9781420060744-5
10. Andreano E, D’Oro U, Rappuoli R, Finco O. Vaccine Evolution and Its Application to Fight Modern Threats. Front Immunol (2019) 10:1722. doi: 10.3389/fimmu.2019.01722
11. Polak D, Wilensky A, Shapira L, Weiss EI, Houri-Haddad Y. Vaccination of Mice With Porphyromonas Gingivalis or Fusobacterium Nucleatum Modulates the Inflammatory Response, But Fails to Prevent Experimental Periodontitis. J Clin Periodontol (2010) 37:812–7. doi: 10.1111/j.1600-051X.2010.01598.x
12. Leone CW, Bokhadhoor H, Kuo D, Desta T, Yang J, Siqueira MF, et al. Immunization Enhances Inflammation and Tissue Destruction in Response to Porphyromonas Gingivalis. Infect Immun (2006) 74:2286–92. doi: 10.1128/IAI.74.4.2286-2292.2006
13. Gibson FC, Genco CA. Prevention of Porphyromonas Gingivalis-Induced Oral Bone Loss Following Immunization With Gingipain R1. Infect Immun (2001) 69:7959–63. doi: 10.1128/IAI.69.12.7959-7963.2001
14. Gonzalez D, Tzianabos AO, Genco CA, Gibson FC. Immunization With Porphyromonas Gingivalis Capsular Polysaccharide Prevents P. Gingivalis-Elicited Oral Bone Loss in a Murine Model. Infect Immun (2003) 71:2283–7. doi: 10.1128/IAI.71.4.2283-2287.2003
15. Han X, LaRosa KB, Kawai T, Taubman MA. DNA-Based Adaptive Immunity Protect Host From Infection-Associated Periodontal Bone Resorption via Recognition of Porphyromonas Gingivalis Virulence Component. Vaccine (2014) 32:297–303. doi: 10.1016/j.vaccine.2013.09.004
16. Miyachp K, Ishihara K, Kimizuka R, Okuda K, Res JD. Arg-Gingipain A DNA Vaccine Prevents Alveolar Bone Loss. J Dent Res (2007) 100:446–50. doi: 10.1177/154405910708600511
17. O’Brien-Simpson NM, Holden JA, Lenzo JC, Tan Y, Brammar GC, Walsh KA, et al. A Therapeutic Porphyromonas Gingivalis Gingipain Vaccine Induces Neutralising IgG1 Antibodies That Protect Against Experimental Periodontitis. NPJ Vaccines (2016) 1:1–11. doi: 10.1038/npjvaccines.2016.22
18. Puth S, Hong SH, Park MJ, Lee HH, Lee YS, Jeong K, et al. Mucosal Immunization With a Flagellin-Adjuvanted Hgp44 Vaccine Enhances Protective Immune Responses in a Murine Porphyromonas Gingivalis Infection Model. Hum Vaccines Immunother (2017) 13:2794–803. doi: 10.1080/21645515.2017.1327109
19. Wilensky A, Potempa J, Houri-Haddad Y, Shapira L. Vaccination With Recombinant RgpA Peptide Protects Against Porphyromonas Gingivalis-Induced Bone Loss. J Periodontal Res (2017) 52:285–91. doi: 10.1111/jre.12393
20. Zhu C, Yang J, Sun J, Shi J, Gou J, Li A. Induction of Immune Response and Prevention of Alveolar Bone Loss With Recombinant Porphyromonas Gingivalis Peptidylarginine Deiminase. Arch Oral Biol (2013) 58:1777–83. doi: 10.1016/j.archoralbio.2013.09.006
21. Evans RT, Klausen B, Sojar HT, Bedi GS, Sfintescu C, Ramamurthy NS, et al. Immunization With Porphyromonas (Bacteroides) Gingivalis Fimbriae Protects Against Periodontal Destruction. Infect Immun (1992) 60:2926–35. doi: 10.1128/iai.60.7.2926-2935.1992
22. Huang N, Shimomura E, Yin G, Tran C, Sato A, Steiner A, et al. Immunization With Cell-Free-Generated Vaccine Protects From Porphyromonas Gingivalis-Induced Alveolar Bone Loss. J Clin Periodontol (2019) 46:197–205. doi: 10.1111/jcpe.13047
23. Rinaudo CD, Telford JL, Rappuoli R, Seib KL. Vaccinology in the Genome Era. J Clin Invest (2009) 11:2515–25. doi: 10.1172/JCI38330
24. Rappuoli R, Black S, Lambert PH. Vaccine Discovery and Translation of New Vaccine Technology. Lancet (2011) 378:360–8. doi: 10.1016/S0140-6736(11)60440-6
25. Delany I, Rappuoli R, Seib KL. Vaccines, Reverse Vaccinology, and Bacterial Pathogenesis. Cold Spring Harb Perspect Med (2013) 3:a012476. doi: 10.1101/cshperspect.a012476
26. Pizza M, Scarlato V, Masignani V, Giuliani MM, Aricò B, Comanducci M, et al. Identification of Vaccine Candidates Against Serogroup B Meningococcus by Whole-Genome Sequencing. Science (2000) 287:1816–20. doi: 10.1126/science.287.5459.1816
27. Naz K, Naz A, Ashraf ST, Rizwan M, Ahmad J, Baumbach J, et al. PanRV: Pangenome-Reverse Vaccinology Approach for Identifications of Potential Vaccine Candidates in Microbial Pangenome. BMC Bioinf (2019) 20:1–10. doi: 10.1186/s12859-019-2713-9
28. Sudha R, Katiyar A, Katiyar P, Singh H, Prasad P. Identification of Potential Drug Targets and Vaccine Candidates in Clostridium Botulinum Using Subtractive Genomics Approach. Bioinformation (2019) 15:18–25. doi: 10.6026/97320630015018
29. Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting Population Coverage of T-Cell Epitope-Based Diagnostics and Vaccines. BMC Bioinf (2006) 7:1–5. doi: 10.1186/1471-2105-7-153
30. Zhang L. Multi-Epitope Vaccines: A Promising Strategy Against Tumors and Viral Infections. Cell Mol Immunol (2018) 15:182–4. doi: 10.1038/cmi.2017.92
31. Lima KM, Dos Santos SA, Rodrigues JM, Silva CL. Vaccine Adjuvant: It Makes the Difference. Vaccine (2004) 22:2374–9. doi: 10.1016/j.vaccine.2003.12.030
32. Marciani DJ. Vaccine Adjuvants: Role and Mechanisms of Action in Vaccine Immunogenicity. Drug Discov Today (2003) 8:934–43. doi: 10.1016/S1359-6446(03)02864-2
33. Du X, Qian J, Wang Y, Zhang M, Chu Y, Li Y. Identification and Immunological Evaluation of Novel TLR2 Agonists Through Structure Optimization of Pam3CSK4. Bioorg Med Chem (2019) 27:2784–800. doi: 10.1016/j.bmc.2019.05.005
34. Wheeler DL, Chappey C, Lash AE, Leipe DD, Madden TL, Schuler GD, et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res (2000) 28:10–4. doi: 10.1093/nar/28.1.10
35. Sanober G, Ahmad S, Azam SS. Identification of Plausible Drug Targets by Investigating the Druggable Genome of MDR Staphylococcus Epidermidis. Gene Rep (2017) 7:147–53. doi: 10.1016/j.genrep.2017.04.008
36. Li W, Godzik A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics (2006) 22:1658–9. doi: 10.1093/bioinformatics/btl158
37. Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, et al. PSORTb V.2.0: Expanded Prediction of Bacterial Protein Subcellular Localization and Insights Gained From Comparative Proteome Analysis. Bioinformatics (2005) 21:617–23. doi: 10.1093/bioinformatics/bti057
38. Ain Q, Ahmad S, Azam SS. Subtractive Proteomics and Immunoinformatics Revealed Novel B-Cell Derived T-Cell Epitopes Against Yersinia Enterocolitica: An Etiological Agent of Yersiniosis. Microb Pathog (2018) 125:336–48. doi: 10.1016/j.micpath.2018.09.042
39. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: A Better Web Interface. Nucleic Acids Res (2008) 36:5–9. doi: 10.1093/nar/gkn201
40. Baseer S, Ahmad S, Ranaghan KE, Azam SS. Towards a Peptide-Based Vaccine Against Shigella Sonnei: A Subtractive Reverse Vaccinology Based Approach. Biologicals (2017) 50:87–99. doi: 10.1016/j.biologicals.2017.08.004
41. Barh D, Barve N, Gupta K, Chandra S, Jain N, Tiwari S, et al. Exoproteome and Secretome Derived Broad Spectrum Novel Drug and Vaccine Candidates in Vibrio Cholerae Targeted by Piper Betel Derived Compounds. PLoS One (2013) 8:1–10. doi: 10.1371/journal.pone.0052773
42. Hassan A, Naz A, Obaid A, Paracha RZ, Naz K, Awan FM, et al. Pangenome and Immuno-Proteomics Analysis of Acinetobacter Baumannii Strains Revealed the Core Peptide Vaccine Targets. BMC Genomics (2016) 17:1–25. doi: 10.1186/s12864-016-2951-4
43. Naz A, Awan FM, Obaid A, Muhammad SA, Paracha RZ, Ahmad J, et al. Identification of Putative Vaccine Candidates Against Helicobacter Pylori Exploiting Exoproteome and Secretome: A Reverse Vaccinology Based Approach. Infect Genet Evol (2015) 32:280–91. doi: 10.1016/j.meegid.2015.03.027
44. Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: A Comparative Pathogenomic Platform With an Interactive Web Interface. Nucleic Acids Res (2019) 47:D687–92. doi: 10.1093/nar/gky1080
45. Doytchinova IA, Flower DR. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinf (2007) 8:1–7. doi: 10.1186/1471-2105-8-4
46. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-Scale Validation of Methods for Cytotoxic T-Lymphocyte Epitope Prediction. BMC Bioinf (2007) 8:1–12. doi: 10.1186/1471-2105-8-424
47. Schmidt ME, Varga SM. The CD8 T Cell Response to Respiratory Virus Infections. Front Immunol (2018) 9:678. doi: 10.3389/fimmu.2018.00678
48. Saha S, Raghava GPS. Prediction of Continuous B-Cell Epitopes in an Antigen Using Recurrent Neural Network. Proteins Struct Funct Genet (2006) 65:40–8. doi: 10.1002/prot.21078
49. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res (2019) 47:D339–43. doi: 10.1093/nar/gky1006
50. Bateman A, Martin MJ, O’Donovan C, Magrane M, Apweiler R, Alpi E, et al. UniProt: A Hub for Protein Information. Nucleic Acids Res (2015) 43:D204–12. doi: 10.1093/nar/gku989
51. Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, et al. Peptide Binding Predictions for HLA DR, DP and DQ Molecules. BMC Bioinf (2010) 11:1–12. doi: 10.1186/1471-2105-11-568
52. Dhanda SK, Vir P, Raghava GPS. Designing of Interferon-Gamma Inducing MHC Class-II Binders. Biol Direct (2013) 8:1–15. doi: 10.1186/1745-6150-8-30
53. Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GPS. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS One (2013) 8:a73957. doi: 10.1371/journal.pone.0073957
54. Guan P, Doytchinova IA, Zygouri C, Flower DR. MHCPred: Bringing a Quantitative Dimension to the Online Prediction of MHC Binding. Appl Bioinf (2003) 31:3621–4. doi: 10.1093/nar/gkg510
55. Chauhan V, Rungta T, Goyal K, Singh MP. Designing a Multi-Epitope Based Vaccine to Combat Kaposi Sarcoma Utilizing Immunoinformatics Approach. Sci Rep (2019) 9:1–15. doi: 10.1038/s41598-019-39299-8
56. Bergmann CC, Yao Q, Ho CK, Buckwold SL. Flanking Residues Alter Antigenicity and Immunogenicity of Multi-Unit CTL Epitopes. J Immunol (1996) 157:3242–9.
57. Dorosti H, Eslami M, Negahdaripour M, Ghoshoon MB, Gholami A, Heidari R, et al. Vaccinomics Approach for Developing Multi-Epitope Peptide Pneumococcal Vaccine. J Biomol Struct Dyn (2019) 37:3524–35. doi: 10.1080/07391102.2018.1519460
58. Livingston B, Crimi C, Newman M, Higashimoto Y, Appella E, Sidney J, et al. A Rational Strategy to Design Multiepitope Immunogens Based on Multiple Th Lymphocyte Epitopes. J Immunol (2002) 168:5499–506. doi: 10.4049/jimmunol.168.11.5499
59. Saadi M, Karkhah A, Nouri HR. Development of a Multi-Epitope Peptide Vaccine Inducing Robust T Cell Responses Against Brucellosis Using Immunoinformatics Based Approaches. Infect Genet Evol (2017) 51:227–34. doi: 10.1016/j.meegid.2017.04.009
60. Makitrynskyy R, Ostash B, Tsypik O, Rebets Y, Doud E, Meredith T, et al. Pleiotropic Regulatory Genes Blda, adpA and absB are Implicated in Production of Phosphoglycolipid Antibiotic Moenomycin. Open Biol (2013) 3:130121. doi: 10.1098/rsob.130121
61. Maurer-Stroh S, Krutz NL, Kern PS, Gunalan V, Nguyen MN, Limviphuvadh V, et al. AllerCatPro-Prediction of Protein Allergenicity Potential From the Protein Sequence. Bioinformatics (2019) 35:3020–7. doi: 10.1093/bioinformatics/btz029
62. Mari A. The Allergome Web Site - a Database of Allergenic Molecules. Aim, Structure, and Data of a Web-Based Resource. J Allergy Clin Immunol (2004) 113:S301. doi: 10.1016/j.jaci.2004.01.574
63. Sever W, Garg VK, Avashthi H, Tiwari A, Jain A, Ramkete PW, et al. MFPPI-Multi FASTA ProtParam Interface. print Bioinforma (2016) 12:74. doi: 10.6026/97320630012074
64. Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM. PDBsum: A Web-Based Database of Summaries and Analyses of All PDB Structures. Trends Biochem Sci (1997) 22:488–90. doi: 10.1016/S0968-0004(97)01140-7
65. Kim DE, Chivian D, Baker D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res (2004) 32:526–31. doi: 10.1093/nar/gkh468
66. Xu D, Zhang J, Roy A, Zhang Y. Automated Protein Structure Modeling in CASP9 by I-TASSER Pipeline Combined With QUARK-Based Ab Initio Folding and FG-MD-Based Structure Refinement. Proteins Struct Funct Bioinforma (2011) 79:147–60. doi: 10.1002/prot.23111
67. Luthy R, Bowei J, Einsenberg D. Verify3D: Assessment of Protein Models With Three-Dimensional Profiles. Methods Enzymol (1997) 277:396–404. doi: 10.1016/S0076-6879(97)77022-8
68. Wiederstein M, Sippl MJ. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res (2007) 35:407–10. doi: 10.1093/nar/gkm290
69. Sheik SS, Sundararajan P, Hussain ASZ, Sekar K. Ramachandran Plot on the Web. Bioinformatics (2002) 18:1548–9. doi: 10.1093/bioinformatics/18.11.1548
70. Craig DB, Dombkowski AA. Disulfide by Design 2.0: A Web-Based Tool for Disulfide Engineering in Proteins. BMC Bioinf (2013) 14:0–6. doi: 10.1186/1471-2105-14-346
71. Schleinkofer K, Wang T, Wade RC. Molecular Docking. Encycl Ref Genomics Proteomics Mol Med (2006) 443:1149–53. doi: 10.1007/3-540-29623-9_3820
72. Sussman JL, Lin D, Jiang J, Manning NO, Prilusky J, Ritter O, et al. Protein Data Bank (PDB): Database of Three-Dimensional Structural Information of Biological Macromolecules. Acta Crystallogr Sect D Biol Crystallogr (1998) 54:1078–84. doi: 10.1107/S0907444998009378
73. Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res (2005) 33:363–7. doi: 10.1093/nar/gki481
74. Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. FireDock: A Web Server for Fast Interaction Refinement in Molecular Docking. Nucleic Acids Res (2008) 36:229–32. doi: 10.1093/nar/gkn186
75. Karplus M, McCammon JA. Molecular Dynamics Simulations of Biomolecules. Nat Struct Biol (2002) 9:646–52. doi: 10.1038/nsb0902-646
76. Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, et al. The Amber Biomolecular Simulation Programs. J Comput Chem (2005) 26:1668–88. doi: 10.1002/jcc.20290
77. Neugebauer J, Reiher M, Kind C, Hess BA. Quantum Chemical Calculation of Vibrational Spectra of Large Molecules - Raman and IR Spectra for Buckminsterfullerene. J Comput Chem (2002) 23:895–910. doi: 10.1002/jcc.10089
78. Jorgensen WL, Blake JF, Buckner JK. Free Energy of TIP4P Water and the Free Energies of Hydration of CH4 and Cl- From Statistical Perturbation Theory. Chem Phys (1989) 129:193–200. doi: 10.1016/0301-0104(89)80004-7
79. Andleeb S, Imtiaz-Ud-Din, Rauf MK, Azam SS, Badshah A, Sadaf H, et al. A One-Pot Multicomponent Facile Synthesis of Dihydropyrimidin-2(1: H)-Thione Derivatives Using Triphenylgermane as a Catalyst and its Binding Pattern Validation. RSC Adv (2016) 6:79651–61. doi: 10.1039/c6ra19162b
80. Kräutler V, Van Gunsteren WF, Hünenberger PH. A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J Comput Chem (2001) 22:501–8. doi: 10.1002/1096-987X(20010415)22:5<501::AID-JCC1021>3.0.CO;2-V
81. Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput (2013) 9:3084–95. doi: 10.1021/ct400341p
82. Miller BR, Mcgee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput (2012) 8:3314–21. doi: 10.1021/ct300418h
83. Seib KL, Dougan G, Rappuoli R. The Key Role of Genomics in Modern Vaccine and Drug Design for Emerging Infectious Diseases. PLoS Genet (2009) 5:e1000612. doi: 10.1371/journal.pgen.1000612
84. Fournier PE, Drancourt M, Raoult D. Bacterial Genome Sequencing and its Use in Infectious Diseases. Lancet Infect Dis (2007) 7:711–23. doi: 10.1016/S1473-3099(07)70260-8
85. Rappuoli R. Reverse Vaccinology, a Genome-Based Approach to Vaccine Development. Vaccine (2001) 19:2688–91. doi: 10.1016/S0264-410X(00)00554-5
86. Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): A Curated non-Redundant Sequence Database of Genomes, Transcripts and Proteins. Nucleic Acids Res (2005) 33:501–4. doi: 10.1093/nar/gki025
87. Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics (2010) 26:680–2. doi: 10.1093/bioinformatics/btq003
88. Gupta R, Pradhan D, Jain AK, Rai CS. TiD: Standalone Software for Mining Putative Drug Targets From Bacterial Proteome. Genomics (2017) 109:51–7. doi: 10.1016/j.ygeno.2016.11.005
89. Bull SC, Doig AJ. Properties of Protein Drug Target Classes. PLoS One (2015) 10:1–44. doi: 10.1371/journal.pone.0117955
90. Grandi G. Bacterial Surface Proteins and Vaccines. F1000 Biol Rep (2010) 2:1–3. doi: 10.3410/B2-36
91. Zachary JF. Mechanisms of Microbial Infections. Pathol Basis Vet Dis (2017) 1:132–241. doi: 10.1016/B978-0-323-35775-3.00004-7
92. Mahram A, Herbordt MC. Fast and Accurate NCBI BLASTP: Acceleration With Multiphase FPGA-Based Prefiltering. Proc Int Conf Supercomput (2010) 24:73–82. doi: 10.1145/1810085.1810099
93. Nalamolu RM, Pasala C, Katari SK, Amineni U. Discovery of Common Putative Drug Targets and Vaccine Candidates for Mycobacterium Tuberculosis Sp. J Drug Deliv Ther (2019) 9:67–71. doi: 10.22270/jddt.v9i2-s.2603
94. Hewitt EW. The MHC Class I Antigen Presentation Pathway: Strategies for Viral Immune Evasion. Immunology (2003) 110:163–9. doi: 10.1046/j.1365-2567.2003.01738.x
95. Jones EY, Fugger L, Strominger JL, Siebold C. MHC Class II Proteins and Disease: A Structural Perspective. Nat Rev Immunol (2006) 6:271–82. doi: 10.1038/nri1805
96. Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK. Peptide Vaccine: Progress and Challenges. Vaccines (2014) 2:515–36. doi: 10.3390/vaccines2030515
97. Reche P, Flower DR, Fridkis-Hareli M, Hoshino Y. Peptide-Based Immunotherapeutics and Vaccines 2015. J Immunol Res (2015) 2015:2–4. doi: 10.1155/2015/349049
98. Bayatzadeh MA, Zare Mirakabadi A, Babaei N, Doulah A, Doosti A. Expression and Purification of Recombinant Alpha-Toxin AnCra1 From the Scorpion Androctonus Crassicauda and its Functional Characterization on Mammalian Sodium Channels. Mol Biol Rep (2021) 9:6303–12. doi: 10.1007/s11033-021-06624-2
99. Young CL, Britton ZT, Robinson AS. Recombinant Protein Expression and Purification: A Comprehensive Review of Affinity Tags and Microbial Applications. Biotechnol J (2012) 5:620–34. doi: 10.1002/biot.201100155
100. Nilsson J, Ståhl S, Lundeberg J, Uhlén M, Nygren PÅ. Affinity Fusion Strategies for Detection, Purification, and Immobilization of Recombinant Proteins. Protein Expr Purif (1997) 11:1–16. doi: 10.1006/prep.1997.0767
101. Bonilla FA, Oettgen HC. Adaptive Immunity. J Allergy Clin Immunol (2010) 125:S33–40. doi: 10.1016/j.jaci.2009.09.017
102. Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B Cell Epitope Predictions: Impacts of Method Development and Improved Benchmarking. PLoS Comput Biol (2012) 8:e1002829. doi: 10.1371/journal.pcbi.1002829
103. McGuffin LJ, Bryson K, Jones DT. The PSIPRED Protein Structure Prediction Server. Bioinformatics (2000) 16:404–5. doi: 10.1093/bioinformatics/16.4.404
104. Colovos C, Yeates TO. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci (1993) 2:1511–9. doi: 10.1002/pro.5560020916
105. Thornton JM. Disulphide Bridges in Globular Proteins. J Mol Biol (1981) 151:261–87. doi: 10.1016/0022-2836(81)90515-5
106. Dombkowski AA, Sultana KZ, Craig DB. Protein Disulfide Engineering. FEBS Lett (2014) 588:206–12. doi: 10.1016/j.febslet.2013.11.024
107. Clarke J, Fersht AR. Engineered Disulfide Bonds as Probes of the Folding Pathway of Barnase: Increasing the Stability of Proteins Against the Rate of Denaturation. Biochemistry (1993) 32:4322–9. doi: 10.1021/bi00067a022
108. Bulaj G. Formation of Disulfide Bonds in Proteins and Peptides. Biotechnol Adv (2005) 23:87–92. doi: 10.1016/j.biotechadv.2004.09.002
109. Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
110. Maiorov VN, Crippen GM. Significance of Root-Mean-Square Deviation in Comparing Three-Dimensional Structures of Globular Proteins. J Mol Biol (1994) 235:625–34. doi: 10.1006/jmbi.1994.1017
111. Joshi H, Cheluvaraja S, Somogyi E, Brown DR, Ortoleva P. A Molecular Dynamics Study of Loop Fluctuation in Human Papillomavirus Type 16 Virus-Like Particles: A Possible Indicator of Immunogenicity. Vaccine (2011) 29:9423–30. doi: 10.1016/j.vaccine.2011.10.039
Keywords: Porphyromonas gingivalis, immunoinformatics, molecular docking, molecular dynamics simulations, epitopes, vaccines
Citation: Shaker B, Ahmad S, Shen J, Kim HW and Na D (2022) Computational Design of a Multi-Epitope Vaccine Against Porphyromonas gingivalis. Front. Immunol. 13:806825. doi: 10.3389/fimmu.2022.806825
Received: 02 November 2021; Accepted: 31 January 2022;
Published: 18 February 2022.
Edited by:
Aldo Tagliabue, Italian National Research Council, ItalyReviewed by:
C. Gopi Mohan, Amrita Vishwa Vidyapeetham University, IndiaBhrugu J. Yagnik, Emory University, United States
Copyright © 2022 Shaker, Ahmad, Shen, Kim and Na. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dokyun Na, Ymxpc3N6ZW5AY2F1LmFjLmty; Hyung Wook Kim, a2ltaHl1bmdAc2Vqb25nLmFjLmty