 Susanne Krammer1†
Susanne Krammer1† Zuqin Yang1†
Zuqin Yang1† Theodor Zimmermann2
Theodor Zimmermann2 Paraskevi Xepapadaki3
Paraskevi Xepapadaki3 Carol I. Geppert4
Carol I. Geppert4 Nikolaos G. Papadopoulos3,5
Nikolaos G. Papadopoulos3,5 Susetta Finotto1*†
Susetta Finotto1*†- 1Department of Molecular Pneumology, Friedrich-Alexander-University (FAU) Erlangen-Nürnberg, Universitätsklinikum Erlangen, Erlangen, Germany
- 2Children’s Hospital, Department of Allergy and Pneumology, Friedrich-Alexander-University (FAU) Erlangen-Nürnberg, Universitätsklinikum Erlangen, Erlangen, Germany
- 3Allergy and Clinical Immunology Unit, 2nd Pediatric Clinic, National and Kapodistrian University of Athens, Athens, Greece
- 4Institute of Pathology, Friedrich-Alexander-University (FAU) Erlangen-Nürnberg, Universitätsklinikum Erlangen, Erlangen, Germany
- 5Centre for Respiratory Medicine & Allergy, Division of Infection, Immunity & Respiratory Medicine, University of Manchester, Manchester, United Kingdom
Background: Allergic asthma is a chronic airway inflammatory disease associated with airway mucus hyper-production. ILC2 cells, which express the Th2 transcription factor GATA3, have been associated with allergic asthma. The cytokine IL-3 is known to support eosinophil, basophil and mucosal mast cell differentiation and survival; however, its role on T regulatory cells as well as on lung ILC2 and in pediatric asthma needs further investigation.
Objectives: To investigate the role of IL-3 in preschool children and to explore its therapeutic role in experimental asthma.
Methods: In a cohort of preschool children with and without asthma, we analyzed the secretion of IL-3 in nasopharyngeal fluid (NPF) and IL-3 receptor (R) alpha chain mRNA expression in peripheral blood mononuclear cells (PBMCs). In a murine model of allergic asthma, we analyzed the phenotype of wild-type untreated and rIL-3 intranasally treated asthmatic mice.
Results: IL-3 was found downregulated in the nasopharyngeal fluid of children with partially controlled asthma, as compared to control children. Moreover, IL-3 was found induced in phytohemagglutinin (PHA)-stimulated PBMCs from children with asthma and treated with steroids. Finally, IL-3 in NPF directly correlated with the anti-inflammatory molecule sST2 in steroid-treated asthmatic children. Intranasal rIL-3 delivery in vivo during the challenge phase decreased airway mucus production and inflammatory eosinophils. Moreover, rIL-3 given during the challenge phase, reduced lung ST2intGATA3+ILC2, accompanied by an induction of T regulatory cells in the airways.
Conclusions: IL-3 was found associated with steroid-resolved asthma. Moreover, treatment with rIL-3 resulted in amelioration of airway eosinophilia and mucus production, two main pathophysiological conditions associated with asthma in a murine model of allergic asthma. Thus, rIL-3 opens new strategies for immunotherapy of this disease.
Introduction
Allergic asthma (AA) is a chronic inflammatory disease of the airways affecting especially children worldwide. The immunopathology of this disease has been associated with the damage induced to the airways by repeated respiratory triggers like allergens, viruses and bacterial infections in a distinct genetic background. The respiratory epithelium represents the first defense barrier of the organism to the external insults (1). The epithelium signals downstream with molecules known as alarmins, which induce the development and recruitment of innate lymphoid cell type 2 (ILC2) (2–6), which initiate a cascade of cellular events resulting in the pathological expansion of a TH2 type immune response to otherwise innocuous airborne allergens (7). However, the events associated with the resolution of AA during acute episodes are yet to be defined. It has been suggested that, allergen-inducible CD4+CD25+Foxp-3+ T regulatory cells limit inflammation and maintain immune homeostasis in the lung. These cells are induced by TGF-beta and release IL-10 (8, 9). The exact mechanism by which, upon allergen challenge or infection, T cells are activated and then drive the airway inflammation seen in allergic asthma needs to be defined (10–12). This airway inflammation encloses the recruitment of cells of the innate immunity, which clear the antigen or infection and finally return to a homeostatic T-immunosuppressive status.
IL-3 promotes the differentiation of multipotent hematopoietic stem cells into myeloid progenitor cells or, with the addition of IL-7, into lymphoid progenitor cells. IL-3 is secreted by basophils and activated T cells. It supports the growth and differentiation of mucosal mast cells as well as T cells from the bone marrow (13). Moreover, although IL-3 was found in the bronchial airways of both asthmatic and control subjects after the allergen challenge, its increase did not correlate with airway hyperresponsiveness, metacromasia or eosinophil activation in allergen-induced asthma (14). Thus, the role of IL-3 in asthma needs further investigation. As mentioned above, another type of infiltrating cells into the site of inflammation in the lung of asthmatic patients is ILC2. Although these cells do not express lineage markers like CD3 or CD4, they show effector functions like Th2 cells expressing IL-5 and IL-13, as well as GATA-binding protein 3 (GATA3) (15–17). ILC2s are developed from common lymphoid precursors (CLP) in the bone marrow upon stimulation mainly with IL-25, IL-33 and thymic stromal lymphopoietin (TLSP), which are released by damaged or allergen- or virus-activated airway epithelial cells (18, 19). In addition, recently, TGF-beta has been shown to support the development of CLP into ILC2 via expression of the IL-33 receptor gene Il1rl1 (encoding IL-1 receptor-like 1, also known as ST2) in ILC2p and common helper-like innate lymphoid progenitors (CHILP) (20). Previous studies have also shown that IL-25, predominantly induces the development of inflammatory (i) ILC2s, while IL-33 preferentially increases tissue-resident natural (n) ILC2s (21, 22). Moreover, we and others have recently demonstrated that 25(OH)VitD3 enhances the production of the soluble (s) ST2 isoform. Because soluble ST2 (sST2) neutralizes the effect of IL33, sST2 is considered an anti-inflammatory factor in asthma (4, 23, 24). However, it is unknown if ILC2s are influenced by IL-3 via IL-3R expression, in general, and in asthma, in particular.
To investigate this hypothesis, we analyzed the secretion of IL-3 in preschool children with and without asthma, correlated its regulation in different asthma phenotypes and treatment, and studied wild-type (WT) mice treated with recombinant IL-3 in an experimental model of allergic asthma.
Methods
Human Study PreDicta
In the European study PreDicta (Post-infectious immune reprogramming and its association with persistence and chronicity of respiratory allergic diseases), we examined a cohort of children with and without asthma at the age of 4 to 6 years. The study was performed in collaboration with the children’s hospital in Erlangen and approved by the local ethics committee of the Friedrich-Alexander University Erlangen-Nürnberg, Germany (Re-No 4435). The study is registered in the German Clinical Trials Register (www.germanctr.de: DRKS00004914). The recruitment of the two cohorts of preschool children, the timescale for the clinical visits, the inclusion and exclusion criteria and the data collection were described recently (4, 5, 25–28) and are reported in other forms in Table 1, Supplementary Tables 1 and 2 along with the relevant clinical aspects and characteristics.
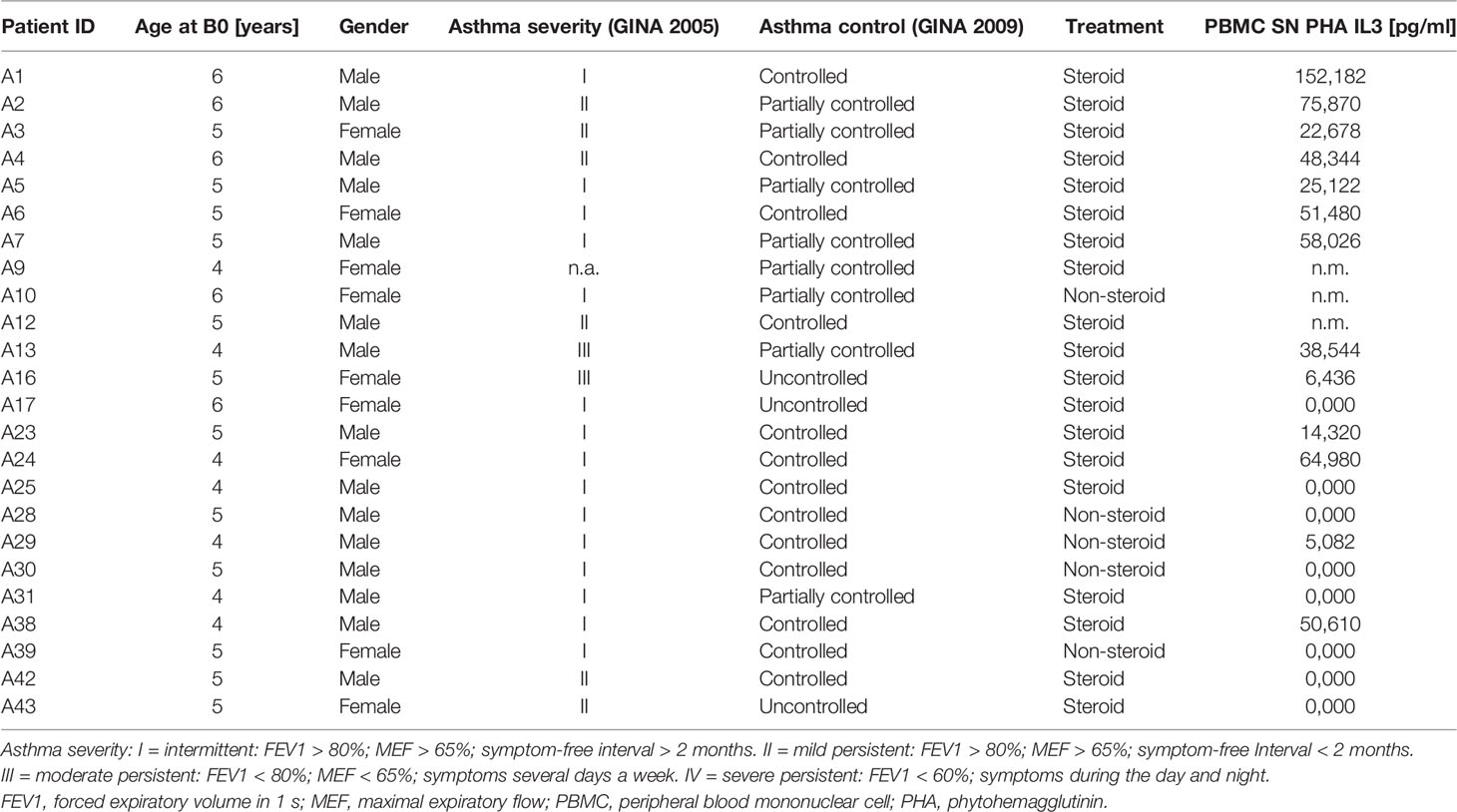
Table 1 Clinical data of the asthma PreDicta cohort WP1-UK-ER analyzed at the baseline visit.
Further, blood was collected for the isolation and culture of peripheral blood mononuclear cells (PBMCs) and subsequent further analysis.
Isolation of Human Peripheral Blood Mononuclear Cells and Cell Culture
Heparinized blood was collected from children with and without asthma at the Baseline Visit of the PreDicta study. Immediately after, PBMCs were isolated with Ficoll using density centrifugation. Afterward, one part of the PBMCs was used for extraction of total mRNA by using QIAzol Lysis Reagent (Qiagen, Hilden, Germany), according to the manufacturer’s protocol and subsequent gene expression analysis, as described below. The remaining cells were cultured at a concentration of 1 × 106 cells/ml for 48 h in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 25 mmol/L of HEPES, 100 IU/ml of penicillin, 100 µg/ml of streptomycin, 1% L-glutamine (200 mmol/L) (all from Anprotec, Bruckberg, Germany), 50 µmol/L of β-mercaptoethanol, 1% MEM Vitamin (all from Sigma-Aldrich, Steinheim, Germany), 1% non-essential amino acids, 1% sodium pyruvate (all from Gibco®, Thermo Fisher Scientific, Waltham, MA, USA), and 10% fetal bovine serum (FBS) (Biochrom GmbH, Berlin, Germany) at 37°C and 5% CO2 and stimulated with 10 µg/ml phytohemagglutinin (PHA) (Sigma-Aldrich, Steinheim, Germany).
Human IL-3 was detected in the cell-culture supernatants by using the IL-3 DuoSet ELISA kit from R&D (Wiesbaden, Germany) according to the manufacturer’s protocol.
Nasopharyngeal Fluid Collection With Swab
For the detection of IL-3 in the upper airways of the children, a nasopharyngeal specimen was collected using a per-nasal applicator swab with a flocked soft nylon fiber tip (E-Swab 482CE; Copan, Brescia, Italy) as described in detail elsewhere (2). ELISA from R&D Systems (Minneapolis, MN, USA) was used to detect IL-3 (DY203) and sST2 (DY523B-05) in human nasopharyngeal fluid (NPF).
Mice
All mice used in this study have Balb/c genetic background. The Balb/c WT mice were obtained from Janvier Labs (Saint-Berthevin, France). All mice were maintained under specific pathogen-free conditions and had free access to food and water. All experiments were performed in accordance with the German and European laws for animal protection and were approved by the government of Unterfranken, Bavaria (Az. 54-2532.1-2/10).
Ovalbumin Sensitization and Challenge
For the induction of ovalbumin (OVA)-dependent allergic asthma, WT female mice at the age of 6–8 weeks were sensitized twice at days 0 and 7 with intraperitoneal (i.p.) injections of 100 µg of OVA (Calbiochem, San Diego, CA, USA) complexed with 10% aluminum potassium sulfate (Sigma Aldrich, Steinheim, Germany). Thereafter, on days 18, 19, and 20, the mice were challenged intranasally (i.n.) with 50 µg of OVA, dissolved in phosphate-buffered saline (PBS). On day 21, the animals were sacrificed to measure the invasive lung function, obtain bronchoalveolar lavage (BAL) and isolate and analyze total lung cells.
rIL-3 Treatment In Vivo
Mice were anesthetized with isoflurane and given 5 µg of murine rIL-3 (PeproTech, Hamburg, Germany) i.n. at different time points. This procedure was done during the sensitization phase (days 0 and 17) in the rIL-3 sensitization group and during the challenge phase on days 19 and 20 in the rIL-3 challenge group. If OVA was applied on the same day; the IL-3 treatment was done 30 min before OVA i.p. injection or 1 h before OVA i.n. application.
Analysis of Airway Hyperresponsiveness
The invasive lung function of the mice was measured by the Flexivent FX System (Scireq, Emka, Montreal, QC, Canada) on day 21. The mice were anesthetized, and a tracheotomy with a 16-G cannula was performed. We challenged the mice with increasing doses (0, 5, 10, 25, and 50 mg/ml) of nebulized methacholine (MCh) to obtain the respiratory resistance (Rrs) values.
Histology
Lungs were removed, then fixed in 4% formaldehyde, dehydrated, and embedded in paraffin. Serial paraffin sections (3 µm) were stained with H&E for semiquantification of inflammation. The scoring of the perivascular and peribronchial inflammation was gathered blindly by a pathologist as previously described in detail (29). For the quantification of mucus production, lung sections were prepared as described above and stained with periodic acid-Schiff (PAS). For the analysis, the PAS score was determined via semiquantified classification of bronchi regarding the size of PAS-positive areas.
Analysis of the Bronchoalveolar Lavage Cells
BAL was obtained on day 21 by intratracheal injection and aspiration of 0.8 ml of Sterofundin (B. Braun Melsungen AG, Melsungen, Germany) twice. BAL cells were harvested for flow cytometry analysis after centrifugation for 5 min at 1.500 rpm. Anti-CD45.2, anti-siglec F, anti-CD62L, anti-CD101, and anti-CD125 were used for the staining of eosinophils in BAL. The staining procedure of the BAL was done in the same way as the lung cell flow cytometry staining.
Total Lung Cell Isolation
Lungs were removed on day 21 of the asthma protocol, and total cells were isolated as previously described (30). Briefly, lung tissue was cut into small pieces by a scalpel and digested with 300 U/ml of Collagenase Type Ia and 0.015% DNase (10 mg/ml) in PBS at 37°C for 1 h. Digested lung was then strained through a 40-µm cell strainer and subsequently lysed with an Ammonium-Chloride-Potassium (ACK)-Lysis buffer (0.15 M of NH4Cl, 0.1 mM of KHCO3, and 0.1 mM of Na2-EDTA dissolved in ddH2O). Finally, the lung cells were washed with PBS and counted using a hemocytometer. Isolated cells were plated at a concentration of 1 × 106 cells/ml in a 48-well plate and cultured in RPMI 1640 medium containing 100 IU/ml of penicillin, 100 µg/ml of streptomycin (all from Anprotec, Bruckberg, Germany), and 10% fetal calf serum (FCS) (Biochrom GmbH, Berlin, Germany) at 37°C and 5% CO2 for 24 h. After 24 h, cell culture supernatants were harvested and analyzed by quantitative ELISA.
Lung ILC2 Skewing
To expand ILC2s in vitro, 1 million lung cells in 1 ml of RPMI 1640 medium containing 100 IU/ml of penicillin, 100 µg/ml of streptomycin (all from Anprotec, Bruckberg, Germany), and 10% FCS (Biochrom GmbH, Berlin, Germany) were stimulated with 20 ng/ml of rmIL-2, 10 ng/ml of rmIL-33 (Immunotools GmbH, Friesoythe, Germany), and 10 ng/ml rmTSLP Invitrogen, Thermo fisher scientific, Waltham, MA, USA); after 5 days, the cells and cell culture supernatants were harvested and analyzed by flow cytometry and ELISA, respectively. In another experimental setting, we differentiated ILC2s as described above. Briefly, we restimulated skewed ILC2 on day 5 with 20 ng/ml of rIL-2, 10 ng/ml of rIL-33, and with or without 5 ng/ml of rIL-3 for further culture until day 12.
Flow Cytometry Analysis (FACS) of Murine Lung Cells
Single-cell suspensions from the murine lung were incubated with the respective mix of surface antibodies diluted in fluorescence-activated cell scanning (FACS) buffer (1× PBS, 1% FCS, 0.1% NaN3) for 30 min in the dark at 4°C. After the cells were washed once in FACS buffer, cells were fixed in 2% paraformaldehyde (PFA) for flow cytometric analysis. For subsequent intracellular staining, cells were fixed and permeabilized with Fixation/Permeabilization buffer (eBioscience, Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s protocol for 35 min in the dark at 4°C followed by intracellular staining with different transcriptions markers diluted in 1× Permeabilization buffer (eBioscience, Thermo Fisher Scientific, Waltham, MA, USA) in the dark at 4°C for 30 min. Afterward, cells were washed and resuspended in FACS buffer for analysis. For ILC2 staining, a premixed lineage cocktail (Miltenyi Biotec, Bergisch Gladbach, Germany) containing biotinylated anti-CD5, anti-CD11b, anti-CD45R (B220), anti-7-4, anti-Gr-1 (Ly-6G/C) and anti-Ter-119 monoclonal antibodies were used according to the manufacturer’s protocol. Streptavidin conjugated to APC was applied in secondary staining together with the APC-conjugated surface markers anti-CD3, anti-CD4, anti-CD11c, anti-Siglec F, and further surface antibodies against anti-KLRG1, anti-Thy1.2, anti-ST2, and anti-IL3Rα followed by intracellular staining with fluorochrome-coupled anti-GATA3 as described above. For Treg staining, anti-CD4 and anti-CD25 antibodies were used for surface staining and anti-Foxp3 for intracellular staining. Flow cytometry was performed on FACS Canto II (BD Biosciences, Heidelberg, Germany), and flow cytometry data were analyzed by FlowJo v10 (TreeStar Inc., San Jose, CA, USA). All of the antibodies used for flow cytometry analysis are shown in Supplementary Table 3.
Enzyme-Linked Immunosorbent Assay
Serum levels of IgE were measured using the OptEIA™ sandwich ELISA kit from BD Bioscience (Heidelberg, Germany). Mouse IL-10 and TGF-β1 from cell culture supernatants were quantified by OptEIA™ sandwich ELISA kit from BD Bioscience (Heidelberg, Germany) and DuoSet sandwich ELISA kits from R&D System (Wiesbaden, Germany), respectively. Each ELISA was performed according to the manufacturer’s instructions.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism v8 for Windows (GraphPad, La Jolla, CA, USA). All datasets were analyzed for normal distribution with the Shapiro–Wilk normality test before statistical analysis was performed. Statistical significances were calculated using a two‐tailed Student’s t-test for the analysis of two-group comparisons and one- or two-way ANOVA for multiple comparisons to generate p‐value data (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001) or the non-parametric equivalent. Correlations were examined by linear regression analysis. The two-tailed Pearson correlation analysis was performed to obtain r and p values. For post-hoc analysis, we used Tukey’s method test (one-way ANOVA) and Sidak’s method test (two-way ANOVA). Unless otherwise indicated, data are presented as mean ± SEM.
Results
IL-3 Is Associated With Controlled and Steroid-Treated Asthma in Preschool Children
To start investigating the function of IL-3 in allergic asthma, we analyzed the IL-3 secretion in the NPF of the cohort of our study PreDicta including 21 healthy controls and 24 asthmatic preschool children (Figure 1A, Table 1 and Supplementary Table 1). Here we found that, IL-3 in the NPF of control preschoolers was comparable to the level measured in asthmatic preschoolers with controlled asthma (A–C) (GINA 2009) (Figure 1B). By contrast, IL-3 levels in the NPF of asthmatic preschoolers with partially controlled asthma (A-PC) were significantly reduced. We also had 3 children with uncontrolled asthma who had scattered levels of IL-3 in the NPF. However, by looking closely at their clinical data (Supplementary Table 2), we found that the child with no IL-3 in the NPF had worse lung function and higher C-reactive protein (CRP), confirming a possible role of IL-3 in the amelioration of asthma. Because IL-3 is known to be released by activated T cells, we challenged the PBMCs with lectin PHA. Here we found an increased secretion of IL-3 in PHA-stimulated PBMCs from asthmatic children treated with steroids (Figure 1C) compared to healthy children. In addition, we analyzed children treated with steroids with controlled and partially controlled asthma and found that IL-3 was induced in this group (Figure 1D).
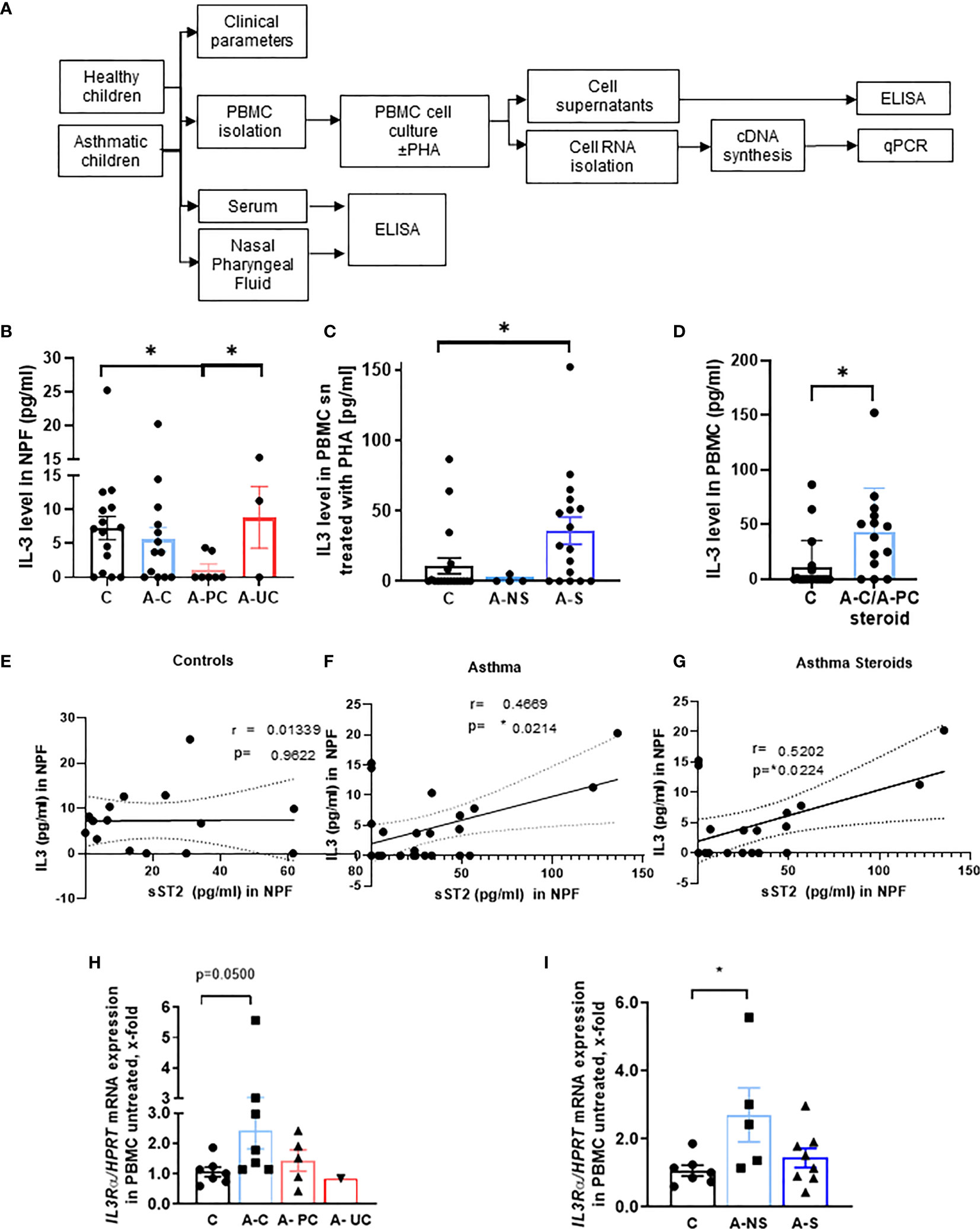
Figure 1 Role of IL-3 in asthmatic patients. (A) Experimental design of the study PreDicta on the children cohorts analyzed in this study. (B) ELISA analysis of IL-3 levels in the nasopharyngeal fluid (NPF) from control preschooler and asthmatic preschooler subgrouped into asthma controlled (A–C), asthma partially controlled (A-PC), and asthma uncontrolled (A-UC) (GINA 2009) (n = 15, 13, 7, 3). (C) ELISA analysis of IL-3 levels in the supernatant from peripheral blood mononuclear cells (PBMCs) isolated from healthy control and asthmatic children and cultured with phytohemagglutinin (PHA) for 24 h. Asthmatic children were subgrouped in accordance to the medications used: non-steroids (NS) and steroids (S) (n = 19, 4, 18; p = 0.028). (D) Analysis (shown in panel C) considering steroid-treated asthmatic children controlled and partially controlled (n = 19, 14). (E–G) IL-3 in NPF from control (E) (n = 15) and asthmatic preschoolers (F) (n = 24) were correlated with the correspondent sST2 in NPF. A significant positive correlation was maintained when only the asthmatic children treated with steroids were correlated (G) (n = 19) (Pearson’s correlation test). (H) IL3Ra/HPRT mRNA expression in PBMCs of controls and asthmatic children subgrouped into asthma controlled (A–C), asthma partially controlled (A-PC), and asthma uncontrolled (A-UC) (n = 7, 7, 5, 1) and (I) in asthmatic children treated with (A-S) or without steroids (A-NS) at the baseline visit (n = 7, 5, 8). Data are presented as means ± SEMs. Two-tailed Student’s t-test or Kruskal–Wallis test was used to calculate statistical significance. *p ≤ 0.05.
IL-3 in Nasopharyngeal Fluid Correlated With the ILC2 Anti-Inflammatory Marker Soluble ST2 in Nasopharyngeal Fluid of Asthmatic Children Treated With Steroids
To analyze the link between IL-3, ILC2, and the amelioration of asthma, we reasoned that IL-3 might be involved in the regulation of the inflammatory and anti-inflammatory properties of ILC2 via the regulation of the anti-inflammatory form of ST2 known as sST2. Here we found that, IL-3 and sST2 directly correlated in the NPF of asthmatics but not control children (Figures 1E, F). Moreover, this correlation was maintained also when only steroid-treated asthmatics were considered (Figure 1G). In summary, we found evidence that IL-3 might be involved in the shedding of the ST2, thus neutralizing IL-33 function in the airways.
IL3Rα Chain Expression Is Associated With Controlled Asthma in Children
We next analyzed the mRNA expression of the specific IL-3 receptor (IL3R) alpha chain in unstimulated PBMCs. Therefore, we wanted to correlate the expression of the IL3R alpha chain in untreated PBMCs and the clinical parameter of the asthmatic preschoolers and found a significant induction of IL-3R alpha chain/HPRT mRNA expression in asthmatic children with controlled asthma as well as in those treated with non-steroid medications (Figures 1H, I). Taken together, we found that both the IL-3 and its specific receptor alpha chain were upregulated in controlled asthma.
Intranasal Treatment With rIL-3 During the Challenge Phase Inhibited Airway Inflammatory Eosinophils and Mucus Production in Asthmatic Mice
Since intranasal steroids induce T regulatory cells (31) and IL-3 was found associated with steroid treatment, we next wanted to investigate the role of recombinant IL-3 intranasal (i.n.) in a murine model of asthma. For this reason, we treated asthmatic WT mice in an OVA-induced asthma model with rIL-3 (Figure 2A). In this model, IL-3 was given i.n., during either the sensitization or the challenge phase of the disease to distinguish the effect of IL-3 on the immune system at different stages after antigen encounter. Here we found that, mice treated with rIL-3 during the sensitization or the challenge phase didn´t show a significant change in airway hyperresponsiveness as compared to OVA alone (Supplementary Figure 1A). In addition, mice treated with rIL-3 showed a trend to reduction of lung inflammation (Supplementary Figures 2B, C) and significant reduction of mucus production (Figures 2B, C) but no change in serum IgE levels (Figure 2D). Finally, in rIL-3-treated mice during the challenge phase, we found a downregulation of inflammatory type and induction of resident type eosinophils in the BAL fluid (BALF) of treated mice (Figures 2E, F and Supplementary Figure 2C).
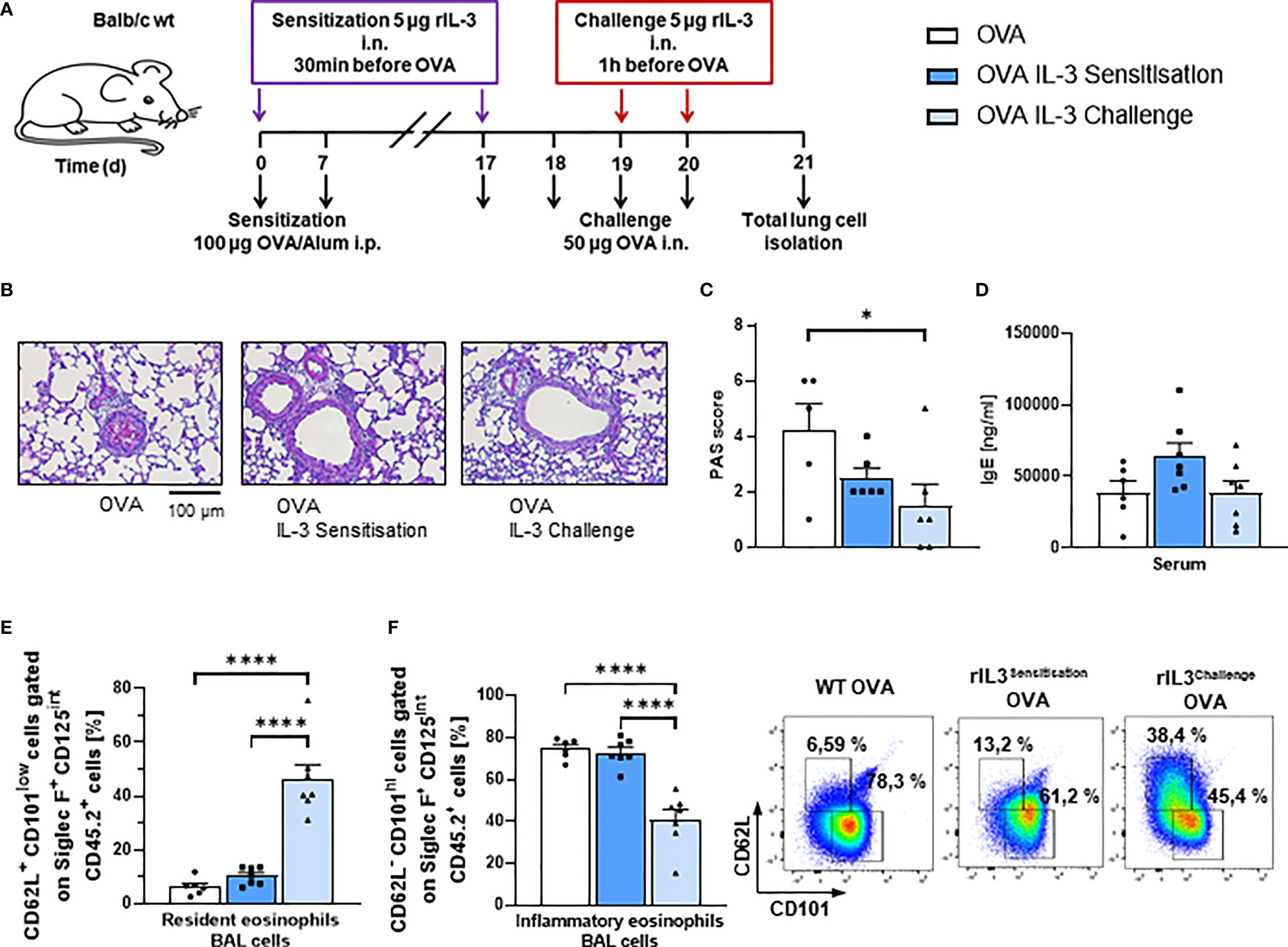
Figure 2 Intranasal treatment with rIL-3 during the challenge phase inhibited airway eosinophil inflammation and mucus production in asthmatic mice. (A) Experimental design for the induction of allergic asthma in wild-type (WT) mice treated with rIL-3 during sensitization or challenge phase for the analysis of lung ILC2s. (B, C) Analysis of the mucus production in the airways. A representative light microscopic picture of the periodic acid-Schiff (PAS) staining of lung sections is shown for each group (n = 6/6/6). (D) Serum IgE level of WT mice with asthma and with and without additional rIL-3 treatment measured by ELISA (n = 6/7/7). (E, F) Flow cytometry analysis of resident (CD62L+CD101low) and inflammatory (CD62L−CD101high) eosinophils in the bronchoalveolar lavage (BAL) cells. A representative dot plot is shown for each group (n = 6/7/7). Data are presented as means ± SEMs. One-way ANOVA or Kruskal–Wallis test was used to calculate statistical significance. *p ≤ 0.05; ****p ≤ 0.0001.
In Vivo Treatment With rIL-3 During the Allergen Challenge Phase and Decreased Lung GATA3+ ST2intILC2 Cell Number
We next asked if rIL-3 given during the allergen sensitization or challenge phase would influence the ILC2 phenotype. We further asked about the direct influence of rIL-3 on lung ILC2. To do so, we treated mice i.n. with rIL-3 during either the allergen sensitization or the allergen challenge phase. At the end of the asthma protocol on day 21, we isolated total lung cells and further differentiated them under ILC2 skewing conditions for 5 days (Figure 3A). As already described, there are two main subgroups of ILC2s, the natural (n) ILC2s and the inflammatory (i) ILC2s, which can be distinguished by the expression of ST2, which is a receptor subunit of IL-33 (32) (Figure 3B). Here, we found that rIL-3 treatment during the allergen sensitization or challenge phase resulted in the reduction of ILC2 cell number (Figures 3C, D). Finally, we found that GATA3+ST2int ILC2 were significantly reduced in mice treated with rIL-3 in vivo during the challenge phase (Figures 3E–G), whereas the ST2int ILC2 and the GATA3+ST2high ILC2 were downregulated only by trend in mice treated in vivo with rIL-3 (Supplementary Figures 1F–H).
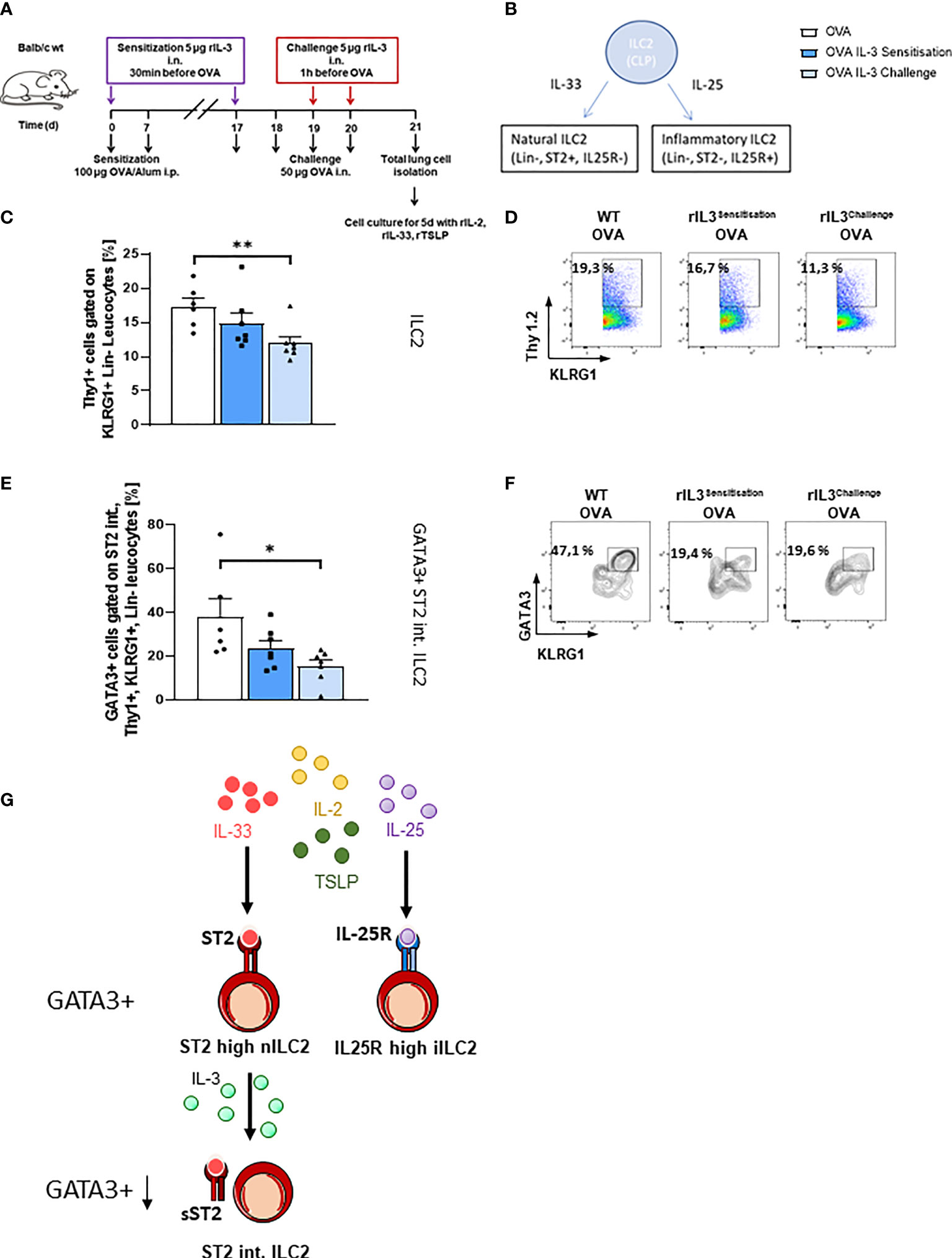
Figure 3 Lung ILC2s in an ovalbumin (OVA)-induced asthma model with intranasal rIL-3 application. (A) Experimental design for the induction of allergic asthma in wild-type (WT) mice treated with rIL-3 for the analysis of lung ILC2s. (B) Cartoon on the differentiation of different ILC2 subpopulations. (C–F) Flow cytometry analysis of ILC2 after in vitro differentiation. Flow cytometry analysis of lung ILC2s (%) (C, D); flow cytometry analysis of GATA3+ ST2int. ILC2 (E, F) in the lung of WT mice with asthma and with and without additional rIL-3 treatment during sensitization or challenge phase (n = 6/7/7). (G) Cartoon on the mechanism of IL-3 involvement in ST2 shedding from ILC2. Data are presented as means ± SEMs. One-way ANOVA or Kruskal–Wallis test was used to calculate statistical significance. *p ≤ 0.05; **p ≤ 0.01.
Intranasal rIL-3 Induced IL-10-Producing ILC2 and Foxp3+ T Regulatory Cells in the Lung
Finally, we measured IL-10 in the supernatants of in vitro skewed ILC2s and found that ILC2s skewed ex vivo from mice that received IL-3 in vivo during the challenge phase released more IL-10 as compared to the those skewed from the lung cells of mice that received IL-3 during the sensitization phase in vivo (Figures 4A, B and Supplementary Figures 1E). We next looked for immunosuppressive molecules like active TGF-beta, which is known to induce Foxp3+ T regulatory cells, and we found that it was upregulated by trend in the lung of in vivo rIL-3-treated mice (Figure 4C).
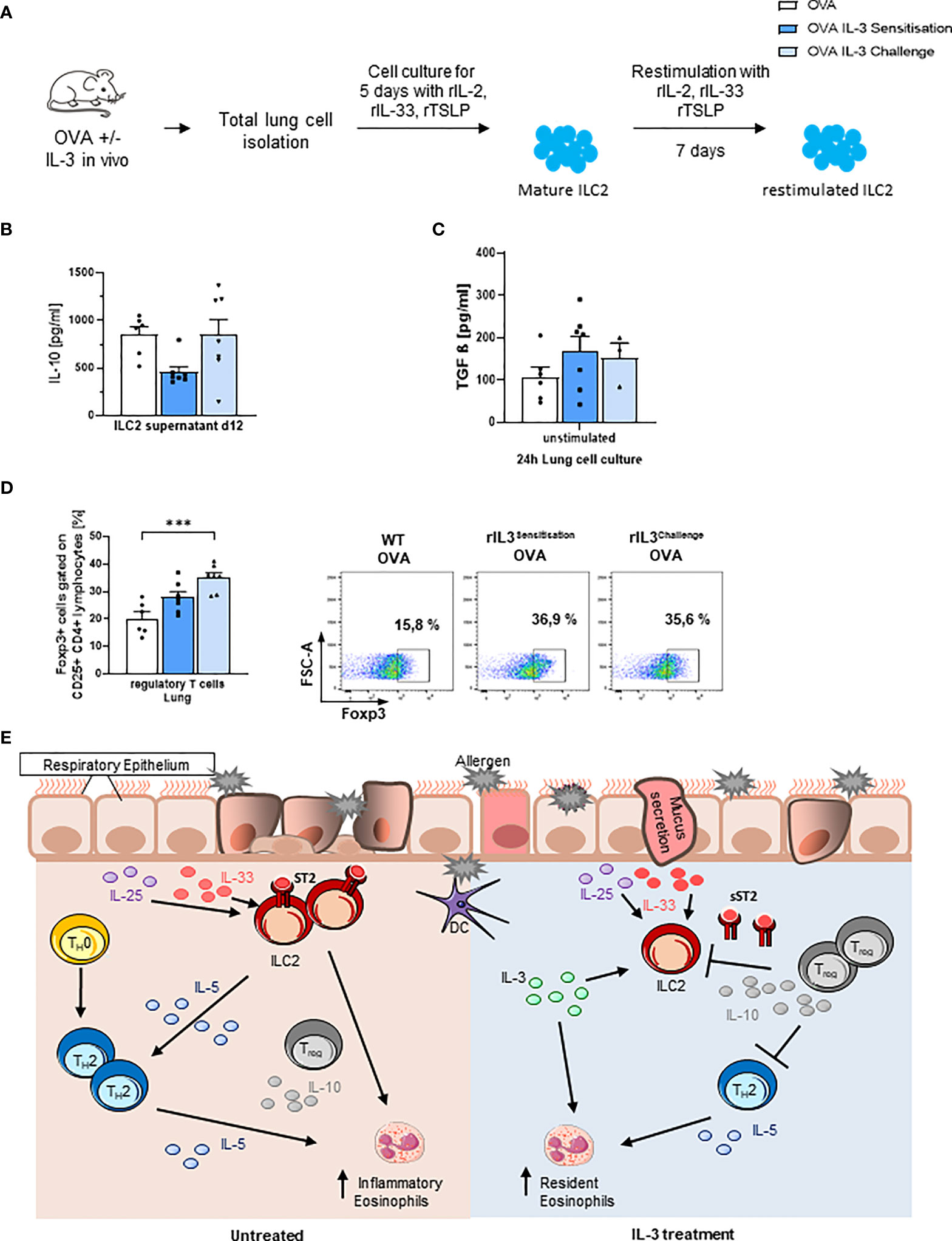
Figure 4 Intranasal rIL-3 application during challenge induces Treg cells. (A) Experimental design for lung ILC2 skewing. (B) After 5 days of in vitro ILC2 differentiation, IL-10 release was measured in the supernatants of ILC2 skewed from the lung of wild-type (WT) asthmatic mice with and without additional rIL-3 treatment during sensitization or challenge phase (n = 6/7/7). (C) TGF-beta level in 24-h unstimulated lung cell culture, measured by ELISA (n = 6/7/4). (D) Flow cytometry analysis of Foxp3+CD25+CD4+ regulatory T cells in total lung cells. A representative dot plot is shown for each group (n = 6/7/7). (E) Summary of the immunoregulatory role of IL-3 in allergic asthma. Data are presented as means ± SEMs. One-way ANOVA or Kruskal–Wallis test was used to calculate statistical significance. ***p ≤ 0.001.
Consistently, rIL-3 given during the challenge phase resulted in induction of CD4+CD25+Foxp3+ T regulatory cells in the lung of treated mice (Figure 4D and Supplementary Figure 2B).
Discussion
In our human study, we describe an increased secretion of IL-3 in the upper airways of children with controlled asthma and PHA-stimulated PBMCs of asthmatic children with A-PC and specifically those children who were treated with steroids. These results, although restricted to a small cohort of children, challenged us to further investigate in detail why IL-3 is regulated under treatment conditions associated with the resolution of this worldwide increasing disease.
To check whether IL-3 is a negative regulator of lung inflammation, we correlated IL-3 production in the airways with the anti-inflammatory marker sST2 and found that IL-3 directly correlated with sST2 in the airways of asthmatic children treated with steroids. Moreover, we treated WT mice i.n. with rIL-3 in our murine model of asthma. Treating asthmatic mice with rIL-3 during the challenge phase reduced lung inflammatory eosinophils, mucus hyperproduction, and the number of ILC2s expressing GATA3 in the lung. The plasticity of ILC2 has been demonstrated in previous studies. Thus, the reduced GATA3+ILC2 found in the mice treated with IL-3 during the challenge phase might be the result of a shift into ILC1 in the presence of IL-12 or into ILC3 in the presence of TGF-β (33–35). However, this point has to be further investigated in the future.
The reduction of eosinophils during the challenge phase is consistent with the literature stating a downregulation of the chemokine receptor CCR3 on eosinophils. Furthermore, IL-3 was shown to negatively regulate the expression of IL-5Rα on eosinophils, resulting in reduced survival and migratory activity of eosinophils (36). In this model, we did not observe induction of basophils upon IL-3 treatment (Supplementary Figure 1I). These results are controversial to other studies describing increased numbers of basophils stimulated by IL-3 (37). Consistent with the decrease of inflammatory lung eosinophils, rIL-3 intranasal administration during the allergen challenge phase increased the release of the immunosuppressive cytokine TGF-beta, which along with IL-10 releasing ILC2 resulted in the induction of the immunosuppressive T regulatory cells. Therefore, treatment with rIL-3 during the challenge phase could offer a possible therapeutic application during seasonal allergen exposure. This is also consistent with the therapeutic use of inhaled steroids during exacerbations of disease.
IL-3 is discussed very controversially in the literature. In our model, we did not find beneficial effects of IL-3 application during the sensitization phase. This might also be due to the different application routes of OVA (i.p.) and rIL-3 (i.n.). In contrast, we see a protective immune regulatory response during the challenge phase, when rIL-3 was applied before challenging the mice i.n. with OVA. On the contrary, another OVA model showed reduced levels of eosinophils and bronchial hyperreactivity, when administering anti-IL-3 on the last day of the protocol, after the challenge (38). These findings imply that IL-3 might only be supportive during a certain time of the experimental model, in our case, before the challenge. It is also possible that IL-3 regulates different processes in a dose-dependent manner. Similar to our group, Srivastava et al. found increased numbers of Tregs upon application of increasing IL-3 doses. Furthermore, IL-3 pretreated Tregs were able to suppress T effector cell proliferation in vitro (39). The underlying mechanism might be that IL-3-dependent Treg induction leads to a suppression of ILC2 via immunosuppressive cytokine IL-10. This effect is only detectable when applying IL-3 prior to antigen challenge, as the Tregs will be boosted simultaneously with the activation of the inflammatory response, enabling them to control this immune reaction.
Another possibility is that IL-3 given during the challenge phase regulated locally the release of sST2 from the surface of ILC2 or other inflammatory cells expressing ST2. This possibility is also supported by our positive correlation between sST2 and IL-3 in the airways of asthmatic children treated with steroids. Consistent with this hypothesis is also the downregulation of the GATA-3 ST2 intermediate population after treatment with IL-3 during the challenge phase. This population might be the result of previous ST2 shedding of the ST2 high population (Figure 3G). Current literature shows that sST2 is able to suppress ILC2-derived IL-5 and IL-13 cytokine production upon IL-33 stimulation (40, 41). Thus, IL-3 might contribute to ST2 shedding in ILC2, which then becomes ST2 intermediate and cannot survive. Further experiments are needed in this direction.
Another interesting finding was on the natural helper cell population (NH cells), which belongs to the ILC family. These innate cells were found to be involved in steroid resistance and are able to produce large numbers of IL-5 and IL-13 upon activation (42). In the lung, these cells are stimulated by IL-33 and a cytokine milieu including IL-2, IL-7, and TSLP (43). IL-3 could be a possible regulator of these NH cells, interfering in their activation and production of TH2 cytokines.
However, in our experiment, we used ILC2s skewed from total lung cells, which are not freshly sorted ILC2s from the lung. Further investigation in this model is needed on the in vivo characteristics of this IL-3-responsive ILC2 population. Additional experiments in house dust mite-induced asthma models would also be of interest. Our human cohort has only a restricted number of participants, and a bigger cohort would certainly strengthen our findings. Further, analysis of IL-3Rα on Treg and ILC2 isolated from control and asthmatic subjects would extend our knowledge on the responsiveness of these cell types to IL-3.
In conclusion, we demonstrate here that IL-3 not only has pro-inflammatory properties but can also regulate the asthmatic burden via inducing T regulatory cell response and reducing ILC2 proliferation. Nevertheless, we are only at the early phase of understanding the possible dual role of rIL-3 for allergic asthma in development and therapy (Figure 4E).
Data Availability Statement
The datasets generated for this study can be accessed upon request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics committee of the Friedrich-Alexander University Erlangen-Nürnberg. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by the Government of Unterfranken, Bavaria.
Author Contributions
The human study PreDicta was performed by previous members of the group at the Molecular Pneumology in Collaboration with the Children Hospital in Erlangen. SF supervised the entire work, analyzed the human data, and created Figure 1. SK and ZY performed the murine experiments, created the respective figures, and contributed with SF to the writing of the manuscript. TZ is the pediatrician who examined most of the children in PreDicta WP1‐UKER and made the medical diagnosis. NP designed the WP1 project PreDicta and was the coordinator of PreDicta. PX helped NP to set up the European PreDicta cohorts. CG coordinated the histology work presented in this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the European Grant PreDicta (Collaborative Project grant 260895) awarded to SF in Erlangen and in the other European Centers to NP in Athens. Furthermore, the work was supported by the Department of Molecular Pneumology and the SFB grant CRC1181/TP08 (Checkpoints for Resolution of Inflammation) awarded to SF. ZY is supported by the SFB grant CRC1181/TP08. SK is supported by an IZKF grant (Project A82) awarded to SF in Erlangen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the whole team at the Department of Molecular Pneumology, especially S. Mittler, S. Trump, A. Geiger, and E. Nendel, for their technical support. Furthermore, we thank all children and their parents/guardians who took part in our Europe‐wide study PreDicta. The authors are grateful to A. Graser and J. Kölle at the Department of Molecular Pneumology and to Dr. A. Kiefer, E. Muschiol, and I. Jawa at the Paediatric Pneumology‐Allergology, Department of Paediatrics and Adolescent Medicine, Universitätsklinikum Erlangen, for their technical support in the PreDicta study. Moreover, the authors thank Drs. M. Wölfel, M. O. Melichar, C. Reinhardt, A. Neubert, and Prof. Dr. W. Rascher from the Department of Paediatrics and Adolescent Medicine, Universitätsklinikum Erlangen, for their assistance in the PreDicta UKER WP‐1 study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.821658/full#supplementary-material
Abbreviations
AHR, airway hyperresponsiveness; BAL(F), bronchoalveolar lavage (fluid); Foxp3, Forkhead-box protein 3; GATA3, GATA-binding protein 3; (n/i) ILC2, (natural/inflammatory) type 2 innate lymphoid cells; i.n., intranasally; i.p., intraperitoneal; IgE, immunoglobulin E; IL, interleukin; IL3Rα, IL-3 receptor α; KLRG1, killer cell lectin-like receptor subfamily G member 1; Lin, lineage; OVA, ovalbumin; PAS, periodic acid-Schiff; PBMCs, peripheral blood mononuclear cells; Penh, enhanced pause; PHA, phytohemagglutinin; PreDicta, post-infectious immune reprogramming and its association with persistence and chronicity of respiratory allergic diseases; r, recombinant; TH, T helper cells; Treg, regulatory T cells; TSLP, thymic stromal lymphopoietin; unst, unstimulated; WT, wild type.
References
1. Tsicopoulos A, de Nadai P, Glineur C. Environmental and Genetic Contribution in Airway Epithelial Barrier in Asthma Pathogenesis. Curr Opin Allergy Clin Immunol (2013) 13(5):495–9. doi: 10.1097/ACI.0b013e328364e9fe
2. Bergauer A, Sopel N, Kroß B, Vuorinen T, Xepapadaki P, Weiss ST, et al. IFN-α/IFN-λ Responses to Respiratory Viruses in Paediatric Asthma. Eur Respir J (2017) 49(2):1600969. doi: 10.1183/13993003.00969-2016
3. Jartti T, Liimatainen U, Xepapadaki P, Vahlberg T, Bachert C, Finotto S, et al. Clinical Correlates of Rhinovirus Infection in Preschool Asthma. Allergy (2021) 76(1):247–54. doi: 10.1111/all.14479
4. Haag P, Sharma H, Rauh M, Zimmermann T, Vuorinen T, Papadopoulos NG, et al. Soluble ST2 Regulation by Rhinovirus and 25(OH)-Vitamin D3 in the Blood of Asthmatic Children. Clin Exp Immunol (2018) 193(2):207–20. doi: 10.1111/cei.13135
5. Hentschke I, Graser A, Melichar VO, Kiefer A, Zimmermann T, Kross B, et al. IL-33/ST2 Immune Responses to Respiratory Bacteria in Pediatric Asthma. Sci Rep (2017) 7:43426. doi: 10.1038/srep43426
6. Krug J, Kiefer A, Koelle J, Vuorinen T, Xepapadaki P, Stanic B, et al. TLR7/8 Regulates Type I and Type III Interferon Signalling in Rhinovirus 1b-Induced Allergic Asthma. Eur Respir J (2021) 57(5):2001562. doi: 10.1183/13993003.01562-2020
7. Finotto S, Glimcher L. T Cell Directives for Transcriptional Regulation in Asthma. Springer Semin Immunopathol (2004) 25(3-4):281–94. doi: 10.1007/s00281-003-0143-1
8. Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, et al. The IL-6R Alpha Chain Controls Lung CD4+CD25+ Treg Development and Function During Allergic Airway Inflammation In Vivo. J Clin Invest (2005) 115(2):313–25. doi: 10.1172/JCI200522433
9. Schramm C, Herz U, Podlech J, Protschka M, Finotto S, Reddehase MJ, et al. TGF-Beta Regulates Airway Responses via T Cells. J Immunol (2003) 170(3):1313–9. doi: 10.4049/jimmunol.170.3.1313
10. Lambrecht BN, Hammad H. The Immunology of Asthma. Nat Immunol (2015) 16(1):45–56. doi: 10.1038/ni.3049
11. Wenzel SE. Asthma Phenotypes: The Evolution From Clinical to Molecular Approaches. Nat Med (2012) 18(5):716–25. doi: 10.1038/nm.2678
12. Taher YA, Henricks PA, van Oosterhout AJ. Allergen-Specific Subcutaneous Immunotherapy in Allergic Asthma: Immunologic Mechanisms and Improvement. Libyan J Med (2010) 5:5303. doi: 10.3402/ljm.v5i0.5303
13. Finotto S, Mekori YA, Metcalfe DD. Glucocorticoids Decrease Tissue Mast Cell Number by Reducing the Production of the C-Kit Ligand, Stem Cell Factor, by Resident Cells: In Vitro and In Vivo Evidence in Murine Systems. J Clin Invest (1997) 99(7):1721–8. doi: 10.1172/JCI119336
14. Woolley KL, Adelroth E, Woolley MJ, Ramis I, Abrams JS, Jordana M, et al. Interleukin-3 in Bronchial Biopsies From Nonasthmatics and Patients With Mild and Allergen-Induced Asthma. Am J Respir Crit Care Med (1996) 153(1):350–5. doi: 10.1164/ajrccm.153.1.8542142
15. Annunziato F, Romagnani C, Romagnani S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J Allergy Clin Immunol (2015) 135(3):626–35. doi: 10.1016/j.jaci.2014.11.001
16. Klose CS, Artis D. Innate Lymphoid Cells as Regulators of Immunity, Inflammation and Tissue Homeostasis. Nat Immunol (2016) 17(7):765–74. doi: 10.1038/ni.3489
17. Schuijs MJ, Halim TYF. Group 2 Innate Lymphocytes at the Interface Between Innate and Adaptive Immunity. Ann N Y Acad Sci (2018) 1417(1):87–103. doi: 10.1111/nyas.13604
18. Walker JA, McKenzie AN. Development and Function of Group 2 Innate Lymphoid Cells. Curr Opin Immunol (2013) 25(2):148–55. doi: 10.1016/j.coi.2013.02.010
19. McKenzie AN. Type-2 Innate Lymphoid Cells in Asthma and Allergy. Ann Am Thorac Soc (2014) 11(Suppl 5):S263–70. doi: 10.1513/AnnalsATS.201403-097AW
20. Wang L, Tang J, Yang X, Zanvit P, Cui K, Ku WL, et al. TGF-Beta Induces ST2 and Programs ILC2 Development. Nat Commun (2020) 11(1):35. doi: 10.1038/s41467-019-13734-w
21. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-Responsive, Lineage-Negative KLRG1(hi) Cells Are Multipotential ‘Inflammatory’ Type 2 Innate Lymphoid Cells. Nat Immunol (2015) 16(2):161–9. doi: 10.1038/ni.3078
22. Koch S, Knipfer L, Kölle J, Mirzakhani H, Graser A, Zimmermann T, et al. Targeted Deletion of NFAT-Interacting-Protein-(NIP) 45 Resolves Experimental Asthma by Inhibiting Innate Lymphoid Cells Group 2 (ILC2). Sci Rep (2019) 9(1):15695. doi: 10.1038/s41598-019-51690-z
23. Pfeffer PE, Chen YH, Woszczek G, Matthews NC, Chevretton E, Gupta A, et al. Vitamin D Enhances Production of Soluble ST2, Inhibiting the Action of IL-33. J Allergy Clin Immunol (2015) 135(3):824–7.e3. doi: 10.1016/j.jaci.2014.09.044
24. Hayakawa H, Hayakawa M, Tominaga SI. Soluble ST2 Blocks Interleukin-33 Signaling in Allergic Airway Inflammation. J Biol Chem (2007) 282(36):26369–80. doi: 10.1074/jbc.M704916200
25. Bergauer A, Sopel N, Kroß B, Vuorinen T, Xepapadaki P, Weiss ST, et al. Rhinovirus Species/Genotypes and Interferon-Lambda: Subtypes, Receptor and Polymorphisms - Missing Pieces of the Puzzle of Childhood Asthma? Eur Respir J (2017) 49(3). doi: 10.1183/13993003.00265-2017
26. Bielor C, Sopel N, Maier A, Blau A, Sharma H, Vuorinen T, et al. Role of TGF-Beta in Anti-Rhinovirus Immune Responses in Asthmatic Patients. J Allergy Clin Immunol (2017) 140(1):283–286.e10. doi: 10.1016/j.jaci.2016.10.049
27. Graser A, Ekici AB, Sopel N, Melichar VO, Zimmermann T, Papadopoulos NG, et al. Rhinovirus Inhibits IL-17A and the Downstream Immune Responses in Allergic Asthma. Mucosal Immunol (2016) 9(5):1183–92. doi: 10.1038/mi.2015.130
28. Übel C, Sopel N, Graser A, Hildner K, Reinhardt C, Zimmermann T, et al. The Activating Protein 1 Transcription Factor Basic Leucine Zipper Transcription Factor, ATF-Like (BATF), Regulates Lymphocyte- and Mast Cell-Driven Immune Responses in the Setting of Allergic Asthma. J Allergy Clin Immunol (2014) 133(1):198–206.e1-9. doi: 10.1016/j.jaci.2013.09.049
29. Doganci A, Karwot R, Maxeiner JH, Scholtes P, Schmitt E, Neurath MF, et al. IL-2 Receptor Beta-Chain Signaling Controls Immunosuppressive CD4+ T Cells in the Draining Lymph Nodes and Lung During Allergic Airway Inflammation In Vivo. J Immunol (2008) 181(3):1917–26. doi: 10.4049/jimmunol.181.3.1917
30. Sauer KA, Scholtes P, Karwot R, Finotto S. Isolation of CD4+ T Cells From Murine Lungs: A Method to Analyze Ongoing Immune Responses in the Lung. Nat Protoc (2006) 1(6):2870–5. doi: 10.1038/nprot.2006.435
31. Mastruzzo C, Greco LR, Nakano K, Nakano A, Palermo F, Pistorio MP. Impact of Intranasal Budesonide on Immune Inflammatory Responses and Epithelial Remodeling in Chronic Upper Airway Inflammation. J Allergy Clin Immunol (2003) 112(1):37–44. doi: 10.1067/mai.2003.1586
32. Duerr CU, Fritz JH. Isolation of Group 2 Innate Lymphoid Cells From Mouse Lungs. Methods Mol Biol (2017) 1656:253–61. doi: 10.1007/978-1-4939-7237-1_16
33. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat Immunol (2016) 17(6):636–45. doi: 10.1038/ni.3444
34. Bernink JH, Ohne Y, Teunissen MBM, Wang J, Wu J, Krabbendam L, et al. C-Kit-Positive ILC2s Exhibit an ILC3-Like Signature That May Contribute to IL-17-Mediated Pathologies. Nat Immunol (2019) 20(8):992–1003. doi: 10.1038/s41590-019-0423-0
35. Golebski K, Ros XR, Nagasawa M, van Tol S, Heesters BA, Aglmous H, et al. IL-1β, IL-23, and TGF-β Drive Plasticity of Human ILC2s Towards IL-17-Producing ILCs in Nasal Inflammation. Nat Commun (2019) 10(1):2162. doi: 10.1038/s41467-019-09883-7
36. Gregory B, Kirchem A, Phipps S, Gevaert P, Pridgeon C, Rankin SM, et al. Differential Regulation of Human Eosinophil IL-3, IL-5, and GM-CSF Receptor α-Chain Expression by Cytokines: IL-3, IL-5, and GM-CSF Down-Regulate IL-5 Receptor α Expression With Loss of IL-5 Responsiveness, But Up-Regulate IL-3 Receptor α Expression. J Immunol (2003) 170(11):5359–66. doi: 10.4049/jimmunol.170.11.5359
37. Ohmori K, Luo Y, Jia Y, Nishida J, Wang Z, Bunting KD, et al. IL-3 Induces Basophil Expansion In Vivo by Directing Granulocyte-Monocyte Progenitors to Differentiate Into Basophil Lineage-Restricted Progenitors in the Bone Marrow and by Increasing the Number of Basophil/Mast Cell Progenitors in the Spleen. J Immunol (2009) 182(5):2835–41. doi: 10.4049/jimmunol.0802870
38. Lloyd CM, Gonzalo J-A, Nguyen T, Delaney T, Tian J, Oettgen H, et al. Resolution of Bronchial Hyperresponsiveness and Pulmonary Inflammation Is Associated With IL-3 and Tissue Leukocyte Apoptosis. J Immunol (2001) 166(3):2033–40. doi: 10.4049/jimmunol.166.3.2033
39. Srivastava RK, Tomar GB, Barhanpurkar AP, Gupta N, Pote ST, Mishra GC, et al. IL-3 Attenuates Collagen-Induced Arthritis by Modulating the Development of Foxp3+ Regulatory T Cells. J Immunol (2011) 186(4):2262–72. doi: 10.4049/jimmunol.1002691
40. Hayakawa H, Hayakawa M, Tominaga SI. Soluble ST2 Suppresses the Effect of Interleukin-33 on Lung Type 2 Innate Lymphoid Cells. Biochem Biophys Rep (2016) 5:401–7. doi: 10.1016/j.bbrep.2016.02.002
41. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The Transcription Factor GATA-3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity (2012) 37(4):634–48. doi: 10.1016/j.immuni.2012.06.020
42. Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic Stromal Lymphopoietin Induces Corticosteroid Resistance in Natural Helper Cells During Airway Inflammation. Nat Commun (2013) 4:2675. doi: 10.1038/ncomms3675
Keywords: allergic asthma, eosinophils, IL-3, ILC2s, T regulatory cells
Citation: Krammer S, Yang Z, Zimmermann T, Xepapadaki P, Geppert CI, Papadopoulos NG and Finotto S (2022) An Immunoregulatory Role of Interleukin-3 in Allergic Asthma. Front. Immunol. 13:821658. doi: 10.3389/fimmu.2022.821658
Received: 24 November 2021; Accepted: 24 January 2022;
Published: 23 February 2022.
Edited by:
Taruna Madan, National Institute for Research in Reproductive Health (ICMR), IndiaReviewed by:
Lakshna Mahajan, University of Delhi, IndiaUday Kishore, Brunel University London, United Kingdom
Copyright © 2022 Krammer, Yang, Zimmermann, Xepapadaki, Geppert, Papadopoulos and Finotto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susetta Finotto, c3VzZXR0YS5maW5vdHRvQHVrLWVybGFuZ2VuLmRl
†These authors have contributed equally to this work and share first authorship