 Kouminin Kanwore1,2
Kouminin Kanwore1,2 Konimpo Kanwore3
Konimpo Kanwore3 Gabriel Komla Adzika4
Gabriel Komla Adzika4 Ayanlaja Abdulrahman Abiola1,2Xiaoxiao Guo1,2
Ayanlaja Abdulrahman Abiola1,2Xiaoxiao Guo1,2 Piniel Alphayo Kambey1,2Ying Xia1,2
Piniel Alphayo Kambey1,2Ying Xia1,2 Dianshuai Gao1,2*
Dianshuai Gao1,2*- 1Xuzhou Key Laboratory of Neurobiology, Department of Neurobiology, Xuzhou Medical University, Xuzhou, China
- 2Xuzhou Key Laboratory of Neurobiology, Department of Anatomy, Xuzhou Medical University, Xuzhou, China
- 3Faculty Mixed of Medicine and Pharmacy, Lomé-Togo, University of Lomé, Lomé, Togo
- 4Department of Physiology, Xuzhou Medical University, Xuzhou, China
Glioma is a type of brain and spinal cord tumor that begins in glial cells that support the nervous system neurons functions. Age, radiation exposure, and family background of glioma constitute are risk factors of glioma initiation. Gliomas are categorized on a scale of four grades according to their growth rate. Grades one and two grow slowly, while grades three and four grow faster. Glioblastoma is a grade four gliomas and the deadliest due to its aggressive nature (accelerated proliferation, invasion, and migration). As such, multiple therapeutic approaches are required to improve treatment outcomes. Recently, studies have implicated the significant roles of immune cells in tumorigenesis and the progression of glioma. The energy demands of gliomas alter their microenvironment quality, thereby inducing heterogeneity and plasticity change of stromal and immune cells via the PI3K/AKT/mTOR pathway, which ultimately results in epigenetic modifications that facilitates tumor growth. PI3K is utilized by many intracellular signaling pathways ensuring the proper functioning of the cell. The activation of PI3K/AKT/mTOR regulates the plasma membrane activities, contributing to the phosphorylation reaction necessary for transcription factors activities and oncogenes hyperactivation. The pleiotropic nature of PI3K/AKT/mTOR makes its activity unpredictable during altered cellular functions. Modification of cancer cell microenvironment affects many cell types, including immune cells that are the frontline cells involved in inflammatory cascades caused by cancer cells via high cytokines synthesis. Typically, the evasion of immunosurveillance by gliomas and their resistance to treatment has been attributed to epigenetic reprogramming of immune cells in the tumor microenvironment, which results from cancer metabolism. Hence, it is speculative that impeding cancer metabolism and/or circumventing the epigenetic alteration of immune cell functions in the tumor microenvironment might enhance treatment outcomes. Herein, from an oncological and immunological perspective, this review discusses the underlying pathomechanism of cell-cell interactions enhancing glioma initiation and metabolism activation and tumor microenvironment changes that affect epigenetic modifications in immune cells. Finally, prospects for therapeutic intervention were highlighted.
Introduction
Gliomas are large brain and spinal cord tumors associated with three types of glial cells (1, 2), astrocytoma (astrocyte, found in cerebrum and cerebelum), ependymomas (ependymal cells found in ventricules and spinal cord), and oligodendrogliomas (oligodendrocytes found in cerebrum). In some cases, these gliomas can combine and affect several glial cells (example, aligodendrocytomas-astrocytomas) (3, 4). Gliomas differ from other brain cancers by their origin (glial cell), aggressiveness with high mortality (15 month maximum), resistance to drugs, and grade scales (one to four) (5). Recent findings have demonstrated that gliomas, especially glioblastomas formations, are initiated by the alteration in the genome and epigenetic modifications of glial cells due to age, radiation, and microenvironment (6, 7). The altered genome and microenvironment trigger glial cells’ excessive proliferation and increased glycolysis via the PI3K/AKT/mTOR pathway and the Warburg effect. These promote epigenetic modification mediated by histone acetylation and DNA methylation, which are characteristics of glioblastoma multiform (the highest grade glioma) (8, 9). Also, upon injury, hypersecretion of proinflammatory cytokines such as interleukins (IL-6) and tumor necrosis factor (TNF) have been shown to contribute to carcinogenesis and impede the destruction of cancerous cells (10, 11). Furthermore, while transforming growth factor-beta (TGF-β) family has been reported to play crucial roles in the embryogenesis and differentiation of the nervous system (12, 13). However, recent works have found TGF-β overexpressed in gliomas, where they are implicated in facilitating the proliferation and regeneration of cancer cells (14–16). Intriguingly, Zhang et al. and others have also demonstrated the essential roles of TGF-β in the polarization of macrophages into tumor-associated macrophages (17, 18). In particular, works from Keshamouni VG’s reported that members of TGF-β family proteins bind to macrophages to induce their reprogramming through the polarization change and alteration of plasma membrane receptors and peptides necessary for the recognition of cancer cells, thereby repressing anti-tumor responses from macrophages (19–21). As such, by promoting the evasion of cancer cells to immune surveillance, cancer cells proliferations are hastened when the aforementioned proinflammatory responses are hyperactive. With evidence that the hypersecretion of IL-6, TNF, and TGF-β scaffolds cascades that promote evasion of immunosurveillance by glioma cells, it is speculated that the development of new therapeutic approaches that combine radiotherapy/chemotherapy and immunotherapy might circumvent immunosurveillance evasion and help target glioma progression effectively.
From an oncological and immunological standpoint, this review briefly discusses cell-cell interactions enhancing gliomas initiation and metabolism activation and tumor microenvironment changes that affect epigenetic modifications in immune cells. Also, perspectives and recent progress in the exploration of therapeutic intervention were highlighted.
Niche and Energy Demands of Gliomas in Brief
The glioma stem cell niche significantly contributes to tumor cell proliferation and propagation and promotes tumor initiation due to their ability to self-renew, regeneration of the transient population of growth cells, and resistance to therapies.
Reportedly, four stem cell niches and microenvironment, namely, perivascular, hypoxic, invasive, and acidic niches, contribute to tumor maintenance and progression. These niches promote the genes that regulate cell stemness. Specifically, hypoxic and perivascular niches have been associated with upregulated expressions of hypoxia-induced factor 1 alpha (HIF-1α) and vascular endothelial growth factor (VEGF) (22). It has been reported that an acidic niche promotes VEGF gene upregulation which tends to support tumor vascularization and growths that promote malignant glioma cell metastasis along neurons propagation directions (23) and tumor progression (24). Xie Q et al. demonstrated that the invasive niche promotes glioma stem cell invasion and reduces their proliferation ability according to the “go or grow” hypothesis (25). Also, they demonstrated that a reduction in glioma cell proliferation significantly improves the glioma cell invasion (25). In consolidation, Hoang-Minh LB et al. also demonstrated that metabolic genes regulate tumor recurrence after clinical resection and that slower glioma-proliferating cells are underlying factors for tumor heterogeneity, progression, and recurrence (26). The ability of these glioma cancer stem cells to regulate progenitor cells in neoplastic tissue contributes to tumor bulk and development. Most tumors generate their energy by glucose fermentation and glycolysis (anaerobic and aerobic glycolysis) in the mitochondria via oxidative phosphorylation reactions to drive tumorigenesis (Warburg effect) (27, 28). Cancer cells prefer this type of metabolism due to the high demand for energy for their rapid proliferation and adaptive capacity to colonize diverse microenvironments. Notably, there are intrinsic heterogeneities in cancer cells (metabolism and gene expression) as well as in the tumor (metabolism, cancer cell types, and plasticity). Also, extrinsic heterogeneity such as abundance metabolic substrates, different types of stromal (fibroblast and astrocytes), and immune cells influences or affect cancerous cells/tumor growth (29, 30).
Incomplete Cell Division: GDNF Interaction With SOX1, SOX2, and Cyclins in Glioma Stem Cell Initiation
Cell division is a crucial and delicate process because any aberrations at any stage might have detrimental effects on the daughter cells (31, 32). As such, three checkpoints (G1, G2, and M phases) are put in place to circumvent any defects in the genetic makeup of daughter cells. At the G1 phases, where cyclins D ensures that cells progressing to the S phase have the essential requirement (33), GDNF has been found to play vital roles in stimulating and regulating the cell to enhance their transition to the S phase (34, 35). Also, during G2, where cyclins and p53 crosscheck the perfection of DNA replication and the absence of any damage before the cell enters the M phase, the involvement of SOX family members (SOX1, SOX2), and signal transduction activator of transcription 3 (STAT3), CREB/CBP has been shown (36, 37). Occasionally, cell division processes in anaphase are incomplete due to underlying idiopathic; however, these cells become dangerous because some retain the ability to multiply (38, 39). Since the cascades in anaphase ensure that the chromosomes are separated and migrated via the mitotic spindle, its incompletion results in unbalanced distribution of genetic codes in the cells. Ultimately, this causes gene mutation in daughter cells and increases the expression of the genes that regulate the cell cycle (40). The overexpression and interaction of cyclins and cyclin-dependent kinases (Cdks) regulate cell cycle progression by promoting the activation of proteins that enable the process to pass designated checkpoints (41). Reportedly, during the cell cycle, the levels of cyclin-dependent kinases remain stable while cyclin levels do change to promote the progression of the cell from one stage to another.
Cells that fail to undergo all the required processing in the anaphase cannot pass the M phase to complete their division (42). Consequently, during the subsequent cell division, G1 and G2 cyclins are overexpressed along with oncogenes that cause irreversible damage such as gene mutation, chromosomes crossing over, and breaking within the cells (33). In this instance, it has been shown that overexpression of growth factors such as GDNF and SOX1 protects the abnormal cells against apoptosis, and the failure of P53 to repair the damage increases their risk of becoming a cancer-initiating cell and cancer stem cell (38). Typically, P53 will halt and repair damages occurring at the G1 phase; as such, it cannot cater to damages occurring at the anaphase and metaphase. Also, oncogenes are able to impede P53 from triggering apoptosis of abnormal cells (38).
Besides gliomas initiations due to the alteration of genetic materials (epigenetics and mutation), other intra- and extra-cellular factors are involved. These include the overexpression of oncogenes during healing, tissue regeneration, nervous system differentiation during embryogenesis, and the development of organs (43, 44). GDNF plays a crucial role in facilitating the aforementioned growth and reparative processes. However, the fluctuation of its expression during inappropriate periods such as immunodeficiency, physiological stress, or during the recovery period preceding a severe illness encourages cell-cell interactions and prompt immune cell activation. During this period, an interaction between the abnormal undivided cell and an activated macrophage stimulates its growth and can lead to cancer initiation (Figure 1). The proliferation of cancer cells and their binding are facilitated by increased fibrin levels, which were meant for supporting immune cell activity. Therefore, the higher expression of fibrin contributes to cancer stem cells colony formations and the initiation of angiogenesis necessary for tumor formation, nutrient supplies, and metastasis. The growth of the tumor mass and its associated cells causes intussusception of the blood vessels, making its membrane fragile and allowing cancer cells to cross into the blood circulation and metastasize within the same or to the different organ(s) (Supplementary Figures 1A, B).
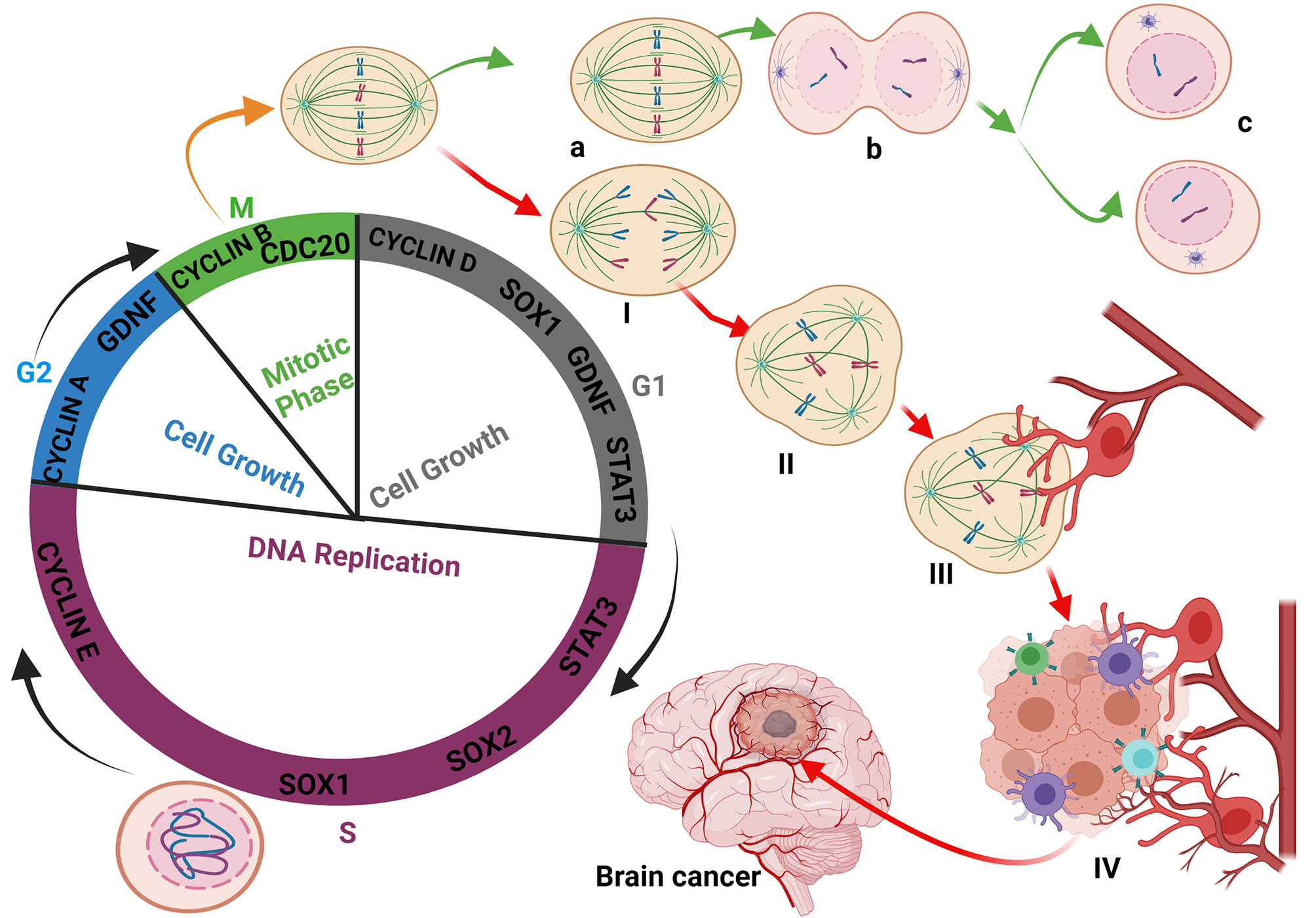
Figure 1 Glioma initiation: Interaction between the abnormal undivided cell and immune cells. During the cell cycle, the defective cell is not allowed to achieve division. Immune cells-like macrophages and T-cells destroyed the defective cell to prevent cancer initiation. Most of the oncogenes expressed during the cell cycle process are mainly Cyclins, GDNF, STAT3, and SOX family members to maintain the checkpoints and respect the timeline of cell division and integrity (A–C). The fact that certain defective cells are blocked and cannot achieve their division stagnates and is continuously exposed and affected by the cell cycle oncogenes (I, II). The exposure of certain immune cells to GDNF reprogrammed and modified their polarity, leading to their activation and massive release of cytokines (III). The reprogramming of macrophages by the bonding of TGF-β family members on their membrane receptor retains the macrophages on their activated form. Sometimes, due to unknown reasons, the defective cell can migrate closer to the blood vessels and benefit from reprogrammed immune cells, which stimulate their multiplication (IV) through cytokine synthesis, activation, and modification of the cell’s metabolism leading to epigenetics modification and glioma initiation.
SOX2 is one of the stem cell markers originating from the sex-determining region Y (SRY) superfamily group and SOXB1 subgroup members with SOX1 and SOX3 (45). SOX2 plays a critical role in neuronal cell formation and holds embryonic stem cells undifferentiated (46). It has been stated that the overexpression of SOX2, Oct4, Klf4, and c-Myc transform differentiated cells into induced pluripotent stem cell and their renewal by activating cell cycle processes (47). The cell cycle is vital for the renewal and proliferation of stem cells (48). In fact, Albright et al. and Zhang et al. have reported that SOX2 gene inhibition promotes neurodegenerative diseases (49, 50). However, when properly expressed, SOX2 actively induces and regulates the cell cycle through its interaction with phosphorylated cyclin-dependent kinases (Cdks) and cyclins (A, B, D, and E) (51). The SOX2/Cdks/cyclin complex can also cause self-renewal and proliferation of stem cells (51). SOX2, SOX1, and SOX3 form part of SOXB1 subgroups and have a similar and redundant genetic sequence (52). The redundancy between members of the SOXB1 subgroup enhances their interactions with other genes involved in the renewal and proliferation of glioma stem cells (SOX1, SOX2, and SOX3)/Cdks/cyclins. Hence, the overexpression of SOXB1 subgroup members in any tumor-initiating cell (differentiated cell) can turn the cell into a reprogrammed induced pluripotent glioma stem cell (IPGSC) that can differentiate easily into any type of brain cell and cause heterogeneity within a tumor. The possible interaction between TGF-β (GDNF) and SOXB1 (SOX1, SOX2, SOX3) family members might explain the difficulties in regulating glioma stem cell recurrence after surgical tumor resection and drug resistance (52, 53). The speculation is based on the fact that SOXB1 subgroup member can reprogram any tumor cell into a tumor stem or progenitor cell (Figure 2). Additionally, GDNF can also compliment this by facilitate their reprogramming into neural stem cells which can differentiate into several types of neurons in the brain and spinal cord. The differentiation of neural stem cells into brain or spinal cord neurons represent a risk factor in the central nervous system cancer progression resulting in cancer cell interaction with the other cells of the central nervous system. The change of neural stem cells micrienvironment modifies its metabolism to facilitate its differentiation (54–56).
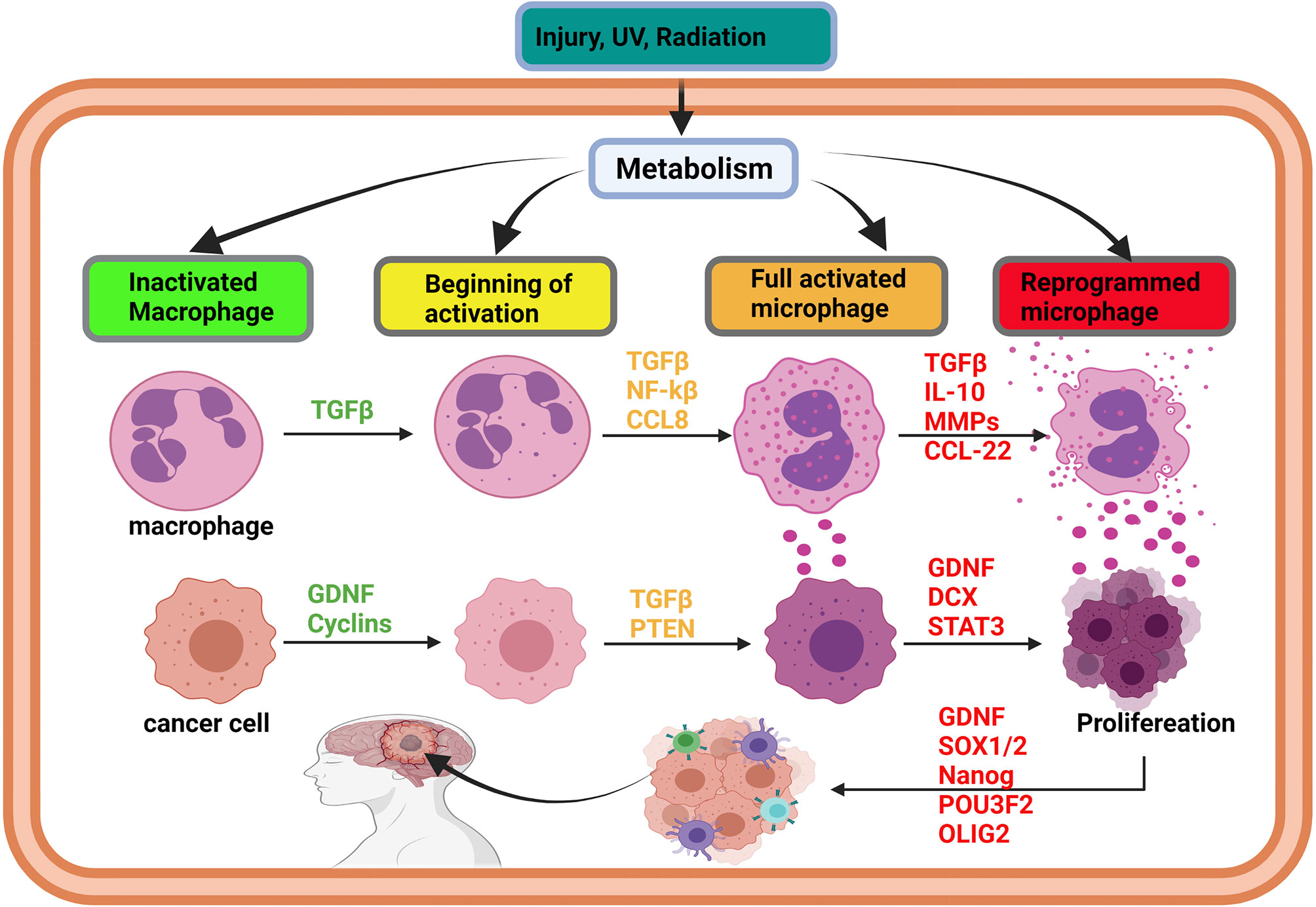
Figure 2 Initiation of glioma via immune cell reprogramming of cancer-associated macrophages. In brain injury, the activation of metabolism can affect the cells’ DNA and promote gene methylation and acetylation. The effects of metabolism on oncogene lead to methylation, acetylation, resulting in their overexpression. Simultaneously, overexpression of oncogenes, such as TGF-beta and IL-10, promotes macrophages’ activation and reprogramming, leading to the overexpression of cytokines by an immune cell. As long as the injury persists, TGF-beta family members and cytokine will be overexpressed to heal the injury. The synergistic effect of TGF-beta and cytokines can induce the transformation of the surrounding cell into a cancer cell and trigger proliferation. Cancer initiating cells can also be reprogrammed into stem-like tumor-propagating cells, spread in the brain, and cause glioma. Glioma initiation is mediated by intrinsic (metabolism, epigenetic modification) and extrinsic factors like U.V. and radiation.
Most research findings report on the role of oncogenes in cancer cell proliferation by enhancing the cancer cell cycle. In some instances, cancerous cells may not be ready to divide, but due to the overexpression of oncogenes, the cells progress in dividing without the necessary preliminary process, thereby resulting in chromosomes and DNA damages. As such, checking the cell cycle via flow cytometry using nucleus staining with propidium iodide (PI) or Dapi to count ploidy (aneuploid, diploid) mighty not enough. It is suggested that the repartition and form of those chromosomes be checked to ensure DNA integrity and predict the possible modifications in the next cell cycle. This will help to mimic tumor growth.
Symbiotic Interaction Between Abnormal Incomplete Divided Cells With Immune Cells: Maladaptations of GDNF
Recent reports have implicated immune cells in playing vital roles during glioma initiations (57, 58). Also, it has been demonstrated that the abnormal symbiosis between cancer and immune cells increases the transformation of both cells (59, 60). For example, during tumor initiation, GDNF (a TGF-β family member) was found to be impeding anti-tumor immune cells activities but instead transforming the cells into tumor-associated immune cells (61, 62). T-cell receptor-mediated immune cell (TCR) nitrosylation inhibition and transformation are achieved by decreasing glycolysis and caspase activity, which regulates immune cell death (63, 64). While caspase activities are decreased, increases in GDNF levels tend to promote abnormal T-cell and tumor-initiating cell survival (65). Meanwhile, the nitrosylation of T-cell receptors, while GDNF is overexpressed, encourages their bindings to epidermal growth factor receptor (EGFR) on T-cell to promote immuno-oncology phenomena, which regulates the proliferation, polarization into M1 or M2 microglia and cytokine (IL-4, IL-10, IL-13) secretions (66). Furthermore, reports have indicated that the synergy of GDNF overexpression and hypersecretion of interleukins trigger the polarization of tumor-associated macrophages into M2 microglia (67). Intriguingly, Zhang et al. had previously demonstrated that M2 microglial cells are found in the tumor microenvironment and were implicated in the participation of cascades facilitating tumor initiation, angiogenesis, and metastasis (68). The interaction between M2 microglia macrophages and abnormal incomplete divided cells highly increases the risk of glioma initiation due to alterations in the brain cell microenvironment resulting from cytokine overexpression. Influenced by the cytokine-altered microenvironment, glioma initiating cells begin to proliferation by raising their metabolism (glycolysis), thereby exacerbating inflammatory responses (69). Tumor-associated macrophages (M2) are capable of reprogramming fibroblasts into cancer-associated fibroblasts (CAFs) by increasing IL-1β, EGF, and VEGF (70–72). Interestingly, the symbiosis between M2 macrophages and tumor-initiating cells is essential for both survival and proliferation (Figure 2 and Supplementary Figure 2).
Change Metabolism Induces Epigenetic Modifications in Immune Cell and Scaffold Cancer-Initiating Cells and Metastasis
Metabolism is one of the essential phenomena contributing to human growth, reproduction, and life in general (73, 74). It basically involves the transformation of the specific substrate required for the activation of several genes and enzymes, which turn nutrients into simple molecules that the body can use. The involved mechanisms correlate cell metabolism, gene expression, and epigenetics modification (75, 76). Hence, cell metabolism and gene expression in healthy/unhealthy cells (such as immune and glioma cells) share many similar signaling pathways, and a minute change in their cell metabolism disproportionally influence several gene expressions (77–79). Among the signaling pathways, PI3K/AKT/mTOR/GSK-3β is the classical metabolism pathway involved in cancer cell growth (80, 81). Depending on the type of metabolic substrate, metabolic signaling pathways can switch from one to another to improve glioma cells’ survival and proliferation abilities. Over recent decades, glucose has been shown to be the essential nutrient for the human brain as its deficiency results in irreversible damage to the brain neurons (82–84). Mitochondria are responsible for glucose conversion into energy (ATP/GTP) (85, 86). Besides signaling via PI3K/AKT/GSK-3β regulating neural cell development and protein phosphorylation (80, 81, 87), it has been shown to regulate mitochondrial activities, specifically, glycolysis (87–90). Also, GSK-3β regulates mitochondrial metabolic function (biogenesis, bioenergetics, and apoptosis), mitochondrial structure (permeability), and mitochondrial dynamic (motility) (87, 90). Interestingly, cancerous cells (gliomas in particular) require all the aforementioned cellular function and makes use of glycolysis to meet energy needs for survival (91–93). Like other cells, alteration in the metabolism of immune cells can drastically change their functions and characteristics (94). Particularly in macrophages, metabolism affects their gene expression through DNA methylation and acetylation (95, 96). Similarly, histones H3K9 and H3K4 acetylation and DNA methylation have been reported to be involved in GDNF and SOX1 overexpression in gliomas (50, 97). Thus, metabolism seems to exert similar effects on gene expressions in macrophages and gliomas. The hypersecretion of cytokines such as interleukins (IL-6) and tumor necrosis factor-alpha (TNF-α) due to hyperglycolysis provides additional evidence that changes in immune cell metabolism affect their gene expression and alter their functions (98, 99).
IL-6 and growth factors (GDNF, SOX1, SOX2) play a critical role during inflammation, damaged tissue repair, cell proliferation, invasion, migration (100, 101). These genes expressions are related to cell metabolism, making the immune cell metabolic pathways the best candidate for the methylation of oncogenes and their overexpression in glioma. Also, the binding of GDNF to immune cell receptors (EGFR) promotes the activation of mTOR, which contributes to the improvement in immune cell metabolism, and increases epigenetic modifications (102). In consolidation of their findings, Yan et al. also recently reported that PI3K/AKT/mTOR regulates HIF-1α, which gets involved in reprogramming immune cell metabolism (99).
Change Metabolism Induces Epigenetic Modifications in Cancer Cells and Scaffold Cancer-Initiating Cells and Metastasis.
Like macrophages, PI3K/AKT/mTOR is the primary signaling pathway that controls the metabolism in glioma cells (Figure 3 and Supplementary Figure 3) (99, 103). Dimitrova et al. and others have further shown that the PI3K/AKT/mTOR pathway controls glioma stem cell proliferation, invasion, angiogenesis, and metastases (104, 105). Additionally, they reported that PI3K/AKT/mTOR/HIF-1α plays a critical role in angiogenesis by inducing overexpression of VEGF. Indicating that PI3K/AKT/mTOR regulates/alters many functions in glioma cells and immune cells by controlling their metabolism (105). It is speculated that the difficulty in eradicating glioma might be due to the complexity of the PI3K/AKT pathway and the multitudes of genes reliant on this pathway. Many genes [including EGF and insulin-like growth factor 1 (IGF-1)] and hormones (including insulin) regulating essential physiological function rely on signaling via PI3K/AKT pathways.
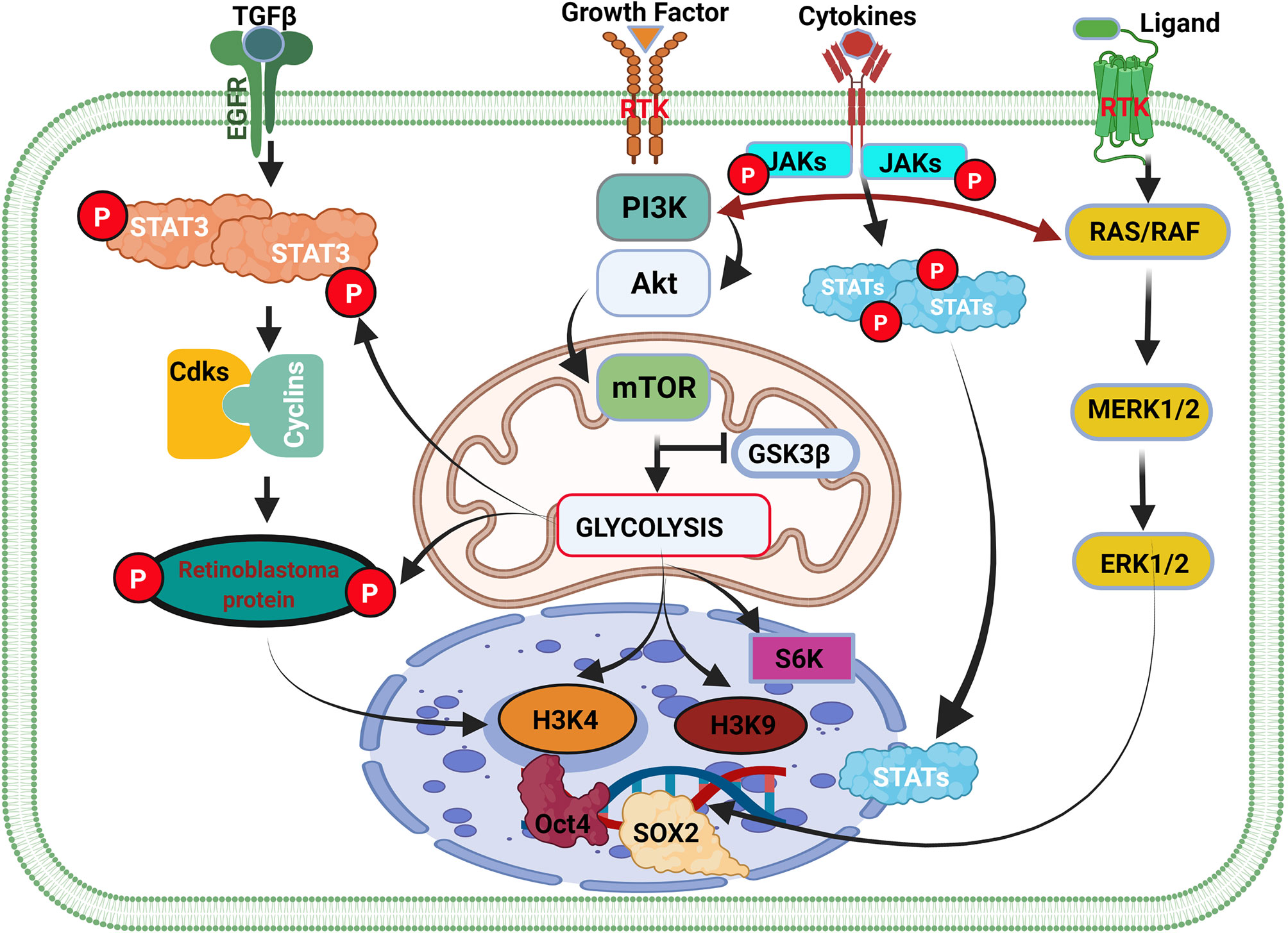
Figure 3 Cell cycle genes and the weaknesses associated with the initiation of glioma. Cell type transition and cancer cell initiation are a succession of events within the cell-mediated by external and internal factors. The overexpression of GDNF in normal cells contributes to the activation of the STAT3 gene and cyclin-dependent kinase (Cdks) binding with cyclin and their respective phosphorylation. The binding of Cdks-cyclin promotes the activation and phosphorylation of retinoblastoma protein (Rb) that stimulates the initiation of the cell cycle and cell division via DNA transcription. In inflammation, the extracellular overexpression of growth factors activates the PI3K/AKT/mTOR pathway, which controls glycolysis by antagonizing GSK3β activity. GSK3β activation promotes glycogen synthesis to promote the accumulation of glycogen that can be used later to produce energy for cell survival and division. GSK3β inhibition causes glycolysis to promote glioma initiation and epigenetic modification. In cancer, overexpression of GDNF or the activation of GDNF family ligands also promotes Ras/Raf signaling pathways leading to ERK1/2 activation and high expression. ERK1/2 translocate in the cell nucleus and regulate SOX1 family members’ overexpression. Activation of the metabolism pathway (PI3K/AKT/mTOR) regulates oncogene expression via gene methylation and chromatin acetylation (H3K4 and H3K9).
Additionally, while PTEN has been said to antagonize PI3K/AKT signaling (106, 107), reports on the effect of GSK-3β on the pathway remain inconclusive. Yadav et al. and others stated that GSK-3β encourages the activation of the PI3K/AKT/mTOR pathway (108, 109). Nonetheless, Spokoini et al. had earlier reported that GSK-3β is antagonistic to the PI3K/AKT pathway (110). Even so, GSK-3β is known for its cell survival function, which aligns with promoting neuron survival and proliferation by PI3K/AKT signaling (111–113). Recently, Vashishtha et al. demonstrated that GSK-3 isoforms (GSK-3α and GSK-3β) were involved in the maintenance and progression of gliomas (114). They further showed that GSK-3 was involved in the survival of glioma cells (114), which apparently contradicted what had been reported earlier (115). Thus, it is speculated that the specific role of GSK-3 might depend on the isoform being expressed and/or their level of expression, as well as the metabolic pathways that might be involved in their genetic activation. Also, the fact that PI3K/AKT can switch among pathways that regulate metabolism and interaction with other proteins in immunes to alter their functions maladaptively just like in glioma, suggests that the pathway might be mediating other multiple underlying cascades leading epigenetic modifications (acetylation and methylation of histone) of immune cells and hastening the initiation and progression of gliomas.
As metabolism is an automatic process, its inhibition can lead to glial cell and neurons death. PI3K/Akt/mTOR inhibition to target cancer cell metabolism through drugs (such as Gleevec) has a side effect of affecting metabolism in a systemic fashion. The reduction of cancer cell metabolism leads to their death and necrosis, which has been shown to contribute to tumor maintenance. Hence, changing tumor metabolism slows its growth speed without reversing tumorigenesis. Moreover, gene sequencing such as the cancer genome atlas (TCGA), next-generation sequence (NGS), single-cell sequencing other transcriptomics only expose gene mutation, expression level, and identification of cancer cell hierarchy highly contribute to cancer therapy theoretically. On the other hand, even after detecting the exact mutations or alterations (amino acid sequences, nucleic base change, epigenetics modifications) that contribute to tumorigenesis and progression, it is difficult to induce any reversion due to the involvement of several factors such as the cell native environment which cannot be exactly mimicked under in vitro conditions. Additionally, the isolation process and tumor dissociation can cause stress to tumor cells alter their behavior. It is also important to acknowledge that cancer cells have multiple complex metabolism preferences. This metabolism heterogeneity makes cancer metabolism difficult to target because it can switch at any time using PI3K/AKT/mTOR or another unknown pathway.
Warburg Effect Regulates Immune Cell Reprogramming to Facilitate Cancer Cell Metastasis Via Angiogenesis
Warburg effect or aerobic glycolysis is one of the metabolic mechanisms cancer cells use to accumulate energy (ATP) and proliferate for survival. Kornberg extensively reviewed the role of the Warburg effect in immune cell metabolic reprogramming (116). By the Warburg effect, oxidation of pyruvate to lactate (oxidative glycolysis) is preferred to oxidative phosphorylation for energy production even in the presence of oxygen. This form of modified cellular metabolism is typical of cancerous cells. The ability of the immune cells to adopt this altered mode of metabolism affects their cell lineage, which modifies immune responses and functions due to the impact of metabolism on regulating gene expressions. During inflammation, the role of immune cell metabolism determines the function of the immune cells in the progression or resolution of inflammation and disorder in autoimmune diseases. Kornberg et al. demonstrated that the anti-inflammatory functions of myeloid and lymphoid cells are regulated by the extent of aerobic glycolysis (117). In that, excessive aerobic glycolysis dampened the activation of anti-inflammatory responses. Evidently, the Warburg effect was shown in a recent work where it prevented inflammatory resolution by reprogramming and sustaining the activation of proinflammatory cytokines secretions and encouraged cancer cells progression (118). Furthermore, the Warburg effect has been shown to promote the methylation of inflammatory genes and the amplification of genes responsible for modifying immune cells’ plasticity and destiny (119, 120).
T lymphocytes such as CD4 and CD8 activation are linked to glycolysis, which regulates cytokine transcription and granule formation (119). The increase of TGF-beta expression in cancer cells and microenvironment affects lymphocyte metabolism via activation of the Warburg effect controlled by glucose transporters (GLUT1 and GLUT4) and pyruvate dehydrogenase. This leads to lymphocyte function inactivation without deactivating the cytokines, which exacerbates inflammation and promotes cancer cell survival and proliferation. The exposure of cancerous cells to cytokine promotes tolerance and activates machinery to modify oncogenes expression necessary for the inactivation of macrophages (121, 122). The activation of PI3K/AKT/mTOR under hypoxic conditions regulates the hyperexpression HIF-1 that regulates oncogenes upregulation through hypermethylation and histone acetylation (H3K4 and H3K9) (123).
In other immune cells (natural killer, B lymphocyte, and dendritic cell) Warburg effect contributes to the activation and increase in oxidative glycolysis with high lactate production. The increase in lactate production and phospholipid metabolism regulates the migration and angiogenetic functions of immune cells as well as cancer cell metastasis (116). As depicted in Figure 1, the association of immune cells with cancer cells stimulates cancer colony growth. Malinarich et al. reported that glycolysis and oxidative phosphorylation (OXPHOS) in fatty acid oxidation modulates immune cell functions to enable them to attack abnormal cells (124). However, metabolic dysregulation might prevent the immune cells from performing their designated function. Also, cancerous cells have been shown to utilize inflammatory responses to facilitate their metastasis along the nerve propagation path via ventriculoperitoneal or blood vessels (23, 125, 126). Previous reports suggested that metastasis rarely occurs in glioma (127). Contrarily, recent findings have shown that glioma stem cells can contribute to glioma metastasis within and out of the brain. The ability of glioma stem cells to divide and differentiate contributes to glioma cell spread in the body even if it is not directly through blood vessels. Evidently, Hao et al. and others demonstrated that glioma cells could metastasize depending on the genes expressions (128, 129) and organelles dysfunctioning (130).
Also, epithelial to mesenchymal transition (EMT) contributes to glioma cell metastasis (131, 132). The ability of glioma cells to switch from one cell type to another highly contributes to tumor invasion, leading to cancer cells metastasis (133). Cancerous cells associate with T-lymphocyte and dendritic cells to enable them to migrate in blood vessels (134). The activation of macrophages and dendritic cell during infection offers the cancerous cell the opportunity to move out of the vessel by employing the same mechanism as before (Figure 4). As such, cancerous cells tend to rely on the activation and inactivation of immune cells to facilitate their crossing in and out of blood vessels to enable them to metastasize (135, 136). Steinmetz et al. reported that radiotherapy could promote the transformation of glioblastoma cells into sarcomatoid metaplasia, which can cross the blood vessel and expedite the metastasize to other organs such as the liver (23, 137).
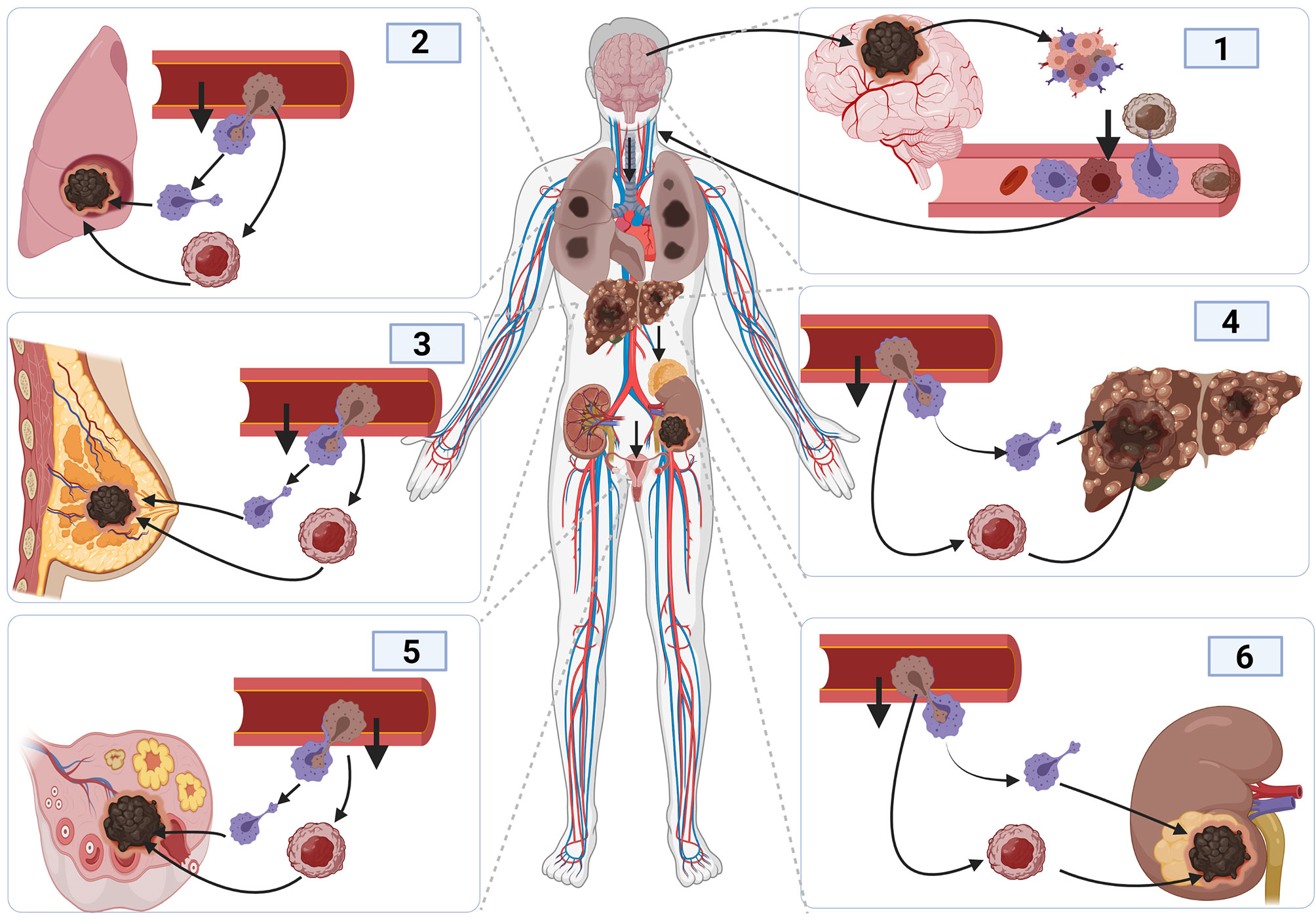
Figure 4 Cancer metastasis mechanism and tumor initiation and spread in other organs. The initiation of brain cancer promotes angiogenesis and tumor development, causing inflammation. The development of inflammation promotes immune response. Cancer cells bind to activated macrophages crossing the blood vessels, facilitating their migration into the blood vessel, and entering the blood circulatory system to reach other organs using the same process used to enter inside blood vessels.
Furthermore, the appearance of new blood vessels facilitates glucose, minerals, and other nutrients supply that support cancer cell proliferation, tumor formation, a metastasis. The molecular mechanisms underlying angiogenesis are complex and still not completely elucidated; however, it has been shown to rely on the epithelial-to-mesenchymal transition (EMT) of immune cells and the polymerization of fibrin present in the tumor microenvironment (138).
Cytoskeleton Polymerization and Epigenetic Modifications Scaffolded by Cell-Cancer Initiation
Hyperactivated metabolism initiated and regulated by the high proliferative effect of cancer cells, in turn, activates microtubule-associated proteins (MAPs). MAPs regulate the cytoskeleton polymerization via the modification of microtubule dynamics (139). The microtubule proteins (actins and tubulins) assembly scaffold a focal adhesion of cancer cells to their new environment to facilitate the transfer of metabolites and substrates necessary for cancer cells development. During glycolysis, the level of acetyl-coA increases with the high synthesis of citrate used for the Krebs cycle (140). Also, elevated citrate levels in the mitochondria induce their passive diffusion into the cytosol, where they are transformed into acetyl-coA that promotes epigenetic modifications (141, 142).
Further, acetyl-coA translocation into the nucleus where acetyl binds to histone (acetylation) to enhance target genes expressions. Additionally, the gene alteration caused by high acetyl-coA limits the activities of P53 and cyclin upregulation, thereby impairing DNA damage repair and cell cycle regulation, respectively (143, 144). The alteration of oncogene leads to high expression of oncogenes and microtubule-associated proteins to enhance the transport of nutrients, substrates, and proteins. The synergy of the aforementioned phenomenon, inhibiting autophagy due to the hyperactivation of growth factor and PI3K/AKT/mTOR signaling pathway, stimulates cancer cells’ proliferation and tumor initiations and progressions. Furthermore, the development of dendrites requires cytoskeleton polymerization to increase the adhesive characteristic that makes phagocytosis easier for them. In addition, the development of microtubule filaments helps dendritic cells to migrate to their intended destination and mediate specific cell-cell interaction during antigen presentation. However, similar to cancerous cells, the cytoskeleton polymerization of dendrites requires energy acquired by the Warburg effect due to reprogramming in the microenvironment of cancerous cells (145). Ultimately, this goes a long way to affect dendritic cell lineage and modifies immune response and function because altered metabolism induces gene expression changes (77–79).
Role of Lipids Metabolism in Tumorigenesis
Phospholipids play a crucial role in endoplasmic reticulum activities and regulate the phosphorylation of several proteins necessary for elevated oncogenes expression (146). Proteins such as phosphatidylinositol (3,4,5)-trisphosphate (PIP3) are associated with the cell membrane. The increase in GDNF (growth factor) is associated with PTEN that regulates phosphatidylinositol 4,5-bisphosphate (PIP2) phosphorylation into PIP3. PIP3 binds to the Golgi apparatus, endoplasmic reticulum, and the nucleus membrane to facilitate transcription factor translocation inside the nucleus (147, 148). The phospholipids can also activate AKT and mTORC2 to regulate glioma cell survival and metabolism, leading to cytoskeletal changes via the regulation of Rho protein and CDC42 (147). Notably, phospholipids have been shown to regulate autophagy, phagosome, and lysosome, contributing to lipid metabolism and trafficking inside the cell (149). Even so, the overuse of phospholipids leads to alteration of the plasma membrane, affecting its permeabilization and reinforcement of the nucleus membrane to protect cancer cells against apoptosis (150).
Similarly, the plasma membrane of brain cells facilitates the flow (inward and outward) of nutrients, hormones, ions that keep the brain neurons alive. Nonetheless, its alteration facilitates massive influxes of calcium which stimulates the excessive synthesis of dopamine and causes overexcitation of the brain neurons (151). Additionally, the increase in intraneural calcium levels contributes to the cancelation of metabolism by interconnecting molecules and oncogenes via electron transfer (152). During the lack of glucose, lipids are transformed into glucose, which contributes to glycolysis and the Warburg effect. Together, these synergic metabolic activities highly contribute to cancer initiation and maintenance.
Due to its metabolic heterogeneity, glioma stem cell plasticity is an example of multiple molecular subclasses that plays several functions in heterogeneous tumors and cell populations (153). The metabolic and molecular heterogeneity is the cause of cellular diversity, morphological modification, adaptation, and resistance to therapy, thereby resulting in tumor recurrence after surgical resection, chemotherapy, and radiotherapy. The proliferation of glioma cancer stem cells requires high energy levels. Hence, the glioma cells can switch to lipid metabolism increasing in fatty acid oxidation to maintain their stemness and ability to differentiate into any cell type (154).
Lipids metabolism also contributes to immune cell reprogramming and immune response (155). TGF-β regulates fatty acid upregulation via lipids metabolism to stimulate cytokine hyperexpression during immune responses (156). The binding of growth factors such as TGF-β to immune cell receptors induces the activation of PI3K/AKT/mTOR and PI3K/AKT/NF-kβ signaling pathway, which contributes to cholesterol synthesis and inflammation via sterol regulatory element-binding protein (SREBP) (157). The activation of the PI3K/AKT pathway promotes the activation of SREBP that regulates the switching between cholesterol synthesis and proinflammatory cytokines such as interleukine and chemokine (157). High cholesterol level regulates immune cell endoplasmic reticulum and Golgi apparatus activities, contributing to epigenetic modification in immune cell DNA (158). Meanwhile, cholesterol synthesis is a fundamental requirement for several hormones and cytokines (159, 160). The conversion of cholesterol into hormones or cytokines contributes to lipid metabolism. Lipid metabolism is essential for tumor-associated macrophages (TAM) that produce more cytokine and chemokine necessary for cancer cell survival (161, 162).
Perspectives and Advancements in Cancer Therapeutic Interventions
Nanotechnology is a new therapy strategy being used in the treatment of many diseases. The use of nanocarriers to support immune responses has become imperative in cancer therapy (163). This is because barriers created by the tumor and the presence of cocktails of protein and chemicals in tumor microenvironments induce immune cell reprogramming, which undermines their ability to fight tumors and reduce cancer cells population (164).
Herein, it is speculated that three therapeutic strategies might help make headways toward the fight and elimination of cancers. In brief, the first therapeutic strategy will be to develop interventions to break the barriers and penetrate tumor machinery. Secondly, delivery drugs or genetically trained immune cells that target inhibition, dysregulation, and/or destruction of cancer cells or functions/processes required for their survival. These trained cells could also be programmed to terminate dysfunctional inflammatory cells supplementing tumors with oncoproteins or facilitating a proinflammatory cytokine-rich microenvironment. Lastly, nanocarrier could be employed in the delivery of therapeutic to specific tumor microenvironment with the aim of effecting the two aforementioned strategies to inhibit their growth and progression. Deployment of these strategies might circumvent the side effects of intravenous chemotherapy and/or radiotherapy and improve treatment outcomes.
Recently, the use of 2-deoxy-D-glucose (2DG), 3-Bromopyruvate (3-BrOP), or dichloroacetic acid (DCA) have been shown to hinder the Warburg effect, thereby serving as blockades to metabolism alterations-induced immune cell reprogramming (165, 166). Also, nanoparticle delivery of temozolomide (TMZ) or other anti-cancer drugs with DNA fragmentation and digestion mechanisms have shown to be potent at inducing cancer cell death by reducing fibrin and collagen level in the cancer cell microenvironment to reduce angiogenesis and nutrient supplies (167, 168) (Figure 5).
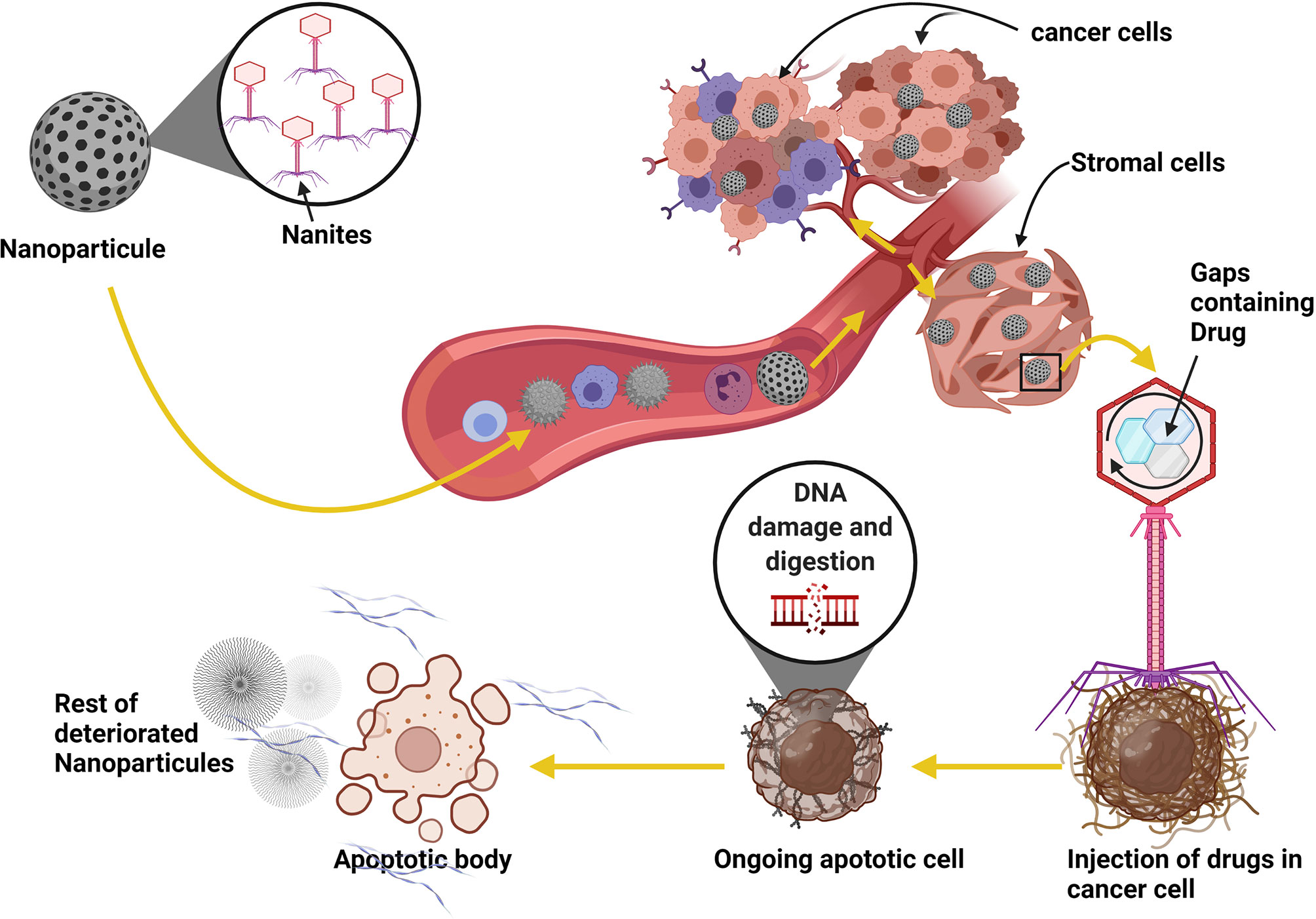
Figure 5 Using nanoparticles as a parasite in cancer therapy strategy. Patients with brain cancer can take the nanites as a pill. The nanites will enter into the blood circulation system using the digestif way. Once in the blood, nanites will target the cancer cells, parasitize them and inject their contents into the cells, resulting in the death of the host cells and the degradation of fibrin necessary for developing blood vessels and the development of cancer. The virus also uses the same process to target its host cell. The use of this method can significantly improve patient life and reduce cancer progression.
The regulation of nutrient supplies to the tumor reduces their proliferation abilities via metabolism (glycolysis) control. Glioma stem cell metabolic heterogeneity (plasticity) accounts for heterogeneous tumors and cell populations. These metabolic and molecular heterogeneity results in cellular diversity, morphological modifications, and adaptations that enhance tumor recurrence after surgical resection, chemotherapy, and radiotherapy. With the knowledge of metabolic substrates preferences of cancer cells, therapeutic strategies can be targeted, impeding metabolites intake/assimilation or deploying ATP decoys such ATPγS or ADPβS, which are hydrolysis-resistant, thereby ultimately inhibiting tumor progression.
Additionally, the glioma cancer stem cell niche significantly contributes to their progression and resistance to therapies. Stroma constituted of nonmalignant cells (fibroblast, T-cells, macrophages, and endothelial cells) in the cancer microenvironment have been shown to affect the metabolic dynamics and tumor heterogeneity (169, 170). Notably, the existence of metabolic competition has been demonstrated between stroma and cancer cells (169, 171, 172). Besides, during migration and metastasis of cancerous cells, they require the association with stroma cells to adapt to the new environment (173–175). As such, it is suggested that pitching stroma against gliomas to compete for metabolites might be a therapeutic approach to inhibit the cancerous cells (176, 177). The use of the therapeutic approach of stromal cells against cancerous cells might reverse glioma phenotype and limit the symbiosis between cancer and stromal cell proliferation.
Furthermore, the fact that metabolic alteration induces reprogramming of immune cells (plasticity and polarization) as well (178, 179), suggests that hindering maladaptive modes of metabolism might utilize the synergy of slowing down tumor growth and enhancing immunosurveillance for detection and destruction of glioma cells. Reportedly, the immune cells (macrophages and T-cells) constituent in the stromal breaks the junctions between the tumors-associated cells and tumor cells and further cleanses the microenvironment, simultaneously avoiding inflammation (180, 181). Therefore, it might be necessary to utilize trained natural killer cells to target polymer between cancer-associated fibroblasts (CAFs) and macrophages and digest abnormal (tumor or tumor-associated) cells to discourage tumor growth. Emphatically, it is imperative to explore trained immune cells to target the strategic functions of glioma. Hence, exploring immunotherapy strategies coupled with other previously mentioned strategies might contribute significantly to the advancements in cancer therapeutics and patient survival.
Conclusion
Immune cell plays an important role in carcinogenesis and hastening its progression. It is evident that the modification of cancer and immune cells’ plasticity contributes to tumor heterogeneity via the regulation of metabolism. Immune cells’ mobility in and out of blood vessels when they are activated scaffolds cancer cells metastasis and angiogenesis. Both immune and cancer cell reprogramming facilitated by TGF alpha and beta family members via hyperactivation of organelles (mitochondria, Golgi apparatus, and reticulum endoplasmic) and metabolism switches from anaerobic to aerobic glycolysis contribute to cancer progression, dampened immune response, and cancer resistance (182). Also, the alteration of cancer cells’ microenvironment constituents tends to create a niche that modifies the functions and induces plasticity of cells within this environment, thereby making the functions of the altered cells contribute to tumor development. The use of nanotechnology in brain cancer therapy has many advantages. The nanites can cross the brain cancer barrier without being repr ogrammed, as nanites do not undergo to metabolic process to contribute to tumor progression.
Author Contributions
KouK drafted and wrote the manuscript. KonK constructed the graphical illustrations. GA and AA proofread the manuscript. PK, XG, XY and DG read and approved the manuscript for submission. All authors contributed to the article and approved the submitted version.
Funding
Research in our laboratories is funded by the National Natural Science Foundation of China (Grant numbers: 81772688 and 81372698). China and Jiangsu Postgraduate Science Foundation-funded project (2013 M540466 and 1301068C), Qing Lan Project, and a project Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
I thank professor GAO Dianshuai for his guidance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.831636/full#supplementary-material
Supplementary Figure 1 | (A) Immune and cancer interaction in brain cancer development and metastasis. In the presence of an abnormal cell or pathogen, the immune cells react to stop the proliferation and spread of the cancer cell or pathogen. The cancer cell develops on their membrane certain receptors that protect them against the immune response. The immune cell did not see cancer cells as a potential danger; instead of destroying the isolated cancer cell, the immune cell aids its tissue colonization to increase its chance to survive. This insertion leads to inflammation of the tissue and the synthesis of cytokines. The synthesis of cytokines leads to cancer cell proliferation and angiogenesis and enhances cancer cell metastasis via intussusception of blood vessels. The cancer cell colony pressure causes intussusception of blood vessels. Weaken blood vessels become more permeable to the immune and cancer cells, thereby facilitating cancer cell metastasis inside the brain. (B) Mechanism used by the cancer cell to escape immune responses. Each cell has many receptors on its membrane important for signal induction and recognition between cells and immune cells. In the normal and immune cell interaction, the immune cell recognizes the normal cell via direct binding of the receptor (A–E). In the interaction with cancer cells, there is an intermediate protein (TGFβ, GDNF, and Calcium), the binding of intermediate protein on cancer cell receptors induces a cascade of reactions inside the cancer cell. The binding of immune cells to cancer cells through the receptor via TGF protein (red) activates the metabolic signaling pathway in both cancer and immune cells leading to tumor progression.
Supplementary Figure 2 | Immune cell lineage and cancer cell reprogramming through the Warburg effect and oncogene upregulation. All immune cell lineage contributes to cancer development. When the immune cells enter the cancer microenvironment, their metabolism changes, and their functions (plasticity and polarization) are altered. The metabolic modification contributes to the infiltration of cancer and immune cell to tumor tissue via the upregulation of cytokines such as interleukins (IL-1α, IL-6, IL-8, IL-10), oncogene such as (TGFα/β, VEGF), and tumor necrosis factor (TNF-α). The combined upregulation of oncogenes and metabolism/Warburg effect promotes cancer cell proliferation, metastasis, and malignancy.
Supplementary Figure 3 | epigenetic alteration and oncogene overexpression due to metabolic changes and activation of PI3K/AKT/mTOR pathways. Metabolism is one of the principal cell dynamics that ensure energy for cell division and survival. During inflammation, the overexpression of certain growth factors activates the PI3K/AKT/mTOR pathway, which controls several genes to perform their tasks. The binding of TGF-β family members on their receptors activates PI3K and aerobic glycolysis in cancer and immune cells. In cancer cells, the binding of TGF-β on their receptor activates PI3K/AKT pathways that simultaneously control the activities of mTOR and GSK3β genes. The activation of AKT modulates the activity of GSK3β and activates the mTOR pathway to regulate mitochondrial glycolysis. When glycolysis is insufficient for glioma cell proliferation and survival, this pathway can be switched using calcium (Ca2+) ions to change from one pathway to another, making the PI3K/AKT pathway complicated. In immune cells, during inflammation, the activation of macrophages also activates the PI3K/AKT/mTOR pathway activating HIF-1β, which causes glycolysis and increases mitochondrial activity. In both cases, the activation of glycolysis significantly affects oncogenes gene expression by encouraging epigenetic modifications such as histones acetylation and DNA methylation (H3K4 for SOX1 and H3K9 for GDNF) to promote glioma.
Abbreviations
GDNF, Glial cell line-derived neurotrophic factor; SOX1, Sex-determining region Y box1; TGF-beta, Transforming Growth Factor beta; EGFR, epidermal growth factor receptor; PI3-K, phosphatidylinositide 3-kinase; STAT3, signal transduction activator of transcription 3; GSK3 beta, Glycogen synthase kinase 3 beta; PTEN, Phosphatase and Tensin, VEGF, Vascular endothelial growth factor; TMZ, temozolomide; CAF, Cancer-Associated Fibroblast; IL-6, interleukins and TNF-α, tumor necrosis factor-alpha.
References
1. Abdalla G, Dixon L, Sanverdi E, Machado PM, Kwong JSW, Panovska-Griffiths J, et al. The Diagnostic Role of Diffusional Kurtosis Imaging in Glioma Grading and Differentiation of Gliomas From Other Intra-Axial Brain Tumours: A Systematic Review With Critical Appraisal and Meta-Analysis. Neuroradiology (2020) 62(7):791–802. doi: 10.1007/s00234-020-02425-9
2. Brandner S, Jaunmuktane Z. Neurological Update: Gliomas and Other Primary Brain Tumours in Adults. J Neurol (2018) 265(3):717–27. doi: 10.1007/s00415-017-8652-3
3. Miller KD, Ostrom QT, Kruchko C, Patil N, Tihan T, Cioffi G, et al. Brain and Other Central Nervous System Tumor Statistic. CA Cancer J Clin (2021) 71(5):381–406. doi: 10.3322/caac.21693
4. Tran B, Rosenthal MA. Survival Comparison Between Glioblastoma Multiforme and Other Incurable Cancers. J Clin Neurosci (2010) 17(4):417–21. doi: 10.1016/j.jocn.2009.09.004
5. Finch A, Solomou G, Wykes V, Pohl U, Bardella C, Watts C. Advances in Research of Adult Gliomas. Int J Mol Sci (2021) 22(2):924. doi: 10.3390/ijms22020924
6. Cahill D, Turcan S. Origin of Gliomas. Semin Neurol (2018) 38(1):5–10. doi: 10.1055/s-0037-1620238
7. Garcia-Fabiani MB, Haase S, Comba A, Carney S, McClellan B, Banerjee K, et al. Genetic Alterations in Gliomas Remodel the Tumor Immune Microenvironment and Impact Immune-Mediated Therapies. Front Oncol (2021) 11:631037. doi: 10.3389/fonc.2021.631037
8. Miranda-Goncalves V, Lameirinhas A, Henrique R, Jeronimo C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front Genet (2018) 9:427. doi: 10.3389/fgene.2018.00427
9. Yu X, Ma R, Wu Y, Zhai Y, Li S. Reciprocal Regulation of Metabolic Reprogramming and Epigenetic Modifications in Cancer. Front Genet (2018) 9:394. doi: 10.3389/fgene.2018.00394
10. Dosquet C, Schaetz A, Faucher C, Lepage E, Wautier JL, Richard F, et al. Tumour Necrosis Factor-Alpha, Interleukin-1 Beta and Interleukin-6 in Patients With Renal Cell Carcinoma. Eur J Cancer (1994) 30A(2):162–7. doi: 10.1016/0959-8049(94)90079-5
11. Grivennikov SI, Karin M. Inflammatory Cytokines in Cancer: Tumour Necrosis Factor and Interleukin 6 Take the Stage. Ann Rheum Dis (2011) 70 Suppl 1:i104–108. doi: 10.1136/ard.2010.140145
12. Liu C, Peng G, Jing N. TGF-Beta Signaling Pathway in Early Mouse Development and Embryonic Stem Cells. Acta Biochim Biophys Sin (Shanghai) (2018) 50(1):68–73. doi: 10.1093/abbs/gmx120
13. Park KS. Tgf-Beta Family Signaling in Embryonic Stem Cells. Int J Stem Cells (2011) 4(1):18–23. doi: 10.15283/ijsc.2011.4.1.18
14. Kaminska B, Cyranowski S. Recent Advances in Understanding Mechanisms of TGF Beta Signaling and Its Role in Glioma Pathogenesis. Adv Exp Med Biol (2020) 1202:179–201. doi: 10.1007/978-3-030-30651-9_9
15. Kaminska B, Kocyk M, Kijewska M. TGF Beta Signaling and its Role in Glioma Pathogenesis. Adv Exp Med Biol (2013) 986:171–87. doi: 10.1007/978-94-007-4719-7_9
16. Roy LO, Poirier MB, Fortin D. Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors. Int J Mol Sci (2018) 19(4):1113. doi: 10.3390/ijms19041113
17. Gratchev A. TGF-Beta Signalling in Tumour Associated Macrophages. Immunobiology (2017) 222(1):75–81. doi: 10.1016/j.imbio.2015.11.016
18. Zhang D, Qiu X, Li J, Zheng S, Li L, Zhao H. TGF-Beta Secreted by Tumor-Associated Macrophages Promotes Proliferation and Invasion of Colorectal Cancer via miR-34a-VEGF Axis. Cell Cycle (2018) 17(24):2766–78. doi: 10.1080/15384101.2018.1556064
19. Chockley PJ, Chen J, Chen G, Beer DG, Standiford TJ, Keshamouni VG. Epithelial-Mesenchymal Transition Leads to NK Cell-Mediated Metastasis-Specific Immunosurveillance in Lung Cancer. J Clin Invest (2018) 128(4):1384–96. doi: 10.1172/jci97611
20. Goswami MT, Reka AK, Kurapati H, Kaza V, Chen J, Standiford TJ, et al. Regulation of Complement-Dependent Cytotoxicity by TGF-β-Induced Epithelial-Mesenchymal Transition. Oncogene (2016) 35(15):1888–98. doi: 10.1038/onc.2015.258
21. Standiford TJ, Kuick R, Bhan U, Chen J, Newstead M, Keshamouni VG. TGF-Beta-Induced IRAK-M Expression in Tumor-Associated Macrophages Regulates Lung Tumor Growth. Oncogene (2011) 30(21):2475–84. doi: 10.1038/onc.2010.619
22. Ferraro F, Celso CL, Scadden D. Adult Stem Cels and Their Niches. Adv Exp Med Biol (2010) 695:155–68. doi: 10.1007/978-1-4419-7037-4_11
23. Steinmetz MP, Barnett GH, Kim BS, Chidel MA, Suh JH. Metastatic Seeding of the Stereotactic Biopsy Tract in Glioblastoma Multiforme: Case Report and Review of the Literature. J Neurooncol (2001) 55(3):167–71. doi: 10.1023/a:1013873431159
24. Yang X, Zhang Y, Hosaka K, Andersson P, Wang J, Tholander F, et al. VEGF-B Promotes Cancer Metastasis Through a VEGF-A-Independent Mechanism and Serves as a Marker of Poor Prognosis for Cancer Patients. Proc Natl Acad Sci USA (2015) 112(22):E2900–9. doi: 10.1073/pnas.1503500112
25. Xie Q, Mittal S, Berens ME. Targeting Adaptive Glioblastoma: An Overview of Proliferation and Invasion. Neuro Oncol (2014) 16(12):1575–84. doi: 10.1093/neuonc/nou147
26. Hoang-Minh LB, Siebzehnrubl FA, Yang C, Suzuki-Hatano S, Dajac K, Loche T, et al. Infiltrative and Drug-Resistant Slow-Cycling Cells Support Metabolic Heterogeneity in Glioblastoma. EMBO J (2018) 37(23):e98772. doi: 10.15252/embj.201798772
27. DeBerardinis RJ, Chandel NS. We Need to Talk About the Warburg Effect. Nat Metab (2020) 2(2):127–9. doi: 10.1038/s42255-020-0172-2
28. Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, et al. A Predictive Model for Selective Targeting of the Warburg Effect Through GAPDH Inhibition With a Natural Product. Cell Metab (2017) 26(4):648–59.e648. doi: 10.1016/j.cmet.2017.08.017
29. Lim ZF, Ma PC. Emerging Insights of Tumor eterogeneity and Drug Resistance Mechanisms in Lung Cancer Targeted Therapy. J Hematol Oncol (2019) 12(1):134. doi: 10.1186/s13045-019-0818-2
30. Zaal EA, Berkers CR. The Influence of Metabolism on Drug Response in Cancer. Front Oncol (2018) 8:500. doi: 10.3389/fonc.2018.00500
31. Bjorklund M. Cell Size Homeostasis: Metabolic Control of Growth and Cell Division. Biochim Biophys Acta Mol Cell Res (2019) 1866(3):409–17. doi: 10.1016/j.bbamcr.2018.10.002
32. Jones AR, Band LR, Murray JAH. Double or Nothing? Cell Division and Cell Size Control. Trends Plant Sci (2019) 24(12):1083–93. doi: 10.1016/j.tplants.2019.09.005
33. Wang Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells (2021) 10(12):3327. doi: 10.3390/cells10123327
34. Fielder GC, Yang TW, Razdan M, Li Y, Lu J, Perry JK, et al. The GDNF Family: A Role in Cancer? Neoplasia (2018) 20(1):99–117. doi: 10.1016/j.neo.2017.10.010
35. Park HJ, Bolton EC. Glial Cell Line-Derived Neurotrophic Factor Induces Cell Proliferation in the Mouse Urogenital Sinus. Mol Endocrinol (2015) 29(2):289–306. doi: 10.1210/me.2014-1312
36. Karnavas T, Mandalos N, Malas S, Remboutsika E. SoxB, Cell Cycle and Neurogenesis. Front Physiol (2013) 4:298. doi: 10.3389/fphys.2013.00298
37. Senturk E, Manfredi JJ. P53 and Cell Cycle Effects After DNA Damage. Methods Mol Biol (2013) 962:49–61. doi: 10.1007/978-1-62703-236-0_4
38. Chen J. The Cell-Cycle Arrest and Apoptotic Functions of P53 in Tumor Initiation and Progression. Cold Spring Harb Perspect Med (2016) 6(3):a026104. doi: 10.1101/cshperspect.a026104
39. Gottlieb SF, Tupper C, Kerndt CC, Tegay DH. Genetics, Nondisjunction. In: StatPearls. Treasure Island, FL: StatPearls (2022).
40. Potapova T, Gorbsky GJ. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biol (Basel) (2017) 6(1):12. doi: 10.3390/biology6010012
41. Lukasik P, Zaluski M, Gutowska I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development-Review. Int J Mol Sci (2021) 22(6):2935. doi: 10.3390/ijms22062935
42. Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, et al. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int J Mol Sci (2020) 21(6):1960. doi: 10.3390/ijms21061960
43. Lisse TS, Sharma M, Vishlaghi N, Pullagura SR, Braun RE. GDNF Promotes Hair Formation and Cutaneous Wound Healing by Targeting Bulge Stem Cells. NPJ Regener Med (2020) 5:13. doi: 10.1038/s41536-020-0098-z
44. Uesaka T, Nagashimada M, Enomoto H. GDNF Signaling Levels Control Migration and Neuronal Differentiation of Enteric Ganglion Precursors. J Neurosci (2013) 33(41):16372–82. doi: 10.1523/JNEUROSCI.2079-13.2013
45. Archer TC, Jin J, Casey ES. Interaction of Sox1, Sox2, Sox3 and Oct4 During Primary Neurogenesis. Dev Biol (2011) 350(2):429–40. doi: 10.1016/j.ydbio.2010.12.013
46. Batool S, Kayani MA, Valis M, Kuca K. Neural Differentiation of Mouse Embryonic Stem Cells-An In Vitro Approach to Profile DNA Methylation of Reprogramming Factor Sox2-Srr2. Front Genet (2021) 12:641095. doi: 10.3389/fgene.2021.641095
47. Muller M, Hermann PC, Liebau S, Weidgang C, Seufferlein T, Kleger A, et al. The Role of Pluripotency Factors to Drive Stemness in Gastrointestinal Cancer. Stem Cell Res (2016) 16(2):349–57. doi: 10.1016/j.scr.2016.02.005
48. Liu L, Michowski W, Kolodziejczyk A, Sicinski P. The Cell Cycle in Stem Cell Proliferation, Pluripotency and Differentiation. Nat Cell Biol (2019) 21(9):1060–7. doi: 10.1038/s41556-019-0384-4
49. Albright JE, Stojkovska I, Rahman AA, Brown CJ, Morrison BE. Nestin-Positive/SOX2-Negative Cells Mediate Adult Neurogenesis of Nigral Dopaminergic Neurons in Mice. Neurosci Lett (2016) 615:50–4. doi: 10.1016/j.neulet.2016.01.019
50. Zhang BL, Liu J, Lei Y, Xiong Y, Li H, Lin X, et al. An Epigenetic Mechanism of High Gdnf Transcription in Glioma Cells Revealed by Specific Sequence Methylation. Mol Neurobiol (2016) 53(7):4352–62. doi: 10.1007/s12035-015-9365-1
51. Lim S, Bhinge A, Bragado Alonso S, Aksoy I, Aprea J, Cheok CF, et al. Cyclin-Dependent Kinase-Dependent Phosphorylation of Sox2 at Serine 39 Regulates Neurogenesis. Mol Cell Biol (2017) 37(16):e00201–17. doi: 10.1128/MCB.00201-17
52. Stevanovic M, Drakulic D, Lazic A, Ninkovic DS, Schwirtlich M, Mojsin M. SOX Transcription Factors as Important Regulators of Neuronal and Glial Differentiation During Nervous System Development and Adult Neurogenesis. Front Mol Neurosci (2021) 14:654031. doi: 10.3389/fnmol.2021.654031
53. Grimm D, Bauer J, Wise P, Kruger M, Simonsen U, Wehland M, et al. The Role of SOX Family Members in Solid Tumours and Metastasis. Semin Cancer Biol (2020) 67(Pt 1):122–53. doi: 10.1016/j.semcancer.2019.03.004
54. Khacho M, Harris R, Slack RS. Mitochondria as Central Regulators of Neural Stem Cell Fate and Cognitive Function. Nat Rev Neurosci (2019) 20(1):34–48. doi: 10.1038/s41583-018-0091-3
55. Kim DY, Rhee I, Paik J. Metabolic Circuits in Neural Stem Cells. Cell Mol Life Sci (2014) 71(21):4221–41. doi: 10.1007/s00018-014-1686-0
56. Lange C, Bifari F. Editorial: Metabolic Regulation of Stem Cells and Tissue Growth. Front Mol Neurosci (2020) 13:625606. doi: 10.3389/fnmol.2020.625606
57. Ma Q, Long W, Xing C, Chu J, Luo M, Wang HY, et al. Cancer Stem Cells and Immunosuppressive Microenvironment in Glioma. Front Immunol (2018) 9:2924. doi: 10.3389/fimmu.2018.02924
58. Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the Glioblastoma Immune Microenvironment as Basis for the Development of New Immunotherapeutic Strategies. Elife (2020) 9:e52176. doi: 10.7554/eLife.52176
59. Gonzalez H, Hagerling C, Werb Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev (2018) 32(19-20):1267–84. doi: 10.1101/gad.314617.118
60. Nandi B, Talluri S, Kumar S, Yenumula C, Gold JS, Prabhala R, et al. The Roles of Homologous Recombination and the Immune System in the Genomic Evolution of Cancer. J Transl Sci (2019) 5(2):10.15761/JTS.1000282. doi: 10.15761/JTS.1000282
61. Rong L, Li R, Li S, Luo R. Immunosuppression of Breast Cancer Cells Mediated by Transforming Growth Factor-Beta in Exosomes From Cancer Cells. Oncol Lett (2016) 11(1):500–4. doi: 10.3892/ol.2015.3841
62. Yoshimura A, Muto G. TGF-Beta Function in Immune Suppression. Curr Top Microbiol Immunol (2011) 350:127–47. doi: 10.1007/82_2010_87
63. Plenchette S, Romagny S, Laurens V, Bettaieb A. S-Nitrosylation in TNF Superfamily Signaling Pathway: Implication in Cancer. Redox Biol (2015) 6:507–15. doi: 10.1016/j.redox.2015.08.019
64. Saligrama PT, Fortner KA, Secinaro MA, Collins CC, Russell JQ, Budd RC. IL-15 Maintains T-Cell Survival via S-Nitrosylation-Mediated Inhibition of Caspase-3. Cell Death Differ (2014) 21(6):904–14. doi: 10.1038/cdd.2014.10
65. Steinkamp M, Gundel H, Schulte N, Spaniol U, Pflueger C, Zizer E, et al. GDNF Protects Enteric Glia From Apoptosis: Evidence for an Autocrine Loop. BMC Gastroenterol (2012) 12:6. doi: 10.1186/1471-230X-12-6
66. Zhao Y, Haney MJ, Gupta R, Bohnsack JP, He Z, Kabanov AV, et al. GDNF-Transfected Macrophages Produce Potent Neuroprotective Effects in Parkinson's Disease Mouse Model. PLoS One (2014) 9(9):e106867. doi: 10.1371/journal.pone.0106867
67. Wang Z, Li S, Wang Y, Zhang X, Chen L, Sun D. GDNF Enhances the Anti-Inflammatory Effect of Human Adipose-Derived Mesenchymal Stem Cell-Based Therapy in Renal Interstitial Fibrosis. Stem Cell Res (2019) 41:101605. doi: 10.1016/j.scr.2019.101605
68. Zhang B, Yao G, Zhang Y, Gao J, Yang B, Rao Z, et al. M2-Polarized Tumor-Associated Macrophages Are Associated With Poor Prognoses Resulting From Accelerated Lymphangiogenesis in Lung Adenocarcinoma. Clinics (Sao Paulo) (2011) 66(11):1879–86. doi: 10.1590/s1807-59322011001100006
69. Mulligan LM. GDNF and the RET Receptor in Cancer: New Insights and Therapeutic Potential. Front Physiol (2018) 9:1873. doi: 10.3389/fphys.2018.01873
70. Chiarugi P. Cancer-Associated Fibroblasts and Macrophages: Friendly Conspirators for Malignancy. Oncoimmunology (2013) 2(9):e25563. doi: 10.4161/onci.25563
71. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-Associated Fibroblasts and M2-Polarized Macrophages Synergize During Prostate Carcinoma Progression. Oncogene (2014) 33(19):2423–31. doi: 10.1038/onc.2013.191
72. Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress NK Cells Function in Colorectal Cancer. Cell Death Dis (2019) 10(4):273. doi: 10.1038/s41419-019-1435-2
73. Folmes CD, Terzic A. Metabolic Determinants of Embryonic Development and Stem Cell Fate. Reprod Fertil Dev (2014) 27(1):82–8. doi: 10.1071/RD14383
74. Miyazawa H, Aulehla A. Revisiting the Role of Metabolism During Development. Development (2018) 145(19):dev131110. doi: 10.1242/dev.131110
75. Bannister AJ, Kouzarides T. Regulation of Chromatin by Histone Modifications. Cell Res (2011) 21(3):381–95. doi: 10.1038/cr.2011.22
76. Etchegaray JP, Mostoslavsky R. Interplay Between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol Cell (2016) 62(5):695–711. doi: 10.1016/j.molcel.2016.05.029
77. Kastenmuller G, Raffler J, Gieger C, Suhre K. Genetics of Human Metabolism: An Update. Hum Mol Genet (2015) 24(R1):R93–101. doi: 10.1093/hmg/ddv263
78. Seldin M, Yang X, Lusis AJ. Systems Genetics Applications in Metabolism Research. Nat Metab (2019) 1(11):1038–50. doi: 10.1038/s42255-019-0132-x
79. Sinkala M, Mulder N, Patrick Martin D. Metabolic Gene Alterations Impact the Clinical Aggressiveness and Drug Responses of 32 Human Cancers. Commun Biol (2019) 2:414. doi: 10.1038/s42003-019-0666-1
80. Lien EC, Lyssiotis CA, Cantley LC. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res (2016) 207:39–72. doi: 10.1007/978-3-319-42118-6_3
81. Yu JS, Cui W. Proliferation, Survival and Metabolism: The Role of PI3K/AKT/mTOR Signalling in Pluripotency and Cell Fate Determination. Development (2016) 143(17):3050–60. doi: 10.1242/dev.137075
82. Benton D, Parker PY, Donohoe RT. The Supply of Glucose to the Brain and Cognitive Functioning. J Biosoc Sci (1996) 28(4):463–79. doi: 10.1017/s0021932000022537
83. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the Brain: The Role of Glucose in Physiological and Pathological Brain Function. Trends Neurosci (2013) 36(10):587–97. doi: 10.1016/j.tins.2013.07.001
84. Nakanishi H, Cruz NF, Adachi K, Sokoloff L, Dienel GA. Influence of Glucose Supply and Demand on Determination of Brain Glucose Content With Labeled Methylglucose. J Cereb Blood Flow Metab (1996) 16(3):439–49. doi: 10.1097/00004647-199605000-00010
85. Chaudhry R, Varacallo M. Biochemistry, Glycolysis. In: StatPearls. Treasure Island, FL: StatPearls (2020).
86. Melkonian EA, Schury MP. Biochemistry, Anaerobic Glycolysis. In: StatPearls. Treasure Island, FL: StatPearls (2020).
87. Martin SA, Souder DC, Miller KN, Clark JP, Sagar AK, Eliceiri KW, et al. GSK3beta Regulates Brain Energy Metabolism. Cell Rep (2018) 23(7):1922–31.e1924. doi: 10.1016/j.celrep.2018.04.045
88. Bijur GN, Jope RS. Rapid Accumulation of Akt in Mitochondria Following Phosphatidylinositol 3-Kinase Activation. J Neurochem (2003) 87(6):1427–35. doi: 10.1046/j.1471-4159.2003.02113.x
89. Jellusova J, Cato MH, Apgar JR, Ramezani-Rad P, Leung CR, Chen C, et al. Gsk3 Is a Metabolic Checkpoint Regulator in B Cells. Nat Immunol (2017) 18(3):303–12. doi: 10.1038/ni.3664
90. Yang K, Chen Z, Gao J, Shi W, Li L, Jiang S, et al. The Key Roles of GSK-3beta in Regulating Mitochondrial Activity. Cell Physiol Biochem (2017) 44(4):1445–59. doi: 10.1159/000485580
91. Chiara F, Rasola A. GSK-3 and Mitochondria in Cancer Cells. Front Oncol (2013) 3:16. doi: 10.3389/fonc.2013.00016
92. Strickland M, Stoll EA. Metabolic Reprogramming in Glioma. Front Cell Dev Biol (2017) 5:43. doi: 10.3389/fcell.2017.00043
93. Zhou W, Wahl DR. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers (Basel) (2019) 11(9):1231. doi: 10.3390/cancers11091231
94. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
95. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol Cell (2012) 48(4):612–26. doi: 10.1016/j.molcel.2012.08.033
96. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-Like Receptor-Induced Changes in Glycolytic Metabolism Regulate Dendritic Cell Activation. Blood (2010) 115(23):4742–9. doi: 10.1182/blood-2009-10-249540
97. Yu ZQ, Zhang BL, Ren QX, Wang JC, Yu RT, Qu DW, et al. Changes in Transcriptional Factor Binding Capacity Resulting From Promoter Region Methylation Induce Aberrantly High GDNF Expression in Human Glioma. Mol Neurobiol (2013) 48(3):571–80. doi: 10.1007/s12035-013-8443-5
98. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida Albicans Infection Affords Protection Against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe (2012) 12(2):223–32. doi: 10.1016/j.chom.2012.06.006
99. Yan C, Koda S, Wu J, Zhang BB, Yu Q, Netea MG, et al. Roles of Trained Immunity in the Pathogenesis of Cholangiopathies: A Novel Therapeutic Target. Hepatology (2020) 72:1838–50. doi: 10.1002/hep.31395
100. Lashinger LM, Ford NA, Hursting SD. Interacting Inflammatory and Growth Factor Signals Underlie the Obesity-Cancer Link. J Nutr (2014) 144(2):109–13. doi: 10.3945/jn.113.178533
101. Wahl SM, Wong H, McCartney-Francis N. Role of Growth Factors in Inflammation and Repair. J Cell Biochem (1989) 40(2):193–9. doi: 10.1002/jcb.240400208
102. Wang M, Guo Y, Wang M, Zhou T, Xue Y, Du G, et al. The Glial Cell-Derived Neurotrophic Factor (GDNF)-Responsive Phosphoprotein Landscape Identifies Raptor Phosphorylation Required for Spermatogonial Progenitor Cell Proliferation. Mol Cell Proteomics (2017) 16(6):982–97. doi: 10.1074/mcp.M116.065797
103. Hoxhaj G, Manning BD. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat Rev Cancer (2020) 20(2):74–88. doi: 10.1038/s41568-019-0216-7
104. Bahmad HF, Mouhieddine TH, Chalhoub RM, Assi S, Araji T, Chamaa F, et al. The Akt/mTOR Pathway in Cancer Stem/Progenitor Cells Is a Potential Therapeutic Target for Glioblastoma and Neuroblastoma. Oncotarget (2018) 9(71):33549–61. doi: 10.18632/oncotarget.26088
105. Dimitrova V, Arcaro A. Targeting the PI3K/AKT/mTOR Signaling Pathway in Medulloblastoma. Curr Mol Med (2015) 15(1):82–93. doi: 10.2174/1566524015666150114115427
106. Giguere V. Canonical Signaling and Nuclear Activity of mTOR-A Teamwork Effort to Regulate Metabolism and Cell Growth. FEBS J (2018) 285(9):1572–88. doi: 10.1111/febs.14384
107. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
108. Hermida MA, Dinesh Kumar J, Leslie NR. GSK3 and Its Interactions With the PI3K/AKT/mTOR Signalling Network. Adv Biol Regul (2017) 65:5–15. doi: 10.1016/j.jbior.2017.06.003
109. Yadav AK, Vashishta V, Joshi N, Taneja P. AR-A 014418 Used Against GSK3beta Downregulates Expression of Hnrnpa1 and SF2/ASF Splicing Factors. J Oncol (2014) 2014:695325. doi: 10.1155/2014/695325
110. Spokoini R, Kfir-Erenfeld S, Yefenof E, Sionov RV. Glycogen Synthase Kinase-3 Plays a Central Role in Mediating Glucocorticoid-Induced Apoptosis. Mol Endocrinol (2010) 24(6):1136–50. doi: 10.1210/me.2009-0466
111. Cao Q, Lu X, Feng YJ. Glycogen Synthase Kinase-3beta Positively Regulates the Proliferation of Human Ovarian Cancer Cells. Cell Res (2006) 16(7):671–7. doi: 10.1038/sj.cr.7310078
112. Duda P, Akula SM, Abrams SL, Steelman LS, Martelli AM, Cocco L, et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells (2020) 9(5):1110. doi: 10.3390/cells9051110
113. Nishimura H, Nakamura O, Yamagami Y, Mori M, Horie R, Fukuoka N, et al. GSK-3 Inhibitor Inhibits Cell Proliferation and Induces Apoptosis in Human Osteosarcoma Cells. Oncol Rep (2016) 35(4):2348–54. doi: 10.3892/or.2016.4565
114. Vashishtha V, Jinghan N, A KY. Antagonistic Role of GSK3 Isoforms in Glioma Survival. J Cancer (2018) 9(10):1846–55. doi: 10.7150/jca.21248
115. Kotliarova S, Pastorino S, Kovell LC, Kotliarov Y, Song H, Zhang W, et al. Glycogen Synthase Kinase-3 Inhibition Induces Glioma Cell Death Through C-MYC, Nuclear Factor-Kappab, and Glucose Regulation. Cancer Res (2008) 68(16):6643–51. doi: 10.1158/0008-5472.CAN-08-0850
116. Kornberg MD. The Immunologic Warburg Effect: Evidence and Therapeutic Opportunities in Autoimmunity. Wiley Interdiscip Rev Syst Biol Med (2020) 12(5):e1486. doi: 10.1002/wsbm.1486
117. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl Fumarate Targets GAPDH and Aerobic Glycolysis to Modulate Immunity. Science (2018) 360(6387):449–53. doi: 10.1126/science.aan4665
118. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The Cancer Metabolic Reprogramming and Immune Response. Mol Cancer (2021) 20(1):28. doi: 10.1186/s12943-021-01316-8
119. Kouidhi S, Elgaaied AB, Chouaib S. Impact of Metabolism on T-Cell Differentiation and Function and Cross Talk With Tumor Microenvironment. Front Immunol (2017) 8:270. doi: 10.3389/fimmu.2017.00270
120. Lin X, Xiao Z, Chen T, Liang SH, Guo H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front Oncol (2020) 10:317. doi: 10.3389/fonc.2020.00317
121. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
122. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
123. Yan C, Koda S, Wu J, Zhang BB, Yu Q, Netea MG, et al. Roles of Trained Immunity in the Pathogenesis of Cholangiopathies: A Therapeutic Target. Hepatology (2020) 72(5):1838–50. doi: 10.1002/hep.31395
124. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High Mitochondrial Respiration and Glycolytic Capacity Represent a Metabolic Phenotype of Human Tolerogenic Dendritic Cells. J Immunol (2015) 194(11):5174–86. doi: 10.4049/jimmunol.1303316
125. Barajas RF Jr, Phelps A, Foster HC, Courtier J, Buelow BD, Gupta N, et al. Metastatic Diffuse Intrinsic Pontine Glioma to the Peritoneal Cavity Via Ventriculoperitoneal Shunt: Case Report and Literature Review. J Neurol Surg Rep (2015) 76(1):e91–6. doi: 10.1055/s-0035-1547365
126. Narayan A, Jallo G, Huisman TA. Extracranial, Peritoneal Seeding of Primary Malignant Brain Tumors Through Ventriculo-Peritoneal Shunts in Children: Case Report and Review of the Literature. Neuroradiol J (2015) 28(5):536–9. doi: 10.1177/1971400915609348
127. Sun Q, Xu R, Xu H, Wang G, Shen X, Jiang H. Extracranial Metastases of High-Grade Glioma: The Clinical Characteristics and Mechanism. World J Surg Oncol (2017) 15(1):181. doi: 10.1186/s12957-017-1249-6
128. Hao B, Chen X, Cao Y. Yes-Associated Protein 1 Promotes the Metastasis of U251 Glioma Cells by Upregulating Jagged-1 Expression and Activating the Notch Signal Pathway. Exp Ther Med (2018) 16(2):1411–6. doi: 10.3892/etm.2018.6322
129. Liu Q, Wang XY, Qin YY, Yan XL, Chen HM, Huang QD, et al. SPOCD1 Promotes the Proliferation and Metastasis of Glioma Cells by Up-Regulating PTX3. Am J Cancer Res (2018) 8(4):624–35.
130. Cai Q, Zhu A, Gong L. Exosomes of Glioma Cells Deliver miR-148a to Promote Proliferation and Metastasis of Glioblastoma via Targeting CADM1. Bull Cancer (2018) 105(7-8):643–51. doi: 10.1016/j.bulcan.2018.05.003
131. Aiello NM, Kang Y. Context-Dependent EMT Programs in Cancer Metastasis. J Exp Med (2019) 216(5):1016–26. doi: 10.1084/jem.20181827
132. Majc B, Sever T, Zaric M, Breznik B, Turk B, Lah TT. Epithelial-To-Mesenchymal Transition as the Driver of Changing Carcinoma and Glioblastoma Microenvironment. Biochim Biophys Acta Mol Cell Res (2020) 1867(10):118782. doi: 10.1016/j.bbamcr.2020.118782
133. Xu A, Wang X, Luo J, Zhou M, Yi R, Huang T, et al. Overexpressed P75CUX1 Promotes EMT in Glioma Infiltration by Activating Beta-Catenin. Cell Death Dis (2021) 12(2):157. doi: 10.1038/s41419-021-03424-1
134. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell (2020) 181(7):1643–60.e1617. doi: 10.1016/j.cell.2020.05.007
135. Clark AG, Vignjevic DM. Modes of Cancer Cell Invasion and the Role of the Microenvironment. Curr Opin Cell Biol (2015) 36:13–22. doi: 10.1016/j.ceb.2015.06.004
136. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct Target Ther (2020) 5(1):28. doi: 10.1038/s41392-020-0134-x
137. Ghous G, Miller D, Doll D, Tuncer T. A Rare Case of Glioblastoma With Extensive Liver Metastases. Oncol (Williston Park) (2021) 35(11):733–40. doi: 10.46883/ONC.2021.3511.0733
138. Chockley PJ, Keshamouni VG. Immunological Consequences of Epithelial-Mesenchymal Transition in Tumor Progression. J Immunol (2016) 197(3):691–8. doi: 10.4049/jimmunol.1600458
139. Rusan NM, Fagerstrom CJ, Yvon AM, Wadsworth P. Cell Cycle-Dependent Changes in Microtubule Dynamics in Living Cells Expressing Green Fluorescent Protein-Alpha Tubulin. Mol Biol Cell (2001) 12(4):971–80. doi: 10.1091/mbc.12.4.971
140. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science (2001) 292(5516):504–7. doi: 10.1126/science.1058079
141. Boon R. Metabolic Fuel for Epigenetic: Nuclear Production Meets Local Consumption. Front Genet (2021) 12:768996. doi: 10.3389/fgene.2021.768996
142. Nieborak A, Schneider R. Metabolic Intermediates - Cellular Messengers Talking to Chromatin Modifiers. Mol Metab (2018) 14:39–52. doi: 10.1016/j.molmet.2018.01.007
143. Simabuco FM, Morale MG, Pavan ICB, Morelli AP, Silva FR, Tamura RE. Correction: P53 and Metabolism: From Mechanism to Therapeutics. Oncotarget (2018) 9(74):34030. doi: 10.18632/oncotarget.26181
144. Simabuco FM, Morale MG, Pavan ICB, Morelli AP, Silva FR, Tamura RE. P53 and Metabolism: From Mechanism to Therapeutics. Oncotarget (2018) 9(34):23780–823. doi: 10.18632/oncotarget.25267
145. Kim J, Cheong JH. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells (2020) 9(7):1691. doi: 10.3390/cells9071691
146. Lagace TA, Ridgway ND. The Role of Phospholipids in the Biological Activity and Structure of the Endoplasmic Reticulum. Biochim Biophys Acta (2013) 1833(11):2499–510. doi: 10.1016/j.bbamcr.2013.05.018
147. Carnero A, Paramio JM. The PTEN/PI3K/AKT Pathway In Vivo, Cancer Mouse Models. Front Oncol (2014) 4:252. doi: 10.3389/fonc.2014.00252
148. Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, et al. Measurement of PIP3 Levels Reveals an Unexpected Role for P110beta in Early Adaptive Responses to P110alpha-Specific Inhibitors in Luminal Breast Cancer. Cancer Cell (2015) 27(1):97–108. doi: 10.1016/j.ccell.2014.11.007
149. Xie Y, Li J, Kang R, Tang D. Interplay Between Lipid Metabolism and Autophagy. Front Cell Dev Biol (2020) 8:431. doi: 10.3389/fcell.2020.00431
150. Preta G. New Insights Into Targeting Membrane Lipids for Cancer Therapy. Front Cell Dev Biol (2020) 8:571237. doi: 10.3389/fcell.2020.571237
151. Gasiorowska A, Wydrych M, Drapich P, Zadrozny M, Steczkowska M, Niewiadomski W, et al. The Biology and Pathobiology of Glutamatergic, Cholinergic, and Dopaminergic Signaling in the Aging Brain. Front Aging Neurosci (2021) 13:654931. doi: 10.3389/fnagi.2021.654931
152. Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and Cancer: Targeting Ca2+ Transport. Nat Rev Cancer (2007) 7(7):519–30. doi: 10.1038/nrc2171
153. Nicolson GL, Poste G. Tumor Cell Diversity and Host Responses in Cancer Metastasis–Part II–host Immune Responses and Therapy of Metastases. Curr Probl Cancer (1983) 7(7):1–42. doi: 10.1016/s0147-0272(83)80005-1
154. Li H, Feng Z, He ML. Lipid Metabolism Alteration Contributes to and Maintains the Properties of Cancer Stem Cells. Theranostics (2020) 10(16):7053–69. doi: 10.7150/thno.41388
155. Hubler MJ, Kennedy AJ. Role of Lipids in the Metabolism and Activation of Immune Cells. J Nutr Biochem (2016) 34:1–7. doi: 10.1016/j.jnutbio.2015.11.002
156. Maizels RM. The Multi-Faceted Roles of TGF-Beta in Regulation of Immunity to Infection. Adv Immunol (2021) 150:1–42. doi: 10.1016/bs.ai.2021.05.001
157. Shimano H, Sato R. SREBP-Regulated Lipid Metabolism: Convergent Physiology - Divergent Pathophysiology. Nat Rev Endocrinol (2017) 13(12):710–30. doi: 10.1038/nrendo.2017.91
158. Dorotea D, Koya D, Ha H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front Pharmacol (2020) 11:265. doi: 10.3389/fphar.2020.00265
159. Niyonzima N, Halvorsen B, Sporsheim B, Garred P, Aukrust P, Mollnes TE, et al. Complement Activation by Cholesterol Crystals Triggers a Subsequent Cytokine Response. Mol Immunol (2017) 84:43–50. doi: 10.1016/j.molimm.2016.09.019
160. Perucha E, Melchiotti R, Bibby JA, Wu W, Frederiksen KS, Roberts CA, et al. The Cholesterol Biosynthesis Pathway Regulates IL-10 Expression in Human Th1 Cells. Nat Commun (2019) 10(1):498. doi: 10.1038/s41467-019-08332-9
161. Bleve A, Durante B, Sica A, Consonni FM. Lipid Metabolism and Cancer Immunotherapy: Immunosuppressive Myeloid Cells at the Crossroad. Int J Mol Sci (2020) 21(16):5845. doi: 10.3390/ijms21165845
162. Fu Y, Zou T, Shen X, Nelson PJ, Li J, Wu C, et al. Lipid Metabolism in Cancer Progression and Therapeutic Strategies. MedComm (2021) 2(1):27–59. doi: 10.1002/mco2.27
163. Yang B, Yao H, Tian H, Yu Z, Guo Y, Wang Y, et al. Intratumoral Synthesis of Nano-Metalchelate for Tumor Catalytic Therapy by Ligand Field-Enhanced Coordination. Nat Commun (2021) 12(1):3393. doi: 10.1038/s41467-021-23710-y
164. Gong L, Yan Q, Zhang Y, Fang X, Liu B, Guan X. Cancer Cell Reprogramming: A Promising Therapy Converting Malignancy to Benignity. Cancer Commun (Lond) (2019) 39(1):48. doi: 10.1186/s40880-019-0393-5
165. Fan T, Sun G, Sun X, Zhao L, Zhong R, Peng Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers (Basel) (2019) 11(3):317. doi: 10.3390/cancers11030317
166. Zhu J, Zheng Y, Zhang H, Sun H. Targeting Cancer Cell Metabolism: The Combination of Metformin and 2-Deoxyglucose Regulates Apoptosis in Ovarian Cancer Cells via P38 MAPK/JNK Signaling Pathway. Am J Transl Res (2016) 8(11):4812–21.
167. Chu L, Wang A, Ni L, Yan X, Song Y, Zhao M, et al. Nose-To-Brain Delivery of Temozolomide-Loaded PLGA Nanoparticles Functionalized With Anti-EPHA3 for Glioblastoma Targeting. Drug Deliv (2018) 25(1):1634–41. doi: 10.1080/10717544.2018.1494226
168. Clemente N, Ferrara B, Gigliotti CL. Solid Lipid Nanoparticles Carrying Temozolomide for Melanoma Treatment. Preliminary In Vitro In Vivo Stud (2018) 19(2):255. doi: 10.3390/ijms19020255
169. Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, et al. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the P62-Deficient Tumor Stroma. Cell Metab (2017) 26(6):817–29.e816. doi: 10.1016/j.cmet.2017.09.001
170. Xiao Z, Dai Z, Locasale JW. Metabolic Landscape of the Tumor Microenvironment at Single Cell Resolution. Nat Commun (2019) 10(1):3763. doi: 10.1038/s41467-019-11738-0
171. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The Pancreas Cancer Microenvironment. Clin Cancer Res (2012) 18(16):4266–76. doi: 10.1158/1078-0432.CCR-11-3114
172. Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic Reprogramming of Stromal Fibroblasts Through P62-Mtorc1 Signaling Promotes Inflammation and Tumorigenesis. Cancer Cell (2014) 26(1):121–35. doi: 10.1016/j.ccr.2014.05.004
173. Aiello NM, Bajor DL, Norgard RJ, Sahmoud A, Bhagwat N, Pham MN, et al. Metastatic Progression Is Associated With Dynamic Changes in the Local Microenvironment. Nat Commun (2016) 7:12819. doi: 10.1038/ncomms12819
174. Hessmann E, Patzak MS, Klein L, Chen N, Kari V, Ramu I, et al. Fibroblast Drug Scavenging Increases Intratumoural Gemcitabine Accumulation in Murine Pancreas Cancer. Gut (2018) 67(3):497–507. doi: 10.1136/gutjnl-2016-311954
175. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res (2015) 21(15):3561–8. doi: 10.1158/1078-0432.CCR-14-1051
176. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
177. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
178. Liu F, Qiu H, Xue M, Zhang S, Zhang X, Xu J, et al. MSC-Secreted TGF-Beta Regulates Lipopolysaccharide-Stimulated Macrophage M2-Like Polarization via the Akt/FoxO1 Pathway. Stem Cell Res Ther (2019) 10(1):345. doi: 10.1186/s13287-019-1447-y
179. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-Beta Induces M2-Like Macrophage Polarization via SNAIL-Mediated Suppression of a Pro-Inflammatory Phenotype. Oncotarget (2016) 7(32):52294–306. doi: 10.18632/oncotarget.10561
180. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front Immunol (2020) 11:940. doi: 10.3389/fimmu.2020.00940
181. Salvador E, Burek M, Förster CY. Tight Junctions and the Tumor Microenvironment. Curr Pathobiol Rep (2016) 4:135–45. doi: 10.1007/s40139-016-0106-6
Keywords: glioma, glioma stem cell, metabolism, immune response, metabolism
Citation: Kanwore K, Kanwore K, Adzika GK, Abiola AA, Guo X, Kambey PA, Xia Y and Gao D (2022) Cancer Metabolism: The Role of Immune Cells Epigenetic Alteration in Tumorigenesis, Progression, and Metastasis of Glioma. Front. Immunol. 13:831636. doi: 10.3389/fimmu.2022.831636
Received: 08 December 2021; Accepted: 28 February 2022;
Published: 22 March 2022.
Edited by:
Sukh Mahendra Singh, Banaras Hindu University, IndiaReviewed by:
Bibekanand Mallick, National Institute of Technology Rourkela, IndiaKalpana Pai, Savitribai Phule Pune University, India
Copyright © 2022 Kanwore, Kanwore, Adzika, Abiola, Guo, Kambey, Xia and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianshuai Gao, Z2RzQHh6aG11LmVkdS5jbg==