 Rachel E. Lamerton1,2†
Rachel E. Lamerton1,2† Edith Marcial-Juarez1†
Edith Marcial-Juarez1† Sian E. Faustini1
Sian E. Faustini1 Marisol Perez-Toledo1
Marisol Perez-Toledo1 Margaret Goodall1
Margaret Goodall1 Siân E. Jossi1Maddy L. Newby3
Siân E. Jossi1Maddy L. Newby3 Iain Chapple4
Iain Chapple4 Thomas Dietrich4Tonny Veenith5
Thomas Dietrich4Tonny Veenith5 Adrian M. Shields1Lorraine Harper6Ian R. Henderson7
Adrian M. Shields1Lorraine Harper6Ian R. Henderson7 Julie Rayes2
Julie Rayes2 David C. Wraith1
David C. Wraith1 Steve P. Watson2
Steve P. Watson2 Max Crispin3Mark T. Drayson1
Max Crispin3Mark T. Drayson1 Alex G. Richter1
Alex G. Richter1 Adam F. Cunningham1*
Adam F. Cunningham1*- 1Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham, United Kingdom
- 2Institute of Cardiovascular Sciences, University of Birmingham, Birmingham, United Kingdom
- 3School of Biological Sciences, University of Southampton, Southampton, United Kingdom
- 4Periodontal Research Group, School of Dentistry, Institute of Clinical Sciences, University of Birmingham, and Birmingham Community Healthcare National Health Service Trust, Birmingham, United Kingdom
- 5Department of Critical Care Medicine, University Hospitals Birmingham National Health Service (NHS) Trust, Birmingham, United Kingdom
- 6Institute of Applied Health Research, University of Birmingham, Birmingham, United Kingdom
- 7Institute for Molecular Bioscience, University of Queensland, St Lucia, QLD, Australia
Antibodies specific for the spike glycoprotein (S) and nucleocapsid (N) SARS-CoV-2 proteins are typically present during severe COVID-19, and induced to S after vaccination. The binding of viral antigens by antibody can initiate the classical complement pathway. Since complement could play pathological or protective roles at distinct times during SARS-CoV-2 infection we determined levels of antibody-dependent complement activation along the complement cascade. Here, we used an ELISA assay to assess complement protein binding (C1q) and the deposition of C4b, C3b, and C5b to S and N antigens in the presence of antibodies to SARS-CoV-2 from different test groups: non-infected, single and double vaccinees, non-hospitalised convalescent (NHC) COVID-19 patients and convalescent hospitalised (ITU-CONV) COVID-19 patients. C1q binding correlates strongly with antibody responses, especially IgG1 levels. However, detection of downstream complement components, C4b, C3b and C5b shows some variability associated with the subject group from whom the sera were obtained. In the ITU-CONV, detection of C3b-C5b to S was observed consistently, but this was not the case in the NHC group. This is in contrast to responses to N, where median levels of complement deposition did not differ between the NHC and ITU-CONV groups. Moreover, for S but not N, downstream complement components were only detected in sera with higher IgG1 levels. Therefore, the classical pathway is activated by antibodies to multiple SARS-CoV-2 antigens, but the downstream effects of this activation may differ depending the disease status of the subject and on the specific antigen targeted.
Introduction
Infection with SARS-CoV-2, the causative agent of COVID-19, results in a spectrum of clinical presentations ranging from asymptomatic infections to severe disease and death. Although some factors that can predict risk of severe disease are known, such as obesity or age, it is clear that other host factors, including immune status, also contribute (1–3). Thus, it is likely that COVID-19 represents a collection of syndromes, caused by one pathogen, where disease severity is influenced by host and pathogen factors.
Two antigens that are common targets of the immune response to SARS-CoV-2 are the spike (S) glycoprotein, which is essential for both binding and entry into host cells, and the nucleocapsid (N) protein, involved in packaging the genomic material (4). Antibodies to these antigens are induced after infection, and antibodies to S glycoprotein can be protective (5–7). Indeed, the S glycoprotein is the sole SARS-CoV-2 viral antigen targeted by all current licensed vaccines (8). After natural infection of non-vaccinated individuals, although the appearance of antibodies to both of these antigens can occur early in mild disease, the presence of such antibodies is usually well-established at times severe disease develops. This means that substantial levels of viral antigen may still be present within the host for these antibodies to bind (9). In contrast, in most vaccinated individuals who have not previously been infected, high levels of antibodies to S are present when they subsequently encounter the pathogen. This means that there can be circumstances when: i) there are concomitant high-levels of antibodies to S and N as well as relatively high-levels of antigen (antibodies induced during infection) and ii) high-levels of antibodies to S and relatively low-levels of antigen (infection of vaccinated, previously naïve individuals).
After antigen binding by antibody, complement activation can occur through the classical pathway (10). This cascade requires C1q binding to antibody and the generation of a C3 convertase derived in part from C4, through the production of C4b. This results in the cleavage of C3 and C5, with C3b and C5b forming a complex proximal to the site of antibody binding. The activation of the complement cascade may have positive or negative effects for the host associated with the timing of its activation and possibly the different pathways involved (11–15).
To improve our understanding of the relationship between SARS-CoV-2-specific antibodies and complement activation, we developed a solid phase C1q-binding assay and C4b, C3b and C5b complement deposition assays using S and N proteins from the SARS-CoV-2 Wuhan strain. These studies identified differences in antibody-associated complement activation that were associated with the stage of infection in the host.
Methods
Ethics and Patient Samples
Sera were obtained from distinct groups of subjects from well-validated cohorts that are described below (3, 5). Group 1: Non-vaccinated individuals without any reported COVID infection (NEG). Sera were obtained from subjects in May 2020, prior to widespread PCR testing and before the introduction of vaccines against SARS-CoV-2 infection. Sera were screened using a clinically validated, CE marked, ELISA assay that measures the IgG, IgA and IgM (IgGAM) response to the S glycoprotein (16, 17) (manufactured by The Binding Site (TBS; product code: MK654), Birmingham). This assay, described below in the section on detecting antibodies to S and N, has been clinically validated and reported to have a sensitivity of 98.6% (95% CI, 92.6-100.0%) and a specificity of 98.3% (95% CI, 96.4-99.4%) (16). Group 2: Individuals without evidence of infection (as determined by an absence of anti-N antibodies), vaccinated 28-35 days previously with BNT162b2 vaccine (VACC). Group 3: Individuals without evidence of infection (as determined by an absence of anti-N antibodies) who had received their second dose of BNT162b2 vaccine at least 28 days previously (DOUBLE VACC). Group 4: These sera were obtained in May 2020 from a cohort of healthcare workers from the University Hospitals Birmingham Foundation Trust, who had previously self-isolated a minimum of 28 days previously because they experienced symptoms suggestive of COVID-19, and had not been hospitalized for any of these symptoms. In May 2020 widespread PCR testing was not available, and thus most of these samples were not from individuals with prior confirmed PCR tests. Since the only predefined exclusion criteria was participation in existing SARS-CoV-2 vaccine trial or current COVID-19 symptomatology and the time was prior to the introduction of vaccines, anti-S antibodies could be used as a reliable surrogate of previous infection. Anti-S IgGAM were determined using the clinically validated anti-S glycoprotein ELISA described above (non-hospitalised convalescents, NHC). Group 5: Non-vaccinated convalescent, PCR confirmed SARS-CoV-2 infection patients who required ITU treatment, samples taken a minimum of 4 months after ITU discharge (ITU-CONV).
Ethical approval for obtaining samples for groups 1 -4 was provided by the London – Camden and Kings Cross Research Ethics Committee reference 20/HRA/1817. Ethical approval for obtaining samples for group 5 was provided by the North West ethics committee, Preston CIA UPH IRAS approval reference REC 20\NW\0240.
Antigens Used in This Study
To generate the spike glycoprotein used in this study, HEK293F cells were transiently transfected with a pα-H plasmid containing the near full-length sequence for the Wuhan SARS-CoV-2 spike (GenBank: MN908947). The spike glycoprotein used here contains 1208 amino acids and includes all the S1 and most of the S2 domain (18, 19). The protein has been modified so that there are an additional four prolines present in addition to the two which are normally expressed (2P) to stabilize recombinant spike (18). The 6 prolines are present at positions 817, 892, 899, 942, 986 and 987 (18). This so-called HexaPro spike glycoprotein expresses as a metastable recombinant SARS-CoV-2 prefusion ectodomain. Extensive comparisons between the native, 2P and HexaPro spike glycoproteins demonstrate that they have comparable native-like protein architecture, have similar antigenic properties including the induction of neutralizing antibodies (20–22) and have similar glycosylation profiles (18, 19, 23, 24).
The HEK293F cells were cultured in Freestyle 293 Expression medium (Fisher Scientific) and maintained at a density of 0.2 x 106 cells/mL at 37°C, 8% CO2 and 125 rpm shaking. Prior to transfection, two solutions of 25 mL Opti-MEM (Fisher Scientific) medium were prepared. The expression plasmid encoding SARS-CoV-2 HexaPro was added to the first solution to give a final concentration of 310 µg/L. To the other solution, 1 mg/mL pH7 polyethylenimine (PEI) max reagent was added to generate a ratio of 3:1 PEI max:plasmid DNA. Both solutions were combined and incubated at room temperature for 30 minutes. Cells were transfected at a density of 1 x 106 cells/mL and incubated for 7 days at 37°C, 8% CO2 and 125 rpm shaking.
Cells were centrifuged at 3041g for 30 minutes at 4°C and supernatant was applied to a 500 mL Stericup-HV sterile vacuum filtration system (Merck) with a pore size of 0.22 µm. Purification of HexaPro S protein was undertaken using an ÄKTA Pure system (Cytiva). A 5 mL HisTrap Excel column (Cytiva) charged with Ni(II) was equilibrated using 10 column volumes (CV) of wash buffer (50 mM Na2PO4, 300 mM NaCl, pH 7). Supernatant was then loaded onto the column at a flow rate of 5 mL/min and washed with 10 CV of washing buffer containing 50 mM imidazole. Protein was eluted from the column in 3 CV of elution buffer (300 mM imidazole in washing buffer) and buffer exchanged to phosphate buffered saline (PBS) and concentrated using a Vivaspin column (MWCO 100 kDa) (Cytiva).
The nickel purified eluate was concentrated to 1 mL in PBS and injected into a Superdex 200 pg 16/600 column (Cytiva) to further purify trimeric S protein using size exclusion chromatography (SEC). The column was washed with PBS at 1 mL/min for 2 hours where fractions corresponding to the correct peak on the size exclusion chromatogram were collected and concentrated to ~1 mL as above.
Nucleocapsid was generated as a recombinant protein from E. coli by the Protein Expression Facility at the University of Birmingham (17).
Detection of Antibodies Specific to S and N
Antibody ELISAs were carried out as previously described (17), with 50 µl per dilution used. In brief, 96 well high-binding plates (Corning) were coated with 0.1 µg S or N protein in PBS and incubated overnight at 4°C. PBS-0.1% Tween 20 was used to wash plates 3 times, and between all subsequent steps. Plates were blocked with 2% (w/v) BSA in PBS-0.1% (v/v) Tween 20 for 1 hr at room temperature (RT). Serum was diluted 1:40 and incubated for 1 hr at RT. HRP-conjugated anti-human secondary antibodies were added for 1 hr at RT. For combined anti-IgG, IgA and IgM (IgGAM) the antibodies came in a combined pre-diluted form from The Binding Site (EACONJ654). The individual constituent HRP-labelled secondary antibodies used in this are polyclonal rabbit anti-human IgG (1:16,000), polyclonal rabbit anti-human IgA (1:2000) and polyclonal rabbit anti-human IgM (1:8000). Individual immunoglobulin isotypes were detected using HRP-conjugated monoclonal antibodies: mouse anti-human IgM (clone AF6, 1:2000), mouse anti-human IgG1 (clone MG6.41, 1:3000), mouse anti-human IgG3 (clone MG5.161, 1:1000). All monoclonal antibodies were produced at the University of Birmingham. Plates were developed for up to 20 minutes using 100 µl TMB Core (Bio-Rad) and the reaction was stopped with 50µl 0.2M H2SO4. Optical density (OD) was read at 450 nm using a SpectraMax ABS Plus plate reader.
Solid Phase C1q-Binding Assay
Plates were coated as above. Plates were washed three times with PBS-0.1% Tween 20 – this wash step was carried out between all subsequent steps. Blocking was carried out for 1 hr at RT with 2% BSA in PBS-0.1% Tween 20. Test serum was heat inactivated at 56°C for 30 minutes, before being diluted 1 in 5 with 2% BSA supplemented with 5 mM calcium chloride and 5 mM magnesium chloride. 50 µl was added to the antigen-coated plate and incubated for 1hr at 37°C. After washing, 50 µl COVID negative normal human serum (same source used throughout all assays, containing no detectable S or N specific antibodies as measured by IgGAM ELISA) at a dilution of 1:40 (in 2% BSA plus 5 mM calcium chloride and 5 mM magnesium chloride) was added to each well for 1hr at RT. 100µl of rabbit anti-C1q FITC antibody (Invitrogen PA5-16601) at a 1:200 dilution in PBS-0.1% Tween 20 was added and incubated at 37°C for 1 hr. HRP conjugated swine anti-rabbit (Dako P0399) at a 1:2000 dilution was then incubated for 1 hr. The assay was amplified using the Perkin Elmer ELAST amplification kit as per manufacturer’s instructions, with an optimised dilution of streptavidin, 1:800, incubated for 20 minutes. Plates were developed using 100 µl TMB Core (Bio-Rad) for 10 minutes, before being stopped with 50 µl 0.2MH2SO4. OD was measured as described above.
C4b, C3b and C5b Complement Deposition Assay
Microtiter plates were coated and washed as described above and blocked with Starting Block (ThermoFisher) for 10 min. Test serum was heat inactivated at 56°C for 30 minutes, before being diluted 1 in 5 with Starting Block supplemented with 5 mM calcium chloride and 5 mM magnesium chloride. 50 µl was added to the antigen-coated plate and incubated for 1hr at 37°C. After washing, 50 µl COVID negative normal human serum (same source used throughout all assays, containing no detectable S or N specific antibodies as measured by IgGAM ELISA) at a dilution of 1:40 (in 2% Starting Block plus 5 mM calcium chloride and 5 mM magnesium chloride) was added to each well for 1 hr at 37°C. The following anti-human monoclonal complement antibodies (100ul, diluted in PBS-0.1% Tween 20) were added and incubated at 37°C for 1 hr: mouse anti-C4b, 1:22,500 (Invitrogen, LF-MA0198); mouse anti-C3b, 1:10,000 (Invitrogen MA1-70053); mouse anti-C5b, 1:10,000 (Invitrogen DIA 011-01-02). HRP conjugated goat anti-mouse at a 1:4000 (Southern Biotech 1010-05) was then incubated at RT for 1 h. Plates were developed and read as described above.
Statistics
Statistical analysis was carried out using GraphPad Prism 9.0. Kruskal-Wallis followed by Dunn’s post-hoc test for multiple groups was used to calculate p values. Statistical significance was accepted at P<0.05. Spearman correlation was carried out on the appropriate data sets.
Results
Anti-S, but Not Anti-N Antibody Responses Differ Between NHC and ITU-CONV Patients
Total IgGAM antibody responses to trimeric S and N were assessed in five different groups: individuals without any reported COVID-19 infection (NEG); post first BNT162b2 vaccine, infection-naïve individuals (VACC); post second BNT162b2 vaccine, infection-naïve individuals (DOUBLE VACC); convalescing non-hospitalised patients (NHC) and convalescing patients who had been hospitalised and required ITU treatment (ITU-CONV). The VACC, DOUBLE VACC, NHC, ITU-CONV groups all had significantly higher anti-S glycoprotein IgGAM responses than the NEG group (Figure 1A), whereas IgGAM levels against N in the two convalescent groups were higher than the NEG, VACC and DOUBLE VACC groups (Figure 1B). There were no significant differences in the anti-N responses between the NEG group and the VACC and DOUBLE VACC groups, consistent with individuals within these groups not having had prior SARS-CoV-2 infections. Similar results were observed when specific IgG1 responses, an IgG isotype efficient at fixing complement, were assessed (Figure 1C). No differences in anti-S IgGAM and IgG1 antibody responses were observed between the VACC and patient groups. Anti-S IgGAM and IgG1 responses were higher in the ITU-CONV group compared to the NHC group, but no differences were observed for anti-N responses in these two groups (Figure 1D). Modest IgM and IgG3 responses to S and N were detected in some individuals (Supplementary Figures 1A, B and Supplementary c 1).
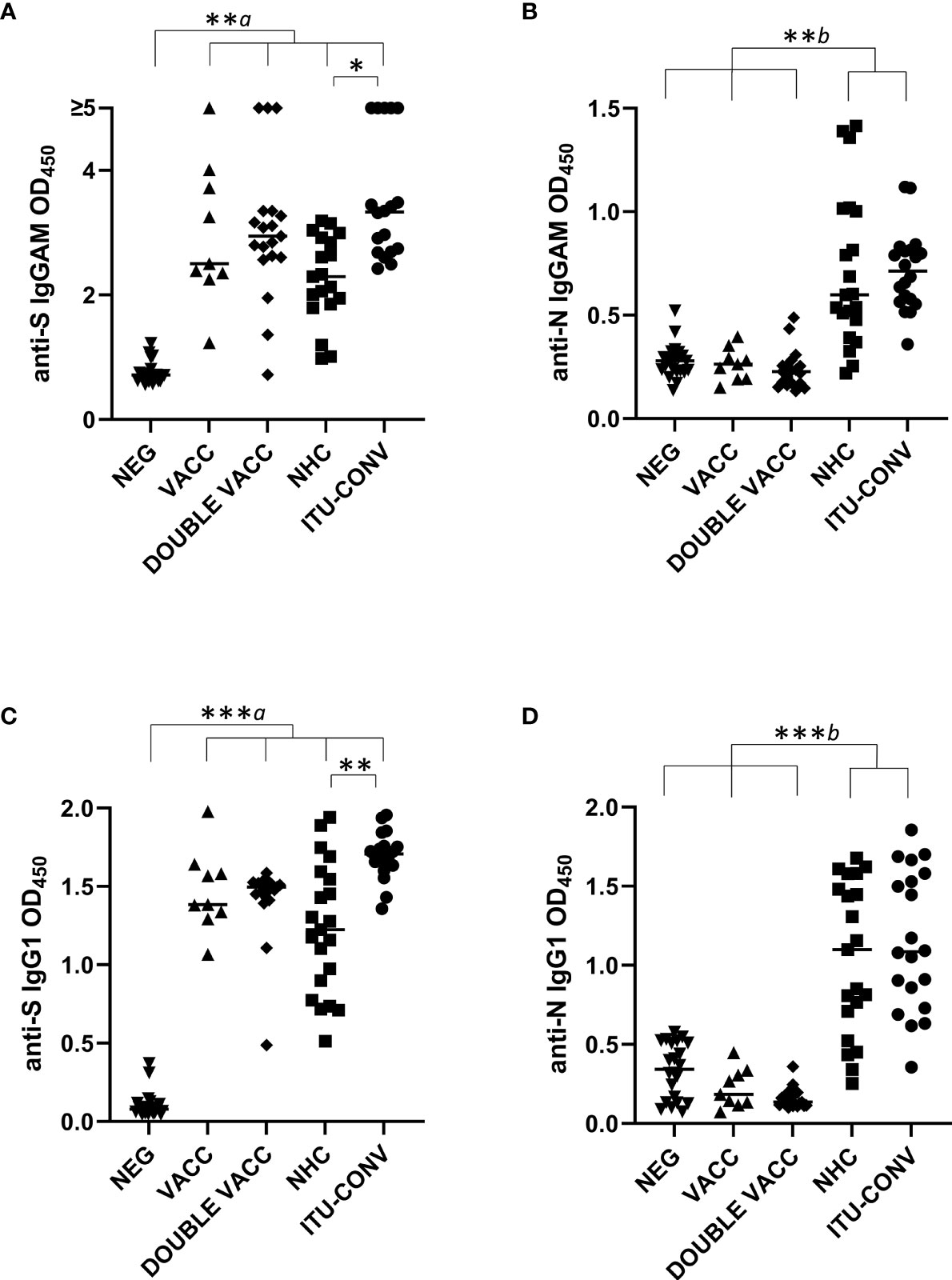
Figure 1 Anti-S, but not anti-N antibody responses differ between NHC and ITU-CONV patients. Using an ELISA against 0.1ug S (A, C) or N (B, D) with HRP-conjugated IgGAM or IgG1 secondary antibodies, GAM and IgG1 levels were assessed in the following subject groups: COVID-19 negatives (NEG, n ≥20), COVID-19 naïve one month post first BNT162b2 vaccine (VACC, n = 9), COVID-19 naïve one month post second BNT162b2 vaccine (DOUBLE VACC, n = 19), COVID-19 positive non-hospitalised convalescents (NHC, n ≥ 19) and COVID-19 positive convalescents who required ITU treatment (ITU-CONV, n ≥ 18) Kruskal-Wallis with Dunn’s multiple comparisons test was used to test significance. a indicates that the four groups bracketed (VACC, DOUBLE VACC, NHC and ITU-CONV) were individually significantly different to the NEG group; b indicates that NHC and ITU-CONV are independently significantly different to NEG, VACC and DOUBLE VACC. ***p < 0.001, **p < 0.01, *p < 0.05. Bars represent median values for each group.
C1q Binding in vitro Correlates With Levels of S- and N-Specific IgGAM and IgG1 Antibodies
To determine if the complement protein C1q can bind to SARS-CoV-2-specific immunoglobulins in vitro we developed a solid phase C1q-binding assay. In these antigen-specific assays, the test serum from COVID-19 patients or vaccinees is heat-inactivated and standardisation of complement is provided by using sera from non-infected, non-vaccinated subjects. Results from this assay showed that C1q binding mirrored IgG1 levels for both S and N antigens, with the lowest signals for S seen in the NEG group (Figure 2A), and for N in the NEG, VACC and DOUBLE VACC groups (Figure 2B). The lack of C1q binding detected for N when sera from the VACC and DOUBLE VACC groups were tested is expected and consistent with a lack of prior infection by SARS-CoV-2 in these groups (Figure 2B), and we have included these two groups in our downstream analyses of the response to N for their value as control groups. No difference in C1q binding was observed between sera from the two convalescent groups (Figures 2A, B). Plotting IgG1 responses against C1q responses shows a positive correlation between the amount of IgG1 antibody and the amount of C1q binding detected (Figures 2C, D). Therefore, C1q binding reflects the serological response to both antigens.
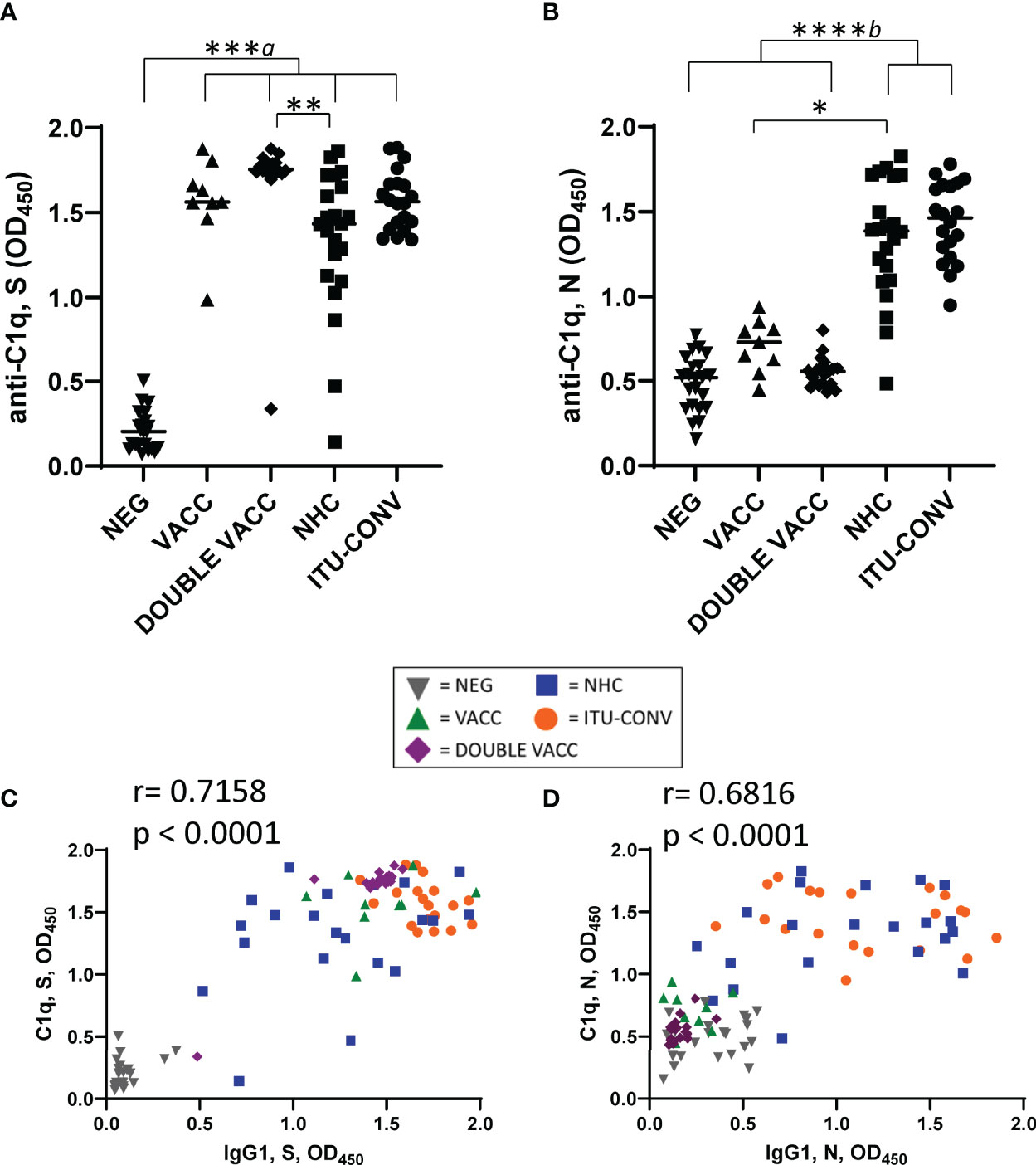
Figure 2 C1q binding to S and N correlates with IgG1 responses. Using an ELISA against 0.1ug S (A) or N (B) with an anti-C1q secondary antibody, followed by an HRP-conjugated tertiary, and the ELAST amplification kit, C1q binding was measured. Correlations of IgG1 OD and C1q OD against S (C) and N (D). NEG, n = 22. VACC, n = 9. DOUBLE VACC n = 19. NHC, n ≥ 21. ITU-CONV n = 20. Kruskal-Wallis with Dunn’s multiple comparisons test was used. a indicates that the four groups bracketed were individually significantly different to the NEG group; b indicates that NHC and ITU-CONV are independently significantly different to NEG and DOUBLE VACC. ****p < 0.0001, ***p <0.001 **p < 0.01, *p <0.05. Bars represent median values for each group. Correlations were determined using the Spearman’s rank correlation test (r and p values presented).
Deposition of C4b, C3b and C5b Varies Dependent Upon Antigen Tested and Subject Group
To determine whether C1q binding reflected downstream activation of the complement cascade, we examined whether complement breakdown products could be detected. Deposition of C4b, a major component of the classical pathway C3 convertase, and the effector molecules C3b and C5b were assessed. In the absence of S or N-specific antibodies, C4b, C3b and C5b breakdown products were not detected, but they were detected in the presence of specific antibodies, indicating involvement of the classical complement pathway (Figure 3). When S was used as the assay antigen, the highest median levels of C4b, C3b and C5b deposition detected were in the DOUBLE VACC and ITU-CONV groups (Figure 3A), whereas the VACC and NHC groups showed similar lower levels of downstream activation. The median levels of C4b, C3b and C5b deposition detected when N was used as the assay antigen were similar between the NHC and ITU-CONV groups (Figure 3B). In contrast, for the NEG, VACC and DOUBLE VACC groups, where anti-N antibody responses and C1q binding were not detected, there was no downstream activation of the complement cascade. Therefore, in this assay differences in complement activation by antibody can be detected dependent upon what patient group and antigen were examined.
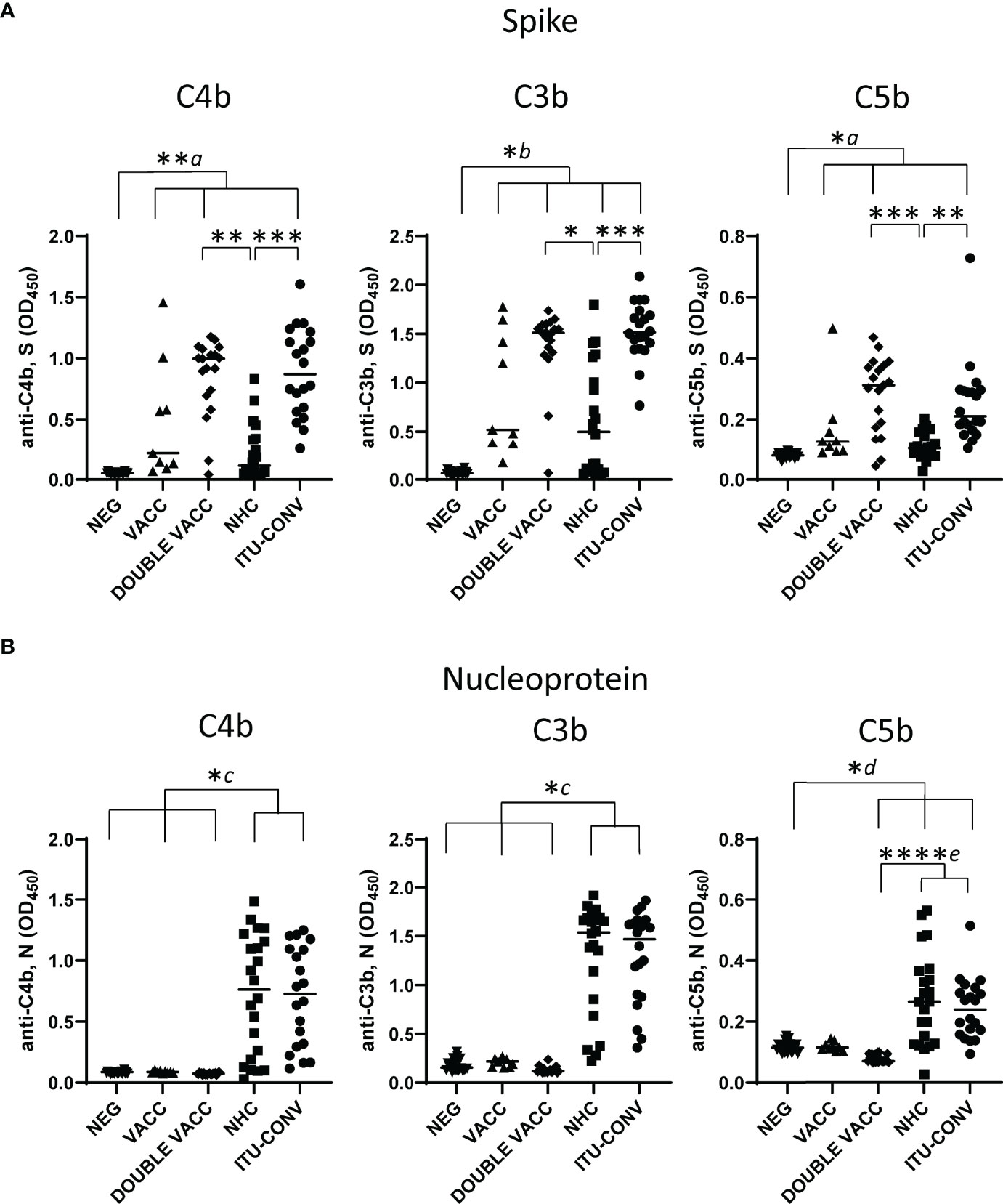
Figure 3 C4b, C3b and C5b show antigen and subject status-dependent variability. Using an ELISA against 0.1ug S (A) or N (B) with either anti-C4b, C3b or C5b secondary antibody, followed by an HRP-conjugated tertiary, downstream complement binding was measured. NEG, n = 22. VACC, n = 9. DOUBLE VACC, n = 19. NHC, n = 22. ITU-CONV n = 20. Kruskal-Wallis with Dunn’s multiple comparisons test was used. a and b indicate that the groups bracketed were individually significantly different to the NEG group; c indicates that NHC and ITU-CONV are independently significantly different to NEG, VACC and DOUBLE VACC; d indicates that DOUBLE VACC, NHC and ITU-CONV are all significantly different to NEG; e indicates that NHC and ITU-CONV are both significantly different to DOUBLE VACC. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05. Bars represent median values for each group.
Downstream Complement Activation Associates With Threshold IgG1 Responses
In contrast to the linear association between anti-S IgG1 and C1q detection, there was a non-linear association between IgG1 and C4b, where C4b was only detectable beyond a threshold level of IgG1. A correlation between C4b and IgG1 to N was also observed, as was a threshold response, although the threshold response was less clear for N than for S (Figure 4A). Similar threshold responses for IgG1 to S and N were also observed if C3b or C5b were plotted against IgG1 (Supplementary Figures 2A, B). When correlations were performed for C1q vs C4b (Figure 4B), C4b vs C3b (Figure 4C) and C3b vs C5b (Figure 4D) for both antigens, then clear correlations were observed. This suggests that in this assay threshold levels of IgG1 are needed to activate downstream complement components for S and N.
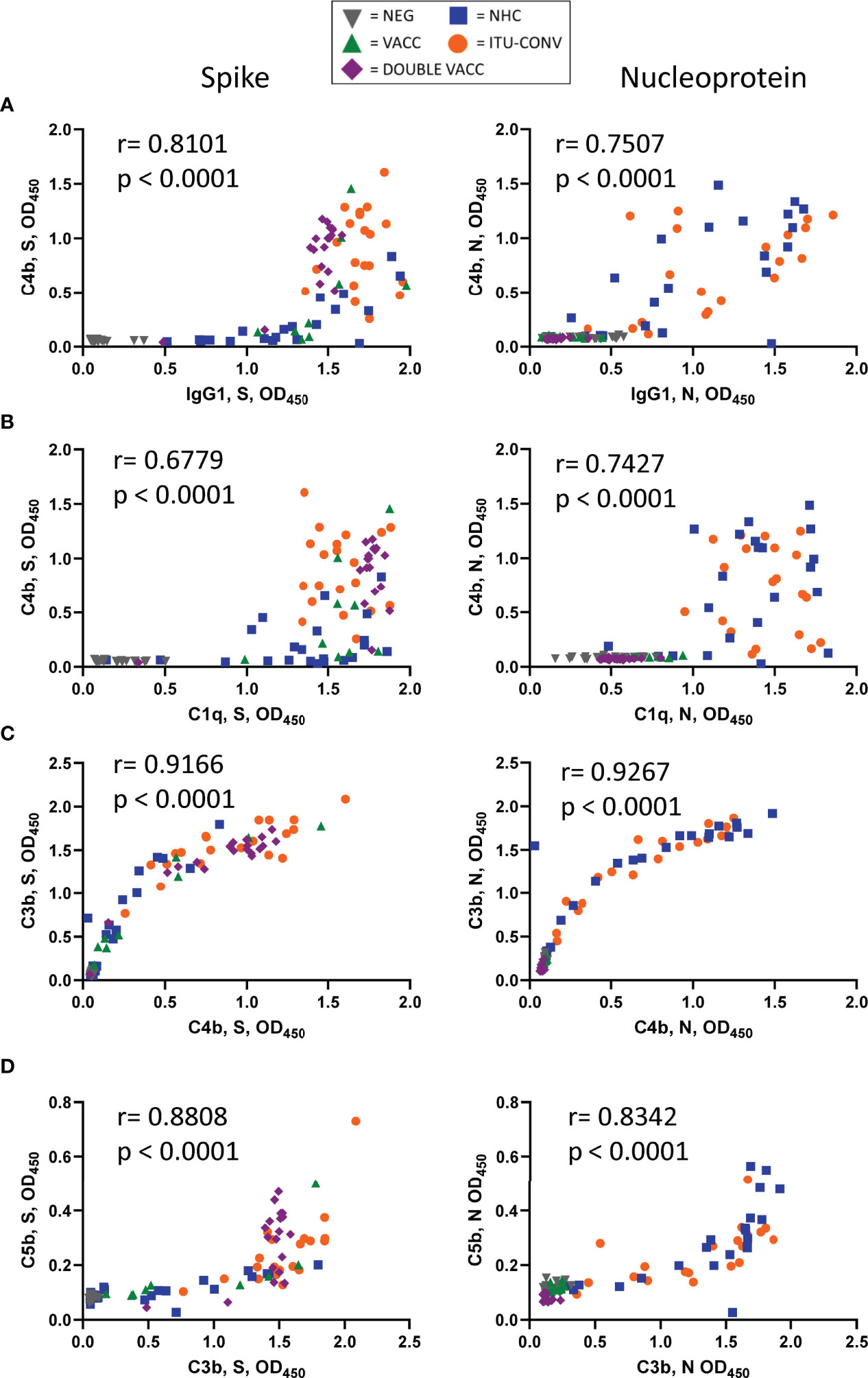
Figure 4 Threshold levels of IgG1 associate with the detection of C4b. (A) XY scatter plots for C4b responses and IgG1 responses, (B) C4b and C1q, (C) C3b and C4b and (D) C5b and C3b against S (left) or N (right). XY pairs, n ≥ 91, each point represents one serum. Correlations were determined using the Spearman's rank correlation test (r and p values presented).
IgG1 Correlates With Complement Activation in Sera With the Lowest IgM Levels Detected
IgM is the most efficient antibody isotype for fixing and activating complement (25) yet most sera tested had only low levels of anti-S or anti-N IgM (Supplementary Figure 1A). This was expected due to the convalescent nature of our infected groups, and the minimum one month period after vaccination that sera were obtained from our vaccinated groups. This suggested IgG1 or IgG3 might compensate for IgM when IgM is not present at high levels. Therefore, we examined the level of complement fixation (C1q binding) and activation (C3b deposition) associated with antigen-specific IgM and how this correlated with antigen-specific IgG1 and IgG3 levels (Figure 5 and Supplementary Figure 3). To do this the IgM to S or N was correlated with C1q and C3b. There were modest correlations between IgM against S and C1q and C3b (r = 0.4613, p < 0.0001 and r = 0.4753, p < 0.0001 respectively; Figures 5A, C). To N protein, no correlation was identified, although only a few sera had elevated levels of IgM (IgM vs C1q, r = 0.1661, p = 0.1155 and IgM vs C3b r = 0.09955, p = 0.3478; Figures 5B, D) and if the 2 vaccinated groups (lacking anti-N responses) were excluded from the analysis for N, then the correlation became stronger (IgM vs C1q, r = 0.3019. p = 0.0162; IgM vs C3b, r = 0.03509, p = 0.0048). These results are consistent with IgM being able to activate complement when present, but not always being present at sufficiently high levels to do so. C1q binding and C3b deposition was detected for many sera in which IgM responses to S or N were low. We hypothesized that sera which exhibited the lowest IgM responses, but which could still fix and activate complement had higher levels of IgG1 or IgG3. We therefore divided the sera with the lower IgM levels into 2 groups, one group in which we detected the highest C1q or C3b levels (termed IgMloC1qhi and IgMloC3bhi respectively) and another group where we detected the lowest C1q and C3b levels (termed IgMloC1qlo and IgMloC3blo respectively). The C1q or C3b response was then plotted against the IgG1 or IgG3 response. In the IgMloC1qhi and IgMloC3bhi groups higher levels of IgG1 were detected than in the IgMloC1qlo and IgMloC3blo groups (Figures 5A–D and Supplementary Figure 3), and this difference was observed for both S and N. IgG3 levels were also higher in the IgMloC1qhi and IgMloC3bhi groups than in the IgMloC1qlo and IgMloC3blo groups (Figures 5A–D and Supplementary Figure 3). Therefore, complement fixation and activation is observed in the presence of IgG1 or IgG3 when IgM levels are low.
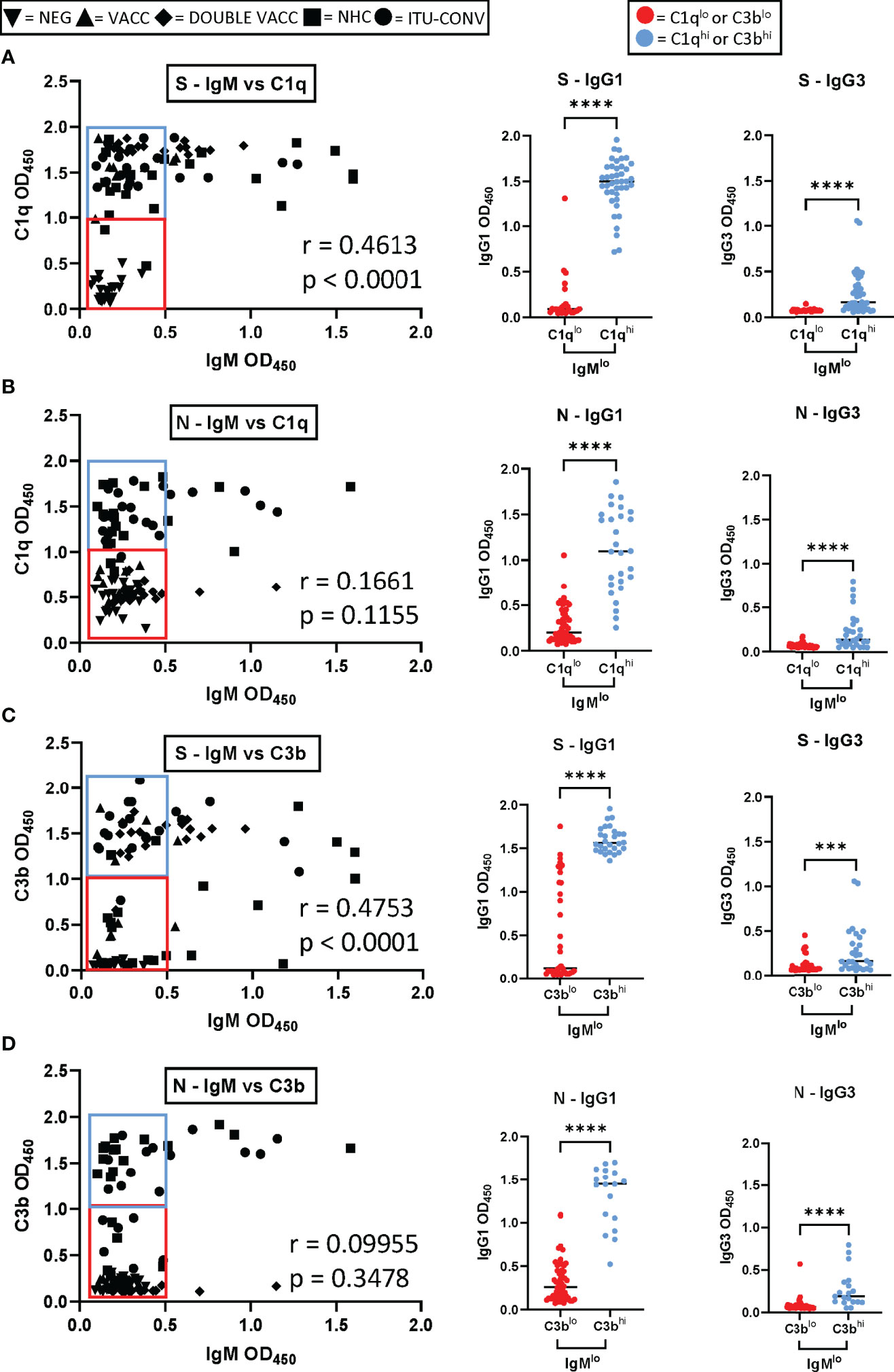
Figure 5 Complement activation by sera with low levels of IgM to S and N. (A) Left hand panel shows the IgM levels plotted against levels of C1q for S (left hand panel; results for 91 sera presented from NEG, VACC, DOUBLE VACC, NHC and ITU-CONV donors). The IgG1 levels for sera presented in the blue box (IgMloC1qhi) or red box (IgMloC1qlo) are shown in the central graph and for IgG3 in the right hand graph (n ≥ 18 for each group). Each coloured dot represents one sera from the corresponding coloured box. (B) As for a, but the anti-N response is presented. (C, D), as for (A, B) respectively but with the results for C3b binding shown rather than C1q. Correlations were determined using the Spearman’s rank correlation test (r and p values presented). Mann-Whitney was used to test significance in the IgG1 and IgG3 column graphs, where ****p < 0.0001 and ***p < 0.001. Bars represent median values for each group.
Discussion
Here we show (i) that antibodies to S and N can activate the classical complement cascade and (ii) the level of activation of the cascade detected vary dependent upon the subject group examined and the antigen used in the test. Therefore, antibodies to two different antigens within the same pathogen can activate the complement cascade in vitro, albeit at levels that depend on the severity of COVID-19. In the presence of specific antibodies to both S and N, similar levels of C1q binding were observed and the greatest variability was detected downstream of C1q binding. In contrast, in the NEG, VACC and DOUBLE VACC groups, where antibodies to N were not detected, activation of the complement cascade was not detected.
In this study, antibody needed to bind antigen in order to detect complement activation. Although threshold IgG1 levels were associated with activation of the complement cascade, the spread in the complement responses observed to spike and nucleoprotein did show some differences (e.g. Figure 3). Such observations may reflect the assay or other factors such as how antibodies themselves interact with these antigens or intrinsic differences in the antigens themselves. For instance, S is a trimeric protein and the approximately 420 kDa trimer is substantially larger than N, which is approximately 46 kDa. This difference may influence how IgG binds to the antigen and affects complement activation. Moreover, the trimeric structure of S means identical epitopes can be juxtaposed to one another in a highly defined manner (18, 26, 27) enabling multiple Fab to bind their target epitopes and promoting cross-linking of antigen. Other factors may also contribute to this process, such as which epitopes are targeted within the S protein. For instance, large-scale changes in the glycosylation pattern of S proteins has a surprisingly limited impact on the level of antibody binding by patient sera as determined by ELISA (19). One interpretation of this is that there are only a limited number of antibody sites available on each S glycoprotein for antibodies to bind, presumably in part because of a combination of relatively few (proteinaceous) epitopes targeted and the steric effects of antibodies themselves. This is similar to what we have observed for epitope recognition by antibodies targeting cell surface proteins on the surface of Salmonella enterica serovar Typhimurium (28). Moreover, studies examining antibody responses to the S glycoprotein of the Omicron variant in sera from vaccinated individuals suggest that amino acid changes in the relatively hypoglycosylated RBD (23, 29, 30) region can have dramatic effects on antibody binding (31) and so may be more likely to affect the level of complement activation. Other factors that may influence complement activation include how antigen is distributed on the surface of the virus (for S) and elsewhere in the host after natural infection. In the context of this assay, technical factors such as how antigen binds to the plate may influence the results observed. Furthermore, the trimeric vs monomeric nature of the antigens tested, the maintenance of native conformation, and the level of antigen denaturing may all influence the results observed using these assays. This highlights the need to develop multiple approaches to study antibody-antigen interactions in vivo to contextualise the in vitro results presented here.
IgM is the most efficient antibody isotype for activating the complement cascade (25). Nevertheless, as seen in many studies, the sera used in this study from individuals’ convalescent post-vaccination or infection, had modest or background levels of antigen-specific IgM detected. Those IgMlo sera that could bind complement had higher IgG1/3 levels than those IgMlo sera that could not activate the complement cascade. Therefore, the results from this study are possibly most relevant for understanding the relationship between IgG isotypes and complement activation, whereas the more pronounced complement-fixing properties of IgM may modulate the strengths of the responses observed. It is likely that in the presence of high levels of IgM, such as during the acute phase of a primary infection, there would be enhanced levels of complement activation. Nevertheless, the activation of the complement cascade in the absence of IgM may be possible and important for the pathophysiology of disease in other scenarios, as well as in the convalescent subjects we have presented here. For instance, many children with severe complications from COVID-19 (Multisystem Inflammatory Syndrome in Children (MIS-C)/paediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 infection (PIMS-TS)) present with disease at times when they have low/negligible levels of IgM, yet high levels of IgG, especially IgG1 and IgG3 (32). This correlates with a strong signature associated with complement activation (33). Collectively, this likely means that IgM can play important roles in activating the complement cascade, but that it is not essential.
The results from these studies lead to further hypotheses to test. For instance, it is known that ITU subjects can have greater activation of the complement cascade (11, 12), and this could be compounding their disease. Indeed, targeting both C3 and C5 within the complement cascade as a way to treat COVID-19 shows promise (34–37). The availability of assays such as those described here will help increase understanding of how these inhibitors act to interfere with the complement cascade in the presence of antibody to different SARS-CoV-2 proteins. Alternatively, since all these subjects survived severe COVID-19 infections it could be hypothesised that the activation of complement is associated with a beneficial outcome. As we did not have sera from individuals who died this is not testable here. One caveat in this argument is that exogenous sources of complement in the form of sera from non-infected individuals were used in these studies and that patients own sera may differ in potency, or polymorphisms in complement components themselves could influence the consequences of complement activation (38, 39). This was not assessed here as the focus was on antibody-mediated activation of complement where we have attempted to standardize the amount of complement available, particularly for the downstream complement components. Additionally, these studies were performed using sera from patients who were infected or immunized weeks previously and the antibodies present may not reflect the antibodies present at the time of infection. Certainly, it could be expected that the affinity of the antibodies would increase over time. One striking feature was the variability in the anti-C4b/C3b response detected in the VACC group. It is unclear why this is the case, but it could simply be that there is variability within the wider population in the ability to activate complement downstream. Ultimately, the results generated by these assays are trying to recapitulate the complexity of the complement cascade in vivo, which is considerably more complex than what is observed or testable in vitro. For these reasons, it is important to interpret the outcomes from these assays as the starting point for further investigations. For instance, in these assays potential inhibition of activation is not attempted. Neither is the separation of competition for epitopes between antibody isotypes that are associated with greater complement-activating potential (e.g. IgM or IgG1) than others (e.g. IgA), or controlling for the absolute levels of each isotype present. Nevertheless, the differences observed may be indicative of differences that can occur in the patients in different tissues or organs or at different stages of infection.
The complement cascade has been reported to be activated through multiple pathways after SARS-CoV-2 infection (40–43). Amongst these, the engagement of the classical pathway is distinct to the non-antibody-dependent pathways due to the potential multiple roles antibody can play during the course of infection. If induced whilst an infection is ongoing, then the activation of the complement cascade by antibody could worsen disease, particularly as antibody responses become detectable concomitant with risk of severe disease. This could happen either through enhanced inflammation (41), such as observed during acute respiratory distress syndrome, or through enhancing the complications of thrombosis and coagulopathy after infection (44, 45). Moreover, antibodies are induced to multiple SARS-CoV-2 antigens and as we show, antibodies to S and N proteins have the capacity to activate complement in vitro. At the sites where complement is activated during primary infection, there could be additional increases in the anaphylatoxins C3a, C4a and C5a, as has been reported (11, 12, 15, 46). These could augment local inflammation through promoting the recruitment of more neutrophils and monocytes leading to tissue damage and worsen disease in patients. In addition, in humans challenged with influenza virus increased levels of C3a and C5a were detected in the upper respiratory tract (47), intriguingly most often during the recovery phase rather than the acute infection phase, presumably concomitant with when antibody responses are established. Other effects of complement activation may include the destruction of host cells due to the formation of the membrane attack complex (MAC) and the activation of the coagulation cascade and effects on the vasculature (38, 46, 48, 49).
Balancing this, positive roles for antibody-mediated complement activation have also been proposed during active infection and vaccination (14). Potentially, the most valuable contribution antibody-mediated complement activation could make to protection is in vaccinated individuals or in those recovered from prior infection. In such individuals, the level of antigen present is likely to be low when antibody encounters its target antigen. This binding of antigen by pre-existing antibody will still result in immune cell recruitment and activation, helping to prevent the wider dissemination of the virus, but the overall magnitude of these sequelae will be lower. This reduced level of inflammation compared to what is observed when antibody is induced during infection could result in reduced levels of immunopathology. Therefore, antibody-mediated activation of complement in this context may be more beneficial for the host because it is contributing to control of infection when the pathogen burden is relatively low and less likely to provoke severe inflammatory responses.
The site of pathogen encounter is likely to influence the type of antibody present, the amount of complement and the outcomes from complement activation. At mucosal surfaces there is an enrichment of anti-pathogen IgA as well as the presence of IgG and IgA is less efficient at activating complement than IgM, IgG1 and IgG3 (50, 51). In saliva, which is often used as a proxy for mucosal responses, both IgA and IgG to SARS-CoV-2 are readily detectable after infection (52, 53), but this is less so after vaccination alone (54). In contrast, IgM is typically not a major component of the antibody repertoire detected in saliva (55). Although there are significant levels of complement in the upper and lower respiratory tract (56, 57), the relative predominance of IgA may mean there is less activation of the complement cascade even between the upper and lower respiratory tracts. Limiting any inflammation or immunopathology associated with complement activation may be beneficial for maintaining barrier integrity. Nevertheless, significant levels of complement and its breakdown components are found in the respiratory tract after many different infections (58–60). This means that complement can be activated in the respiratory tract and so in some circumstances may have negative impacts on the host.
In summary, we have identified activation of the classical complement pathway after vaccination against COVID-19, or after COVID-19 infection. Future studies will help us further understand how complement is activated in the presence of antibodies and how this may contribute to protection and harm in those who encounter this pathogen.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by London – Camden and Kings Cross Research Ethics Committee reference 20/HRA/1817 and North West ethics committee, Preston CIA UPH IRAS approval reference REC 20\NW\0240. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Conceptualization: EM-J, AS, LH, IH, JR, DW, MC, MD, AR, AC. Methodology: RL, EM-J, MP-T, SJ. Investigation: RL. Data analysis: RL. Resources: SF, MG, MN, IC, TD, TV, AS, AR. Supervision: SW, AC. Writing – original draft: RL, AC. All authors reviewed and edited the final manuscript.
Funding
This work was supported by the Wellcome Trust Mechanisms of Inflammatory Disease (MIDAS) PhD Programme [grant number 222389/Z/21/Z, part of 108871/B/15/Z] to RL; The Royal Society Newton International Fellowship [grant number NIF\R1\192061] to E.M.J and AC; a British Heart Foundation Intermediate Fellowship [grant number FS/IBSRF/20/25039] to JR; The University of Southampton Coronavirus Response Fund to MC and Medical Research Council [grant number MR/W010011/1] to LH, AR and AC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the staff of the Clinical Immunology Service, University of Birmingham for their invaluable work in sample collection and processing. We also gratefully acknowledge the University of Birmingham Protein Expression Facility for the production of nucleoprotein. We thank Jason McLellan for providing the expression plasmid for HexaPro. The content of this manuscript has appeared online as a preprint at https://doi.org/10.1101/2021.11.22.21266681 (61).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.838780/full#supplementary-material
References
1. Docherty AB, Harrison EM, Green CA, Hardwick HE, Pius R, Norman L, et al. Features of 20133 UK Patients in Hospital With Covid-19 Using the ISARIC WHO Clinical Characterisation Protocol: Prospective Observational Cohort Study. BMJ-British Med J (2020) 369:12. doi: 10.1136/bmj.m1985
2. Petrilli CM, Jones SA, Yang J, Rajagopalan H, O'Donnell L, Chernyak Y, et al. Factors Associated With Hospital Admission and Critical Illness Among 5279 People With Coronavirus Disease 2019 in New York City: Prospective Cohort Study. BMJ-British Med J (2020) 369:15. doi: 10.1136/bmj.m1966
3. Shields AM, Faustini SE, Perez-Toledo M, Jossi S, Allen JD, Al-Taei S, et al. Serological Responses to SARS-CoV-2 Following non-Hospitalised Infection: Clinical and Ethnodemographic Features Associated With the Magnitude of the Antibody Response. BMJ Open Respir Res (2021) 8(1):e000872. doi: 10.1136/bmjresp-2020-000872
4. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell (2020) 181(2):281–92. doi: 10.1016/j.cell.2020.02.058
5. Shields AM, Faustini SE, Kristunas CA, Cook AM, Backhouse C, Dunbar L, et al. COVID-19: Seroprevalence and Vaccine Responses in UK Dental Care Professionals. J Dental Res (2021) 100(11):1220–7. doi: 10.1177/00220345211020270
6. Lumley SF, O'Donnell D, Stoesser NE, Matthews PC, Howarth A, Hatch SB, et al. Antibody Status and Incidence of SARS-CoV-2 Infection in Health Care Workers. New Engl J Med (2021) 384(6):533–40. doi: 10.1056/NEJMoa2034545
7. Hanrath AT, Payne BAI, Duncan CJA, , Newcastle Hosp HCWTG. Prior SARS-CoV-2 Infection is Associated With Protection Against Symptomatic Reinfection. J Infect (2021) 82(4):E29–30. doi: 10.1016/j.jinf.2020.12.023
8. Darby AC, Hiscox JA. COVID-19: Variants and Vaccination. BMJ-British Med J (2021) 372:2. doi: 10.1136/bmj.n771
9. To KKW, Tsang OTY, Leung WS, Tam AR, Wu TC, Lung DC, et al. Temporal Profiles of Viral Load in Posterior Oropharyngeal Saliva Samples and Serum Antibody Responses During Infection by SARS-CoV-2: An Observational Cohort Study. Lancet Infect Dis (2020) 20(5):565–74. doi: 10.1016/S1473-3099(20)30196-1
10. Dunkelberger JR, Song WC. Complement and its Role in Innate and Adaptive Immune Responses. Cell Res (2010) 20(1):34–50. doi: 10.1038/cr.2009.139
11. Carvelli J, Demaria O, Vély F, Batista L, Benmansour NC, Fares J, et al. Association of COVID-19 Inflammation With Activation of the C5a–C5aR1 Axis. Nature (2020) 588(7836):146. doi: 10.1038/s41586-020-2600-6
12. Holter JC, Pischke SE, de Boer E, Lind A, Jenum S, Holten AR, et al. Systemic Complement Activation is Associated With Respiratory Failure in COVID-19 Hospitalized Patients. Proc Natl Acad Sci United States America (2020) 117(40):25018–25. doi: 10.1073/pnas.2010540117
13. Ng N, Powell CA. Targeting the Complement Cascade in the Pathophysiology of COVID-19 Disease. J Clin Med (2021) 10(10):13. doi: 10.3390/jcm10102188
14. Kurtovic L, Beeson JG. Complement Factors in COVID-19 Therapeutics and Vaccines. Trends Immunol (2021) 42(2):94–103. doi: 10.1016/j.it.2020.12.002
15. Ma L, Sahu SK, Cano M, Kuppuswamy V, Bajwa J, McPhatter J, et al. Increased Complement Activation is a Distinctive Feature of Severe SARS-CoV-2 Infection. Sci Immunol (2021) 6(59). doi: 10.1126/sciimmunol.abh2259
16. Cook AM, Faustini SE, Williams LJ, Cunningham AF, Drayson MT, Shields AM, et al. Validation of a Combined ELISA to Detect IgG, IgA and IgM Antibody Responses to SARS-CoV-2 in Mild or Moderate non-Hospitalised Patients. J Immunol Methods (2021) 494:7. doi: 10.1016/j.jim.2021.113046
17. Faustini SE, Jossi SE, Perez-Toledo M, Shields AM, Allen JD, Watanabe Y, et al. Development of a High-Sensitivity ELISA Detecting IgG, IgA and IgM Antibodies to the SARS-CoV-2 Spike Glycoprotein in Serum and Saliva. Immunology (2021) 164(1):135–47. doi: 10.1111/imm.13349
18. Hsieh CL, Goldsmith JA, Schaub JM, DiVenere AM, Kuo HC, Javanmardi K, et al. Structure-Based Design of Prefusion-Stabilized SARS-CoV-2 Spikes. Science (2020) 369(6510):1501–+. doi: 10.1126/science.abd0826
19. Chawla H, Jossi SE, Faustini SE, Samsudin F, Allen JD, Watanabe Y, et al. Glycosylation and Serological Reactivity of an Expression-Enhanced SARS-CoV-2 Viral Spike Mimetic. J Mol Biol (2021) 167332. doi: 10.1016/j.jmb.2021.167332
20. Seephetdee C, Buasri N, Bhukhai K, Srisanga K, Manopwisedjaroen S, Lertjintanakit S, et al. Mice Immunized With the Vaccine Candidate HexaPro Spike Produce Neutralizing Antibodies Against SARS-CoV-2. Vaccines (2021) 9(5):498. doi: 10.3390/vaccines9050498
21. McMillan CLD, Choo JJY, Idris A, Supramaniam A, Modhiran N, Amarilla AA, et al. Complete Protection by a Single Dose Skin Patch Delivered SARS-CoV-2 Spike Vaccine. bioRxiv (2021) 2021(05):30.446357. doi: 10.1126/sciadv.abj8065
22. Walls AC, Miranda MC, Schäfer A, Pham MN, Greaney A, Arunachalam PS, et al. Elicitation of Broadly Protective Sarbecovirus Immunity by Receptor-Binding Domain Nanoparticle Vaccines. Cell (2021) 184(21):5432–5447.e16. doi: 10.1016/j.cell.2021.09.015
23. Watanabe Y, Allen Joel D, Wrapp D, McLellan Jason S, Crispin M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science (2020) 369(6501):330–3. doi: 10.1126/science.abb9983
24. Allen JD, Chawla H, Samsudin F, Zuzic L, Shivgan AT, Watanabe Y, et al. Site-Specific Steric Control of SARS-CoV-2 Spike Glycosylation. Biochemistry (2021) 60(27):2153–69. doi: 10.1021/acs.biochem.1c00279
25. Keyt BA, Baliga R, Sinclair AM, Carroll SF, Peterson MS. Structure, Function, and Therapeutic Use of IgM Antibodies. Antibodies (2020) 9(4):53. doi: 10.3390/antib9040053
26. Bangaru S, Ozorowski G, Turner HL, Antanasijevic A, Huang D, Wang X, et al. Structural Analysis of Full-Length SARS-CoV-2 Spike Protein From an Advanced Vaccine Candidate. Science (2020) 370(6520):1089–94. doi: 10.1126/science.abe1502
27. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM Structure of the 2019-Ncov Spike in the Prefusion Conformation. Science (2020) 367(6483):1260–3. doi: 10.1126/science.abb2507
28. Domínguez-Medina CC, Pérez-Toledo M, Schager AE, Marshall JL, Cook CN, Bobat S, et al. Outer Membrane Protein Size and LPS O-Antigen Define Protective Antibody Targeting to the Salmonella Surface. Nat Commun (2020) 11(1):851. doi: 10.1038/s41467-020-14655-9
29. Grant OC, Montgomery D, Ito K, Woods RJ. Analysis of the SARS-CoV-2 Spike Protein Glycan Shield Reveals Implications for Immune Recognition. Sci Rep (2020) 10(1):14991. doi: 10.1038/s41598-020-71748-7
30. Wu L, Zhou L, Mo M, Liu T, Wu C, Gong C, et al. SARS-CoV-2 Omicron RBD Shows Weaker Binding Affinity Than the Currently Dominant Delta Variant to Human Ace2. Signal Transduct Targeted Ther (2022) 7(1):8. doi: 10.1038/s41392-021-00863-2
31. Faustini S, Shields A, Banham G, Wall N, Al-Taei S, Tanner C, et al. Cross Reactivity of Spike Glycoprotein Induced Antibody Against Delta and Omicron Variants Before and After Third SARS-CoV-2 Vaccine Dose in Healthy and Immunocompromised Individuals. J Infect (2022) 84(4):579–613. doi: 10.1016/j.jinf.2022.01.002
32. Perez-Toledo M, Faustini SE, Jossi SE, Shields AM, Marcial-Juarez E, Kanthimathinathan HK, et al. SARS-CoV-2-Specific IgG1/IgG3 But Not IgM in Children With Pediatric Inflammatory Multi-System Syndrome. Pediatr Allergy Immunol (2021) 32(5):1125–9. doi: 10.1111/pai.13504
33. Syrimi E, Fennell E, Richter A, Vrljicak P, Stark R, Ott S, et al. The Immune Landscape of SARS-CoV-2-Associated Multisystem Inflammatory Syndrome in Children (MIS-C) From Acute Disease to Recovery. iScience (2021) 24(11):103215. doi: 10.1016/j.isci.2021.103215
34. Annane D, Heming N, Grimaldi-Bensouda L, Frémeaux-Bacchi V, Vigan M, Roux A-L, et al. Eculizumab as an Emergency Treatment for Adult Patients With Severe COVID-19 in the Intensive Care Unit: A Proof-Of-Concept Study. EClinicalMedicine (2020) 28:100590. doi: 10.1016/j.eclinm.2020.100590
35. Zelek WM, Cole J, Ponsford MJ, Harrison RA, Schroeder BE, Webb N, et al. Complement Inhibition With the C5 Blocker LFG316 in Severe COVID-19. Am J Respir Crit Care Med (2020) 202(9):1304–8. doi: 10.1164/rccm.202007-2778LE
36. Mastellos DC, Pires da Silva BGP, Fonseca BAL, Fonseca NP, Auxiliadora-Martins M, Mastaglio S, et al. Complement C3 vs C5 Inhibition in Severe COVID-19: Early Clinical Findings Reveal Differential Biological Efficacy. Clin Immunol (2020) 220:108598. doi: 10.1016/j.clim.2020.108598
37. Vlaar APJ, de Bruin S, Busch M, Timmermans SAMEG, van Zeggeren IE, Koning R, et al. Anti-C5a Antibody IFX-1 (Vilobelimab) Treatment Versus Best Supportive Care for Patients With Severe COVID-19 (PANAMO): An Exploratory, Open-Label, Phase 2 Randomised Controlled Trial. Lancet Rheumatol (2020) 2(12):e764–73. doi: 10.1016/S2665-9913(20)30341-6
38. Ramlall V, Thangaraj PM, Meydan C, Foox J, Butler D, Kim J, et al. Immune Complement and Coagulation Dysfunction in Adverse Outcomes of SARS-CoV-2 Infection. Nat Med (2020) 26(10):1609–15. doi: 10.1038/s41591-020-1021-2
39. Delanghe JR, De Buyzere ML, Speeckaert MM. Genetic Polymorphisms in the Host and COVID-19 Infection, in Coronavirus Disease - COVID-19. Rezaei N, editor. Cham: Springer International Publishing (2021) p. 109–18.
40. Ali YM, Ferrari M, Lynch NJ, Yaseen S, Dudler T, Gragerov S, et al. Lectin Pathway Mediates Complement Activation by SARS-CoV-2 Proteins. Front Immunol (2021) 12:8. doi: 10.3389/fimmu.2021.714511
41. Jarlhelt I, Nielsen SK, Jahn CXH, Hansen CB, Pérez-Alós L, Rosbjerg A, et al. SARS-CoV-2 Antibodies Mediate Complement and Cellular Driven Inflammation. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.767981
42. Medjeral-Thomas NR, Troldborg A, Hansen AG, Gisby J, Clarke CL, Prendecki M, et al. Plasma Lectin Pathway Complement Proteins in Patients With COVID-19 and Renal Disease. Front Immunol (2021) 12:11. doi: 10.3389/fimmu.2021.671052
43. Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct Activation of the Alternative Complement Pathway by SARS-CoV-2 Spike Proteins is Blocked by Factor D Inhibition. Blood (2020) 136(18):2080–9. doi: 10.1182/blood.2020008248
44. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Trans Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
45. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and Tissue Factor-Enriched Neutrophil Extracellular Traps are Key Drivers in COVID-19 Immunothrombosis. J Clin Invest (2020) 130(11):6151–7. doi: 10.1172/JCI141374
46. Lipcsey M, Persson B, Eriksson O, Blom AM, Fromell K, Hultström M, et al. The Outcome of Critically Ill COVID-19 Patients is Linked to Thromboinflammation Dominated by the Kallikrein/Kinin System. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.627579
47. Bjornson AB, Mellencamp MA, Schiff GM. Complement is Activated in the Upper Respiratory Tract During Influenza Virus Infection. Am Rev Respir Dis (1990) 143(5):1062–6. doi: 10.1164/ajrccm/143.5_Pt_1.1062
48. Lo MW, Kemper C, Woodruff TM. COVID-19: Complement, Coagulation, and Collateral Damage. J Immunol (2020) 205(6):1488–95. doi: 10.4049/jimmunol.2000644
49. Martínez-Salazar B, Holwerda M, Stüdle C, Piragyte I, Mercader N, Engelhardt B, et al. COVID-19 and the Vasculature: Current Aspects and Long-Term Consequences. Front Cell Dev Biol (2022) 10. doi: 10.3389/fcell.2022.824851
50. Woof JM, Russell MW. Structure and Function Relationships in IgA. Mucosal Immunol (2011) 4(6):590–7. doi: 10.1038/mi.2011.39
51. Daha NA, Banda NK, Roos A, Beurskens FJ, Bakker JM, Daha MR, et al. Complement Activation by (Auto-) Antibodies. Mol Immunol (2011) 48(14):1656–65. doi: 10.1016/j.molimm.2011.04.024
52. Cervia C, Nilsson J, Zurbuchen Y, Valaperti A, Schreiner J, Wolfensberger A, et al. Systemic and Mucosal Antibody Responses Specific to SARS-CoV-2 During Mild Versus Severe COVID-19. J Allergy Clin Immunol (2021) 147(2):545–557.e9. doi: 10.1016/j.jaci.2020.10.040
53. Costantini VP, Nguyen K, Lyski Z, Novosad S, Bardossy AC, Lyons AK, et al. Development and Validation of an Enzyme Immunoassay for Detection and Quantification of SARS-CoV-2 Salivary IgA and IgG. J Immunol (2022) 208(6):1500–8. doi: 10.4049/jimmunol.2100934
54. Azzi L, Dalla Gasperina D, Veronesi G, Shallak M, Ietto G, Iovino D, et al. Mucosal Immune Response in BNT162b2 COVID-19 Vaccine Recipients. eBioMedicine (2022) 75:103788. doi: 10.1016/j.ebiom.2021.103788
55. Klingler J, Lambert GS, Itri V, Liu S, Bandres JC, Enyindah-Asonye G, et al. Detection of Antibody Responses Against SARS-CoV-2 in Plasma and Saliva From Vaccinated and Infected Individuals. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.759688
56. Bolger MS, Ross DS, Jiang H, Frank MM, Ghio AJ, Schwartz DA, et al. Complement Levels and Activity in the Normal and LPS-Injured Lung. Am J Physiol Lung Cell Mol Physiol (2007) 292(3):L748–59. doi: 10.1152/ajplung.00127.2006
57. Varsano S, Kaminsky M, Kaiser M, Rashkovsky L. Generation of Complement C3 and Expression of Cell Membrane Complement Inhibitory Proteins by Human Bronchial Epithelium Cell Line. Thorax (2000) 55(5):364. doi: 10.1136/thorax.55.5.364
58. Mateu-Borrás M, González-Alsina A, Doménech-Sánchez A, Querol-García J, Fernández FJ, Vega MC, et al. Pseudomonas Aeruginosa Adaptation in Cystic Fibrosis Patients Increases C5a Levels and Promotes Neutrophil Recruitment. Virulence (2022) 13(1):215–24. doi: 10.1080/21505594.2022.2028484
59. Wells TJ, Whitters D, Sevastsyanovich YR, Heath JN, Pravin J, Goodall M, et al. Increased Severity of Respiratory Infections Associated With Elevated Anti-LPS IgG2 Which Inhibits Serum Bactericidal Killing. J Exp Med (2014) 211(9):1893–904. doi: 10.1084/jem.20132444
60. Satyam A, Tsokos MG, Brook OR, Hecht JL, Moulton VR, Tsokos GC. Activation of Classical and Alternative Complement Pathways in the Pathogenesis of Lung Injury in COVID-19. Clin Immunol (2021) 226:108716. doi: 10.1016/j.clim.2021.108716
Keywords: complement, COVID-19, SARS-CoV-2, vaccine, antibodies
Citation: Lamerton RE, Marcial-Juarez E, Faustini SE, Perez-Toledo M, Goodall M, Jossi SE, Newby ML, Chapple I, Dietrich T, Veenith T, Shields AM, Harper L, Henderson IR, Rayes J, Wraith DC, Watson SP, Crispin M, Drayson MT, Richter AG and Cunningham AF (2022) SARS-CoV-2 Spike- and Nucleoprotein-Specific Antibodies Induced After Vaccination or Infection Promote Classical Complement Activation. Front. Immunol. 13:838780. doi: 10.3389/fimmu.2022.838780
Received: 18 December 2021; Accepted: 07 June 2022;
Published: 04 July 2022.
Edited by:
Shailendra Saxena, King George’s Medical University, IndiaReviewed by:
Ranjan Ramasamy, Id-Fish Technology Inc, United StatesKshitij Srivastava, Clinical Center (NIH), United States
Giuseppe Portella, University of Naples Federico II, Italy
Copyright © 2022 Lamerton, Marcial-Juarez, Faustini, Perez-Toledo, Goodall, Jossi, Newby, Chapple, Dietrich, Veenith, Shields, Harper, Henderson, Rayes, Wraith, Watson, Crispin, Drayson, Richter and Cunningham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adam F. Cunningham, QS5GLkN1bm5pbmdoYW1AYmhhbS5hYy51aw==
†These authors have contributed equally to this work