 Teding Chang
Teding Chang Jingzhi Yang
Jingzhi Yang Hai Deng1
Hai Deng1 XiangPing Yang
XiangPing Yang Zhao-Hui Tang
Zhao-Hui Tang- 1Division of Trauma & Surgical Critical Care, Department of Surgery, Tongji Hospital, Tongji, China
- 2Department of Immunology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Uncontrolled severe acute respiratory syndrome-coronavirus (SARS-CoV)-2 infection is closely related to disorders of the innate immune and delayed adaptive immune systems. Dendritic cells (DCs) “bridge” innate immunity and adaptive immunity. DCs have important roles in defending against SARS-CoV-2 infection. In this review, we summarize the latest research concerning the role of DCs in SARS-CoV-2 infection. We focus on the complex interplay between DCs and SARS-CoV-2: pyroptosis-induced activation; activation of the renin–angiotensin–aldosterone system; and activation of dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin. We also discuss the decline in DC number, the impaired antigen-presentation capability, and the reduced production of type-I interferon of DCs in severe SARS-CoV-2 infection. In addition, we discuss the potential mechanisms for pathological activation of DCs to understand the pattern of SARS-CoV-2 infection. Lastly, we provide a brief overview of novel vaccination and immunotherapy strategies based on DC targeting to overcome SARS-CoV-2 infection.
1 Introduction
The coronavirus disease 2019 (COVID-19) pandemic poses a serious threat to public health and economic systems worldwide (1). As of January 27, 2022, more than 356.95 million people had been infected with the virus that causes COVID-19 [severe acute respiratory syndrome-coronavirus (SARS-CoV)-2] and 5.61 million individuals had died (data from WHO Coronavirus Dashboard).
Coronaviruses can cause intestinal and respiratory infections in animals and humans (2–6). SARS-CoV-2 is a genetically diverse virus found in a range of host species, including birds and mammals. It is transmitted mainly through the respiratory tract (7). The “spike” glycoprotein of SARS-CoV-2 binds to angiotensin-converting enzyme 2 (ACE2) and mediates membrane fusion and virus entry (8). SARS-CoV-2 infection induces pyroptosis (a highly inflammatory form of programmed cell death seen in cytopathic viruses), which leads to release of SARS-CoV-2, pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs) (7, 9, 10). Innate immune cells are recruited by these products to respond to SARS-CoV-2 invasion and then release proinflammatory cytokines and “prime” the adaptive immune response via T cells and B cells. Due to unrestrained infiltration of inflammatory cells, the products induced by immune cells mediate lung injury by activating excess innate immune cells and oversecreting protease and reactive oxygen species (11). Therefore, understanding how innate immune cells respond to SARS-CoV-2 infection is critical for the prevention and treatment of COVID-19.
Dendritic cells (DCs) act as a “bridge” between innate immunity and adaptive immunity. They have important roles in viral infection. Pattern recognition receptors (PRRs) expressed on the membrane of DCs, such as Toll-like receptor (TLR)7, retinoic acid-inducible gene-I, melanoma differentiation-associated protein-5, and the cyclic guanosine monophosphate–adenosine monophosphate synthase–stimulator of interferon genes pathway, can recognize SARS-CoV-2-induced PAMPs and DAMPs (12–14). If these receptors are activated, a range of signaling pathways [e.g., interferon regulatory factor 3 (IRF3), IRF7, nuclear factor-kappa B (NF-κB)] are activated to regulate proinflammatory cytokines (e.g., tumor necrosis factor α (TNFα), interleukin (IL)-6, monocyte chemoattractant-1, macrophage inflammatory protein (MIP)1α, MIP1β) in the nucleus and induce interferon type I (IFN-I; the main cytokine responsible for producing a strong antiviral response) production (15, 16). DCs are also responsible for ingesting, transporting, processing, and presenting antigens to T cells, inducing the adaptive immune response, and have important roles in virus infection (17–19). However, the number of DCs, the ability to secrete antiviral cytokines (especially IFN-I), and the capability of antigen presentation decline unexpectedly in patients with severe COVID-19. This review focuses on explaining the (i) interrelationship between DCs and SARS-CoV-2 and (ii) reduced number and dysfunction of DCs. In this way, we hope to create a breakthrough in immunomodulation therapy and an IFN strategy against COVID-19.
2 Direct and Indirect Interaction of SARS-CoV-2 With DCs
2.1 Pyroptosis: A Major Inducer for SARS-CoV-2 to Attract DCs
There is growing evidence of cytolysis (mostly pyroptosis but not necroptosis) in patients infected with SARS-CoV-2 (7, 20). “Pyroptosis” refers to the biological process that relies on caspase-1-dependent activation of gasdermin D to form membrane pores. “Necroptosis” is dependent upon the intracellular signal transduction of receptor-interacting protein kinases, and membrane pores are formed through phosphorylation of mixed lineage kinase domain-like pseudokinase, which results in cytolysis (21). In vitro studies have shown that caspase-1 is activated in patients with severe COVID-19, downstream secretion of IL-1β increased, and decomposition of gasdermin D accelerated (22). Those data show that cytolysis during SARS-CoV-2 infection is mainly pyroptosis rather than necroptosis. Nucleotide-binding oligomerization domain-like receptors (NLRs) recognize the danger signals (which are derived from homologous hosts or microorganisms) and form large supramolecular chemical inflammasomes (9). Caspase-1 activation may be related to SARS-CoV-2 recognition by NLRs in cells. After caspase-1 has been activated, it initiates the pyroptosis pathway, activates gasdermin D to form plasma-membrane pores, and allows water influx, cell swelling, and osmotic lysis. Activated caspase-1 can also activate IL-1β and IL-18, which are released mainly through plasma-membrane pores (9). Many PAMPs, DAMPs, and virus products, which are released by SARS-CoV-2-induced pyroptosis, recruit various types of immune cells (including DCs) to infiltrate lung tissue and promote secretion of cytokines, particularly IL-6, IL-1β, IL-10, TNF, granulocyte macrophage-colony stimulating factor, IFN-induced protein-10, IL-17, monocyte chemoattractant-3, and IL-1ra (23).
ACE2 is the main target of SARS-CoV-2 (11). We postulate that ACE2 is also the inducer of pyroptosis caused by SARS-CoV-2. ACE2 is a transmembrane protease and is the main receptor for virus invasion (24). The N-terminal extracellular domain of ACE2 comprises a “claw-like” protease domain (PD). The receptor-binding domain (RBD) of SARS-CoV-2 combines with the PD of ACE2 to form an RBD–PD complex (24). The C-terminus is a transmembrane domain, also known as the collectrin-like domain (24). Studies have suggested that ACE2 is the major receptor of SARS-CoV and SARS-CoV-2 and binds to transmembrane serine protease (TMPRSS)2-activated spike proteins to induce virus entry into endosomes (25). ACE2 and TMPRSS2 are expressed primarily in cells of the upper respiratory tract and lungs (11, 25–27). These respiratory-tract cells with special cell-membrane receptors are ideal targets for SARS-CoV-2 invasion. This phenomenon may explain why SARS-CoV-2 is transmitted through aerosols and is very infectious (7).
The increased levels of lactate dehydrogenase (LDH) (28) and D-dimer in the plasma of patients infected with SARS-CoV-2 suggest that there is a high level of tissue injury in patients with severe COVID-19 (22, 23, 29, 30). Patients with severe COVID-19 with high LDH levels and leukopenia have impaired integrity of cell membranes (23, 31–33). In addition, the full blood count and biochemical findings of patients with severe COVID-19 reveal that the leukopenia observed in such patients appears to precede the “cytokine storm” (31, 34). SARS-CoV-2 infects human primary monocytes in vitro, resulting in their lysis and death. Flow cytometry and fluorescence microscopy have demonstrated the membrane disruption triggered by SARS-CoV-2 (22). In conclusion, we believe that SARS-CoV-2 invades cells through the ACE2, induces pyroptosis, and finally, causes the subsequent cytokine storm, leading to disease progression.
Products which are induced by SARS-CoV-2 attract DCs selectively into inflammatory sites (35–38). DC aggregation has been observed in the bronchoalveolar lavage fluid of COVID-19 patients, which suggests that DCs infiltrate into lung tissues by the products of SARS-CoV-2-induced pyroptosis (35–38) (Figure 1). Flow cytometry of the bronchoalveolar lavage fluid of COVID-19 patients has revealed that only type-2 conventional dendritic cells (cDC2) accumulate in the lungs (35, 36). cDC1 and plasmacytoid dendritic cells (pDCs), which are involved in IFN-I secretion, are absent in infected lungs (35, 36). cDC2 support the cluster of differentiation (CD)4+ T-cell response, stimulate follicular T-helper (Th) cells (which activate a humoral antiviral adaptive immune response), and produce inefficient antiviral proinflammatory cytokines (39–41).
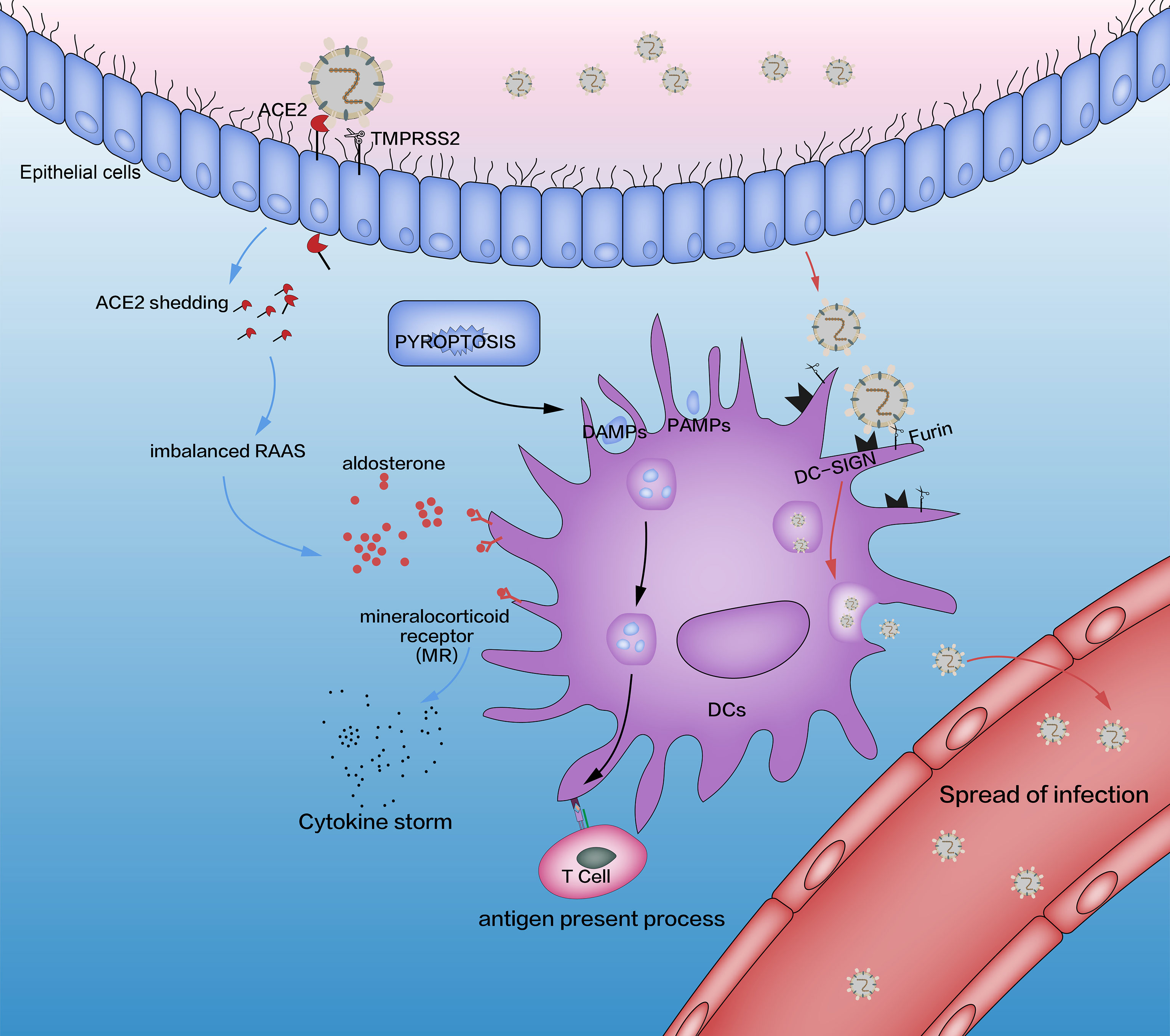
Figure 1 Interaction between SARS-CoV-2 and DCs (schematic). The three pathways affected by SARS-CoV-2 are pyroptosis (black lines), imbalanced RAAS (blue lines), and DC-SIGN (red lines). The adaptive immune system is induced by pyroptosis, which is activated by SARS-CoV-2. In addition, SARS-CoV-2 combines with the ACE2 receptor through its spike protein (S), which is activated by TMPRSS2. This process causes an imbalance in the RAAS through the shedding of ACE2 and releases excessive amounts of aldosterone, which promotes the release of proinflammatory cytokines in DCs through MRs. SARS-CoV-2 impacts DCs directly by DC-SIGN, a receptor which has critical roles in the recognition of viruses (e.g., HIV, Ebola, dengue, cytomegalovirus) and other pathogens (e.g., Leishmania species, Candida albicans, Mycobacterium tuberculosis, Streptococcus pneumoniae, Aspergillus fumigatus). Although SARS-CoV-2 replication in lung cells is well-documented, a similar process has not been confirmed in alveolar DCs. Some researchers have suggested such a replication based on triggering aberrant production of proinflammatory cytokines/chemokines and inducing the spread of SARS-CoV-2 infection, as is the case with SARS-CoV and MERS-CoV, but other scholars have ruled out SARS-CoV replication in human DCs. RAAS, renin–angiotensin–aldosterone system; DCs, dendritic cells; TMPRSS2, transmembrane serine protease 2; MRs, mineralocorticoid receptors; DC-SIGN, dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin.
SARS-CoV-2 attacks nasal cells and lung cells through ACE2, activates pyroptosis, and leads to the production of many DAMPs, PAMPs, and progeny viruses. These products activate innate immunity (Figure 1).
2.2 Imbalanced Renin–Angiotensin–Aldosterone System: Potential Regulation of DCs
SARS-CoV-2 enters cells through the ACE2. ACE2 is a key factor in the renin–angiotensin–aldosterone system (RAAS) (42, 43). SARS-CoV-2 may affect the RAAS and lead to disease development by affecting the shedding of ACE2. The RAAS mediates blood-pressure control, inflammation, sodium reabsorption, and fibrosis (44). RAAS disorders can lead to heart failure, low blood pressure, atherosclerosis, and diabetes mellitus (45). The factor related most closely with ACE2 in the RAAS is angiotensin (Ang)II. The latter stimulates vascular contraction, secondary inflammation, and atherosclerosis through the type-1 angiotensin II receptor (AT1R) (46). Another receptor, type-2 angiotensin II (AT2R), in contrast to AT1R, is activated by AngII to promote vascular dilation, platelet aggregation, and promotion of insulin action. However, AT2R is rarely expressed in healthy adults (47). Therefore, the regulation and balance of AngII are dependent mainly on ACE2. The latter can convert AngII to Ang-(1–7), which is similar to AT2R stimulation (44, 47). An excess of AngII promotes pulmonary vascular contraction, inflammation, and cytokine-induced organ damage, increases the permeability of cell membranes and apoptosis, and induces acute kidney injury and acute respiratory distress syndrome (ARDS). AngII overactivation is associated with an increase in ACE2 shedding in patients with COVID-19. In a cohort of 12 COVID-19 patients, the circulating level of AngII was significantly higher than that in healthy controls (linearly correlated with viral load), which suggested a direct link between RAAS imbalance and multiorgan damage caused by SARS-CoV-2 infection (1, 48, 49).
Among hospitalized COVID-19 patients, the increase in AngII level is accompanied by an increased level of IL-6, with the highest mortality rate (29). This phenomenon has also been found in the patients with avian influenza A (H7N9) infection. Within 4 weeks of H7N9 infection, AngII levels increase gradually, which is associated with a worse clinical prognosis (50). In COVID-19 patients, AngII levels have been found to be closely related to viral titer and partial pressure of oxygen in arterial blood/fraction of inspired oxygen (49). COVID-19-induced AngII aggregation has been shown to promote acute lung injury (ALI) by activating cytokine-induced inflammation, activating the nicotinamide adenine dinucleotide hydrogen (NADH)/nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidation system and vasoconstriction (49, 51, 52). Researchers found that COVID-19 was more severe and carried a worse prognosis in older people and males, if ACE2 expression declined (49, 53, 54). Coincidentally, patients with COVID-19 with diseases associated with RAAS overactivation (e.g., hypertension, diabetes mellitus) have been shown to carry a higher risk of transfer to the intensive care unit and mortality (55). RAAS overactivation appears to be closely related to a poor prognosis in COVID-19 patients, and the loss of ACE2 (which can regulate the effects of an overactive RAAS during the disease) will lead to worse consequences.
The ACE expressed on the DCs is still able to participate in the RAAS disorders (56–58). It has been proved that the ACE expression increased was correlated with the differentiation and stimulation of DCs (59). Elevated ACE is involved in the production of AngII and the peptide repertoire trimming as part of the MHC-II complex (59, 60). Accumulation of AngII due to the shedding of ACE2 leads to the phosphorylation of the ERK and promotes the secretion of IL-6 and TNFα (61). In the meantime, AngII has the ability to improve the migration, maturation, and antigen presentation of DCs to improve Th1 cells (62).
The increase in the AngII level is accompanied by an increase in the aldosterone level because AngII can stimulate the adrenal cortex to secrete aldosterone. DCs are key immune cells involved in severe COVID-19-induced lung damage mediated by aldosterone, which can stimulate DCs to produce IL-6 and transforming growth factor-β1 via the mineralocorticoid receptor (Figure 1) (63, 64). AngII and aldosterone can alter the proliferation and maturation of DCs, leading to DC dysfunction (36). Supplementation of ACE2 may remedy this problem. Recombinant human (rh)ACE2 has a protective effect in SARS-CoV-induced ALI, and injection of rnACE2 can reduce inflammation and improve lung function (65–67). Therefore, rhACE2 may be a potential treatment for SARS-CoV-2-induced ALI.
2.3 Dendritic Cell-Specific Intercellular Adhesion Molecule 3-Grabbing Non-Integrin: Direct Interaction Between SARS-CoV-2 and DCs
Yang and colleagues (68) showed that SARS-CoV-2 enters DCs and macrophages through dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) and furin rather than through ACE2 and TMPRSS2. DC-SIGN (CD209) is a C-type calcium-dependent lectin and a type-II membrane protein. DC-SIGN consists of three domains: extracellular, transmembrane, and intracellular (69–72). The intracellular domain is an N-terminal domain responsible for the binding, phagocytosis, and intracellular transport of molecules associated with signal transduction. The transmembrane domain anchors DC-SIGN to the cell membrane. The extracellular domain consists of two portions: a neck domain (which forms a tetramer that stabilizes the extracellular part of the molecule) and a c-type carbohydrate-recognition domain (a calcium-dependent receptor with a highly conserved sequence) (43, 69, 73). The extracellular domain is essential for binding and recognizing high-mannose oligosaccharides (43, 74, 75). DC-SIGN is expressed exclusively by mature and immature DCs in the skin, mucosa, and lymphoid organs (76). It is a PRR/adhesion receptor in DCs that promotes migration and adhesion and mediates the inflammatory response by activating innate immune and adaptive immune systems. Remarkably, DC-SIGN has a pivotal role in the “immune escape” of pathogens and tumor cells (70, 71). As an antigen-capture receptor, DC-SIGN recognizes, internalizes, and decomposes antigens and, eventually, presents them to CD4+ T cells to trigger an immune response (77). However, virus-infected DCs, through DC-SIGN, also spread SARS-CoV-2 to other tissues and organs (Figure 1). This phenomenon is typical in human immunodeficiency virus (HIV) infection, where the DC-SIGN signal is activated by the HIV and induces its migration to lymph nodes. In lymph nodes, DC-SIGN promotes the migration of HIV-1 from DCs to CD4+ T cells, thereby promoting cell infection and virus transmission (78–80). Hence, whether this “Trojan horse” phenomenon exists in SARS-CoV-2 infection is worthy of further study.
The role of DC-SIGN in SARS-CoV-2 infection is not known. Recent studies have shown that SARS-CoV binds DC-SIGN through its activated spike protein and takes part in the inflammatory response of DCs (Figure 1) (43, 81, 82). DC-SIGN also plays an important part in Middle East respiratory syndrome-related coronavirus (MERS-CoV) infection and is too crucial to neglect in SARS-CoV-2 infection (83). DC-SIGN could become an alternative receptor for SARS-CoV-2 to invade DCs. It has been suggested that the interaction of SARS-CoV-2 or its envelope proteins (including the products of pyroptosis) with DC-SIGN result in the loss of a specific DC subpopulation (36). Moreover, activated DC-SIGN receptors downregulate the expression of major histocompatibility class (MHC)-II molecules (84), which is a key factor of impaired presentation of antigens. In addition, the combination between DC-SIGN and HIV can promote a Th2 cell-based immune response, thereby reducing levels of IL-12 and IFNs (42), which helps to explain the innate immunosuppression in patients with severe COVID-19.
The DC-SIGNR (L-SIGN, CD209L), which is the homology receptor of the DC-SIGN, is broadly expressed in both endothelial cells and epithelial cells (85, 86). The DC-SIGN/L-SIGN may be an alternative receptor, due to the fact that ACE2-deficient endothelial cells but not DC-SIGNR-deficient endothelial cells allow SARS-CoV-2 entry (85, 86). Michel et al. confirm the important role of DC-SIGN in the Trojan Horse model of DCs during SARS-CoV-2 infection (87). They think that SARS-CoV-2 adheres to the surface of DCs through DC-SIGN and secondly present SARS-CoV-2 to susceptible cells in the process recognized as trans-infection which relies on the characteristics of DCs’ migration (87). Intriguingly, the DC-SIGN gene expression is interestingly decreased in lung DCs but increased in circulation DCs which would undoubtedly increase the amount of SARS-CoV-2 carried by DCs and enhance its global transmission ability (88).
As in HIV infection, DC-SIGN-mediated virus internalization is a critical mechanism for immune escape in SARS-CoV-2 infection (75). DC-SIGN has become another potential invasion portal for SARS-CoV-2, and its function deserves further exploration in COVID-19 patients. However, after SARS-CoV-2 infection, the depletion and dysfunction of DCs result in persistent virus infection. The exact mechanism by which SARS-CoV-2 infection reduces the number and function of DCs is discussed below.
3 Depletion and Dysfunction of DCs in COVID-19
3.1 Decline in the Number of DCs in COVID-19 Patients
cDC1, cDC2, and pDCs are recruited into lung tissue during infection (39). cDC1s (also known as CD141+ DC) are found in peripheral blood and among resident DCs of the lymph nodes, bone marrow, and spleen (39, 89–93). They participate in cross-presentation of antigens via MHC-I molecules to activate CD8+ T cells and promote Th1 cells and natural-killer-cell responses though IL-12 (39). cDC1s also secrete IFN-III (including IFN-λ) (39, 94). cDC2s (also known as CD1c+ DC) are present mainly in peripheral blood, lymphoid organs, and tissues. They are activated to become robust producers of IL-12 and are excellent cross-presentation cells. cDC2s are also major producers of IL-23, IL-1, TNF-α, IL-8, and IL-10 (39, 95–97). pDCs (which were identified first in peripheral blood and tonsils) “sense” and respond to viral infection through rapid production of IFN-I (39, 98, 99). These three types of DCs are activated by PAMPs and DAMPs to promote the innate immune response and to activate the adaptive immune response.
The number of DCs in the blood of COVID-19 patients is reduced. Among the three types of DCs, only cDC2s accumulate in the lungs of COVID-19 patients (35, 100–103). The mechanisms that result in a decline in the number of DCs may be caused by an alteration in distribution of DCs, increased apoptosis of DCs, and the inhibitory effects of myeloid-derived suppressor cells (MDSCs) (Figure 2).
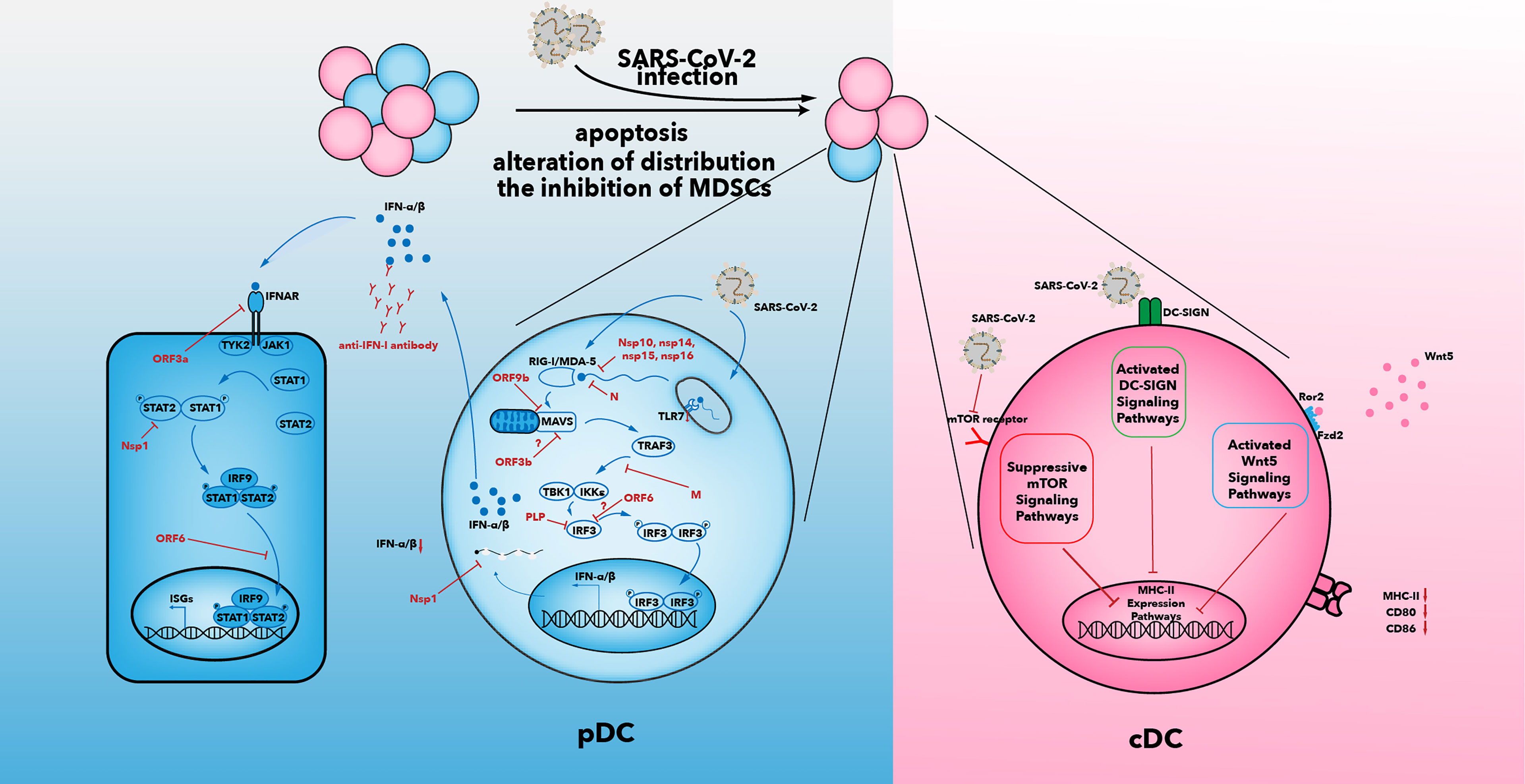
Figure 2 Multiple suppressive mechanisms in SARS-CoV-2-infected DCs from patients with severe COVID-19. The number of DCs in patients decreases after SARS-CoV-2 infection. Increased apoptosis, alterations in distribution of DCs, and inhibition of MDSCs may be associated with a decrease in DC number. IFN-I secretion is inhibited by various viral proteins that have been shown to be effective against IFN-I signaling in SARS-CoV infection (104). In addition to the effects of viral proteins, anti-IFN antibody and reduced expression of TLR7 have been observed in some patients with severe COVID-19. The capability of antigen presentation is impaired in cDC1 and cDC2. Inhibition of the mTOR signaling pathway, activated DC-SIGN pathway, and activated Wnt5 pathway could contribute to downregulation of MHC-II and co-stimulatory molecules. IFN, interferon; IFNAR, interferon alpha and beta receptor; mTOR, mammalian target of rapamycin; IκB, inhibitor of nuclear factor κB; IRF, IFN regulatory factor; ISG, IFN-stimulated gene; JAK, Janus kinase; IKKϵ, IκB kinase-ϵ; M, membrane; MAVs, mitochondrial antiviral signaling proteins; N, nucleocapsid; Nsp, non-structural protein; ORF, open reading frame; P, phosphate; DC-SIGN, dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin; PLP, papain-like protease; RIG-I, retinoic acid-inducible gene-I; MDA5, melanoma differentiation-associated gene 5; SARS-CoV-2, severe acute respiratory syndrome-coronavirus 2; TANK, TRAF family member-associated NF-κB activator; TBK1, TANK-binding kinase 1; TRAF3, tumor necrosis factor receptor-associated factor 3; STAT, signal transducer and activator of transcription; TYK2, tyrosine kinase 2; Wnt5, Wnt oncogene analog 5.
3.1.1 Alteration in the Distribution of DCs
SARS-CoV-2 causes pyroptosis through infection of respiratory epithelial cells via ACE2 and TMPRSS2 (11, 27) and leads to production of plentiful cytokines and chemokines that attract DCs to migrate from peripheral blood into the lungs (7, 20, 21). Sanchez-Cerrillo and coworkers (35) analyzed DC profiles in the blood and lungs of COVID-19 patients. They found that pDCs and cDC1s showed a significant depletion and that cDC2s migrated from the blood to the lungs. Xiong and colleagues (38) observed abundant mature DCs in bronchoalveolar lavage fluid, which indicated DC accumulation in SARS-CoV-2-infected lungs. Insufficient recruitment of pDCs and cDC1s to the lungs may be due to downregulation of chemokine receptors such as C–C chemokine receptor type-2 and C–X–C motif chemokine receptor-3 (100). A reduction in the circulation of pDCs and cDC1 may be a result of migration to or sequestration in lymphoid tissues (105, 106). Changes in the distribution of cDC2 may contribute to the reduction in circulation of DCs.
3.1.2 Incremental Apoptosis of DCs
The circulation of DCs (especially pDCs) is diminished significantly in COVID-19 (35). pDCs are key factors involved in the antiviral efficacy of IFN-I (39). Hence, SARS-CoV-2 may target pDCs and reduce their number. Saichi and colleagues found p53 signaling to be upregulated in pDCs in COVID-19 cases (105). However, Onodi and colleagues found that pDCs, which were isolated from healthy donors, are improved during SARS-CoV-2 infection (107), thereby suggesting that SARS-CoV-2 could not kill pDCs directly. In sum, other mechanisms must be involved in pDC apoptosis.
Recently, “immunometabolism reprogramming” has been postulated to explain why DC apoptosis is increased during SARS-CoV-2 infection (108). Immunometabolism reprogramming is characterized by changes in the metabolic stages of immune cells from homeostasis to an inflammatory environment or infectious environment (108). Recent studies have revealed that DC apoptosis in COVID-19 patients was because of the TNF-related apoptosis-inducing ligand or apolipoprotein-2 ligand but was not dependent on the Fas ligand (108–111). The displacement of hexokinase-II from mitochondrial-associated membranes increases glycolysis to produce glucose-6-phosphate. This action can lead to excess release of Ca2+ from the endoplasmic reticulum to the cytosol to activate calpains, mitochondrial depolarization, and apoptosis (112).
3.1.3 Inhibitory Effect of MDSCs
MDSCs are induced in pathological conditions such as inflammation, cancer, or some autoimmune disorders. They inhibit immature myeloid cells from differentiating into mature DCs, upregulate expression of immunosuppressive factors (e.g., inducible nitric oxide synthase, arginase), and promote the production of nitric oxide and reactive oxygen species. Those productions result in expansion of an immunosuppressive population of immature myeloid cells and lead to DC depletion (113, 114). Recent studies have found that the increased number of MDSCs in COVID-19 patients might be key to the decline in DC number (101, 115).
3.2 Impaired Antigen Presentation of SARS-CoV-2-Infected DCs
Not only does the number of DCs decline, but also the ability of DCs to present antigens is impaired during SARS-CoV-2 infection (101, 105, 115, 116). Zhou et al. studied the antigen-presentation capacity of circulating DCs in the acute phase of COVID-19. They found that expression of the co-stimulatory molecule (CD80, CD86) and antigen-presentation molecule (human leukocyte antigen-DR) decreased after stimulation with a “cytokine cocktail” (IL-1β, IL-6, TNF-α, prostaglandin E2) (101). DCs in the acute phase of COVID-19 failed to stimulate the proliferation of CD4+ and CD8+ T cells according to a mixed lymphocyte reaction assay with allogeneic CD4+ and CD8+ T cells (101). The antigen-presentation capability of DCs could act as a bridge between innate immunity and adaptive immunity. Determining the reason for the decline in the antigen-presentation capacity of DCs is crucial, and some reasons are discussed in the following (Figure 2).
3.2.1 Impaired Mammalian Target of Rapamycin Signaling
Impaired mammalian target of rapamycin (mTOR) is a crucial regulator of the development, maturation, and function of DCs. mTOR can dictate and “shape” the inflammatory immune response of DCs (117). Inhibition of mTOR expression hinders DC maturation and reduces the antigen uptake in the early stage of infection (117, 118). Prabhu et al. found a decrease in the pS6 (mTOR marker) level in blood, thereby indicating inhibition of the mTOR signal in COVID-19 patients (115). Suppression of the mTOR signaling pathway may contribute to the impaired antigen presentation of DCs in COVID-19 patients.
3.2.2 Upregulation of Wnt Oncogene Analog Expression
Wnt oncogene analog (Wnt)5 can impair the function and maturation of DCs (104, 119). Wnt5a is activated in ARDS and sepsis as an inhibitor of the repair process (119). Wnt5a could be a candidate biomarker of SARS-CoV-2 progression (116, 120). The Wnt5 signaling pathway may also impair the function of DCs to present antigens to T cells.
3.2.3 Summary
The mechanism by which the antigen-presentation capability of DCs is impaired is not clear. A greater focus on the SARS-CoV-2-induced microenvironment (including alteration of signaling pathways such as DC-SIGN, mTOR, and Wnt5) may be needed to ascertain the underlying mechanism.
3.3 IFN-I Deficiency in SARS-CoV-2-Infected DCs
IFN-I is a powerful weapon for DCs in their fight against viral infection. However, SARS-CoV-2 often leads to the effects of IFN-I being hampered. Zhou et al. found that the ability of infected cDC1 and cDC2 to produce proinflammatory cytokines (IL-1β, IL-6, TNF-α) was not influenced, but the ability of infected pDCs to secrete IFN-I was suppressed significantly, in COVID-19 patients (101). Achille et al. found that the ability of cDC1 to produce IFN-λ was not impaired, which may contribute to disruption of the epithelial barrier in the lungs upon SARS-CoV-2 recognition (121). Various studies have shown that SARS-CoV-2 inhibits only pDCs from secreting IFN-I but does not affect cDC production of other proinflammatory cytokines (68, 101, 115, 116). An IFN-I deficiency promotes SARS-CoV-2 to escape recognition by the immune system. Such an escape by SARS-CoV-2 leads to the body producing many inefficient antiviral proinflammatory cytokines that will initiate the cytokine storm that damages lung tissues and results in ARDS or death. Three main reasons have been postulated as to how SARS-CoV-2 may specifically impair the production and transformation of IFN-I (Figure 2).
First, the IFN-I level is reduced significantly in coronavirus infections (e.g., SARS-CoV, MERS-CoV) and such inhibition is a hallmark of coronavirus infections (122–124). Little is known about how SARS-CoV-2 suppresses IFN signaling, but clues can be obtained from SARS-CoV. IFN-I production in SARS-CoV-2 infection is even less than that observed in SARS-CoV infection (124). SARS-CoV-2 encodes various proteins that have been shown to inhibit IFN-I signaling in SARS-CoV infection (122, 124) (Figure 2).
Second, some researchers have found congenital deletion of TLR7 in critically ill patients and that the pDCs of patients with a TLR7 deletion cannot produce IFN-I (125). In addition, anti-IFN antibodies have been found in a small number of critically ill patients; anti-IFN antibodies bind to IFN and prevent IFN transformation (126). The failure of the immunometabolism reprogramming such as the impairment of the glycogenolysis-mediated glycolysis which is stored as a source of energy of DCs will impair the IFN-I generation in DCs (108, 127, 128). SARS-CoV-2 inhibits the early induction of glycolysis via suppression of IFN-I generation, and in turn, the impaired induction of glycolysis inhibits IFN-I generation in DCs (129). This positive feedback loop further reduces IFN-I generation.
Third, the high viremia of COVID-19 is associated with impaired generation of IFN-I in peripheral blood mononuclear cells (130). Supplementation of IFN in the early stage of SARS-CoV-2 infection is beneficial to patients. IFN-β combined with lopinavir and ribavirin can improve physical status (131) and alter the production of proinflammatory cytokines in the latter stages of COVID-19 (97, 132). There is growing recognition that inappropriate, excessive, and mistimed IFN treatments are deleterious in viral infections (131, 133). In the early stage of COVID-19, IFN treatment could improve patient outcomes (122, 131, 134–136), but immunomodulation therapy is recommended at the latter stage of COVID-19 (122, 137, 138). The delayed response to IFN-I leads to accumulation of pathogenic mononuclear macrophages, which cause vascular leakage, lung disorders, and inappropriate T-cell responses (139, 140). Understanding the exact immunopathogenesis of impaired IFN production and restoring the IFN system at the early stage of COVID-19 is a top priority.
4 Conclusions
As a bridge between innate immunity and adaptive immunity, DCs have important roles during virus invasion. The impaired function and reduced numbers of DCs are a catastrophe for the immune system during SARS-CoV-2 infection. The deficiency and dysfunction of DCs persist for several months after SARS-CoV-2 infection (113). Seven months after SARS-CoV-2 infection, the function of cDC2, as well as the number and IFNα production in pDCs, remains abnormal (113). This prolonged deficiency and dysfunction of DCs are associated with “post-acute COVID-19 syndrome” (“long-hauler syndrome”) in COVID-19 patients. It is characterized by persistent symptoms and/or delayed or long-term complications of SARS-CoV-2 infection beyond 4 weeks from symptom onset (141, 142). Scholars have reported that SARS-CoV-2 can persist in the intestines 7 months after symptom resolution (143). We postulate that the persistent tissue damage and presence of viral antigens which are hard to eliminate due to the deficiency and dysfunction of DCs may contribute to long-hauler syndrome in COVID-19 patients.
The morbidity and mortality of older patients are very high if they have severe SARS-CoV-2 infection (144). Age-associated DC dysfunction has a critical role in the mortality prevalence of COVID-19 patients (103). Age may contribute to the reduction and IFN dysfunction of pDCs and antigen-presentation inhibition of cDC to CD8+ T cells, which hinders the transition from naïve CD8+ T cells to cytotoxic CD8+ T cells (145). In older patients, DC dysfunction leads to an increased proinflammatory response and decreased anti-inflammatory and immunomodulatory responses, which result in a chronic inflammatory state (103). In SARS-CoV-2 infection, DC dysfunction could lead to uncontrolled infection and exacerbate the cytokine storm in older patients (103).
As important antigen-presenting cells, DCs have a critical role in the immunotherapy of SARS-CoV-2. Development of immunotherapies against SARS-CoV-2 includes classic platforms and next-generation platforms. The ongoing vaccine research on classic platforms includes whole-inactive viruses, live-attenuated viruses, protein subunits, and virus-like particles. Next-generation platforms include viral vectors, DNA, RNA, and antigen-presenting cells. LV-SMENP-DC and pathogen-specific artificial antigen-presenting cells (aAPC) from Shenzhen Genoimmune Medical Institute (Shenzhen, China) have recently moved into phase-I clinical development. LV-SMENP-DC is an antigen-presenting-cell vaccine based on DCs. In LV-SMENP-DC, DCs are modified with a lentiviral vector expressing a “synthetic minigene” based on the domains of selected viral proteins, and LV-SMENP-DC is administered with antigen-specific cytotoxic T-cells (NCT04276896) (146, 147).
This review shows that DCs have vital roles in SARS-CoV-2 infection. Studying the relationship between DCs and SARS-CoV-2 is very important. Novel DC-induced immunotherapy strategies for COVID-19 may be discovered in the near future.
Author Contributions
T-DC and Z-HT participated in the design and drafting of the manuscript. JZY, HD, and DC participated in critical discussions and revised the manuscript. TC prepared the figures. Z-HT supervised the project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the National Natural Science Foundation of China (81873870).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579(7798):270. doi: 10.1038/s41586-020-2012-7
2. Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. Identification of a Novel Coronavirus in Patients With Severe Acute Respiratory Syndrome. N Engl J Med (2003) 348(20):1967–76. doi: 10.1056/NEJMoa030747
3. Zhong NS, Zheng BJ, Li YM, Poon LLM, Xie ZH, Chan KH, et al. Epidemiology and Cause of Severe Acute Respiratory Syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet (2003) 362(9393):1353–8. doi: 10.1016/s0140-6736(03)14630-2
4. Arabi YM, Balkhy HH, Hayden FG, Bouchama A, Luke T, Baillie JK, et al. Middle East Respiratory Syndrome. N Engl J Med (2017) 376(6):584–94. doi: 10.1056/NEJMsr1408795
5. Resta S, Luby JP, Rosenfeld CR, Siegel JD. Isolation and Propagation of a Human Enteric Coronavirus. Science (New York NY) (1985) 229(4717):978–81. doi: 10.1126/science.2992091
6. Sun J, He W-T, Wang L, Lai A, Ji X, Zhai X, et al. COVID-19: Epidemiology, Evolution, and Cross-Disciplinary Perspectives. Trends Mol Med (2020) 26(5):483–95. doi: 10.1016/j.molmed.2020.02.008
7. Tu Y-F, Chien C-S, Yarmishyn AA, Lin Y-Y, Luo Y-H, Lin Y-T, et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int J Mol Sci (2020) 21(7):2567. doi: 10.3390/ijms21072657
8. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity With SARS-CoV. Nat Commun (2020) 11(1):1620. doi: 10.1038/s41467-020-15562-9
9. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host Cell Death and Inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi: 10.1038/nrmicro2070
10. Fink SL, Cookson BT. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect Immun (2005) 73(4):1907–16. doi: 10.1128/iai.73.4.1907-1916.2005
11. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat Rev Immunol (2020) 20(6):363–74. doi: 10.1038/s41577-020-0311-8
12. Yamada T, Sato S, Sotoyama Y, Orba Y, Sawa H, Yamauchi H, et al. RIG-I Triggers a Signaling-Abortive Anti-SARS-CoV-2 Defense in Human Lung Cells. Nat Immunol (2021) 22(7):820. doi: 10.1038/s41590-021-00942-0
13. Yang DM, Geng TT, Harrison AG, Wang PH. Differential Roles of RIG-I Like Receptors in SARS-CoV-2 Infection. Military Med Res (2021) 8(1). doi: 10.1186/s40779-021-00340-5
14. van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. Jama-J Am Med Assoc (2020) 324(7):663–73. doi: 10.1001/jama.2020.13719
15. Schneider WM, Chevillotte MD, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
16. Rouse BT, Sehrawat S. Immunity and Immunopathology to Viruses: What Decides the Outcome? Nat Rev Immunol (2010) 10(7):514–26. doi: 10.1038/nri2802
17. Hart DNJ. Dendritic Cells: Unique Leukocyte Populations Which Control the Primary Immune Response. Blood (1997) 90(9):3245–87. doi: 10.1182/blood.V90.9.3245
18. Matzinger P. Tolerance, Danger, and the Extended Family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
19. Steinman RM. The Dendritic Cell System and Its Role in Immunogenicity. Annu Rev Immunol (1991) 9:271–96. doi: 10.1146/annurev.iy.09.040191.001415
20. Vardhana SA, Wolchok JD. The Many Faces of the Anti-COVID Immune Response. J Exp Med (2020) 217(6):e20200678. doi: 10.1084/jem.20200678
21. Frank D, Vince JE. Pyroptosis Versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ (2019) 26(1):99–114. doi: 10.1038/s41418-018-0212-6
22. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, SdSG D, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 Engages Inflammasome and Pyroptosis in Human Primary Monocytes. Cell Death Discov (2021) 7(1):43. doi: 10.1038/s41420-021-00477-1
23. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine Storm and Leukocyte Changes in Mild Versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J Leukocyte Biol (2020) 108(1):17–41. doi: 10.1002/jlb.3covr0520-272r
24. Gheblawi M, Wang K, Viveiros A, Quynh N, Zhong J-C, Turner AJ, et al. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System Celebrating the 20th Anniversary of the Discovery of ACE2. Circ Res (2020) 126(10):1456–74. doi: 10.1161/circresaha.120.317015
25. Hoffmann M, Kleine-Weber H, Schroeder S, Krueger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271. doi: 10.1016/j.cell.2020.02.052
26. Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, et al. Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PloS One (2012) 7(4):e35876. doi: 10.1371/journal.pone.0035876
27. Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T, et al. SARS-CoV-2 Receptor ACE2 and TMPRSS2 Are Primarily Expressed in Bronchial Transient Secretory Cells. EMBO J (2020) 39(10):e105114. doi: 10.15252/embj.20105114
28. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of Oxidative Stress and Toll-Like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell (2008) 133(2):235–49. doi: 10.1016/j.cell.2008.02.043
29. Zhou F, Yu T, Du R. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study (Vol 395, Pg 1054, 2020). Lancet (2020) 395(10229):1038–. doi: 10.1016/s0140-6736(20)30638-3
30. Woelfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Mueller MA, et al. Virological Assessment of Hospitalized Patients With COVID-2019. Nature (2020) 581(7809):465. doi: 10.1038/s41586-020-2196-x
31. Han Y, Zhang H, Mu S, Wei W, Jin C, Tong C, et al. Lactate Dehydrogenase, an Independent Risk Factor of Severe COVID-19 Patients: A Retrospective and Observational Study. Aging-Us (2020) 12(12):11245–58. doi: 10.18632/aging.103372
32. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of COVID-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/s2213-2600(20)30076-x
33. Chen Z, John Wherry E. T Cell Responses in Patients With COVID-19. Nat Rev Immunol (2020) 20(9):529–36. doi: 10.1038/s41577-020-0402-6
34. Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological Findings and Complications of COVID-19. Am J Hematol (2020) 95(7):834–47. doi: 10.1002/ajh.25829
35. Sanchez-Cerrillo I, Landete P, Aldave B, Sanchez-Alonso S, Sanchez-Azofra A, Marcos-Jimenez A, et al. COVID-19 Severity Associates With Pulmonary Redistribution of CD1c(+) DCs and Inflammatory Transitional and Nonclassical Monocytes. J Clin Invest (2020) 130(12):6290–300. doi: 10.1172/jci140335
36. Campana P, Parisi V, Leosco D, Bencivenga D, Della Ragione F, Borriello A. Dendritic Cells and SARS-CoV-2 Infection: Still an Unclarified Connection. Cells (2020) 9(9):2046. doi: 10.3390/cells9092046
37. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe (2020) 27(6):883. doi: 10.1016/j.chom.2020.04.017
38. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in COVID-19 Patients. Emerg Microbes Infections (2020) 9(1):761–70. doi: 10.1080/22221751.2020.1747363
39. Collin M, Bigley V. Human Dendritic Cell Subsets: An Update. Immunology (2018) 154(1):3–20. doi: 10.1111/imm.12888
40. Padoan A, Sciacovelli L, Basso D, Negrini D, Zuin S, Cosma C, et al. IgA-Ab Response to Spike Glycoprotein of SARS-CoV-2 in Patients With COVID-19: A Longitudinal Study. Clin Chimica Acta (2020) 507:164–6. doi: 10.1016/j.cca.2020.04.026
41. Martin-Gayo E, Gao C, Chen HR, Ouyang ZY, Kim D, Kolb KE, et al. Immunological Fingerprints of Controllers Developing Neutralizing HIV-1 Antibodies. Cell Rep (2020) 30(4):984. doi: 10.1016/j.celrep.2019.12.087
42. Law HKW, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine Up-Regulation in SARS-Coronavirus-Infected, Monocyte-Derived Human Dendritic Cells. Blood (2005) 106(7):2366–74. doi: 10.1182/blood-2004-10-4166
43. Guo Y, Feinberg H, Conroy E, Mitchell DA, Alvarez R, Blixt O, et al. Structural Basis for Distinct Ligand-Binding and Targeting Properties of the Receptors DC-SIGN and DC-SIGNR. Nat Struct Mol Biol (2004) 11(7):591–8. doi: 10.1038/nsmb784
44. Ghazi L, Drawz P. Advances in Understanding the Renin-Angiotensin-Aldosterone System (RAAS) in Blood Pressure Control and Recent Pivotal Trials of RAAS Blockade in Heart Failure and Diabetic Nephropathy. F1000Research (2017) 6:F10000 Faculty Rev-297. doi: 10.12688/f1000research.9692.1
45. Tikellis C, Thomas MC. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int J Peptides (2012) 2012:256294. doi: 10.1155/2012/256294
46. Balakumar P, Jagadeesh G. A Century Old Renin-Angiotensin System Still Grows With Endless Possibilities: AT(1) Receptor Signaling Cascades in Cardiovascular Physiopathology. Cell Signalling (2014) 26(10):2147–60. doi: 10.1016/j.cellsig.2014.06.011
47. Dandona P, Dhindsa S, Ghanim H, Chaudhuri A. Angiotensin II and Inflammation: The Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockade. J Hum Hypertension (2007) 21(1):20–7. doi: 10.1038/sj.jhh.1002101
48. Yan T, Xiao R, Lin G. Angiotensin-Converting Enzyme 2 in Severe Acute Respiratory Syndrome Coronavirus and SARS-CoV-2: A Double-Edged Sword? FASEB J (2020) 34(5):6017–26. doi: 10.1096/fj.202000782
49. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and Biochemical Indexes From 2019-Ncov Infected Patients Linked to Viral Loads and Lung Injury. Sci China-Life Sci (2020) 63(3):364–74. doi: 10.1007/s11427-020-1643-8
50. Yang PH, Gu HJ, Zhao ZP, Wang W, Cao B, Lai CC, et al. Angiotensin-Converting Enzyme 2 (ACE2) Mediates Influenza H7N9 Virus-Induced Acute Lung Injury. Sci Rep (2014) 4:7027. doi: 10.1038/srep07027
51. Bavishi C, Maddox TM, Messerli FH. Coronavirus Disease 2019 (COVID-19) Infection and Renin Angiotensin System Blockers. JAMA Cardiol (2020) 5(7):745–7. doi: 10.1001/jamacardio.2020.1282
52. Pioli MR, de Faria AP. Pro-Inflammatory Cytokines and Resistant Hypertension: Potential for Novel Treatments? Curr Hypertens Rep (2019) 21(12):95. doi: 10.1007/s11906-019-1003-2
53. Xie XD, Chen JZ, Wang XX, Zhang FR, Liu YR. Age-And Gender-Related Difference of ACE2 Expression in Rat-Lung (Vol 79, Pg 2499, 2006). Life Sci (2006) 79(26):2499–. doi: 10.1016/j.lfs.2006.09.028
54. Cai H. Sex Difference and Smoking Predisposition in Patients With COVID-19. Lancet Respir Med (2020) 8(4):e20. doi: 10.1016/S2213-2600(20)30117-X
55. Driggin E, Madhavan MV, Bikdeli B, Chuich T, Laracy J, Biondi-Zoccai G, et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems During the COVID-19 Pandemic. J Am Coll Cardiol (2020) 75(18):2352–71. doi: 10.1016/j.jacc.2020.03.031
56. Danilov SM, Sadovnikova E, Scharenborg N, Balyasnikova IV, Svinareva DA, Semikina EL, et al. Angiotensin-Converting Enzyme (CD143) Is Abundantly Expressed by Dendritic Cells and Discriminates Human Monocyte-Derived Dendritic Cells From Acute Myeloid Leukemia-Derived Dendritic Cells. Exp Hematol (2003) 31(12):1301–9. doi: 10.1016/j.exphem.2003.08.018
57. Eisenlohr LC, Bacik I, Bennink JR, Bernstein K, Yewdell JW. Expression of a Membrane Protease Enhances Presentation of Endogenous Antigens to MHC Class I-Restricted T Lymphocytes. Cell (1992) 71(6):963–72. doi: 10.1016/0092-8674(92)90392-p
58. Cao D-Y, Saito S, Veiras LC, Okwan-Duodu D, Bernstein EA, Giani JF, et al. Role of Angiotensin-Converting Enzyme in Myeloid Cell Immune Responses. Cell Mol Biol Lett (2020) 25(1):31. doi: 10.1186/s11658-020-00225-w
59. Shen XZ, Billet S, Lin C, Okwan-Duodu D, Chen X, Lukacher AE, et al. The Carboxypeptidase ACE Shapes the MHC Class I Peptide Repertoire. Nat Immunol (2011) 12(11):1078–85. doi: 10.1038/ni.2107
60. Shen XZ, Lukacher AE, Billet S, Williams IR, Bernstein KE. Expression of Angiotensin-Converting Enzyme Changes Major Histocompatibility Complex Class I Peptide Presentation by Modifying C Termini of Peptide Precursors. J Biol Chem (2008) 283(15):9957–65. doi: 10.1074/jbc.M709574200
61. Nie W, Yan H, Li S, Zhang Y, Yu F, Zhu W, et al. Angiotensin-(1-7) Enhances Angiotensin II Induced Phosphorylation of ERK1/2 in Mouse Bone Marrow-Derived Dendritic Cells. Mol Immunol (2009) 46(3):355–61. doi: 10.1016/j.molimm.2008.10.022
62. Lapteva N, Ide K, Nieda M, Ando Y, Hatta-Ohashi Y, Minami M, et al. Activation and Suppression of Renin-Angiotensin System in Human Dendritic Cells. Biochem Biophys Res Commun (2002) 296(1):194–200. doi: 10.1016/s0006-291x(02)00855-0
63. Liaudet L, Szabo C. Blocking Mineralocorticoid Receptor With Spironolactone may Have a Wide Range of Therapeutic Actions in Severe COVID-19 Disease. Crit Care (2020) 24(1):318. doi: 10.1186/s13054-020-03055-6
64. Brown NJ. Contribution of Aldosterone to Cardiovascular and Renal Inflammation and Fibrosis. Nat Rev Nephrol (2013) 9(8):459–69. doi: 10.1038/nrneph.2013.110
65. Jia H. PULMONARY ANGIOTENSIN-CONVERTING ENZYME 2 (ACE2) AND INFLAMMATORY LUNG DISEASE. Shock (2016) 46(3):239–48. doi: 10.1097/shk.0000000000000633
66. Kuba K, Imai Y, Rao SA, Gao H, Guo F, Guan B, et al. A Crucial Role of Angiotensin Converting Enzyme 2 (ACE2) in SARS Coronavirus-Induced Lung Injury. Nat Med (2005) 11(8):875–9. doi: 10.1038/nm1267
67. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-Converting Enzyme 2 Protects From Severe Acute Lung Failure. Nature (2005) 436(7047):112–6. doi: 10.1038/nature03712
68. Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC-Y, et al. Attenuated Interferon and Proinflammatory Response in SARS-CoV-2-Infected Human Dendritic Cells Is Associated With Viral Antagonism of STAT1 Phosphorylation. J Infect Dis (2020) 222(5):734–45. doi: 10.1093/infdis/jiaa356
69. Bashirova AA, Wu L, Cheng J, Martin TD, Martin MP, Benveniste RE, et al. Novel Member of the CD209 (DC-SIGN) Gene Family in Primates. J Virol (2003) 77(1):217–27. doi: 10.1128/jvi.77.1.217-227.2003
70. Geijtenbeek TBH, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GCF, Middel J, et al. DC-SIGN, a Dendritic Cell-Specific HIV-1-Binding Protein That Enhances Trans-Infection of T Cells. Cell (2000) 100(5):587–97. doi: 10.1016/s0092-8674(00)80694-7
71. Geijtenbeek TBH, Torensma R, van Vliet SJ, van Duijnhoven GCF, Adema GJ, van Kooyk Y, et al. Identification of DC-SIGN, a Novel Dendritic Cell-Specific ICAM-3 Receptor That Supports Primary Immune Responses. Cell (2000) 100(5):575–85. doi: 10.1016/s0092-8674(00)80693-5
72. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YT, et al. Immunobiology of Dendritic Cells. Annu Rev Immunol (2000) 18:767. doi: 10.1146/annurev.immunol.18.1.767
73. van Kooyk Y, Geijtenbeek TBH. DC-Sign: Escape Mechanism for Pathogens. Nat Rev Immunol (2003) 3(9):697–709. doi: 10.1038/nri1182
74. Bogoevska V, Horst A, Klampe B, Lucka L, Wagener C, Nollau P. CEACAM1, an Adhesion Molecule of Human Granulocytes, Is Fucosylated by Fucosyltransferase IX and Interacts With DC-SIGN of Dendritic Cells via Lewis X Residues. Glycobiology (2006) 16(3):197–209. doi: 10.1093/glycob/cwj057
75. Zhou T, Chen Y, Hao L, Zhang Y. DC-SIGN and Immunoregulation. Cell Mol Immunol (2006) 3(4):279–83.
76. Bleijs DA, Geijtenbeek TBH, Figdor CG, van Kooyk Y. DC-SIGN and LFA-1: A Battle for Ligand. Trends Immunol (2001) 22(8):457–63. doi: 10.1016/s1471-4906(01)01974-3
77. Engering A, Geijtenbeek TBH, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, et al. The Dendritic Cell-Specific Adhesion Receptor DC-SIGN Internalizes Antigen for Presentation to T Cells. J Immunol (2002) 168(5):2118–26. doi: 10.4049/jimmunol.168.5.2118
78. Engering A, Geijtenbeek TBH, van Kooyk Y. Immune Escape Through C-Type Lectins on Dendritic Cells. Trends Immunol (2002) 23(10):480–5. doi: 10.1016/s1471-4906(02)02296-2
79. McDonald D. Dendritic Cells and HIV-1 Trans-Infection. Viruses-Basel (2010) 2(8):1404–717. doi: 10.3390/v2081704
80. Jan M, Upadhyay C, Hioe CE. HIV-1 Envelope Glycan Composition as a Key Determinant of Efficient Virus Transmission via DC-SIGN and Resistance to Inhibitory Lectins. Iscience (2019) 21:413. doi: 10.1016/j.isci.2019.10.030
81. Marzi A, Gramberg T, Simmons G, Moller P, Rennekamp AJ, Krumbiegel M, et al. DC-SIGN and DC-SIGNR Interact With the Glycoprotein of Marburg Virus and the S Protein of Severe Acute Respiratory Syndrome Coronavirus. J Virol (2004) 78(21):12090–5. doi: 10.1128/jvi.78.21.12090-12095.2004
82. Feinberg H, Mitchell DA, Drickamer K, Weis WI. Structural Basis for Selective Recognition of Oligosaccharides by DC-SIGN and DC-SIGNR. Science (2001) 294(5549):2163–6. doi: 10.1126/science.1066371
83. Chu H, Zhou J, Wong BH-Y, Li C, Cheng Z-S, Lin X, et al. Productive Replication of Middle East Respiratory Syndrome Coronavirus in Monocyte-Derived Dendritic Cells Modulates Innate Immune Response. Virology (2014) 454:197–205. doi: 10.1016/j.virol.2014.02.018
84. Li L, Dong L, Zhao D, Gao F, Yan J. Classical Dendritic Cells Regulate Acute Lung Inflammation and Injury in Mice With Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome. Int J Mol Med (2019) 44(2):617–29. doi: 10.3892/ijmm.2019.4208
85. Amraei R, Yin W, Napoleon MA, Suder EL, Berrigan J, Zhao Q, et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent Sci (2021) 7(7):1156–65. doi: 10.1021/acscentsci.0c01537
86. Deng H, Tang TX, Chen D, Tang LS, Yang XP, Tang ZH. Endothelial Dysfunction and SARS-CoV-2 Infection: Association and Therapeutic Strategies. Pathogens (2021) 10(5):582. doi: 10.3390/pathogens10050582
87. Cai G, Du M, Bosse Y, Albrecht H, Qin F, Luo X, et al. SARS-CoV-2 Impairs Dendritic Cells and Regulates DC-SIGN Gene Expression in Tissues. Int J Mol Sci (2021) 22(17):9228. doi: 10.3390/ijms22179228
88. Thepaut M, Luczkowiak J, Vives C, Labiod N, Bally I, Lasala F, et al. Dc/L-SIGN Recognition of Spike Glycoprotein Promotes SARS-CoV-2 Trans-Infection and can be Inhibited by a Glycomimetic Antagonist. PloS Pathog (2021) 17(5):e1009576. doi: 10.1371/journal.ppat.1009576
89. Segura E, Valladeau-Guilemond J, Donnadieu M-H, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of Resident and Migratory Dendritic Cells in Human Lymph Nodes. J Exp Med (2012) 209(4):653–60. doi: 10.1084/jem.20111457
90. Mittag D, Proietto AI, Loudovaris T, Mannering SI, Vremec D, Shortman K, et al. Human Dendritic Cell Subsets From Spleen and Blood Are Similar in Phenotype and Function But Modified by Donor Health Status. J Immunol (2011) 186(11):6207–17. doi: 10.4049/jimmunol.1002632
91. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141(+) (BDCA-3)(+) Dendritic Cells (DCs) Represent a Unique Myeloid DC Subset That Cross-Presents Necrotic Cell Antigens. J Exp Med (2010) 207(6):1247–60. doi: 10.1084/jem.20092140
92. Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, et al. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics Over Human Life. Immunity (2017) 46(3):504–15. doi: 10.1016/j.immuni.2017.02.019
93. Franz Poulin L, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen J-L, et al. Characterization of Human DNGR-1(+) BDCA3(+) Leukocytes as Putative Equivalents of Mouse CD8 Alpha(+) Dendritic Cells. J Exp Med (2010) 207(6):1261–71. doi: 10.1084/jem.20092618
94. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science (1999) 284(5421):1835–7. doi: 10.1126/science.284.5421.1835
95. Sittig SP, Bakdash G, Weiden J, Skold AE, Tel J, Figdor CG, et al. A Comparative Study of the T Cell Stimulatory and Polarizing Capacity of Human Primary Blood Dendritic Cell Subsets. Mediators Inflammation (2016) 2016::3605643. doi: 10.1155/2016/3605643
96. Nizzoli G, Larghi P, Paroni M, Crosti MC, Moro M, Neddermann P, et al. IL-10 Promotes Homeostatic Proliferation of Human CD8(+) Memory T Cells and, When Produced by CD1c(+) DCs, Shapes Naive CD8(+) T-Cell Priming. Eur J Immunol (2016) 46(7):1622–32. doi: 10.1002/eji.201546136
97. Chen D, Tang TX, Deng H, Yang XP, Tang ZH. Interleukin-7 Biology and Its Effects on Immune Cells: Mediator of Generation, Differentiation, Survival, and Homeostasis. Front Immunol (2021) 12:747324. doi: 10.3389/fimmu.2021.747324
98. Swiecki M, Colonna M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat Rev Immunol (2015) 15(8):471–85. doi: 10.1038/nri3865
99. Bao M, Liu Y-J. Regulation of TLR7/9 Signaling in Plasmacytoid Dendritic Cells. Protein Cell (2013) 4(1):40–52. doi: 10.1007/s13238-012-2104-8
100. Winheim E, Rinke L, Lutz K, Reischer A, Leutbecher A, Wolfram L, et al. Impaired Function and Delayed Regeneration of Dendritic Cells in COVID-19. PloS Pathog (2021) 17(10):e1009742. doi: 10.1371/journal.ppat.1009742
101. Zhou R, To KK-W, Wong Y-C, Liu L, Zhou B, Li X, et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity (2020) 53(4):864. doi: 10.1016/j.immuni.2020.07.026
102. Laing AG, Lorenc A, del Molino del Barrio I, Das A, Fish M, Monin L, et al. A Dynamic COVID-19 Immune Signature Includes Associations With Poor Prognosis (Vol 56, Pg 512, 2020). Nat Med (2020) 26(10):1663–. doi: 10.1038/s41591-020-1079-x
103. Borges RC, Hohmann MS, Borghi SM. Dendritic Cells in COVID-19 Immunopathogenesis: Insights for a Possible Role in Determining Disease Outcome. Int Rev Immunol (2021) 40(1-2):108–25. doi: 10.1080/08830185.2020.1844195
104. Lopez-Bergami P, Barbero G. The Emerging Role of Wnt5a in the Promotion of a Pro-Inflammatory and Immunosuppressive Tumor Microenvironment. Cancer Metastasis Rev (2020) 39(3):933–52. doi: 10.1007/s10555-020-09878-7
105. Saichi M, Ladjemi MZ, Korniotis S, Rousseau C, Ait Hamou Z, Massenet-Regad L, et al. Single-Cell RNA Sequencing of Blood Antigen-Presenting Cells in Severe COVID-19 Reveals Multi-Process Defects in Antiviral Immunity. Nat Cell Biol (2021) 23(5):538. doi: 10.1038/s41556-021-00681-2
106. Liu C, Martins AJ, Lau WW, Rachmaninoff N, Chen J, Imberti L, et al. Time-Resolved Systems Immunology Reveals a Late Juncture Linked to Fatal COVID-19. Cell (2021) 184(7):1836. doi: 10.1016/j.cell.2021.02.018
107. Onodi F, Bonnet-Madin L, Meertens L, Karpf L, Poirot J, Zhang S-Y, et al. SARS-CoV-2 Induces Human Plasmacytoid Predendritic Cell Diversification via UNC93B and IRAK4. J Exp Med (2021) 218(4):e20201387. doi: 10.1084/jem.20201387
108. Kumar V. How Could We Forget Immunometabolism in SARS-CoV2 Infection or COVID-19? Int Rev Immunol (2021) 40(1-2):72–107. doi: 10.1080/08830185.2020.1840567
109. Lau YL, Peiris JSM, Law HKW. Role of Dendritic Cells in SARS Coronavirus Infection. Hong Kong Med J Xianggang Yi Xue Za Zhi (2012) 18 Suppl 3:28–30.
110. Kumar V. Dendritic Cells in Sepsis: Potential Immunoregulatory Cells With Therapeutic Potential. Mol Immunol (2018) 101:615–26. doi: 10.1016/j.molimm.2018.07.007
111. Falschlehner C, Schaefer U, Walczak H. Following Trail’s Path in the Immune System. Immunology (2009) 127(2):145–54. doi: 10.1111/j.1365-2567.2009.03058.x
112. Ciscato F, Filadi R, Masgras I, Pizzi M, Marin O, Damiano N, et al. Hexokinase 2 Displacement From Mitochondria-Associated Membranes Prompts Ca2+-Dependent Death of Cancer Cells. EMBO Rep (2020) 21(7):e49117. doi: 10.15252/embr.201949117
113. Perez-Gomez A, Vitalle J, Gasca-Capote C, Gutierrez-Valencia A, Trujillo-Rodriguez M, Serna-Gallego A, et al. Dendritic Cell Deficiencies Persist Seven Months After SARS-CoV-2 Infection. Cell Mol Immunol (2021) 18(9):2128–39. doi: 10.1038/s41423-021-00728-2
114. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
115. Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, et al. Systems Biological Assessment of Immunity to Mild Versus Severe COVID-19 Infection in Humans. Science (2020) 369(6508):1210. doi: 10.1126/science.abc6261
116. Parackova Z, Zentsova I, Bloomfield M, Vrabcova P, Smetanova J, Klocperk A, et al. Disharmonic Inflammatory Signatures in COVID-19: Augmented Neutrophils’ But Impaired Monocytes’ and Dendritic Cells’ Responsiveness. Cells (2020) 9(10):2206. doi: 10.3390/cells9102206
117. Sukhbaatar N, Hengstschlaeger M, Weichhart T. mTOR-Mediated Regulation of Dendritic Cell Differentiation and Function. Trends Immunol (2016) 37(11):778–89. doi: 10.1016/j.it.2016.08.009
118. Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory Functions of mTOR Inhibition. Nat Rev Immunol (2009) 9(5):324–37. doi: 10.1038/nri2546
119. Zhou Z, Chen H, Xie R, Wang H, Li S, Xu Q, et al. Epigenetically Modulated FOXM1 Suppresses Dendritic Cell Maturation in Pancreatic Cancer and Colon Cancer. Mol Oncol (2019) 13(4):873–93. doi: 10.1002/1878-0261.12443
120. Choi EY, Park HH, Kim H, Kim HN, Kim I, Jeon S, et al. Wnt5a and Wnt11 as Acute Respiratory Distress Syndrome Biomarkers for Severe Acute Respiratory Syndrome Coronavirus 2 Patients. Eur Respir J (2020) 56(5):2001531. doi: 10.1183/13993003.01531-2020
121. Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, et al. Type III Interferons Disrupt the Lung Epithelial Barrier Upon Viral Recognition. Science (2020) 369(6504):706. doi: 10.1126/science.abc3545
122. Ribero MS, Jouvenet N, Dreux M, Nisole S. Interplay Between SARS-CoV-2 and the Type I Interferon Response. PloS Pathog (2020) 16(7):e1008737. doi: 10.1371/journal.ppat.1008737
123. Chan RWY, Chan MCW, Agnihothram S, Chan LLY, Kuok DIT, Fong JHM, et al. Tropism of and Innate Immune Responses to the Novel Human Betacoronavirus Lineage C Virus in Human Ex Vivo Respiratory Organ Cultures. J Virol (2013) 87(12):6604–14. doi: 10.1128/jvi.00009-13
124. Chan JF-W, Kok K-H, Zhu Z, Chu H, To KK-W, Yuan S, et al. Genomic Characterization of the 2019 Novel Human-Pathogenic Coronavirus Isolated From a Patient With Atypical Pneumonia After Visiting Wuhan. Emerg Microbes Infections (2020) 9(1):221–36. doi: 10.1080/22221751.2020.1719902
125. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn Errors of Type I IFN Immunity in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):422. doi: 10.1126/science.abd4570
126. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Autoantibodies Against Type I IFNs in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):423. doi: 10.1126/science.abd4585
127. Thwe PM, Pelgrom L, Cooper R, Beauchamp S, Reisz JA, D’Alessandro A, et al. Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses. Cell Metab (2017) 26(3):558. doi: 10.1016/j.cmet.2017.08.012
128. Curtis KD, Smith PR, Despres HW, Snyder JP, Hogan TC, Rodriguez PD, et al. Glycogen Metabolism Supports Early Glycolytic Reprogramming and Activation in Dendritic Cells in Response to Both TLR and Syk-Dependent CLR Agonists. Cells (2020) 9(3):715. doi: 10.3390/cells9030715
129. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-Driven Early Glycolytic Reprogramming via the Kinases TBK1-IKK Epsilon Supports the Anabolic Demands of Dendritic Cell Activation. Nat Immunol (2014) 15(4):323. doi: 10.1038/ni.2833
130. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science (2020) 369(6504):718. doi: 10.1126/science.abc6027
131. Hung IF-N, Lung K-C, Tso EY-K, Liu R, Chung TW-H, Chu M-Y, et al. Triple Combination of Interferon Beta-1b, Lopinavir-Ritonavir, and Ribavirin in the Treatment of Patients Admitted to Hospital With COVID-19: An Open-Label, Randomised, Phase 2 Trial. Lancet (2020) 395(10238):1695–704. doi: 10.1016/s0140-6736(20)31042-4
132. Buttenschoen J, Mattner J. The Interplay Between Dendritic Cells and CD8 T Lymphocytes Is a Crucial Component of SARS-CoV-2 Immunity. Cell Mol Immunol (2021) 18(2):247–9. doi: 10.1038/s41423-020-00624-1
133. Davidson S, Maini MK, Wack A. Disease-Promoting Effects of Type I Interferons in Viral, Bacterial, and Coinfections. J Interferon Cytokine Res (2015) 35(4):252–64. doi: 10.1089/jir.2014.0227
134. Zhou Q, Chen V, Shannon CP, Wei X-S, Xiang X, Wang X, et al. Interferon-Alpha 2b Treatment for COVID-19. Front Immunol (2020) 11:1061. doi: 10.3389/fimmu.2020.01061
135. Laube BL. The Expanding Role of Aerosols in Systemic Drug Delivery, Gene Therapy and Vaccination: An Update. Trans Respir Med (2014) 2:3–. doi: 10.1186/2213-0802-2-3
136. Dhanani J, Fraser JF, Chan H-K, Rello J, Cohen J, Roberts JA. Fundamentals of Aerosol Therapy in Critical Care. Crit Care (2016) 20(1):269. doi: 10.1186/s13054-016-1448-5
137. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, et al. Endothelial Cells Are Central Orchestrators of Cytokine Amplification During Influenza Virus Infection. Cell (2011) 146(6):980–91. doi: 10.1016/j.cell.2011.08.015
138. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans With COVID-19 Disease and Unexposed Individuals. Cell (2020) 181(7):1489. doi: 10.1016/j.cell.2020.05.015
139. Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, et al. IFN-I Response Timing Relative to Virus Replication Determines MERS Coronavirus Infection Outcomes. J Clin Invest (2019) 129(9):3625–39. doi: 10.1172/jci126363
140. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe (2016) 19(2):181–93. doi: 10.1016/j.chom.2016.01.007
141. Szekanecz Z, Valyi-Nagy I. Post-Acute COVID-19 Syndrome. Orvosi Hetilap (2021) 162(27):1067–78. doi: 10.1556/650.2021.32282
142. Rubin R. As Their Numbers Grow, COVID-19 “Long Haulers” Stump Experts. Jama-J Am Med Assoc (2020) 324(14):1381–3. doi: 10.1001/jama.2020.17709
143. Tokuyama M LM, Jha D, Cossarini F, Livanos AE, Reidy J, Tankelevich M, et al. (2021). SARS-CoV-2 Persists in Intestinal Enterocytes Up to 7 Months After Symptom Resolution. In: Proceedings of the Abstracts from the Virtual CROI 2021 Conference on Retroviruses and Opportunistic Infections. San Francisco, CA, USA (2021) Abstract 115. p. 31.
144. O’Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-Specific Mortality and Immunity Patterns of SARS-CoV-2. Nature (2021) 590(7844):140–5. doi: 10.1038/s41586-020-2918-0
145. Zacca ER, Crespo MI, Acland RP, Roselli E, Nunez NG, Maccioni M, et al. Aging Impairs the Ability of Conventional Dendritic Cells to Cross-Prime CD8(+) T Cells Upon Stimulation With a TLR7 Ligand. PloS One (2015) 10(10):e0140672. doi: 10.1371/journal.pone.0140672
146. van Riel D, de Wit E. Next-Generation Vaccine Platforms for COVID-19. Nat Mater (2020) 19(8):810–2. doi: 10.1038/s41563-020-0746-0
Keywords: COVID-19, SARS-CoV-2, dendritic cells, immunopathogenesis, severe acute respiratory syndrome coronavirus 2
Citation: Chang T, Yang J, Deng H, Chen D, Yang X and Tang Z-H (2022) Depletion and Dysfunction of Dendritic Cells: Understanding SARS-CoV-2 Infection. Front. Immunol. 13:843342. doi: 10.3389/fimmu.2022.843342
Received: 25 December 2021; Accepted: 31 January 2022;
Published: 21 February 2022.
Edited by:
Julia Kzhyshkowska, Heidelberg University, GermanyReviewed by:
Mankgopo Magdeline Kgatle, Nuclear Medicine Research Infrastructure, South AfricaEnrique Martin-Gayo, Fundación de la Universidad Autónoma de Madrid, Spain
Copyright © 2022 Chang, Yang, Deng, Chen, Yang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhao-Hui Tang, dGFuZ3poQHRqaC50am11LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship