 Jack Mellors1,2,3*
Jack Mellors1,2,3* Tom Tipton3Sarah Katharina Fehling4Joseph Akoi Bore5,6Fara Raymond Koundouno6,7Yper Hall1Jacob Hudson1,8,9
Tom Tipton3Sarah Katharina Fehling4Joseph Akoi Bore5,6Fara Raymond Koundouno6,7Yper Hall1Jacob Hudson1,8,9 Frances Alexander1Stephanie Longet3
Frances Alexander1Stephanie Longet3 Stephen Taylor1
Stephen Taylor1 Andrew Gorringe1N’Faly Magassouba10
Andrew Gorringe1N’Faly Magassouba10 Mandy Kader Konde5
Mandy Kader Konde5 Julian Hiscox2
Julian Hiscox2 Thomas Strecker4
Thomas Strecker4 Miles Carroll3*
Miles Carroll3*- 1Department of Research and Evaluation, United Kingdom (UK) Health Security Agency, Salisbury, United Kingdom
- 2Department of Infection Biology, Institute of Infection and Global Health, University of Liverpool, Liverpool, United Kingdom
- 3Wellcome Centre for Human Genetics and the Pandemic Sciences Institute, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- 4Institute of Virology, Philipps University Marburg, Marburg, Germany
- 5Center for Training and Research on Priority Diseases including Malaria in Guinea, Conakry, Guinea
- 6Department of Research, Ministry of Health Guinea, Conakry, Guinea
- 7Department of Virology, Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany
- 8School of Biological Sciences, Faculty of Environmental and Life Sciences, University of Southampton, Southampton, United Kingdom
- 9Department of Biochemical Sciences, School of Biosciences and Medicine, University of Surrey, Surrey, United Kingdom
- 10Viral Haemorrhagic Fever Reference Department, Projet Laboratoire Fièvres Hémorragiques, Conakry, Guinea
The 2013–2016 Ebola virus (EBOV) epidemic in West Africa was unprecedented in case numbers and fatalities, and sporadic outbreaks continue to arise. Antibodies to the EBOV glycoprotein (GP) are strongly associated with survival and their use in immunotherapy is often initially based on their performance in neutralisation assays. Other immune effector functions also contribute to EBOV protection but are more complex to measure. Their interactions with the complement system in particular are comparatively under-researched and commonly excluded from cellular immunoassays. Using EBOV convalescent plasma samples from the 2013–2016 epidemic, we investigated antibody and complement-mediated neutralisation and how these interactions can influence immunity in response to EBOV-GP and its secreted form (EBOV-sGP). We defined two cohorts: one with low-neutralising titres in relation to EBOV-GP IgG titres (LN cohort) and the other with a direct linear relationship between neutralisation and EBOV-GP IgG titres (N cohort). Using flow cytometry antibody-dependent complement deposition (ADCD) assays, we found that the LN cohort was equally efficient at mediating ADCD in response to the EBOV-GP but was significantly lower in response to the EBOV-sGP, compared to the N cohort. Using wild-type EBOV neutralisation assays with a cohort of the LN plasma, we observed a significant increase in neutralisation associated with the addition of pooled human plasma as a source of complement. Flow cytometry ADCD was also applied using the GP of the highly virulent Sudan virus (SUDV) of the Sudan ebolavirus species. There are no licensed vaccines or therapeutics against SUDV and it overlaps in endemicity with EBOV. We found that the LN plasma was significantly less efficient at cross-reacting and mediating ADCD. Overall, we found a differential response in ADCD between LN and N plasma in response to various Ebolavirus glycoproteins, and that these interactions could significantly improve EBOV neutralisation for selected LN plasma samples. Preservation of the complement system in immunoassays could augment our understanding of neutralisation and thus protection against infection
Introduction
Since the 2013–2016 Zaire ebolavirus (EBOV) epidemic in West Africa, outbreaks have continued to arise in Guinea and the Democratic Republic of the Congo (DRC), including the second largest on record in eastern DRC during 2018 which affected over 3,000 people. Protection against EBOV infection is strongly associated with the presence of anti-EBOV-GP neutralising antibodies and this knowledge has supported the development of animal models, vaccines, and therapeutics (1–5). The research efforts in this field have contributed to the FDA licensure of the Ervebo® vaccine (6), EMA marketing authorisation and use of a two-dose heterologous vaccine regimen of Ad26.ZEBOV (Zabdeno®) boosted with MVA-BN-Filo (Mvabea®) (7, 8), and two licensed antibody treatments against EBOV (9). However, the emergence of new variants puts pressure on developing new interventions particularly with the current use of monoclonal antibody treatments, and other highly-virulent Ebolaviruses such as Sudan virus (SUDV) and Bundibugyo virus (BDBV) currently have no licensed therapeutics. A clearer understanding of what determines protection can expedite this process. Much of our current knowledge of neutralising antibodies is first based on their performance in immunoassays, but this method neglects the wider interactions with other aspects of immunity known to influence EBOV pathogenesis, such as the complement system.
The complement system is a network of plasma and membrane-bound proteins that can be divided into three pathways (classical, lectin, alternative) which converge at a single point; the cleavage of the C3 protein (Figure 1). The classical pathway typically requires IgM and/or IgG in complex with the target antigen for C1q binding and pathway activation to occur. The lectin pathway is activated by the interaction of lectins with glycosylated regions of foreign antigens in an antibody-independent manner. The alternative pathway is spontaneously activated through the cleavage of C3 and primarily works to augment the lectin and classical pathways (10).
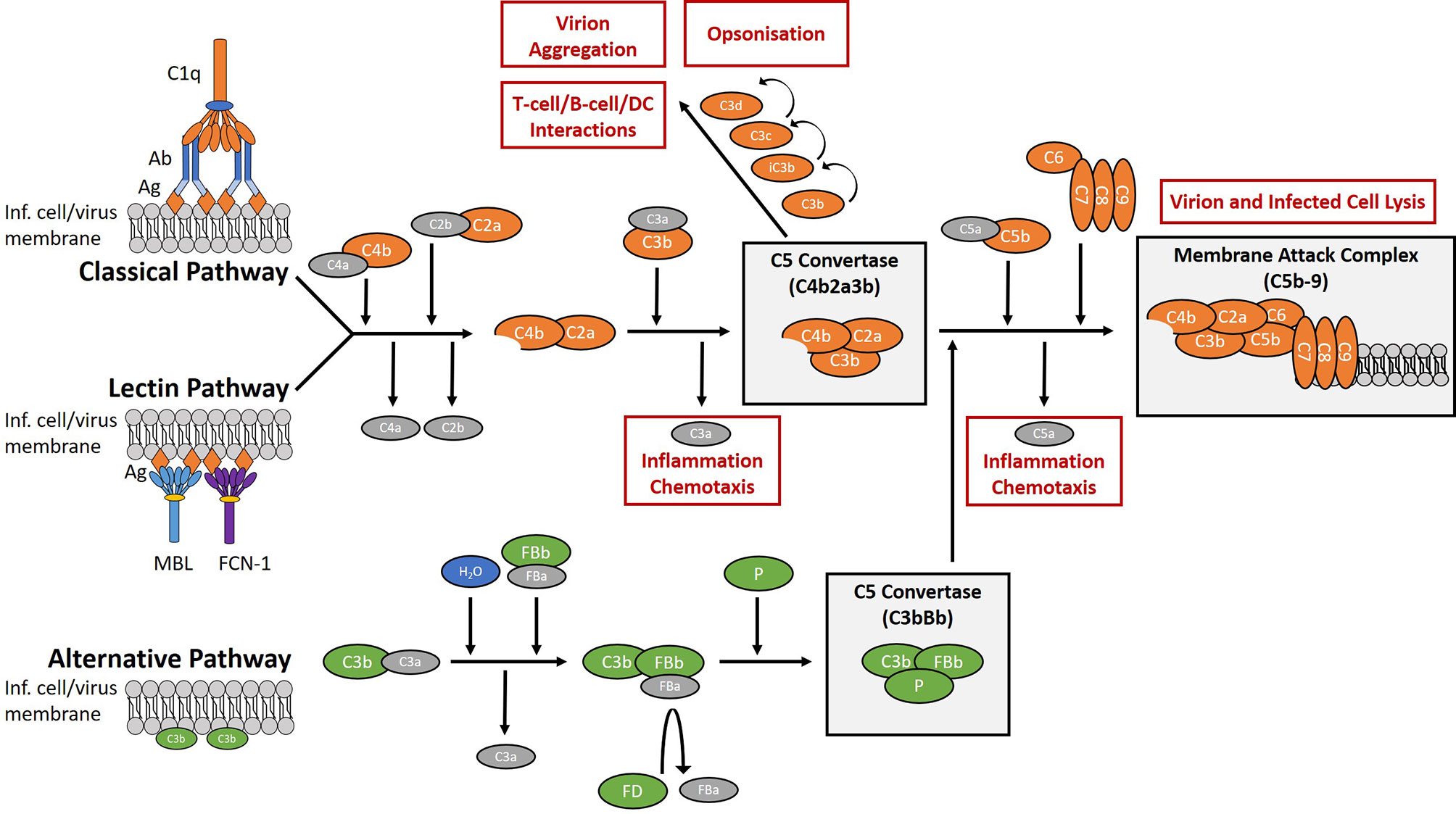
Figure 1 Overview of the complement system. The complement system is a collection of plasma and membrane-bound proteins which form part of the innate immune response against invading pathogens as well as performing other immunological roles. The system can be divided into three pathways (classical, lectin, alternative). The classical pathway is typically antibody-dependent and relies on the binding of C1q protein. The lectin pathway is antibody-independent and utilises lectins such as mannose-binding lectin (MBL) and ficolin-1 (FCN-1) to bind glycosylated regions on the surface antigens of pathogens. The alternative pathway is activated from spontaneous hydrolysis of the C3 protein and an absence of regulatory proteins on the microbial surface. In the context of viral infection, complement activation aims to limit infection through the promotion of inflammation and chemotaxis, the opsonisation of virions, the aggregation of virions, the direct lysis of virion and infected cell membranes, and by aiding the development of the adaptive immune response. (Ab), antibody; (Ag), antigen; (DC), dendritic cells; (FB), factor B; (FD), factor D; (FCN-1), ficolin-1; (Inf.), infected; (MBL), mannose-binding lectin; (P), properdin.
Several studies have reported on the effects of the complement system and EBOV infection. Mannose-binding lectin (MBL) of the lectin pathway has been shown to bind the EBOV-glycoprotein (EBOV-GP), resulting in the cleavage of C4 and inhibition of viral interactions with host receptor DC-SIGN in vitro (11). In vivo, recombinant MBL treatment rescued 40% of mice infected with a lethal dose of mouse-adapted EBOV (12). However, in vitro studies using relatively high concentrations of MBL compared to other complement proteins (13), or ficolin-1 (14), another activator of the lectin pathway, showed an enhancement of EBOV infection into various cell lines including human monocyte-derived macrophages. C1q of the classical pathway has previously been described as a possible mediator of antibody-dependent enhancement (ADE) in EBOV infections, where four distinct epitopes on the EBOV-GP were shown to mediate ADE with the use of monoclonal antibodies and C1q (15, 16), although there have been no reports of ADE following the use of the rVSV-based vaccine Ervebo®. Lastly, some anti-EBOV-GP monoclonal antibodies were shown to only be capable of virus neutralisation in the presence of complement (17). Furthermore, the administration of monoclonal antibodies as therapeutics in mice showed that complete protection against EBOV favoured the more efficient complement-activating murine IgG2a subclass over IgG1 or IgG3 (17). For other pathogenic Ebolaviruses, one report shows the risk of hearing loss in long-term sequelae post-BDBV infection is reduced with antibodies mediating antibody-dependent complement deposition (ADCD) and other polyfunctional responses (18). To our knowledge, there are currently no reports on the interactions with, or the effects of, the complement system with SUDV.
The extent of classical pathway activation in response to pathogens is known to significantly vary between antibodies, depending on factors such as antibody isotype (19–21) and epitope diversity (22–24). Common practices in handling plasma prior to use in immunoassays such as heat-inactivation or EDTA treatment inactivate the complement system and so this aspect of immunity is commonly overlooked (25–29). The importance of antibody-mediated immune effector functions independent from neutralisation have been shown for EBOV infection (30, 31) and polyfunctional immunity strongly correlates with protection (32, 33). Engineering of Fc variants has shown that the ability to mediate ADCD was crucial for complete protection against EBOV infection in an in vivo mouse model (34). The role of complement in EBOV infection is complex and ADCD has implications for pathogenesis and protection, yet remains largely under-researched.
In this study we assess the potential of low-neutralising plasma to engage the complement system as a possible factor in EBOV immunity. We first identified a cohort with low-neutralisation titres in relation to EBOV-GP IgG titres determined via ELISA (LN cohort) and another with a direct linear relationship between neutralisation and EBOV-GP IgG titres (N cohort) to reflect low-neutralising plasma and a control set of plasma, respectively. We used flow cytometry assays to determine the relationship of IgG and ADCD for LN and N EBOV convalescent plasma samples in response to EBOV-GP and its secreted form (EBOV-sGP). To demonstrate a functional effect of the LN plasma with complement, a sub-set of this cohort was used in wild-type EBOV (Makona variant) neutralisation assays supplemented with pooled human plasma (PHP) as a source of complement. Lastly, we adapted these flow cytometry methods to determine the extent of ADCD following cross-reactivity with SUDV-GP. This virus is the second-most virulent species of the Ebolavirus genus that infects humans and has overlapping endemicity to EBOV, yet remains comparatively under-researched.
Methods
Sample Collection and Identification
West African plasma samples (n = 206) were obtained as part of a longitudinal study (2015–2017) from survivors of the 2013–2016 EBOV outbreak and from EBOV negative individuals within the same region who did not come into contact with EBOV patients nor show any symptoms of EBOV disease (EVD) (35). Data collected in this longitudinal study included wild-type EBOV neutralisation assays and anti-EBOV-GP IgG ELISAs, which were correlated using 145 plasma samples available from the year 2017. From this 2017 data set we identified the LN cohort using a maximum neutralisation score cut-off of 130 geometric mean titre (GMT) of four replicates, a minimum antibody titre cut-off of 0.35 optical density (O.D.) at 405 nm, and a maximum residual cut-off from the line of best fit of -100 GMT. The N cohort was selected using a neutralisation titre cut-off greater than 200 GMT and the closest possible residual to the line of best fit to obtain matching cohort numbers. Two additional plasma samples for each cohort were identified using 2017 historical data collected prior to this study and the flow cytometry assays used within this study. Correlations were defined as follows: no correlation (R2 = < 0.200 and P value > 0.050), weak correlation (R2 = 0.210–0.400 and P value < 0.050), moderate correlation (R2 = 0.410–0.700 and P value < 0.050), strong correlation (R2 = 0.710–1.000 and P value < 0.050).
Ethical approval was obtained from the National Ethics Committee for Health Research, Guinea (33/CNERS/15) and from the National Research Ethics Service, UK for the collection and use of West African EBOV negative and convalescent plasma. All volunteers were informed of the purpose and procedures of the study and only consenting participants were included. PHP anti-coagulated with hirudin to preserve complement activity was collected from volunteers in the UK as previously described (36) and used as the exogenous source of complement for the flow cytometry and neutralisation assays described in this study.
Protein Conjugation to Fluorescent Beads
EBOV-GP (Makona strain sourced from Nuffield Department of Medicine, Oxford University, Oxford, UK. GenBank Accession: AHX24649.1) (35), EBOV-sGP (Mayinga strain sourced from IBT Bioservices. GenBank Accession: AHC70242.1), and SUDV-GP (Gulu strain sourced from SinoBiological. GenBank Accession: YP_138523.1) proteins expressed in HEK 293 cells were covalently coupled to SPHERO™ Magnetic Flow Cytometry Multiplex Bead Assay Particles (Spherotech) at saturation levels using a modification of a previously established protocol (37). Modifications were the substitution of centrifugation steps for magnetic bead retention with the EasyEights™ EasySep™ Magnet (STEMCELL Technologies) and blocking with phosphate-buffered saline (PBS) solution containing 2% Bovine Serum Albumin (BSA), and 0.05% sodium azide (pH 7.4). Successful conjugation was determined via IgG detection with a known positive EBOV convalescent sample.
Flow Cytometry Data Acquisition
For all flow cytometry experiments, samples were analysed as previously described (38), with the additional use of the PE channel for IgG, C1q, and C5b-9 detection. The gating method is demonstrated in Supplementary Figure 1. All samples were acquired with a CytoFLEX S flow cytometer (Beckman Coulter) collecting a minimum of 100 beads per sample, analysed using FlowJo software (version 10.8.0.), and presented using GraphPad software (Version 9).
Flow Cytometry IgG Binding Assays
Heat-inactivated plasma (heat block at 56°C for 30 min) was diluted 1:50 in blocking buffer (Hank’s Balanced Salt Solution (HBSS), 2% BSA) for a final volume of 40 µl and titrated 1:2 for a 3-point dilution series. The final plasma dilutions (1:100, 1:500, 1:2500) were made by adding 20 µl of the EBOV-GP, EBOV-sGP, or SUDV-GP conjugated beads (50 beads per µl) into each plasma dilution. Samples were incubated for 1 h at RT whilst shaking at 550 rpm, then washed twice in 200 µl of wash buffer (HBSS, 0.05% tween-20) and resuspended in 100 µl (0.5 µg/ml) PE-conjugated anti-human IgG (Cambridge Bioscience) in blocking buffer. Samples were again incubated for 1 h at RT whilst shaking at 550 rpm, washed twice in 200 µl of wash buffer, and resuspended in 50 µl HBSS.
For all IgG assays, three plasma dilutions with the EBOV-GP conjugated beads were used as quality controls (QCs) for assay performance and were all below 30% CV (Supplementary Figure 3A). Further controls were included for the SUDV-GP and EBOV-sGP assays using their respective bead conjugates to monitor the bead integrity at a single dilution point (Supplementary Figure 3B) and were all below 15% CV. The final results were reported using a single dilution point which avoided assay saturation and subtracted the relevant negative plasma sample from each plate.
Flow Cytometry C1q Binding Assay
Each bead conjugate (1000 beads per sample) was incubated with heat-inactivated EBOV survivor plasma with known IgG binding to EBOV-GP, EBOV-sGP, and SUDV-GP, along with anti-EBOV-GP negative plasma, at a final 1:20 plasma dilution. The bead and plasma mixture was then incubated for 30 min at 25°C whilst shaking at 900 rpm, washed twice in 200 µl of wash buffer, and resuspended in 100 µl of purified C1q protein (Sigma Aldrich) in blocking buffer at 5 µg/ml, 2.5 µg/ml, 1.25 µg/ml or with blocking buffer only. The samples were then incubated at 25°C for 1 h whilst shaking at 900 rpm, washed twice in 200 µl of wash buffer, resuspended in 100 µl (1 µg/ml) anti-C1q monoclonal antibody (Quidel) and incubated at 25°C for 30 min whilst shaking at 900 rpm. The samples were washed again, resuspended in 100 µl (1 µg/ml) PE-anti-mouse IgG (ThermoFisher Scientific) and incubated at 25°C for 30 min whilst shaking at 900 rpm. The final wash step was carried out and the samples resuspended in 50 µl HBSS. A negative cut-off was determined using an average of all bead and plasma controls which excluded the primary antibody step, plus three standard deviations.
Flow Cytometry C3c and C5b-9 Deposition Assays
The methods used in this study have previously been utilised and published for detecting antibody-dependent C3c deposition on the SARS-CoV-2 spike protein (38) and used again in a current preprint (39). EBOV-GP, EBOV-sGP, or SUDV-GP conjugated beads (50 beads per µl) were mixed with heat-inactivated EBOV survivor plasma diluted four times at a 1:2 dilution starting from 1:10 (SUDV-GP) or 1:20 (EBOV-GP and EBOV-sGP) and incubated for 30 min at 25°C whilst shaking at 900 rpm. The beads were washed twice with 200 µl wash buffer and resuspended in 50 µl PHP (diluted 1:10 in blocking buffer) as a source of complement, then incubated for 15 min at 37°C with shaking at 900 rpm. For C3c detection, the beads were washed twice with 200 µl wash buffer and resuspended in 100 µl FITC-conjugated rabbit anti-human C3c polyclonal antibody (Abcam) diluted 1:500 in blocking buffer and incubated for 20 min in the dark. For detection of C5b-9 deposition, the C3c antibody was replaced with a monoclonal C5b-9 antibody (SantaCruz Biotechnology) at a concentration of 1 µg/ml in 100 µl blocking buffer. A further wash step and incubation with 100 µl PE-conjugated anti-mouse polyclonal antibody (ThermoFisher Scientific) at 1 µg/ml in blocking buffer for 20 min in the dark was required for C5b-9 detection. For both C3c and C5b-9 deposition assays, the beads were washed a final two times in 200 µl wash buffer before re-suspension in 50 µl HBSS.
Each test plate included a heat-inactivated negative plasma control (EBOV naïve Guinean plasma sample), a PHP-only control, a conjugate-only control, a plate QC using EBOV-GP beads with a fixed plasma dilution (C3c: Supplementary Figure 4A, C5b-9: Supplementary Figure 5A), and a bead QC using either EBOV-GP, EBOV-sGP, or SUDV-GP beads at a fixed plasma dilution (C3c: Supplementary Figure 4B, C5b-9: Supplementary Figure 5B). All plate and bead QCs were within 30% CV. Where the dilutions for some samples saturated the assay, linear regression was used from larger dilutions to predict these values. Where multiple bead batches were used (EBOV-GP C3c assay), the negative sample median fluorescence intensity (MFI) on each plate was subtracted from all samples to best normalise the data based on the QCs. All other assays used a single bead batch where the PHP-only control was subtracted.
Wild-Type EBOV Neutralisation Assay
Wild-type EBOV neutralisation assays were performed in the BSL4 laboratory at the Institute of Virology, Philipps University of Marburg, Germany. Virus neutralisation assays were a modification of methods previously described using the EBOV Makona variant (40, 41). Briefly, eight plasma samples randomly selected from the LN cohort and one high neutralising control sample were serially diluted 1/23 to 1/28 in 50 µl Dulbecco’s modified Eagle’s medium (DMEM) supplemented with penicillin (100 U/ml), streptomycin (100 mg/ml), L-glutamine (2 mmol/l), and exogenous PHP for a final concentration of 20%, 10% or 0%. 100 TCID50 units (50 µl) of EBOV (Makona isolate, GenBank accession No. KJ660347) in DMEM with 2% FCS were added to the plasma dilutions and incubated at 37°C for 1 h. Vero cells diluted in DMEM containing 2% FCS (9.4x103 cells) were then added to each well and the plates were incubated at 37°C with 5% CO2 and cytopathic effects analysed at nine days post infection. Neutralisation titres were calculated as geometric mean titres of four replicates. Each plate included PHP-only, cell-only, and heat-inactivated PHP controls.
Individual samples were analysed via a log2 fold-change of GMT compared to the plasma-only condition with a significance threshold of +/- 1.5. Each group (plasma-only, 10% PHP, 20% PHP) was compared in a pairwise manner using a one-tailed, Wilcoxon signed-rank test as an increase was expected with PHP, and a significance threshold of P < 0.050. Samples were analysed and presented using GraphPad (Version 9).
Results
Selection of Low-Neutralising Plasma Samples and Confirmation of IgG/C1q Binding
Low-neutralising plasma samples against EBOV were of particular interest in this study as we aimed to characterise their possible interaction with the complement system and whether this interaction enhanced neutralisation. Previous studies successfully demonstrating a complement-mediated enhancement of antibody neutralisation were performed using non or low-neutralising antibodies (17, 42–44) and so the same hypothesis was applied here.
Analysis of the correlation between historic anti-EBOV-GP IgG and neutralisation data (35) (Figure 2A) showed a similar distinction of cohorts to anti-EBOV-GP IgG determined via flow cytometry in this study (including four additional plasma samples) and the same historic neutralisation data (Supplementary Figure 2). Using flow cytometry, we observed IgG binding to EBOV-GP (Supplementary Figure 3C), EBOV-sGP (Supplementary Figure 3D), and SUDV-GP (Supplementary Figure 3E) with all convalescent plasma samples. No binding was observed when using EBOV negative plasma. These proteins belong to the two most virulent species of the Ebolavirus genus and were selected as they are presented on the surface of the virions and infected cells or are actively secreted into the extracellular space. Therefore, they are the most likely EBOV proteins to encounter the complement system, where the first step in conventional activation of the classical pathway is the binding of IgG.
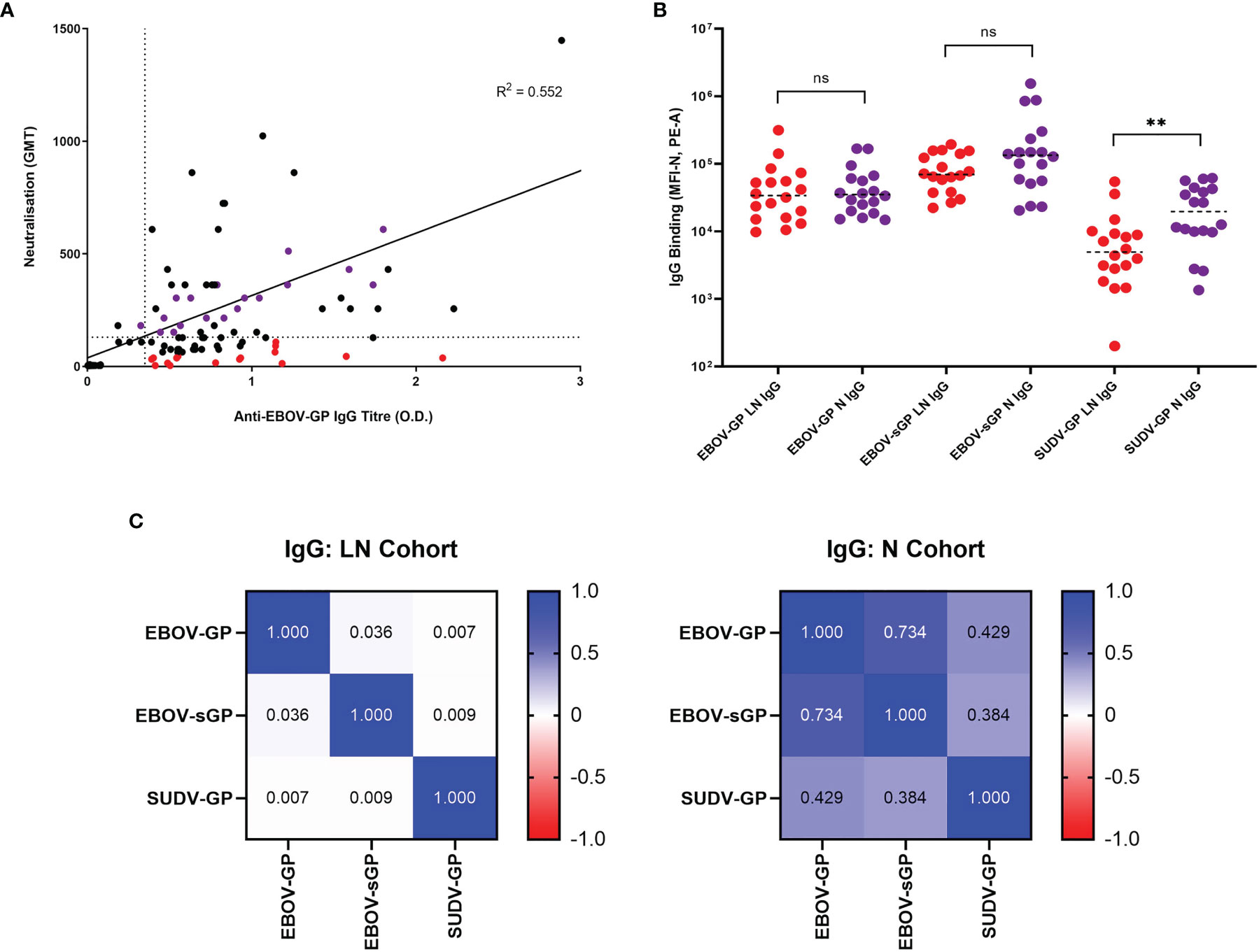
Figure 2 Selection of EBOV-GP plasma samples and their abilities to bind EBOV-GP, EBOV-sGP, and SUDV-GP. (A) 145 samples from historic EBOV-GP ELISAs and EBOV neutralisation assays were correlated and analysed via linear regression. Samples greater than the axis limits were excluded from the graph for the purpose of clarity, but still included in the analysis. The LN cohort (red dots, n = 16) was selected using a neutralisation cut-off < 130 GMT (horizontal dotted line) and an antibody titre > 0.35 O.D. (vertical dotted line), with a maximum residual from the line of best fit (< -100 GMT). The N cohort (purple dots, n = 16) was selected using a neutralisation cut-off > 200 GMT and the closest possible residual to the line of best fit. (B) EBOV-GP, EBOV-sGP, and SUDV-GP conjugated beads were incubated with plasma from the LN (n = 18) or N (n = 18) cohorts and analysed via flow cytometry. Mean values are represented by horizontal dotted lines and significance determined using a Mann-Whitney U test. (C) A pairwise linear regression analysis was performed for each bead conjugate with LN (n = 18) and N (n = 18) plasma cohorts and the R2 values for IgG binding were presented in the form of a heatmap. ** = significant (P < 0.01), ns, Not significant.
After subtracting the MFI of the negative plasma sample on each plate from all samples at the chosen dilutions, the total IgG binding of the LN and N cohort was compared for each protein (Figure 2B). For EBOV-GP and EBOV-sGP, the overall IgG titres showed no significant difference when using a Mann-Whitney U test (P < 0.050). For SUDV-GP, the N cohort demonstrated a significantly higher level of IgG binding compared to the LN cohort (P = 0.005) with a 1.4 log2 fold increase. The final MFI for IgG binding of the LN and N cohorts to each protein were then analysed via linear regression (Figure 2C). Differences in the relationship of IgG binding to Ebolavirus proteins for each cohort might indicate variations in antibody epitopes, antibody diversity, or cross-reactivity, which are important factors for ADCD. For the N cohort, there was a strong correlation between EBOV-GP and EBOV-sGP (R2 = 0.734), a moderate correlation between SUDV-GP and EBOV-GP (R2 = 0.429) and a weak correlation between SUDV-GP and EBOV-sGP (R2 = 0.384). For the LN cohort, there was no correlation between any of the proteins tested: EBOV-GP and EBOV-sGP (R2 = 0.036), SUDV-GP and EBOV-GP (R2 = 0.007), and SUDV-GP and EBOV-sGP (R2 = 0.009).
Following antibody binding, the next step in the activation cascade of the classical complement pathway would typically be the binding of the C1q protein to IgG/IgM in complex with the target antigen. In this study, the detection of C1q binding was only observed following addition of purified C1q to plasma containing anti-EBOV-GP IgG bound to EBOV-GP or EBOV-sGP. We did not observe C1q binding in the presence of anti-EBOV-GP IgG negative plasma nor with the use of SUDV-GP conjugated beads (Figure 3 and Supplementary Figure 6). C1q binding may occur in the absence of antibodies by directly binding the antigen or utilising acute phase proteins against some pathogens. However, our results indicate that C1q binding to EBOV-GP and EBOV-sGP was dependent on the presence of IgG.
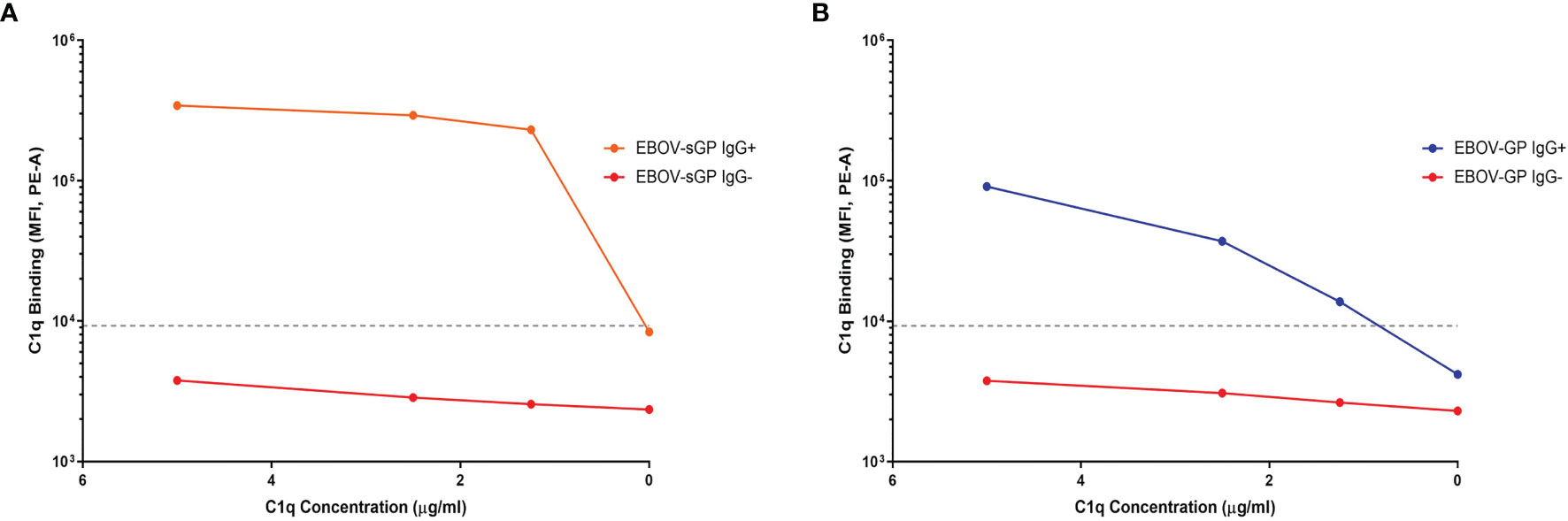
Figure 3 The binding of purified C1q protein to EBOV-GP and EBOV-sGP in antibody complexes. Purified C1q protein was titrated against EBOV-GP (A) and EBOV-sGP (B) conjugated beads with human plasma in the presence or absence of EBOV-GP IgG. A negative cut-off (grey dotted line) was determined using the mean value for all control samples where the primary antibody was excluded, plus three standard deviations.
In summary, we identified plasma from two cohorts based on their ability to neutralise EBOV relative to their anti-EBOV-GP IgG titres. The LN cohort showed significantly lower IgG binding to the SUDV-GP compared to the N cohort despite similar anti-EBOV-GP and anti-EBOV-sGP titres. Furthermore, the LN cohort showed no clear relationship in IgG titres between the Ebolavirus proteins, whereas IgG binding in the N cohort correlated as expected (Figure 2). C1q binding could be detected following the binding of IgG to EBOV-GP and EBOV-sGP (Figure 3).
ADCD and Its Relationship With IgG Binding
After confirmation of IgG and C1q binding, the extent of ADCD via the classical pathway for the N and LN cohort was determined by the levels of C3c and C5b-9 deposition. Complement activation can have both local and systemic effects on a wide range of immune functions. The extent of this activation varies depending on antibody characteristics and so provides a mechanism through which LN plasma samples could influence EBOV pathogenesis.
For the EBOV-GP LN and N cohort there was no significant difference in IgG binding (P = 0.673) (previously shown in Figure 2B), C3c deposition (P = 0.239), nor C5b-9 deposition (P = 0.181) using a Mann-Whitney U test (Figure 4A). A linear regression analysis was also performed to assess the relationship between these three parameters and the R2 values presented as a heatmap in Figure 4B. Both the N and LN cohort showed a strong correlation for C3c and C5b-9 deposition with R2 = 0.914 and R2 = 0.938, respectively. IgG values also strongly correlated with C3c for the N cohort (R2 = 0.856) and the LN cohort (R2 = 0.788), and again with C5b-9 for the N (R2 = 0.940) and LN (R2 = 0.881) cohorts.
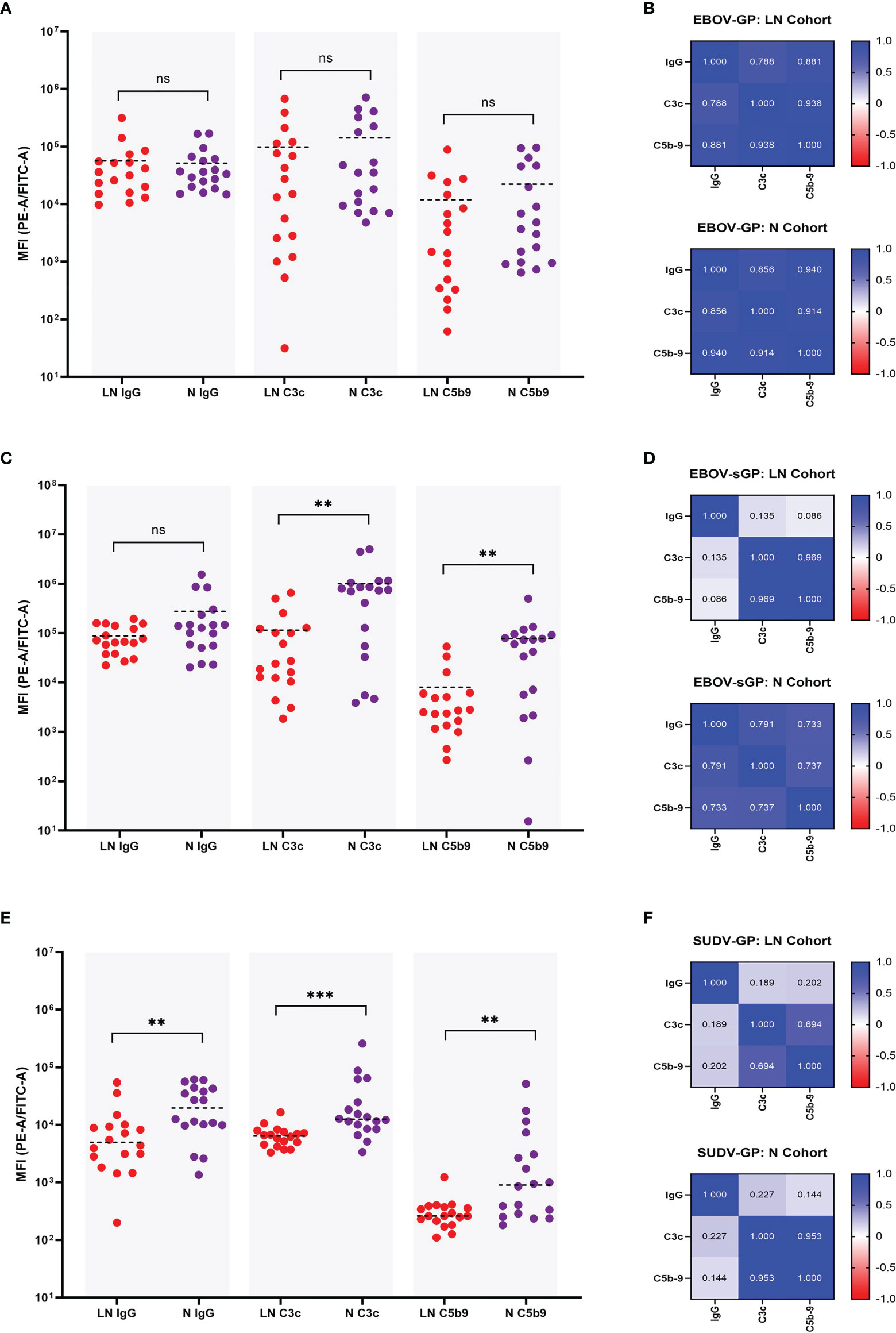
Figure 4 Comparison of IgG binding, C3c deposition, and C5b-9 deposition for EBOV-GP, EBOV-sGP, and SUDV-GP. Plasma samples in the N cohort (purple dots, n = 18) and LN cohort (red dots, n = 18) were compared using a Mann-Whitney U test for IgG binding, C3c deposition and C5b-9 deposition against EBOV-GP (A), EBOV-sGP (C), and SUDV-GP (E). The pairwise relationship between each parameter for both cohorts was then analysed via linear regression and the R2 values reported as a heatmap for EBOV-GP (B), EBOV-sGP (D), and SUDV-GP (F). Grey shaded areas group the samples based on assay type. ** = significant (P < 0.01), *** = significant (P < 0.001), ns, not significant.
EBOV-sGP total IgG binding showed no significant difference (P = 0.239) between the LN and N cohorts (previously shown in Figure 2B, yet the N cohort showed significantly higher levels of C3c deposition (P = 0.002) and C5b-9 deposition (P = 0.003) (Figure 4C). The linear regression analysis (Figure 4D) showed a strong correlation when analysing the N cohort and LN cohorts for C3c and C5b-9 deposition of R2 = 0.737 and R2 = 0.969, respectively. For C3c and IgG, a strong correlation was observed for the N cohort (R2 = 0.791), but no correlation was observed for the LN cohort (R2 = 0.135). Similar findings were observed for IgG and C5b-9 deposition, with a strong correlation for the N cohort (R2 = 0.733) but no relationship with the LN cohort (R2 = 0.086).
For SUDV-GP, the N cohort showed significantly higher IgG binding (P = 0.005) (previously shown in Figure 2B), C3c deposition (P = < 0.001), and C5b-9 deposition (P = 0.004) (Figure 4E) compared to the LN cohort. The linear regression analysis (Figure 4F) showed a strong correlation for C3c and C5b-9 deposition for the N cohort (R2 = 0.953) and a moderate correlation for the LN cohort (R2 = 0.694). For C3c and IgG, a weak correlation was observed for the N cohort (R2 = 0.227) but no correlation for the LN cohort (R2 = 0.189), and no correlation was observed for IgG and C5b-9 deposition for the N cohort (R2 = 0.144) or the LN cohort (R2 = 0.202).
In summary, the levels of ADCD varied depending on whether the plasma was from the LN or N cohort, and depended on the Ebolavirus protein present (Figure 4). For EBOV-GP, both plasma cohorts showed similar levels of ADCD and this response correlated strongly with IgG titres. For EBOV-sGP, the LN cohort was less efficient at mediating ADCD despite similar IgG binding titres to the N cohort. Whilst ADCD with the N cohort appeared dependent on IgG titre, ADCD and IgG titre did not correlate for the LN cohort. Results using SUDV-GP were also different, as the LN cohort was significantly lower than the N cohort for all parameters tested, and ADCD in neither cohort correlated with IgG titre.
The Effect of PHP on Wild-Type EBOV Antibody Neutralisation Assays
As mentioned previously, antibodies can activate the complement system and influence a number of immune effects, both local and systemic. One of these effects is the enhancement or development of neutralisation in low or non-neutralising antibodies, respectively. Since the LN cohort demonstrated a clear ability to mediate ADCD, we investigated the effect on wild-type EBOV neutralisation with the addition of exogenous PHP.
Eight samples from the LN cohort were selected at random (Figure 5A) for wild-type EBOV neutralisation assays with or without the addition of exogenous PHP, and a strongly neutralising positive control (sample C147). The antibody-with-PHP group had a significantly higher (P = 0.031) neutralisation titre than the antibody-only group as part of the same assay (Figure 5B). The significance was also higher when comparing the antibody-with-PHP group to historic data from the 2017 assay (P = 0.012), whilst the antibody-only groups in this study and from 2017 showed no significant difference (P = 0.500). The addition of PHP at 20% resulted in a significant increase but remains below the median value of the 132 survivor samples tested in the historic 2017 cohort (35).
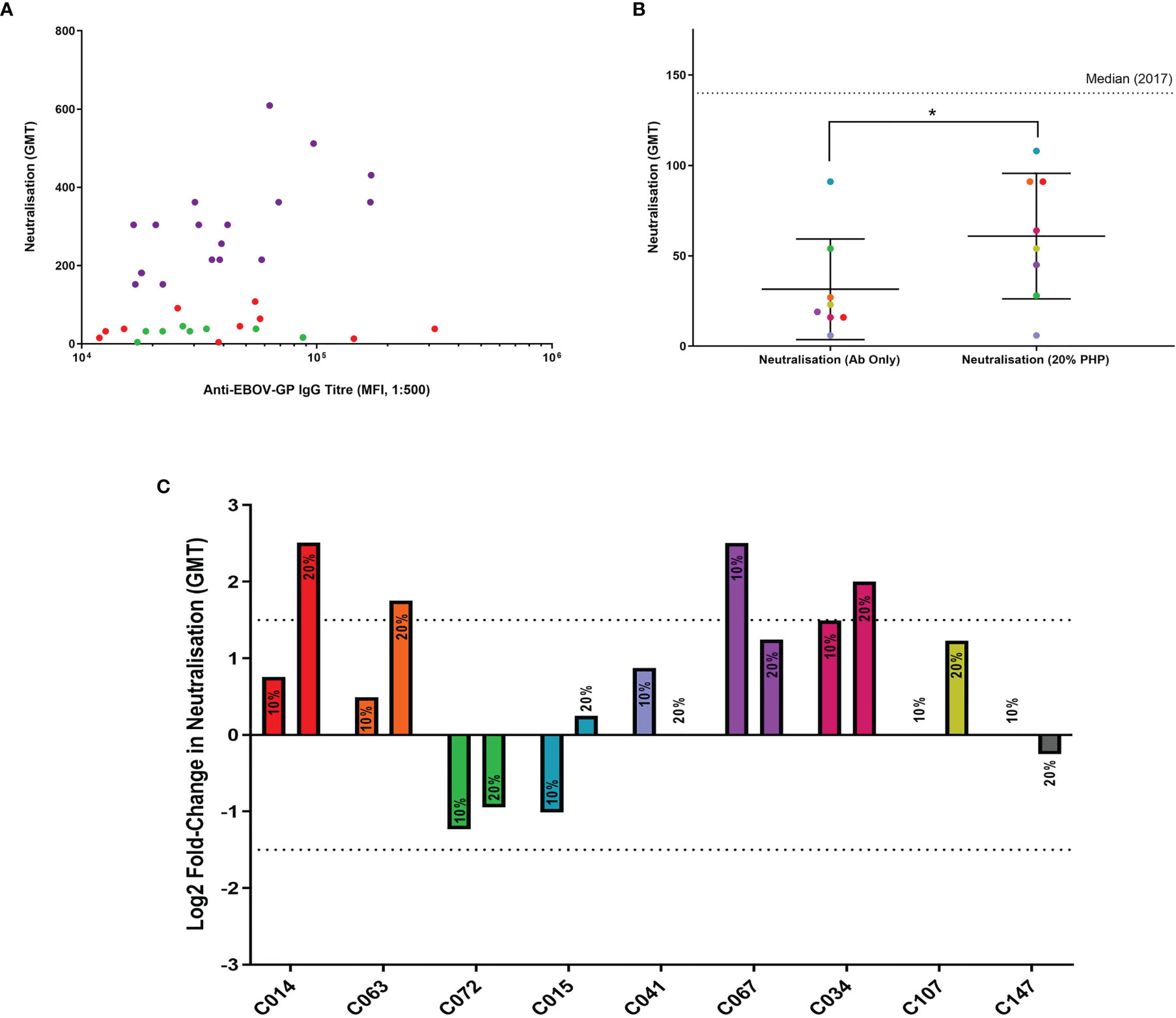
Figure 5 Antibody neutralisation assays with wild-type EBOV, supplemented with PHP as a source of complement. (A) Eight samples from the LN cohort were selected for use in wild-type EBOV neutralisation assays (green dots). The remaining samples represent the LN cohort (red dots) and the N cohort (purple dots). All samples are illustrated using IgG titres determined in this study via flow cytometry and compared to historic 2017 neutralisation data. (B) Comparison of LN cohort neutralisation with or without 20% PHP, analysed using a one-tailed Wilcoxon signed-rank test (P = 0.031). (C) Comparison of individual samples analysed via a log2 fold-change with 10% and 20% PHP compared to antibody-only samples, with a negative cut-off below 1.5 (dotted line). * = significant (P < 0.05).
When the log2 fold change for each sample was analysed with 10% and 20% PHP compared to their antibody-only controls, one sample showed a significant increase in neutralisation with 10% PHP (CO67) and three samples significantly increased when using 20% PHP (Figure 5C). No samples showed a significant decrease in neutralisation with the addition of PHP. No cytotoxic effects were observed when using PHP at 10% and 20% concentrations. However, when increasing PHP to 40%, evidence of cell cytotoxicity emerged and this data was subsequently excluded from the study (Supplementary Figure 7).
In summary, the addition of PHP as a source of complement was able to significantly increase the level of neutralisation for selected LN plasma samples. This effect was most noticeable with the highest PHP concentration tested at 20%.
Discussion
In this study we describe a potential role of the complement system in the context of Ebolavirus infections mediated by convalescent EBOV plasma. The use of LN plasma expands on our current knowledge regarding Fc-mediated EBOV antibody functions. First, we demonstrated the potential for convalescent EBOV plasma samples to mediate ADCD and showed that this response significantly varied depending on whether the samples were from the LN or N cohort. Furthermore, ADCD was dependent on the type of EBOV protein present in the assay (EBOV-GP, EBOV-sGP, or SUDV-GP), indicating a possible function in Ebolavirus cross-reactivity. These interactions resulted in a significant enhancement of neutralisation for selected samples when tested against wild-type EBOV. These findings show a capacity for LN antibodies to mediate ADCD in the context of EBOV infection, influencing neutralisation and potentially further local and systemic immune responses including pro-inflammatory responses, chemotaxis, opsonisation, agglutination, and immune cell regulation. This highlights an acute need for further research to fully determine the role of these mechanisms in immunity.
We found that plasma samples from the LN cohort were less capable of cross-reacting with SUDV-GP and that there was no relationship of IgG binding between all proteins, unlike the N cohort (Figure 2). The significantly lower level of LN IgG binding to SUDV-GP suggests that either the epitopes recognised by plasma in the N cohort are better conserved amongst these proteins, or that the N cohort plasma may have a more diverse antibody response to recognise a broader range of targets. This could also explain the lack of correlation of LN IgG binding to SUDV-GP compared with EBOV-GP and EBOV-sGP. The lack of correlation of LN plasma IgG bound to EBOV-GP and EBOV-sGP may be because different plasma antibodies target certain conformational epitopes of either the sGP or whole EBOV-GP such that overall binding is not affected (45). It’s possible that the bead conjugation process restricts certain epitopes. However, this method utilises abundant and regularly distributed free amine groups on these glycoproteins and so this is unlikely. Whilst care was taken to ensure all proteins were obtained from HEK 293 mammalian cell expression systems to reduce differences in glycosylation and protein processing, they originated from different suppliers and relied upon different methods of purification.
Activation of the classical complement pathway typically requires IgG/IgM binding prior to engagement of the C1q protein, although in rare instances C1q may bind viral antigens directly to activate the complement system (46–48). In our observations, ADCD in response to EBOV-GP and EBOV-sGP was dependent on the binding of anti-EBOV-GP antibodies (Figure 3). The absence of C1q:IgG binding for SUDV-GP was likely an assay sensitivity issue due to the lower levels of IgG binding. This would decrease the number of targets for the C1q protein and have a lower epitope density, thus reducing the formation of antibody clusters required for efficient C1q binding (24) and complement activation (22, 23). Based on previous studies regarding antibody kinetics (49, 50), we do not anticipate that these samples, collected at least 1-year post-exposure, would contain significant levels of IgM. These results highlight some important functional differences in the initial stages of ADCD between the LN and N cohorts.
We report a significant difference in the levels of ADCD between the LN and N cohorts and the relationship of deposition to IgG, depending on the EBOV protein present (Figure 4). For EBOV-GP (Figures 4A, B), the LN plasma IgG was equally efficient at binding the target protein and mediating ADCD compared to the N cohort plasma, and the level of deposition for both cohorts was dependent on IgG titre. Therefore, both LN and N plasma could play a role in EBOV infection through the activation of complement, which in turn can promote inflammation and chemotaxis (10, 51) and reduce viral load (47, 52–54).
For EBOV-sGP (Figures 4C, D), despite similar IgG titres, the LN cohort was significantly less efficient at mediating ADCD compared to the N cohort. This could be the result of antibody isotypes involved in binding for each cohort, as IgG1 and IgG3 activate complement most efficiently, followed by IgG2, whilst IgG4 has no activity and may even be inhibitory (19–21). Epitope density, antibody recognition, and antibody clustering for efficient C1q binding may also influence activation as discussed previously. Interestingly, and unlike the N cohort, IgG binding and ADCD did not correlate for the LN plasma. This may be explained by the isotype ratio and epitope specificity leading to varying efficiencies in complement activation. Acute-phase reactive proteins that facilitate C1q binding in place of antibodies (10) might explain this phenomenon. However, the N cohort correlated positively with IgG as expected, we did not observe C1q binding with EBOV negative plasma, and we only observed negative results for the PHP-only controls. Furthermore, the EBOV-GP did not show a similar trend which might otherwise be expected. It is possible that the difference in strains used for the EBOV-GP (Makona) and EBOV-sGP (Mayinga) might affect some of the comparisons being made, as the survivor cohort were infected with the Makona variant. It is therefore possible that antibodies targeted to a non-homologous region of the EBOV-GP could be missed when comparing binding and subsequent ADCD on the EBOV-sGP. A comparison of the full-length genomes of representative EBOV isolates shows an estimated 97% sequence identity between the Makona and Mayinga variants used in this study (55).
The sGP is the primary transcript from the EBOV GP gene (56) and is actively secreted during infection to levels detectable in the blood of acutely infected patients (57). The sGP is purportedly an antibody decoy molecule capable of subverting the immune response away from GP1,2 and inhibiting neutralisation (58, 59). Antibody-mediated complement activation in response to high levels of sGP could be an interesting focus for future studies, as complement depletion (60) and the production of decoy molecules for complement evasion (61) are disease mechanisms described for other pathogens that may be relevant to sGP. Excessive complement activation has been associated with fatal EVD outcome based on transcriptomic signatures (62) and has been shown to exacerbate other viral infections (63–67). It is therefore possible that the ADCD we describe here in response to EBOV-sGP could influence EBOV immunity.
For SUDV-GP (Figures 4E, F), the LN cohort had significantly lower levels of bound IgG, C3c deposition, and C5b-9 deposition compared to the N cohort. Whilst C3c and C5b-9 deposition showed a clear correlation for both cohorts, the association of IgG compared to C3c and C5b-9 deposition was either weak or not significant. As mentioned previously, the IgG isotypes and/or the antibody epitopes to enable clustering and C1q binding may account for some of this variation. The involvement of complement in cross-reactivity with SUDV from EBOV convalescent plasma could have implications for cross-protection, resulting in complement deposition, the production of C3a and C5a anaphylatoxins, and the formation of the membrane attack complex (Figure 1). Our results suggest the level of this response would vary depending on the capacity for neutralisation of plasma samples. However, it is not clear how these levels would translate in vivo and whether the resulting effects would be beneficial or detrimental to immunity.
Previous reported outbreaks of EBOV and SUDV have occurred in geographically similar areas, with EBOV causing repeated outbreaks in DRC and on one occasion a spillover into neighbouring Uganda, whilst SUDV has the been the cause of multiple outbreaks in Uganda and the neighbouring South Sudan (68). These three countries also provide suitable habitats for putative EBOV bat reservoirs based on a MaxEnt niche modelling approach (69). Furthermore, the added complexity of human-to-human transmission (70), viral persistence in immune privileged sites including ocular fluid (71), semen (72, 73), breast milk (74, 75) and cerebrospinal fluid (76), the potential for recrudescence (71, 76), and a general lack of resources for viral surveillance in the affected areas complicate the spread, transmission, and possible overlap of these viruses.
The addition of PHP to LN plasma in wild-type EBOV neutralisation assays resulted in a significant enhancement to their neutralisation (Figure 5). In a previous study, the presence of complement has been shown to enable neutralisation with otherwise non-neutralising purified anti-EBOV-GP antibodies (17), although the addition of complement to human plasma in a different study showed no significant difference (77). An important distinction between the latter study and ours may be the use of human instead of guinea pig complement which shows some key functional differences (78–80), or their use of historical samples 40 years after infection as the non-complement activating IgG-4 isotype reportedly starts developing 1–2 years post-EBOV infection (81). In our study, a significant increase in wild-type EBOV neutralisation was observed using 20% PHP compared to the antibody-only controls. The increase in neutralisation was considered modest as the neutralisation values remained below the median neutralisation value of all 132 survivor samples from the 2017 historic data (35). However, it should be noted that EBOV is a blood-borne pathogen and would encounter high concentrations of complement during infection. One previous study investigating the effects of Zika virus and complement from normal human serum (NHS) used concentrations up to 50% (with EDTA) with an increasingly positive trend between neutralisation and NHS concentration (47). We increased the PHP concentration to 40% to see if the trend in increasing neutralisation would continue, but evidence of cell cytotoxicity emerged (Supplementary Figure 7). To test these higher concentrations, the use of EDTA may be required post-virus incubation and pre-cell infection. This would preserve cell integrity but potentially overlook complement interactions with the sGP which is secreted during infection and the possible lysis of infected cells (52, 82) or prevention of spread into neighbouring cells (17).
When analysed individually, we found a subset of samples that were significantly influenced by the presence of complement (Figure 5). This is in agreement with similar studies (42–44, 54) and this difference has previously been attributed to the antibody subclass (17). Compared to the latter study by Wilson et al, our observed increase in neutralisation was lower. This may be explained by our use of native plasma which better represents the polyclonal antibody response of natural immunity compared to the use of a single purified antibody. Sample C067 showed a significant increase in neutralisation with 10% PHP but fell just below the threshold when PHP was increased to 20%. This observation could be the result of inherent assay variance as the difference falls within the 1.5 log2 fold-change threshold. It is unlikely that our observations are explained by saturation with 10% PHP, as the majority of samples show a positive trend between 10% and 20% PHP concentration and neutralisation. The different antibody repertoires between plasma samples will affect how they engage complement proteins and thus impact on immunity, however our sample size was restricted due to limited resources. These results show that the commonly used neutralisation immunoassays can be limited by excluding complement and this should be noted when considering such assays, including in vivo experiments or screening for therapeutics.
In conclusion, we show a potential for complement-mediated enhancement of antibody-mediated immunity in both the LN and N cohorts and highlight where they significantly differ depending on IgG binding, neutralising ability, and the Ebolavirus protein used. This may have implications for wider immune responses important to EVD such as inflammation and chemotaxis that could be pursued in future studies. We have also shown how LN plasma can neutralise wild-type EBOV more efficiently in the presence of complement at relatively low concentrations. Future investigations of antibody-mediated neutralisation may benefit from the addition of complement to immunoassays and should consider the use of EDTA when testing higher complement concentrations. As our findings are assumed to be IgG-mediated, they hold most relevance to re-exposure, recrudescence, vaccination, and cross-reactivity with Ebolaviruses. Future studies may consider similar investigations with IgM or the possibility of acute-phase reaction proteins to engage complement. Flow cytometry methods used in this study have previously been applied to the SARS-CoV-2 spike protein where multifunctional antibody responses beyond neutralisation could be important for protection (38) and similarly, investigations on variants of concern demonstrate that neutralisation as classically defined should not be considered as the sole determinant of protection (83).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by National Ethics Committee for Health Research, Guinea (33/CNERS/15) and from the National Research Ethics Service, UK. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Authors MC, JHi, ST, NM, and MK conceptualized the study. Plasma sample collection and processing was conducted by TT, YH, JA, FK, and FA. Authors JM, TT, TS, SKF, FA, and JHu contributed to the experimental work and authors JM, TS, SKF, and MC were involved in data analysis. Lastly, authors JM, TT, TS, SKF, ST, AG, JHu, YH, SL, and MC contributed to the writing of the manuscript.
Funding
This work was funded via a UK Health Security Agency studentship programme and by the US Food and Drug Administration Medical Countermeasures Initiative contract 75F40120C00085 and contract HHSF223201510104C. Author T.S. received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Projektnummer 197785619 - SFB1021.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge the Ebola virus disease survivor’s association and all participants involved in this study from Coyah and Guéckédou for their ongoing support and commitment. We are also grateful for the continued support from the Guinean Ministry of Health and local health agencies. We thank Gotthard Ludwig and Michael Schmidt from the biosafety level 4 facility at the Philipps-University of Marburg for technical assistance. Finally, we would like to thank Rowan Curtis and Bethany White for their contributions to the final editing of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.857481/full#supplementary-material
References
1. Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, et al. Isolation of Potent Neutralizing Antibodies From a Survivor of the 2014 Ebola Virus Outbreak. Science (2016) 351(6277):1078–83. doi: 10.1126/science.aad5788
2. Warfield KL, Howell KA, Vu H, Geisbert J, Wong G, Shulenin S, et al. Role of Antibodies in Protection Against Ebola Virus in Nonhuman Primates Immunized With Three Vaccine Platforms. J Infect Dis (2018) 218(suppl_5):S553–64. doi: 10.1093/infdis/jiy316
3. Rijal P, Elias SC, Machado SR, Xiao J, Schimanski L, O’Dowd V, et al. Therapeutic Monoclonal Antibodies for Ebola Virus Infection Derived From Vaccinated Humans. Cell Rep (2019) 27(1):172–186.e7. doi: 10.1016/j.celrep.2019.03.020
4. Fuentes S, Ravichandran S, Coyle EM, Klenow L, Khurana S. Human Antibody Repertoire Following Ebola Virus Infection and Vaccination. iScience (2020) 23(3):100920. doi: 10.1016/j.isci.2020.100920
5. Longet S, Mellors J, Carroll MW, Tipton T. Ebolavirus: Comparison of Survivor Immunology and Animal Models in the Search for a Correlate of Protection. Front Immunol (2021) 11:3871. doi: 10.3389/fimmu.2020.599568
6. Centers for Disease Control and Prevention. Ebola Vaccine: Information About Ervebo® | Clinicians | Ebola (Ebola Virus Disease). Atlanta: CDC (2021). Available at: https://www.cdc.gov/vhf/ebola/clinicians/vaccine/index.html.
7. European Medicines Agency. New Vaccine for Prevention of Ebola Virus Disease Recommended Approval in the European Union. Amsterdam: European Medicines Agency (2020). Available at: https://www.ema.europa.eu/en/news/new-vaccine-prevention-ebola-virus-disease-recommended-approval-european-union.
8. Ishola D, Manno D, Afolabi MO, Keshinro B, Bockstal V, Rogers B, et al. Safety and Long-Term Immunogenicity of the Two-Dose Heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccine Regimen in Adults in Sierra Leone: A Combined Open-Label, Non-Randomised Stage 1, and a Randomised, Double-Blind, Controlled Stage 2 Trial. Lancet Infect Dis (2021) 97–109. doi: 10.1016/S1473-3099(21)00125-0
9. Centers for Disease Control and Prevention. Treatment | Ebola (Ebola Virus Disease). Atlanta: CDC (2021). Available at: https://www.cdc.gov/vhf/ebola/treatment/index.html.
10. Mellors J, Tipton T, Longet S, Carroll M. Viral Evasion of the Complement System and Its Importance for Vaccines and Therapeutics. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01450
11. Ji X, Olinger GG, Aris S, Chen Y, Gewurz H, Spear GT. Mannose-Binding Lectin Binds to Ebola and Marburg Envelope Glycoproteins, Resulting in Blocking of Virus Interaction With DC-SIGN and Complement-Mediated Virus Neutralization. J Gen Virol (2005) 86(Pt 9):2535–42. doi: 10.1099/vir.0.81199-0
12. Michelow IC, Lear C, Scully C, Prugar LI, Longley CB, Yantosca LM, et al. High-Dose Mannose-Binding Lectin Therapy for Ebola Virus Infection. J Infect Dis (2011) 203(2):175–9. doi: 10.1093/infdis/jiq025
13. Brudner M, Karpel M, Lear C, Chen L, Yantosca LM, Scully C, et al. Lectin-Dependent Enhancement of Ebola Virus Infection via Soluble and Transmembrane C-Type Lectin Receptors. PloS One (2013) 8(4):e60838. doi: 10.1371/journal.pone.0060838
14. Favier A-L, Gout E, Reynard O, Ferraris O, Kleman J-P, Volchkov V, et al. Enhancement of Ebola Virus Infection via Ficolin-1 Interaction With the Mucin Domain of GP Glycoprotein. J Virol (2016) 90(11):5256–69. doi: 10.1128/JVI.00232-16
15. Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-Dependent Enhancement of Ebola Virus Infection. J Virol (2003) 77(13):7539–44. doi: 10.1128/JVI.77.13.7539-7544.2003
16. Takada A, Ebihara H, Feldmann H, Geisbert TW, Kawaoka Y. Epitopes Required for Antibody-Dependent Enhancement of Ebola Virus Infection. J Infect Dis (2007) 196(Supplement_2):S347–56. doi: 10.1086/520581
17. Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, et al. Epitopes Involved in Antibody-Mediated Protection From Ebola Virus. Science (2000) 287(5458):1664–6. doi: 10.1126/science.287.5458.1664
18. Paquin-Proulx D, Gunn BM, Alrubayyi A, Clark DV, Creegan M, Kim D, et al. Associations Between Antibody Fc-Mediated Effector Functions and Long-Term Sequelae in Ebola Virus Survivors. Front Immunol (2021) 12:1917. doi: 10.3389/fimmu.2021.682120
19. Bindon CI, Hale G, Brüggemann M, Waldmann H. Human Monoclonal IgG Isotypes Differ in Complement Activating Function at the Level of C4 as Well as C1q. J Exp Med (1988) 168(1):127–42. doi: 10.1084/jem.168.1.127
20. Kaul M, Loos M. Dissection of C1q Capability of Interacting With IgG Time-Dependent Formation of a Tight and Only Partly Reversible Association. J Biol Chem (1997) 272(52):33234–44. doi: 10.1074/jbc.272.52.33234
21. Lilienthal G-M, Rahmöller J, Petry J, Bartsch YC, Leliavski A, Ehlers M. Potential of Murine IgG1 and Human IgG4 to Inhibit the Classical Complement and Fcγ Receptor Activation Pathways. Front Immunol (2018) 9:958. doi: 10.3389/fimmu.2018.00958
22. Garred P, Michaelsen TE, Aase A. The IgG Subclass Pattern of Complement Activation Depends on Epitope Density and Antibody and Complement Concentration. Scand J Immunol (1989) 30(3):379–82. doi: 10.1111/j.1365-3083.1989.tb01225.x
23. Michaelsen TE, Garred P, Aase A. Human IgG Subclass Pattern of Inducing Complement-Mediated Cytolysis Depends on Antigen Concentration and to a Lesser Extent on Epitope Patchiness, Antibody Affinity and Complement Concentration. Eur J Immunol (1991) 21(1):11–6. doi: 10.1002/eji.1830210103
24. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science (2014) 343(6176):1260–3. doi: 10.1126/science.1248943
25. Tuck MK, Chan DW, Chia D, Godwin AK, Grizzle WE, Krueger KE, et al. Standard Operating Procedures for Serum and Plasma Collection: Early Detection Research Network Consensus Statement Standard Operating Procedure Integration Working Group. J Proteome Res (2009) 8(1):113–7. doi: 10.1021/pr800545q
26. Namekar M, Kumar M, O’Connell M, Nerurkar VR. Effect of Serum Heat-Inactivation and Dilution on Detection of Anti-WNV Antibodies in Mice by West Nile Virus E-Protein Microsphere Immunoassay. PloS One (2012) 7(9):e45851. doi: 10.1371/journal.pone.0045851
27. Pastorino B, Touret F, Gilles M, de Lamballerie X, Charrel RN. Heat Inactivation of Different Types of SARS-CoV-2 Samples: What Protocols for Biosafety, Molecular Detection and Serological Diagnostics? Viruses (2020) 12(7):735. doi: 10.3390/v12070735
28. Bewley KR, Coombes NS, Gagnon L, McInroy L, Baker N, Shaik I, et al. Quantification of SARS-CoV-2 Neutralizing Antibody by Wild-Type Plaque Reduction Neutralization, Microneutralization and Pseudotyped Virus Neutralization Assays. Nat Protoc (2021) 16(6):3114–40. doi: 10.1038/s41596-021-00536-y
29. Sotelo-Orozco J, Chen S-Y, Hertz-Picciotto I, Slupsky CM. A Comparison of Serum and Plasma Blood Collection Tubes for the Integration of Epidemiological and Metabolomics Data. Front Mol Biosci (2021) 8:650. doi: 10.3389/fmolb.2021.682134
30. Takada A, Ebihara H, Jones S, Feldmann H, Kawaoka Y. Protective Efficacy of Neutralizing Antibodies Against Ebola Virus Infection. Vaccine (2007) 25(6):993–9. doi: 10.1016/j.vaccine.2006.09.076
31. Liu Q, Fan C, Li Q, Zhou S, Huang W, Wang L, et al. Antibody-Dependent-Cellular-Cytotoxicity-Inducing Antibodies Significantly Affect the Post-Exposure Treatment of Ebola Virus Infection. Sci Rep (2017) 7:45552. doi: 10.1038/srep45552
32. Saphire EO, Schendel SL, Fusco ML, Gangavarapu K, Gunn BM, Wec AZ, et al. Systematic Analysis of Monoclonal Antibodies Against Ebola Virus GP Defines Features That Contribute to Protection. Cell (2018) 174(4):938–952.e13. doi: 10.1016/j.cell.2018.07.033
33. Saphire EO, Schendel SL, Gunn BM, Milligan JC, Alter G. Antibody-Mediated Protection Against Ebola Virus. Nat Immunol (2018) 19(11):1169–78. doi: 10.1038/s41590-018-0233-9
34. Gunn BM, Lu R, Slein MD, Ilinykh PA, Huang K, Atyeo C, et al. A Fc Engineering Approach to Define Functional Humoral Correlates of Immunity Against Ebola Virus. Immunity (2021) 54(4):815–828.e5. doi: 10.1016/j.immuni.2021.03.009
35. Thom R, Tipton T, Strecker T, Hall Y, Akoi Bore J, Maes P, et al. Longitudinal Antibody and T Cell Responses in Ebola Virus Disease Survivors and Contacts: An Observational Cohort Study. Lancet Infect Dis (2021) 21(4):507–16. doi: 10.1016/S1473-3099(20)30736-2
36. Alexander F, Brunt E, Humphries H, Cavell B, Leung S, Allen L, et al. Generation of a Universal Human Complement Source by Large-Scale Depletion of IgG and IgM From Pooled Human Plasma. Methods Mol Biol Clifton NJ (2022) 2414:341–62. doi: 10.1007/978-1-0716-1900-1_18
37. Brown EP, Licht AF, Dugast A-S, Choi I, Bailey-Kellogg C, Alter G, et al. High-Throughput, Multiplexed IgG Subclassing of Antigen-Specific Antibodies From Clinical Samples. J Immunol Methods (2012) 386(1–2):117–23. doi: 10.1016/j.jim.2012.09.007
38. Barrett JR, Belij-Rammerstorfer S, Dold C, Ewer KJ, Folegatti PM, Gilbride C, et al. Phase 1/2 Trial of SARS-CoV-2 Vaccine ChAdOx1 Ncov-19 With a Booster Dose Induces Multifunctional Antibody Responses. Nat Med (2020) 27(2):1–10. doi: 10.1038/s41591-020-01179-4
39. Tomic A, Skelly DT, Ogbe A, O’Connor D, Pace M, Adland E, et al. Divergent Trajectories of Antiviral Memory After SARS-Cov-2 Infection. Nat Commun (2021) 13(1):1251. doi: 10.21203/rs.3.rs-612205/v1
40. Agnandji ST, Huttner A, Zinser ME, Njuguna P, Dahlke C, Fernandes JF, et al. Phase 1 Trials of rVSV Ebola Vaccine in Africa and Europe. N Engl J Med (2016) 374(17):1647–60. doi: 10.1056/NEJMoa1502924
41. Dowall SD, Callan J, Zeltina A, Al-Abdulla I, Strecker T, Fehling SK, et al. Development of a Cost-Effective Ovine Polyclonal Antibody-Based Product, EBOTAb, to Treat Ebola Virus Infection. J Infect Dis (2016) 213(7):1124–33. doi: 10.1093/infdis/jiv565
42. Johnson JB, Capraro GA, Parks GD. Differential Mechanisms of Complement-Mediated Neutralization of the Closely Related Paramyxoviruses Simian Virus 5 and Mumps Virus. Virology (2008) 376(1):112–23. doi: 10.1016/j.virol.2008.03.022
43. Li F, Freed DC, Tang A, Rustandi RR, Troutman MC, Espeseth AS, et al. Complement Enhances In Vitro Neutralizing Potency of Antibodies to Human Cytomegalovirus Glycoprotein B (Gb) and Immune Sera Induced by Gb/MF59 Vaccination. NPJ Vaccines (2017) 2:1. doi: 10.1038/s41541-017-0038-0
44. Golden JW, Shoemaker CJ, Lindquist ME, Zeng X, Daye SP, Williams JA, et al. GP38-Targeting Monoclonal Antibodies Protect Adult Mice Against Lethal Crimean-Congo Hemorrhagic Fever Virus Infection. Sci Adv (2019) 5(7):eaaw9535. doi: 10.1126/sciadv.aaw9535
45. Pallesen J, Murin CD, de Val N, Cottrell CA, Hastie KM, Turner HL, et al. Structures of Ebola Virus GP and sGP in Complex With Therapeutic Antibodies. Nat Microbiol (2016) 1(9):1–9. doi: 10.1038/nmicrobiol.2016.128
46. Ebenbichler CF, Thielens NM, Vornhagen R, Marschang P, Arlaud GJ, Dierich MP. Human Immunodeficiency Virus Type 1 Activates the Classical Pathway of Complement by Direct C1 Binding Through Specific Sites in the Transmembrane Glycoprotein Gp41. J Exp Med (1991) 174(6):1417–24. doi: 10.1084/jem.174.6.1417
47. Schiela B, Bernklau S, Malekshahi Z, Deutschmann D, Koske I, Banki Z, et al. Active Human Complement Reduces the Zika Virus Load via Formation of the Membrane-Attack Complex. Front Immunol (2018) 9:2177. doi: 10.3389/fimmu.2018.02177
48. Kunnakkadan U, Nag J, Kumar NA, Mukesh RK, Suma SM, Johnson JB. Complement-Mediated Neutralization of a Potent Neurotropic Human Pathogen, Chandipura Virus, Is Dependent on C1q. J Virol (2019) 93(19):e00994–19. doi: 10.1128/JVI.00994-19
49. Ksiazek TG, West CP, Rollin PE, Jahrling PB, Peters CJ. ELISA for the Detection of Antibodies to Ebola Viruses. J Infect Dis (1999) 179(Supplement_1):S192–8. doi: 10.1086/514313
50. Liu Y, Sun Y, Wu W, Li Aq, Yang X, Zhang S, et al. Serological Investigation of Laboratory-Confirmed and Suspected Ebola Virus Disease Patients During the Late Phase of the Ebola Outbreak in Sierra Leone. Virol Sin (2018) 33(4):323–34. doi: 10.1007/s12250-018-0044-z
51. Markiewski MM, Lambris JD. The Role of Complement in Inflammatory Diseases From Behind the Scenes Into the Spotlight. Am J Pathol (2007) 171(3):715–27. doi: 10.2353/ajpath.2007.070166
52. Harris SL, Frank I, Yee A, Cohen GH, Eisenberg RJ, Friedman HM. Glycoprotein C of Herpes Simplex Virus Type 1 Prevents Complement-Mediated Cell Lysis and Virus Neutralization. J Infect Dis (1990) 162(2):331–7. doi: 10.1093/infdis/162.2.331
53. Huber M, Fischer M, Misselwitz B, Manrique A, Kuster H, Niederöst B, et al. Complement Lysis Activity in Autologous Plasma Is Associated With Lower Viral Loads During the Acute Phase of HIV-1 Infection. PloS Med (2006) 3(11):e441. doi: 10.1371/journal.pmed.0030441
54. Terajima M, Cruz J, Co MDT, Lee J-H, Kaur K, Wilson PC, et al. Complement-Dependent Lysis of Influenza A Virus-Infected Cells by Broadly Cross-Reactive Human Monoclonal Antibodies. J Virol (2011) 85(24):13463. doi: 10.1128/JVI.05193-11
55. Albariño CG, Wiggleton Guerrero L, Lo MK, Nichol ST, Towner JS. Development of a Reverse Genetics System to Generate a Recombinant Ebola Virus Makona Expressing a Green Fluorescent Protein. Virology (2015) 484:259–64. doi: 10.1016/j.virol.2015.06.013
56. de La Vega M-A, Wong G, Kobinger GP, Qiu X. The Multiple Roles of sGP in Ebola Pathogenesis. Viral Immunol (2015) 28(1):3–9. doi: 10.1089/vim.2014.0068
57. Sanchez A, Trappier SG, Mahy BW, Peters CJ, Nichol ST. The Virion Glycoproteins of Ebola Viruses Are Encoded in Two Reading Frames and Are Expressed Through Transcriptional Editing. Proc Natl Acad Sci USA (1996) 93(8):3602–7. doi: 10.1073/pnas.93.8.3602
58. Ito H, Watanabe S, Takada A, Kawaoka Y. Ebola Virus Glycoprotein: Proteolytic Processing, Acylation, Cell Tropism, and Detection of Neutralizing Antibodies. J Virol (2001) 75(3):1576–80. doi: 10.1128/JVI.75.3.1576-1580.2001
59. Mohan GS, Li W, Ye L, Compans RW, Yang C. Antigenic Subversion: A Novel Mechanism of Host Immune Evasion by Ebola Virus. PloS Pathog (2012) 8(12):e1003065. doi: 10.1371/journal.ppat.1003065
60. Hovingh ES, van den Broek B, Kuipers B, Pinelli E, Rooijakkers SHM, Jongerius I. Acquisition of C1 Inhibitor by Bordetella Pertussis Virulence Associated Gene 8 Results in C2 and C4 Consumption Away From the Bacterial Surface. PloS Pathog (2017) 13(7):e1006531. doi: 10.1371/journal.ppat.1006531
61. Thiemmeca S, Tamdet C, Punyadee N, Prommool T, Songjaeng A, Noisakran S, et al. Secreted NS1 Protects Dengue Virus From Mannose-Binding Lectin-Mediated Neutralization. J Immunol Baltim Md (2016) 197(10):4053–65. doi: 10.4049/jimmunol.1600323
62. Liu X, Speranza E, Muñoz-Fontela C, Haldenby S, Rickett NY, Garcia-Dorival I, et al. Transcriptomic Signatures Differentiate Survival From Fatal Outcomes in Humans Infected With Ebola Virus. Genome Biol (2017) 18. doi: 10.1186/s13059-016-1137-3
63. Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, et al. Vascular Leakage in Severe Dengue Virus Infections: A Potential Role for the Nonstructural Viral Protein NS1 and Complement. J Infect Dis (2006) 193(8):1078–88. doi: 10.1086/500949
64. Sun S, Zhao G, Liu C, Wu X, Guo Y, Yu H, et al. Inhibition of Complement Activation Alleviates Acute Lung Injury Induced by Highly Pathogenic Avian Influenza H5N1 Virus Infection. Am J Respir Cell Mol Biol (2013) 49(2):221–30. doi: 10.1165/rcmb.2012-0428OC
65. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio (2018) 9(5):e01753–18. doi: 10.1128/mBio.01753-18
66. Gunn BM, Jones JE, Shabman RS, Whitmore AC, Sarkar S, Blevins LK, et al. Ross River Virus Envelope Glycans Contribute to Disease Through Activation of the Host Complement System. Virology (2018) 515:250–60. doi: 10.1016/j.virol.2017.12.022
67. Jiang Y, Zhao G, Song N, Li P, Chen Y, Guo Y, et al. Blockade of the C5a–C5aR Axis Alleviates Lung Damage in Hdpp4-Transgenic Mice Infected With MERS-CoV. Emerg Microbes Infect (2018) 7:1–12. doi: 10.1038/s41426-018-0063-8
68. Centers for Disease Control and Prevention. Ebola Virus Disease Distribution Map: Cases of Ebola Virus Disease in Africa Since 1976 (2021). Available at: https://www.cdc.gov/vhf/ebola/history/distribution-map.html.
69. Koch LK, Cunze S, Kochmann J, Klimpel S. Bats as Putative Zaire Ebolavirus Reservoir Hosts and Their Habitat Suitability in Africa. Sci Rep (2020) 10(1):14268. doi: 10.1038/s41598-020-71226-0
70. World Health Organization. Ebola Virus Disease (2021). Available at: https://www.who.int/westernpacific/health-topics/ebola.
71. Varkey JB, Shantha JG, Crozier I, Kraft CS, Lyon GM, Mehta AK, et al. Persistence of Ebola Virus in Ocular Fluid During Convalescence. N Engl J Med (2015) 372(25):2423–7. doi: 10.1056/NEJMoa1500306
72. Deen GF, Broutet N, Xu W, Knust B, Sesay FR, McDonald SLR, et al. Ebola RNA Persistence in Semen of Ebola Virus Disease Survivors — Final Report. PloS Med (2015) 377(15):1428–37. doi: 10.1056/NEJMoa1511410
73. Thorson AE, Deen GF, Bernstein KT, Liu WJ, Yamba F, Habib N, et al. Persistence of Ebola Virus in Semen Among Ebola Virus Disease Survivors in Sierra Leone: A Cohort Study of Frequency, Duration, and Risk Factors. N Engl J Med (2021) 18(2):e1003273. doi: 10.1371/journal.pmed.1003273
74. Bausch DG, Towner JS, Dowell SF, Kaducu F, Lukwiya M, Sanchez A, et al. Assessment of the Risk of Ebola Virus Transmission From Bodily Fluids and Fomites. J Infect Dis (2007) 196(Supplement_2):S142–7. doi: 10.1086/520545
75. Sissoko D, Keïta M, Diallo B, Aliabadi N, Fitter DL, Dahl BA, et al. Ebola Virus Persistence in Breast Milk After No Reported Illness: A Likely Source of Virus Transmission From Mother to Child. Clin Infect Dis Off Publ Infect Dis Soc Am (2017) 64(4):513–6. doi: 10.1093/cid/ciw793
76. Jacobs M, Rodger A, Bell DJ, Bhagani S, Cropley I, Filipe A, et al. Late Ebola Virus Relapse Causing Meningoencephalitis: A Case Report. Lancet (2016) 388(10043):498–503. doi: 10.1016/S0140-6736(16)30386-5
77. Rimoin AW, Lu K, Bramble MS, Steffen I, Doshi RH, Hoff NA, et al. Ebola Virus Neutralizing Antibodies Detectable in Survivors of Theyambuku, Zaire Outbreak 40 Years After Infection. J Infect Dis (2018) 217(2):223–31. doi: 10.1093/infdis/jix584
78. Bartholomew RM, Esser AF. Differences in Activation of Human and Guinea Pig Complement by Retroviruses. J Immunol (1978) 121(5):1748–51.
79. van der Zee JS, Beuvery EC, van Ree R, Aalberse RC. Human IgM Antibodies do Not Activate Guinea-Pig Complement After Interaction With Soluble Antigen. Mol Immunol (1986) 23(6):669–73. doi: 10.1016/0161-5890(86)90105-7
80. Collins C, Tsui FWL, Shulman MJ. Differential Activation of Human and Guinea Pig Complement by Pentameric and Hexameric IgM. Eur J Immunol (2002) 32(6):1802–10. doi: 10.1002/1521-4141(200206)32:6<1802::AID-IMMU1802>3.0.CO;2-C
81. Davis CW, Jackson KJL, McElroy AK, Halfmann P, Huang J, Chennareddy C, et al. Longitudinal Analysis of the Human B Cell Response to Ebola Virus Infection. Cell (2019) 177(6):1566–82.e17. doi: 10.1016/j.cell.2019.04.036
82. Devaux P, Christiansen D, Plumet S, Gerlier D. Cell Surface Activation of the Alternative Complement Pathway by the Fusion Protein of Measles Virus. J Gen Virol (2004) 85(6):1665–73. doi: 10.1099/vir.0.79880-0
Keywords: complement system, immunology, virology, neutralisation, ebola virus, antibodies, protection, pathogenesis
Citation: Mellors J, Tipton T, Fehling SK, Akoi Bore J, Koundouno FR, Hall Y, Hudson J, Alexander F, Longet S, Taylor S, Gorringe A, Magassouba N’F, Konde MK, Hiscox J, Strecker T and Carroll M (2022) Complement-Mediated Neutralisation Identified in Ebola Virus Disease Survivor Plasma: Implications for Protection and Pathogenesis. Front. Immunol. 13:857481. doi: 10.3389/fimmu.2022.857481
Received: 18 January 2022; Accepted: 21 March 2022;
Published: 12 April 2022.
Edited by:
Pierre Roques, CEA Saclay, FranceReviewed by:
Axel T. Lehrer, University of Hawaii at Manoa, United StatesJohn Bernet Johnson, Rajiv Gandhi Centre for Biotechnology, India
Copyright © 2022 Mellors, Tipton, Fehling, Akoi Bore, Koundouno, Hall, Hudson, Alexander, Longet, Taylor, Gorringe, Magassouba, Konde, Hiscox, Strecker and Carroll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jack Mellors, am1lbGxvcnNAd2VsbC5veC5hYy51aw==; Miles Carroll, bWlsZXMuY2Fycm9sbEBuZG0ub3guYWMudWs=