 Charles E. Booth Jr
Charles E. Booth Jr Alexandra D. Powell-Pierce
Alexandra D. Powell-Pierce Jon T. Skare
Jon T. Skare Brandon L. Garcia
Brandon L. Garcia- 1Department of Microbiology and Immunology, Brody School of Medicine, East Carolina University, Greenville, NC, United States
- 2Department of Microbial Pathogenesis and Immunology, College of Medicine, Texas A&M University, Bryan, TX, United States
Pathogens that traffic in the blood of their hosts must employ mechanisms to evade the host innate immune system, including the complement cascade. The Lyme disease spirochete, Borreliella burgdorferi, has evolved numerous outer membrane lipoproteins that interact directly with host proteins. Compared to Lyme disease-associated spirochetes, relatively little is known about how an emerging tick-borne spirochetal pathogen, Borrelia miyamotoi, utilizes surface lipoproteins to interact with a human host. B. burgdorferi expresses the multifunctional lipoprotein, BBK32, that inhibits the classical pathway of complement through interaction with the initiating protease C1r, and also interacts with fibronectin using a separate intrinsically disordered domain. B. miyamotoi encodes two separate bbk32 orthologs denoted fbpA and fbpB; however, the activities of these proteins are unknown. Here, we show that B. miyamotoi FbpA binds human fibronectin in a manner similar to B. burgdorferi BBK32, whereas FbpB does not. FbpA and FbpB both bind human complement C1r and protect a serum-sensitive B. burgdorferi strain from complement-mediated killing, but surprisingly, differ in their ability to recognize activated C1r versus zymogen states of C1r. To better understand the observed differences in C1r recognition and inhibition properties, high-resolution X-ray crystallography structures were solved of the C1r-binding regions of B. miyamotoi FbpA and FbpB at 1.9Å and 2.1Å, respectively. Collectively, these data suggest that FbpA and FbpB have partially overlapping functions but are functionally and structurally distinct. The data presented herein enhances our overall understanding of how bloodborne pathogens interact with fibronectin and modulate the complement system.
Introduction
In the United States, tick-borne relapsing fever (TBRF) is caused by the spirochetes Borrelia hermsii and Borrelia turicatae. Whereas these spirochetes are vectored by the soft tick of the Ornithodoros genus, a related TBRF pathogen, Borrelia miyamotoi, is transmitted by ixodid ticks that also vector the agent of Lyme disease (LD), Borreliella burgdorferi (1–6). A recent study suggests that B. miyamotoi is more widespread in some areas of the U.S. than previously recognized (7). Human infection by B. miyamotoi results in overlapping but differing pathology from TBRF, and is known as B. miyamotoi disease (BMD) (4, 6). In immunocompetent hosts, BMD presents as a recurrent influenza-like illness that is treatable with doxycycline, ceftriaxone, azithromycin, and potentially amoxicillin (8, 9). Although BMD-, TBRF-, and LD-causing spirochetes are tick-transmitted, their lifestyles within vertebrate hosts differ (10, 11). In the context of vertebrate infection, LD Borreliella are thought to survive within the skin and briefly in the bloodstream as a means to disseminate to deeper distal tissues (12–14), while TBRF and BMD Borrelia predominantly exist in host blood, indicating a heightened need to evade both soluble and cellular blood-borne immune components (1, 15).
To survive in immunocompetent hosts, TBRF and BMD spirochetes exhibit a robust antigenic variation mechanism via a surface-exposed lipoprotein termed variable major protein (Vmp), which allows them to evade clearance from the adaptive immune response (4, 16–18). Each Vmp-associated serotype leads to high bacteremia until a targeted antibody response lowers the relapsing fever Borrelia load, after which a new serotype arises, resulting in a “relapse” (19, 20). TBRF symptoms are correlated with the presence of blood-borne spirochetes and the gene conversion event that produces unique Vmp proteins is associated with relapses for TBRF, though less prevalent in BMD (1, 4). In addition to their Vmp-associated immune evasive strategy, B. miyamotoi uses other surface lipoproteins to target innate immune mechanisms, including the complement system (21–25). The complement system serves as a first line of defense against invading pathogens. It is initiated through three pathways known as the alternative (AP), lectin (LP), and classical pathway (CP). The CP plays a critical role in elimination of foreign cells and is initiated by antibody-antigen complexes bound by the circulating complement component, C1 (26, 27). Upon activation of C1, proteolytic cleavage of downstream complement components leads to a self-amplifying cascade, resulting in opsonization and phagocytosis of the target cell, modulation of adaptive immunity, neutrophil synergy, and bactericidal activity through formation of the terminal complement complex (TCC), also known as the membrane attack complex (MAC) (27). To avoid destruction of healthy self-cells, host regulators bind complement components, preventing their activation in circulation, such as C1 esterase inhibitor (C1-INH), or on the surface of cells, such as Factor H (FH) (27, 28). A number of bacterial complement inhibitors have been characterized, from inhibitors of complement activation in Gram-positive pathogens, to inhibitors of the MAC in susceptible Gram-negative pathogens, as well as factors that reduce opsonization and phagocytosis by host immune cells (29, 30).
While the complement/complement-evasion axis is better understood for Lyme disease-associated spirochetes (31), recent reports highlight the likely importance of complement evasion by B. miyamotoi. CbiA, a surface-exposed FH, C3, C3b, C4b, and C5-binding lipoprotein, inhibits both the CP and AP (21). BOM1093 is a B. miyamotoi vitronectin-binding protein that inhibits complement via vitronectin’s endogenous TCC-inhibitory activity (25). Additionally, Vlp15/16 and Vlp18 were shown to inhibit the AP, though the mechanism of this activity has not yet been determined (22). The identification of a range of complement inhibitors, despite the relative nascent research into B. miyamotoi pathogenesis, is consistent with its blood-borne lifecycle in the vertebrate host.
We have previously shown that the B. burgdorferi BBK32 surface-exposed lipoprotein inhibits the CP by directly binding and inhibiting C1r, a serine protease required for activation of the CP (31–34). Separately, we demonstrated that BBK32 provides resistance to a serum-sensitive strain of B. burgdorferi, indicating a biologically relevant role for BBK32 in complement resistance (32–34). In addition, BBK32 is multifunctional, also acting as an adhesin to fibronectin and glycosaminoglycans (GAGs) (35–40). Additional work showed that BBK32 is required for optimal B. burgdorferi infection (35, 40). Phylogenetic analyses revealed orthologous protein families to BBK32 in both BMD and TBRF spirochetes, designated FbpA, FbpB, and FbpC (41). Both BMD and TBRF spirochetes harbor different sets of Fbp genes, with Borrelia miyamotoi encoding only fbpA and fbpB. In the TBRF pathogen B. hermsii, FbpC has been shown to bind fibronectin, however whether B. miyamotoi Fbp proteins retain functional similarity to BBK32 has not previously been explored (41, 42). In this study, we characterized the ability of B. miyamotoi FbpA and FbpB to bind human fibronectin and the complement protease C1r. Our detailed structure-function investigation reveals that these proteins exhibit differential interactions with both fibronectin and C1r, differences in the three-dimensional structures of their complement inhibitory domains, and demonstrates their ability to protect a serum-sensitive strain of B. burgdorferi from serum-mediated killing. Collectively, our data show that Fbp proteins from B. miyamotoi harbor partially overlapping, but distinct functions from one another. The novel finding that FbpB specifically recognizes active C1r may inform future efforts to develop therapeutics that are specific for the classical pathway of complement. The need for such complement-directed drugs is highlighted by the association of dysregulated or overactivated complement in important human pathologies (43–45).
Materials and Methods
Growth of Bacterial Strains
Borreliella burgdorferi B31 strain B314 (46) (Table 1) was grown to mid-log phase in BSK-II and 6% normal rabbit serum (Pel-Freez Biologicals, Rogers, AR) under conventional conditions (1% CO2, 32°C, pH 7.6). Genetically transformed B. burgdorferi strains were grown with kanamycin at 300 μg/mL. Borrelia miyamotoi strain FR64b was grown in MKP-F media (24) under the same conditions listed above for B. burgdorferi. Escherichia coli strain NEB-5α (New England Biolabs) was grown in Luria broth media under aerobic conditions at 37°C and, as appropriate, with kanamycin at 50 μg/mL. E. coli strain BL21 (DE3) (Thermo Fisher) was grown in Terrific Broth (Fisher Bioreagents) with kanamycin (50 μg/mL) at 37°C until an OD600 between 0.6-0.8 was achieved, then moved to 16°C overnight for production of recombinant Fbp proteins.

Table 1 Bacterial Strains and Plasmid Constructs used in this study.
Plasmid Constructs
The following constructs for B. miyamotoi FR64b fbpA (UNIPROT#: AHH06014.1) and fbpB (UNIPROT#: AHH05635.1) were subcloned into pT7HMT or a modification of pT7HMT encoding maltose binding protein (MBP) (48). All plasmid constructs are listed in Table 1. E. coli codon-optimized DNA fragments were purchased from IDT Technologies gBlock Gene Fragment Service or produced using PCR amplification templated from previously cloned constructs using methodology and constructs previously described (32–34, 48): FbpA-C residues 222-371; FbpA-C-R264A-K343A; FbpB-C residues 264-432; FbpB-Δ5C residues 264-427; FbpA-N residues 152-201; and FbpB-N residues 207-248. Oligonucleotides used are listed in Table 2. Residue numbering were based on UNIPROT numbering. The expression constructs for B. burgdorferi BBK32-C and C1r-CCP2-SP originated from previous studies (32, 33).
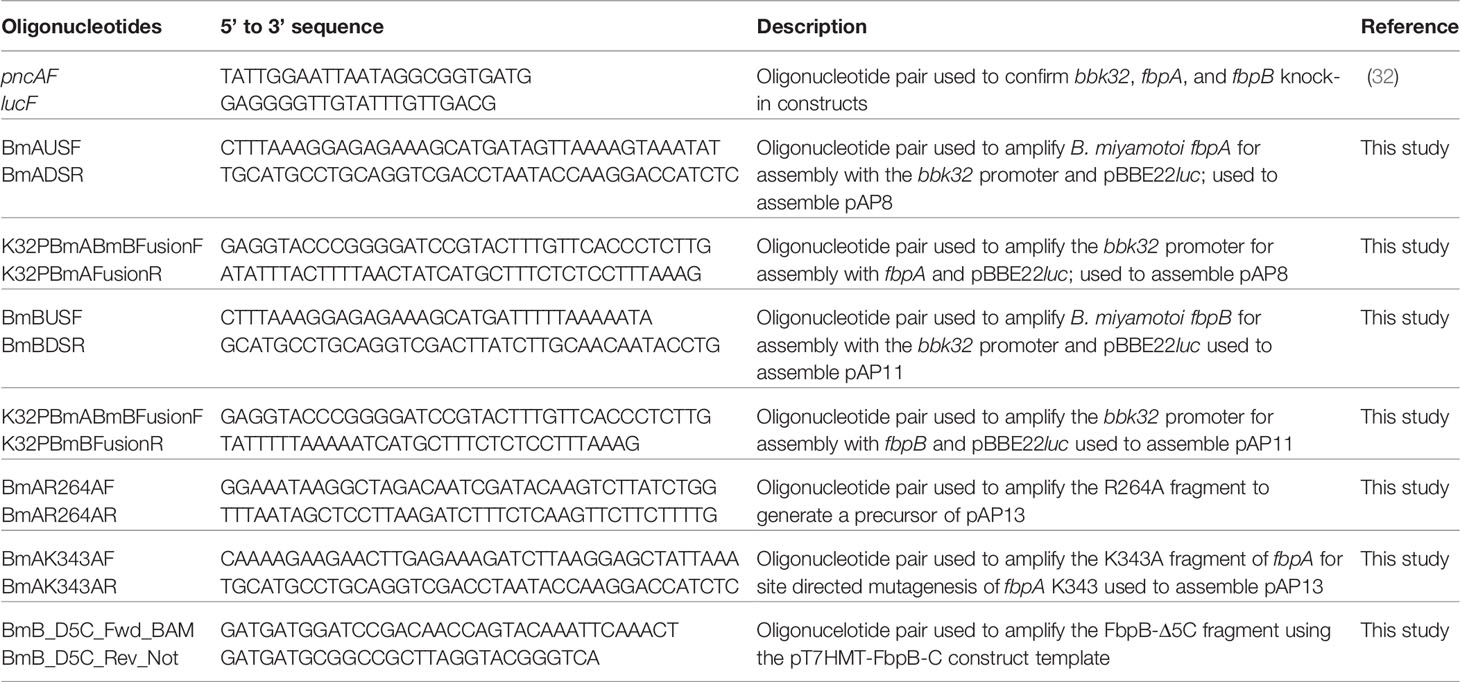
Table 2 Oligonucleotides used in this report.
Expression constructs for B. miyamotoi fbpA and fbpB were assembled using the NEBBuilderHiFi DNA Assembly Cloning Kit as described in (34). The B. miyamotoi fbpA and fbpB genes were transcriptionally linked with the bbk32 promoter from B. burgdorferi to promote equal expression in B314 relative to bbk32. The R264A and K343A mutations in fbpA were constructed sequentially by synthesizing a 194 bp fragment of fbpA from nucleotide 918 to 1112 containing the R264A mutation, and then once the R264A construct was obtained, with a 254 bp fragment from nucleotide 1112 to 1366 containing the K343A mutation. Both DNA fragments were synthesized by IDT gBlocks using the native B. miyamotoi strain FR64b fbpA sequences. The fragments were each assembled into the fbpA gene using primers that added overhangs complementing the fbpA gene adjacent to the synthesized fragments. These fragments were then assembled into the fbpA-expressing vector using NEBuilderHiFi DNA Assembly Cloning Kit. All plasmids were sequenced to ensure the desired mutations were obtained and that no additional mutations were introduced during the assembly process.
Transformation of B. burgdorferi
Transformation of strain B314 with the plasmid constructs pAP8, pAP11, and pAP13 was done as previously described (33, 49).
Protein Expression and Purification
Recombinant proteins were expressed and purified as in (32, 33). Briefly, after elution of the Ni column using the Elution buffer (20 mM Tris (pH 8.0), 500 mM NaCl, 500 mM Imidazole), the proteins were exchanged into 20 mM Tris (pH 8.0), 500 mM NaCl, 10 mM Imidazole using a Desalting 26/10 column (GE Healthcare). The His-tag was then removed by incubation with the tobacco etch virus (TEV) protease and 5 mM β-mercaptoethanol overnight at room temperature. The TEV digested proteins were separated from the His-tag by passing over a 5 mL HisTrap-FF column on the FPLC with the captured flowthrough further purified using a HiLoad Superdex 75 PG gel filtration column (GE Healthcare). The single peak after gel filtration was examined using SDS-PAGE analysis, then pooled and exchanged into HBS buffer (10 mM HEPES [pH 7.3], 140 mM NaCl). The C1r-CCP2-SP construct was purified according to previously published protocols with an added final gel filtration chromatography step using a HiLoad Superdex 75 PG column (GE Healthcare) and exchanged into HBS buffer (33, 34, 50, 51). Purified full-length complement proteins of both the zymogen and enzymatically active forms of C1r were obtained from Complement Technology, Inc. (Tyler, TX). Purified full-length human fibronectin was purchased from Millipore Sigma. Sequence alignments of the proteins were generated using Clustal alignment server from EMBL-EBI.
Surface Plasmon Resonance
Surface plasmon resonance (SPR) was performed on a Biacore T200 instrument set to 25°C with a flowrate of 30 μL/min. The running buffer was HBS-T (10 mM HEPES (pH 7.3), 140 mM NaCl, and 0.005% Tween 20) or HBS-T containing 5 mM CaCl2. Proteins of interest were immobilized on CMD200 sensor chips (Xantec Bioanalytics) via standard amine coupling as before (32–34). Immobilization quantities were as follows: human plasma fibronectin over MBP-FbpA-C (1015.3 RU), MBP-FbpB-C (1074.1 RU); C1r-CCP2-SP over FbpA-C (1061.2 RU), FbpB-C (877.7 RU), FbpA-C-R264A-K343A (FbpA DA-C) (1591.8 RU); C1r zymogen or active C1r over FbpA-C (852.4 RU), FbpB-C (1116.6 RU). C1r-CCP2-SP over Fbps was performed in multi-cycle using a twofold dilution series of the C1r-CCP2-SP ranging from 0-100 nM. Data was obtained in triplicate with each cycle having an association time of 2 min, a dissociation time of 3 min, and three 1 min injections of regeneration buffer (0.1 M Glycine [pH 2.0], 2.5 M NaCl). Equilibrium dissociation constants (KD) were obtained from the resulting kinetic fits generated by the Biacore T200 Evaluation Software (GE Healthcare). Zymogen or active C1r binding data for FbpA-C and FbpB-C was obtained in single cycle using a fivefold dilution series of either C1r zymogen or active C1r ranging from 0-100 nM. Data was obtained in triplicate with each cycle having an association time of 5 min, a final dissociation time of 1 hour, and two 1 min regeneration injections with regeneration buffer (0.1 M Glycine [pH 2.0], 2.5 M NaCl). Equilibrium dissociation constants (KD) were obtained from the resulting kinetic fits generated by the Biacore T200 Evaluation Software (GE Healthcare). Human plasma fibronectin was buffer exchanged into HBS-T and then injected over MBP-FbpA-N or MBP-FbpB-N. Data was obtained in multicycle using a twofold dilution series ranging from 0-500 nM. Data was obtained in triplicate with each cycle having an association time of 2 min, a dissociation time of 3 min, and two 1 min injections of regeneration buffer (10 mM CAPS [pH 10.0]). Steady-state affinity fits were performed using Biacore T200 Evaluation Software.
Surface Plasmon Resonance Competition Experiments
SPR was performed using conditions described for the direct binding experiments. FbpA-C (935.4 RU), FbpB-C (1082.9 RU), and FbpB-Δ5C (1115.4 RU) were immobilized on a new CMD200 sensor chip (Xantec Bioanalytics) via standard amine coupling as before (32–34). 25 nM C1r-CCP2-SP alone, or with 25 nM FbpA-C, FbpB-B, FbpA DA-C, or FbpB-Δ5C were injected over the Fbp biosensors. Injections were performed in triplicate with each cycle having an association time of 2 min, a dissociation time of 3 min, and two 1 min injections of regeneration buffer (0.1 M Glycine [pH 2.0], 2.5 M NaCl). The binding response just prior to injection stop was used to determine differences for the ability of the soluble Fbp proteins to compete with immobilized Fbps for C1r-CCP2-SP binding.
Generation of Polylonal Antibodies Against FbpA and FbpB
Polyclonal antibodies reactive with FbpA and FbpB were generated by separately immunizing female C57BL/6 mice intradermally with 25 µg either FbpA-C or FbpB-C in an equal volume of PBS and TiterMax® Gold Adjuvant (Sigma Aldrich) as outlined (52). Two weeks after the initial immunization, mice were boosted with 25 µg of the same protein. Two weeks after boosting, mice were euthanized and blood was immediately isolated by exsangunation, allowed to clot, and serum removed after low speed centrifugation. Reactivity and specificity of each serum sample to B. miyamotoi FbpA and FbpB was evaluated by Western Blot using purified recombinant protein. All animal work was reviewed and approved by the Texas A&M University Institutional Animal Care and Use Committee (protocol number 2019-0422).
Far Western Overlay Analysis With Full-Length B. miyamotoi Proteins
Far Western overlays were carried out as described in (32, 35). Briefly, B. burgdorferi strain B314 derivatives were cultured in BSKII media supplemented with 300 μg/mL kanamycin. Cultures were harvested and B. burgdorferi whole-cell lysates were generated for B314 strains producing FbpA, FbpB, FbpA DA, or from the pBBE22luc vector-only control. 2.5 x107 whole cell equivalents were resolved by SDS-PAGE with 0.625 μg of recombinant Fbp-C and MBP-Fbp-N proteins used as controls for C1r and fibronectin, respectively. The SDS-PAGE resolved proteins were transferred to a PVDF membrane and then blocked overnight in 5% non-fat milk. The blots were washed and incubated with 20 μg of zymogen or active C1r for 1 hour. PVDF membranes were probed for C1r binding using a goat antibody to human C1r (R&D Systems) at a 1:3,000 dilution followed by a rabbit anti-goat antibody conjugated to HRP (Invitrogen) at a 1:5,000 dilution. Similar experimentation was done after incubation with 20 μg of human fibronectin with subsequent binding determined using a mouse monoclonal antibody specific for human fibronectin conjugated to HRP (Santa Cruz Biotechnology) at a 1:1,000 dilution. All blots were then visualized using the SuperSignal™ West Pico Plus chemiluminescent kit (Thermo Scientific). Control Western blots were performed as above with 2.5 x107 whole cell equivalents to monitor Fbp expression and to serve as loading controls. To probe for FbpA and FbpB expression, mouse antibodies against FbpA or FbpB were used at a 1:5,000 dilution, respectively. To examine the flagellar protein FlaB, a mouse monoclonal antibody specific for FlaB (US Biologicals, Inc.) was used at a 1:20,000 dilution. Detection of membrane bound immune complexes was facilitated with a goat anti-mouse immunoglobulin HRP conjugate diluted 1:10,000 (Invitrogen). Visualization was the same as above. All Far Western blots were performed in duplicate.
Crystallization, Structure Determination, Refinement, and Analysis
FbpA-Cwas concentrated to 12.8 mg/mL total protein in a buffer of (10 mM HEPES [pH 7.3], 50 mM NaCl). Crystals were obtained by vapor diffusion of sitting drops at 20°C by mixing 1 µL of protein with 1 µL of precipitant solution (0.1 M Tris [pH 8.5], 2 M ammonium sulfate). Large crystal plate clusters generally appeared after 9-10 d. Sequential rounds of micro-seeding produced samples of sufficient size and quality for harvesting. Samples were cryopreserved in precipitant solution supplemented with an additional 20% glycerol.
Crystallization trials using FbpB-C failed to produce diffraction quality crystals. Constructs removing residues from the N-terminus and C-terminus were screened resulting in identification of crystallization conditions for a construct of FbpB where five residues were removed from the C-terminus (i.e., FbpB-Δ5C). FbpB-Δ5C was concentrated to 19.1 mg/mL and 8.69 mg/mL in 10 mM Tris (pH 8.0), 50 mM NaCl buffer. Crystals of FbpB-Δ5C were obtained by vapor diffusion of sitting drops at 4°C for both concentrations. Initial crystals grew in a condition containing 0.2M MgCl2, 0.1M Tris (pH 8.5), 30% PEG 4,000 and an optimized condition was identified by additive screening 4 mg/mL in 0.2 M MgCl2, 0.1 M Tris (pH 8.5), 26.25% PEG 4,000, 35 mM spermidine followed by microseeding. Crystals were harvested and cryoprotected by supplementing the crystallization buffer with 10% glycerol.
Monochromatic X-ray diffraction data were collected at 0.975Å (FbpA-C) or 1.00Å (FbpB-Δ5C) wavelength using beamline 22-ID of the Advanced Photon Source (Argonne National Laboratory). Diffraction data were integrated, scaled, and reduced using the HKL2000 software suite (53) and assessed for data quality (54) (Table 3). FbpA-C crystals grew in the space group P21 and contained two copies in the asymmetric unit which were related by twofold non-crystallographic symmetry. FbpB-Δ5C crystals grew in space group C2 and contained a single copy of FbpB-Δ5C in the asymmetric unit. Initial phases for FbpA-C were obtained by molecular replacement using a single copy of a poly-alanine model of BBK32-C (PBD:6N1L) generated with the program CHAINSAW via the CCP4 software package (55). Initial phases for FbpB-Δ5C were obtained by molecular replacement using a FbpA-C polyalanine model that was created using CHAINSAW from CCP4 (55). Following an initial round of refinement, models for FbpA-C and FbpB-Δ5C were completed by a combination of automated chain tracing using PHENIX.AUTOBUILD (56–58) and manual building into electron density maps using COOT (59). The final model was completed upon iterative cycles of manual rebuilding and refinement using PHENIX.REFINE (56–58). The N-terminal cloning artifact sequence for FbpA-C (Residues G-S-T-G-S) and FbpB-Δ5C, and residues 264-268, 355, 401-403, and 427 of FbpB-Δ5C were not modeled in the final refined structure due to poor electron density. Refined coordinates and structure factors have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University (www.rcsb.org/) under PDB ID codes 7RPR for FbpA-C and 7RPS for FbpB-Δ5C. A description of crystal cell constants, diffraction data quality, and properties of the final models for FbpA-C and FbpB-Δ5C can be found in Table 3. Representations of the protein structures and electron density maps were generated by PyMol (www.pymol.org/).
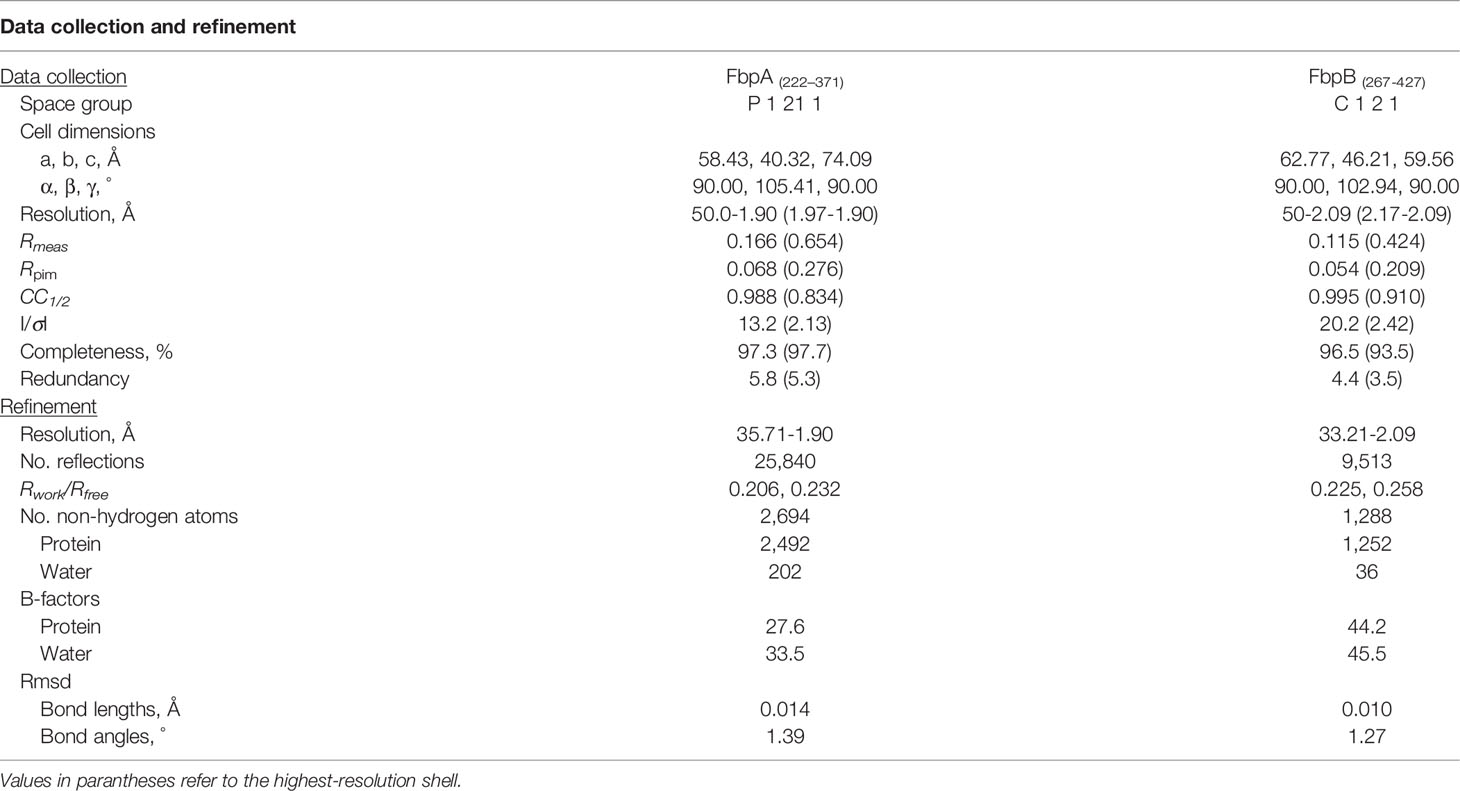
Table 3 Refinement data for the solved FbpA and FbpB crystal structures.
Circular Dichroism
Far-UV spectra were collected to assess the secondary structure for FbpA-C, FbpB-C, FbpA DA-C, and FbpB-Δ5C. Samples were diluted to 10 μM in 10 mM Na3PO4 based on previous protocols (60). Spectra for a buffer control were used to subtract background noise. Spectra were collected across a wavelength range of 180-300 nm, at 120 nm min-1, using 1 nm step, 0.5 sec response, and 1 nm bandwidth. Data was collected using a Chirascan V100 instrument with a square small volume quartz cuvette with a path length of 0.05 cm (Applied Photophysics, UK).
Analytical Gel Filtration Chromatography
Analytical gel filtration was performed as before (34). 100 μg of FbpB-C and FbpB-Δ5C in a total volume of 500 μL were injected onto a Superdex 75 10/300 GL column (GE Healthcare) at a flowrate of 0.5 mL/min. The running buffer used was 10 mM HEPES (pH 7.3), 140 mM NaCl.
C1r-CCP2-SP Enzyme Inhibition Assay
A colorimetric assay was used to monitor dose-dependent inhibition of C1r-CCP2-SP enzyme activity based off (33, 34). 1 μM Fbps-C were incubated with 15 nM C1r-CCP2-SP enzyme and 300 μM of the serine protease substrate Z-Gly-Arg-thiobenzyl (MP Biomedical) in HBS buffer (10 mM HEPES [pH 7.3] and 140 mM NaCl). Enzyme cleavage of Z-Gly-Arg-thiobenzyl was determined by incubating the reaction with 100 μM DTNB (5,5’-Dithiobis-[2-nitrobenzoic acid], Ellman’s reagent). The EnSight Multimode Plate Reader (PerkinElmer) was used to read the absorbance values for the reaction at 412 nm. Data were obtained in triplicate and normalized with 15 nM C1r-CCP2-SP lacking inhibitor as a positive control that represented the 100% amount of Z-Gly-Arg-thiobenzyl cleaved by C1r-CCP2-SP in 30 min at 25°C. Using a DTNB and Z-Gly-Arg-thiobenzyl only negative control allowed for subtraction of any non-specific colorimetric change.
ELISA-Based Complement Inhibition Assay
An ELISA-based assay that utilizes IgM as a specific activator of the classical pathway as described (32–34, 50, 61), was used to determine the pathway’s downstream effect while in the presence of the Fbps. Data were obtained in triplicate and normalized using no inhibitor with 2% normal human serum (Innovative Research) as a positive control that represented the 100% amount of C4b deposition in 1 hour at 37°C. Non-specific interactions were subtracted out using a negative control with no serum. An EnSight Multimode Plate Reader (PerkinElmer) was used to read the absorbance values at 450 nm for the reaction.
Hemolysis Assay
A hemolysis assay that uses antibody-opsonized sheep red blood cells to specifically activate the classical pathway was performed as previously described (32, 33, 62) to determine if B. miyamotoi Fbp-C proteins protected erythrocytes from complement-mediated hemolysis. A serial dilution series of Fbp-C proteins was incubated with buffer-exchanged pre-sensitized sheep erythrocytes (Complement Technology) and 2% normal human serum (Innovative Research). After a 1 hour incubation at 37°C the reaction was clarified via centrifugation at 500 x g and the absorbance read using an EnSight Multimode Plate Reader (PerkinElmer) at 412 nm. Data were obtained in triplicate normalized using a no inhibitor with serum positive control that represented the 100% cell lysis at 37°C in 1 hour. Background hemolysis was subtracted out using a negative control with only buffer and red blood cells.
Quantitative ELISA to Assess B. miyamotoi Lipoproteins Binding to Serum C1r
The interaction of B. miyamotoi Fbp proteins with C1r in human serum was measured using an ELISA-type binding assay based on (63). Fbp proteins were immobilized overnight onto high-binding 96-well plates (Greiner Bio-One) at 0.2 mg/mL in 100 mM NaCO3 (pH 9.6). A twofold serial dilution of normal human serum (Innovative Research) ranging from 0.0012-10% serum in CP Assay buffer (10 mM HEPES [pH 7.3], 140 mM NaCl, 2 mM CaCl2, 0.5 mM MgCl2, 0.1% gelatin) was incubated with immobilized Fbp-C proteins at 37°C for 1 hour. The amount of serum C1r bound to immobilized Fbps was found using a goat antibody to human C1r (R&D Systems) diluted 1:2,000 and immune complexes detected with rabbit anti-goat Ig conjugated to HRP (Invitrogen) at a 1:3,000 dilution. Data were obtained in at least duplicate and read at 450 nm using the EnSight Multimode Plate Reader (PerkinElmer) with non-specific interactions subtracted out using a negative control with no serum.
Proteinase K Accessibility Assays
B. burgdorferi strain B314 was grown to mid-log phase and harvested by centrifugation at 4,500 x g for 10 minutes at 4°C, and washed once with PBS. The cell pellet was resuspended in 0.8 mL of either PBS alone, or PBS with proteinase K to a final concentration of 200 μg/mL, and incubated at 20°C for 40 min. Reactions were terminated by the addition of phenylmethylsulfonyl fluoride (PMSF) to a final concentration of 1 mM. Cells were centrifuged (4,500 x g for 10 min at 4°C), washed twice with PBS, and resuspended in sample buffer. Samples corresponding to 5x107 whole cell equivalents were run on SDS-PAGE gel, transferred to PVDF membranes and immunoblotted with mouse polyclonal antibodies to FbpA and FbpB or monoclonal antibodies directed against BBK32 or borrelial FlaB (US Biologicals Inc.) and imaged with chemiluminescence as previously described (33).
Serum Complement Sensitivity Assay
Assays were performed as in (33, 34). Briefly, B. burgdorferi strain B314 producing BBK32 (pCD100), B. miyamotoi FbpA (pAP8) or FbpA-DA (pAP13), and B. miyamotoi FbpB (pAP11) (see Table 1), as well as the vector-only control B314 pBBE22luc, were grown at 32°C in 1% CO2 to early- to mid-log phase and diluted in complete BSK-II media to a final cell density of 1x106 cells/mL. B. burgdorferi cells were then incubated in 15% normal human serum (Complement Technology) or heat inactivated serum (55°C for 30 min.), with thermal inactivation of complement proteins as a positive control for survival, for 1.5 hours at 37°C under gentle agitation. Cell survival was assessed by dark field microscopy based on cell motility and overt membrane damage or lysis.
Soluble Protein Inhibition Rescue Assay
The ability of exogenous B. miyamotoi FbpA-C, FbpA DA-C, and FbpB-C to rescue serum-sensitive B. burgdorferi B314 pBBE22luc was measured similarly to the serum sensitivity assay. Exogenous complement inhibitory proteins were added at a final concentration of 48 nM, 240 nM, and 1.2 μM, approximately five-fold less than, equal to, and five-fold greater than the concentration of C1r in the human serum sample used. Survival was compared to that of B314 pBBE22luc with no exogenous protein addition, and of B314 pCD100, which produces BBK32, as described for the serum sensitivity assays.
Statistics
Data is shown as the average of the replicates with 95% confidence intervals. Half-maximal inhibitory concentrations (IC50) values for the CP inhibitory ELISA and CP hemolysis assays were determined using a non-linear variable slope regression fit. The half-maximal effective concentration (EC50) for the serum C1r binding ELISA was determined using a non-linear one-site total regression fit. One-way ANOVA was used for the C1r enzyme and competition SPR experiments to determine differences among the means, followed by a post-hoc Tukey analysis for pairwise comparison of each Fbp protein. For serum sensitivity and B314 “rescue” assays, two-way ANOVA with a Šidák correction for multiple comparisons was used. All statistical analysis was performed using GraphPad Prism version 9.3.
Results
B. miyamotoi Encodes Two Orthologous Proteins to B. burgdorferi BBK32
B. burgdorferi BBK32 interacts with multiple host proteins through non-overlapping protein domains. The N-terminus of BBK32 (BBK32-N) binds to fibronectin (35–37, 40, 64–70), while the C-terminal domain (BBK32-C) interacts with complement serine protease C1r (32, 33). Relapsing fever spirochetes encode a combination of up to three separate BBK32 orthologs termed fibronectin binding proteins (Fbps) (41) (Figure 1A). Fbp genes organize into three protein families denoted FbpA, FbpB, and FbpC and vary in their sequence identity to BBK32 between 56-62% (FbpAs), 25-30% (FbpBs), and 22-27% (FbpCs) (Figure S1A). A BLASTp search of the B. miyamotoi strain FR64b genomic sequence, using B. burgdorferi strain B31 BBK32 as a query, identified only two orthologous sequences; one belonging to the FbpA family and one to the FbpB family (Figure 1A). Unlike B. miyamotoi FbpA, FbpB lacks significant amino acid sequence homology for a key motif that is important for BBK32’s interaction with fibronectin (Figure 1B) (i.e., gelatin-binding domain sequence [GBD]) (69). Similarly, while a critical residue involved in the BBK32/C1r interaction is conserved in both FbpA and FbpB (i.e. BBK32-R248/FbpA-R264/FbpB-R309), another important ‘hot-spot’ lysine residue is conserved only in FbpA (BBK32-K327/FbpA-K343) (Figure 1B) (34).
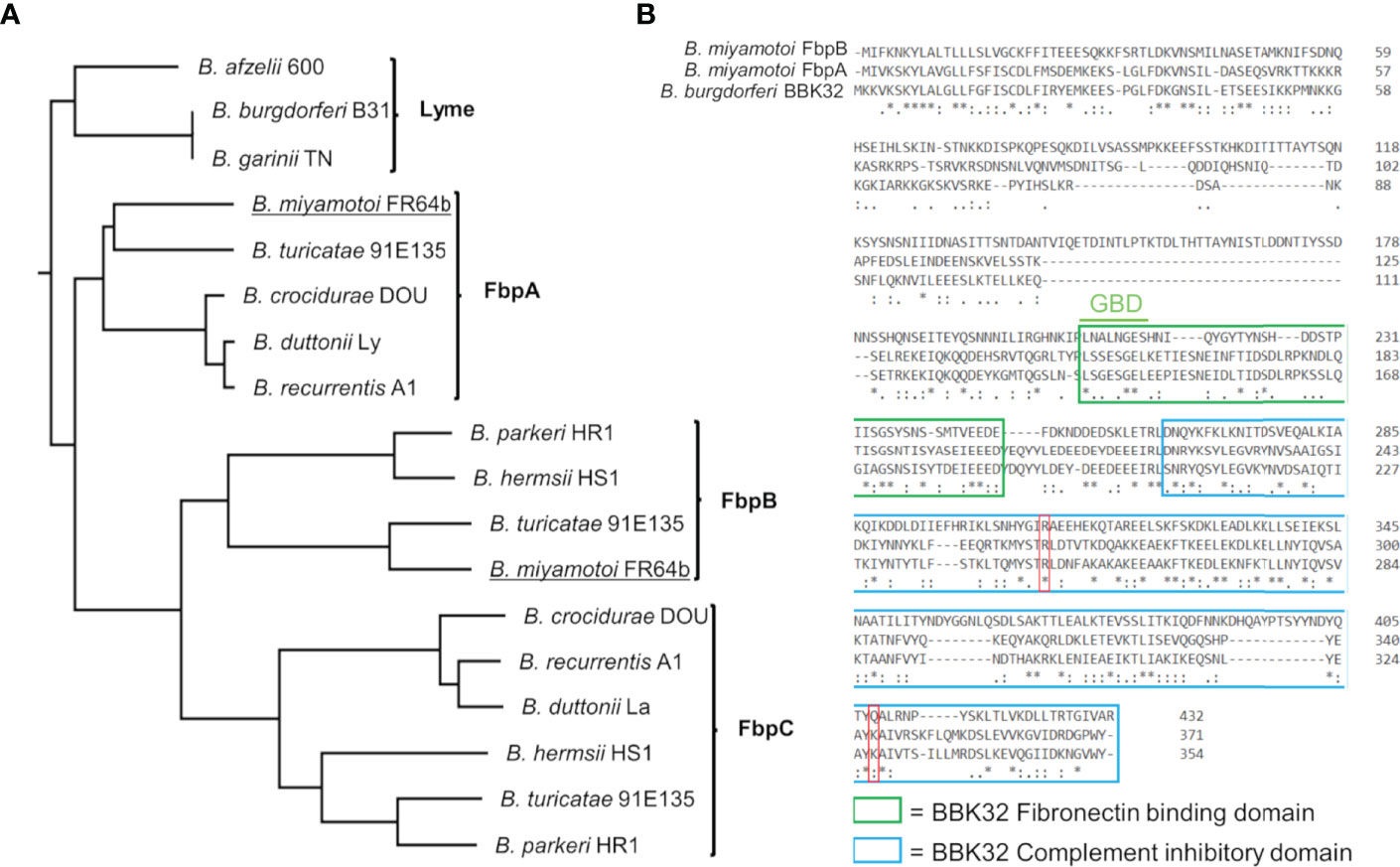
Figure 1 B. miyamotoi encodes two orthologous genes to B. burgdorferi BBK32. (A) BBK32 orthologs are found in relapsing fever (RF)-associated and B. miyamotoi spirochetes and are denoted FbpA, FbpB, and FbpC (41). B. miyamotoi FR64b FbpA and FbpB are underlined. (B) An alignment of B. miyamotoi strain FR64b FbpA and FbpB to B. burgdorferi strain B31 BBK32 shows differences of the amino acid sequences within the fibronectin binding (green box) and complement inhibitory domains (blue box). The gelatin-binding domain (GBD) of BBK32 is denoted. The key residues R248 and K327 of BBK32 involved in complement C1r binding are indicated by a red box. * conserved, : strongly similar, . weakly similar.
Differential Interactions of B. miyamotoi Fbps With Human Fibronectin
We hypothesized that the sequence variation in predicted functional sites of FbpB, and to a lesser degree FbpA, may result in altered function of B. miyamotoi Fbps relative to BBK32. To test this, we first began by evaluating the direct interaction of each protein with human fibronectin. We expressed and purified recombinant maltose-binding protein (MBP) fusion proteins that encoded the predicted fibronectin-binding site based on alignment to BBK32 (termed MBP-FbpA-N and MBP-FbpB-N) (Figure 1B, green box). Surface plasmon resonance (SPR) experiments were carried out by immobilizing each fusion protein on the surface of an SPR sensorchip and evaluating purified human fibronectin as an analyte in a dose-dependent manner. MBP-FbpA-N bound dose-dependently to soluble human fibronectin (KD = 170 nM) (Figures 2A, C), whereas MBP-FbpB-N exhibited no detectable interaction with fibronectin at concentrations up to 500 nM (Figures 2B, D).
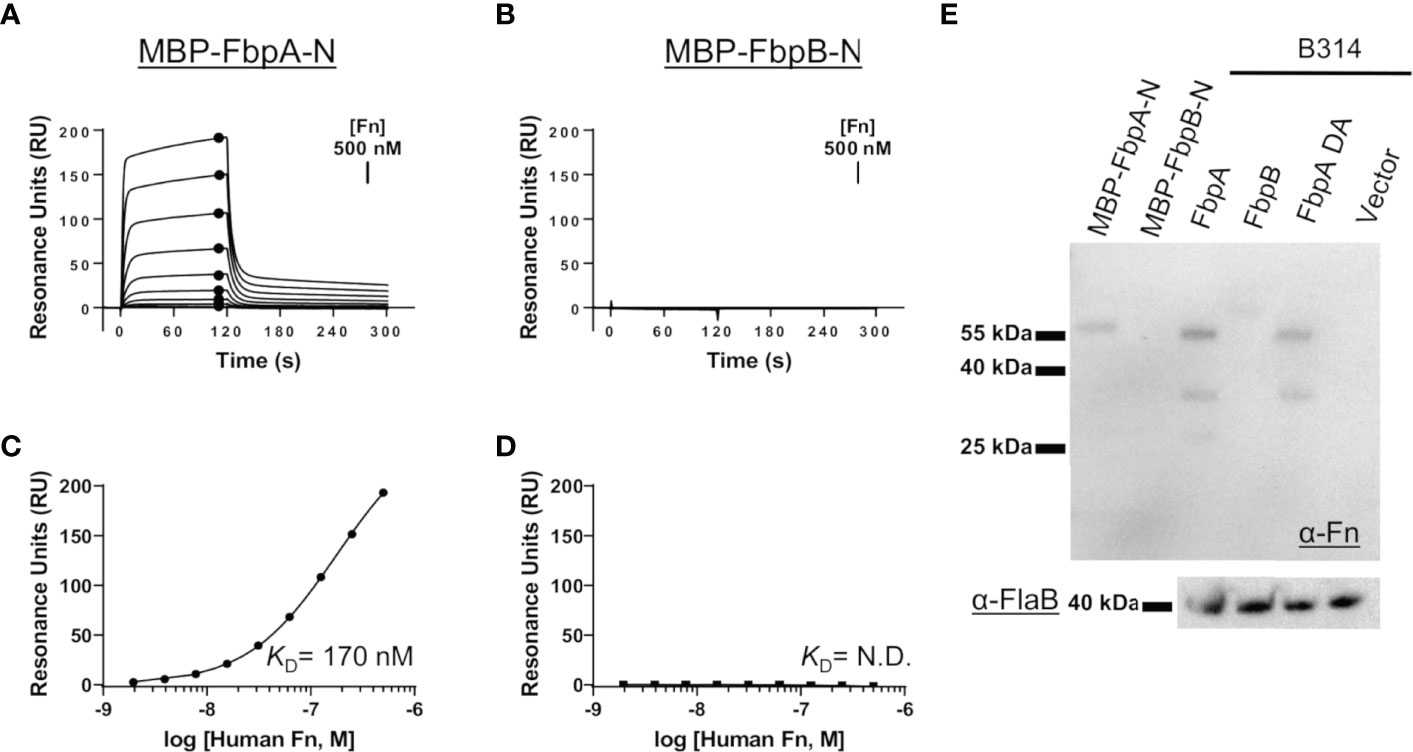
Figure 2 Assessing the interaction of human fibronectin with B. miyamotoi FbpA and FbpB. (A, B) SPR was used to quantitatively measure the interaction between FbpA-N (A) and FbpB-N (B) with human fibronectin. MBP-Fbp-N fusion proteins (Fbp sequences denoted by green box in Figure 1B) were immobilized on a sensorchip using amine coupling, then a twofold dilution series of purified human fibronectin ranging from 1.95-500 nM was injected. A representative sensorgram from the three replicates for each biosensor surface is shown. (C, D) Equilibrium dissociation constants (KD) were determined using steady state fits and are shown as the mean +/- standard deviation of three replicates in Table 4 for FbpA (C) and FbpB (D). Closed circles in (A) correspond to the steady-state response used for calculation of the steady state fits in (C), respectively. A KD value for the fibronectin interactions with MBP-FbpB-N was not determined (N.D.). (E) To assess if full-length Fbp proteins were capable of binding fibronectin, a Far Western approach was implemented by probing full-length Fbps from B. burgdorferi strain B314 “knock-in” lysates with fibronectin. Full-length FbpB fails to interact with fibronectin, whereas FbpA and FbpA DA do. A spirochete transformed with shuttle vector only (pBBE22luc) and recombinant MBP-FbpA-N/MBP-FbpB-N were used as controls. A Western blot against the flagellar protein FlaB (α-FlaB) was used as a loading control.
To exclude the possibility that FbpB may utilize a site outside of the predicted fibronectin-binding region to engage fibronectin, and to evaluate the interaction in the context of a full-length lipoprotein, we expressed intact fbpA and fbpB ectopically in B. burgdorferi strain B314. B. burgdorferi B314 is a serum-sensitive, high-passaged strain that has lost all linear plasmids including the 36 kilobase linear plasmid (lp36) that encodes bbk32 (46). Equivalent protein lysates from B. burgdorferi strain B314 derivatives were tested for their ability to produce native B. miyamotoi FbpA, a double alanine mutant of FbpA at residues 264 and 343—R264A and K343A—denoted FbpA-DA (discussed below), and FbpB, by SDS-PAGE and Western immunoblot analysis (Figures S2A, B). Whereas the antibody to FbpA recognized only the FbpA protein, antisera to FbpB recognized both FbpB and, to a lesser extent, FbpA (Figure S2A). We then tested these same samples using a Far Western approach with human fibronectin as the probe. Both FbpA and FbpA-DA in B314 bound human fibronectin whereas samples containing FbpB or the pBBE22luc shuttle vector control spirochetes exhibited no detectable binding (Figure 2E). Consistent with the SPR experiments (Figures 2A–D), recombinant MBP-FbpA-N fusion protein bound human fibronectin in this assay format, whereas the MBP-FbpB-N protein did not (Figure 2E). Analysis of total lysate samples by SDS-PAGE (Figure S2B) and FlaB protein by Western blot (Figure 2E) demonstrated equivalent protein levels were used for comparison. These results show that while B. miyamotoi FbpA retains BBK32-like fibronectin-binding activity, FbpB does not.
B. miyamotoi FbpA and FbpB Bind to Activated C1r in a BBK32-Like Manner
Next, we examined if B. miyamotoi FbpA and FbpB bound human C1r in a manner similar to B. burgdorferi BBK32 (32–34). BBK32 binds C1r via a C-terminal domain where residues R248 and K327 are critical in mediating the interaction with the S1 site of the C1r serine protease (SP) domain and the B-loop of C1r, respectively (34). Each of these key residues are conserved in FbpA (i.e., R264 and K343), whereas in FbpB, only the arginine is conserved (i.e., R309) (Figure 1B). We expressed and purified recombinant C-terminal truncation constructs of B. miyamotoi FbpA (FbpA-C), a site-directed double alanine mutant of FbpA-C (FbpA-R264A-K343A, denoted FbpA DA-C) (Figure S2C), and FbpB (FbpB-C), and assessed their direct interaction with a recombinant activated C1r construct encompassing the complement control protein 2 and serine protease domains (C1r-CCP2-SP). SPR binding assays showed that FbpA-C and FbpB-C bound with high affinity to C1r-CCP2-SP with KD = 0.90 nM and 1.4 nM, respectively (Figures 3A, B and Table 4), whereas FbpA DA-C exhibited no detectable C1r-CCP2-SP binding (Figure 3C).
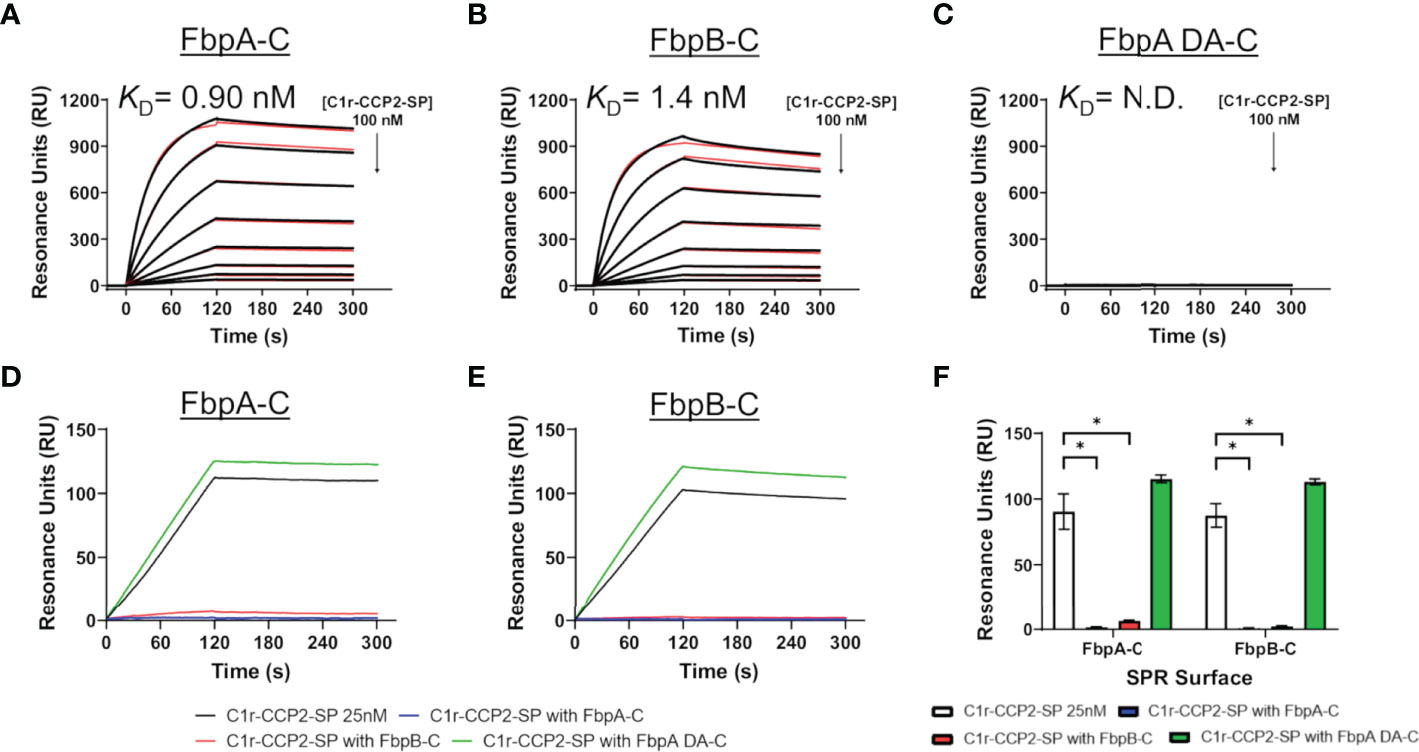
Figure 3 Assessing the interaction of human C1r with B. miyamotoi FbpA and FbpB. SPR was used to assess protein-protein interactions between the C-terminal regions of each Fbp protein and activated C1r-CCP2-SP. Immobilized (A) FbpA-C, (B) FbpB-C, and (C) FbpA-C-R264A-K343A (referred to as FbpA DA-C throughout) were subjected to an injection series of serially diluted C1r-CCP2-SP (0.78 - 100 nM). A representative sensorgram from the three replicates for each Fbp is shown with the black curve being the sensorgram and the red curve the associated kinetic fit. KD values were determined using kinetic fits and are shown as the mean +/- standard deviation of three replicates in Table 4. A KD value for C1r-CCP2-SP interactions with FbpA DA-C was not determined (N.D.). (D, E) Competition experiments were carried out using 25 nM C1r-CCP2-SP alone or mixed with 25 nM soluble Fbp proteins injected over FbpA-C (D) or FbpB-C (E). (F) Comparisons of statistical significance were performed for data shown in panels (D, E) using a one-way ANOVA followed by a multiple comparison Tukey test (* = p < 0.05).
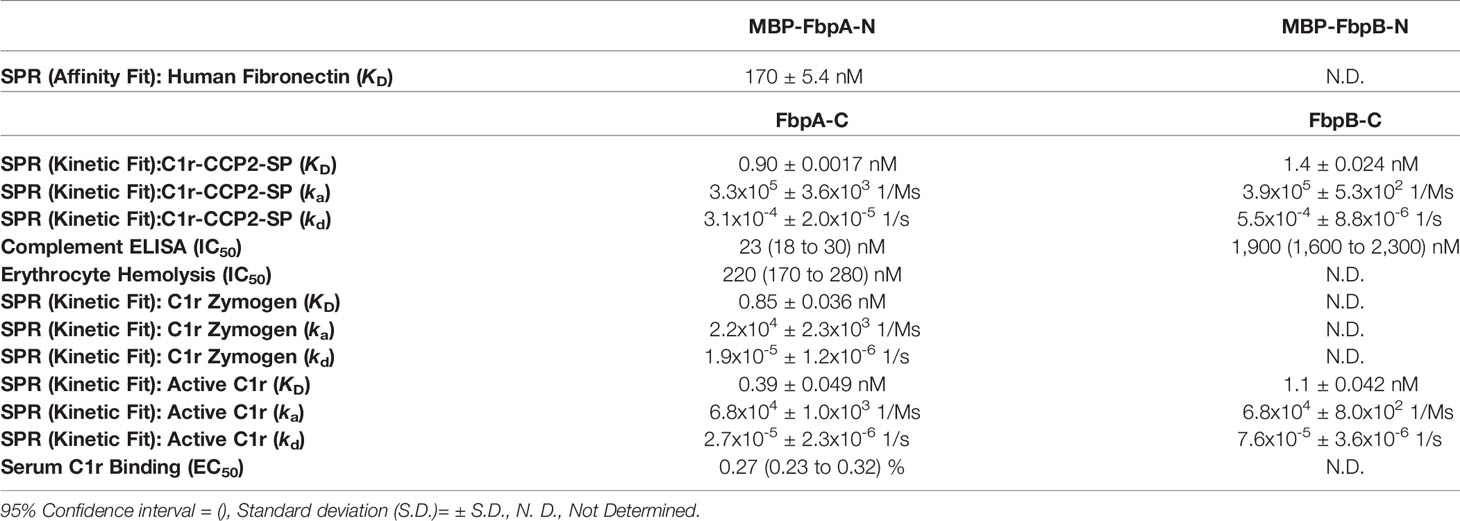
Table 4 SPR and complement assay results.
Next, we determined if FbpA-C and FbpB-C could compete with one another for binding to C1r-CCP2-SP. Equimolar mixtures of C1r-CCP2-SP with soluble FbpA-C, FbpA DA-C or FbpB-C were injected over immobilized FbpA-C and FbpB-C. Significantly reduced binding signals were observed when soluble FbpA-C or FbpB-C was used, but not for FbpA DA-C, indicating that active FbpA and FbpB proteins are capable of competing with one another for binding to activated C1r (Figures 3D–F). These results suggest that both FbpA and FbpB bind to activated C1r in a manner similar to what has been reported for BBK32 and underscores the importance of the conserved R248 and K327 residues (BBK32 numbering) in the FbpA/C1r interaction (34). Interestingly, although BBK32-K327 is not conserved in FbpB (Figure 1B), this did not preclude high-affinity interaction with activated C1r-CCP2-SP.
Differential Inhibition of Human Classical Pathway Complement Activation by B. miyamotoi FbpA and FbpB
B. miyamotoi FbpA-C and FbpB-C were next assessed for the ability to inhibit the classical complement cascade due ostensibly to the inhibition of C1r. A classical pathway-specific ELISA-based assay was implemented with Fbp-based inhibitors and normal human serum (32–34). As expected, in light of the C1r-CCP2-SP binding data (Figure 3), FbpA-C demonstrated potent inhibition of classical pathway-mediated complement activation (IC50 = 23 nM), whereas the FbpA DA-C mutant lost all inhibitory activity (Figure 4A and Table 4). FbpB-C blocked complement activation but, surprisingly, was ~80-fold less potent than FbpA (IC50 = 1,900 nM) despite having similar high affinity for activated C1r-CCP2-SP. To confirm these activities, each protein was serially diluted and incubated with opsonized sheep erythrocytes and normal human serum. Consistent with the ELISA-based assay, FbpA, but not FbpA DA prevented hemolysis (Figure 4B). In this assay format, FbpB-C had even lower activity, showing no inhibition at concentrations up to 4µM.
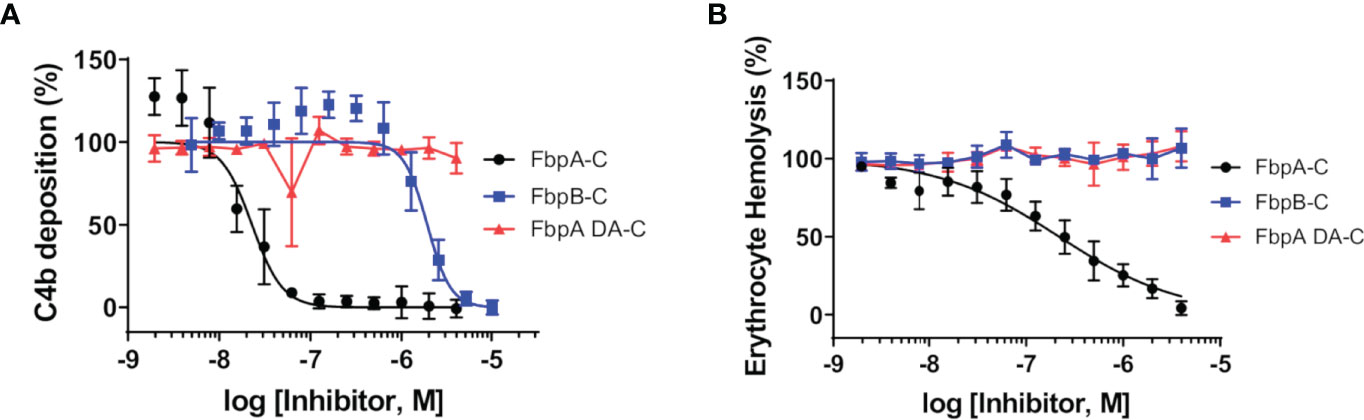
Figure 4 Assessing the complement inhibitory properties of B. miyamotoi FbpA and FbpB in vitro. (A) An ELISA-based assay specific for CP complement activation was used to determine if FbpA-C and FbpB-C were able to inhibit human CP progression. FbpA-C, FbpA DA-C, and FbpB-C, were serially diluted two-fold then incubated with normal human serum, followed by detection of deposited C4b. IC50 values were determined using normalized non-linear regression analysis and reported in Table 4 along with 95% confidence intervals. (B) A CP hemolysis assay was carried out using a two-fold serial dilution of Fbp proteins incubated with pre-sensitized sheep erythrocytes and 2% normal human serum. MAC formation and progression of the CP was measured by detecting MAC-mediated erythrocyte hemolysis. IC50 values were determined using normalized non-linear regression analysis and reported in Table 4 along with 95% confidence intervals.
Structural Differences Between B. miyamotoi FbpA-C and FbpB-C
While differences in the ability of B. miyamotoi FbpA and FbpB to bind fibronectin are likely attributed to low sequence conservation of critical fibronectin-binding motifs in FbpB (Figure 1B) (36, 37, 66, 69, 70), the basis for differences in the complement inhibitory activities of FbpA-C and FbpB-C were less clear. To determine if FbpA and FbpB have altered structures in their C1r-binding C-terminal regions, we solved the crystal structures of each protein. FbpA-C crystals diffracted to a limiting resolution of 1.9Å (Rfree (%)/Rwork (%) = 20.6/23.2) (PDB: 7RPR) (Figure 5A, Figures S3A, B and Table 3). FbpB-C was recalcitrant to crystallization; however, an active FbpB-C construct lacking five C-terminal residues (Figure S4, referred to as FbpB-Δ5C hereafter), was crystallized and the structure resolved to a limiting resolution of 2.1Å (Rfree (%)/Rwork (%) = 22.5/25.8) (PDB: 7RPS) (Figure 5B, Figures S3C, D and Table 3).
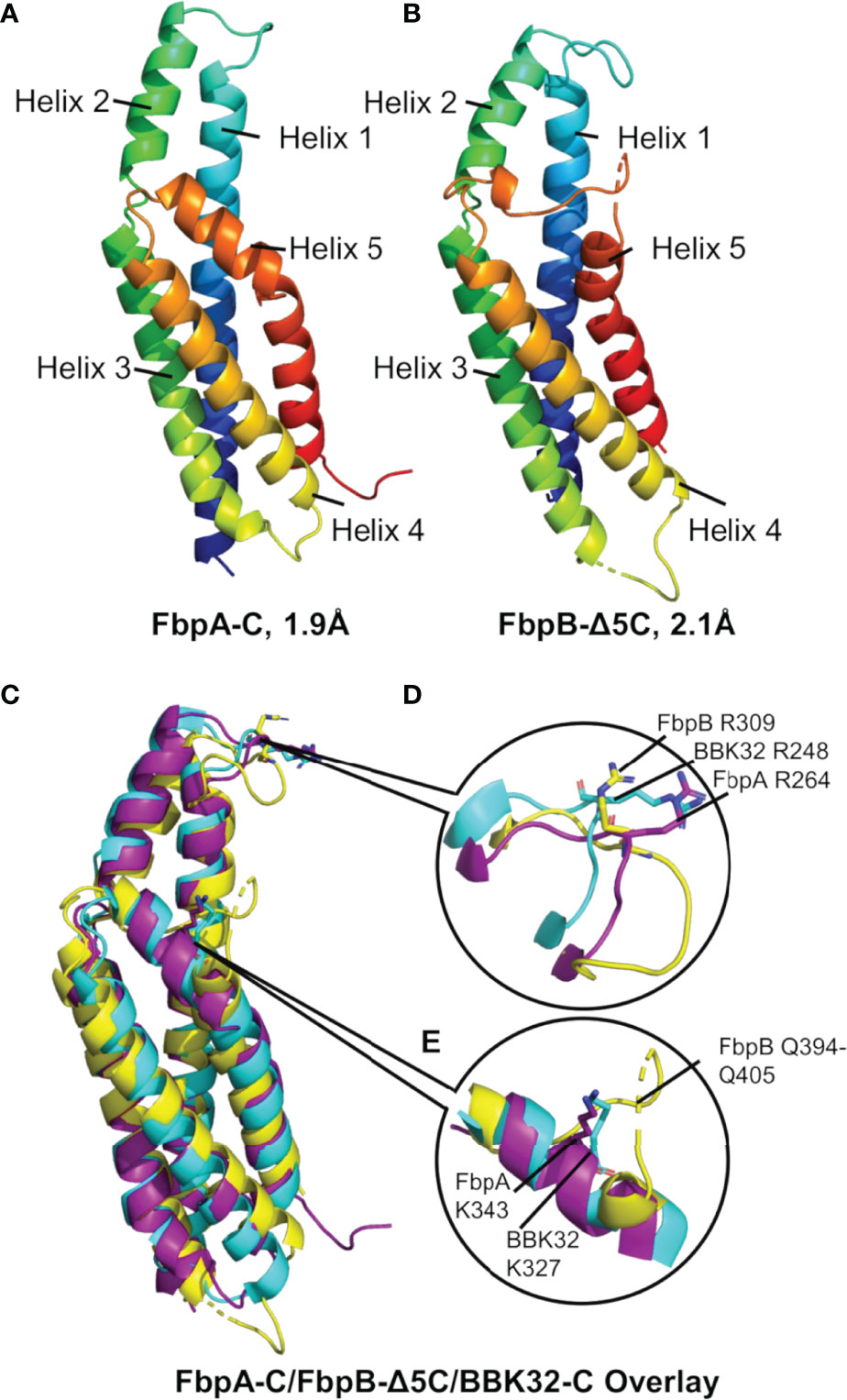
Figure 5 The crystal structures of B. miyamotoi FbpA-C and FbpB-Δ5C. Crystal structures of (A) FbpA-C (PDB Code: 7RPR) and (B) FbpB-Δ5C (PDB Code: 7RPS) were solved to a limiting resolution of 1.9Å and 2.1Å, respectively. Both FbpA-C and FbpB-Δ5C are shown as a color spectrum where blue represents the N-terminus and red represents the C-terminus. (C) A structural alignment of FbpA-C (purple), FbpB-Δ5C (yellow), and B) burgdorferi BBK32-C (cyan, PDB Code: 6N1L). (D) Each protein is structurally similar near the functionally critical BBK32-R248 residue (i.e., FbpA-R264 and FbpB-R309). (E) Differences between BBK32-C, FbpA-C, and FbpB-C are found at the functionally critical BBK32-K327 residue (K343 in B miyamotoi FbpA). FbpB-Δ5C shows an altered secondary structure with an extended loop structure corresponding to residues Q394-Q405 that aligns to the alpha helical structures found in both FbpA-C and BBK32-C.
A structural overlay with BBK32-C (PDB: 6N1L) and B. miyamotoi FbpA-C and FbpB-Δ5C reveals that all three proteins have similar folds involving a core four-helix bundle (helices 1, 3, 4, and 5) with alpha helix 2 extending away from the core (Figure 5C). BBK32-C was previously shown to interact with activated C1r using two primary binding sites: i) the active site of C1r involving several BBK32 residues, including R248, and ii) the B-loop of C1r involving residues on the fifth alpha helix, including K327 (34). A structural overlay of FbpA-C, FbpB-Δ5C, and BBK32-C shows that the three proteins retain an overall similar structure around the conserved R248 residue (BBK32 numbering) (Figure 5D). However, we noted that the area near BBK32-K327 involves a minor structural alteration in FbpA-C and a more major alteration in FbpB-Δ5C (Figure 5E). In FbpA-C, the kinked portion of alpha helix 5 extends one turn longer than in BBK32, resulting in FbpA-C containing an additional surface-exposed residue on this helix (Figure 5E). More strikingly, the structure of FbpB-Δ5C has a significantly altered secondary structure in this region when compared to both FbpA-C and BBK32-C (Figure 5E). Specifically, FbpB-Δ5C shows a loss of secondary structure within alpha helix 5 that accommodates a loop insertion sequence (i.e., residues Q394-Q405), which are not conserved in both BBK32-C or FbpA-C. Furthermore, a proline residue (P397) in this sequence interrupts the alpha helical secondary structure in FbpB-Δ5C. A portion of this unique loop structure in FbpB has apparent increased disorder in the crystal structure, indicated by weak electron density surrounding residues 401-403 in this region of the protein. Collectively, these data show that FbpA and FbpB are structurally divergent from BBK32 at a functionally critical region of the protein on the fifth alpha helix (Figures 5C–E).
B. miyamotoi FbpA and FbpB Differentially Recognize C1r Activation States
C1r is a serine protease subcomponent of the C1 complex which circulates in blood as an inactive zymogen, and is converted to an activated enzyme by autocatalysis when C1 binds to an activating ligand (71–75). This transition involves a conformational change in the SP domain of C1r resulting in a rearrangement of several loop structures, including the C1r B-loop (75) (Figure S5). We have previously shown that BBK32 binds to both active and zymogen forms of C1r in a manner that relies on R248 and K327 (34). Given the involvement of B-loop residues in the BBK32-K327 binding site, the observed structural differences at this site in the Fbp crystal structures (Figure 5), and the potential for different conformations of this loop between zymogen and active states of C1r (Figure S5) (75), we hypothesized that FbpA and FbpB may have altered recognition properties for zymogen C1r.
To assess this, B. miyamotoi FbpA-C and FbpB-C were immobilized on an SPR chip and a five-fold dilution series of either zymogen or active forms of full-length purified C1r were injected over each surface. Interestingly, while FbpA-C was able to bind to both zymogen and active forms of C1r readily (C1r zymogen KD = 0.85 ± 0.036 nM, active C1r KD = 0.39 ± 0.049 nM), FbpB selectively interacted with active C1r only (active C1r KD= 1.1 ± 0.042 nM) (Figure 6A). We next confirmed that the selective nature of FbpB was observed with full-length proteins expressed in B. burgdorferi B314 strains in an approach identical to that presented for fibronectin in Figure 2E. Whereas FbpA could recognize both zymogen and activated forms of purified C1r, FbpB preferentially bound activated C1r. The negative controls, FbpA DA and vector-only lysates, did not bind either form of C1r as expected (Figure 6B), although we note that FbpA DA retained fibronectin binding (Figure 2E).
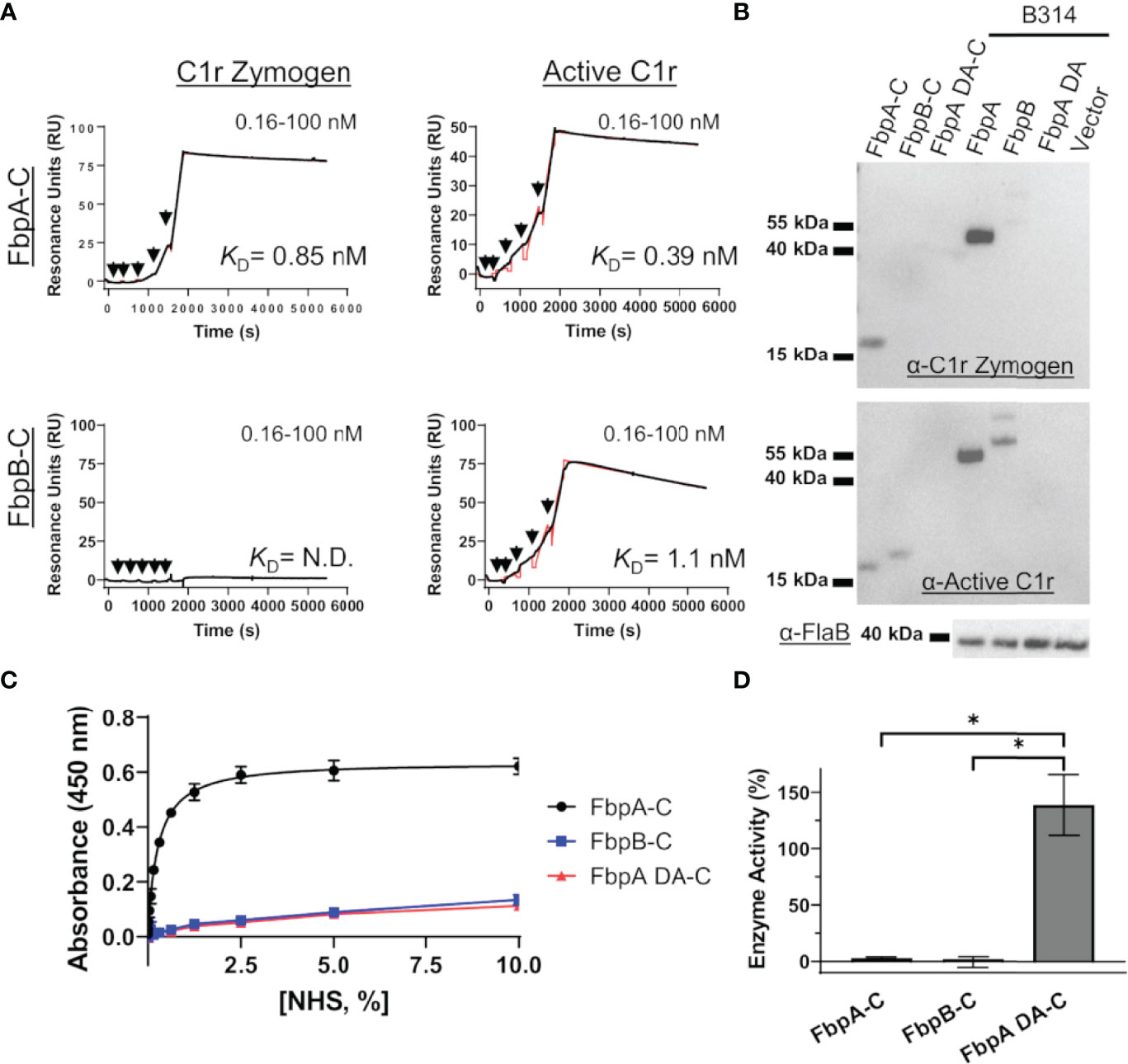
Figure 6 FbpA-C and FbpB-C interact differentially with zymogen and activated forms of human C1r. (A) Single cycle SPR was used to determine binding affinities of C1r zymogen or active C1r ranging from 0.16 - 100 nM injected over immobilized Fbps. A representative sensorgram from a three-injection series for each Fbp-C and C1r active state is shown with the black curve being the sensorgram and the red curve the associated kinetic fit. KD’s were determined using kinetic fits and are shown as the average and standard deviation of three replicates in Table 4. (B) Far Western overlays using B. burgdorferi B314 lysates probed with zymogen or enzymatic forms of purified full-length C1r, followed by an antibody to C1r. (C) An ELISA-type binding assay was used to determine the ability of FbpA-C, FbpA DA-C and FbpB-C to interact with C1r in human serum. FbpA-C (black), FbpB-C (blue), and FbpA DA-C (red) were immobilized on an ELISA plate then incubated with a two-fold dilution series of normal human serum ranging from 0.0012-10%. Corresponding EC50 values for FbpA-C, FbpB-C, and FbpA DA-C are reported in Table 4. (D) Fbp-mediated inhibition of purified active C1r-CCP2-SP enzymatic activity was assessed by incubating 15 nM C1r-CCP2-SP with a 1 μM of FbpA-C (white), FbpB-C (striped), and FbpA DA-C (grey). C1r-CCP2-SP enzymatic activity was determined by incubation with a C1r substrate (Z-Gly-Arg-sBzl) that, once cleaved, reacts with DTNB resulting in a colorimetric change. No statistical difference was found between FbpA-C and FbpB-C inhibitory activity, whereas FbpA DA-C exhibited significant loss in inhibitory activity. Statistical analysis was performed using a one-way ANOVA followed by a multiple comparison Tukey test (*=p<0.05).
We considered whether the apparent zymogen/active C1r selectivity differences between FbpA and FbpB could be observed in serum where C1r is in a zymogen form (71–74). To test this, we implemented a serum-based ELISA-type binding assay. B. miyamotoi FbpA-C, FbpB-C, and FbpA DA-C were immobilized onto an ELISA plate and incubated with a dilution series of normal human serum followed by detection of C1r binding using a C1r antibody. FbpA-C readily bound serum C1r (half-maximal effective concentration (EC50) = 0.27% (v/v) serum concentration), while FbpB-C and FbpA DA-C did not (Figure 6C). However, consistent with the ability of FbpB-C to bind tightly to previously activated forms of C1r (Figures 3, 6A), FbpB-C inhibited the enzymatic activity of pre-activated C1r-CCP2-SP enzyme similar to FbpA-C (Figure 6D). Collectively, these results show that, like BBK32, B. miyamotoi FbpA can bind and inhibit both the zymogen and active forms of C1r, whereas B. miyamotoi FbpB selectively interacts with and inhibits only the active form of the C1r protease.
B. miyamotoi Fbp Proteins Protect a Serum-Sensitive Strain of B. burgdorferi From Complement-Mediated Killing
Next, we assessed the protection B. miyamotoi FbpA and FbpB conferred to B. burgdorferi B314 when ectopically expressed on the cell surface. Since B. miyamotoi has not yet been genetically manipulated, we modeled these proteins’ surface expression and activity in B314 relative to a pBBE22luc vector-only control and cells producing BBK32, given that BBK32 confers serum resistance to strain B314 (32–34). As a prelude to the serum sensitivity assay, we first tested whether B. miyamotoi FbpA, FbpA DA or FbpB were surface exposed in B. burgdorferi strain B314 relative to a B314 BBK32 control, as well as expressed on the surface of B. miyamotoi strain FR64b, using the well-established proteinase K accessibility assay (32, 76). The results clearly show that FbpA, FbpA DA, FbpB, and BBK32 were all produced and are all protease-sensitive, indicating surface exposure in strain B314. However, only FbpA is detected on the surface of B. miyamotoi strain FR64b, potentially due to the in vitro growth conditions not being conducive for expression of fbpB (Figure 7A). Importantly, the subsurface endoflagellar protein, FlaB, was unaffected by the treatment in both B. miyamotoi strain FR64b and B. burgdorferi B314, indicating the preserved structural integrity of the borrelial cells tested (Figure 7A). As observed in Figure S2A, antibody to B. miyamotoi FbpB also recognized FbpA and FbpA DA, albeit to a lesser extent (Figure 7A).
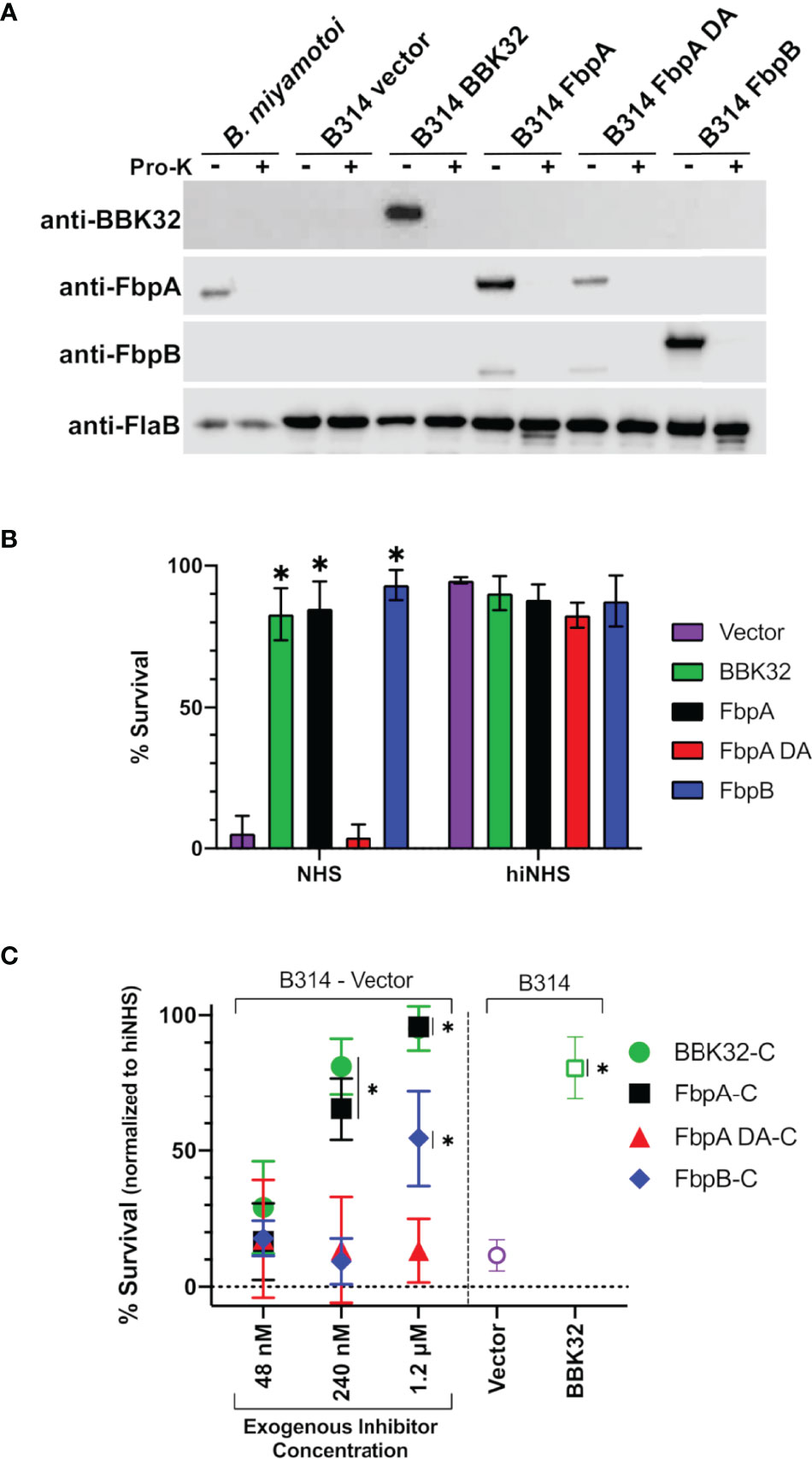
Figure 7 Determining the ability of surface-expressed B. miyamotoi FbpA and FbpB to protect a serum-sensitive strain of B. burgdorferi. (A) Western blots of lysates from B. miyamotoi strain FR64b and B burgdorferi B314 isolates expressing fbpA, fbpA-DA, fbpB, and bbk32 were probed with antibodies to FbpA (anti-FbpA), FbpB (anti-FbpB), BBK32 (anti-BBK32) and FlaB (anti-FlaB), the latter as a loading control and a control for a subsurface target. Vector refers to the plasmid-only backbone sample (B314/pBBE22luc). Samples from each population were subjected to proteinase K accessibility treatments to determine the surface expression of the proteins. Due to its periplasmic location, the flagellar protein, FlaB, is unaffected by proteinase K in intact cells and depicts structural integrity in the treated B. miyamotoi and B314 derivatives listed. (B) B. miyamotoi Fbp proteins were tested for their ability to confer resistance to normal human serum (NHS) sensitive B. burgdorferi strain B314 relative to vector-only and BBK32-expressing controls (negative and positive controls, respectively). Asterisks depict a significant increase in survival relative to FbpA DA and the vector control (*=p<0.0001). All strains exposed to heat inactivated NHS (hiNHS), rendering complement proteins inactive, were largely unaffected. (C) Rescue assays were used to determine if soluble exogenous recombinant proteins could promote survival of serum-sensitive vector-containing B. burgdorferi B314. Increasing five-fold concentrations of BBK32-C, FbpA-C, FbpA DA-C, and FbpB-C were added in serum sensitivity assays. The middle samples (i.e., 240 nM) have added recombinant protein that is roughly equivalent to the concentration of C1r in the assay conditions employed. Cells were assessed via darkfield microscopy. Included as controls are a vector-only strain of B314 with no exogenous protein addition, as well as a BBK32-producing B314 isolate (right side). Asterisks depict a significant increase in survival relative to the vector control (*=p<0.015). Significance for serum sensitivity and rescue assays was determined using two-way ANOVA with a Šidák correction for multiple comparisons.
We then exposed each B314 knock-in strain to normal human serum and assessed the viability of the cells. B. miyamotoi FbpA conferred levels of resistance comparable to BBK32, while the FbpA DA strain was as sensitive to serum killing as the vector-only control (Figure 7B). In contrast to the lower apparent inhibitory activity of recombinant FbpB-C (Figure 4), B. burgdorferi strain B314 producing FbpB on the surface also conferred significant protection against human serum (Figure 7B). In order to more fully address differences in the resistance observed for FbpB in the prior assays, we incubated serum sensitive B314 pBBE22luc with differential levels of B. miyamotoi FbpA-C, FbpB-C, FbpA DA-C, and B. burgdorferi BBK32-C to ask whether these soluble components could provide resistance to complement-dependent killing with normal human serum and thus “rescue” this sensitive B. burgdorferi strain. Exogenous addition of the C-terminus of the B. miyamotoi Fbps and B. burgdorferi BBK32-C resulted in dose-dependent rescue for BBK32-C, FbpA-C, and FbpB-C (Figure 7C). However, the inhibitory effect was weaker for FbpB-C, with significant inhibition observed only at the 1.2 µM treatment, which is comparable to the IC50 value observed for FbpB-C in the ELISA-based inhibition assay (Figure 4). Consistent with its inability to inhibit complement, addition of FbpA DA-C was unable to rescue serum-based killing of B314 pBBE22luc cells at any of the protein concentrations used (Figure 7C). Collectively, these data demonstrate that ectopically-produced B. miyamotoi FbpA and FbpB confer resistance to human serum.
Discussion
Like other tick-borne microbial agents, B. miyamotoi spirochetes must survive host immune defenses, including complement, beginning at the tick bloodmeal stage and throughout host infection. B. miyamotoi has been reported to evade the alternative pathway (AP) of complement through recruitment of endogenous complement regulators or plasmin, and produces lipoproteins that directly bind to and inhibit complement proteins, like CbiA, to protect the spirochete from complement-mediated killing (21, 22, 77). However, little is known about how B. miyamotoi defends against the classical pathway (CP), which may be particularly important during later stages of infection when an adaptive immune response has been developed. Interestingly, the Lyme disease spirochete B. burgdorferi has evolved at least two mechanistically distinct inhibitors of the classical complement pathway, i) BBK32 (32–34) and ii) ElpB/ElpQ (78). As elp genes with CP inhibitory activity have yet to be identified in B. miyamotoi genomes, we hypothesized that B. miyamotoi may use orthologs of BBK32 to neutralize the classical complement cascade and/or exert BBK32-like fibronectin-binding activity (32–34).
B. miyamotoi strain FR64b encodes two BBK32 orthologs, FbpA and FbpB, that share 56% and 25% amino acid sequence identity to B. burgdorferi strain B31 BBK32, respectively (Figures 1B, S1). B. miyamotoi strain LB-2001, a North American strain of B. miyamotoi (24), also encodes FbpA and FbpB proteins with 85% and 81% identity to FR64b FbpA and FbpB, respectively. We found that B. miyamotoi strain FR64b FbpA exhibits similar activity to BBK32 for both fibronectin-binding and C1r-binding/inhibition. The results presented herein highlight that FbpA and BBK32 are multifunctional proteins capable of interacting with more than one host protein via separate domains (32–37, 40, 64–68). Similar to what has been proposed for B. burgdorferi BBK32, the anti-C1r activity found here for FbpA may selectively protect B. miyamotoi from CP-mediated complement killing. This is supported by the ability of FbpA to protect previously serum-sensitive spirochetes from complement-mediated bacteriolysis (Figure 7B). The functional consequence of the fibronectin-binding activity of B. miyamotoi FbpA interacting with fibronectin may be related to extravasation as has been shown for BBK32 (35–37, 40, 64–68), and/or it may serve other functions including acting as an adhesin to the vascular endothelium or binding to plasma fibronectin, another activity reported for BBK32 (38, 65, 79).
The functional consequence of B. miyamotoi FbpA and FbpB production within a host will need to be investigated in vivo. A current limitation is the lack of an immunocompetent mouse model for B. miyamotoi to determine the temporal expression of fbpA and fbpB during mammalian infection. The culturing of B. miyamotoi is possible (24), but challenging, and allows for only limited experimentation. Additionally, genetic manipulation of B. miyamotoi has not yet been reported. To overcome these challenges, we used an established human serum sensitivity assay in which serum resistance can be quantitatively measured and correlated with protein production of B. miyamotoi FbpA and FbpB on the surface of the serum-sensitive B. burgdorferi strain B314, e.g., as a “knock-in” construct (Figure 7B) (32–34). Alternatively, we tested whether the addition of exogenous FbpA-C, FbpA DA-C, or FbpB-C could “rescue” normal human serum-dependent killing of a sensitive B314 derivative (Figure 7C). The “knock-in” system is a minimalist approach that measures the complement inhibitory activity of surface-exposed proteins due to the absence of all linear plasmid-encoded lipoproteins and the concomitant absence of redundant immune evasion mechanisms. Proteinase K accessibility assays confirmed the surface localization of B. miyamotoi FbpA, FbpA DA, and FbpB in the surrogate B. burgdorferi B314 knock-in background (Figure 7A). Interestingly, only FbpA was produced following in vitro cultivation of B. miyamotoi strain FR64b and surface-expression was also confirmed; no FbpB was detectable by Western immunoblotting (Figure 7A). Limited cultivation conditions were tested here for B. miyamotoi and additional experimentation using mammalian-like growth conditions may also induce FbpB production comparable to that observed for pathogenesis-associated proteins in B. burgdorferi (80).
In contrast to B. miyamotoi FbpA, we found that FbpB fails to interact with human fibronectin in a BBK32-like manner and lacks a key motif that is known to be important for BBK32 recognition of fibronectin (37, 66) (Figure 2). The C1r-binding activity for FbpB proved to be more complex. Unlike BBK32/FbpA, FbpB fails to interact with zymogen forms of human C1r but instead selectively binds and inhibits the activated form (Figures 3, 6A, B, D). The crystal structures presented here of FbpA-C and FbpB-C show that a defining feature of the C1r-binding domains for BBK32 and B. miyamotoi Fbps is a four-helix core with an extended second alpha helix that presents a functional arginine residue on a small surface-loop (Figures 5C, D). However, the crystal structure of FbpB revealed structural differences in the fifth alpha helix relative to FbpA and BBK32. FbpB has a divergent loop structure in this region and therefore lacks the same secondary structure seen in both FbpA and BBK32 (Figure 5E). We have previously shown that residues on the fifth alpha helix of BBK32 interact with a non-active site loop on C1r known as the B-loop (34). Despite these structural differences, FbpB-C was still capable of saturable inhibition of human complement activation, although at several orders of magnitude weaker potency in the ELISA-based assay (Figure 4A). Recombinant FbpB-C also protected serum sensitive spirochetes from complement-mediated killing, but was significantly weaker in its ability to do so relative to FbpA-C (Figure 7C). Initially, these results pointed to FbpA being the more relevant classical pathway complement inhibitor in B. miyamotoi and suggested that targeting of the zymogen form of human C1r is an important functional feature of this class of inhibitors. However, in our serum sensitivity assays, where B. miyamotoi FbpA and FbpB were produced ectopically on the surface of B. burgdorferi, both proteins conferred significantly greater protection to B314 than FbpA DA or the vector-only control (Figure 7B). These data suggest that within the physiological context of complement activation on the spirochete surface, FbpB’s inhibition of activated C1r is sufficient to prevent downstream complement activation. Thus, the selective recognition of activated C1r by FbpB implies that it functions at a different temporal or spatial level than FbpA. Combined with the differences in relative fibronectin-binding activities of FbpA and FbpB, our data suggest that Fbp proteins have evolved to have partially overlapping, but non-identical functions compared to the Lyme disease-associated BBK32 orthologs.
Nonetheless, B. miyamotoi FbpB’s conferred resistance in the human serum sensitivity assays was initially surprising based on in vitro biochemical data showing very limited inhibition of serum-based complement activation (Figure 4B). The lack of consensus between FbpB activity in the human serum sensitivity assay relative to FbpB-C likely reflects differences in the truncated soluble form of the recombinant protein relative to the full-length lipoprotein-tethered surface-exposed configuration (Figure 7B). Given that soluble FbpB-C significantly protected serum-sensitive spirochetes at high concentrations (Figure 7C), one explanation may be that differences observed in the B314 background are related to sufficient protein concentrations of surface-exposed FbpB that can confer complement resistance to these borrelial cells (Figure 7B). Another possibility is suggested by the observation that soluble FbpB-C does not interact with zymogen C1r in serum (Figure 6C), but rather interacts and inhibits C1r only once activated (Figure 6). Thus soluble forms of FbpA, but not FbpB, are capable of binding to C1r prior to surface activation. This difference likely makes the inhibitory activity of FbpB more dependent on surface localization compared to FbpA, which binds tightly to zymogen C1r in solution (Figure 6C).
In summary, we have characterized the structure and function of two bbk32 orthologs within the emerging pathogen B. miyamotoi, termed FbpA and FbpB. Our investigation showed that FbpA and FbpB share some activities with one another and with B. burgdorferi BBK32. However, important differences – both structurally and at the functional level – were found to influence their relative ability to interact with both human fibronectin and complement C1r. Surprisingly, our data suggest that B. burgdorferi BBK32 and B. miyamotoi Fbp proteins have evolved distinct molecular mechanisms across the family of inhibitors. These observations have significance for understanding how B. miyamotoi modulates the host immune response and provides novel insight into the diverse structure-function relationships of this multifunctional class of borrelial lipoproteins.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.wwpdb.org/, 7RPR, http://www.wwpdb.org/, 7RPS.
Ethics Statement
The animal work described herein was reviewed and approved by Texas A&M IACUC.
Author Contributions
CB and AP-P are equal contributors. JS and BG are co-corresponding authors. JS and BG designed research. CB and AP-P conducted research. AP-P, JS, BG CB analyzed data. CB, AP-P, JS, and BG wrote the paper.
Funding
Support was provided by Public Health Service Grant R01-AI146930 from the National Institute of Allergy and Infectious Diseases (to JS and BG). X-ray diffraction data were collected at Southeast Regional Collaborative Access Team 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.886733/full#supplementary-material
References
1. Lopez J, Hovius JW, Bergström S. Pathogenesis of Relapsing Fever Curr Issues Mol Biol (2021) 42:519–50. doi: 10.21775/cimb.042.519.
2. Lopez JE, Krishnavahjala A, Garcia MN, Bermudez S. Tick-Borne Relapsing Fever Spirochetes in the Americas. Vet Sci (2016) 3:1–18. doi: 10.3390/vetsci3030016
3. Wagemakers A, Staarink PJ, Sprong H, Hovius JWR. Borrelia miyamotoi: A Widespread Tick-Borne Relapsing Fever Spirochete. Trends Parasitol (2015) 31:260–9. doi: 10.1016/j.pt.2015.03.008
4. Cutler S, Vayssier-Taussat M, Estrada-Peña A, Potkonjak A, Mihalca AD, Zeller H. A New Borrelia on the Block: Borrelia miyamotoi - a Human Health Risk? Euro Surveill (2019) 24:1–14. doi: 10.2807/1560-7917.ES.2019.24.18.1800170
5. Telford SR, Goethert HK, Molloy PJ, Berardi VP, Chowdri HR, Gugliotta JL, et al. Borrelia miyamotoi Disease: Neither Lyme Disease Nor Relapsing Fever. Clin Lab Med (2015) 35:867–82. doi: 10.1016/j.cll.2015.08.002
6. Talagrand-Reboul E, Boyer PH, Bergström S, Vial L, Boulanger N. Relapsing Fevers: Neglected Tick-Borne Diseases. Front Cell Infect Microbiol (2018) 8:98. doi: 10.3389/fcimb.2018.00098
7. Johnston D, Kelly JR, Ledizet M, Lavoie N, Smith RP, Parsonnet J, et al. Frequency and Geographic Distribution of Borrelia miyamotoi, Borrelia burgdorferi, and Babesia microti Infections in New England Residents. Clin Infect Dis (2022), 1–6. doi: 10.1093/cid/ciac107
8. Krause PJ, Narasimhan S, Wormser GP, Rollend L, Fikrig E, Lepore T, et al. Human Borrelia miyamotoi Infection in the United States. N Engl J Med (2013) 368:291–3. doi: 10.1056/nejmc1215469
9. Koetsveld J, Draga ROP, Wagemakers A, Manger A, Oei A, Visser CE, et al. In Vitro Susceptibility of the Relapsing-Fever Spirochete Borrelia miyamotoi to Antimicrobial Agents. Antimicrob Agents Chemother (2017) 61:1–9. doi: 10.1128/AAC.00535-17
10. Lynn GE, Breuner NE, Eisen L, Hojgaard A, Replogle AJ, Eisen RJ. An Immunocompromised Mouse Model to Infect Ixodes scapularis Ticks With the Relapsing Fever Spirochete, Borrelia miyamotoi. Ticks Tick Borne Dis (2019) 10:352–9. doi: 10.1016/j.ttbdis.2018.11.017
11. Barbour AG, Bunikis J, Travinsky B, Hoen AG, Diuk-Wasser MA, Fish D, et al. Niche Partitioning of Borrelia burgdorferi and Borrelia miyamotoi in the Same Tick Vector and Mammalian Reservoir Species. Am J Trop Med Hyg (2009) 81:1120–31. doi: 10.4269/ajtmh.2009.09-0208
12. Kullberg BJ, Vrijmoeth HD, van de Schoor F, Hovius JW. Lyme Borreliosis: Diagnosis and Management. BMJ (2020) 369:1–20. doi: 10.1136/bmj.m1041
13. Steere AC, Strle F, Wormser GP, Hu LT, Branda JA, Hovius JWR, et al. Lyme Borreliosis. Nat Rev Dis Primers (2016) 2:1–19. doi: 10.1038/nrdp.2016.90
14. Radolf JD, Strle K, Lemieux JE, Strle F. Lyme Disease in Humans Curr. Issues Mol Biol (2021) 42:333–84. doi: 10.21775/cimb.042.333
15. Dworkin MS, Schwan TG, Anderson DE, Borchardt SM. Tick-Borne Relapsing Fever. Infect Dis Clin North Am (2008) 22:449–68. doi: 10.1016/j.idc.2008.03.006
16. Hamase A, Takahashi Y, Nohgi K, Fukunaga M. Homology of Variable Major Protein Genes Between Borrelia hermsii and Borrelia miyamotoi. FEMS Microbiol Lett (1996) 140:131–7. doi: 10.1016/0378-1097(96)00168-1
17. Barbour AG, Dai Q, Restrepo BI, Stoenner HG, Frank SA. Pathogen Escape From Host Immunity by a Genome Program for Antigenic Variation. Proc Natl Acad Sci USA (2006) 103:18290–5. doi: 10.1073/pnas.0605302103
18. Barbour AG. Multiple and Diverse Vsp and Vlp Sequences in Borrelia miyamotoi, a Hard Tick-Borne Zoonotic Pathogen. PloS One (2016) 11:1–16. doi: 10.1371/journal.pone.0146283
19. Barbour AG. Antigenic Variation of Surface Proteins of Borrelia Species. Rev Infect Dis (1988) 10:399–402. doi: 10.1093/cid/10.Supplement_2.S399
20. Wagemakers A, Koetsveld J, Narasimhan S, Wickel M, Deponte K, Bleijlevens B, et al. Variable Major Proteins as Targets for Specific Antibodies Against Borrelia miyamotoi. J Immunol (2016) 196:4185–95. doi: 10.4049/jimmunol.1600014
21. Röttgerding F, Wagemakers A, Koetsveld J, Fingerle V, Kirschfink M, Hovius JW, et al. Immune Evasion of Borrelia miyamotoi: CbiA, a Novel Outer Surface Protein Exhibiting Complement Binding and Inactivating Properties. Sci Rep (2017) 7:1–15. doi: 10.1038/s41598-017-00412-4
22. Schmidt FL, Sürth V, Berg TK, Lin Y-P, Hovius JW, Kraiczy P. Interaction Between Borrelia miyamotoi Variable Major Proteins Vlp15/16 and Vlp18 With Plasminogen and Complement. Sci Rep (2021) 11:1–13. doi: 10.1038/s41598-021-84533-x
23. Teegler A, Herzberger P, Margos G, Fingerle V, Kraiczy P. The Relapsing Fever Spirochete Borrelia miyamotoi Resists Complement-Mediated Killing by Human Serum. Ticks Tick Borne Dis (2014) 5:898–901. doi: 10.1016/j.ttbdis.2014.07.011
24. Wagemakers A, Oei A, Fikrig MM, Miellet WR, Hovius JW. The Relapsing Fever Spirochete Borrelia miyamotoi Is Cultivable in a Modified Kelly-Pettenkofer Medium, and is Resistant to Human Complement. Parasites Vectors (2014) 7:1–6. doi: 10.1186/1756-3305-7-418
25. Sato K, Kumagai Y, Sekizuka T, Kuroda M, Hayashi T, Takano A, et al. Vitronectin Binding Protein, BOM1093, Confers Serum Resistance on Borrelia miyamotoi. Sci Rep (2021) 11:1–13. doi: 10.1038/s41598-021-85069-w
26. Varela JC, Tomlinson S. Complement: An Overview for the Clinician. Hematol/Oncol Clin North Am (2015) 29:409–27. doi: 10.1016/j.hoc.2015.02.001
27. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
28. Bjanes E, Nizet V. More Than a Pore: Nonlytic Antimicrobial Functions of Complement and Bacterial Strategies for Evasion. Microbiol Mol Biol Rev (2021) 85:1–27. doi: 10.1128/mmbr.00177-20
30. Lambris JD, Ricklin D. Geisbrecht B V. Complement Evasion by Human Pathogens. Nat Rev Microbiol (2008) 6:132–42. doi: 10.1038/nrmicro1824
31. Skare JT, Garcia BL. Complement Evasion by Lyme Disease Spirochetes. Trends Microbiol (2020) 28:1–11. doi: 10.1016/j.tim.2020.05.004
32. Garcia BL, Zhi H, Wager B, Höök M, Skare JT. Borrelia burgdorferi BBK32 Inhibits the Classical Pathway by Blocking Activation of the C1 Complement Complex. PloS Pathog (2016) 12:1–28. doi: 10.1371/journal.ppat.1005404
33. Xie J, Zhi H, Garrigues RJ, Keightley A, Garcia BL, Skare JT. Structural Determination of the Complement Inhibitory Domain of Borrelia burgdorferi BBK32 Provides Insight Into Classical Pathway Complement Evasion by Lyme Disease Spirochetes. PloS Pathog (2019) 15:1–29. doi: 10.1371/journal.ppat.1007659
34. Garrigues RJ, Powell-Pierce AD, Hammel M, Skare JT, Garcia BL. A Structural Basis for Inhibition of the Complement Initiator Protease C1r by Lyme Disease Spirochetes. J Immunol (2021) 207:2856–67. doi: 10.4049/JIMMUNOL.2100815
35. Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Höök M, et al. Inactivation of the Fibronectin-Binding Adhesin Gene bbk32 Significantly Attenuates the Infectivity Potential of Borrelia burgdorferi. Mol Microbiol (2006) 59:1591–601. doi: 10.1111/j.1365-2958.2005.05042.x
36. Probert WS, Kim JH, Höök M, Johnson BJB. Mapping the Ligand-Binding Region of Borrelia burgdorferi Fibronectin-Binding Protein BBK32. Infect Immun (2001) 69:4129–33. doi: 10.1128/IAI.69.6.4129-4133.2001
37. Harris G, Ma W, Maurer LM, Potts JR, Mosher DF. Borrelia burgdorferi Protein BBK32 Binds to Soluble Fibronectin Via the N-terminal 70-kDa Region, Causing Fibronectin to Undergo Conformational Extension. J Biol Chem (2014) 289:22490–9. doi: 10.1074/jbc.M114.578419
38. Kim JH, Singvall J, Schwarz-Linek U, Johnson BJB, Potts JR, Höök M. BBK32, a Fibronectin Binding MSCRAMM From Borrelia burgdorferi, Contains a Disordered Region That Undergoes a Conformational Change on Ligand Binding. J Biol Chem (2004) 279:41706–14. doi: 10.1074/jbc.M401691200
39. Lin YP, Chen Q, Ritchie JA, Dufour NP, Fischer JR, Coburn J, et al. Glycosaminoglycan Binding by Borrelia burgdorferi Adhesin BBK32 Specifically and Uniquely Promotes Joint Colonization. Cell Microbiol (2015) 17:860–75. doi: 10.1111/cmi.12407
40. Hyde JA, Weening EH, Chang M, Trzeciakowski JP, Höök M, Cirillo JD, et al. Bioluminescent Imaging of Borrelia burgdorferi In Vivo Demonstrates That the Fibronectin-Binding Protein BBK32 Is Required for Optimal Infectivity. Mol Microbiol (2011) 82:99–113. doi: 10.1111/j.1365-2958.2011.07801.x
41. Lewis ERG, Marcsisin RA, Campeau Miller SA, Hue F, Phillips A, AuCoin DP, et al. Fibronectin-Binding Protein of Borrelia hermsii Expressed in the Blood of Mice With Relapsing Fever. Infect Immun (2014) 82:2520–31. doi: 10.1128/IAI.01582-14
42. Brenner C, Bomans K, Habicht J, Simon MM, Wallich R. Mapping the Ligand-Binding Region of Borrelia hermsii Fibronectin-Binding Protein. PloS One (2013) 8:26–8. doi: 10.1371/journal.pone.0063437
43. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The Renaissance of Complement Therapeutics. Nat Rev Nephrol (2017) 14:26–47. doi: 10.1038/nrneph.2017.156
44. Ricklin D, Lambris JD. Complement in Immune and Inflammatory Disorders: Pathophysiological Mechanisms. J Immunol (2013) 190:3831–8. doi: 10.4049/jimmunol.1203487
45. Mastellos DC, Ricklin D, Lambris JD. Clinical Promise of Next-Generation Complement Therapeutics. Nat Rev Drug Discov (2019) 18:707–29. doi: 10.1038/s41573-019-0031-6
46. Sadziene A, Wilske B, Ferdows MS, Barbour AG. The Cryptic ospC Gene of Borrelia burgdorferi B31 Is Located on a Circular Plasmid. Infect Immun (1993) 61:2192–5. doi: 10.1128/iai.61.5.2192-2195.1993
47. Fukunaga M, Takahashi Y, Tsuruta Y, Matsushita O, Ralph D, McClelland M, et al. Genetic and Phenotypic Analysis of Borrelia miyamotoi Sp. Nov., Isolated From the Ixodid Tick Ixodes persulcatus, the Vector for Lyme Disease in Japan. Int J Syst Bacteriol (1995) 45:804–10. doi: 10.1099/00207713-45-4-804
48. Geisbrecht BV, Bouyain S, Pop M. An Optimized System for Expression and Purification of Secreted Bacterial Proteins. Protein Expr Purif (2006) 46:23–32. doi: 10.1016/j.pep.2005.09.003
49. Samuels DS, Drecktrah D, Hall LS. Genetic Transformation and Complementation. Methods Mol Biol (2018) 1690:183–200. doi: 10.1007/978-1-4939-7383-5_15
50. Rushing BR, Rohlik DL, Roy S, Skaff DA, Garcia BL. Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery. Molecules (2020) 25:1–20. doi: 10.3390/molecules25174016
51. Kardos J, Gál P, Szilágyi L, Thielens NM, Szilágyi K, Lõrincz Z, et al. The Role of the Individual Domains in the Structure and Function of the Catalytic Region of a Modular Serine Protease, C1r. J Immunol (2001) 167:5202–8. doi: 10.4049/jimmunol.167.9.5202
52. Harlow E, Lane D. “Antibodies: A Laboratory Manual”, 2nd ed. Cold Spring Harbor, New York: Cold Spring Harbor (1998). pp. 103–19.
53. Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol (1997) 276:307–26. doi: 10.1016/S0076-6879(97)76066-X
54. Karplus PA, Diederichs K. Linking Crystallographic Model and Data Quality. Science (2012) 336:1030–3. doi: 10.1126/science.1218231
55. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr (2011) 67:235–42. doi: 10.1107/S0907444910045749
56. Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, et al. Automated Structure Solution With the PHENIX Suite. Methods Mol Biol (2008) 426:419–35. doi: 10.1007/978-1-60327-058-8_28
57. Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. Phenix: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr (2002) 58:1948–54. doi: 10.1107/S0907444902016657
58. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. Phenix: A Comprehensive Python-based System for Macromolecular Structure Solution. Acta Crystallogr (2010) 66:213–21. doi: 10.1107/S0907444909052925
59. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of COOT. Acta Crystallogr (2010) 66:486–501. doi: 10.1107/S0907444910007493
60. Summers BJ, Garcia BL, Woehl JL, Ramyar KX, Yao X. Geisbrecht B V. Identification of Peptidic Inhibitors of the Alternative Complement Pathway Based on Staphylococcus aureus SCIN Proteins. Mol Immunol (2015) 67:193–205. doi: 10.1016/j.molimm.2015.05.012
61. Roos A, Bouwman LH, Munoz J, Zuiverloon T, Faber-Krol MC, Fallaux-van den Houten FC, et al. Functional Characterization of the Lectin Pathway of Complement in Human Serum. Mol Immunol (2003) 39:655–68. doi: 10.1016/S0161-5890(02)00254-7
62. Nilsson UR, Nilsson B. Simplified Assays of Hemolytic Activity of the Classical and Alternative Complement Pathways. J Immunol Methods (1984) 72:49–59. doi: 10.1016/0022-1759(84)90432-0
63. Grosskinsky S, Schott M, Brenner C, Cutler SJ, Kraiczy P, Zipfel PF, et al. Borrelia recurrentis Employs a Novel Multifunctional Surface Protein With Anti-Complement, Anti-Opsonic and Invasive Potential to Escape Innate Immunity. PloS One (2009) 4:1–12. doi: 10.1371/journal.pone.0004858
64. Ebady R, Niddam AF, Boczula AE, Kim YR, Gupta N, Tang TT, et al. Biomechanics of Borrelia burgdorferi Vascular Interactions. Cell Rep (2016) 16:2593–604. doi: 10.1016/j.celrep.2016.08.013
65. Moriarty TJ, Shi M, Lin YP, Ebady R, Zhou H, Odisho T, et al. Vascular Binding of a Pathogen Under Shear Force Through Mechanistically Distinct Sequential Interactions With Host Macromolecules. Mol Microbiol (2012) 86:1116–31. doi: 10.1111/mmi.12045
66. Probert WS, Johnson BJB. Identification of a 47 kDa Fibronectin-Binding Protein Expressed by Borrelia burgdorferi Isolate B31. Mol Microbiol (1998) 30:1003–15. doi: 10.1046/j.1365-2958.1998.01127.x
67. Liang X, Garcia BL, Visai L, Prabhakaran S, Meenan NAG, Potts JR, et al. Allosteric Regulation of Fibronectin/α5β1 Interaction by Fibronectin-Binding MSCRAMMS. PloS One (2016) 11:1–17. doi: 10.1371/journal.pone.0159118
68. Niddam AF, Ebady R, Bansal A, Koehler A, Hinz B, Moriarty TJ. Plasma Fibronectin Stabilizes Borrelia burgdorferi-Endothelial Interactions Under Vascular Shear Stress by a Catch-Bond Mechanism. Proc Natl Acad Sci USA (2017) 114:3490–8. doi: 10.1073/pnas.1615007114
69. Raibaud S, Schwarz-Linek U, Kim JH, Jenkins HT, Baines ER, Gurasiddappa S, et al. Borrelia burgdorferi Binds Fibronectin Through a Tandem β-Zipper, a Common Mechanism of Fibronectin Binding in Staphylococci, Streptococci, and Spirochetes. J Biol Chem (2005) 280:18803–9. doi: 10.1074/jbc.M501731200
70. Prabhakaran S, Liang X, Skare JT, Potts JR, Höök M. A Novel Fibronectin Binding Motif in MSCRAMMs Targets F3 Modules. PloS One (2009) 4:e5412. doi: 10.1371/journal.pone.0005412
71. Lepow IH, Ratnoff OD, Levy LR. Studies on the Activation of a Proesterase Associated With Partially Purified First Component of Human Complement. J Exp Med (1958) 107:451–74. doi: 10.1084/jem.107.3.451
73. Ziccardi RJ. The First Component of Human Complement (C1): Activation and Control. Springer Semin Immunopathol (1983) 6:213–30. doi: 10.1007/BF00205874
74. Ziccardi RJ. Spontaneous Activation of the First Component of Human Complement (C1) by an Intramolecular Autocatalytic Mechanism. J Immunol (1982) 128:1–5.
75. Budayova-Spano M, Grabarse W, Thielens NM, Hillen H, Lacroix M, Schmidt M, et al. Monomeric Structures of the Zymogen and Active Catalytic Domain of Complement Protease C1r: Further Insights Into the C1 Activation Mechanism. Structure (2002) 10:1509–19. doi: 10.1016/S0969-2126(02)00881-X
76. Probert WS, Allsup KM, LeFebvre RB. Identification and Characterization of a Surface-Exposed, 66-Kilodalton Protein From Borrelia burgdorferi. Infect Immun (1995) 63:1933–9. doi: 10.1128/iai.63.5.1933-1939.1995
77. Nguyen NTT, Röttgerding F, Devraj G, Lin YP, Koenigs A, Kraiczy P. The Complement Binding and Inhibitory Protein CbiA of Borrelia miyamotoi Degrades Extracellular Matrix Components by Interacting With Plasmin(Ogen). Front Cell Infect Microbiol (2018) 8:23. doi: 10.3389/fcimb.2018.00023
78. Pereira MJ, Wager B, Garrigues RJ, Gerlach E, Quinn JD, Osburne MS, et al. Lipoproteome Screening of the Lyme Disease Agent Identifies Inhibitors of Antibody-Mediated Complement Killing. Proc Nat Acad Sci USA (2022) 119(13):e2117770119. doi: 10.1073/pnas.2117770119
79. Moriarty TJ, Norman MU, Colarusso P, Bankhead T, Kubes P, Chaconas G. Real-Time High Resolution 3D Imaging of the Lyme Disease Spirochete Adhering to and Escaping From the Vasculature of a Living Host. PloS Pathog (2008) 4:17–9. doi: 10.1371/journal.ppat.1000090
Keywords: complement C1r, classical pathway of complement, Borrelia miyamotoi, complement evasion, spirochete, BBK32
Citation: Booth CE Jr, Powell-Pierce AD, Skare JT and Garcia BL (2022) Borrelia miyamotoi FbpA and FbpB Are Immunomodulatory Outer Surface Lipoproteins With Distinct Structures and Functions. Front. Immunol. 13:886733. doi: 10.3389/fimmu.2022.886733
Received: 28 February 2022; Accepted: 19 April 2022;
Published: 27 May 2022.
Edited by:
Péter Gál, Research Centre for Natural Sciences, HungaryReviewed by:
Christine Gaboriaud, UMR5075 Institut de Biologie Structurale (IBS), FranceCláudia Vilhena, Leibniz Institute for Natural Product Research and Infection Biology, Germany
Copyright © 2022 Booth, Powell-Pierce, Skare and Garcia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brandon L. Garcia, Z2FyY2lhYnIxOEBlY3UuZWR1; Jon T. Skare, anNrYXJlQHRhbXUuZWR1
†These authors have contributed equally to this work