 Walaa K. Mousa
Walaa K. Mousa Fadia Chehadeh1
Fadia Chehadeh1 Shannon Husband
Shannon Husband- 1Biology Department, Whitman College, Walla Walla, WA, United States
- 2College of Pharmacy, Al Ain University, Abu Dhabi, United Arab Emirates
- 3College of Pharmacy, Mansoura University, Mansoura, Egypt
Trillions of microbes survive and thrive inside the human body. These tiny creatures are crucial to the development and maturation of our immune system and to maintain gut immune homeostasis. Microbial dysbiosis is the main driver of local inflammatory and autoimmune diseases such as colitis and inflammatory bowel diseases. Dysbiosis in the gut can also drive systemic autoimmune diseases such as type 1 diabetes, rheumatic arthritis, and multiple sclerosis. Gut microbes directly interact with the immune system by multiple mechanisms including modulation of the host microRNAs affecting gene expression at the post-transcriptional level or production of microbial metabolites that interact with cellular receptors such as TLRs and GPCRs. This interaction modulates crucial immune functions such as differentiation of lymphocytes, production of interleukins, or controlling the leakage of inflammatory molecules from the gut to the systemic circulation. In this review, we compile and analyze data to gain insights into the underpinning mechanisms mediating systemic autoimmune diseases. Understanding how gut microbes can trigger or protect from systemic autoimmune diseases is crucial to (1) tackle these diseases through diet or lifestyle modification, (2) develop new microbiome-based therapeutics such as prebiotics or probiotics, (3) identify diagnostic biomarkers to predict disease risk, and (4) observe and intervene with microbial population change with the flare-up of autoimmune responses. Considering the microbiome signature as a crucial player in systemic autoimmune diseases might hold a promise to turn these untreatable diseases into manageable or preventable ones.
Introduction
The gastrointestinal tract is home to trillions of diverse microbial species including bacteria, viruses, and fungi (1–3). To colonize the human body, these microbes interact with the immune system to develop tolerance and maintain gut homeostasis. Gut microbes, or their secreted metabolites, directly interact with gut-associated lymphoid tissues (GALTs), which include Peyer’s patches, mesenteric lymph nodes, intraepithelial lymphocytes, lamina propria, and isolated lymphoid follicles (4, 5). This interaction is mediated through Toll-like receptors (TLRs), leading to induction of immune cell differentiations, setting the balance between helper T cells and regulatory T cells (6). Moreover, gut microbes modulate genes involved in maintaining the mucosal barrier function such as those involved in the synthesis of the tight junctions (6, 7) and formation of mucin barrier (8, 9).
Gut microbes could also modulate host genes through microRNAs (miRNAs), short non-coding RNA sequences that silence gene expression (10, 11). Host-derived miRNAs serve an essential function to control microbial population abundance by directly affecting microbial gene expression and mucosal colonization (12–15). Microbes, or their secreted metabolites, modulate miRNAs affecting host genes. The affected host genes include those linked to differentiation of T cells, production of interleukins, proliferation of intestinal epithelial cells, gut permeability, and autophagy process (16–21). Selected examples of miRNAs mediating host–microbe interactions leading to or preventing inflammation are noted and summarized (21–34) (Table 1).
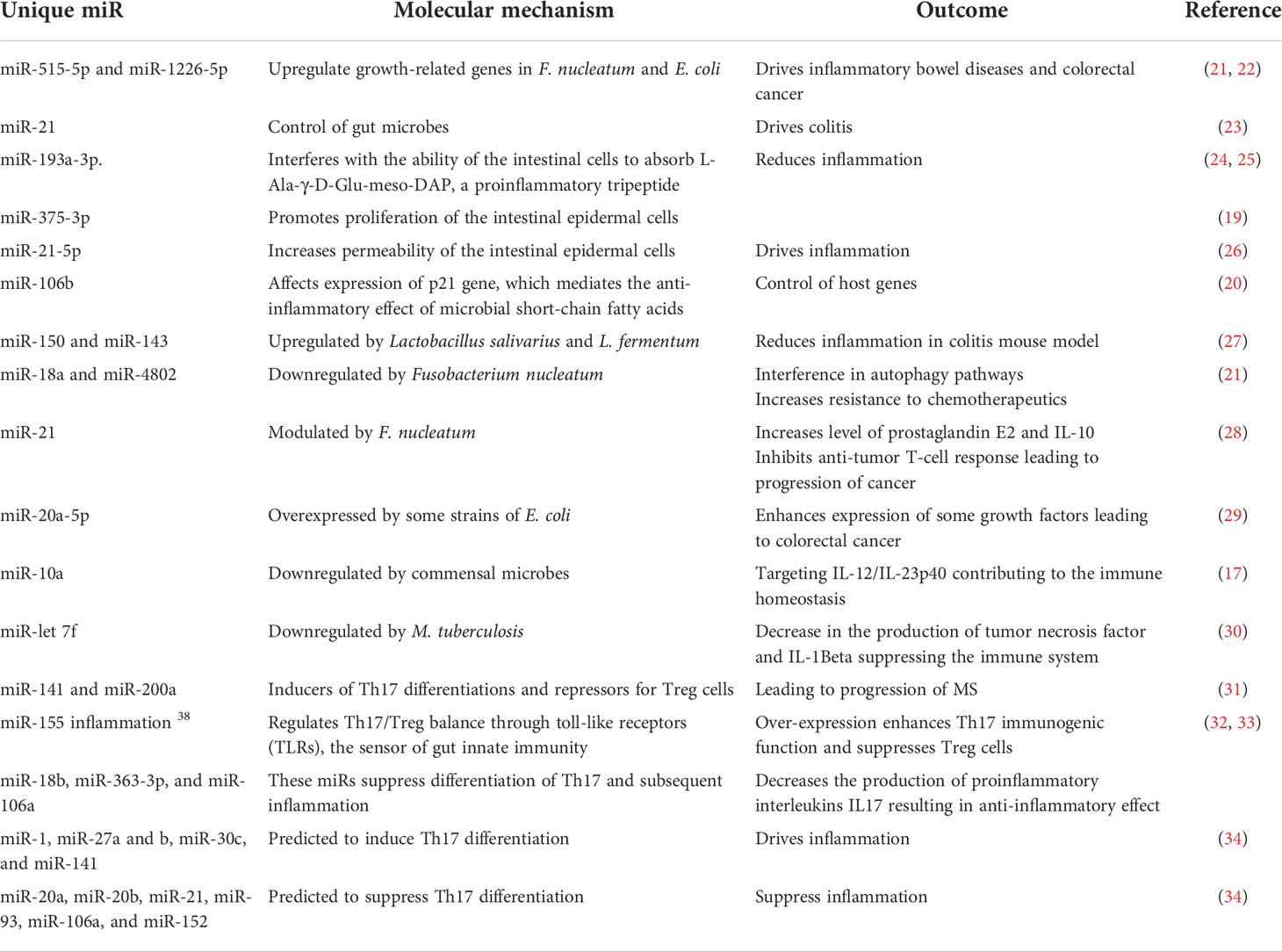
Table 1 The role of selected miRNAs in shaping the microbiome structure and mediating inflammatory and autoimmune diseases.
Microbial dysbiosis disturbs the immune function leading to inflammation and sensitization of the immune system and causing autoimmune diseases (35, 36). Many factors influence microbial dysbiosis such as diet, stress, drugs, diseases, age, and lifestyle. The imbalance in helper T cells/regulatory T cells drives autoimmune diseases such as colitis and multiple sclerosis (MS) (18, 33). Leakage of metabolites such as lipopolysaccharides sensitizes the immune system, leading to a higher production of pro-inflammatory interleukins, and degradation of mucin resulting in irritation of the gut lining and microbial invasion (37). Figure 1 illustrates the role of some microbial taxa in maintaining gut barrier function and how microbial dysbiosis results in a leaky gut. Each microbe prevents or drives inflammation by a unique mechanism. For example, Faecalibacterium prausnitzii prevents inflammation by inducing Treg differentiation, leading to the subsequent higher production of IL-10 (an anti-inflammatory interleukin) (38, 39). In contrast, Fusobacterium nucleatum drives inflammation by inhibiting cytotoxic T cells and modulation of miRNAs, leading to suppression of autophagy (28). Several examples of individual microbes that modulate host immune response to prevent or drive inflammation and autoimmune reaction are noted (40–72) and summarized (Table 2).
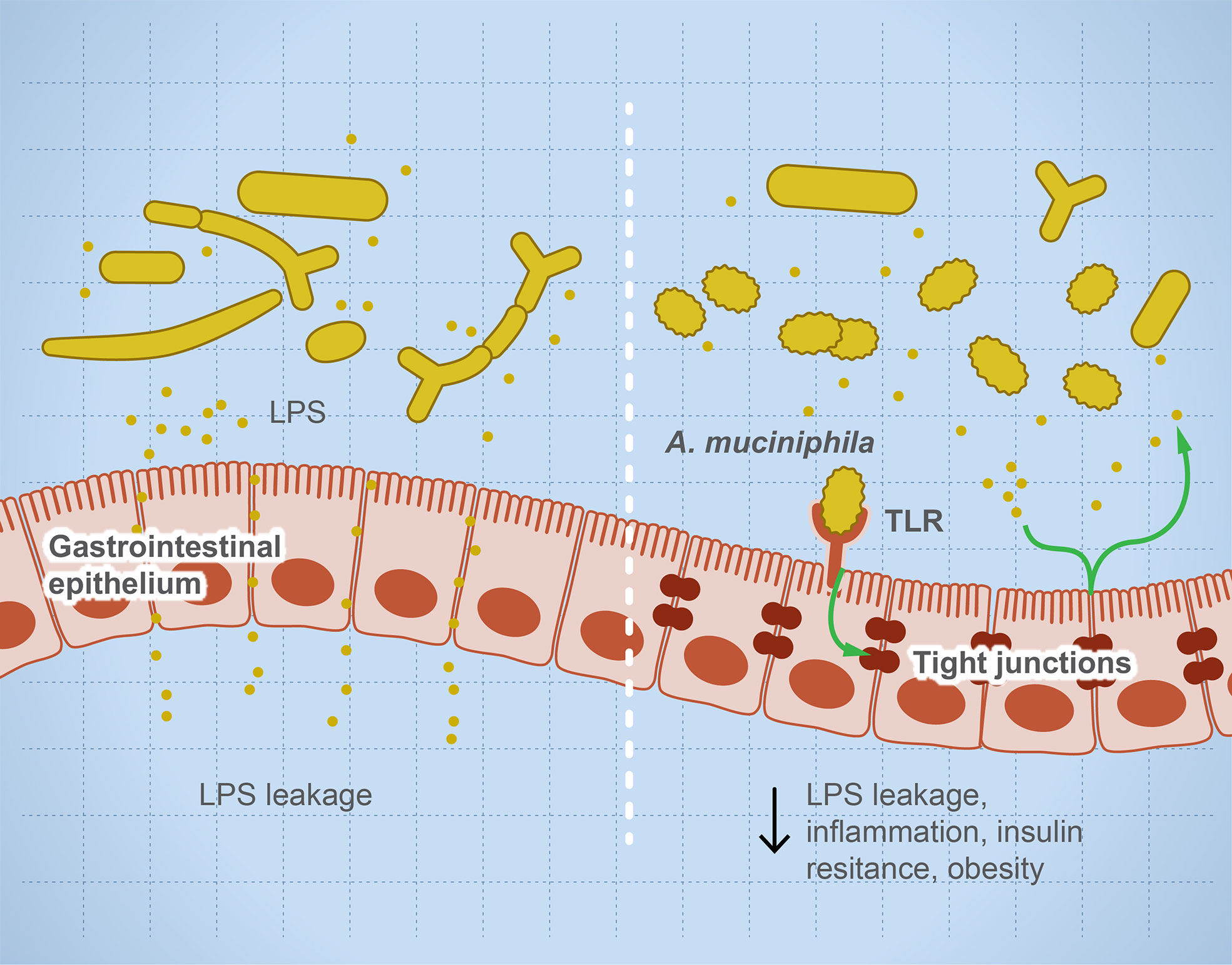
Figure 1 Microbial dysbiosis drives systemic inflammation by targeting the mucosal barrier. The illustration represents that, during microbial dysbiosis, the gut barrier is leaky, which results in diffusion of microbial metabolites such as lipopolysaccharides into the circulation, causing systemic inflammation. However, healthy microbiome has a balanced microbial composition including potential anti-inflammatory microbes such as Akkermansia muciniphilia and Faecalibacterium prausnitzii. These microbes or their metabolites activate TLRs leading to overexpression of tight junction proteins and prevent gut leakage. This leakage is thought to drive systemic inflammatory and autoimmune diseases such as insulin resistance and obesity.

Table 2 Unique gut microbes as drivers or inhibitors of autoimmune diseases.
Microbial dysbiosis drives systemic autoimmune diseases
Microbial dysbiosis is strongly linked to local inflammation and autoimmune diseases in the gut such as Crohn’s and inflammatory bowel diseases. However, much less is known about the link between dysbiosis in the gut and systemic autoimmune diseases. Evidence suggests that gut microbes exert some control over the systemic immune response, particularly innate immunity. To induce immune tolerance to commensal microbes, the antigens of the gut microbes are sampled by dendritic cells (DCs) and presented to T cells in the pancreatic lymph nodes (PLNs) (77, 78). Sometimes, T cells activated by microbial antigen fragments spread and trigger a systemic immune response. Additionally, some microbial metabolites might leak from the gut barrier to other tissues or organs. Short-chain fatty acids (SCFAs) suppress neutrophil function via binding to GPCRs (79) while microbial peptidoglycans stimulate neutrophil function via Nod1 (40). Here, we discuss links between microbial dysbiosis and some systemic autoimmune diseases including type 1 diabetes (T1D), Multiple Sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE).
The role of microbial dysbiosis in triggering type 1 diabetes
T1D is a systemic autoimmune disease that is linked to microbial dysbiosis. Preclinical and clinical T1D is mostly associated with GIT pathogenesis, such as celiac disease or increased intestinal leakage potentially due to microbial dysbiosis (41–43). Multiple studies report significant shifts in gut microbes including bacteria, viruses, and fungi before the onset of T1D as reviewed (44). Figure 2 illustrates the balanced microbial interaction that contributes to glucose metabolism and suppresses hyperglycemia.
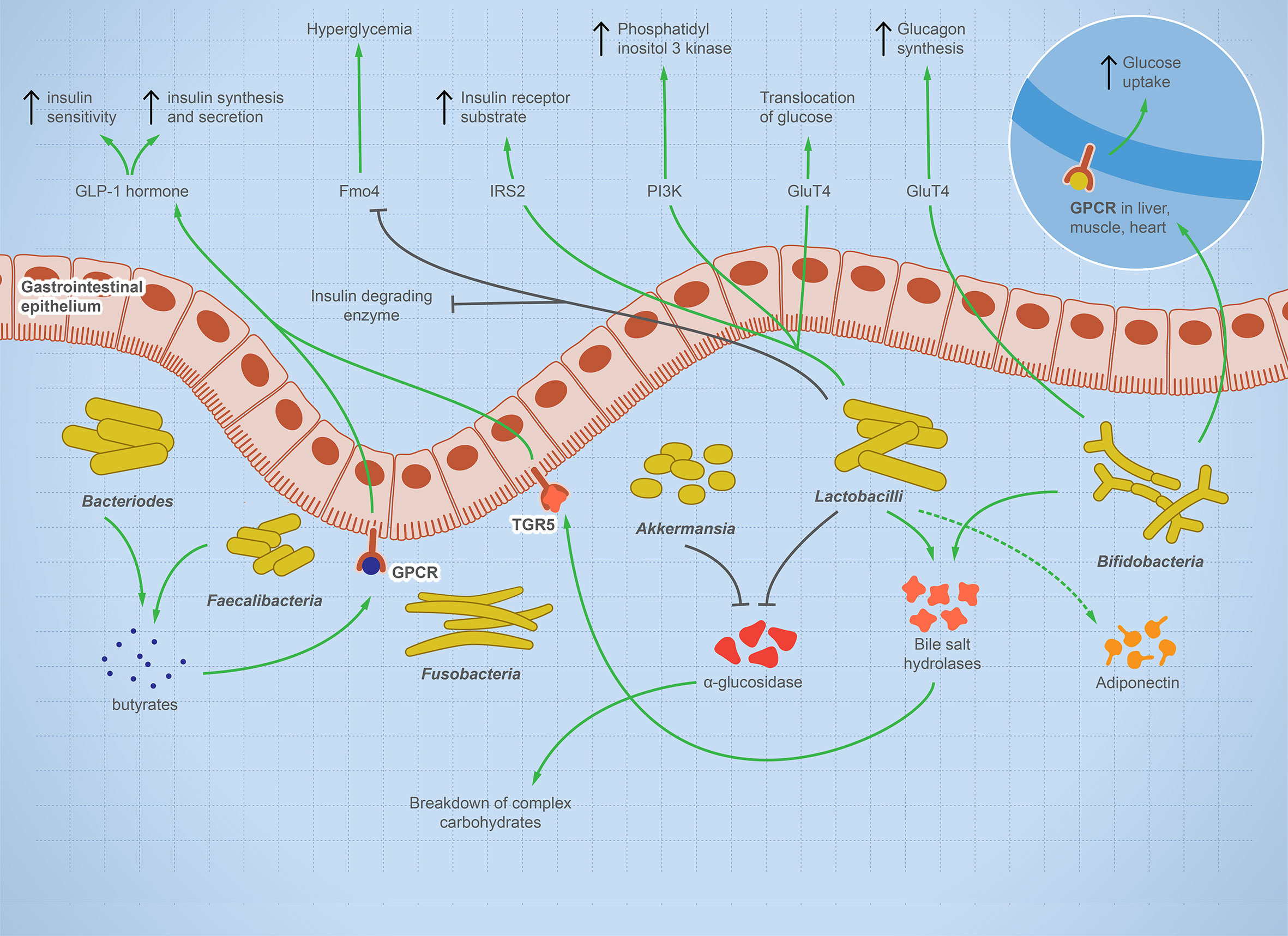
Figure 2 The integral role of the balanced gut microbes in maintaining glucose level and suppressing hyperglycemia. The illustration shows the fundamental role of some representative gut microbes in glucose metabolism. The onset of T1D is characterized by a change in microbial composition characterized by decrease in specific taxa such as Bifidobacterium, Lactobacilli, Bacteroides, Faecalibacterium, and Akkermansia—while other taxa such as Fusobacterium is enriched. Bifidobacterium increases glucose uptake in heart, muscle, and liver tissues and stimulates glucagon synthesis. Lactobacilli stimulates cellular receptors involved in translocation of glucose, production of phosphatidyl inositol-3-phosphate, production of insulin receptor substrate, and suppression of hyperglycemia. Bacteroides and Faecalibacterium produce butyrate, which binds to GPCR, activating the production of GLP-1 hormone, which is involved in insulin synthesis, secretion, and sensitivity. Lactobacilli and Akkermansia have an inhibitory effect on alpha-glucosidase enzyme, which helps in the breakdown of complex carbohydrate, raising sugar level. Lactobacilli and Bifidobacterium increase bile salt hydrolyses, leading to GLP-1 hormone stimulation. If this microbial role is disturbed, inflammation and autoimmune diseases arise.
A study on mice reported that cytophaga-flavobacter-bacteroidetes (CFB) affects Th17/Treg balance, which is crucial for immune homeostasis, leading to autoimmune diseases (58). CFB induces Th17 differentiation, and its absence is associated with induction of Treg cells in the lamina propria, and this effect is diminished after selective antibiotic treatment (58). Another study reported an enrichment in Bacteriodetes dorei with preclinical or clinical T1D. B. dorei produces LPS that induces the innate immune response (58). The abundance of Desulfovibrio piger is associated with a higher level of plasma 1-arachidonoyl-GPC, a metabolite known to negatively affect CD4+, CXCR3+, CD8+, and CXCR3+ T cells (50), preventing further progression of autoimmunity (50). A similar effect of this metabolite is reported in mice (80). D. piger is also known to produce hydrogen sulfide, which affects T cells and immune response (81–83).
A study reported that T1D patients show enrichment in Proteobacteria, Actinobacteria, and Bacteroidetes and lack of butyrate-producing bacteria (84). Knowing that butyrate induces mucin formation and assembly of tight junction explains the increased gut permeability in T1D patients. T1D microbiota is also deficient in Provetella species that break down mucin and enriched in bacteria that produce propionates and acetates known to impair neutrophil functions observed in T1D patients (60, 61). Other evidence suggests that microbial dysbiosis alters the gut immunity, resulting in excessive stimulation of inflammatory response leading to T1D even without an observable change in gut permeability. For example, a study reported that T1D patients show a high expression level of intercellular adhesion molecule-1, HLA-DR, HLA-DP, IL-4, and IL-1α-positive cells (60, 85). Another study showed that DCs are not able to induce FoxP3+Treg cell differentiation in T1D patients (86). The authors claimed that the deficiency of Treg cells in the gut decreases the ability of the immune system to tolerate and discriminate self-antigens in the pancreatic β-cells (86).
A mouse model of T1D has a reduction in IL-22, IL-17A, and IL-23A that is associated with loss of segmented filamentous bacteria (87). This alteration is reversed by treatment with anti-inflammatory drugs, suggesting that dysbiosis is linked to inflammation rather than to T1D (87). Another study shows that the immunomodulatory compound, indole-3-carbinol, binds to aryl hydrocarbon receptor (AhR) in NOD mice (88). AhR is a transcription factor that prevents T1D. Interestingly, this activation was mainly localized to the small intestine (88). Meanwhile, no alteration was observed in the differentiation of T cells in the spleen or PLNs (88). These changes were associated with a signature trans-kingdom network characterized by a reduction in ruminiclostridium, intestinimonas, and lachnospiraceae mediated by an increase in CD25 (88). A study found that prediabetes in rats is associated with enrichment in Bifidobacterium, Lactobacillus, and Bacteroides species and antibiotic treatment decreased the incidence of T1D (89, 90). Other research shows that Lactobacillus bifidus can activate the autoimmune response in IL-1 receptor antagonist-knockout mice (74, 75).
Mice that are deficient in MyD88, a specific adapter molecule involved in TLRs signaling pathways, are protected from T1D in the presence of a microbiome signature characterized by a richness in Lactobacillus genera and low Firmicutes/Bacteroidetes ratio (91). Interestingly, Lactobacillus shows protection against T1D while Bacillus cereus delayed the onset of T1D in NOD mice (51–53). Metagenomic analysis revealed that stool samples from T1D patients are enriched in genes responsible for bacterial adhesion and flagella formation that are possibly involved in triggering systemic immune response (84). T1D patients are also deficient in butyrate producers and mucin-degrading bacteria (84).
Preclinical T1D is also associated with a reduction in butyrate-producing genera such as Roseburia (48). When dietary fibers (DF) intake is limited, gut microbes degrade mucin to increase the supply of butyrate. Mucin degradation increases gut permeability and immunogenicity leading to T1D (92, 93). Butyrate modulates susceptibility to T1D through a variety of possible mechanisms such as upregulation of tight junction proteins (94). Butyrate enhances differentiation of Treg cells through histone H3 acetylation (95, 96) and induces apoptosis of proinflammatory T cells in the murine cell line (97). Another study shows that Akkermansia sp., a butyrate producer, protects against pancreatic autoimmunity (54). Butyrate promotes Th1 differentiation through induction of IFN-γ and T-bet expression by inhibition of histone deacetylase. Butyrate inhibits Th17 differentiation through suppression of Rorα, Rorγt, and IL-17 (98). The literature shows mixed results for the effect of oral administrations of butyrate on the development of T1D in humans versus experimental animals (99).
Several reports suggest that viral infections can trigger or attenuate the development of T1D or pancreatic autoimmunity, particularly infection with rotavirus, cytomegalovirus (CMV), and enterovirus, although the underpinning mechanisms are not well-understood (100–104). Several findings in experimental animals, particularly NOD mice, suggest that the viruses affect T1D development by molecular mimicry or bystander inflammation. Molecular mimicry is reported for molecules such as glutamic acid decarboxylase enzyme, tyrosine phosphatase IA-2/IAR, and heat shock protein 60 (105, 106). For example, Coxsackievirus B4 (CVB4) triggers T1D by mimicry to host-related molecules such as beta-cell glutamic acid decarboxylase enzyme 65 (105, 106). Interestingly, the adequate response of pancreatic B cells to interferon-gamma reduced CVB4-induced T1D (107). Another hypothesis is that the virome induces pancreatic bystander inflammation, which involves the inability of CD8+ cytotoxic T lymphocytes to recognize self-antigens of the pancreatic beta cells. Interestingly, there are some reports of viral infections linked to less incidence of T1D such as CXADR rs6517774, Mastadeno-virus C, and Norovirus-4 (103, 108). In the NOD mouse model, infection with mouse Norovirus-4 (MNV4) is associated with enrichment in the alpha diversity of gut bacteria and expansion of Treg cells (108). To examine if the effect of MNV4 on immunity is due to indirect modulation of the gut microbes, the authors repeated the experiment on germ-free mice. Interestingly, germ-free mice infected with MNV4 still show modulation of the immune response including an increase in Treg and alteration in cellular and secreted components of the immune system such as B cells, T cells, macrophages, and cytokine biomarkers (108).
Dysbiosis in the virome is also associated with several autoimmune diseases including T1D (109, 110). The virome is the collection of endogenous retroviruses, eukaryotic viruses, and bacteriophages inhabiting the gut microbiota (110, 111). Most of the gut viruses are sourced from lysogens; the latter is defined as bacteria containing dormant phages (prophages) inserted in the bacterial genomes. These phages are released from the lysogens upon receiving particular signals, including a change in diet, and further affect microbiome structure and susceptibility to autoimmune diseases (112). Prophages might encode immune-modulatory molecules that directly affect the immune system response. For example, the novel prophage ΦHKU.vir encodes toxin with superantigens that enable colonization of S. pyogenes by inducing nonspecific differentiation of T cells (113). The virome and host–disease associations are much less studied compared to the gut microbiome. Moreover, the literature shows mixed results and a non-confirmed link between virome shift and T1D. The main challenge in understanding virome interaction is that it directly affects bacterial population dynamics and diversity, increasing confounding factors and making it hard to draw a confirmed conclusion (111, 114). Phage infection of gnotobiotic mice, inoculated with defined human microbes, resulted in direct reduction of the susceptible species and indirect effect on the other species likely through interspecies interactions (114). Another study reported an association between the initial abundance of amyloid-producing E. coli and E. coli bacteriophage/E. coli ratio, which results in depletion of E. coli, in the development of T1D (115).
In summary, many studies investigate the role of microbial dysbiosis in triggering T1D through a variety of mechanisms such as modulation of Th17/Treg balance, variation in interleukin production, and change in gut permeability. These effects are mediated by some microbial metabolites such as butyrate, LPS, and arachidonoyl-GPC. Activation of latent viral infections can also drive microbial dysbiosis leading to T1D. However, less is known about microbial dysbiosis in late-onset T1D and if diet management can modulate disease severity or progression (116). A study reported an enrichment in Veillonella and Clostridium genera coupled with a reduction in Bifidobacterium, Lactobacillus, and Prevotella in the pediatric T1D compared to healthy children (117). Another study reported similar results in adults, particularly the Bifidobacterium signature (118). However, all studies investigating the association between late-onset T1D and microbiota shift lack directionality as T1D is thought to drive microbiota change as the diseases progress.
The role of microbial dysbiosis in triggering multiple sclerosis
MS is a chronic autoimmune disease affecting 2.8 million people around the world (119). MS causes demyelination of neurons, leading to neuroaxonal degeneration in the brain and spinal cord, resulting in an unpredictable outcome that can result in a permanent disability (120, 121). Although there is no cure for MS, some interventions can improve the quality of life and reduce complications such as anti-CD20 monoclonal antibodies, which destroy circulating memory B cells and subsequently weaken the immune system and increase the risk of infection (122). The underlying causes are largely unknown and thought to be linked to genetic factors and/or viral infection (123, 124). Figure 3 illustrates the potential role of gut dysbiosis in MS, although the directionality of this interaction is not clear. Gut microbes are definitely altered in MS patients, and this alteration is associated with the varied severity of the disease (125–128). The microbiome signature in MS patients is characterized by the lower abundance of F. prausnitzii, Prevotella, and Bacteroides, and a higher abundance of Akkermansia muciniphila (126, 128). Pediatric MS patients show a signature reduction in SCFA-producing Ruminococcaceae compared to healthy children (129). However, the directionality of this association is not clear. Moreover, MS-modifying drugs can alter the microbiota composition. Administration of dimethyl fumarate and glatiramer acetate resulted in a significant reduction in Lachnospiraceae and Veillonellaceae (130).
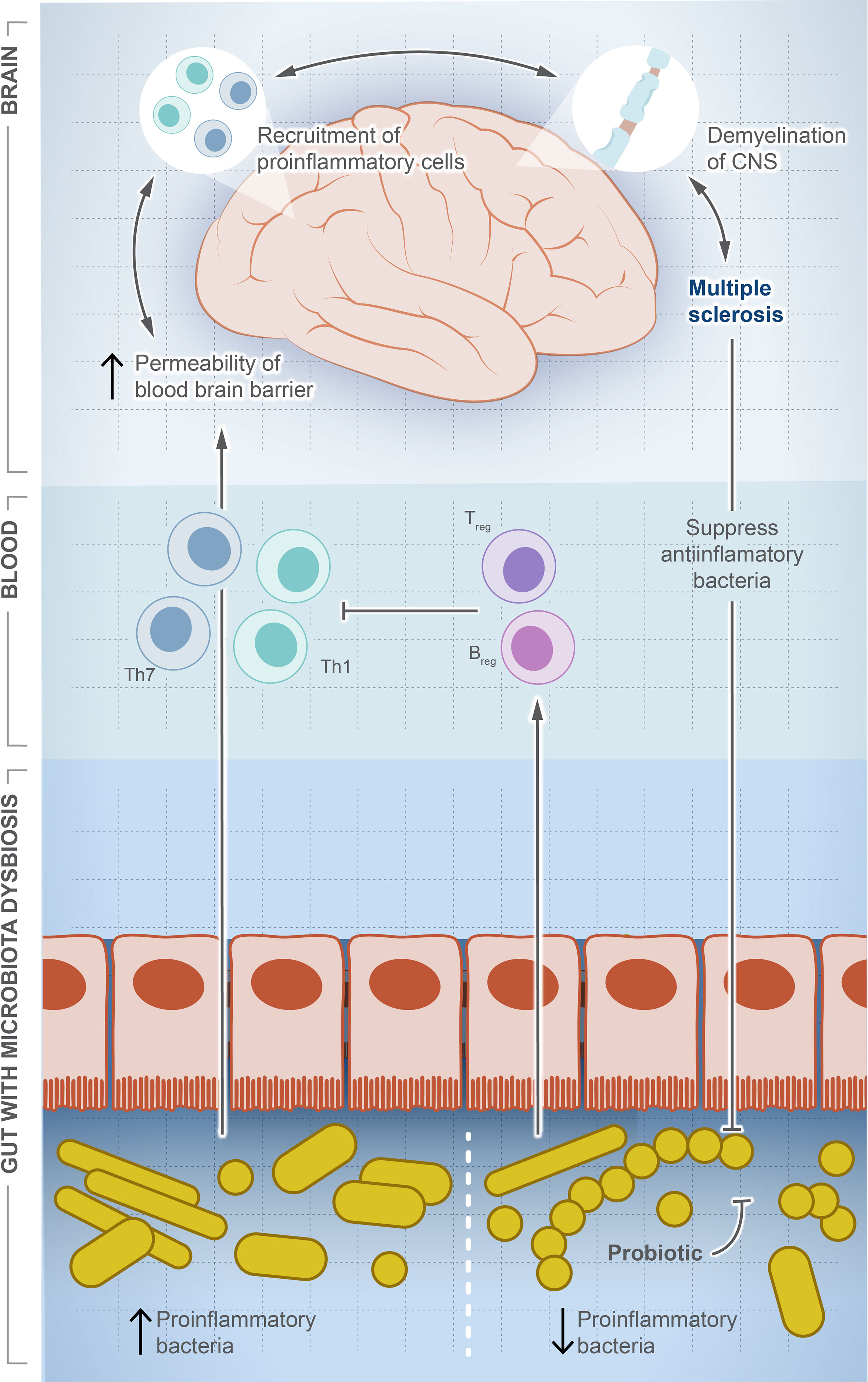
Figure 3 Microbial dysbiosis is a potential factor driving multiple sclerosis. In this illustration, we show how the change in population dynamic of gut microbes suppresses or drives multiple sclerosis. The increase in proinflammatory bacteria induces differentiation of Th1/Th17, which travel systemically to the brain and recruit more proinflammatory cells producing inflammatory cytokines. In contrast, the decrease in proinflammatory bacteria induces differentiation of Treg cells and production of anti-inflammatory cytokines, which balance or counteract Th1/Th17. Interestingly, once MS is developed, a significant decrease in anti-inflammatory community is observed, but the exact signaling mechanism is unknown.
Mechanistically, some evidence suggests the role of miRNAs such as miR-141, miR-200a, and miR-155 in driving MS by shifting Th17/Treg balance towards the Th17 side, promoting production of the proinflammatory mediator IL17 (31) (Table 1). These miRNAs are implicated in interfering with repressor proteins that regulate Th17 differentiation (31-33). As microbes regulate miRNAs, there is a hope that probiotics, antibiotics, or specialized microbial metabolites might suppress Th17 production or induce Treg differentiation by checking MS progression. Another possible mechanism of how microbial dysbiosis drives MS might be through molecular mimicry. Studies on neuromyelitis optica, a degenerative autoimmune disease that results in inflammation, demyelination, and nerve necrosis, suggest a possible molecular mimicry between brain and microbial antigens (131). Neuromyelitis optica is characterized by the presence of IgG1 autoantibodies that attack aquaporin 4 (AQP4), a predominant water channel in the CNS (132). This attack results in deposition of immunoglobulin complements causing demyelination and tissue damage (133, 134). The mechanism involves a molecular mimicry between AQP4 and ABC transporter permease in the gut microbe, Clostridium perfringens (131).
Transfer of microbiota from MS patients to mice elicits an immune response and inflammation (6). Opposing this finding, another research shows that the transfer of microbiota from a mouse model of MS to another healthy mouse resulted in disease protection thanks to miR-30d (135). The unexpected effects of this miR could be attributed to its stimulatory effect on the growth of Akkermansia muciniphila, which exhibits an anti-inflammatory role (135). Another possible mechanism for MS protection might be the induction of anti-inflammatory Treg cells (135). Other cohort studies show enrichment in Eggerthella lenta and Akkermansia muciniphila in MS patients (70, 71). MS patients show an increase in anti-A muciniphila immunoglobulin G while no difference in IgG is noted for other gut microbiota such as Bacteroides fragilis, Fusobacterium, and Acinetobacter baumannii (72). Other studies suggest that certain gut microbiota such as A. muciniphila interacts with spore-forming bacteria to escalate the inflammation leading to MS through a direct effect on T lymphocytes (73). Breakthrough research reported that some gut microbiome taxa enriched in MS patients directly interact with IgA-producing cells at the gut lining; the latter translocate to the brain cells and locally produce immunoglobulin A (IgA), which mediates severe inflammation. MS patients show a signature decrease in Prevotella genera compared to healthy control. Although this association lacks directionality, it varies depending on the disease severity. This finding raises the possibility of using probiotics to manage brain inflammation in MS (136). A study shows that supplementation of Prevotella histicola suppressed autoimmune encephalomyelitis (EAE) in the HLA-DR3.DQ8 transgenic mouse model (137). This model expresses HLA-DR3 and DQ8 genes and can develop EAE, a severe spinal cord and brain inflammation that is very comparable to MS in human (138). In a follow-up study, the authors found that treatment with P. histicola yielded a similar disease-suppression effect as the MS drug Copaxone. However, co-administration of both P. histicola and Copaxone does not provide a synergic effect. Copaxone acts by decreasing the response of antigenic T cells in the brain (139). Data show that treatment with P. histicola increased the level of regulatory T cells and decreased proinflammatory cells, particularly those producing IL-17 and IFN-γ (140). Interestingly, a study shows that microbial transplant from MS patients developed EAE in transgenic mice (141). These results are a promising development to microbiome-based therapeutics for autoimmune diseases. However, some preclinical data claim that the beneficial effects of probiotics in delaying MS progression (142) are likely through indirect anti-inflammatory and immune-modulatory activity.
A recent study suggests a strong link between lung microbiome dysbiosis and MS in rats. Microbiome shift to a less lipopolysaccharide-producing phyla escalates MS while its enrichment decreases the proinflammatory response (143). The mechanistic underpinning is impairment in the responsiveness of microglial cells in the brain to type II interferons, resulting in a reduced recruitment of immune cells and further clinical manifestations (143).
Activation of latent Epstein–Barr virus (EBV) infection is linked to the development of MS (144, 145). EBV is a common virus that is considered part of the commensal microbiome (146). EBV infects B cells and epithelial cells, and because it shares molecular mimicry to some host protein, the viral genome integrates within the host DNA. When triggered, by yet unknown signals, it can lead to systemic autoimmune diseases. A recent study shows that EBV antibodies are associated with 99% of MS cases with the US military (145). The authors identified a strong positive association between MS and EBV where EBV infection increases the risk of developing MS by 32% (145) and MS only develops after EBV infection. If EBV is truly a prerequisite to MS, this discovery holds the promise of turning these untreatable diseases into vaccine-preventable ones.
The role of microbial dysbiosis in triggering rheumatoid arthritis
RA is a systemic autoimmune disease affecting joints, and sometimes other internal organs, causing inflammation and swelling. One of the first reports of the connection between microbial dysbiosis and RA dates back to 1979 with the discovery that germ-free rats are 100% susceptible to developing RA upon injection of an intradermal adjuvant (147), while conventional rats are only 0 to 20% susceptible and further develop weak or delayed inflammation (147). Interestingly, this induced inflammation is resolved by inoculation of E. coli and slightly worsens by inoculation of Lactobacilli (148). The authors claimed a possible role of the LPS of E. coli in resolving RA (138). Studies show that the microbiome in RA patients is enriched in specific microbial taxa such as Provetella, Lactobacillus sabotage, and segmented filamentous bacteria (62). Higher abundances of these microbes increase their proinflammatory metabolites such as serum amyloid protein A, which increases the production of Th1 and Th17 that migrate systemically and contribute to the diseases (62). Some members of the Provetella genus are implicated in several inflammatory and autoimmune conditions that range from low-grade inflammation to periodontitis, bacterial vaginosis, and RA (67).
A growing body of research suggests a possible role of Provetella copri in the development of RA (63–67). Mechanistically, the ability of Prevotella copri to induce mucosal inflammation is due to the activation of TLR-2 stimulating the differentiation of Th17 and leading to the excessive production of proinflammatory interleukins such as IL-23 and IL-1 and the recruitment of neutrophils (67, 149). This inflammation increases gut permeability and leakage of microbes and proinflammatory molecules, leading to more systemic inflammation and immune reaction. A higher abundance of P. copri is also associated with RA in mice with knocked out NLRP6 gene, a proinflammatory gene that is part of the inflammasome (63). These mice are genetically modified to produce lower levels of proinflammatory cytokines. Co-housing of wild-type and knockout mice resulted in inflammatory symptoms in the wild type and suggests that gut microbes can induce autoimmune diseases even without a genetic predisposition. This finding also suggests a possible role of Provetella in arthritis etiology as a pathobiont, and it could be potentially used as a diagnostic biomarker for RA (150). Prevotella intestinalis is another member of the family implicated in intestinal inflammation and particularly colitis through a reduction in SCFAs and the anti-inflammatory interleukin IL-18 in mice (68). Other studies show that P. histicola has anti-inflammatory activity and can protect mice from arthritis (55). Interestingly, the mechanism of inflammation suppression looks opposite to that induced by other pathobionts from the same genus. The authors show that P. histicola regulates DCs (CD103+), resulting in the generation of Treg cells, which suppresses Th17, decreasing proinflammatory interleukins, and increasing anti-inflammatory interleukins such as IL-10 (55). In addition, P. histicola upregulates the production of the tight junction protein, which decreases gut permeability (55). These findings suggest a potential application of P. histicola as a probiotic for arthritis and possibly other autoimmune diseases (151). Figure 4 illustrates the paradoxical activities of P. copri and P. histicola in mediating RA.
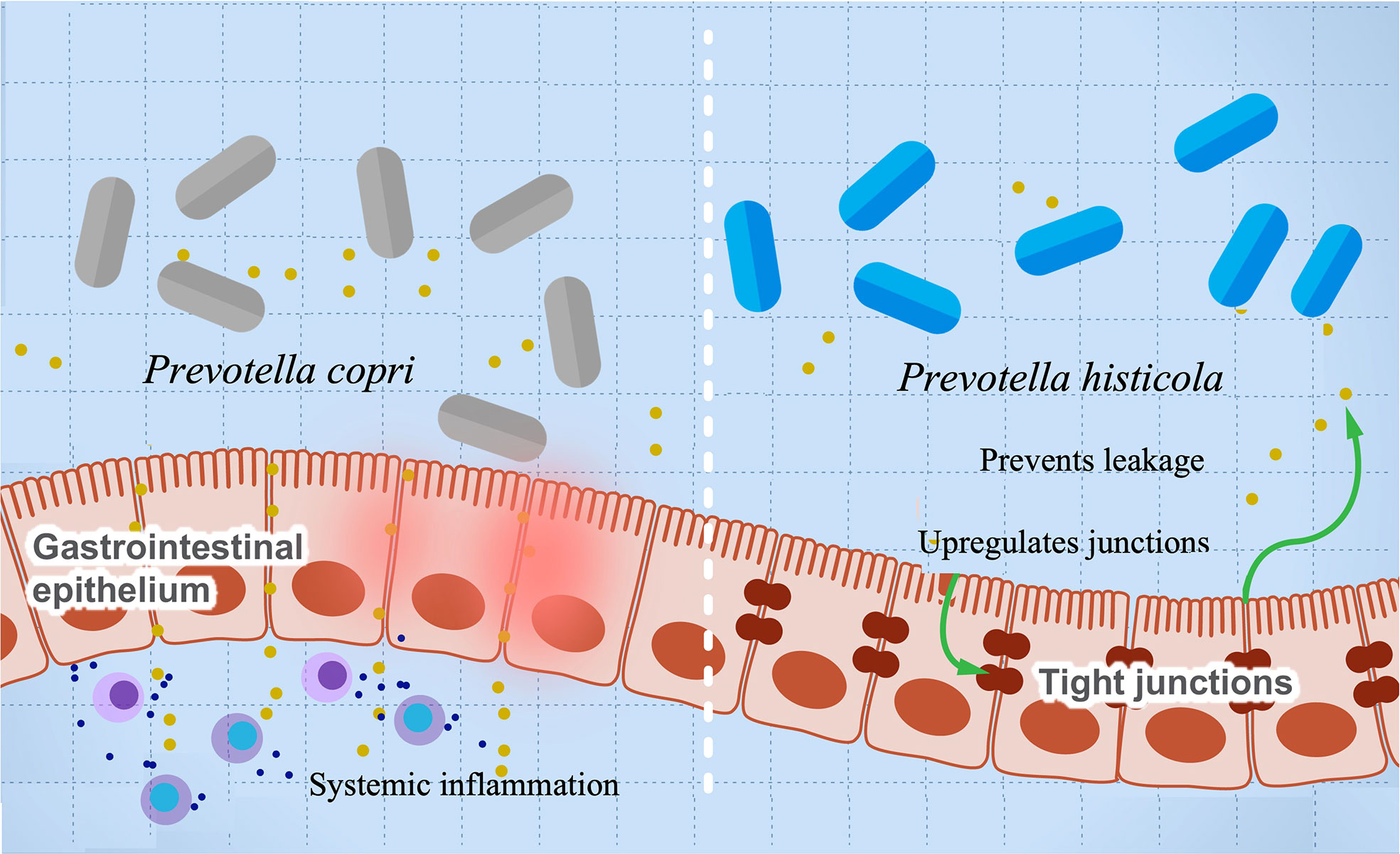
Figure 4 The influence of Prevotella species in preventing or mediating RA. The figure illustrates the role of P. histicola in upregulating the tight junctions, which prevent leakage of proinflammatory metabolites and subsequently prevents inflammation (right side). On the contrary, P. copri stimulates differentiation of Th17, leading to upregulation of proinflammatory cytokines’ production and systemic inflammation.
The role of microbial dysbiosis in triggering systemic lupus erythematosus
SLE is an autoimmune disease that affects joints, blood, kidney, and other organs with yet an elusive etiology. The hallmark of SLE is the formation and deposition of immune complexes from the production of autoantibodies directed towards nuclear antigen and could be detected several years before the onset of the disease (152–154). This autoimmune attack results in inflammation and organ failure (155). The mechanism beyond stimulation of autoreactive T cells and autoantibody production is still unclear, but various theories exist such as genetic deposition or environmental factors exhibiting molecular mimicry (153–155). This inflammation is increasingly believed to be attributed to an imbalance in Th17 and Treg cells, leading to a higher production of proinflammatory cytokines such as IL-17, IL-22, and IL-23 (156, 157) that drive systemic inflammation. Evidently, a high level of IL-17-producing cells is reported to infiltrate tissues of kidney, lung, and skin of SLE patients, resulting in organ damage (158). Understanding the role of the human microbiota in driving, intensifying, or preventing SLE is gaining intriguing interest (159). A unique gut microbiota signature is reported with the inflammatory flares and as the disease progresses (160, 161). This signature is characterized by a reduction in microbiota diversity with enrichment in some genera such as Campylobacter, Streptococcus, and Veillonella, and depletion of others such as Bifidobacterium (159). A significantly low abundance of Lactobacillus and an increase in Lachnospiraceae are associated with SLE in mice (162). The enrichment in Lachnospiraceae is gender-specific, with more abundance in females, which increases the disease’s severity. Interestingly, lupus-susceptible mice showed enrichment in the metabolic pathways of motility and sporulation genes (162), which might be linked to the ability of microbes to cause systemic inflammation. Restoration of lactobacilli abundance by feeding retinoic acid resulted in improved symptoms of lupus, suggesting a potential role of lactobacilli in preventing or counteracting inflammation. Studies reported a shift in Firmicutes/Bacteroidetes ratio accompanied by a change in SCFAs and Th17 levels in serum of SEL patients (56, 163). The shifted microbiota in SEL is characterized by an imbalance in the Treg/Th17/Th1 ratio. Specifically, two strains from the genus Clostridium drive an imbalance in Th17/Th1, promoting differentiation of CD4+ lymphocytes into Th17 and resulting in inflammation (56). Bifidobacterium bifidum inhibits the excessive stimulation of CD4+ lymphocytes (56). Other studies show that microbiota translocation might be implemented in triggering autoimmune diseases including SLE (69, 164). A study showed that translocation of Enterococcus gallinarum from the gut to the liver resulted in overproduction of autoimmune antibodies, inflammation, and mortality in genetically susceptible mice (69). While antibiotic treatment aimed to eradicate E. gallinarum, it also eliminated the autoantibodies in mice. Interestingly DNA of E. gallinarum recovered from the liver of autoimmune patients induced proinflammation in human hepatocytes mimicking the interaction in mice (69). Activation of latent EBV infection is also linked to the development of SLE (165–167). The tumorigenic activity of EBV might resonate with its ability to evade the immune system. Antigens of EBV share molecular mimicry to SLE antigens, which leads to an autoimmune response during EBV activation (168, 169). Furthermore, EBV suppresses the anti-inflammatory interleukins, resulting in more systemic inflammation (168, 169). A trial EBV peptide vaccine in experimental animals generated cross-reactive antibodies and caused SLE-like symptoms (170, 171). Although EBV is known to induce a transit increase in autoantibodies and inflammation, some studies show that this inflammation can further escalate and spread systemically (172, 173).
Microbiome-based therapeutics for tackling autoimmune diseases
The prevalence of autoimmune diseases and allergies especially in children increased 40% over the last decade (174), resulting from the change in lifestyles such as diet, stress, and pollution. These changes result in a significant shift in microbiome composition (175). A recent study reported a strain-level significant microbiome signature in pediatric allergy characterized by a higher abundance of Ruminococcus gnavus, which is enriched in genes for the production of proinflammatory polysaccharides and lowers the potential for fiber-degrading enzymes (76). An interesting study shows that gut microbes drive sex-biased regulation of autoimmunity by directly regulating serum testosterone in NOD mice (176, 177). The authors show that an elevated testosterone level protects against T1D in male mice, and this protection is transferable to immature female mice by microbiome transplant (177). Another interesting study suggests that gut fungi induce behavioral change in mice through stimulation of immune response mediated by IL17, which binds to receptors in the brain (178).
Manipulating the virome to control autoimmune diseases
One of the striking findings is the ability of some gut viruses of the Iridoviridae family to produce insulin-like molecules that mimic host insulin by 50% and can form the critical 3D molecules needed to bind and activate the insulin receptors (179). Interestingly, these viruses are primarily sequenced from fish (180) and recently have been identified in human fecal genomes (181). Recent studies show that the presence of LCDV-Sa is a risk factor for developing T1D in children (182). However, it is unclear if the presence of these insulin-mimic molecules helps to trigger diabetes or protect from diabetes. Previous studies show that microbial metabolites that mimic the host-derived molecules can trigger an immune reaction against insulin-producing cells resulting in T1D. Although we know much about the structural diversity of the microbiome bacteria, relatively much less research has been done on the human virome. A recent study identified 1,700 viral species in the gut microbiome. However, to date, only 2% of viruses are sequenced. This makes the virome research and its association with human diseases a very exciting area of development that will certainly advance our understanding of the microbe’s host interaction and association with human diseases. The use of phages to modulate the microbiome is still an unexplored avenue with unpredicted interspecies interactions (114).
Tackling autoimmune diseases by microbial transplant or microbial metabolites
A study shows that germ-free mice remain immune-compromised even if they are colonized with animal or human microbiota (183). An interesting approach is the fecal transplant of gut microbes from a healthy donor to patients with autoimmune diseases. A randomized controlled clinical trial shows that fecal microbial transplant in newly diagnosed T1D patients prevents the further decline of insulin production by stabilizing the pancreatic B cells’ function (50). This effect is associated with a shift in plasma microbial metabolites, autoreactive T cells, and gene expression of the intestine (50). This trial not only provides hope for microbiome-based interventions for the treatment of autoimmune diseases but also provides solid evidence that microbiota dysbiosis drives T1D. Another possible scenario is the use of critical microbial metabolites such as butyrate as a supplement. Since butyrate has a protective role against autoimmunity (97), a diet rich in butyrate might dial down the autoimmune reaction. A study in NOD mice shows that butyrate and acetate supplementation decreased inflammation and might be a good candidate for therapeutic interventions to control autoimmune diseases such as T1D.
Conclusion
Each living organism requires specific microbial species that are coevolved to prime the immune system (183). Changing the microbiome structure impairs important functions such as (1) gut permeability leading to leakage of antigens and inflammatory mediators to the blood circulation, (2) inability to produce anti-inflammatory microbial metabolites or to degrade food, and (3) loss of immune homeostasis leading to allergy and autoimmune reactions (6, 184–186). Understanding how gut microbes drive or suppress autoimmune diseases is crucial to developing innovative microbiome-based diagnostic tools and therapeutics.
Author contributions
SH and FC collected data on multiple sclerosis. WM curated all data; reviewed the literature; critically analyzed the current knowledge, gaps, challenges, and future directions; designed/developed tables and figures; and wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by a start-up fund from Whitman College and the Murdock Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ursell LK, Metcalf JL, Parfrey LW, Knight R. Defining the human microbiome. Nutr Rev (2012) 70(Suppl 1):S38–44. doi: 10.1111/j.1753-4887.2012.00493.x
2. Li M, Chen W-D, Wang Y-D. The roles of the gut microbiota–MiRNA interaction in the host pathophysiology. Mol Med (2020) 26(1):101. doi: 10.1186/s10020-020-00234-7
3. Hornung B, Martins dos Santos VAP, Smidt H, Schaap PJ. Studying microbial functionality within the gut ecosystem by systems biology. Genes Nutr (2018) 13(1):5. doi: 10.1186/s12263-018-0594-6
4. He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity (2007) 26(6):812–26. doi: 10.1016/j.immuni.2007.04.014
5. Wu H-J, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes (2012) 3(1):4–14. doi: 10.4161/gmic.19320
6. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res (2020) 30(6):492–506. doi: 10.1038/s41422-020-0332-7
7. Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science (2001) 291(5505):881–4. doi: 10.1126/science.291.5505.881
8. Johansson MEV, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA (2008) 105(39):15064–9. doi: 10.1073/pnas.0803124105
9. Lindén SK, Florin THJ, McGuckin MA. Mucin dynamics in intestinal bacterial infection. PloS One (2008) 3(12):e3952. doi: 10.1371/journal.pone.0003952
10. Rupaimoole R, Slack FJ. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov (2017) 16(3):203–22. doi: 10.1038/nrd.2016.246
11. Okamura K, Ladewig E, Zhou L, Lai EC. Functional small RNAs are generated from select MiRNA hairpin loops in flies and mammals. Genes Dev (2013) 27(7):778–92. doi: 10.1101/gad.211698.112
12. Tarallo S, Ferrero G, Gallo G, Francavilla A, Clerico G, Realis LA, et al. Altered fecal small RNA profiles in colorectal cancer reflect gut microbiome composition in stool samples. mSystems 4(5):e00289-19. doi: 10.1128/mSystems.00289-19
13. Thomas H. Gut microbiota: Host faecal MiRNA regulates gut microbiota. Nat Rev Gastroenterol Hepatol (2016) 13(3):122–3. doi: 10.1038/nrgastro.2016.19
14. Viennois E, Chassaing B, Tahsin A, Pujada A, Wang L, Gewirtz AT, et al. Host- derived fecal MicroRNAs can indicate gut microbiota healthiness and ability to induce inflammation. Theranostics (2019) 9(15):4542–57. doi: 10.7150/thno.35282
15. Bi K, Zhang X, Chen W, Diao H. MicroRNAs regulate intestinal immunity and gut microbiota for gastrointestinal health: A comprehensive review. Genes (2020) 11. doi: 10.3390/genes11091075
16. Singh N, Shirdel EA, Waldron L, Zhang R-H, Jurisica I, Comelli EM. The murine caecal MicroRNA signature depends on the presence of the endogenous microbiota. Int J Biol Sci (2012) 8(2):171–86. doi: 10.7150/ijbs.8.171
17. Xue X, Feng T, Yao S, Wolf KJ, Liu C-G, Liu X, et al. Microbiota downregulates dendritic cell expression of MiR-10a, which targets IL-12/IL-23p40. J Immunol Baltim. Md 1950 (2011) 187(11):5879–86. doi: 10.4049/jimmunol.1100535
18. Bi K, Zhang X, Chen W, Diao H. MicroRNAs regulate intestinal immunity and gut microbiota for gastrointestinal health: A comprehensive review. Genes (2020) 11(9). doi: 10.3390/genes11091075
19. Peck BCE, Mah AT, Pitman WA, Ding S, Lund PK, Sethupathy P. FunctionalTranscriptomics in diverse intestinal epithelial cell types reveals robust MicroRNA sensitivity in intestinal stem cells to microbial status. J Biol Chem (2017) 292(7):2586–600. doi: 10.1074/jbc.M116.770099
20. Hu S, Dong TS, Dalal SR, Wu F, Bissonnette M, Kwon JH, et al. The microbe-derived short chain fatty acid butyrate targets MiRNA-dependent P21 gene expression in human colon cancer. PloS One (2011) 6(1):e16221. doi: 10.1371/journal.pone.0016221
21. Li M, Chen W-D, Wang Y-D. The roles of the gut microbiota–MiRNA interaction in the host pathophysiology. Mol Med (2020) 26(1):101. doi: 10.1186/s10020-020-00234-7
22. Ji Y, Li X, Zhu Y, Li N, Zhang N, Niu M. Faecal MicroRNA as a biomarker of the activity and prognosis of inflammatory bowel diseases. Biochem Biophys Res Commun (2018) 503(4):2443–50. doi: 10.1016/j.bbrc.2018.06.174
23. Johnston DGW, Williams MA, Thaiss CA, Cabrera-Rubio R, Raverdeau M, McEntee C, et al. Loss of MicroRNA-21 influences the gut microbiota, causing reduced susceptibility in a murine model of colitis. J Crohns Colitis (2018) 12(7):835–48. doi: 10.1093/ecco-jcc/jjy038
24. Dalmasso G, Nguyen HTT, Charrier-Hisamuddin L, Yan Y, Laroui H, Demoulin B, et al. PepT1 mediates transport of the proinflammatory bacterial tripeptide l-Ala-γ-d-Glu-Meso-DAP in intestinal epithelial cells. Am J Physiol.- Gastrointest. Liver Physiol (2010) 299(3):G687–96.
25. Ayyadurai S, Charania MA, Xiao B, Viennois E, Merlin D. PepT1 expressed in immune cells has an important role in promoting the immune response during experimentally induced colitis. Lab Investig J Tech. Methods Pathol (2013) 93(8):888–99. doi: 10.1038/labinvest.2013.77
26. Rodríguez-Nogales A, Algieri F, Garrido-Mesa J, Vezza T, Utrilla MP, Chueca N, et al. Differential intestinal anti- inflammatory effects of lactobacillus fermentum and lactobacillus salivarius in DSS mouse colitis: Impact on MicroRNAs expression and microbiota composition. Mol Nutr Food Res (2017) 61(11). doi: 10.1002/mnfr.201700144
27. Rodríguez-Nogales A, Algieri F, Garrido-Mesa J, Vezza T, Utrilla MP, Chueca N, et al. Differential intestinal anti- inflammatory effects of lactobacillus fermentum and lactobacillus salivarius in DSS mouse colitis: Impact on MicroRNAs expression and microbiota composition. Mol Nutr Food Res (2017) 61(11):1700144.
28. Nosho K, Sukawa Y, Adachi Y, Ito M, Mitsuhashi K, Kurihara H, et al. Association of fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J Gastroenterol (2016) 22(2):557–66. doi: 10.3748/wjg.v22.i2.557
29. Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes (2014) 5(5):675–80. doi: 10.4161/19490976.2014.969989
30. Kumar M, Sahu SK, Kumar R, Subuddhi A, Maji RK, Jana K, et al. MicroRNA let-7 modulates the immune response to mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-κB pathway. Cell Host Microbe (2015) 17(3):345–56. doi: 10.1016/j.chom.2015.01.007
31. Naghavian R, Ghaedi K, Kiani-Esfahani A, Ganjalikhani-Hakemi M, Etemadifar M, Nasr-Esfahani MH. MiR-141 and MiR-200a, revelation of new possible players in modulation of Th17/Treg differentiation and pathogenesis of multiple sclerosis. PloS One (2015) 10(5):e0124555. doi: 10.1371/journal.pone.0124555
32. Seddiki N, Brezar V, Ruffin N, Lévy Y, Swaminathan S. Role of MiR-155 in the regulation of lymphocyte immune function and disease. Immunology (2014) 142(1):32–8. doi: 10.1111/imm.12227
33. Zhou H, Li J, Gao P, Wang Q, Zhang J. MiR-155: A novel target in allergic asthma. Int J Mol Sci (2016) 17(10). doi: 10.3390/ijms17101773
34. Honardoost MA, Naghavian R, Ahmadinejad F, Hosseini A, Ghaedi K. Integrative computational MRNA–MiRNA interaction analyses of the autoimmune-deregulated MiRNAs and well-known Th17 differentiation regulators: An attempt to discover new potential MiRNAs involved in Th17 differentiation. Gene (2015) 572(2):153–62. doi: 10.1016/j.gene.2015.08.043
35. Stanisavljević S, Lukić J, Momčilović M, Miljković M, Jevtić B, Kojić M, et al. Gut-associated lymphoid tissue, gut microbes and susceptibility to experimental autoimmune encephalomyelitis. Benef. Microbes (2016) 7(3):363–73. doi: 10.3920/BM2015.0159
36. Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients (2012) 4(8):1095–119. doi: 10.3390/nu4081095
37. Jiao Y, Wu L, Huntington ND, Zhang X. Crosstalk between gut microbiota and innate immunity and its implication in autoimmune diseases. Front Immunol (2020) 11:282. doi: 10.3389/fimmu.2020.00282
38. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux J.-, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of crohn disease patients. Proc Natl Acad Sci USA (2008) 105(43):16731–6. doi: 10.1073/pnas.0804812105
39. Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, et al. Genomic view of the human-bacteroides thetaiotaomicron symbiosis. Science (2003) 299(5615):2074–6. doi: 10.1126/science.1080029
40. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med (2010) 16(2):228–31. doi: 10.1038/nm.2087
41. Secondulfo M, Iafusco D, Carratù R, deMagistris L, Sapone A, Generoso M, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig. Liver Dis Off J Ital. Soc Gastroenterol Ital. Assoc Study Liver (2004) 36(1):35–45. doi: 10.1016/j.dld.2003.09.016
42. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia (2006) 49(12):2824–7. doi: 10.1007/s00125-006-0465-3
43. Singh P, Rawat A, Al-Jarrah B, Saraswathi S, Gad H, Elawad M, et al. Distinctive microbial signatures and gut-brain crosstalk in pediatric patients with coeliac disease and type 1 diabetes mellitus. Int J Mol Sci (2021) 22(4). doi: 10.3390/ijms22041511
44. Fuhri Snethlage CM, Nieuwdorp M, van Raalte DH, Rampanelli E, Verchere BC, Hanssen NMJ. Auto-immunity and the gut microbiome in type 1 diabetes: Lessons from rodent and human studies. Best Pract Res Clin Endocrinol Metab (2021) 35(3):101544. doi: 10.1016/j.beem.2021.101544
45. Baba N, Samson S, Bourdet-Sicard R, Rubio M, Sarfati M. Selected commensal-related bacteria and toll-like receptor 3 agonist combinatorial codes synergistically induce interleukin-12 production by dendritic cells to trigger a T helper type 1 polarizing programme. Immunology (2009) 128(1 Suppl):e523–531. doi: 10.1111/j.1365-2567.2008.03022.x
46. Zhang M, Zhou L, Wang Y, Dorfman RG, Tang D, Xu L, et al. Faecalibacterium prausnitzii produces butyrate to decrease c- myc-related metabolism and Th17 differentiation by inhibiting histone deacetylase 3. Int Immunol (2019) 31(8):499–514. doi: 10.1093/intimm/dxz022
47. Imhann F, Vich Vila A, Bonder MJ, Fu J, Gevers D, Visschedijk MC, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut (2018) 67(1):108–19. doi: 10.1136/gutjnl-2016-312135
48. de Groot PF, Belzer C, Aydin Ö., Levin E, Levels JH, Aalvink S, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PloS One (2017) 12(12):e0188475. doi: 10.1371/journal.pone.0188475
49. Rodríguez-Nogales A, Algieri F, Garrido-Mesa J, Vezza T, Utrilla MP, Chueca N, et al. Differential intestinal anti- inflammatory effects of lactobacillus fermentum and lactobacillus salivarius in DSS mouse colitis: Impact on MicroRNAs expression and microbiota composition. Mol Nutr Food Res (2017) 61(11):1700144. doi: 10.1002/mnfr.201700144
50. de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal microbiota transplantation halts progression of human new-onset type 1 diabetes in a randomised controlled trial. Gut (2021) 70(1):92–105. doi: 10.1136/gutjnl-2020-322630
51. Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia (2005) 48(8):1565–75. doi: 10.1007/s00125-005-1831-2
52. Hansen AK, Ling F, Kaas A, Funda DP, Farlov H, Buschard K. Diabetes preventive gluten-free diet decreases the number of caecal bacteria in non-obese diabetic mice. Diabetes Metab Res Rev (2006) 22(3):220–5. doi: 10.1002/dmrr.609
53. King C, Sarvetnick N. The incidence of type-1 diabetes in NOD mice is modulated by restricted flora not germ-free conditions. PloS One (2011) 6(2):e17049. doi: 10.1371/journal.pone.0017049
54. Endesfelder D, Engel M, Davis-Richardson AG, Ardissone AN, Achenbach P, Hummel S, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome (2016) 4:17. doi: 10.1186/s40168-016-0163-4
55. Marietta EV, Murray JA, Luckey DH, Jeraldo PR, Lamba A, Patel R, et al. Suppression of inflammatory arthritis by human gut-derived prevotella histicola in humanized mice. Arthritis Rheumatol Hoboken NJ (2016) 68(12):2878–88. doi: 10.1002/art.39785
56. López P, de Paz B, Rodríguez-Carrio J, Hevia A, Sánchez B, Margolles A, et al. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep (2016) 6(1):24072. doi: 10.1038/srep24072
57. Tierney BT, Tan Y, Kostic AD, Patel CJ. Gene-level metagenomic architectures across diseases yield high-resolution microbiome diagnostic indicators. Nat Commun (2021) 12(1):2907. doi: 10.1038/s41467-021-23029-8
58. Ivanov II, Frutos R, de L, Manel N, Yoshinaga K, Rifkin DB, et al. Specific microbiota direct the differentiation of IL-17-Producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe (2008) 4(4):337–49. doi: 10.1016/j.chom.2008.09.009
59. Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM, et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol (2014) 5:678. doi: 10.3389/fmicb.2014.00678
60. Bilgic S, Aktas E, Salman F, Ersahin G, Erten G, Yilmaz MT, et al. Intracytoplasmic cytokine levels and neutrophil functions in early clinical stage of type 1 diabetes. Diabetes Res Clin Pract (2008) 79(1):31–6. doi: 10.1016/j.diabres.2007.06.011
61. Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes (2013) 62(6):2072–7. doi: 10.2337/db12-1345
62. Flannigan KL, Denning TL. Segmented filamentous bacteria-induced immune responses: A balancing act between host protection and autoimmunity. Immunology (2018) 154(4):537–46. doi: 10.1111/imm.12950
63. Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal prevotella copri correlates with enhanced susceptibility to arthritis. eLife (2013) 2:e01202. doi: 10.7554/eLife.01202
64. Bernard NJ. Rheumatoid arthritis: Prevotella copri associated with new-onset untreated RA. Nat Rev Rheumatol (2014) 10(1):2. doi: 10.1038/nrrheum.2013.187
65. Moreno J. Prevotella copri and the microbial pathogenesis of rheumatoid arthritis. Reumatol. Clin (2015) 11(2):61–3. doi: 10.1016/j.reuma.2014.11.001
66. Kim D, Kim W-U. Editorial: Can prevotella copri be a causative pathobiont in rheumatoid arthritis? arthritis rheumatol. Hoboken NJ (2016) 68(11):2565–7. doi: 10.1002/art.39807
67. Larsen JM. The immune response to prevotella bacteria in chronic inflammatory disease. Immunology (2017) 151(4):363–74. doi: 10.1111/imm.12760
68. Iljazovic A, Roy U, Gálvez EJC, Lesker TR, Zhao B, Gronow A, et al. Perturbation of the gut microbiome by prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol (2021) 14(1):113–24. doi: 10.1038/s41385-020-0296-4
69. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (2018) 359(6380):1156–61. doi: 10.1126/science.aar7201
70. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA (2017) 114(40):10713–8. doi: 10.1073/pnas.1711235114
71. Schepici G, Silvestro S, Bramanti P, Mazzon E. The gut microbiota in multiple sclerosis: An overview of clinical trials. Cell Transplant. (2019) 28(12):1507–27. doi: 10.1177/0963689719873890
72. Vallino A, Dos Santos A, Mathé CV, Garcia A, Morille J, Dugast E, et al. Gut bacteria akkermansia elicits a specific IgG response in CSF of patients with MS. Neurol.-Neuroimmunol. Neuroinflamm (2020) 7(3):e688. doi: 10.1212/NXI.0000000000000688
73. Cekanaviciute E, Pröbstel A-K, Thomann A, Runia TF, Casaccia P, Katz Sand I, et al. Multiple sclerosis-associated changes in the composition and immune functions of spore-forming bacteria. mSystems (2018) 3(6):e00083-18. doi: 10.1128/mSystems.00083-18
74. Abdollahi-Roodsaz S, Joosten LAB, Koenders MI, Devesa I, Roelofs MF, Radstake TRDJ, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. (2008) 118(1):205–16. doi: 10.1172/JCI32639
75. Sorini C, Falcone M. Shaping the (Auto) immune response in the gut: The role of intestinal immune regulation in the prevention of type 1 diabetes. Am J Clin Exp Immunol (2013) 2(2):156–71.
76. De Filippis F, Paparo L, Nocerino R, Della Gatta G, Carucci L, Russo R, et al. Specific gut microbiome signatures and the associated pro- inflamatory functions are linked to pediatric allergy and acquisition of immune tolerance. Nat Commun (2021) 12(1):5958. doi: 10.1038/s41467-021-26266-z
77. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science (2004) 303(5664):1662–5. doi: 10.1126/science.1091334
78. Turley SJ, Lee J-W, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci USA (2005) 102(49):17729–33. doi: 10.1073/pnas.0509006102
79. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature (2009) 461(7268):1282–6. doi: 10.1038/nature08530
80. Blahnik G, Uchtenhagen H, Chow I-T, Speake C, Greenbaum C, Kwok WW, et al. Analysis of pancreatic beta cell specific CD4+ T cells reveals a predominance of proinsulin specific cells. Cell Immunol (2019) 335:68–75. doi: 10.1016/j.cellimm.2018.11.004
81. Rey FE, Gonzalez MD, Cheng J, Wu M, Ahern PP, Gordon JI. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc Natl Acad Sci USA (2013) 110(33):13582–7. doi: 10.1073/pnas.1312524110
82. Schicho R, Krueger D, Zeller F, Von Weyhern CWH, Frieling T, Kimura H, et al. Hydrogen sulfide is a novel prosecretory neuromodulator in the Guinea-pig and human colon. Gastroenterology (2006) 131(5):1542–52. doi: 10.1053/j.gastro.2006.08.035
83. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, et al. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity (2015) 43(2):251–63. doi: 10.1016/j.immuni.2015.07.017
84. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PloS One (2011) 6(10):e25792. doi: 10.1371/journal.pone.0025792
85. Westerholm-Ormio M, Vaarala O, Pihkala P, Ilonen J, Savilahti E. Immunologic activity in the small intestinal mucosa of pediatric patients with type 1 diabetes. Diabetes (2003) 52(9):2287–95. doi: 10.2337/diabetes.52.9.2287
86. Badami E, Sorini C, Coccia M, Usuelli V, Molteni L, Bolla AM, et al. Defective differentiation of regulatory FoxP3+ T cells by small-intestinal dendritic cells in patients with type 1 diabetes. Diabetes (2011) 60(8):2120–4. doi: 10.2337/db10-1201
87. Rouland M, Beaudoin L, Rouxel O, Bertrand L, Cagninacci L, Saffarian A, et al. Gut mucosa alterations and loss of segmented filamentous bacteria in type 1 diabetes are associated with inflammation rather than hyperglycaemia. Gut (2022) 71(2):296. doi: 10.1136/gutjnl-2020-323664
88. Kahalehili HM, Newman NK, Pennington JM, Kolluri SK, Kerkvliet NI, Shulzhenko N, et al. Dietary indole-3-Carbinol activates AhR in the gut, alters Th17-microbe interactions, and exacerbates insulitis in NOD mice. Front Immunol (2020) 11:606441. doi: 10.3389/fimmu.2020.606441
89. Roesch LFW, Lorca GL, Casella G, Giongo A, Naranjo A, Pionzio AM, et al. Culture- independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J (2009) 3(5):536–48. doi: 10.1038/ismej.2009.5
90. Brugman S, Klatter FA, Visser JTJ, Wildeboer-Veloo ACM, Harmsen HJM, Rozing J, et al. Antibiotic treatment partially protects against type 1 diabetes in the bio-breeding diabetes-prone rat. is the gut flora involved in the development of type 1 diabetes? Diabetologia (2006) 49(9):2105–8. doi: 10.1007/s00125-006-0334-0
91. Pozzilli P, Signore A, Williams AJ, Beales PE. NOD mouse colonies around the world–recent facts and figures. Immunol Today (1993) 14(5):193–6. doi: 10.1016/0167-5699(93)90160-M
92. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. Dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell (2016) 167(5):1339–1353.e21. doi: 10.1016/j.cell.2016.10.043
93. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia (2006) 49(12):2824–7. doi: 10.1007/s00125-006-0465-3
94. Ohata A, Usami M, Miyoshi M. Short-chain fatty acids alter tight junction permeability in intestinal monolayer cells via lipoxygenase activation. Nutr Burbank Los Angel. Cty. Calif (2005) 21(7–8):838–47. doi: 10.1016/j.nut.2004.12.004
95. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
96. Chang Pamela V, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci (2014) 111(6):2247–52. doi: 10.1073/pnas.1322269111
97. Zimmerman MA, Singh N, Martin PM, Thangaraju M, Ganapathy V, Waller JL, et al. Butyrate suppresses colonic inflammation through HDAC1-dependent fas upregulation and fas-mediated apoptosis of T cells. am. j. physiol. gastrointest. Liver Physiol (2012) 302(12):G1405–1415. doi: 10.1152/ajpgi.00543.2011
98. Chen L, Sun M, Wu W, Yang W, Huang X, Xiao Y, et al. Microbiota metabolite butyrate differentially regulates Th1 and Th17 cells’ differentiation and function in induction of colitis. Inflamm Bowel Dis (2019) 25(9):1450–61. doi: 10.1093/ibd/izz046
99. de Groot PF, Nikolic T, Imangaliyev S, Bekkering S, Duinkerken G, Keij FM, et al. Oral butyrate does not affect innate immunity and islet autoimmunity in individuals with longstanding type 1 diabetes: A randomised controlled trial. Diabetologia (2020) 63(3):597–610. doi: 10.1007/s00125-019-05073-8
100. Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, et al. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia (2004) 47(2):225–39. doi: 10.1007/s00125-003-1297-z
101. Ekman I, Vuorinen T, Knip M, Veijola R, Toppari J, Hyöty H, et al. Early childhood CMV infection may decelerate the progression to clinical type 1 diabetes. Pediatr Diabetes (2019) 20(1):73–7. doi: 10.1111/pedi.12788
102. Rogers MAM, Basu T, Kim C. Lower incidence rate of type 1 diabetes after receipt of the rotavirus vaccine in the united states, 2001-2017. Sci Rep (2019) 9(1):7727. doi: 10.1038/s41598-019-44193-4
103. Vehik K, Lynch KF, Wong MC, Tian X, Ross MC, Gibbs RA, et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat Med (2019) 25(12):1865–72. doi: 10.1038/s41591-019-0667-0
104. Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: Role of anti-self (Virus) immune response. Cell (1991) 65(2):319–31. doi: 10.1016/0092-8674(91)90165-u
105. Härkönen T, Paananen A, Lankinen H, Hovi T, Vaarala O, Roivainen M. Enterovirus infection may induce humoral immune response reacting with islet cell autoantigens in humans. J Med Virol (2003) 69(3):426–40. doi: 10.1002/jmv.10306
106. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvennick N. Diabetes induced by coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat Med (1998) 4(7):781–5. doi: 10.1038/nm0798-781
107. Flodström M, Maday A, Balakrishna D, Cleary MM, Yoshimura A, Sarvetnick N. Target cell defense prevents the development of diabetes after viral infection. Nat Immunol (2002) 3(4):373–82. doi: 10.1038/ni771
108. Pearson JA, Tai N, Ekanayake-Alper DK, Peng J, Hu Y, Hager K, et al. Norovirus changes susceptibility to type 1 diabetes by altering intestinal microbiota and immune cell functions. Front Immunol (2019) 10:2654. doi: 10.3389/fimmu.2019.02654
109. Zhao G, Vatanen T, Droit L, Park A, Kostic Aleksandar D, Poon Tiffany W, et al. Intestinal virome changes precede autoimmunity in type I diabetes- susceptible children. Proc Natl Acad Sci (2017) 114(30):E6166–75. doi: 10.1073/pnas.1706359114
110. Santiago-Rodriguez TM, Hollister EB. Human virome and disease: High-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses (2019) 11(7). doi: 10.3390/v11070656
v111. Sausset R, Petit MA, Gaboriau-Routhiau V, De Paepe M. New insights into intestinal phages. Mucosal Immunol (2020) 13(2):205–15. doi: 10.1038/s41385-019-0250-5
112. Oh J-H, Alexander LM, Pan M, Schueler KL, Keller MP, Attie AD, et al. Dietary fructose and microbiota-derived short-chain fatty acids promote bacteriophage production in the gut symbiont lactobacillus reuteri. Cell Host Microbe (2019) 25(2):273–284.e6. doi: 10.1016/j.chom.2018.11.016
113. Brouwer S, Barnett TC, Ly D, Kasper KJ, De Oliveira DMP, Rivera-Hernandez T, et al. Prophage exotoxins enhance colonization fitness in epidemic scarlet fever-causing streptococcus pyogenes. Nat Commun (2020) 11(1):5018. doi: 10.1038/s41467-020-18700-5
114. Hsu BB, Gibson TE, Yeliseyev V, Liu Q, Lyon L, Bry L, et al. Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe (2019) 25(6):803–814.e5. doi: 10.1016/j.chom.2019.05.001
115. Tetz G, Brown SM, Hao Y, Tetz V. Type 1 diabetes: An association between autoimmunity, the dynamics of gut amyloid-producing e. coli and their phages. Sci Rep (2019) 9(1):9685. doi: 10.1038/s41598-019-46087-x
116. Jamshidi P, Hasanzadeh S, Tahvildari A, Farsi Y, Arbabi M, Mota JF, et al. Is there any association between gut microbiota and type 1 diabetes? a systematic review. Gut Pathog (2019) 11(1):49. doi: 10.1186/s13099-019-0332-7
117. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: A case-control study. BMC Med (2013) 11:46. doi: 10.1186/1741-7015-11-46
118. Salamon D, Sroka-Oleksiak A, Kapusta P, Szopa M, Mrozińska S, Ludwig-Słomczyńska AH, et al. Characteristics of gut microbiota in adult patients with type 1 and type 2 diabetes based on next−generation sequencing of the 16S RRNA gene fragment. Pol Arch Intern Med (2018) 128(6):336–43. doi: 10.20452/pamw.4246
119. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med (2018) 378(2):169–80. doi: 10.1056/NEJMra1401483
120. Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol (2018) 14(10):577–89. doi: 10.1038/s41582-018-0058-z
121. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med (2018) 378(2):169–80. doi: 10.1056/NEJMra1401483
122. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung H-P, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med (2017) 376(3):221–34. doi: 10.1056/NEJMoa1601277
123. Fransen NL, Hsiao C-C, van der Poel M, Engelenburg HJ, Verdaasdonk K, Vincenten MCJ, et al. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain J Neurol (2020) 143(6):1714–30. doi: 10.1093/brain/awaa117
124. Dolei A. The aliens inside us: HERV-W endogenous retroviruses and multiple sclerosis. Mult. Scler. Houndmills Basingstoke Engl (2018) 24(1):42–7. doi: 10.1177/1352458517737370
125. Troci A, Zimmermann O, Esser D, Krampitz P, May S, Franke A, et al. B-Cell-Depletion reverses dysbiosis of the microbiome in multiple sclerosis patients. Sci Rep (2022) 12(1):3728. doi: 10.1038/s41598-022-07336-8
126. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun (2016) 7:12015. doi: 10.1038/ncomms12015
127. Budhram A, Parvathy S, Kremenchutzky M, Silverman M. Breaking down the gut microbiome composition in multiple sclerosis. Mult. Scler. Houndmills Basingstoke Engl (2017) 23(5):628–36. doi: 10.1177/1352458516682105
128. Mirza A, Forbes JD, Zhu F, Bernstein CN, Van Domselaar G, Graham M, et al. The multiple sclerosis gut microbiota: A systematic review. Mult. Scler. Relat Disord (2020) 37:101427. doi: 10.1016/j.msard.2019.101427
129. Tremlett H, Zhu F, Arnold D, Bar-Or A, Bernstein CN, Bonner C, et al. US Network of pediatric MS centers, the c. p. d. d. n. the gut microbiota in pediatric multiple sclerosis and demyelinating syndromes. Ann Clin Transl Neurol (2021) 8(12):2252–69. doi: 10.1002/acn3.51476
130. Katz Sand I, Zhu Y, Ntranos A, Clemente JC, Cekanaviciute E, Brandstadter R, et al. Disease-modifying therapies alter gut microbial composition in MS. Neurol Neuroimmunol Neuroinflamm (2018) 6(1):e517. doi: 10.1212/NXI.0000000000000517
131. Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, C. Cree BA, et al. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize clostridium ABC transporter. Ann Neurol (2012) 72(1):53–64. doi: 10.1002/ana.23651
132. Dujmovic I, Mader S, Schanda K, Deisenhammer F, Stojsavljevic N, Kostic J, et al. Temporal dynamics of cerebrospinal fluid anti- aquaporin-4 antibodies in patients with neuromyelitis optica spectrum disorders. J Neuroimmunol (2011) 234. doi: 10.1016/j.jneuroim.2011.01.007
133. Phuan P-W, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement- dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. J Biol Chem (2012) 287(17):13829–39. doi: 10.1074/jbc.M112.344325
134. Vaishnav RA, Liu R, Chapman J, Roberts AM, Ye H, Rebolledo-Mendez JD, et al. Aquaporin 4 molecular mimicry and implications for neuromyelitis optica. J Neuroimmunol (2013) 260(1–2):92–8. doi: 10.1016/j.jneuroim.2013.04.015
135. Liu S, Rezende RM, Moreira TG, Tankou SK, Cox LM, Wu M, et al. Oral administration of MiR-30d from feces of MS patients suppresses MS-like symptoms in mice by expanding akkermansia muciniphila. Cell Host Microbe (2019) 26(6):779–794.e8. doi: 10.1016/j.chom.2019.10.008
136. Pröbstel A-K, Zhou X, Baumann R, Wischnewski S, Kutza M, Rojas OL, et al. Gut microbiota-specific IgA(+) b cells traffic to the CNS in active multiple sclerosis. Sci Immunol (2020) 5(53). doi: 10.1126/sciimmunol.abc7191
137. Mangalam A, Shahi SK, Luckey D, Karau M, Marietta E, Luo N, et al. Human gut-derived commensal bacteria suppress CNS inflammatory and demyelinating disease. Cell Rep (2017) 20(6):1269–77. doi: 10.1016/j.celrep.2017.07.031
138. Mangalam A, Luckey D, Basal E, Jackson M, Smart M, Rodriguez M, et al. HLA-DQ8 (DQB1*0302)-restricted Th17 cells exacerbate experimental autoimmune encephalomyelitis in HLA-DR3-Transgenic mice. J Immunol Baltim. Md 1950 (2009) 182(8):5131–9. doi: 10.4049/jimmunol.0803918
139. Teitelbaum D, Aharoni R, Arnon R, Sela M. Specific inhibition of the T-cell response to myelin basic protein by the synthetic copolymer cop 1. Proc Natl Acad Sci USA (1988) 85(24):9724–8. doi: 10.1073/pnas.85.24.9724
140. Shahi SK, Freedman SN, Murra AC, Zarei K, Sompallae R, Gibson-Corley KN, et al. Is as potent as COPAXONE® in an animal model of multiple sclerosis. Front Immunol (2019) 10:462. doi: 10.3389/fimmu.2019.00462
141. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA (2017) 114(40):10719–24. doi: 10.1073/pnas.1711233114
142. Jiang J, Chu C, Wu C, Wang C, Zhang C, Li T, et al. Efficacy of probiotics in multiple sclerosis: A systematic review of preclinical trials and meta-analysis of randomized controlled trials. Food Funct (2021) 12(6):2354–77. doi: 10.1039/d0fo03203d
143. Hosang L, Canals RC, van der Flier FJ, Hollensteiner J, Daniel R, Flügel A, et al. The lung microbiome regulates brain autoimmunity. Nature (2022) 603(7899):138–44. doi: 10.1038/s41586-022-04427-4
144. Yaron JR, Ambadapadi S, Zhang L, Chavan RN, Tibbetts SA, Keinan S, et al. Immune protection is dependent on the gut microbiome in a lethal mouse gammaherpesviral infection. Sci Rep (2020) 10(1):2371. doi: 10.1038/s41598-020-59269-9
145. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science (2022) 375(6578):296–301. doi: 10.1126/science.abj8222
146. Fugl A, Andersen CL. Epstein-Barr Virus and its association with disease - a review of relevance to general practice. BMC Fam. Pract (2019) 20(1):62–2. doi: 10.1186/s12875-019-0954-3
147. Kohashi O, Kuwata J, Umehara K, Uemura F, Takahashi T, Ozawa A. Susceptibility to adjuvant-induced arthritis among germfree, specific-Pathogen-Free, and conventional rats. Infect Immun (1979) 26(3):791–4. doi: 10.1128/iai.26.3.791-794.1979
148. Kohashi O, Kohashi Y, Takahashi T, Ozawa A, Shigematsu N. Suppressive effect of escherichia coli on adjuvant-induced arthritis in germ-free rats. Arthritis Rheumatol (1986) 29(4):547–53. doi: 10.1002/art.1780290413
149. Huang Y, Tang J, Cai Z, Zhou K, Chang L, Bai Y, et al. Prevotella induces the production of Th17 cells in the colon of mice. J Immunol Res (2020) 2020:9607328. doi: 10.1155/2020/9607328
150. Taneja V. Arthritis susceptibility and the gut microbiome. FEBS Lett (2014) 588(22):1060 4244–4249. doi: 10.1016/j.febslet.2014.05.034
151. Shahi SK, Jensen SN, Murra AC, Tang N, Guo H, Gibson-Corley KN, et al. Human commensal prevotella histicola ameliorates disease as effectively as interferon-beta in the experimental autoimmune encephalomyelitis. Front Immunol (2020) 11:578648. doi: 10.3389/fimmu.2020.578648
152. Sanz I, Lee F. E.-H. B. Cells as therapeutic targets in SLE. Nat Rev Rheumatol (2010) 6(6):326–37. doi: 10.1038/nrrheum.2010.68
153. Lewis JE, Fu SM, Gaskin F. Autoimmunity, end organ damage, and the origin of autoantibodies and autoreactive T cells in systemic lupus erythematosus. Discov Med (2013) 15(81):85–92.
154. Jog NR, James JA. Epstein Barr Virus and autoimmune responses in systemic lupus erythematosus. Front Immunol (2021) 11.
155. Hagberg N, Lundtoft C, Rönnblom L. Immunogenetics in systemic lupus erythematosus: Transitioning from genetic associations to cellular effects. Scand J Immunol (2020) 92(4):e12894. doi: 10.1111/sji.12894
156. Alunno A, Bartoloni E, Bistoni O, Nocentini G, Ronchetti S, Caterbi S, et al. Balance between regulatory T and Th17 cells in systemic lupus erythematosus: The old and the new. Clin Dev Immunol (2012) 2012:823085. doi: 10.1155/2012/823085
157. Qu N, Xu M, Mizoguchi I, Furusawa J, Kaneko K, Watanabe K, et al. Pivotal roles of T-helper 17-related cytokines, IL-17, IL-22, and IL-23, in inflammatory diseases. Clin Dev Immunol (2013) 2013:968549. doi: 10.1155/2013/968549
158. Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol Baltim Md 1950 (2008) 181(12):8761–6. doi: 10.4049/jimmunol.181.12.8761
159. Li Y, Wang H-F, Li X, Li H-X, Zhang Q, Zhou H-W, et al. Disordered intestinal microbes are associated with the activity of systemic lupus erythematosus. Clin Sci Lond Engl 1979 (2019) 133(7):821–38. doi: 10.1042/CS20180841
160. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol (2018) 84(4). doi: 10.1128/AEM.02288-17
161. Chen B, Jia X-M, Xu J-Y, Zhao L-D, Ji J-Y, Wu B-X, et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol Hoboken NJ (2021) 73(2):232–43. doi: 10.1002/art.41511
162. Zhang H, Liao X, Sparks JB, Luo XM. Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol (2014) 80(24):7551–60. doi: 10.1128/AEM.02676-14
163. Rodríguez-Carrio J, López P, Sánchez B, González S, Gueimonde M, Margolles A, et al. Intestinal dysbiosis is associated with altered short- chain fatty acids and serum-free fatty acids in systemic lupus erythematosus. Front Immunol (2017) 8:23. doi: 10.3389/fimmu.2017.00023
164. McHugh J. Systemic lupus erythematosus: Escape of gut microbe to the liver drives autoimmunity. Nat Rev Rheumatol (2018) 14(5):247. doi: 10.1038/nrrheum.2018.53
165. Moon UY, Park SJ, Oh ST, Kim W-U, Park S-H, Lee S-H, et al. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Res Ther (2004) 6(4):R295–302. doi: 10.1186/ar1181
166. Gross AJ, Hochberg D, Rand WM, Thorley-Lawson DA. EBV and systemic lupus erythematosus: A new perspective. J Immunol Baltim. Md 1950 (2005) 174(11):6599–6607. doi: 10.4049/jimmunol.174.11.6599
167. James JA, Harley JB, Scofield RH. Epstein-Barr Virus and systemic lupus erythematosus. Curr Opin Rheumatol (2006) 18(5):462–7. doi: 10.1097/01.bor.0000240355.37927.94
168. James JA, Harley JB, Scofield RH. Epstein-Barr Virus and systemic lupus erythematosus. Curr Opin Rheumatol (2006) 18(5):462–7. doi: 10.1097/01.bor.0000240355.37927.94
169. McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med (2005) 11(1):85–9. doi: 10.1038/nm1167
170. James JA, Gross T, Scofield RH, Harley JB. Immunoglobulin epitope spreading and autoimmune disease after peptide immunization: Sm B/B’-derived PPPGMRPP and PPPGIRGP induce spliceosome autoimmunity. J Exp Med (1995) 181(2):453–61. doi: 10.1084/jem.181.2.453
171. Sundar K, Jacques S, Gottlieb P, Villars R, Benito M-E, Taylor DK, et al. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-DsDNA and anti-Sm antibodies. J Autoimmun (2004) 23(2):127–40. doi: 10.1016/j.jaut.2004.06.001
172. Papesch M, Watkins R. Epstein-Barr Virus infectious mononucleosis. Clin Otolaryngol Allied Sci (2001) 26(1):3–8. doi: 10.1046/j.1365-2273.2001.00431.x
173. James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest (1997) 100(12):3019–26. doi: 10.1172/JCI119856
174. Loh W, Tang MLK. The epidemiology of food allergy in the global context. int. j. environ. res. public. Health (2018) 15(9). doi: 10.3390/ijerph15092043
175. Berni Canani R, Paparo L, Nocerino R, Di Scala C, Della Gatta G, Maddalena Y, et al. Gut microbiome as target for innovative strategies against food allergy. Front Immunol (2019) 10.
176. Markle JGM, Frank DN, Adeli K, von Bergen M, Danska JS. Microbiome manipulation modifies sex-specific risk for autoimmunity. Gut Microbes (2014) 5(4):485–93. doi: 10.4161/gmic.29795
177. Markle JGM, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle- Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339(6123):1084–8. doi: 10.1126/science.1233521
178. Leonardi I, Gao IH, Lin W-Y, Allen M, Li XV, Fiers WD, et al. Mucosal fungi promote gut barrier function and social behavior via type 17 immunity. Cell (2022) 185(5):831–46.e14. doi: 10.1016/j.cell.2022.01.017
179. Altindis E, Cai W, Sakaguchi M, Zhang F, GuoXiao W, Liu F, et al. Viral insulin-like peptides activate human insulin and IGF-1 receptor signaling: A paradigm shift for host–microbe interactions. Proc Natl Acad Sci (2018) 115(10):2461. doi: 10.1073/pnas.1721117115
180. Valverde EJ, Borrego JJ, Sarasquete MC, Ortiz-Delgado JB, Castro D. Target organs for lymphocystis disease virus replication in gilthead seabream (Sparus aurata). Vet Res (2017) 48(1):21. doi: 10.1186/s13567-017-0428-3
181. Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell (2015) 160(3):447–60. doi: 10.1016/j.cell.2015.01.002
182. Kramná L, Kolářová K, Oikarinen S, Pursiheimo J-P, Ilonen J, Simell O, et al. Gut virome sequencing in children with early islet autoimmunity. Diabetes Care (2015) 38(5):930–3. doi: 10.2337/dc14-2490
183. Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell (2012) 149(7):1578–93. doi: 10.1016/j.cell.2012.04.037
184. Iweala OI, Nagler CR. The microbiome and food allergy. Annu Rev Immunol (2019) 37:377–403. doi: 10.1146/annurev-immunol-042718-041621
185. Zimmermann P, Messina N, Mohn WW, Finlay BB, Curtis N. Association between the intestinal microbiota and allergic sensitization, eczema, and asthma: A systematic review. J Allergy Clin Immunol (2019) 143(2):467–85. doi: 10.1016/j.jaci.2018.09.025
Keywords: microbiome, autoimmune diseases, dysbiosis, T1D, MS, SLE, arthrities
Citation: Mousa WK, Chehadeh F and Husband S (2022) Microbial dysbiosis in the gut drives systemic autoimmune diseases. Front. Immunol. 13:906258. doi: 10.3389/fimmu.2022.906258
Received: 28 March 2022; Accepted: 20 September 2022;
Published: 20 October 2022.
Edited by:
Allen D. Smith, Agricultural Research Service (USDA), United StatesReviewed by:
Javier Ochoa-Repáraz, Boise State University, United StatesRajesh Pandey, CSIR-Institute of Genomics and Integrative Biology (CSIR-IGIB), India
Copyright © 2022 Mousa, Chehadeh and Husband. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walaa K. Mousa, V2FsYWEubW91c2FAYWF1LmFjLmFl
†ORCID: Walaa K. Mousa, orcid.org/0000-0003-3229-4499