 Yuqing Liu1,2
Yuqing Liu1,2 Xin Cheng
Xin Cheng Shan Hui
Shan Hui Zheyu Zhang
Zheyu Zhang Yang Xiao
Yang Xiao Weijun Peng
Weijun Peng- 1Department of Integrated Traditional Chinese and Western Medicine, The Second Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Mental Disorder, Changsha, China
- 3Department of Geratology, Hunan Provincial People’s Hospital, The First Affiliated Hospital of Hunan Normal University, Changsha, China
- 4National Clinical Research Center for Metabolic Diseases, Key Laboratory of Diabetes Immunology, Ministry of Education, Changsha, China
- 5Department of Metabolism and Endocrinology, The Second Xiangya Hospital, Central South University, Changsha, China
Alzheimer’s disease (AD) is one of the most common causes of dementia. Although significant breakthroughs have been made in understanding the progression and pathogenesis of AD, it remains a worldwide problem and a significant public health burden. Thus, more efficient diagnostic and therapeutic strategies are urgently required. The latest research studies have revealed that neuroinflammation is crucial in the pathogenesis of AD. Non-coding RNAs (ncRNAs), including long noncoding RNAs (lncRNAs), microRNAs (miRNAs), circular RNAs (circRNAs), PIWI-interacting RNAs (piRNAs), and transfer RNA-derived small RNAs (tsRNAs), have been strongly associated with AD-induced neuroinflammation. Furthermore, several ongoing pre-clinical studies are currently investigating ncRNA as disease biomarkers and therapeutic interventions to provide new perspectives for AD diagnosis and treatment. In this review, the role of different types of ncRNAs in neuroinflammation during AD are summarized in order to improve our understanding of AD etiology and aid in the translation of basic research into clinical practice.
1 Introduction
Alzheimer’s disease (AD) is a degenerative disease characterized by progressive deterioration of memory and cognitive function, leading to loss of autonomy and accounting for at least two-thirds of dementia cases in patients aged ≥65 years (1, 2). More than 3 out of every 10 elderly people aged >85 years suffer from AD worldwide (3). Age and gender both influence AD occurrence, with approximately two-thirds of AD patients being women (4).Gonadal hormones, environment, society, and culture have different effects on women and men, and there is growing evidence that gender influences the cause, presentation, and treatment outcomes of many diseases. At present, there are about 50 million AD patients in the world, with about one-third of these cases occurring in China. However, research on the mechanisms driving the pathogenesis of AD is far behind expectations.
Several hypotheses have been proposed to explain the actual pathological development of AD (5). One of the earliest of these was the cholinergic hypothesis. Characteristic symptoms, such as cognitive impairment, were initially thought to be caused by cholinergic deficiency due to cholinergic neuron degeneration. Schumacher et al. (6) found that patients with mild cognitive impairment exhibited degeneration of the Meynert basal cholinergic nucleus accompanied by an early reduction in the integrity of white matter projections originating in this structure. This degeneration of the Meynert’s basal cholinergic nucleus was positively correlated with cognitive impairment. With advancing research, two proteins deposited in the brain—beta-amyloid (Aβ) and hyperphosphorylated tau protein—were identified and are considered to play crucial roles in the development of AD (7). Aβ is abnormally deposited in the brain of AD patients, forming plaques that attenuate neuronal function. Cleavage of the transmembrane amyloid precursor protein (APP) produces Aβ (8). Senile plaques are formed when Aβ is deposited outside of the cell (9, 10) (Figure 1). Aβ is a neurotoxic protein that plays a key role in neuronal hypofunction and the progression of AD. Hyperphosphorylated tau protein aggregates to form insoluble fibrous tangles that block synaptic transmission (11). The progressive development of neuritis plaques is a crucial driving factor of AD.
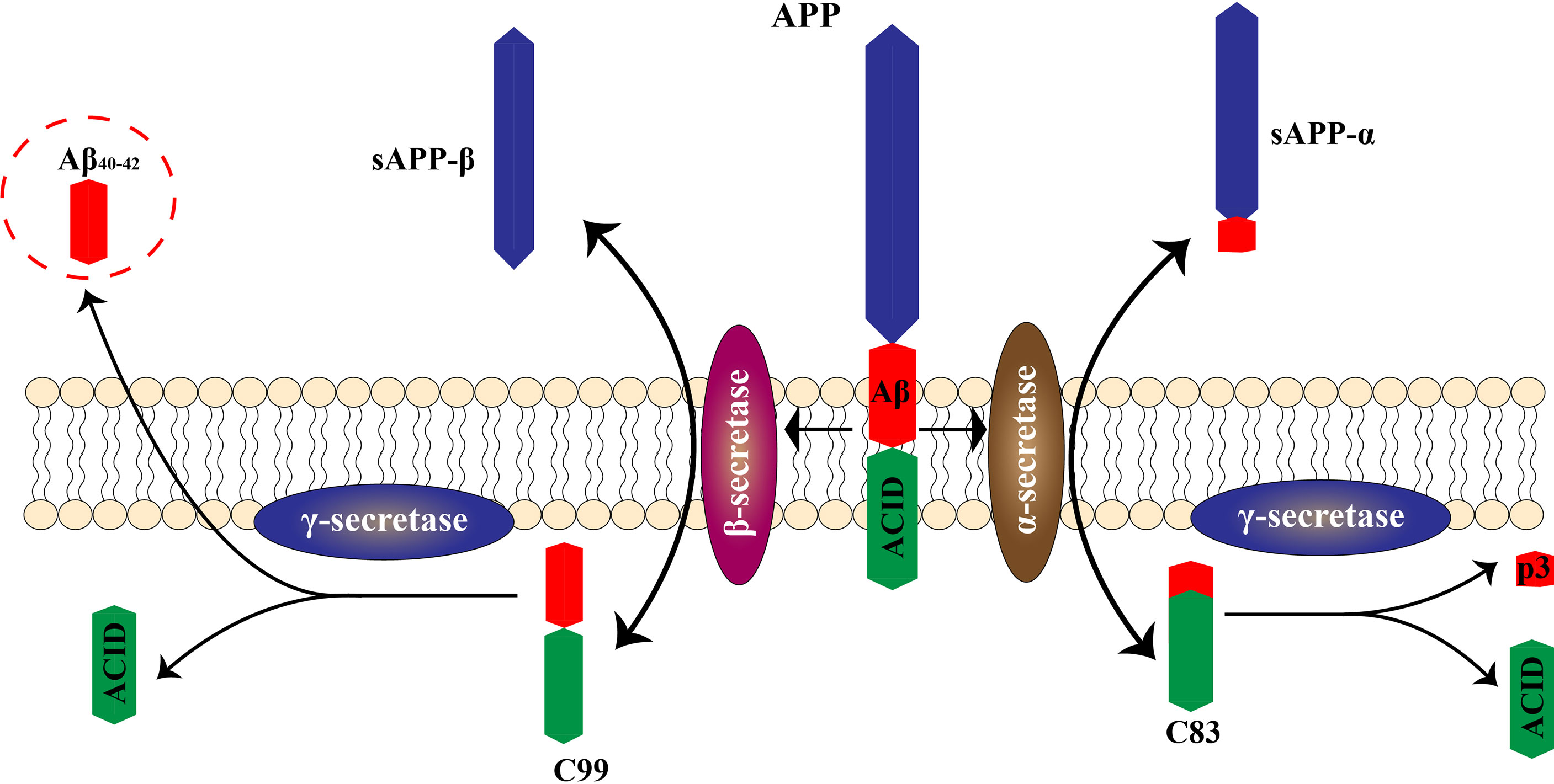
Figure 1 Pathological and physiological APP metabolism. APP is cleaved by two enzymes, α secretase and β secretase, resulting in different outcomes. α secretase excises the amyloid region, preventing Aβ production. The co-produced C83 protein and sAPPα enter the cytoplasm without causing neurotoxicity. The β-secretase cleavage site bypasses the amyloid region, producing sAPPβ and C99. Further processing of C99 leads to production of intracellular binding domain of amyloid precursor protein (AICD) fragments and Aβ. Aβ molecules bind each other to form oligomers, eventually forming fibers that are neurotoxic.
More recently, neuroinflammation has emerged as another key cause of AD. Many studies have tentatively shown a persistent inflammatory response within the brain of AD patients (12). Aβ may act as a neurotoxic and inflammation-related protein that activates microglia and astrocytes and promotes the inflammatory response (13, 14). In addition, glial proliferation has been found clustered around pathological neurofibrillary tangles. Furthermore, microglia release inflammatory cytokines in response to Aβ (15–17). Thus, the occurrence and progression of AD may be due to persistent inflammation in the brain that leads to nerve damage and neuronal death (18, 19). Studies have shown that the neuroinflammatory response plays a dual role in the pathological development of AD (20). On one hand, inflammation promotes Aβ phagocytosis by microglia to reduce Aβ deposition. On the other hand, excessive inflammation may cause irreversible damage to nerve tissue (3). This dual effect may be an important manifestation of Aβ neurotoxicity and may contribute to the development and progression of AD.
2 Role of Neuroinflammation in the Course of AD
Neuroinflammation is regarded as an inflammatory response occurring in the central nervous system that is triggered by ischemia, hypoxia, shock, or other injuries. Inflammation in nerve tissue appears to play a dual role in the development of AD, counteracting neurotoxic effects in the initial stages of the acute response and becoming harmful in later stages that are characterized by a persistent inflammatory response (21). Activated microglia and astrocytes are central to neuroinflammation (Figure 2). In AD, it has been hypothesized that the presence of Aβ is the main driver of microglial activation. Moreover, activated microglia respond to Aβ, and numerous studies have shown that activated microglia engulf Aβ (20). This increased immune response leads to Aβ clearance early in the onset of AD (22). However, continued immune response activation or overactivation leads to reduced microglia binding and phagocytosis of Aβ, as well as reduced microglia antibody-degrading enzyme activity, resulting in decreased phagocytosis of Aβ plaques. Although some microglia functions are inhibited during continued activation or overactivation, the ability of microglia to produce pro-inflammatory cytokines is unaffected, leading to neurodegeneration and additional microglia activation (3, 12).
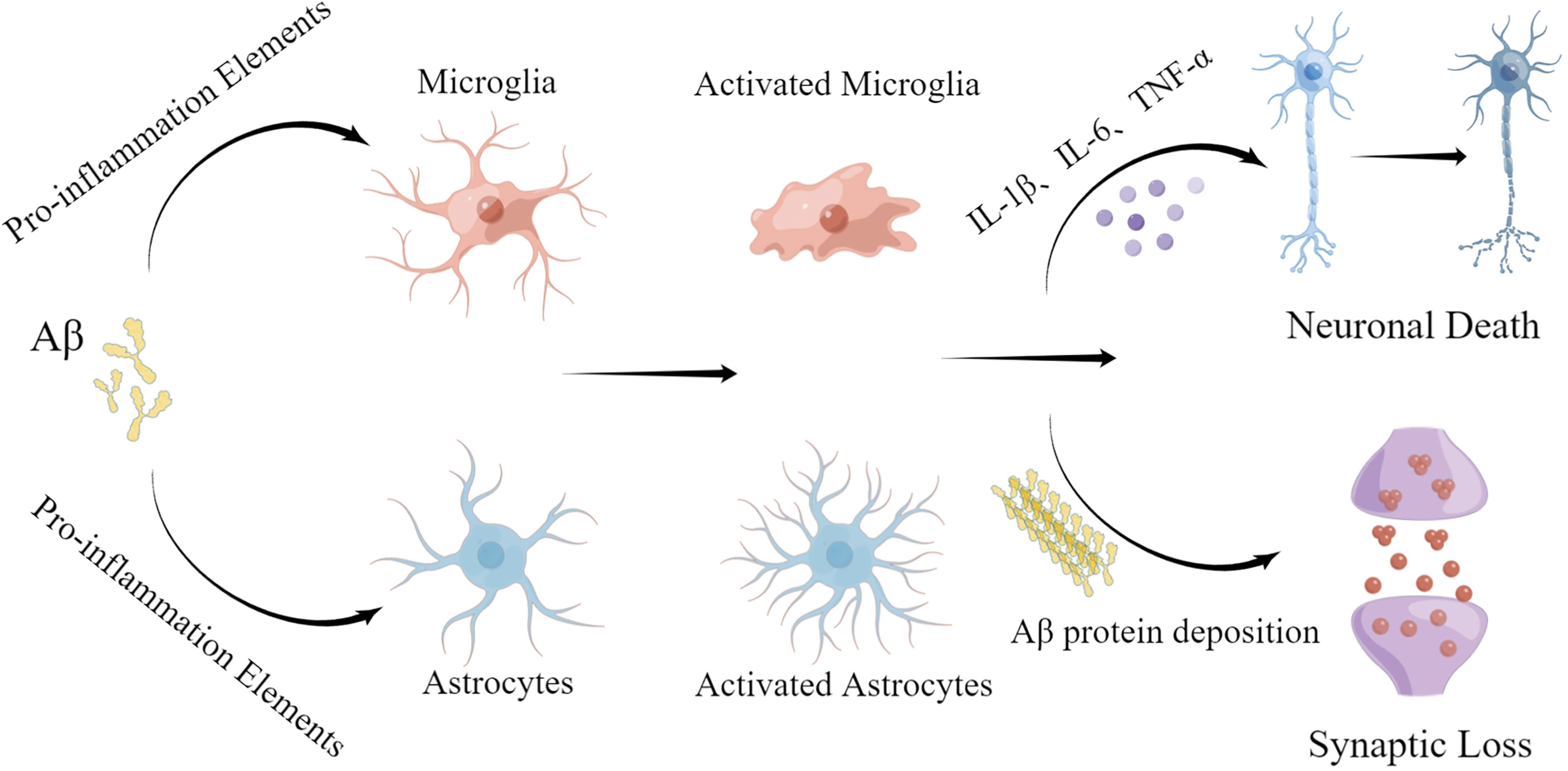
Figure 2 Roles of microglia and astrocytes in AD. The neurotoxic effects of Aβ protein promote activation of microglia and astrocytes, leading to a neuroinflammatory response. Excessive activation promotes deposition of Aβ protein, leading to further nerve damage.
Microglia and astrocytes mainly release interleukin 1 beta (IL-1β), interleukin 6 (IL-6), tumor necrosis factor (TNF), nitric oxide (NO), and reactive oxygen species (23). Damaged capillary endothelial cells and invading blood cells may also promote neuroinflammation, especially when the blood-brain barrier (BBB) is continuously damaged. When the BBB is disrupted, pro-inflammatory molecules are more prone to contact nerve tissues, causing synaptic damage and neuronal death (24, 25). TNF binds to the tumor necrosis factor receptor 1 (TNFR1), leading to caspase 8 aggregation and neuronal death (26). The complement system is also activated as part of the inflammatory response. Once activated, it promotes phagocytosis by microglia. Thus, its overactivation may lead to inappropriate synaptic pruning. In neuroinflammatory responses, anti-inflammatory factors such as IL-1 receptor antagonists, interleukin 4 (IL-4), interleukin 10 (IL-10), and interleukin 11 (IL-11) are also produced, possibly as part of a protective mechanism against neuroinflammatory overactivation (27). In conclusion, in neurodegenerative diseases, neuroinflammation is often a persistent process that causes irreversible damage and is considered to be an important driver of the disease state.
A 2019 meta-analysis (28) of 170 studies that measured peripheral or cerebrospinal fluid inflammatory markers combined results from 9,842 AD patients, 3,526 mild cognitive impairment (MCI) patients, and 9,002 control patients and found statistically significant changes in both blood and cerebrospinal fluid inflammatory markers in AD patients. These results indicate the presence of peripheral and neuroinflammatory responses in patients with AD. Studies have shown that peripheral and cerebrospinal fluid inflammation may mediate early cognitive impairment in neurodegeneration. Possible pathogenic processes that contribute to the progressive development of AD include inhibition of angiogenesis and neurotrophic and neuroprotective mechanisms, induction of neuron loss, and activation of myelin sheath injury (29). Although inflammation has a protective effect on the body, excessive inflammation may lead to or contribute to tissue damage and disease pathology. Studies have found that the occurrence, transmission, and resolution of inflammatory responses in central nervous system diseases depend on soluble factors including cytokines and various ncRNAs. Microglia, for example, participate in the inflammatory reaction by releasing vesicles enriched in IL-1β and miR-155 or TNF-α and IL-6 in response to lipopolysaccharides (LPS) stimulation. In addition, miR-146a is secreted by hippocampal-located exosomes in AD mouse and human brains and binds to the toll-like receptor (TLR) to activate the pro-inflammatory response and cytokine release, and this response is related to disease severity (30). Most studies believe that ncRNA plays a dual role in AD. Maintaining the balance between the beneficial and harmful effects of exosomes on neuropathological progression and the ability to exogenously alter this balance for the treatment of AD remains to be determined (31–33).
3 Role of Non-Coding RNAS in AD Neuroinflammation
ncRNAs are a widely diversified family of non-protein-coding transcripts found in various tissues that comprise at least 98% of the total genome, based on human transcriptome sequencing (34). ncRNAs regulate heredity, epigenetic inheritance and translation (35), and can be divided into long non-coding RNA (lncRNA, >200 nt), small ncRNA (sncRNA, < 200 nt), and circRNA. Moreover, sncRNA can be further classified as microRNA (miRNA), piwi-associated small RNA (piRNA), or transfer RNA-derived small RNA (tsRNA), among others. ncRNA networks regulate a variety of physiological processes, such as gene transcription, mRNA translation, and protein modification through their unique structural and functional roles (36). Pathological neuroinflammation underlies many central nervous system diseases, including AD (20). Determining the specific mechanisms that initiate and maintain neuroinflammation will undoubtedly help slow the progression of AD and allow development of new treatment strategies. ncRNA is one of the important molecules that regulate neuroinflammatory signals (37). In fact, specific ncRNAs may be common to many diseases in which they play a crucial role in controlling inflammation (38). While some ncRNAs may be disease-specific, others exert cumulative effects by interacting with multiple ncRNAs to influence neuroinflammation during disease. In neuroinflammatory diseases, ncRNAs also circulate in biological fluids and exhibit specific patterns of altered expression. Identifying the specific mechanisms of ncRNA action in these diseases, including AD, may allow for the development and research of their diagnostic and therapeutic applications.
3.1 MicroRNAs (MiRNAs) in AD
miRNAs are small single-stranded ncRNAs, ranging from 19–25 nucleotides long, that post-transcriptionally regulate gene expression in plants and animals by binding to the 3’ untranslated region (3’UTR) of their mRNA target (39). Data suggest that miRNAs contribute to synaptic plasticity, neuronal differentiation, and development. In addition, >50% of mRNAs are predicted to host miRNAs, enabling them to regulate most biological processes, including neurogenesis, apoptosis, inflammation, and oxidative stress (40). Studies have found that miRNA is specifically expressed in immune cells, where it may regulate the activation and function of these immune cells. Later, it was found that miR-155, miR-146, and miR-223 can bind TLRs to regulate the acute inflammatory response (41). There is evidence that miRNAs play a central role in regulating AD progression. Furthermore, many miRNAs target several genes related to post-AD inflammation. Transcription of several important miRNAs involved in neuroinflammation is regulated by nuclear factor-kappa B (NF-кB). NF-кB is an immune- and stress-induced transcription factor (42). Acetylcholine blocks inflammation-induced NF-кB activation under physiological conditions, which is an important pathway in miRNA-related neuroinflammatory regulation (43, 44).
3.1.1 MiR-155
miRNA-155 is a multifunctional miRNA with a unique expression profile that is related to hematopoietic, inflammatory, and immune processes (45). Clinical trials have shown that miR-155 regulation may be efficacious in treating diseases such as cancer (46). Experiments using target site blockers and RNA immunoprecipitation have shown that miR-155 binds to peptidylarginine deiminase type4 (PAD4) to regulate PAD4 transcription. Targeting miR-155 may help inhibit overproduction of neutrophil extracellular traps (NETs) in inflammatory diseases (47). In central neurodegenerative diseases, pro-inflammatory miR-155 overexpression reduces Aβ40-42 catabolism (48) (Table 1). In the mouse brain, miR-155 alters the BBB permeability in neuroinflammatory disorders in the central nervous system by regulating cell-interacting molecules. Moreover, miR-155 is involved in T cell immune function and increases formation of inflammatory cytokines, such as IL-6 and interferon-β (IFN-β) (49) (Figure 3). Further studies involving the regulation of miR-155 levels may promote innovative, effective therapies for AD.
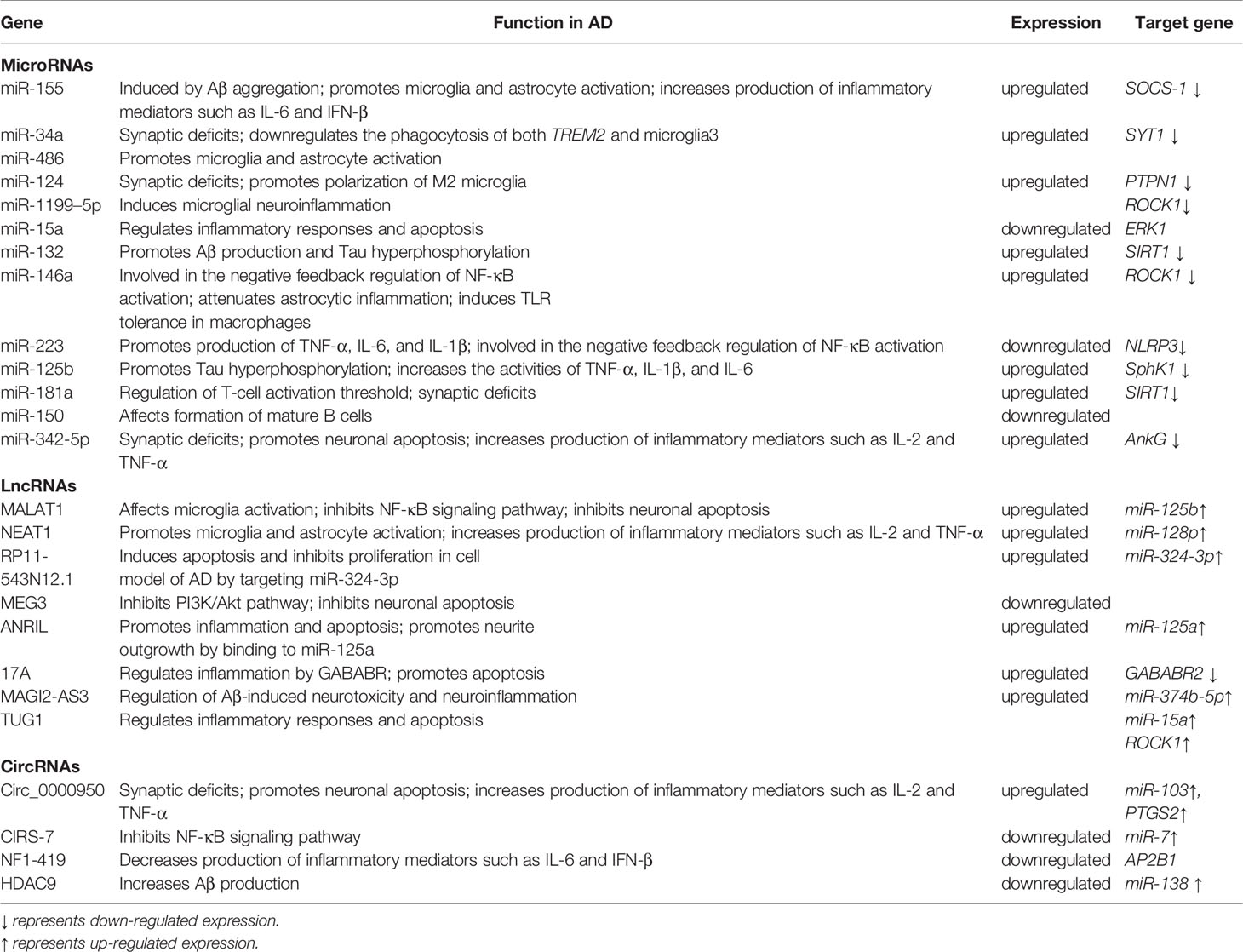
Table 1 The main inflammatory ncRNAs for AD.
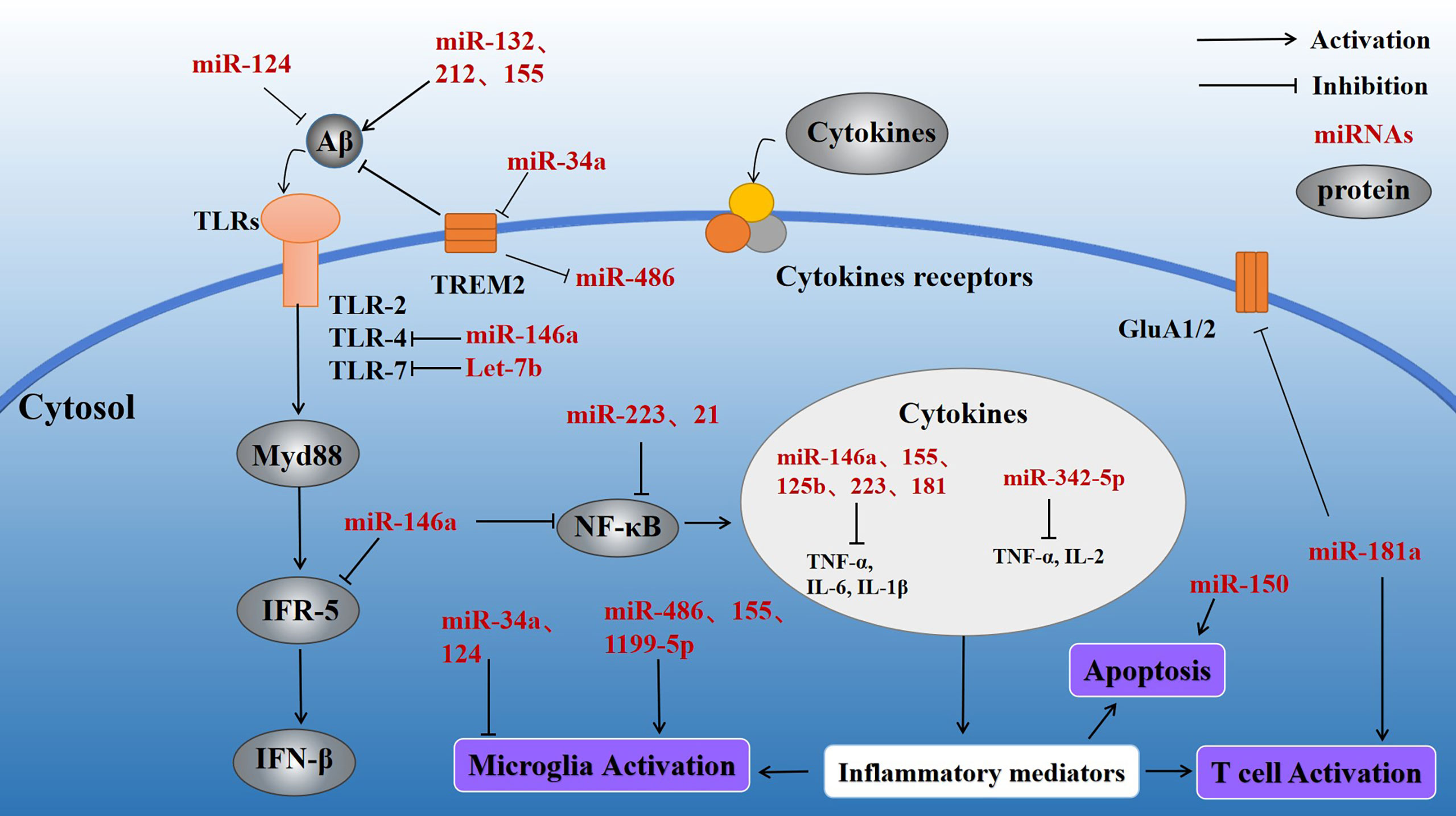
Figure 3 Effect of inflammatory miRNAs on AD inflammation. Multiple inflammatory miRNAs may play a synergistic or antagonistic role in different inflammatory pathways. For example, mir-146a targets the MyD88-related pathway by binding to TLR-4. At the same time, NF-κB is inhibited, preventing release of inflammatory factors. Meanwhile, miR-155 and miR-181a regulate a variety of inflammatory mediators. Microglial activation is important in various inflammatory processes and is stimulated by miR-34a and miR-486. Apoptosis, as the terminal link in the inflammatory response, also plays a significant part in the progression of AD and is regulated by miR-150.
3.1.2 MiR-34a
miR-34a is a tumor-suppressing transcript whose deletion has been associated with a variety of human cancers, including brain malignancies (50). Interestingly, miR-34a is highly expressed in the adult mammalian brain and may be closely related to neuronal survival. miR-34a is primarily involved in the TP53 tumor suppressor network. miR-34a transcriptional activation is promoted by TP53 and may lead to cell cycle arrest and apoptosis (51). Meanwhile, in the brain of AD patients, miR-34a targets several genes that maintain normal physiological activity of neural tissues, such as genes related to synaptic plasticity, nerve repair, and memory (52). miR-34a is upregulated in the brain of AD patients (Table 1), and bioinformatics/transfected luciferase reporter assays show that miRNA-34a targets 299 nucleotides in the trem2-mRNA-3’UTR, leading to downregulation of triggering receptor expressed on myeloid cells 2 (TREM2). TREM2 is a transmembrane sensing receptor that plays a significant role in Aβ clearance. Aβ42 activates microglia to enhance Aβ phagocytosis, whereas miR-34a inhibits TREM2 activation of microglial phagocytosis (53) (Figure 3), and bioinformatics/transfected luciferase reporter assays show that miRNA-34a targets 299 nucleotides in the trem2-mRNA-3’UTR, leading to downregulation of triggering receptor expressed on myeloid cells 2 (TREM2). TREM2 is a transmembrane sensing receptor that plays a significant role in Aβ clearance. Aβ42 activates microglia to enhance Aβ phagocytosis, whereas miR-34a inhibits TREM2 activation of microglial phagocytosis (54).
3.1.3 MiR-486
miR-486-5p is a miRNA with broad bioregulatory functions that may serve as a potential biomarker for the diagnosis of various cancers, including chronic myeloid leukemia (CML) (55). miR-486-5p affects the angiogenic activity of endothelial cells by regulating its target gene, matrix metalloproteinase 19 (MMP19) (56). It has also been found that the severity of the inflammatory response in sepsis is positively correlated with miR-486-5p expression in cells, making miR-486-5p a potential diagnostic biomarker of sepsis (57). Overexpression of miR-486 has been observed in myocardial ischemia cell models (58). miR-486 may be an important regulatory factor of the inflammatory response (59) (Table 1). In neural tissues, miR-486 expression is regulated by TREM2 (60, 61), which acts to alter the miRNA structural activity and physiological functions. This process results in altered levels of protein kinase B (Akt) and promotes microglial activation, leading to a persistent inflammatory response (Figure 2).
3.1.4 MiR-124
miR-124 regulates myeloid cell activity and hematopoietic function and has been found to be involved in cardiovascular diseases. In addition, miR-124 is downregulated in a wide range of human cancers. As an abundantly expressed miRNA in the brain, miR-124 promotes formation of synapses and plays an important regulatory role in many neurodegenerative diseases (62). miR-124 is specifically expressed in microglia, and miR-124 upregulation leads to microglial and CNS macrophage phenotypic switching to an anti-inflammatory state. miR-124 is an important target of many lncRNAs (63–65). Yang et al. (66) found that exogenous miR-124 promotes the transformation of microglia into an anti-inflammatory phenotype and promotes synaptic connection in the hippocampal region, as well as the recovery of neurological function after brain injury (Table 1, Figure 3). miR-124 promotes microglia transformation by inhibiting TLR4, as validated in microglia models (66). miR-124 is significantly elevated in mouse hippocampal neurons, and similar changes have been observed in the nerve tissues of AD patients, suggesting that miR-124 plays a significant role in AD nerve injury. Moreover, Wang et al. found that miR-124 affects synaptic function and consciousness disorders in AD patients by regulating protein tyrosine phosphatase non-receptor type 1 (PTPN1) (67). Other researchers have also found that miR-124 upregulation decreases coordination complement component 1q-like 3 (C1ql3) expression in the brains of APP/PS1 transgenic mice. Conversely, downregulation of miR-124 expression is also accompanied by changes in C1ql3, leading to Aβ deposition in the brain and cerebrovascular damage, evidenced by reductions in microvessel density and angiogenesis. Treatment of BBB damage in APP/PS1 mice with lentivirus-mediated miR-124 overexpression or C1q inhibitor (C1INH) promotes angiogenesis, reduces Aβ deposition, and, ultimately, alleviates impairments in consciousness and memory (68). Collectively, these data suggest that miR-124 regulates the neuroinflammatory response by regulating expression of complement component C1ql3 (45).
3.1. 5 MiR-1199–5p
miR-1199-5p is a tumor metastasis suppressor that may be used in tumor therapy and research (69, 70). miR199 targets the RHO-associated protein kinase 1 (ROCK1) gene (Table 1), and ROCKs are related to cell motility and apoptosis. Alterations in miR-1199-5p levels have not been found in AD models, although research has shown that miR-1199-5p participates in regulation of the neuroinflammatory response in AD. LPS inhibits AK148321 expression in BV2 cells (71). miR-1199-5p acts as a sponge to reduce AK148321 function, and AK148321 inhibits microglia activation and reduces inflammatory cytokine release (Figure 3). In addition, miR-1199-5p may directly target heat-shock protein family A (HSPA5) in LPS-stimulated BV2 cells. AK148321 overexpression in BV2 cells inhibits apoptosis of hippocampal neurons, and HSPA5 downregulation prevents this inhibition (72).
3.1.6 MiR-15a
miR-15a is a miRNA that is downregulated in many cancers. As a tumor suppressor gene, miR-15a loss leads to an imbalance in the expression of oncogenic bcl-2 and onco-suppressor p53 proteins (73). miR-15a is involved in regulating the inflammatory response and apoptosis and is increasingly important in neurological diseases (74). miR-15a is downregulated in the brain of AD (Table 1). Extracellular signal regulated kinase 1 (ERK1) is an enzyme that participates in tau phosphorylation. ERK1 expression is regulated by miR-15 in neural tissues (75). Studies on other neurological diseases have also found that miR-15 may participate in regulating neuroinflammatory responses. For example, Cai et al. (76) found that, compared with the control group, rats with chronic constriction injury (CCI) showed significantly decreased expression of miR-15a and this decrease further promoted neuroinflammation in the spinal cord tissue. Conversely, exogenous miR-15a significantly reduced the levels of CCI-induced pro-inflammatory cytokines (Figure 3). miR-15a also regulates protein kinase B 3 (AKT3) in a microglia model. In addition, miR-15a upregulation induces the expression of autophagy-related proteins, indicating that miR-15a is involved in AKT3-mediated autophagy via AKT3 inhibition. Moreover, miR-15a-5P targets NR2B in epileptic patients, negatively regulating its expression and inhibiting neuronal apoptosis in the hippocampus (77). The specific mechanism of miR-15a’s action in AD is unclear and requires further study. miR-15a may become an important target for AD.
3.1.7 MiR-132
miR-132, a miRNA enriched in various tissues, is a regulatory RNA universally expressed in the cardiovascular system and specifically expressed in various cardiac diseases, for which it may serve as a potential diagnostic biomarker (78). miR-132 is involved in neuronal genesis and synaptic plasticity (79). miR-132 was found to be significantly lower in the brains of patients with AD than in normal controls (Table 1). Additionally, miR-132 deletion in a mouse model of AD leads to more severe Aβ deposition and more severe memory impairment when compared to AD mice with normal miR-132. Xu et al. (80) found a possible downstream target of miR-132, namely C1q, through silicon analysis. C1q, a major protein of the typical complement cascade, is highly expressed in the nerve tissue of AD patients. Further studies showed that miR-132 or C1INH treatment increases levels of synaptic protein expression when compared to controls. Moreover, subsequent studies suggested that miR-132 may regulate C1q function. Another study (81) found that miR-132 inhibits the expression of inducible NO synthase (iNOS) in the hippocampus and prevents oxidative stress via mitogen-activated protein kinase 1 (MAPK1) inhibition. These alterations improved the cognitive function of AD rats. miR-132 may participate in regulating the neuroinflammatory response (Figure 3).
miR-132 interacts other miRNAs in AD. Pascal et al. (82) found that insufficient miR-132/212 increased tau expression. Studies based on cellular genetics have shown that miR-132 regulates tau mRNA expression. Mouse models with miR-132/212 deletion exhibit greater tau protein aggregation compared to control mice. In contrast, treatment with exogenous miR-132 mimics improve partial memory function and enhance tau decomposition in AD mice. In addition, expression of miR-132 and miR-212 in the hippocampus are positively correlated with cognitive score in AD patients. These results suggest that highly expressed miR-132 and miR-212 in the brain may protect the elderly from neurological disorders (79).
3.1 8 MiR-146a
miR-146a, an miRNA with abundant expression in different tissues (83), inhibits doxorubicin-induced cardiotoxicity in the myocardium (84), and its downregulation inhibits the effects of LPS on angiogenesis (85). In neural tissue studies, co-culture of miR-146a with mesenchymal stem cells has been found to reduce the astrocyte-associated inflammatory response and synaptic formation. miR-146a also improved cognitive dysfunction and memory loss in an AD mouse model (86). Further analysis showed that the levels of both miR-146a and NF-κB were decreased in the brain of AD mice treated with stem cells compared with the control mice (Table 1; Figure 3). NF-κB is an important regulator of inflammation and cancer progression (87). miR-146a has been used as a myeloid-selective NF-κB inhibitor in other diseases, such as leukemia, and has been demonstrated to inhibit NF-κB expression in AD models (30). miR-146a may participate in AD by inhibiting NF-κB and regulating the neuroinflammatory response and synaptic formation.
3.1.9 MiR-223
miR-223 is expressed in a variety of tissues where it plays different roles and participates in several pathological states, such as autoimmune and inflammatory diseases (88). miR-223-3p is thought to antagonize the function of NOD-like receptor thermal protein domain associated protein 3 (NLRP3), an inflammatory microprotein (89). In myeloid bone marrow cells, downregulation of miR-223 induced NF-κB and MAPK activity, which subsequently promoted the production of inflammatory factors (90) (Table 1; Figure 3). Chronic inflammasome activation is a potential feature of neurodegenerative diseases. Studies have evaluated the serum concentrations of miR-223-3p in AD patients and healthy controls (HC) and found that miR-223-3p plasma concentrations in AD groups were reduced compared with HC (91). miR-223 may participate in AD by affecting the neuroinflammatory response, making it a potential therapeutic target.
3.1.10 MiR-125b
mir-125b is an important cancer regulator that acts through multiple signaling pathways, such as the Wnt, PI3K/Akt, STAT-3, MAPK, NF-κB and p53 pathways (92). Moreover, miR-125b is an important inflammation-related miRNA that is richly expressed in neural tissues (93). miR-125b upregulation induces tau deposition in rat brain, whereas inhibition of miR-125b expression targets beta amyloid cleaving enzyme 1 (BACE1) to reduce apoptosis and oxidative stress, thereby inhibiting Aβ-induced neurotoxicity (94) (Table 1). miR-125b is increased in AD patients (95). Moreover, miR-125b overexpression inhibits the proliferation of AD model cells in vitro, induces apoptosis, and enhances inflammation and oxidative stress (96) (Figure 3). On the contrary, IL-10 activity is significantly reduced in in vitro models of AD (97). Therefore, the proinflammatory microRNA, miR-125b, may promote development of AD.
3.1.11 MiR-181a
The target genes of miR-181a contain multiple apoptosis-related genes, and dysregulation of miR-181a expression is closely related to the occurrence and development of a variety of cancers. miR-181a is abundant in neural tissues and affects synaptic regeneration and functional repair (83, 98). It has previously been reported that miR-181a is upregulated in the hippocampus of an AD mouse model (Table 1; Figure 3). Inhibition of miR-181a in this AD mouse model reduced memory deficits and increased glutamic acid (GluA) 2 and GluA1 levels, which regulate cognitive impairment. miR-181a is expressed in T cells, where it regulates their activation threshold (99). Interestingly, miR-181a has been verified as being involved in age-related diseases, as the expression of miR-181a in naive T cells decreases with age (100). Although the exact mechanism is unclear, miR-181a likely participates in regulating the AD neuroinflammatory response.
There are many other miRNAs that have been implicated in regulating inflammatory responses, including miR-150, miR-21, and Let-7 (Figure 3). These same miRNAs may be involved in AD-related neuroinflammation. For example, miR-150 is downregulated in AD patients (Table 1), and miR-150 knockdown enhances the activity of hippocampal neurons in a model of consciousness disorder. Conversely, miR-150 overexpression significantly increases apoptosis in the PC12 cell line (101). In addition, miR-150 was found to influence the formation of mature B cells. miR-150 is abundant in lymphocytes, and its expression changes with differentiation and maturation of B cells (102). miR-150 may be involved in regulating the inflammatory response via B cell regulation (Figure 3). miR-21 is an anti-inflammatory regulatory factor that is abundant in microglia (103, 104) regulatory factor that is abundant in microglia (105). miR-21 expression is induced by MyD88 and NF-κB, resulting in NF-κB downregulation (106). In addition, miR-21 reduces TNF-α secretion and, thus, promotes macrophage switching to an anti-inflammatory phenotype and inhibits microglial activation (107). Let-7, an important regulator of neuroinflammation, is an evolutionarily conserved family of miRNAs (108, 109). Let-7 affects TLR-7 function to regulate the neuroinflammatory response, making it a potential regulator of AD. Although an increasing number of miRNAs are being discovered and studied, the role of many miRNAs remains unknown.
3.2 LncRNAs in AD
lncRNAs are larger than 200 nt, ranging up to 100 kb in size. lncRNAs participate in many important physiological processes (110) and regulate protein formation at the transcriptional and post-transcriptional levels. Further, lncRNAs interact with other RNAs as part of the RNA network. For example, lncRNAs act as miRNA sponges to regulate miRNA functions, and, in turn, miRNAs regulate lncRNA function, activation, and stability. lncRNA is highly expressed in mammalian neural tissue, enabling it to respond quickly to environmental and molecular changes. In addition, lncRNAs participate in the physiological processes of a variety of neurodegenerative diseases such as AD, Parkinson’s disease, and Huntington’s disease (111–113). Neuroinflammation is an important factor driving deterioration in various neurodegenerative diseases. With disease progression, inflammatory cells are constantly stimulated, and the formation of persistent neuroinflammation can cause damage to the nerve tissue. Many studies have shown that lncRNAs participate in various neuroinflammatory processes.
3.2.1 LncRNA MALAT1
Metastasis-associated transcript 1 (MALAT1) was first identified in human lung adenocarcinoma and then found to be expressed in a variety of tissues. MALAT1 plays an important role in regulating cellular proliferation, apoptosis, autophagy, and other physiological processes (114). Upregulation of MALAT1 inhibits neuronal apoptosis and neuroinflammation in patients with AD. Some scholars believe that the mechanism of MALATI-induced inhibition may be related to microglia activation via NF-κB inhibition. Peizhi et al. (97) found that in an AD mouse model, MALAT1 upregulation reduced neuronal apoptosis, promoted neuronal functional repair and regeneration, and downregulated IL-6 and TNF-α levels (Figure 4) while upregulating IL-10 levels compared to control animals. Downregulation of MALAT1 produced the opposite effects. Additionally, MALAT1 was found to inhibit miR-125b expression. In an AD model overexpressing MALAT1, miR-125b was downregulated and this downregulation was associated with reduced inflammatory cytokine release.
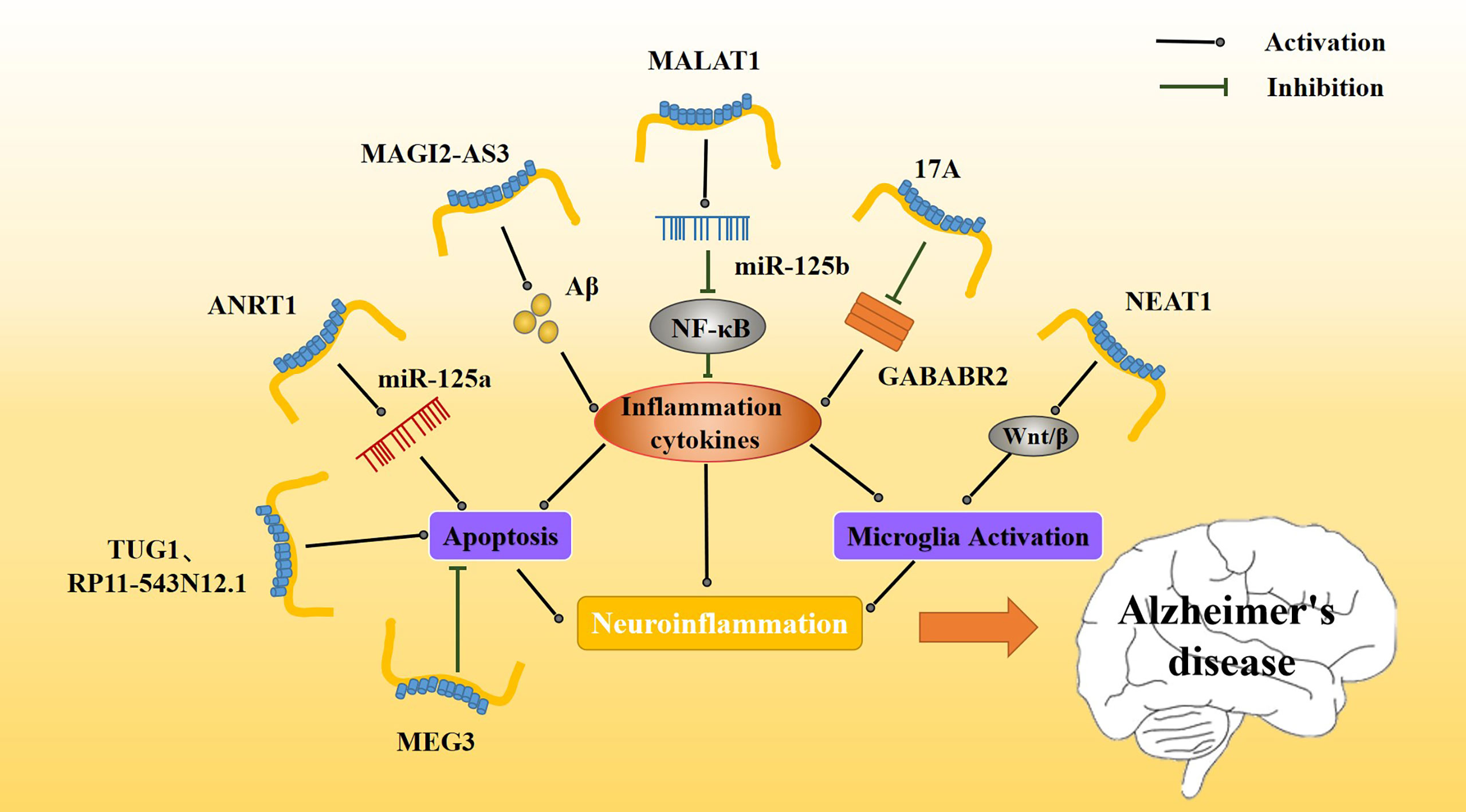
Figure 4 Effect of inflammatory lncRNAs on AD inflammation. Multiple inflammatory lncRNAs may be involved in different inflammatory pathways. MAGI2-AS3 promotes the Aβ-induced inflammatory response, whereas MALAT1 regulates release of inflammatory cytokines via NF-κB. The release of inflammatory mediators further affects the activation and apoptosis of microglia. Together, these constitute a persistent neuroinflammatory response that accelerates the progression of AD.
3.2.2 LncRNA NEAT1
Abnormal overexpression of long non-coding RNA nuclear paraspeckle assembly transcript 1 (NEAT1) has been found in several types of solid tumors, such as lung cancer, colorectal cancer, and hepatocellular carcinoma. As an important nuclear gene, NEAT1 is closely related to physiological processes, such as cell replication, growth, and apoptosis (115). NEAT1 is upregulated in the nerve tissue of AD patients (Table 1), and it has been found that amyloid deposition in AD patients causes NEAT1 upregulation. Upregulation of NEAT1 also affects AD progression by increasing the apoptosis rate and decreasing the activity of nerve cells. Moreover, NEAT1 upregulation increases expression of amyloid, BACE1, APP, and tau and increases phosphorylation levels of these proteins. It also increases caspase 3. Increases in these proteins or in their phosphorylation levels may be related to NEAT1 triggering neuroinflammation (116). A number of experiments have shown that NEAT1 activates microglia, promotes neuroinflammation, upregulates inflammatory mediators such as IL-2 and IL-6, and promotes the progression of various neurological diseases (Figure 4). NEAT1 may contribute to neuroinflammatory injury through the Wnt/β signaling pathway during brain hypoxia injury (117).
3.2.3 LncRNA RP11-543N12.1
lncRNA RP11-543N12.1 is highly expressed in AD patients (Table 1). RP11-543N12.1 expression is higher in the nucleus than in the cytoplasm, suggesting that RP11-543N12.1 may regulate transcription in the nucleus. In addition, RP11-543N12.1 and Aβ25-35 synergistically promote miR-324-3p expression and promote cell apoptosis during AD progression (Figure 4). Upregulation of RP11-543N12.1 and miR-324-3p may cause cellular apoptosis by promoting continuous release of inflammation-related factors. lncRNA RP11-543N12.1 acts as a miR-324–3p sponge in microglia and neuron models, promoting neuroinflammation and tau deposition and phosphorylated (118).
3.2.4 LncRNA MEG3
Maternally expressed gene 3 (MEG3) was first found to be an important tumor suppressor, specifically expressed in lung cancer and hepatocellular carcinoma. MEG3 inhibits the biological functions of tumor cells by regulating major tumor suppressor genes p53 and Rb (119). lncRNA MEG3 expression is decreased in AD patients (120) (Table 1). In addition, upregulation of MEG3 in AD rat models improves AD prognosis, reduces memory loss, inhibits neuronal injury and apoptosis, and reduces Aβ deposition. The mechanism of lncRNA MEG3 action is believed to be related to a neuroinflammatory response to MEG3. Jiping et al. (121) reported that MEG3 upregulation is associated with decreased levels of apoptosis (Figure 4), PI3/Akt signaling pathway-related proteins, and inflammation-related proteins. MEG3 is likely to play a neuroprotective role by inactivating the PI3/Akt pathway to decrease oxidative damage and inflammation.
3.2.5 LncRNA ANRIL
lncRNA at the INK4 locus (ANRIL), a newly discovered lncRNA, is closely associated with regulation of cell growth and apoptosis in various cancers, and has also been associated with vascular endothelial injury in cardiovascular diseases (122). ANRIL is associated with many inflammatory and neurological disorders, including various ischemic injury diseases, and may participate in regulating the inflammatory response in vivo (123, 124). Recently, lncANRIL was found to be upregulated in the brain of AD patients (Table 1), and this upregulation may also be related to the progression of the AD neuroinflammatory response. In an AD model, lncANRIL binds mir-125a. In terms of inflammatory response, lncANRIL downregulation reduces the production of many inflammatory mediators, such as TNF-α and IL-1β, and inhibits apoptosis. In addition, upregulation of lncANRIL increases neuronal growth and miR-125a expression, whereas miR-125a inhibition weakens the neuroprotective effect of lncANRIL downregulation in cellular AD models (Figure 4). Luciferase reporter gene detection has shown that lncANRIL directly binds to miR-125a in an AD mouse model (125). lncANRIL may interfere with neuroinflammatory responses by acting on miR-125a. This specific mechanism requires further study and may represent a possible new breakthrough for AD and other inflammatory diseases in the future.
3.2.6 LncRNA 17A
lncRNA 17A is upregulated in the brain of AD patients (126) (Table 1), and it may affect the progression and prognosis of AD by inducing formation of Aβ. The proportion of apoptotic cells decreases when 17A is inhibited and increases when 17A is overexpressed. Studies have found that 17A blocks GABAB2, affecting cAMP production and K(+) channel activation to inhibit its signal transduction (127). The expression of 17A in different cells is associated with GABAB receptor (GABABR) 2 expression. Specifically, when 17A is overexpressed, GABABR2 expression is downregulated (Figure 4). The GABABR is involved in regulating microglia function and neuroinflammation, and GABABR2 is involved in the inhibition of synaptic transmission. It has been reported that upregulation of GABABR2 inhibits the release of pro-inflammatory cytokines. 17A may participate in AD by regulating neuroinflammation via GABABR (128). Recently, it was found that the role of 17A in determining AD progression may also be regulated by Wnt/β-catenin (126). The mechanism whereby 17A regulates neuroinflammation requires additional study, as it has the potential to become a novel target for AD.
3.2.7 LncRNA MAGI2-AS3
MAGI2-AS3 is a lncRNA transcribed from the antisense strand, near the MAGI2 gene. MAGI2-AS3 plays an important regulatory role in many cancers, including lung cancer (129–132). MAGI2-AS3 also acts as a miR-374b-5p sponge in many cancers (133, 134). MAGI2-AS3 is upregulated in patients with AD. MAGI2-AS3 acts as a sponge affecting the function of miR-374b-5p and BACE1, an enzyme important for degrading Aβ protein. In addition, some scholars have found that MAGI2-AS3 may play a regulatory role in chronic inflammation (Table 1, Figure 4). Studies have shown that upregulation of MAGI2-AS3 promotes Aβ deposition and inflammation, leading to speculation that MAGI2-AS3 may be related to the neuroinflammatory response in AD. Moreover, it downregulation of MAGI2-AS3 enhances neuronal activity and reduces neuroinflammation, and miR-374b-5P overexpression causes similar results (135). The MAGI2-AS3/miR-374b-5p axis may be a novel target for the treatment for AD.
3.2.8 LncRNA TUG1
lncRNA taurine upregulated gene 1 (TUG1) (136), a novel lncRNA, participates in ischemic disorders, including ischemic spinal cord injury and ischemic myocardial injury (137, 138). It also promotes neuronal apoptosis through the absorption of miR-9 by sponges. It was found that small interference TUG1 (Si-TUG1) mitigates neuroinflammation in ketamine-induced hippocampal neurons in rats. Hence, some scholars believe that TUG1 may also regulate neuroinflammation in AD (Table 1). Li et al. found that TUG1 silencing and miR-15a up-regulation alleviated memory loss, improved pathological damage, inhibited apoptosis, and enhanced antioxidant capacity of the nerve tissue in an AD rat model (139). In an in vitro model, hippocampal neurons treated with Aβ25-35 exhibited inhibition of neuronal activity and enhanced apoptosis (Figure 4). TUG1 silencing and miR-15a upregulation inhibit the activity of Aβ, but the underlying mechanism for these results needs further study.
3.3 CircRNAs in AD
circRNA is a type of RNA molecule characterized by a single stranded ring, found in a variety of species from lower to higher organisms (140). circRNA effectively prevents transcription of microRNAs, and through this microRNA transcription regulation, it affects downstream mRNA expression (141, 142). In addition, emerging evidence suggests that circRNA participates in various protein modifications. circRNAs are formed in higher eukaryotes by reverse splicing loops of pre-mRNA, creating a covalent linkage between the downstream 5’ end of the exon and the upstream 3’ end. circRNAs are resistant to exonuclease degradation due to the 5’ and 3’ linkages and are, therefore, more stable than linear mRNAs (with a half-life of greater than 48 h). circRNAs exhibit a variety of physiological functions, including acting as miRNAs and protein sponges, regulating parental gene transcription, and participating in mRNA splicing. circRNAs play a role in AD, heart failure, and hypertrophy. They are also abundant in neural tissues and are involved in the production of inflammatory factors in physiological and pathophysiological processes. For example, circ_0000950 acts as an miR-103 sponge to stabilize mir-103 function and increase the inflammatory response. CIRS-7, circ NF1-419, circHDAC9, and circAβ-a have also been confirmed to be involved in the pathogenesis of AD, with specific changes observed in AD patients. However, the potential role of circRNAs in AD is still largely unknown.
3.3.1 Circ_0000950
circ_0000950 is overexpressed in AD patients (Table 1). The circRNAs act as a sponge for miR-103 and may regulate lipid metabolism through miR-103. Yang et al. found that circ_0000950 overexpression promoted neuronal apoptosis, inhibited neuronal growth, and increased the levels of many inflammatory factors, such as Il-1β and TNF-α in an AD model (Figure 5). Luciferase assay confirmed that circ_0000950 achieves its effects by direct sponge absorption of miR-103 (143).
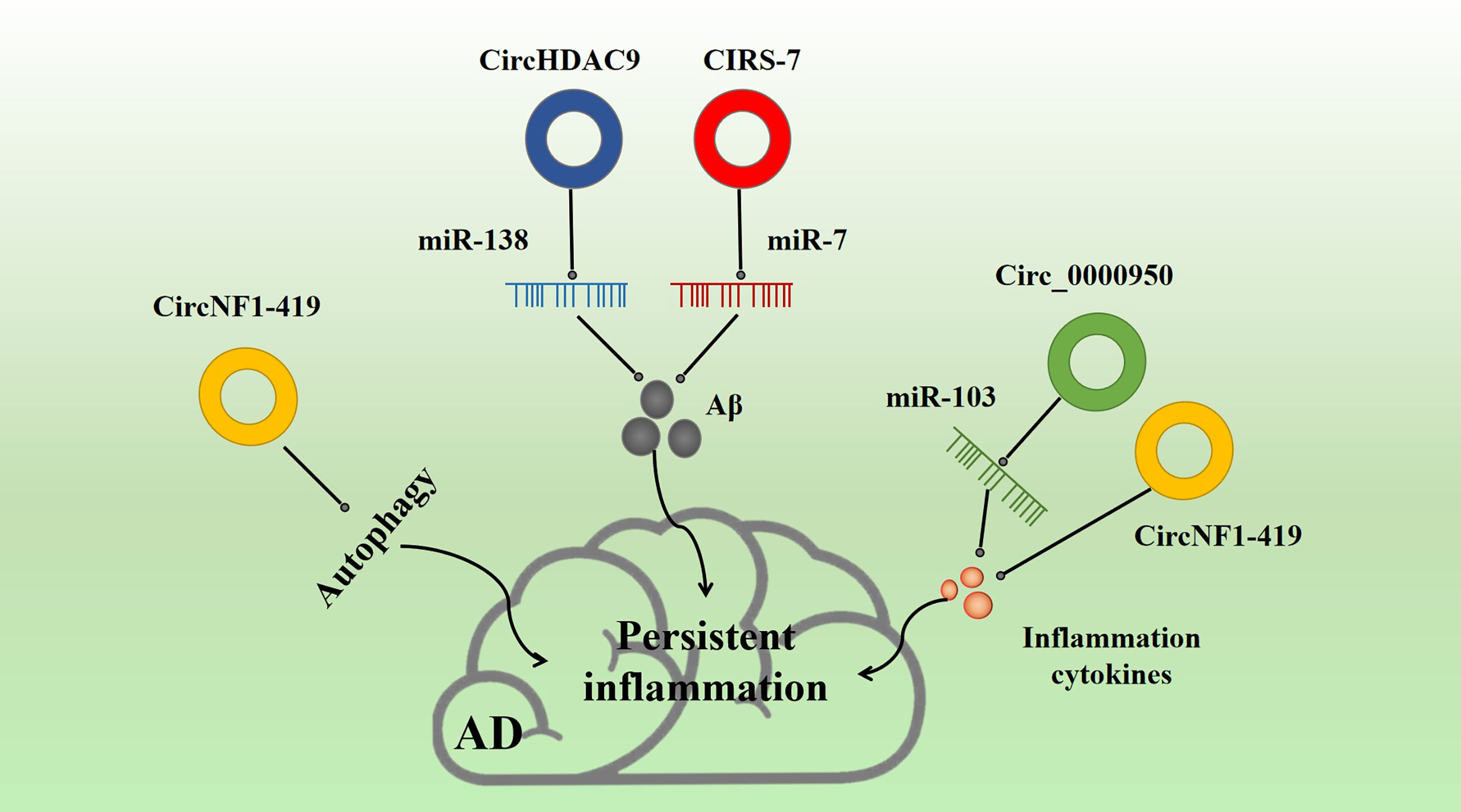
Figure 5 Effect of inflammatory circRNA on AD inflammation. Multiple inflammatory circRNAs participate in various inflammation-related pathways. For example, CIRS-7 and NF1-419 promote Aβ-induced inflammation. circ_0000950 and circNF1-419 are associated with the release of inflammation cytokines, and circNF1-419 is associated with autophagy.
3.3.2 CIRS-7
CIRS-7 is a common endogenous circRNA found in many tissues. Hansen et al. (144) proved for the first time in 2013 that CIRS-7 acts as a miR-7 sponge, regulating gene translation and protein modification. In particular, CIRS-7 can regulate cancer progression through competitive inhibition of miR-7 (145), which has been proven to be related to the occurrence and development of renal cell cancer (146), cervical cancer (147), oral cell cancer (148), and thyroid cancer (149). Moreover, CIRS-7, as a miR-7 sponge, is involved in determining BACE1 and APP concentrations by promoting proteasome and lysosomal degradation of BACE1 and APP. Thus, CIRS-7 is a major regulatory factor in the progression of AD (150) (Table 1, Figure 5). CIRS-7 overexpression reduces Aβ deposition, suggesting that circRNAs may possess a neuroprotective function. CIRS-7 inhibits NF-κB, a key regulatory target of inflammation. CIRS-7 has also been implicated in regulating inflammation, and the CIRS-7/miR-7 axis may play a role in inducing apoptosis and inflammation via IL-1β (151). CIRS-7 has also been found to promote autophagy and inflammation in various neurological diseases (152).
3.3.3 CircNF1-419
circNF1-419 is involved in tumor development and downregulated in AD (Table 1) (153). In mouse astrocytes, it has been shown that NF1-419 regulates astrocyte autophagy through the PI3K-Akt-MTOR signaling pathway. circNF1-419 overexpression in the mouse brain promotes autophagy by binding AP2B1. This interaction regulates the aging biomarkers P21, P35/25, and P16 and the pro-inflammatory factors TNF-α and NF-κB, and leads to downregulation of tau, P-tau, Aβ-1, and APOE, thereby delaying the onset of AD (154, 155) (Figure 5).
3.3.4 CircHDAC9
circHDAC9 is downregulated in parallel to miR-138 upregulation in AD (Table 1) (156). miR-138 plays a tumor suppressive role by targeting genes that regulate the proliferation and apoptosis of tumor cells, thus affecting the development and prognosis of cancer. circHDAC9 binds mir-138 to produce similar effects. Moreover, circHDAC9 enhances Sirt1 inhibition and reduces miR-138-related Aβ deposition. More importantly, circHDAC9 expression is downregulated in both AD and MCI patients. Moreover, Aβ42 has been found to induce significant downregulation of circHDAC9 and overexpression of miR-142-5p in neurons (Figure 5). In addition, circHDAC9 acts as a sponge for miR-142-5p. Upregulation of circHDAC9 mitigates Aβ42-induced neuroinflammation and neurotoxicity in HN cells via miR-142-5p (157).
Other circRNAs such as circRNA-017963 also participate in the pathological development of AD. Huang et al. (158) found that the expression of circRNA-017963 was greatly decreased in 10-month-old SAMP8 mice, suggesting that circRNA-017963 may have a significant role in AD. Furthermore, miR-7033-3p may show the greatest relationship with circRNA-017963 and is likely to be involved in autophagosome formation and synaptic transmission, which have been proven to be related to AD pathogenesis.
3.4 PiRNAs in AD
Piwi-interacting RNAs (piRNAs) have a length of 24–30 nt and represent a unique class of RNA that are important in governing stem cell (SC) function (159, 160). piRNA is not conserved among species, and there is a considerable difference in its biological origin when compared to miRNAs and siRNAs (161). The piRNA source is not dsRNA, so Dicer shearing is not involved. However, mature piRNAs can target transposers in the genome and silence them (162). piRNAs are also involved in regulating epigenetic function and stabilizing mRNA. In general, piRNA is highly expressed in reproductive tissues and acts as a guardian of the genome through stable transposons.
More recently, however, piRNA has been found in other tissues, including nerve tissue (163), suggesting that it may be involved in neuro-related diseases. Kim et al. have reported that the piwi-piRNA pathway may govern neuronal function in many animals, affecting axonal regeneration and memory loss (164). In addition, there is evidence that the piwi/piRNA complex promotes synaptic repair and reduces memory loss via cAMP response element-binding protein (CREB) (165). Analysis of piRNAs in cerebrospinal fluid exosomes may improve the antecedent diagnosis of AD and may also help with patient stratification in clinical trials. From a therapeutic perspective, it is of importance to find a biomarker that helps predict the transition to AD dementia in patients with MCI (166). In addition, piRNAs have been shown to be important for maintaining genomic integrity and inhibiting transposer activity, and piRNAs may be involved in the origination of single nucleotide polymorphisms (SNPs) associated with AD (167). With further research on piRNA still ongoing, the mechanism of its involvement in AD will become clearer; although extensive research is still needed.
3.5 TsRNAs in AD
Transfer RNA-derived small RNAs (tsRNAs) can be divided into two types based on biological origin: tRNA-derived fragments (tRFs) and half of tRNA (tiRNAs) (168, 169). The length of tRF ranges from 14 to 36 nt. tRFs can be divided into tRF-1, tRF-2, tRF-3, tRF-5, and i-tRF, according to their cleavage site and origin. The formation of tiRNAs involves the specific cleavage of angiopoietin on the anticodon ring of mature tRNA. These tsRNAs regulate a range of molecular processes, from gene translation and RNA stability to cellular proliferation and differentiation (170, 171).tsRNA is a new RNA species found abundantly in many tissues including neural tissue (172). Although it is largely unstudied in the context of neurological diseases, Magistri et al. (173) demonstrated that the upregulation of try-tRNA and arg-tRNA is involved in altering RNA kinase in CLP1 knockout mice, leading to the gradual loss of motor nerve function in affected mice. Shortly thereafter, another study found that accumulation of tRNA fragments caused by tRNA processing defects may lead to the development of neurodegenerative diseases (174). These tsRNAs make motor neurons more susceptible to oxidative stress, indicating that tsRNAs may be involved in neurodegenerative diseases via neuronal oxidative stress response regulation. The TRF expression from tRNA subsets is altered in the hippocampus of AD patients. Changes in TRF expression may be related to age and disease stage. Decreased NOP2/Sun RNA methyltransferase 2 (NSun2) expression has been observed in AD patients aged <65 years, suggesting that decreases in NSun2 may reduce the degree of tRNA methylation to enhance tRF production (175). As a newly discovered sncRNA, many mechanisms of tsRNA action in the human body are still unclear. In the future, they may become important targets for AD.
In conclusion, comprehensive investigations of non-coding RNAs, such as miRNAs, lncRNAs, circRNAs, piRNA, and tsRNA, that includes interactions with other RNAs, DNAs, and proteins, will be helpful in fully elucidating the cause/consequence relationships of AD dysfunction. It is of significant importance to develop a valid therapy to treat this devastating disease.
4 Potential Therapeutic Targets
There is no doubt that ncRNAs play a significant role in several neuroinflammatory diseases (37). For many diseases with unclear molecular mechanisms of pathogenesis and progression, ncRNAs may serve as therapeutic targets in the future (120, 176). In fact, ncRNA-based therapies have been developed for many diseases with complex mechanisms that are difficult to cure, such as various cardiomyopathies, diabetes, and neurodegenerative diseases (177, 178). Thus, many novel strategies have been invented to control the function of ncRNAs. Oligonucleotides have several advantages in targeting ncRNAs in contrast to traditional therapies (e.g., small molecules and antibodies) (179). Oligonucleotides complement their target RNA precisely, making screening and detection of targeting tools relatively simple. In addition, oligonucleotides can enter cells and specifically target RNAs that are inaccessible to many factors acting on cell membranes (180, 181). Because oligonucleotides are more targeted than other subclasses of drugs, potential side effects are reduced (182).
Abnormalities in the modulation of miRNA expression have been observed in a large number of diseases and are regarded as feasible therapeutic targets and regulators with a wide variety of applications for preclinical and clinical trials (183–185). miRNA mimics represent a novel approach to miRNA therapy. miRNA mimics are small double-stranded RNA molecules derived from the exogenous synthesis of relevant miRNAs that are modified into functional miRNAs (186). Other miRNA mimics may act to inhibit the function of endogenous miRNAs, and their synthesis is usually based on the complementary sequence of the relevant miRNA. These miRNA mimics block the binding of endogenous miRNAs to their target and include anti-antibodies, miRNA sponges, and small molecule antisense oligonucleotides (187).
Current therapies targeting lncRNAs include oligonucleotide compounds (e.g., thiocarcinogenic main chains and various sugar modifications). Targeting of lncRNAs can also be achieved by blocking or knocking out their expression via antisense oligonucleotides (ASOs) or small interfering RNA (siRNA) (120, 188). These methods have been tested clinically with promising results. CRISPR-Cas9 is an effective tool for gene mapping and modification (189). Cas9 endonucleases can be targeted for DNA editing or gene knockout. In a novel modification to the approach, Cas9 binds to transcriptional activators such as NF-κB to upregulate genes. Crispr-cas9-based therapies are already used to treat some autoimmune diseases such as cystic fibrosis (190).
The major obstacle in AD treatment is providing ncRNA-based therapies that can cross the BBB (191). Promising strategies currently under investigation include using non-viral pathways and lipid or polymer nanoparticle delivery systems (192, 193). Focused ultrasound is another technique that has been found to temporarily block the BBB, promising the application of targeted therapy in brain tissue (25, 194). Adeno-associated virus (AAV) vectors have been shown to target ncRNAs in the CNS (195, 196). Therefore, identifying ideal ncRNA-targeted drugs may help improve the management and treatment of pathologic neuroinflammation. The discovery of ncRNAs provides new perspectives for understanding genetic treatments (176). The promise of ncRNAs is that they affect multiple genes or biological processes, although attempting to understand their full potential is not without challenges.
5 Conclusions
Dysregulation of neuroinflammation-related ncRNAs leads to dysfunction of certain signaling pathways via regulation of target genes that act as inflammatory pathologic factors in the development and progression of AD. As described above, these ncRNAs regulate key genes that may play a significant role in the development of AD. Existing studies have shown that these ncRNAs influence the progression of AD in cell and animal models and in human patients. Thus, by studying the physicochemical properties of these ncRNAs and their specific mechanisms related to disease, effective treatments for AD may be discovered. For example, induction of anti-inflammatory ncRNAs or inhibition of pro-inflammatory ncRNAs may represent a therapeutic strategy to improve central nervous system tissue damage following neuroinflammation (197–199). Oligonucleotide therapy is an emerging tool for ncRNA targeting and has the benefits of specific targeting and low toxicity. Thus, oligonucleotide therapy is currently the most widely researched method in ncRNA-targeting drug discovery and application (182). Despite advances in determining how ncRNAs act in neuroinflammation, the characteristics and role of many ncRNAs remain unknown.
There are limitations that need to be addressed. First, most ncRNAs are found in low concentrations in bodily fluids, and the ncRNA extraction and separation methods are complicated. Moreover, owing to the easily degradable nature of ncRNAs, it is difficult to ensure the quality of preservation. Second, the four existing ncRNA detection methods all have inherent defects. For example, it is difficult to construct cDNA libraries for ncRNA molecules with low expression levels. In addition, the results of qRT-PCR are uncertain, making primer design crucial. The reproducibility of gene chip technology is poor, and the results are closely related to the sample size. Finally, high-throughput sequencing is costly and wasteful. Further research is needed to develop accurate diagnostic criteria for clinical use. With more extensive research, the understanding of the mechanisms of ncRNA will become more substantial, and it will be an important means for the research and treatment of AD.
Although significant advances have been made in the physiological characteristics of ncRNA and its role in neuroinflammation associated with AD, this field remains largely unknown. Future investigation should give top priority to targets and biological processes that are involved in the key processes of AD pathology, validate them in model organisms that are more closely related to humans, and develop new experimental paradigms to improve research efficiency. Because the ultimate goal of these studies is to prevent the occurrence of AD or improve the prognosis of AD patients, differences in clinical responses based on patient age and gender are also crucial for the study of AD. It is important that this effort be combined with more attention to clinical application in the clinical transformation of AD.
Author Contributions
Conceiving, YL, ZZ, YX, and WP. Searching literature, YL, XC, SH, and ZZ. Manuscript drafting, YL, XC, HL, and SH. Manuscript revising, YL, HL, ZZ, YX, and WP. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 81873169, 82104967), Hunan Provincial Natural Science Foundation of China (No. 2020JJ4803), Hunan Scientific Research Program of Traditional Chinese Medicine (No.2021192).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kumar A, Sidhu J, Goyal A, Tsao JW. “Alzheimer Disease,” In: StatPearls, vol. 2022. Treasure Island FL: StatPearls Publishing Copyright (2022).
2. Tatulian SA. Challenges and Hopes for Alzheimer’s Disease. Drug Discov Today (2022) 27(4):1027–43. doi: 10.1016/j.drudis.2022.01.016
3. Leng F, Edison P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where do We Go From Here? Nat Rev Neurol (2021) 17:157–72. doi: 10.1038/s41582-020-00435-y
4. Nebel RA, Aggarwal NT, Barnes LL, Gallagher A, Goldstein JM, Kantarci K, et al. Understanding the Impact of Sex and Gender in Alzheimer’s Disease: A Call to Action. Alzheimers Dement (2018) 14:1171–83. doi: 10.1016/j.jalz.2018.04.008
5. Crestini A, Santilli F, Martellucci S, Carbone E, Sorice M, Piscopo P, et al. Prions and Neurodegenerative Diseases: A Focus on Alzheimer’s Disease. J Alzheimers Dis (2022) 85:503–18. doi: 10.3233/JAD-215171
6. Schumacher J, Ray NJ, Hamilton CA, Donaghy PC, Firbank M, Roberts G, et al. Cholinergic White Matter Pathways in Dementia With Lewy Bodies and Alzheimer’s Disease. Brain (2021) 4:awab372. doi: 10.1093/brain/awab372
7. Kang MJ, Abdelmohsen K, Hutchison ER, Mitchell SJ, Grammatikakis I, Guo R, et al. HuD Regulates Coding and Noncoding RNA to Induce APP→Aβ Processing. Cell Rep (2014) 7:1401–9. doi: 10.1016/j.celrep.2014.04.050
8. Yan R. Physiological Functions of the β-Site Amyloid Precursor Protein Cleaving Enzyme 1 and 2. Front Mol Neurosci (2017) 10:97. doi: 10.3389/fnmol.2017.00097
9. Montesinos J, Pera M, Larrea D, Guardia-Laguarta C, Agrawal RR, Velasco KR, et al. The Alzheimer’s Disease-Associated C99 Fragment of APP Regulates Cellular Cholesterol Trafficking. EMBO J (2020) 39:e103791. doi: 10.15252/embj.2019103791
10. Pulina MV, Hopkins M, Haroutunian V, Greengard P, Bustos V. C99 Selectively Accumulates in Vulnerable Neurons in Alzheimer’s Disease. Alzheimers Dement (2020) 16:273–82. doi: 10.1016/j.jalz.2019.09.002
11. Chen C, Zhou Y, Wang H, Alam A, Kang SS, Ahn EH, et al. Gut Inflammation Triggers C/Ebpβ/δ-Secretase-Dependent Gut-to-Brain Propagation of Aβ and Tau Fibrils in Alzheimer’s Disease. EMBO J (2021) 40:e106320. doi: 10.15252/embj.2020106320
12. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimers Dement (N Y) (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
13. Akhter R, Shao Y, Formica S, Khrestian M, Bekris LM. TREM2 Alters the Phagocytic, Apoptotic and Inflammatory Response to Aβ(42) in HMC3 Cells. Mol Immunol (2021) 131:171–9. doi: 10.1016/j.molimm.2020.12.035
14. Kong C, Jia L, Jia J. γ-Mangostin Attenuates Amyloid-β42-Induced Neuroinflammation and Oxidative Stress in Microglia-Like BV2 Cells via the Mitogen-Activated Protein Kinases Signaling Pathway. Eur J Pharmacol (2022) 917:174744. doi: 10.1016/j.ejphar.2022.174744
15. Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science (2002) 297:353–6. doi: 10.1126/science.1072994
16. Cai Z, Hussain MD, Yan LJ. Microglia, Neuroinflammation, and Beta-Amyloid Protein in Alzheimer’s Disease. Int J Neurosci (2014) 124:307–21. doi: 10.3109/00207454.2013.833510
17. Jiao H, Jia J. Ginsenoside Compound K Acts via LRP1 to Alleviate Amyloid β(42)-Induced Neuroinflammation in Microglia by Suppressing NF-κb. Biochem Biophys Res Commun (2022) 590:14–9. doi: 10.1016/j.bbrc.2021.12.071
18. Ozben T, Ozben S. Neuro-Inflammation and Anti-Inflammatory Treatment Options for Alzheimer’s Disease. Clin Biochem (2019) 72:87–9. doi: 10.1016/j.clinbiochem.2019.04.001
19. Xie L, Zhang N, Zhang Q, Li C, Sandhu AF, Iii GW, et al. Inflammatory Factors and Amyloid β-Induced Microglial Polarization Promote Inflammatory Crosstalk With Astrocytes. Aging (Albany NY) (2020) 12:22538–49. doi: 10.18632/aging.103663
20. Onyango IG, Jauregui GV, Čarná M, Bennett JP Jr., Stokin GB. Neuroinflammation in Alzheimer’s Disease. Biomedicines (2021) 9(5):524. doi: 10.3390/biomedicines9050524
21. Bowman GL, Dayon L, Kirkland R, Wojcik J, Peyratout G, Severin IC, et al. Blood-Brain Barrier Breakdown, Neuroinflammation, and Cognitive Decline in Older Adults. Alzheimers Dement (2018) 14:1640–50. doi: 10.1016/j.jalz.2018.06.2857
22. Mahdiabadi S, Momtazmanesh S, Perry G, Rezaei N. Immune Modulations and Immunotherapies for Alzheimer’s Disease: A Comprehensive Review. Rev Neurosci (2021) 10. doi: 10.1515/revneuro-2021-0092
23. Su F, Bai F, Zhang Z. Inflammatory Cytokines and Alzheimer’s Disease: A Review From the Perspective of Genetic Polymorphisms. Neurosci Bull (2016) 32:469–80. doi: 10.1007/s12264-016-0055-4
24. Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J Alzheimers Dis (2018) 63:1223–34. doi: 10.3233/JAD-180098
25. Rezai AR, Ranjan M, D’haese PF, Haut MW, Carpenter J, Najib U, et al. Noninvasive Hippocampal Blood-Brain Barrier Opening in Alzheimer’s Disease With Focused Ultrasound. Proc Natl Acad Sci USA (2020) 117:9180–2. doi: 10.1073/pnas.2002571117
26. Richter F, Williams SK, John K, Huber C, Vaslin C, Zanker H, et al. The TNFR1 Antagonist Atrosimab Is Therapeutic in Mouse Models of Acute and Chronic Inflammation. Front Immunol (2021) 12:705485. doi: 10.3389/fimmu.2021.705485
27. Wang X, Wang L, Yu X, Li Y, Liu Z, Zou Y, et al. Glutaminyl Cyclase Inhibitor Exhibits Anti-Inflammatory Effects in Both AD and LPS-Induced Inflammatory Model Mice. Int Immunopharmacol (2019) 75:105770. doi: 10.1016/j.intimp.2019.105770
28. Shen XN, Niu LD, Wang YJ, Cao XP, Liu Q, Tan L, et al. Inflammatory Markers in Alzheimer’s Disease and Mild Cognitive Impairment: A Meta-Analysis and Systematic Review of 170 Studies. J Neurol Neurosurg Psychiatry (2019) 90:590–8. doi: 10.1136/jnnp-2018-319148
29. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
30. Su YL, Wang X, Mann M, Adamus TP, Wang D, Moreira DF, et al. Myeloid Cell-Targeted miR-146a Mimic Inhibits NF-κb-Driven Inflammation and Leukemia Progression In Vivo. Blood (2020) 135:167–80. doi: 10.1182/blood.2019002045
31. Kim KM, Abdelmohsen K, Mustapic M, Kapogiannis D, Gorospe M. RNA in Extracellular Vesicles. Wiley Interdiscip Rev RNA (2017) 8(4):10. doi: 10.1002/wrna.1413
32. Noonin C, Thongboonkerd V. Exosome-Inflammasome Crosstalk and Their Roles in Inflammatory Responses. Theranostics (2021) 11:4436–51. doi: 10.7150/thno.54004
33. Xu YZ, Cheng MG, Wang X, Hu Y. The Emerging Role of non-Coding RNAs From Extracellular Vesicles in Alzheimer’s Disease. J Integr Neurosci (2021) 20:239–45. doi: 10.31083/j.jin.2021.01.360
34. Akbari Dilmaghnai N, Shoorei H, Sharifi G, Mohaqiq M, Majidpoor J, Dinger ME, et al. Non-Coding RNAs Modulate Function of Extracellular Matrix Proteins. BioMed Pharmacother (2021) 136:111240. doi: 10.1016/j.biopha.2021.111240
35. Virciglio C, Abel Y, Rederstorff M. Regulatory Non-Coding RNAs: An Overview. Methods Mol Biol (2021) 2300:3–9. doi: 10.1007/978-1-0716-1386-3_1
36. Saw PE, Xu X, Chen J, Song EW. Non-Coding RNAs: The New Central Dogma of Cancer Biology. Sci China Life Sci (2021) 64:22–50. doi: 10.1007/s11427-020-1700-9
37. Ma N, Tie C, Yu B, Zhang W, Wan J. Identifying lncRNA-miRNA-mRNA Networks to Investigate Alzheimer’s Disease Pathogenesis and Therapy Strategy. Aging (Albany NY) (2020) 12:2897–920. doi: 10.18632/aging.102785
38. Zingale VD, Gugliandolo A, Mazzon E. MiR-155: An Important Regulator of Neuroinflammation. Int J Mol Sci (2021) 23(1):90. doi: 10.3390/ijms23010090
39. Kumar S, Vijayan M, Bhatti JS, Reddy PH. MicroRNAs as Peripheral Biomarkers in Aging and Age-Related Diseases. Prog Mol Biol Transl Sci (2017) 146:47–94. doi: 10.1016/bs.pmbts.2016.12.013
40. Mushtaq G, Greig NH, Anwar F, Zamzami MA, Choudhry H, Shaik MM, et al. miRNAs as Circulating Biomarkers for Alzheimer’s Disease and Parkinson’s Disease. Med Chem (2016) 12:217–25. doi: 10.2174/1573406411666151030112140
41. Fabbri M. TLRs as miRNA Receptors. Cancer Res (2012) 72:6333–7. doi: 10.1158/0008-5472.CAN-12-3229
42. Sivandzade F, Prasad S, Bhalerao A, Cucullo L. NRF2 and NF-κb Interplay in Cerebrovascular and Neurodegenerative Disorders: Molecular Mechanisms and Possible Therapeutic Approaches. Redox Biol (2019) 21:101059. doi: 10.1016/j.redox.2018.11.017
43. Slota JA, Booth SA. MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Noncoding RNA (2019) 5(2):35. doi: 10.3390/ncrna5020035
44. Nunomura A, Perry G. RNA and Oxidative Stress in Alzheimer’s Disease: Focus on microRNAs. Oxid Med Cell Longev (2020) 2020:2638130. doi: 10.1155/2020/2638130
45. Guo Y, Hong W, Wang X, Zhang P, Körner H, Tu J, et al. MicroRNAs in Microglia: How do MicroRNAs Affect Activation, Inflammation, Polarization of Microglia and Mediate the Interaction Between Microglia and Glioma? Front Mol Neurosci (2019) 12:125. doi: 10.3389/fnmol.2019.00125
46. Bayraktar R, Van Roosbroeck K. miR-155 in Cancer Drug Resistance and as Target for miRNA-Based Therapeutics. Cancer Metastasis Rev (2018) 37:33–44. doi: 10.1007/s10555-017-9724-7
47. Hawez A, Al-Haidari A, Madhi R, Rahman M, Thorlacius H. MiR-155 Regulates PAD4-Dependent Formation of Neutrophil Extracellular Traps. Front Immunol (2019) 10:2462. doi: 10.3389/fimmu.2019.02462
48. Aloi MS, Prater KE, Sopher B, Davidson S, Jayadev S, Garden GA. The Pro-Inflammatory microRNA miR-155 Influences Fibrillar β-Amyloid(1) (-42) Catabolism by Microglia. Glia (2021) 69:1736–48. doi: 10.1002/glia.23988
49. Song J, Lee JE. miR-155 Is Involved in Alzheimer’s Disease by Regulating T Lymphocyte Function. Front Aging Neurosci (2015) 7:61. doi: 10.3389/fnagi.2015.00061
50. Chua CEL, Tang BL. miR-34a in Neurophysiology and Neuropathology. J Mol Neurosci (2019) 67:235–46. doi: 10.1007/s12031-018-1231-y
51. Adams BD, Parsons C, Slack FJ. The Tumor-Suppressive and Potential Therapeutic Functions of miR-34a in Epithelial Carcinomas. Expert Opin Ther Targets (2016) 20:737–53. doi: 10.1517/14728222.2016.1114102
52. Van Den Berg MMJ, Krauskopf J, Ramaekers JG, Kleinjans JCS, Prickaerts J, Briedé JJ. Circulating microRNAs as Potential Biomarkers for Psychiatric and Neurodegenerative Disorders. Prog Neurobiol (2020) 185:101732. doi: 10.1016/j.pneurobio.2019.101732
53. Bhattacharjee S, Zhao Y, Dua P, Rogaev EI, Lukiw WJ. microRNA-34a-Mediated Down-Regulation of the Microglial-Enriched Triggering Receptor and Phagocytosis-Sensor TREM2 in Age-Related Macular Degeneration. PloS One (2016) 11:e0150211. doi: 10.1371/journal.pone.0150211
54. Cieślik M, Czapski GA, Wójtowicz S, Wieczorek I, Wencel PL, Strosznajder RP, et al. Alterations of Transcription of Genes Coding Anti-Oxidative and Mitochondria-Related Proteins in Amyloid β Toxicity: Relevance to Alzheimer’s Disease. Mol Neurobiol (2020) 57:1374–88. doi: 10.1007/s12035-019-01819-y
55. Ninawe A, Guru SA, Yadav P, Masroor M, Samadhiya A, Bhutani N, et al. miR-486-5p: A Prognostic Biomarker for Chronic Myeloid Leukemia. ACS Omega (2021) 6:7711–8. doi: 10.1021/acsomega.1c00035
56. Li Q, Xu Y, Lv K, Wang Y, Zhong Z, Xiao C, et al. Small Extracellular Vesicles Containing miR-486-5p Promote Angiogenesis After Myocardial Infarction in Mice and Nonhuman Primates. Sci Transl Med (2021) 13(584):eabb0202. doi: 10.1126/scitranslmed.abb0202
57. Sun B, Guo S. miR-486-5p Serves as a Diagnostic Biomarker for Sepsis and Its Predictive Value for Clinical Outcomes. J Inflammation Res (2021) 14:3687–95. doi: 10.2147/JIR.S323433
58. Wang N, Yu YB. MiR-486 Alleviates Hypoxia/Reoxygenation-Induced H9c2 Cell Injury by Regulating Forkhead Box D3. Eur Rev Med Pharmacol Sci (2022) 26:422–31. doi: 10.26355/eurrev_202201_27866
59. Morris G, Berk M, Maes M, Puri BK. Could Alzheimer’s Disease Originate in the Periphery and If So How So? Mol Neurobiol (2019) 56:406–34. doi: 10.1007/s12035-018-1092-y
60. Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated With Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron (2018) 98:1141–54.e7. doi: 10.1016/j.neuron.2018.05.008
61. Safieh M, Korczyn AD, Michaelson DM. ApoE4: An Emerging Therapeutic Target for Alzheimer’s Disease. BMC Med (2019) 17:64. doi: 10.1186/s12916-019-1299-4
62. Ghafouri-Fard S, Shoorei H, Bahroudi Z, Abak A, Majidpoor J, Taheri M. An Update on the Role of miR-124 in the Pathogenesis of Human Disorders. BioMed Pharmacother (2021) 135:111198. doi: 10.1016/j.biopha.2020.111198
63. An F, Gong G, Wang Y, Bian M, Yu L, Wei C. MiR-124 Acts as a Target for Alzheimer’s Disease by Regulating BACE1. Oncotarget (2017) 8:114065–71. doi: 10.18632/oncotarget.23119
64. Zhao MY, Wang GQ, Wang NN, Yu QY, Liu RL, Shi WQ. The Long-Non-Coding RNA NEAT1 Is a Novel Target for Alzheimer’s Disease Progression via miR-124/BACE1 Axis. Neurol Res (2019) 41:489–97. doi: 10.1080/01616412.2018.1548747
65. Yue D, Guanqun G, Jingxin L, Sen S, Shuang L, Yan S, et al. Silencing of Long Noncoding RNA XIST Attenuated Alzheimer’s Disease-Related BACE1 Alteration Through miR-124. Cell Biol Int (2020) 44:630–6. doi: 10.1002/cbin.11263
66. Yang Y, Ye Y, Kong C, Su X, Zhang X, Bai W, et al. MiR-124 Enriched Exosomes Promoted the M2 Polarization of Microglia and Enhanced Hippocampus Neurogenesis After Traumatic Brain Injury by Inhibiting TLR4 Pathway. Neurochem Res (2019) 44:811–28. doi: 10.1007/s11064-018-02714-z
67. Wang X, Liu D, Huang HZ, Wang ZH, Hou TY, Yang X, et al. A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol Psychiatry (2018) 83:395–405. doi: 10.1016/j.biopsych.2017.07.023
68. Li AD, Tong L, Xu N, Ye Y, Nie PY, Wang ZY, et al. miR-124 Regulates Cerebromicrovascular Function in APP/PS1 Transgenic Mice via C1ql3. Brain Res Bull (2019) 153:214–22. doi: 10.1016/j.brainresbull.2019.09.002
69. Diepenbruck M, Tiede S, Saxena M, Ivanek R, Kalathur RKR, Lüönd F, et al. miR-1199-5p and Zeb1 Function in a Double-Negative Feedback Loop Potentially Coordinating EMT and Tumour Metastasis. Nat Commun (2017) 8:1168. doi: 10.1038/s41467-017-01197-w
70. Zhao Z, Liu G, Zhang H, Ruan P, Ge J, Liu Q. BIRC5, GAJ5, and lncRNA NPHP3-AS1 Are Correlated With the Development of Atrial Fibrillation-Valvular Heart Disease. Int Heart J (2021) 62:153–61. doi: 10.1536/ihj.20-238
71. Kong J, Du Z, Dong L. Pinitol Prevents Lipopolysaccharide (LPS)-Induced Inflammatory Responses in BV2 Microglia Mediated by TREM2. Neurotox Res (2020) 38:96–104. doi: 10.1007/s12640-020-00187-z
72. Gao S, Cheng QC, Hu YG, Tan ZZ, Chen L, Liu SW, et al. LncRNA AK148321 Alleviates Neuroinflammation in LPS-Stimulated BV2 Microglial Cell Through Regulating microRNA-1199-5p/HSPA5 Axis. Life Sci (2021) 266:118863. doi: 10.1016/j.lfs.2020.118863
73. Braga TV, Evangelista FCG, Gomes LC, Araújo S, Carvalho MDG, Sabino AP. Evaluation of MiR-15a and MiR-16-1 as Prognostic Biomarkers in Chronic Lymphocytic Leukemia. BioMed Pharmacother (2017) 92:864–9. doi: 10.1016/j.biopha.2017.05.144
74. Chen Y, Lian YJ, Ma YQ, Wu CJ, Zheng YK, Xie NC. LncRNA SNHG1 Promotes α-Synuclein Aggregation and Toxicity by Targeting miR-15b-5p to Activate SIAH1 in Human Neuroblastoma SH-SY5Y Cells. Neurotoxicology (2018) 68:212–21. doi: 10.1016/j.neuro.2017.12.001
75. Hébert SS, Papadopoulou AS, Smith P, Galas MC, Planel E, Silahtaroglu AN, et al. Genetic Ablation of Dicer in Adult Forebrain Neurons Results in Abnormal Tau Hyperphosphorylation and Neurodegeneration. Hum Mol Genet (2010) 19:3959–69. doi: 10.1093/hmg/ddq311
76. Cai L, Liu X, Guo Q, Huang Q, Zhang Q, Cao Z. MiR-15a Attenuates Peripheral Nerve Injury-Induced Neuropathic Pain by Targeting AKT3 to Regulate Autophagy. Genes Genomics (2020) 42:77–85. doi: 10.1007/s13258-019-00881-z
77. Liu X, Geng J, Guo H, Zhao H, Ai Y. Propofol Inhibited Apoptosis of Hippocampal Neurons in Status Epilepticus Through miR-15a-5p/NR2B/ERK1/2 Pathway. Cell Cycle (2020) 19:1000–11. doi: 10.1080/15384101.2020.1743909
78. Xu K, Chen C, Wu Y, Wu M, Lin L. Advances in miR-132-Based Biomarker and Therapeutic Potential in the Cardiovascular System. Front Pharmacol (2021) 12:751487. doi: 10.3389/fphar.2021.751487
79. Hadar A, Milanesi E, Walczak M, Puzianowska-Kuźnicka M, Kuźnicki J, Squassina A, et al. SIRT1, miR-132 and miR-212 Link Human Longevity to Alzheimer’s Disease. Sci Rep (2018) 8:8465. doi: 10.1038/s41598-018-26547-6
80. Xu N, Li AD, Ji LL, Ye Y, Wang ZY, Tong L. miR-132 Regulates the Expression of Synaptic Proteins in APP/PS1 Transgenic Mice Through C1q. Eur J Histochem (2019) 63(2):3008. doi: 10.4081/ejh.2019.3008
81. Deng Y, Zhang J, Sun X, Ma G, Luo G, Miao Z, et al. miR-132 Improves the Cognitive Function of Rats With Alzheimer’s Disease by Inhibiting the MAPK1 Signal Pathway. Exp Ther Med (2020) 20:159. doi: 10.3892/etm.2020.9288
82. Cha DJ, Mengel D, Mustapic M, Liu W, Selkoe DJ, Kapogiannis D, et al. miR-212 and miR-132 Are Downregulated in Neurally Derived Plasma Exosomes of Alzheimer’s Patients. Front Neurosci (2019) 13:1208. doi: 10.3389/fnins.2019.01208
83. Ansari A, Maffioletti E, Milanesi E, Marizzoni M, Frisoni GB, Blin O, et al. miR-146a and miR-181a Are Involved in the Progression of Mild Cognitive Impairment to Alzheimer’s Disease. Neurobiol Aging (2019) 82:102–9. doi: 10.1016/j.neurobiolaging.2019.06.005
84. Pan JA, Tang Y, Yu JY, Zhang H, Zhang JF, Wang CQ, et al. miR-146a Attenuates Apoptosis and Modulates Autophagy by Targeting TAF9b/P53 Pathway in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis (2019) 10:668. doi: 10.1038/s41419-019-1901-x
85. Li Y, Zhu H, Wei X, Li H, Yu Z, Zhang H, et al. LPS Induces HUVEC Angiogenesis In Vitro Through miR-146a-Mediated TGF-β1 Inhibition. Am J Transl Res (2017) 9:591–600.
86. Nakano M, Kubota K, Kobayashi E, Chikenji TS, Saito Y, Konari N, et al. Bone Marrow-Derived Mesenchymal Stem Cells Improve Cognitive Impairment in an Alzheimer’s Disease Model by Increasing the Expression of microRNA-146a in Hippocampus. Sci Rep (2020) 10:10772. doi: 10.1038/s41598-020-67460-1
87. Sundaravinayagam D, Kim HR, Wu T, Kim HH, Lee HS, Jun S, et al. Mir146a-Mediated Targeting of FANCM During Inflammation Compromises Genome Integrity. Oncotarget (2016) 7:45976–94. doi: 10.18632/oncotarget.10275
88. Haneklaus M, Gerlic M, O’neill LA, Masters SL. miR-223: Infection, Inflammation and Cancer. J Intern Med (2013) 274:215–26. doi: 10.1111/joim.12099
89. Long FQ, Kou CX, Li K, Wu J, Wang QQ. MiR-223-3p Inhibits Rtp17-Induced Inflammasome Activation and Pyroptosis by Targeting NLRP3. J Cell Mol Med (2020) 24:14405–14. doi: 10.1111/jcmm.16061
90. Zhang N, Fu L, Bu Y, Yao Y, Wang Y. Downregulated Expression of miR-223 Promotes Toll-Like Receptor-Activated Inflammatory Responses in Macrophages by Targeting RhoB. Mol Immunol (2017) 91:42–8. doi: 10.1016/j.molimm.2017.08.026
91. Mancuso R, Agostini S, Hernis A, Zanzottera M, Bianchi A, Clerici M. Circulatory miR-223-3p Discriminates Between Parkinson’s and Alzheimer’s Patients. Sci Rep (2019) 9:9393. doi: 10.1038/s41598-019-45687-x
92. Peng B, Theng PY, Le MTN. Essential Functions of miR-125b in Cancer. Cell Prolif (2021) 54:e12913. doi: 10.1111/cpr.12913
93. Xie C, Zhang LZ, Chen ZL, Zhong WJ, Fang JH, Zhu Y, et al. A Hmtr4-PDIA3P1-miR-125/124-TRAF6 Regulatory Axis and Its Function in NF Kappa B Signaling and Chemoresistance. Hepatology (2020) 71:1660–77. doi: 10.1002/hep.30931
94. Li P, Xu Y, Wang B, Huang J, Li Q. miR-34a-5p and miR-125b-5p Attenuate Aβ-Induced Neurotoxicity Through Targeting BACE1. J Neurol Sci (2020) 413:116793. doi: 10.1016/j.jns.2020.116793
95. Tan L, Yu JT, Liu QY, Tan MS, Zhang W, Hu N, et al. Circulating miR-125b as a Biomarker of Alzheimer’s Disease. J Neurol Sci (2014) 336:52–6. doi: 10.1016/j.jns.2013.10.002
96. Jin Y, Tu Q, Liu M. MicroRNA−125b Regulates Alzheimer’s Disease Through SphK1 Regulation. Mol Med Rep (2018) 18:2373–80. doi: 10.3892/mmr.2018.9156
97. Ma P, Li Y, Zhang W, Fang F, Sun J, Liu M, et al. Long Non-Coding RNA MALAT1 Inhibits Neuron Apoptosis and Neuroinflammation While Stimulates Neurite Outgrowth and Its Correlation With MiR-125b Mediates PTGS2, CDK5 and FOXQ1 in Alzheimer’s Disease. Curr Alzheimer Res (2019) 16:596–612. doi: 10.2174/1567205016666190725130134
98. Rodriguez-Ortiz CJ, Prieto GA, Martini AC, Forner S, Trujillo-Estrada L, Laferla FM, et al. miR-181a Negatively Modulates Synaptic Plasticity in Hippocampal Cultures and Its Inhibition Rescues Memory Deficits in a Mouse Model of Alzheimer’s Disease. Aging Cell (2020) 19:e13118. doi: 10.1111/acel.13118
99. Kim C, Ye Z, Weyand CM, Goronzy JJ. miR-181a-Regulated Pathways in T-Cell Differentiation and Aging. Immun Ageing (2021) 18:28. doi: 10.1186/s12979-021-00240-1
100. Wang H, Wang X, Zhang Y, Zhao J. LncRNA SNHG1 Promotes Neuronal Injury in Parkinson’s Disease Cell Model by miR-181a-5p/CXCL12 Axis. J Mol Histol (2021) 52:153–63. doi: 10.1007/s10735-020-09931-3
101. Min JW, Lee J, Mun HJ, Kim DH, Park BG, Yoon B, et al. Diagnostic and Therapeutic Biomarkers for Alzheimer’s Disease in Human-Derived Platelets. Genes Genomics (2020) 42:1467–75. doi: 10.1007/s13258-020-01015-6
102. Ying W, Tseng A, Chang RC, Wang H, Lin YL, Kanameni S, et al. miR-150 Regulates Obesity-Associated Insulin Resistance by Controlling B Cell Functions. Sci Rep (2016) 6:20176. doi: 10.1038/srep20176
103. Barnett RE, Conklin DJ, Ryan L, Keskey RC, Ramjee V, Sepulveda EA, et al. Anti-Inflammatory Effects of miR-21 in the Macrophage Response to Peritonitis. J Leukoc Biol (2016) 99:361–71. doi: 10.1189/jlb.4A1014-489R
104. Zhou W, Su L, Duan X, Chen X, Hays A, Upadhyayula S, et al. MicroRNA-21 Down-Regulates Inflammation and Inhibits Periodontitis. Mol Immunol (2018) 101:608–14. doi: 10.1016/j.molimm.2018.05.008
105. Yin Z, Han Z, Hu T, Zhang S, Ge X, Huang S, et al. Neuron-Derived Exosomes With High miR-21-5p Expression Promoted Polarization of M1 Microglia in Culture. Brain Behav Immun (2020) 83:270–82. doi: 10.1016/j.bbi.2019.11.004
106. Olivieri F, Prattichizzo F, Giuliani A, Matacchione G, Rippo MR, Sabbatinelli J, et al. miR-21 and miR-146a: The microRNAs of Inflammaging and Age-Related Diseases. Ageing Res Rev (2021) 70:101374. doi: 10.1016/j.arr.2021.101374
107. Ning SL, Zhu H, Shao J, Liu YC, Lan J, Miao J. MiR-21 Inhibitor Improves Locomotor Function Recovery by Inhibiting IL-6r/JAK-STAT Pathway-Mediated Inflammation After Spinal Cord Injury in Model of Rat. Eur Rev Med Pharmacol Sci (2019) 23:433–40. doi: 10.26355/eurrev_201901_16852
108. Roush S, Slack FJ. The Let-7 Family of microRNAs. Trends Cell Biol (2008) 18:505–16. doi: 10.1016/j.tcb.2008.07.007
109. Bernstein DL, Jiang X, Rom S. Let-7 microRNAs: Their Role in Cerebral and Cardiovascular Diseases, Inflammation, Cancer, and Their Regulation. Biomedicines (2021) 9(6):606. doi: 10.3390/biomedicines9060606
110. Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA Localization and Function. J Cell Biol (2021) 220(2):e202009045. doi: 10.1083/jcb.202009045
111. Wu P, Zuo X, Deng H, Liu X, Liu L, Ji A. Roles of Long Noncoding RNAs in Brain Development, Functional Diversification and Neurodegenerative Diseases. Brain Res Bull (2013) 97:69–80. doi: 10.1016/j.brainresbull.2013.06.001
112. Riva P, Ratti A, Venturin M. The Long Non-Coding RNAs in Neurodegenerative Diseases: Novel Mechanisms of Pathogenesis. Curr Alzheimer Res (2016) 13:1219–31. doi: 10.2174/1567205013666160622112234
113. Zhou S, Yu X, Wang M, Meng Y, Song D, Yang H, et al. Long Non-Coding RNAs in Pathogenesis of Neurodegenerative Diseases. Front Cell Dev Biol (2021) 9:719247. doi: 10.3389/fcell.2021.719247
114. Yan Y, Song D, Song X, Song C. The Role of lncRNA MALAT1 in Cardiovascular Disease. IUBMB Life (2020) 72:334–42. doi: 10.1002/iub.2210
115. Yu X, Li Z, Zheng H, Chan MT, Wu WK. NEAT1: A Novel Cancer-Related Long Non-Coding RNA. Cell Prolif (2017) 50(2):e12329. doi: 10.1111/cpr.12329
116. Dong LX, Zhang YY, Bao HL, Liu Y, Zhang GW, An FM. LncRNA NEAT1 Promotes Alzheimer’s Disease by Down Regulating Micro-27a-3p. Am J Transl Res (2021) 13:8885–96.
117. Han D, Zhou Y. YY1-Induced Upregulation of lncRNA NEAT1 Contributes to OGD/R Injury-Induced Inflammatory Response in Cerebral Microglial Cells via Wnt/β-Catenin Signaling Pathway. In Vitro Cell Dev Biol Anim (2019) 55:501–11. doi: 10.1007/s11626-019-00375-y
118. Cai M, Wang YW, Xu SH, Qiao S, Shu QF, Du JZ, et al. Regulatory Effects of the Long Non−Coding RNA RP11−543N12.1 and microRNA−324−3p Axis on the Neuronal Apoptosis Induced by the Inflammatory Reactions of Microglia. Int J Mol Med (2018) 42:1741–55. doi: 10.3892/ijmm.2018.3736
119. Al-Rugeebah A, Alanazi M, Parine NR. MEG3: An Oncogenic Long Non-Coding RNA in Different Cancers. Pathol Oncol Res (2019) 25:859–74. doi: 10.1007/s12253-019-00614-3
120. Chen Y, Li Z, Chen X, Zhang S. Long Non-Coding RNAs: From Disease Code to Drug Role. Acta Pharm Sin B (2021) 11:340–54. doi: 10.1016/j.apsb.2020.10.001
121. Yi J, Chen B, Yao X, Lei Y, Ou F, Huang F. Upregulation of the lncRNA MEG3 Improves Cognitive Impairment, Alleviates Neuronal Damage, and Inhibits Activation of Astrocytes in Hippocampus Tissues in Alzheimer’s Disease Through Inactivating the PI3K/Akt Signaling Pathway. J Cell Biochem (2019) 120:18053–65. doi: 10.1002/jcb.29108
122. Chen L, Qu H, Guo M, Zhang Y, Cui Y, Yang Q, et al. ANRIL and Atherosclerosis. J Clin Pharm Ther (2020) 45:240–8. doi: 10.1111/jcpt.13060
123. Guo F, Tang C, Li Y, Liu Y, Lv P, Wang W, et al. The Interplay of LncRNA ANRIL and miR-181b on the Inflammation-Relevant Coronary Artery Disease Through Mediating NF-κb Signalling Pathway. J Cell Mol Med (2018) 22:5062–75. doi: 10.1111/jcmm.13790
124. Wei JC, Shi YL, Wang Q. LncRNA ANRIL Knockdown Ameliorates Retinopathy in Diabetic Rats by Inhibiting the NF-κb Pathway. Eur Rev Med Pharmacol Sci (2019) 23:7732–9. doi: 10.26355/eurrev_201909_18982
125. Zhou B, Li L, Qiu X, Wu J, Xu L, Shao W. Long non-Coding RNA ANRIL Knockdown Suppresses Apoptosis and Pro-Inflammatory Cytokines While Enhancing Neurite Outgrowth via Binding microRNA-125a in a Cellular Model of Alzheimer’s Disease. Mol Med Rep (2020) 22:1489–97. doi: 10.3892/mmr.2020.11203
126. Zhang C, Li B. The Correlation Between LncRNA-17A Expression in Peripheral Blood Mononuclear Cells and Wnt/β-Catenin Signaling Pathway and Cognitive Function in Patients With Alzheimer Disease. Am J Transl Res (2021) 13:11981–6.
127. Massone S, Vassallo I, Fiorino G, Castelnuovo M, Barbieri F, Borghi R, et al. 17A, a Novel Non-Coding RNA, Regulates GABA B Alternative Splicing and Signaling in Response to Inflammatory Stimuli and in Alzheimer Disease. Neurobiol Dis (2011) 41:308–17. doi: 10.1016/j.nbd.2010.09.019
128. Wang X, Zhang M, Liu H. LncRNA17A Regulates Autophagy and Apoptosis of SH-SY5Y Cell Line as an In Vitro Model for Alzheimer’s Disease. Biosci Biotechnol Biochem (2019) 83:609–21. doi: 10.1080/09168451.2018.1562874
129. Li D, Wang J, Zhang M, Hu X, She J, Qiu X, et al. LncRNA MAGI2-AS3 Is Regulated by BRD4 and Promotes Gastric Cancer Progression via Maintaining ZEB1 Overexpression by Sponging miR-141/200a. Mol Ther Nucleic Acids (2020) 19:109–23. doi: 10.1016/j.omtn.2019.11.003
130. Sui Y, Chi W, Feng L, Jiang J. LncRNA MAGI2-AS3 Is Downregulated in non-Small Cell Lung Cancer and may be a Sponge of miR-25. BMC Pulm Med (2020) 20:59. doi: 10.1186/s12890-020-1064-7
131. Tang C, Cai Y, Jiang H, Lv Z, Yang C, Xu H, et al. LncRNA MAGI2-AS3 Inhibits Bladder Cancer Progression by Targeting the miR-31-5p/TNS1 Axis. Aging (Albany NY) (2020) 12:25547–63. doi: 10.18632/aging.104162
132. Xu X, Yuan X, Ni J, Guo J, Gao Y, Yin W, et al. MAGI2-AS3 Inhibits Breast Cancer by Downregulating DNA Methylation of MAGI2. J Cell Physiol (2021) 236:1116–30. doi: 10.1002/jcp.29922
133. Chang H, Zhang X, Li B, Meng X. MAGI2-AS3 Suppresses MYC Signaling to Inhibit Cell Proliferation and Migration in Ovarian Cancer Through Targeting miR-525-5p/MXD1 Axis. Cancer Med (2020) 9:6377–86. doi: 10.1002/cam4.3126
134. Fang G, Wang J, Sun X, Xu R, Zhao X, Shao L, et al. LncRNA MAGI2-AS3 is Downregulated in the Distant Recurrence of Hepatocellular Carcinoma After Surgical Resection and Affects Migration and Invasion via ROCK2. Ann Hepatol (2020) 19:535–40. doi: 10.1016/j.aohep.2020.04.007
135. Zhang J, Wang R. Deregulated lncRNA MAGI2-AS3 in Alzheimer’s Disease Attenuates Amyloid-β Induced Neurotoxicity and Neuroinflammation by Sponging miR-374b-5p. Exp Gerontol (2021) 144:111180. doi: 10.1016/j.exger.2020.111180
136. Guo C, Qi Y, Qu J, Gai L, Shi Y, Yuan C. Pathophysiological Functions of the lncRNA Tug1. Curr Pharm Des (2020) 26:688–700. doi: 10.2174/1381612826666191227154009
137. Su Q, Liu Y, Lv XW, Dai RX, Yang XH, Kong BH. LncRNA TUG1 Mediates Ischemic Myocardial Injury by Targeting miR-132-3p/HDAC3 Axis. Am J Physiol Heart Circ Physiol (2020) 318:H332–h344. doi: 10.1152/ajpheart.00444.2019
138. Jia H, Li Z, Chang Y, Fang B, Zhou Y, Ma H. Downregulation of Long Noncoding RNA TUG1 Attenuates MTDH-Mediated Inflammatory Damage via Targeting miR-29b-1-5p After Spinal Cord Ischemia Reperfusion. J Neuropathol Exp Neurol (2021) 80:254–64. doi: 10.1093/jnen/nlaa138
139. Li X, Wang SW, Li XL, Yu FY, Cong HM. Knockdown of Long Non-Coding RNA TUG1 Depresses Apoptosis of Hippocampal Neurons in Alzheimer’s Disease by Elevating microRNA-15a and Repressing ROCK1 Expression. Inflammation Res (2020) 69:897–910. doi: 10.1007/s00011-020-01364-8
140. Zhou WY, Cai ZR, Liu J, Wang DS, Ju HQ, Xu RH. Circular RNA: Metabolism, Functions and Interactions With Proteins. Mol Cancer (2020) 19:172. doi: 10.1186/s12943-020-01286-3
141. Xu X, Zhang J, Tian Y, Gao Y, Dong X, Chen W, et al. CircRNA Inhibits DNA Damage Repair by Interacting With Host Gene. Mol Cancer (2020) 19:128. doi: 10.1186/s12943-020-01246-x
142. Zang J, Lu D, Xu A. The Interaction of circRNAs and RNA Binding Proteins: An Important Part of circRNA Maintenance and Function. J Neurosci Res (2020) 98:87–97. doi: 10.1002/jnr.24356
143. Yang H, Wang H, Shang H, Chen X, Yang S, Qu Y, et al. Circular RNA Circ_0000950 Promotes Neuron Apoptosis, Suppresses Neurite Outgrowth and Elevates Inflammatory Cytokines Levels via Directly Sponging miR-103 in Alzheimer’s Disease. Cell Cycle (2019) 18:2197–214. doi: 10.1080/15384101.2019.1629773
144. Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in Cancer. Cancer Res (2013) 73:5609–12. doi: 10.1158/0008-5472.CAN-13-1568
145. Chen J, Yang J, Fei X, Wang X, Wang K. CircRNA ciRS-7: A Novel Oncogene in Multiple Cancers. Int J Biol Sci (2021) 17:379–89. doi: 10.7150/ijbs.54292
146. Zhao YH, Wang Z, Zhang N, Cui T, Zhang YH. Effect of ciRS-7 Expression on Clear Cell Renal Cell Carcinoma Progression. Chin Med J (Engl) (2020) 133:2084–9. doi: 10.1097/CM9.0000000000000867
147. Zhou Y, Shen L, Wang YZ, Zhou CC. The Potential of ciRS-7 for Predicting Onset and Prognosis of Cervical Cancer. Neoplasma (2020) 67:312–22. doi: 10.4149/neo_2019_190415N334
148. Dou Z, Gao L, Ren W, Zhang H, Wang X, Li S, et al. CiRS-7 Functions as a ceRNA of RAF-1/PIK3CD to Promote Metastatic Progression of Oral Squamous Cell Carcinoma via MAPK/AKT Signaling Pathways. Exp Cell Res (2020) 396:112290. doi: 10.1016/j.yexcr.2020.112290
149. Han JY, Guo S, Wei N, Xue R, Li W, Dong G, et al. ciRS-7 Promotes the Proliferation and Migration of Papillary Thyroid Cancer by Negatively Regulating the miR-7/Epidermal Growth Factor Receptor Axis. BioMed Res Int (2020) 2020:9875636. doi: 10.1155/2020/9875636
150. Shi Z, Chen T, Yao Q, Zheng L, Zhang Z, Wang J, et al. The Circular RNA ciRS-7 Promotes APP and BACE1 Degradation in an NF-κb-Dependent Manner. FEBS J (2017) 284:1096–109. doi: 10.1111/febs.14045
151. Zhou X, Jiang L, Fan G, Yang H, Wu L, Huang Y, et al. Role of the ciRS-7/miR-7 Axis in the Regulation of Proliferation, Apoptosis and Inflammation of Chondrocytes Induced by IL-1β. Int Immunopharmacol (2019) 71:233–40. doi: 10.1016/j.intimp.2019.03.037
152. Cai W, Zhang Y, Su Z. ciRS-7 Targeting miR-135a-5p Promotes Neuropathic Pain in CCI Rats via Inflammation and Autophagy. Gene (2020) 736:144386. doi: 10.1016/j.gene.2020.144386
153. Li R, Tang X, Xu C, Guo Y, Qi L, Li S, et al. Circular RNA NF1-419 Inhibits Proliferation and Induces Apoptosis by Regulating Lipid Metabolism in Astroglioma Cells. Recent Pat Anticancer Drug Discov (2021) 17(2):162–77. doi: 10.2174/1574892816666210729125802
154. Diling C, Yinrui G, Longkai Q, Xiaocui T, Yadi L, Xin Y, et al. Circular RNA NF1-419 Enhances Autophagy to Ameliorate Senile Dementia by Binding Dynamin-1 and Adaptor Protein 2 B1 in AD-Like Mice. Aging (Albany NY) (2019) 11:12002–31. doi: 10.18632/aging.102529
155. Diling C, Longkai Q, Yinrui G, Yadi L, Xiaocui T, Xiangxiang Z, et al. CircNF1-419 Improves the Gut Microbiome Structure and Function in AD-Like Mice. Aging (Albany NY) (2020) 12:260–87. doi: 10.18632/aging.102614
156. Lu Y, Tan L, Wang X. Circular HDAC9/microRNA-138/Sirtuin-1 Pathway Mediates Synaptic and Amyloid Precursor Protein Processing Deficits in Alzheimer’s Disease. Neurosci Bull (2019) 35:877–88. doi: 10.1007/s12264-019-00361-0
157. Zhang N, Gao Y, Yu S, Sun X, Shen K. Berberine Attenuates Aβ42-Induced Neuronal Damage Through Regulating Circhdac9/miR-142-5p Axis in Human Neuronal Cells. Life Sci (2020) 252:117637. doi: 10.1016/j.lfs.2020.117637
158. Huang JL, Qin MC, Zhou Y, Xu ZH, Yang SM, Zhang F, et al. Comprehensive Analysis of Differentially Expressed Profiles of Alzheimer’s Disease Associated Circular RNAs in an Alzheimer’s Disease Mouse Model. Aging (Albany NY) (2018) 10:253–65. doi: 10.18632/aging.101387
159. Klattenhoff C, Theurkauf W. Biogenesis and Germline Functions of piRNAs. Development (2008) 135:3–9. doi: 10.1242/dev.006486
160. Samji T. PIWI, piRNAs, and Germline Stem Cells: What’s the Link? Yale J Biol Med (2009) 82:121–4.
161. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
162. Cornes E, Bourdon L, Singh M, Mueller F, Quarato P, Wernersson E, et al. piRNAs Initiate Transcriptional Silencing of Spermatogenic Genes During C. Elegans Germline Development. Dev Cell (2022) 57:180–196.e187. doi: 10.1016/j.devcel.2021.11.025
163. Ow MC, Hall SE. piRNAs and endo-siRNAs: Small Molecules With Large Roles in the Nervous System. Neurochem Int (2021) 148:105086. doi: 10.1016/j.neuint.2021.105086
164. Kim KW. PIWI Proteins and piRNAs in the Nervous System. Mol Cells (2019) 42:828–35. doi: 10.14348/molcells.2019.0241
165. Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, et al. A Role for Neuronal piRNAs in the Epigenetic Control of Memory-Related Synaptic Plasticity. Cell (2012) 149:693–707. doi: 10.1016/j.cell.2012.02.057
166. Jain G, Stuendl A, Rao P, Berulava T, Pena Centeno T, Kaurani L, et al. A Combined miRNA-piRNA Signature to Detect Alzheimer’s Disease. Transl Psychiatry (2019) 9:250. doi: 10.1038/s41398-019-0579-2
167. Mao Q, Fan L, Wang X, Lin X, Cao Y, Zheng C, et al. Transcriptome-Wide piRNA Profiling in Human Brains for Aging Genetic Factors. Jacobs J Genet (2019) 4(1):14.
168. Li S, Xu Z, Sheng J. tRNA-Derived Small RNA: A Novel Regulatory Small Non-Coding RNA. Genes (Basel) (2018) 9(5):246. doi: 10.3390/genes9050246
169. Oberbauer V, Schaefer MR. tRNA-Derived Small RNAs: Biogenesis, Modification, Function and Potential Impact on Human Disease Development. Genes (Basel) (2018) 9(12):607. doi: 10.3390/genes9120607
170. Di Fazio A, Schlackow M, Pong SK, Alagia A, Gullerova M. Dicer Dependent tRNA Derived Small RNAs Promote Nascent RNA Silencing. Nucleic Acids Res (2022) 50:1734–52. doi: 10.1093/nar/gkac022
171. Peng R, Santos HJ, Nozaki T. Transfer RNA-Derived Small RNAs in the Pathogenesis of Parasitic Protozoa. Genes (Basel) (2022) 13(2):286. doi: 10.3390/genes13020286
172. Xu XJ, Yang MS, Zhang B, Ge QQ, Niu F, Dong JQ, et al. Genome-Wide Interrogation of Transfer RNA-Derived Small RNAs in a Mouse Model of Traumatic Brain Injury. Neural Regener Res (2022) 17:386–94. doi: 10.4103/1673-5374.314315
173. Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, et al. CLP1 Links tRNA Metabolism to Progressive Motor-Neuron Loss. Nature (2013) 495:474–80. doi: 10.1038/nature11923
174. Weitzer S, Hanada T, Penninger JM, Martinez J. CLP1 as a Novel Player in Linking tRNA Splicing to Neurodegenerative Disorders. Wiley Interdiscip Rev RNA (2015) 6:47–63. doi: 10.1002/wrna.1255
175. Wu W, Lee I, Spratt H, Fang X, Bao X. tRNA-Derived Fragments in Alzheimer’s Disease: Implications for New Disease Biomarkers and Neuropathological Mechanisms. J Alzheimers Dis (2021) 79:793–806. doi: 10.3233/JAD-200917
176. Matsui M, Corey DR. Non-Coding RNAs as Drug Targets. Nat Rev Drug Discovery (2017) 16:167–79. doi: 10.1038/nrd.2016.117
177. Guo C, Liu J, Zhou Q, Song J, Zhang Z, Li Z, et al. Exosomal Noncoding RNAs and Tumor Drug Resistance. Cancer Res (2020) 80:4307–13. doi: 10.1158/0008-5472.CAN-20-0032
178. Li Y, Wang R, Zhang S, Xu H, Deng L. LRGCPND: Predicting Associations Between ncRNA and Drug Resistance via Linear Residual Graph Convolution. Int J Mol Sci (2021) 22(19):10508. doi: 10.3390/ijms221910508
179. Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA-Targeted Therapeutics. Cell Metab (2018) 27:714–39. doi: 10.1016/j.cmet.2018.03.004
180. Chi X, Gatti P, Papoian T. Safety of Antisense Oligonucleotide and siRNA-Based Therapeutics. Drug Discovery Today (2017) 22:823–33. doi: 10.1016/j.drudis.2017.01.013
181. Yu AM, Choi YH, Tu MJ. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacol Rev (2020) 72:862–98. doi: 10.1124/pr.120.019554
182. Hammond SM, Aartsma-Rus A, Alves S, Borgos SE, Buijsen R, Collin RWJ, et al. Delivery of Oligonucleotide-Based Therapeutics: Challenges and Opportunities. EMBO Mol Med (2021) 13:e13243. doi: 10.15252/emmm.202013243
183. Vishnoi A, Rani S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol Biol (2017) 1509:1–10. doi: 10.1007/978-1-4939-6524-3_1
184. Faravelli I, Corti S. MicroRNA-Directed Neuronal Reprogramming as a Therapeutic Strategy for Neurological Diseases. Mol Neurobiol (2018) 55:4428–36. doi: 10.1007/s12035-017-0671-7
185. Gandhi G, Abdullah S, Foead AI, Yeo WWY. The Potential Role of miRNA Therapies in Spinal Muscle Atrophy. J Neurol Sci (2021) 427:117485. doi: 10.1016/j.jns.2021.117485
186. Kranick JC, Chadalavada DM, Sahu D, Showalter SA. Engineering Double-Stranded RNA Binding Activity Into the Drosha Double-Stranded RNA Binding Domain Results in a Loss of microRNA Processing Function. PloS One (2017) 12:e0182445. doi: 10.1371/journal.pone.0182445
187. Zhou X, Dai E, Song Q, Ma X, Meng Q, Jiang Y, et al. In Silico Drug Repositioning Based on drug-miRNA Associations. Brief Bioinform (2020) 21:498–510. doi: 10.1093/bib/bbz012
188. Blokhin I, Khorkova O, Hsiao J, Wahlestedt C. Developments in lncRNA Drug Discovery: Where are We Heading? Expert Opin Drug Discovery (2018) 13:837–49. doi: 10.1080/17460441.2018.1501024
189. Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med (2021) 385:493–502. doi: 10.1056/NEJMoa2107454
190. Liu C, Zhang L, Liu H, Cheng K. Delivery Strategies of the CRISPR-Cas9 Gene-Editing System for Therapeutic Applications. J Control Release (2017) 266:17–26. doi: 10.1016/j.jconrel.2017.09.012
191. Min HS, Kim HJ, Naito M, Ogura S, Toh K, Hayashi K, et al. Systemic Brain Delivery of Antisense Oligonucleotides Across the Blood-Brain Barrier With a Glucose-Coated Polymeric Nanocarrier. Angew Chem Int Ed Engl (2020) 59:8173–80. doi: 10.1002/anie.201914751
192. Cullis PR, Hope MJ. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol Ther (2017) 25:1467–75. doi: 10.1016/j.ymthe.2017.03.013
193. Dong Y, Siegwart DJ, Anderson DG. Strategies, Design, and Chemistry in siRNA Delivery Systems. Adv Drug Delivery Rev (2019) 144:133–47. doi: 10.1016/j.addr.2019.05.004
194. Di Ianni T, Bose RJC, Sukumar UK, Bachawal S, Wang H, Telichko A, et al. Ultrasound/microbubble-Mediated Targeted Delivery of Anticancer microRNA-Loaded Nanoparticles to Deep Tissues in Pigs. J Control Release (2019) 309:1–10. doi: 10.1016/j.jconrel.2019.07.024
195. Kimura T, Ferran B, Tsukahara Y, Shang Q, Desai S, Fedoce A, et al. Production of Adeno-Associated Virus Vectors for In Vitro and In Vivo Applications. Sci Rep (2019) 9:13601. doi: 10.1038/s41598-019-49624-w
196. Fu YW, Dai XY, Wang WT, Yang ZX, Zhao JJ, Zhang JP, et al. Dynamics and Competition of CRISPR-Cas9 Ribonucleoproteins and AAV Donor-Mediated NHEJ, MMEJ and HDR Editing. Nucleic Acids Res (2021) 49:969–85. doi: 10.1093/nar/gkaa1251
197. Kou X, Chen D, Chen N. The Regulation of microRNAs in Alzheimer’s Disease. Front Neurol (2020) 11:288. doi: 10.3389/fneur.2020.00288
198. Giunti D, Marini C, Parodi B, Usai C, Milanese M, Bonanno G, et al. Role of miRNAs Shuttled by Mesenchymal Stem Cell-Derived Small Extracellular Vesicles in Modulating Neuroinflammation. Sci Rep (2021) 11:1740. doi: 10.1038/s41598-021-81039-4
199. Lee CY, Ryu IS, Ryu JH, Cho HJ. miRNAs as Therapeutic Tools in Alzheimer’s Disease. Int J Mol Sci (2021) 22(23):13012. doi: 10.3390/ijms222313012
Glossary

Keywords: non-coding RNAs, Alzheimer’s disease, neuroinflammation, regulators, therapeutic targets
Citation: Liu Y, Cheng X, Li H, Hui S, Zhang Z, Xiao Y and Peng W (2022) Non-Coding RNAs as Novel Regulators of Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 13:908076. doi: 10.3389/fimmu.2022.908076
Received: 30 March 2022; Accepted: 09 May 2022;
Published: 02 June 2022.
Edited by:
Isabella Laura Simone, University of Bari Aldo Moro, ItalyReviewed by:
Hermona Soreq, Hebrew University of Jerusalem, IsraelErica Costantini, University of Studies G. d’Annunzio Chieti and Pescara, Italy
Copyright © 2022 Liu, Cheng, Li, Hui, Zhang, Xiao and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Xiao, eGlhb3lhbmcyOUBjc3UuZWR1LmNu; Weijun Peng, cGVuZ3dlaWp1bjg3QGNzdS5lZHUuY24=