 Alejandro Mendoza-Alvarez1
Alejandro Mendoza-Alvarez1 Eva Tosco-Herrera1
Eva Tosco-Herrera1 Adrian Muñoz-Barrera2
Adrian Muñoz-Barrera2 Luis A. Rubio-Rodríguez2
Luis A. Rubio-Rodríguez2 Aitana Alonso-Gonzalez1,3Almudena Corrales1,4Antonio Iñigo-Campos2
Aitana Alonso-Gonzalez1,3Almudena Corrales1,4Antonio Iñigo-Campos2 Lourdes Almeida-Quintana5Elena Martin-Fernandez6Dara Martinez-Beltran7Eva Perez-Rodriguez8
Lourdes Almeida-Quintana5Elena Martin-Fernandez6Dara Martinez-Beltran7Eva Perez-Rodriguez8 Ariel Callero8
Ariel Callero8 Jose C. Garcia-Robaina8
Jose C. Garcia-Robaina8 Rafaela González-Montelongo2
Rafaela González-Montelongo2 Itahisa Marcelino-Rodriguez1,9
Itahisa Marcelino-Rodriguez1,9 Jose M. Lorenzo-Salazar2
Jose M. Lorenzo-Salazar2 Carlos Flores1,2,4,10*
Carlos Flores1,2,4,10*- 1Research Unit, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, Spain
- 2Genomics Division, Instituto Tecnológico y de Energías Renovables, Santa Cruz de Tenerife, Spain
- 3Universidad de Santiago de Compostela, Santiago de Compostela, Spain
- 4CIBER de Enfermedades Respiratorias, Instituto de Salud Carlos III, Madrid, Spain
- 5Allergy Service, Hospital Universitario de Gran Canaria Dr. Negrín, Las Palmas de Gran Canaria, Spain
- 6Allergy Service, Hospital Universitario Dr. Molina Orosa, Las Palmas de Gran Canaria, Spain
- 7Allergy Service, Hospital Universitario Insular-Materno Infantil, Las Palmas de Gran Canaria, Spain
- 8Allergy Service, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, Spain
- 9Public Health and Preventive Medicine Area, Universidad de La Laguna, Santa Cruz de Tenerife, Spain
- 10Facultad de Ciencias de la Salud, Universidad Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain
Hereditary angioedema (HAE) is a rare disease where known causes involve C1 inhibitor dysfunction or dysregulation of the kinin cascade. The updated HAE management guidelines recommend performing genetic tests to reach a precise diagnosis. Unfortunately, genetic tests are still uncommon in the diagnosis routine. Here, we characterized for the first time the genetic causes of HAE in affected families from the Canary Islands (Spain). Whole-exome sequencing data was obtained from 41 affected patients and unaffected relatives from 29 unrelated families identified in the archipelago. The Hereditary Angioedema Database Annotation (HADA) tool was used for pathogenicity classification and causal variant prioritization among the genes known to cause HAE. Manual reclassification of prioritized variants was used in those families lacking known causal variants. We detected a total of eight different variants causing HAE in this patient series, affecting essentially SERPING1 and F12 genes, one of them being a novel SERPING1 variant (c.686-12A>G) with a predicted splicing effect which was reclassified as likely pathogenic in one family. Altogether, the diagnostic yield by assessing previously reported causal genes and considering variant reclassifications according to the American College of Medical Genetics guidelines reached 66.7% (95% Confidence Interval [CI]: 30.1-91.0) in families with more than one affected member and 10.0% (95% CI: 1.8-33.1) among cases without family information for the disease. Despite the genetic causes of many patients remain to be identified, our results reinforce the need of genetic tests as first-tier diagnostic tool in this disease, as recommended by the international WAO/EAACI guidelines for the management of HAE.
Introduction
Hereditary angioedema (HAE) is a rare genetic dominant condition with incomplete penetrance characterized by recurrent swellings (edema) affecting the skin, internal organs, mucosa or the upper airways (1, 2). Where genetic pathogenesis is known, symptoms are caused by dysfunction of the C1 esterase inhibitor (C1-INH) or dysregulation of the kinin cascade, leading to bradykinin release and resulting in inflammation episodes. Bradykinin is a vasoactive peptide and the main activator of the bradikinin receptor B2 expressed in endothelial cells (3) whose activation leads to increased vascular permeability and edema, causing the HAE symptoms (4). HAE attacks can turn into a life-threatening episode if the edema develops in the laryngeal track, which leads to the obstruction of the upper respiratory airways (5). HAE attacks are unpredictable and often develop in response to triggering factors such as mental stress (6), use of contraception hormones (7), infections (8), injuries or surgery interventions (9), and weather changes (6), among others. HAE prevalence has been estimated in over 1:50,000 worldwide (10) and has been reported in all ethnic groups (11).
This genetic condition is typically divided in two main groups attending to the C1-INH plasmatic levels. The most frequent form (85% of the cases described) is caused by decreased levels of C1-INH (11, 12), also known as HAE-C1-INH of type I. The rest of cases are found either with normal levels of non-functional C1-INH protein (also known as HAE-C1-INH of type II) or with normal levels of a functional C1-INH protein (HAE-nC1-INH). At the moment, more than 450 disease causing variants have been reported in SERPING1 gene leading to HAE-C1-INH forms (11, 13–15). However, causal variants affecting other genes of the bradykinin pathway underlie HAE-nC1-INH cases (11).
The first genetic study in HAE families with normal levels of C1-INH detected a mutation in the exon nine of F12 (16). FXII activation increases the bradykinin accumulation, driving to increased vascular permeability. Surprisingly, only 20% of patients with HAE-nC1-INH in Europe carry causal variants in the F12 gene (17). Until 2018, only SERPING1 and F12 were known to cause HAE, which explains why HAE diagnosis has been mostly based on plasmatic determinations of complement proteins or the activity. The decreasing costs of Next Generation Sequencing (NGS) prompted the first whole-exome sequencing (WES) which allowed to detect variants affecting function in HAE-nC1-INH patients in angiopoietin (ANGPT1) and plasminogen (PLG) genes (18, 19). Using the same approach, Bork et al. also identified variants in the kininogen 1 (KNG1) gene as another cause of HAE (20). Most recently, two other causal genes were found by applying WES in HAE-nC1-INH families, encoding the heparan sulfate 3-O-sulfotransferase 6 (HS3ST6), and myoferlin (MYOF) (21, 22). The latter being causal strongly suggests a key role of VEGF-mediated signaling in HAE pathophysiology, although the mechanism triggering the symptoms remains unclear.
Because of the nonspecific signs, HAE remains a poorly recognized clinical entity, resulting in delayed diagnoses and deficient treatment conditions for long periods. In fact, a reported mean diagnosis delay of 7.9 years, ranging from months to 50 years, has been recently exposed in the HAE cases from the Canary Islands (23). Those patients lacking a diagnosis have an increased risk for morbidities and mortality compared to those that had been diagnosed, especially if attacks affect the airways (24). Advances in testing procedures and disease recognition have not improved HAE diagnosis, still representing a challenge for professionals (25). Currently, genetic testing is not part of the clinical diagnosis of HAE routine in many health care systems despite the updated HAE management guidelines recommend a genetic-based diagnosis in order to increase the diagnostic yield and reduce the diagnosis odyssey (11, 26, 27). Furthermore, the debilitating nature of HAE attacks makes early and precise diagnosis critical to rapidly establish the actions and treatments for short and long-term prophylaxis (28). Because of the importance of establishing an accurate diagnosis and to prescribe the optimal treatment, many countries have developed their own HAE patient registry. This has fostered the development of standards and specialized facilities to help managing and diagnosing patients while reducing the diagnosis delay. Here we present the first genetic characterization of HAE patients from the Canary Islands (Spain).
Material and methods
Patient population and setting
The study was approved by the Hospital Universitario Nuestra Señora de Candelaria (HUNSC; PI 57–17) Ethics Committee and written informed consent was obtained from all patients and relatives. The patient cohort is composed by affected individuals that had compatible clinical history of angioedema attacks without urticaria, and a differential diagnosis by blood molecular assays. We excluded patients using medication that were known to trigger angioedema attacks (i.e., angiotensin-converting enzyme inhibitors) and patients suffering other pathologies that can potentially cause angioedema attacks (i.e., hepatitis, HIV, hepatic/renal disorders, immunological deficiencies, and infections due to Helicobacter pylori).
HAE diagnosis was based on plasmatic determination of C1-INH and C4, and C1-INH activity, according to the international WAO/EAACI guidelines for the management of HAE (29).
Whole-exome sequencing and variant calling
DNA was extracted from 4 mL of peripheral blood with Illustra™ blood genomicPrep kit (GE Healthcare; Chicago, IL). Alternatively, for pediatric patients and adults where blood sample extraction was not available, DNA from saliva samples were obtained with the OG-250 kit (DNA Genotek, Ontario, CA) following manufacturer’s instructions. DNA concentration was evaluated using the dsDNA BroadRange Assay Kit for the Qubit® 3.0 Fluorometer (Termo Fisher Scientific, Waltham, MA).
Libraries were prepared using the TruSeq Rapid Exome Library Prep Kit (Illumina, San Francisco, CA). Library sizes and concentrations were assesed on a TapeStation 4200 (Agilent Technologies, Santa Clara, CA) and sequences were obtained with a HiSeq 4000 Sequencing System (Illumina, San Francisco, CA) with paired-end 75-base reads. PhiX was loaded and sequenced at 1% as an internal control of the experiments (Figure 1).
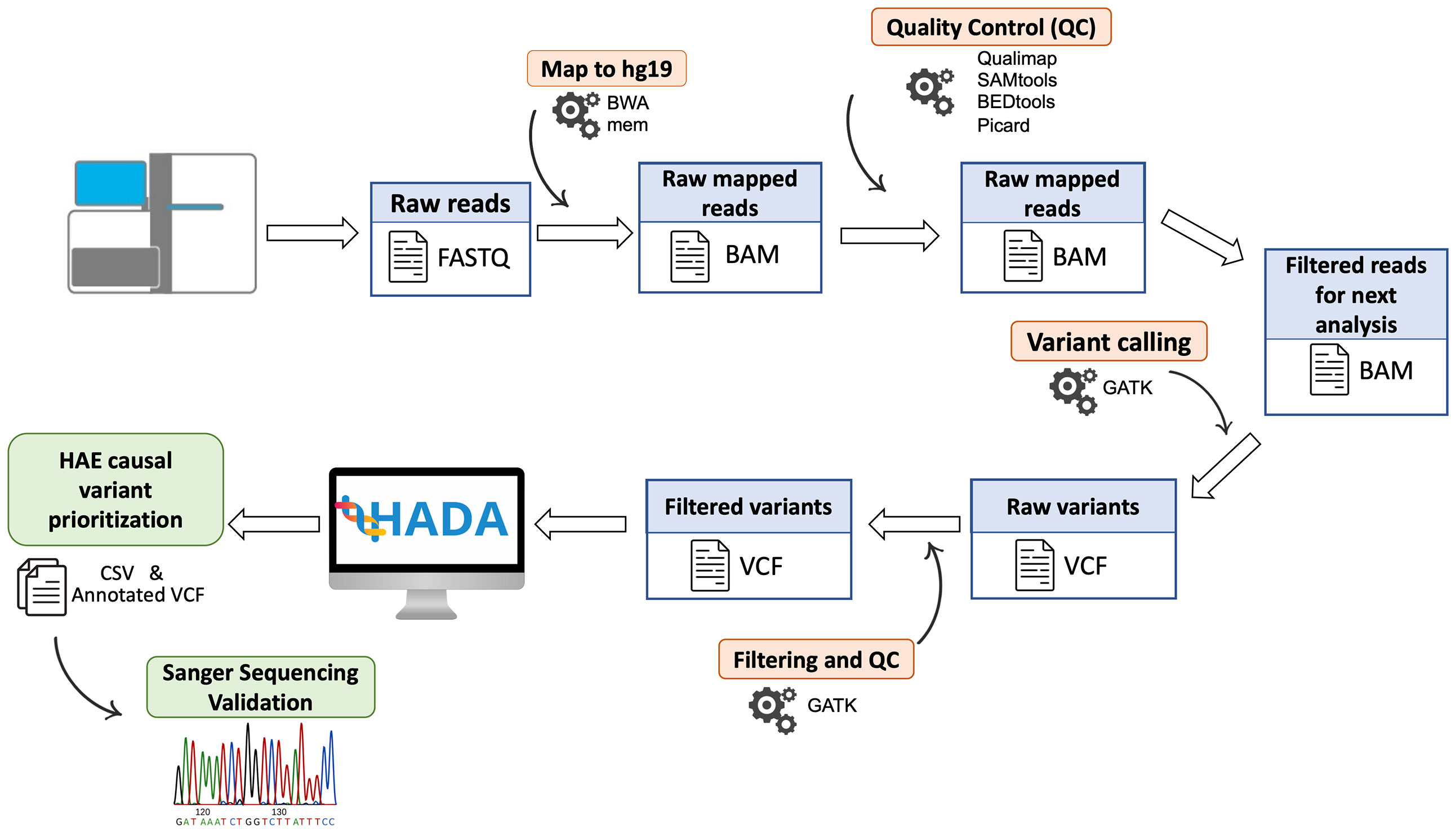
Figure 1 Schematic representation of the steps followed for patient DNA sequencing data. Algorithms are shown in orange and processed files in blue. The HAE causal variants prioritized by HADA and the Sanger validation step are shown in green.
Sequencing reads were preprocessed with bcl2fastq v2.18 and mapped to hg19/GRCh37 with Burrows-Wheeler Aligner v0.7.15 (30), and BAM files were processed with Qualimap v2.2.1 (31), SAMtools v1.3 (32), BEDTools (33), and Picard v2.10.10 (http://broadinstitute.github.io/picard) for quality control steps. Variant calling of germline variants was performed using an in‐house bioinformatics pipeline based on the Genome Analysis Toolkit (GATK) v.3.8 (34). The pipeline has been designed for the detection of nucleotide substitutions (SNVs) and small indels (<50 bp) following the GATK best practices (35) and its description is publicly available (https://github.com/genomicsITER/benchmarking/tree/master/WES).
Subsequently, the identified variation was filtered by means of SAMtools and VCFtools (36) based on “PASS” filter, depth of coverage per position (≥ 20×), genotype quality (≥ 100), and mapping quality (≥ 50).
The analysis was carried out at the Teide-HPC Supercomputing facility (http://teidehpc.iter.es/en).
Variant annotation and causal variant prioritization
The resulting variant calls were annotated with available information (Figure 1). For that, ANNOVAR (37) was used to include the allele frequency in reference populations, gene location, known functional consequences, links with disease based on ClinVar (38) and The Human Gene Mutation Database (39), and several pathogenicity scores including the Combined Annotation-Dependent Depletion (CADD, 40), among others. The classification of pathogenic potential of variants was obtained and annotated using InterVar software (41) following the American College of Medical Genetics and Genomics (ACMG) guidelines (42).
The annotated variant calls were individually processed for each patient by Hereditary Angioedema Database Annotation tool (HADA, http://hada.hpc.iter.es/), an in-house designed variant prioritization server of HAE genes to facilitate the identification of the variants affecting function as well as other accompanying information from the literature (13). In those patients where causal variants were not detected by HADA, Exomiser v12.1 was used to aim to identify the variants affecting function causing HAE (43). SQUIRLs v1.3.0 was used to specifically analyze candidate splicing variants and their predicted effect, prioritizing them by the internal pathogenicity score (44).
Finally, the variants that were classified as Variants of Uncertain Significance (VUS) that could be reclassified as Pathogenic/Likely Pathogenic variants based on complimentary information were validated by direct Sanger sequencing of PCR amplicons from the two strands. This assessment was performed by Macrogen Spain sequencing services with a customized design of primers (Supplementary Table 1).
Assessment of potential structural variants in sequencing data
ExomeDepth v1.1.15 was used to identify putative structural variants (SVs) that could constitute possible variants affecting function causing HAE (45). Candidate variants obtained from BAM files were filtered to remove false positives calls by Bayes factor (BF) higher than 20.
To provide a clinical interpretation of the candidate SVs, filtered calls were processed with ClassifyCNV (46), based on the ACMG guidelines to determine pathogenicity potential. Complementarily, StrVCTURE (47) was used to distinguish pathogenic SVs from benign SVs that overlap exons based on CADD scores. The classification was carried out with the consensus of both algorithms.
Screening for mobile elements in sequencing data
At least 17 Alu elements which are in SERPING1 gene have been reported in the scientific literature. As such, pathogenic rearrangements associated with this repetitive element have been estimated to be responsible for approximately 15% of HAE cases (48). Because of that, the Soft Clipped Read Alignment Mapper v1.0.1 (SCRAMble) (49) was used to screen for mobile element insertions (MEIs) in the exome data, with a particular emphasis on SERPING1.
Results
Patient population and sequencing summary
Forty-one patients and nine healthy relatives from 29 unrelated families with HAE diagnoses residing in the Canary Islands were included in the study. Thirty-seven patients self-declared European ancestry, two declared ancestry from Colombia (family 3), and two from Israel (family 5). The study sample included eight males (19.5%) and 33 females (80.5%). The protective role of male hormones and the well-known influence of estrogens as one of the main triggers of HAE attacks could explain this sexual disbalance (50–52). The patients were aged between four and 72 years (mean: 36.8 ± 16.9 years) and a positive family history for HAE attacks was reported for 23 (56.1%) of them. Based on biochemical analysis, all HAE types are present in the study sample, where HAE-C1-INH was predominant, with 23 patients diagnosed for HAE type I (56.1%) and four patients for HAE type II (9.8%). The rest of patients (14, 34.1%) were diagnosed as HAE-nC1-INH, and most of them were females (92.8%).
WES of this case series yielded an average of 8.36 Gb per patient, with an average of 100% of on-target reads and a median depth of 57×, and a transition/transversion ratio in the range of 3.1 to 3.3.
Causal variant identification and concordance between clinical and genetic assessments
Using HADA, we detected a total of six variants affecting function linked to different HAE types in the study (Table 1 and Figure 2). SERPING1 was the most commonly affected gene (n=5). Two unrelated families clinically classified as HAE type I were carriers of the synonymous variant affecting function c.751C>T; p.Leu251= (families 4 and 11), which was previously reported in Spain (53). The missense variant c.613T>C; p.Cys205Arg, previously described in Spanish cases, was also found in another family with HAE type I (family 8). We detected a likely pathogenic frameshift variant affecting function located in exon 3 of SERPING1 gene, c.143_144delCA; p.Thr48SerfsTer9, which was associated with decreased C1-INH plasmatic levels and leading to HAE type I (family 9). We also detected the causative missense variant c.1396C>T; p.Arg466Ser in the index patient of family 1. In this family, the index patient [with normal levels of C1-INH (47 mg/dL), reduced C1-INH activity (10%) and low C4 levels (1.8 mg/dL) had similar biochemical findings as his asymptomatic sister (with normal) levels of C1-INH (65 mg/dl); reduced C1-INH activity (< 10%), and low C4 levels (7.6 mg/dl)]. Sequence analysis of this asymptomatic relative also releveled the missense variant c.1396C>T; p.Arg466Ser. This variant has been widely reported in Spanish patients in the literature, although linked to HAE type II instead (54).
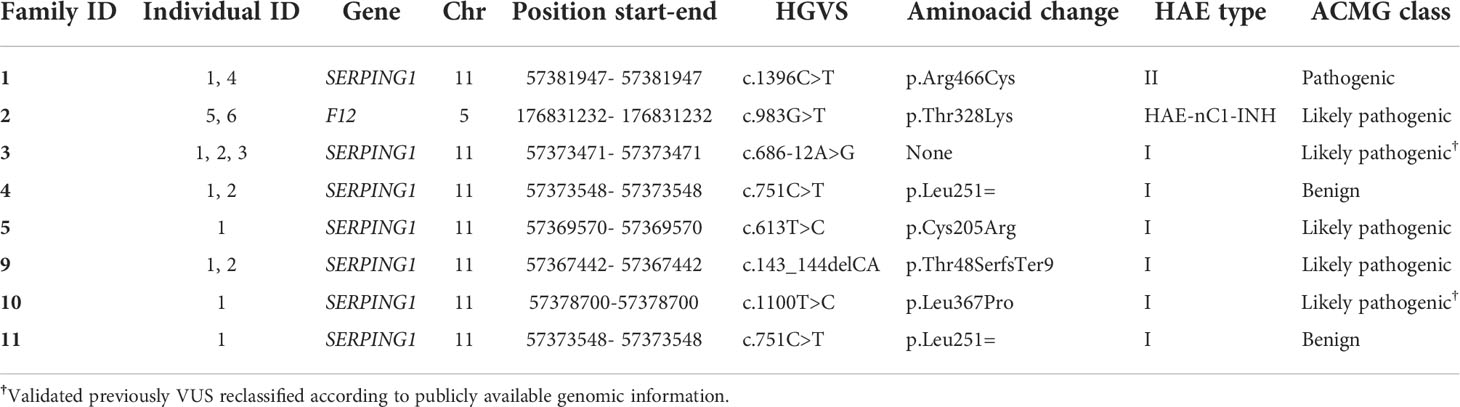
Table 1 Detected causal variants affecting function of HAE in this study.
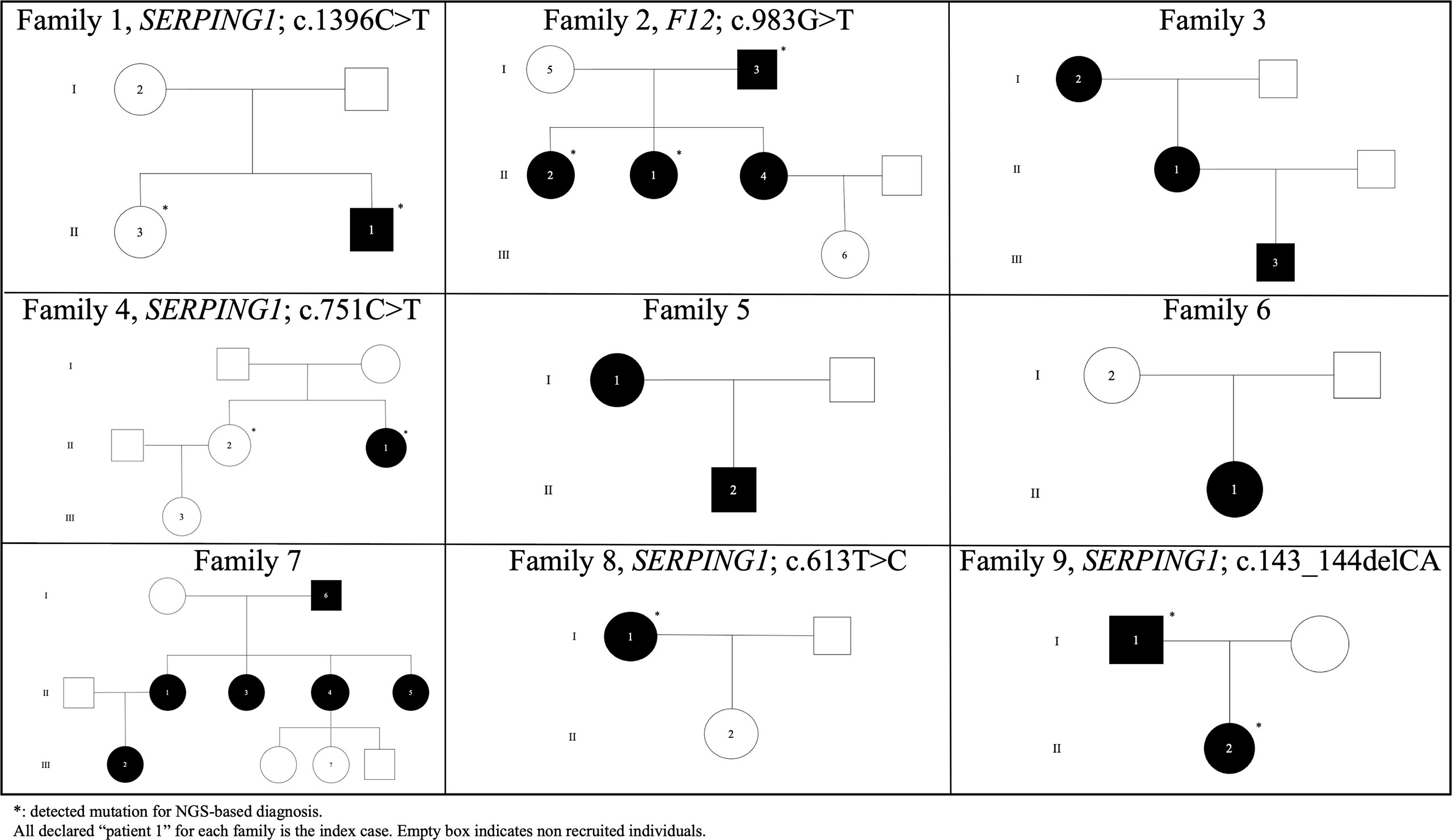
Figure 2 Pedigrees of unrelated HAE families in the Canary Islands.
Furthermore, a well-known causal variant of F12 (c.983G>T; p.Thr328Lys) was detected in members of the family 2 with HAE-nC1-INH (Figure 2). This variant affecting function has not been previously reported in Spanish patients and is absent from ExAC (55) and gnomAD (56).
We also identified a missense variant affecting function in SERPING1 gene that was previously classified as a VUS. The variant c.1100T>C; p.Leu367Pro was detected in one patient for which information from relatives was lacking (Figure 3). Although this variant was described in previous genetic studies with HAE patients, it lacks population frequency data or prediction scores which could supporta a damaging effect, including individual predictions (SIFT, PolyPhen2, CADD, M-CAP, among others) and meta scores (MetaLR, MetaSVM, MetaRNN and REVEL). Despite that, we reclassified it as a likely pathogenic variant according to the ACMG guidelines using VarSome (57) based on the classification criteria PM1 (moderate), PM2 (moderate), PM5 (moderate), PP2 (supporting), and PP3 (supporting).
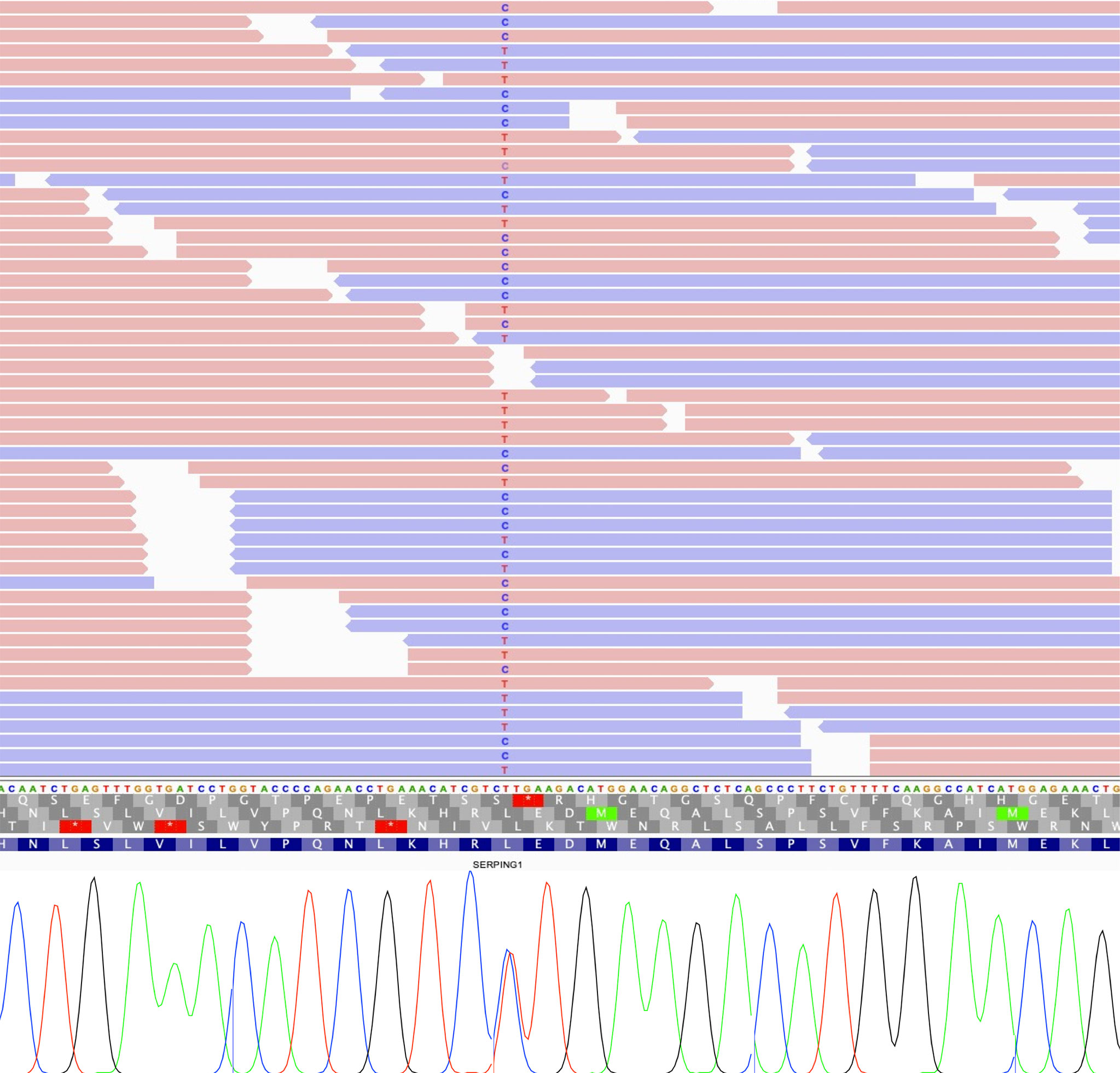
Figure 3 IGV view of sequencing reads supporting each allele of SERPING1 variant (up), and Sanger sequencing validation showing both alleles (down).
The manual inspection of sequencing data in the positions with candidate calls for SVs and MEIs revealed that all the prioritized findings were false positives due to low depth of coverage in the inspected regions, therefore, offering insufficient support to declare variants.
Splicing defects detected
The splicing variant defect c.686-12A>G in SERPING1 was identified in all the affected patients of family 3 (Figure 4). This variant was previously reported in two unrelated Italian patients (58) and in one case of a Serbian HAE type I family (59). However, despite the authors did not assess the pathogenicity of the variant, its function was assessed using a minigene in vitro model (60). According to that study, the c.686-12A>G variant was found to provoke an aberrant splicing effect possibly triggering transcript degradation as deduced by the minimal amount of the transcript detected in the patient’s blood. Based on this extended information, we then scored this variant with the following ACMG criteria: BP4 (supporting), PS2 (strong), PM2 (moderate), PP1 (supporting), PP3 (supporting), and PP4 (supporting). As a result, VarSome reclassified the c.686-12A>G variant from VUS to Likely Pathogenic.
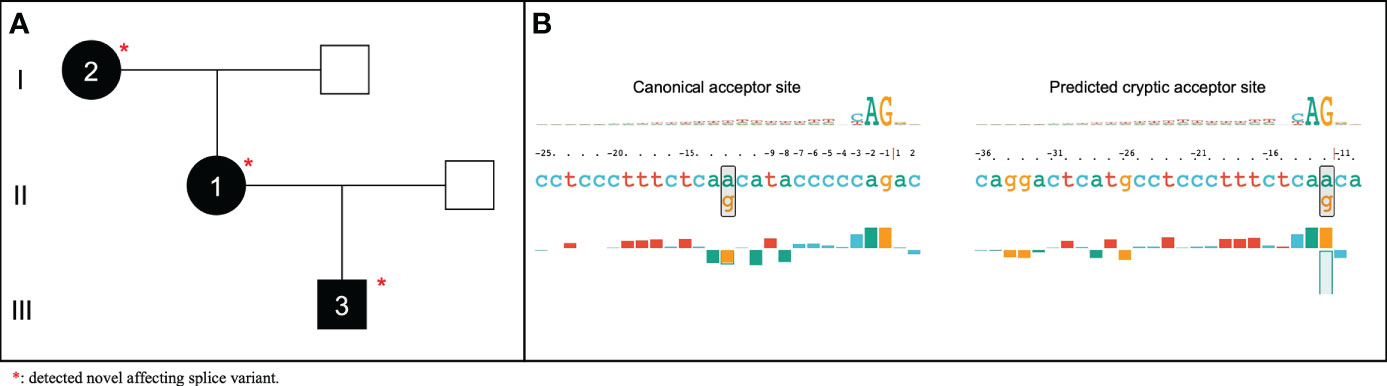
Figure 4 Pedigree of HAE affected family without previously reported causal variant (A) and representation generated by SQUIRLs of predicted acceptor site for the novel SERPING1 c.686-12A>G affecting splice variant (B).
Diagnostic yield
Aggregating all the described evidence for the variants affecting function detected across study patients from families with different affected members, we obtained a genetic diagnostic yield of 66.7% (95% Confidence Interval [CI]: 30.1-91.0%; 6/9 families) when considering only the previously known causal variants (Figure 2). However, the genetic diagnosis yield among proband-only cases decreased to 10.0% (95% CI: 1.8-33.1%; 2/20 cases). This finding supports the recommendation of the WAO/EAACI current guidelines for HAE management to recruit unaffected family members for causal variant screening.
Discussion
In recent years, the widely use of NGS in the clinical field has significantly contributed to the achievement of early and accurate diagnosis. In this context, we aimed to characterize for the first time the underlying genetic causes of HAE in the Canary Islands, where clinical studies have estimated a prevalence of around 1.90:100,000 (23). This prevalence is higher than the that reported for overall Spain and is closer to that observed in other European studies, estimated in 2:100,000 (53). The isolation and the recent demographic history of the Canary Islands population could explain the HAE prevalence in the archipelago. Genetic studies have highlighted that the genes from the regulation of inflammatory response and the complement cascade are significantly enriched in the genomic regions that harbor distinctive genetic variation of this population (61). These findings, together with the estimated higher prevalence of HAE in the Canary Islands, may add to the underlying genetic factors involved in angioedema that have not been identified so far. In this context, NGS-based studies with HAE affected individuals and families are being constantly implemented in several countries with the aim of increasing the diagnostic yield, such as Denmark (62), Saudi Arabia (63), Turkey (64), Japan (65), Portugal (66), Greece (67), Norway (68), Switzerland (69), Croatia (70), China (71), Romania (72), Austria (73), and Puerto Rico (74), among others. However, NGS-based genetic studies have not been common in HAE studies until recently (17). Part of the explanation may reside in the fact that only two causal genes were identified until 2018 and that clinical diagnosis was essentially based on clinical symptoms and biochemical measurements of C4 and C1-INH levels/C1-INH activity in plasma (11). In certain cases, the clinicians could not refine the diagnosis in HAE borderline patients. In these cases, WES brings new and efficient opportunities to clearly identify the underlaying causes, especially in those without C1-INH deficiency (11). Besides, obtaining WES from all patients offers the possibility to unravel novel disease genes, as well as to identify the underlying causes in the known HAE genes in a single test. In this context, this study describes the first step in the genetic characterization of HAE patients from the Canary Islands. Subsequent studies based on phenotype-driven prioritizations of causal variants from the whole-exome and genome-wide studies will aim to identify novel genetic factors involved in HAE in this population.
Following the international WAO/EAACI guidelines for the management of HAE (26), we have obtained two remarkable results in one of the families. First, we amended the initial diagnosis based on the clinical and biochemical studies, which supported a HAE type I in family 1. However, the prioritization of genetic variants revealed a well-known variant of HAE type II (SERPING1; c.1396C>T). Secondly, this variant was also detected in one asymptomatic relative, allowing a diagnostic anticipation to the manifestation of symptoms in this family member. The development of bioinformatics tools focused on variant interpretation in the context of rare diseases accelerates diagnosis accuracy, which translates into a great benefit for patients and their families. However, we face a global problem with the high proportion of VUS detected in NGS-based studies, since it complicates an accurate clinical diagnosis identifying the genetic causes, as recommended by current international guidelines for HAE patient management. In addition, there is also a widespread conflict in the pathogenic classification rules for genetic variants. In family 4, we identified the synonymous variant SERPING1; c.751C>T, which has been classified as benign according to the ACMG guidelines. However, this variant has been declared as causal of the disease in Spanish patients with HAE (53). Synonymous variants can contribute to diseases, affecting gene function by splicing (75), binding of transcription factors (76), or mRNA stability (77), among others. They are common in the human genome, but their role in diseases have often been underestimated (78). It is well-known that variants affecting splicing are significant contributors to human diseases and are often missed in standard variant filtering approaches, which tend to focus on protein coding regions (79). In this study, we detected the SERPING1 c.686-12A>G variant in all affected members of a recruited HAE type I family, previously reported as VUS and here reclassified as likely pathogenic. As indicated before, the increased number of sequencing studies in rare disorders have overcome the main limitations to the interpretation of the pathogenic potential of detected variants and their association with the disease (80, 81). In this context, the diagnostic yield of NGS-based solutions has improved to 35-60% nowadays (44). However, the genetic cause of most cases of this study is lacking (21/29 independent cases, 72.4%, 95% CI: 52.5-86.6). The diagnostic process of HAE is also a stressful period due to confusing signs with histaminergic angioedema attacks, which could be delayed for many years. To reduce the clinical odyssey, we previously designed HADA, a variant prioritization tool for DNA sequencing studies focused on HAE diagnosis (13). HADA retrieves previously reported causal variants in HAE patients in a few minutes, integrating extended genetic variant information from public updated databases used for pathogenic classification according to ACMG guidelines. Taken all prioritized variants by HADA in our study, we have estimated a NGS diagnostic yield of 66.7% (6/9 families). Unfortunately, the diagnosis yield decreases to 10.0% in proband-only analyses (2/20 cases). Remarkably, despite the capabilities of using a WES approach, we did not find variants affecting function in PLG, ANGPT1, KNG1, MYOF, and HS3ST6 genes (involved in HAE-nC1-INH) in any of the studied families.
Our study has some strengths and limitations. Among limitations, we acknowledge that the number of patients where a causal variant was identified is limited, precluding for now a robust study of the relationships between phenotypes and genotypes, as well as the links with drug responses or the patient management. We also acknowledge a relatively low breath of coverage of some exons of the F12 gene compared with that of SERPING1 gene (66.49% of bases covered at least 10× in F12 compared with 88.33% for SERPING1), which may hinder the identification of causal genetic variants in patients with clinical suspicion of HAE-nC1-INH. Another limitation was the low genetic diagnosis yield obtained among proband-only cases, which could be increased with the recruitment of family members for genetic causative variant identification and determining its origin (inherited vs de novo), as is recommended by WAO/EAACI guidelines. Although WES is not the optimal approach, we have aimed to identify and prioritize candidate SVs and MEIs. However, the results did not support the possibility that the cases could be caused by SVs in the known HAE genes. Even though HADA assist in the genetic testing of HAE patients, the tool was designed to prioritize genetic variants within the literature identified genes of HAE, thus lacking the function to identify novel HAE genes. To fill this gap, complementary studies relying on phenotype-driven approaches and genomic scans, among others, are needed to allow the identification of novel genetic factors of HAE, especially in those affected patients who remain without genetic diagnosis. Among the strengths of our study, we highlight the widespread recruitment of HAE patients and their relatives in the archipelago by the Allergy Services of the main hospitals from each island, allowing us to carry out an early detection of new cases of HAE and study the genetic causes through DNA sequencing. The detection of the SERPING1 c.1396C>T; p.Arg466Ser variant led to a diagnosis anticipation in an asymptomatic relative considering both genetic results and the biochemical findings, allowing to achieve a precise diagnosis of HAE type II. Our study highlights the benefits of a first-tier genetic testing for HAE diagnosis and reinforces the need to clearly identify underlying genetic causes, as recommended by the international WAO/EAACI guidelines for the management of HAE and enhance the benefits of NGS as first-tier genetic testing tool for its diagnosis (26).
Conclusions
To characterize the underlying genetic causes of HAE in the Canary Islands, we have performed the first genomic study of affected families from the region. A rapid detection of the causal variants in sequencing data from whole-exome was carried out based on HADA, providing in some families a refined diagnosis, and revealing the genetic affectation in asymptomatic members. Our results reinforce the necessity of NGS based studies on HAE patients to identify and characterize novel genetic factors involved in this disease. Complementary and more exhaustive analysis are required to identify the genetic cause in all the families.
Data availability statement
The data presented in the study are deposited in the EGA repository (https://ega-archive.org/datasets/EGAD00001009318). Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Hospital Universitario Nuestra Señora de Candelaria Ethics Committee (HUNSC; PI 57–17). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
Conceptualization, CF and ACa; methodology, AM-A, ET-H and IM-R; patient and sample recruitment, LA-Q, EM-F, DM-B, EP-R, JG-R, and ACa; sample preparation and DNA sequencing, ACo, AI-C, and RG-M; formal analysis, AM-A, AM-B, LR-R, AA-G and JL-S; writing—original draft preparation, AM-A and CF; writing—review and editing, AM-A, IM-R, JL-S, ACa and CF; supervision, CF; funding acquisition, ACa, IM-R and CF. All authors have read and agreed to the published version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Ministerio de Ciencia e Innovación (RTC-2017-6471-1; AEI/FEDER, UE) and the Instituto de Salud Carlos III (CD19/00231), which were co-financed by the European Regional Development Funds ‘A way of making Europe’ from the European Union; Cabildo Insular de Tenerife (CGIEU0000219140); Agreement OA17/008 with the Instituto Tecnológico y de Energías Renovables (ITER) to strengthen scientific and technological education, training, research, development, and innovation in genomics, personalized medicine, and biotechnology; Fundación Canaria Instituto de Investigación Sanitaria de Canarias (PIFIISC19/48); and SEAIC Foundation (18_A01). A.M.-A. and E.T.-H. were supported by a fellowship from Agencia Canaria De Investigación Innovación Y Sociedad De La Información Gobierno De Canarias (TESIS2020010002 and TESIS2021010046, respectively) co-funded by European Social Fund. AAG was supported by Ministerio de Universidades (modality Margarita Salas). The content of this publication is solely the responsibility of the authors and does not necessarily reflect the views or policies of the funding agencies.
Acknowledgments
We would like to thank the patients and nurses involved in the design of this study. AM-A, ET-H, AM-B, and LR-R. acknowledge the University of La Laguna for the training support during their PhD studies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.997148/full#supplementary-material
References
1. Nzeako UC, Frigas E, Tremaine WJ. Hereditary angioedema: a broad review for clinicians. Arch Intern Med (2001) 161:2417–29. doi: 10.1001/archinte.161.20.2417
2. Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bork K, Bouillet L, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol (2004) 114:S51–131. doi: 10.1016/j.jaci.2004.06.047
3. Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation and inflammatory disease. J Allergy Clin Immunol (2002) 109:195–209. doi: 10.1067/mai.2002.121316
4. Banday AZ, Kaur A, Jindal AK, Rawat A, Singh S. An update on the genetics and pathogenesis of hereditary angioedema. Genes Dis (2019) 7:75–83. doi: 10.1016/j.gendis.2019.07.002
5. Caballero T, Cabañas R, Campos A, Cimbollek S, Gómez-Traseira C, González-Quevedo T, et al. Consensus statement on the diagnosis, management, and treatment of angioedema mediated by bradykinin. part i. classification, epidemiology, pathophysiology, genetics, clinical symptoms, and diagnosis. J Investig Allergol Clin Immunol (2011) 21:333–47.
6. Zotter Z, Csuka D, Szabó E, Czaller I, Nébenführer Z, Temesszentandrási G, et al. The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis (2014) 9:44. doi: 10.1186/1750-1172-9-44
7. Gompel A, Fain O, Boccon-Gibod I, Gobert D, Bouillet L. Exogenous hormones and hereditary angioedema. Int Immunopharmacol (2019) 78:106080. doi: 10.1016/j.intimp.2019.106080
8. Campos R de A, Valle SOR, Toledo EC. Hereditary angioedema: a disease seldom diagnosed by pediatricians. J Pediatr (2021) 97(Suppl 1):S10–6. doi: 10.1016/j.jped.2020.10.011
9. Campos RA, Valle SOR, Toledo EC. Hereditary angioedema: a disease seldom diagnosed by pediatricians. J Pediatr (2021) 97:S10–16. doi: 10.1016/j.jped.2020.10.011
10. Zuraw BL. Clinical practice. hereditary angioedema. N Engl J Med (2008) 359:1027–36. doi: 10.1056/NEJMcp0803977
11. Marcelino I, Callero A, Mendoza-Alvarez A, Pérez-Rodríguez E, Barrios-Recio J, García-Robaina J, et al. Bradykinin-mediated angioedema: an update of the genetic causes and the impact of genomics. Front Genet (2019) 10:900. doi: 10.3389/fgene.2019.00900
12. Caballero T, Farkas H, Bouillet L, Bowen T, Gompel A, Fagerberg C, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol (2012) 129:308–20. doi: 10.1016/j.jaci.2011.11.025
13. Mendoza-Alvarez A, Muñoz-Barrera A, Rubio-Rodríguez LA, Marcelino-Rodriguez I, Corrales A, Iñigo-Campos A, et al. Interactive web-based resource for annotation of genetic variants causing hereditary angioedema (HADA): Database development, implementation, and validation. J Med Internet Res (2020) 22:e19040. doi: 10.2196/19040
14. Patel G, Pongracic JA. Hereditary and acquired angioedema. Allergy Asthma Proc (2019) 40:441–5. doi: 10.2500/aap.2019.40.4267
15. Ponard D, Gaboriaud C, Charignon D, Ghannam A, Wagenaar-Bos IGA, Roem D, et al. SERPING1 mutation update: Mutation spectrum and C1 inhibitor phenotypes. Hum Mutat (2019) 41:38– 57. doi: 10.1002/humu.23917
16. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun (2006) 343:1286–9. doi: 10.1016/j.bbrc.2006.03.092
17. Zuraw BL. Hereditary angioedema with normal C1 inhibitor: Four types and counting. J Allergy Clin Immunol (2018) 141:884–5. doi: 10.1016/j.jaci.2018.01.015
18. Bafunno V, Firinu D, D’Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol (2018) 141:1009–17. doi: 10.1016/j.jaci.2017.05.020
19. Bork K, Wulff K, Steinmüller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy (2018) 73:442–50. doi: 10.1111/all.13270
20. Bork K, Wulff K, Rossmann H, Steinmüller-Magin L, Braenne I, Witzke G, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the n-terminal cleavage site of bradykinin. Allergy (2019) 74:2479–81. doi: 10.1111/all.13869
21. Ariano A, D’Apolito M, Bova M, Bellanti F, Loffredo S, D’Andrea G, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy (2020) 75:2989–92. doi: 10.1111/all.14454
22. Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-o-sulfotransferase 6 gene mutation. J Allergy Clin Immunol (2021) 148:1041–8. doi: 10.1016/j.jaci.2021.01.011
23. Mendoza-Alvarez A, Marcelino-Rodriguez I, Almeida-Quintana L, Martin-Fernandez E, Martinez-Beltran D, Almeida-Sanchez Z, et al. First census of patients with hereditary angioedema in the canary islands. J Clin Med Res (2021) 10:4711. doi: 10.3390/jcm10204711
24. Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol (2012) 130:692–7. doi: 10.1016/j.jaci.2012.05.055
25. Bernstein JA. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am J Manage Care (2018) 24:S292–8.
26. Maurer M, Magerl M, Ansotegui I, Aygören-Pürsün E, Betschel S, Bork K, et al. The international WAO/EAACI guideline for the management of hereditary angioedema – the 2017 revision and update. World Allergy Organ J (2018) 11:5. doi: 10.1186/s40413-017-0180-1
27. Germenis AE, Cicardi M. Driving towards precision medicine for angioedema without wheals. J Autoimmun (2019) 104:102312. doi: 10.1016/j.jaut.2019.102312
28. Henry Li H, Zuraw B, Longhurst HJ, Cicardi M, Bork K, Baker J, et al. Subcutaneous C1 inhibitor for prevention of attacks of hereditary angioedema: additional outcomes and subgroup analysis of a placebo-controlled randomized study. Allergy Asthma Clin Immunol (2019) 15:1–8. doi: 10.1186/s13223-019-0362-1
29. Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema – the 2021 revision and update. World Allergy Organ J (2022) 15:100627. doi: 10.1016/j.waojou.2022.100627
30. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
31. García-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics (2012) 28:2678–9. doi: 10.1093/bioinformatics/bts503
32. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
33. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (2010) 26:841–2. doi: 10.1093/bioinformatics/btq033
34. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res (2010) 20:1297–303. doi: 10.1101/gr.107524.110
35. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet (2011) 43:491–8. doi: 10.1038/ng.806
36. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics (2011) 27:2156–8. doi: 10.1093/bioinformatics/btr330
37. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res (2010) 38:e164. doi: 10.1093/nar/gkq603
38. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res (2014) 42:D980–5, 42. doi: 10.1093/nar/gkt1113
39. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet (2017) 136:665–77. doi: 10.1007/s00439-017-1779-6
40. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet (2014) 46:310–5. doi: 10.1038/ng.2892
41. Li Q, Wang K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
42. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
43. Robinson PN, Köhler S, Oellrich A, Sanger Mouse Genetics Project, Wang K, Mungall CJ, et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res (2014) 24:340–8. doi: 10.1101/gr.160325.113
44. Danis D, Jacobsen JOB, Carmody L, Gargano M, McMurry JA, Hegde A, et al. Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Cold Spring Harbor Lab (2021) 108:1564–77. doi: 10.1101/2021.01.28.428499. 2021.01.28.428499.
45. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics (2012) 28:2747–54. doi: 10.1093/bioinformatics/bts526
46. Gurbich TA, Ilinsky VV. ClassifyCNV: a tool for clinical annotation of copy-number variants. Sci Rep (2020) 10:20375. doi: 10.1038/s41598-020-76425-3
47. Sharo AG, Hu Z, Sunyaev SR, Brenner SE. StrVCTVRE: A supervised learning method to predict the pathogenicity of human genome structural variants. Am J Hum Genet (2022) 109:195–209. doi: 10.1016/j.ajhg.2021.12.007. 2020.05.15.097048.
48. Carter PE, Duponchel C, Tosi M, Fothergill JE. Complete nucleotide sequence of the gene for human C1 inhibitor with an unusually high density of alu elements. Eur J Biochem (1991) 197:301–8. doi: 10.1111/j.1432-1033.1991.tb15911.x
49. Torene RI, Galens K, Liu S, Arvai K, Borroto C, Scuffins J, et al. Mobile element insertion detection in 89,874 clinical exomes. Genet Med (2020) 22:974–8. doi: 10.1038/s41436-020-0749-x
50. Bygum A, Aygören-Pürsün E, Caballero T, Beusterien K, Gholizadeh S, Musingarimi P, et al. The hereditary angioedema burden of illness study in Europe (HAE-BOIS-Europe): background and methodology. BMC Dermatol (2012) 12:4. doi: 10.1186/1471-5945-12-4
51. Craig T. Triggers and short-term prophylaxis in patients with hereditary angioedema. Allergy Asthma Proc (2020) 41:S30–4. doi: 10.2500/aap.2020.41.200058
52. Kulkarni M, Travers JB, Rohan C. High Estrogen States in Hereditary Angioedema: a Spectrum. Clin Rev Allergy Immunol (2021) 60:396–403. doi: 10.1007/s12016-021-08863-4
53. Roche O, Blanch A, Duponchel C, Fontán G, Tosi M, López-Trascasa M. Hereditary angioedema: the mutation spectrum of SERPING1/C1NH in a large Spanish cohort. Hum Mutat (2005) 26:135–44. doi: 10.1002/humu.20197
54. López-Lera A, Garrido S, Roche O, López-Trascasa M. SERPING1 mutations in 59 families with hereditary angioedema. Mol Immunol (2011) 49:18–27. doi: 10.1016/j.molimm.2011.07.010
55. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature (2016) 536:285–91. doi: 10.1038/nature19057
56. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
57. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: The human genomic variant search engine. Bioinformatics (2018) 35:1978–80. doi: 10.1093/bioinformatics/bty897
58. Pappalardo E, Caccia S, Suffritti C, Tordai A, Zingale LC, Cicardi M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: functional and structural correlates. Mol Immunol (2008) 45:3536–44. doi: 10.1016/j.molimm.2008.05.007
59. Andrejević S, Korošec P, Šilar M, Košnik M, Mijanović R, Bonači-Nikolić B, et al. Hereditary Angioedema Due to C1 Inhibitor Deficiency in Serbia: Two Novel Mutations and Evidence of Genotype-Phenotype Association. PloS One (2015) 10:e0142174. doi: 10.1371/journal.pone.0142174
60. Grodecká L, Hujová P, Kramárek M, Kršjaková T, Kováčová T, Vondrášková K, et al. Systematic analysis of splicing defects in selected primary immunodeficiencies-related genes. Clin Immunol (2017) 180:33–44. doi: 10.1016/j.clim.2017.03.010
61. Guillen-Guio B, Lorenzo-Salazar JM, González-Montelongo R, Díaz-de Usera A, Marcelino-Rodríguez I, Corrales A, et al. Genomic analyses of human European diversity at the southwestern edge: Isolation, African influence and disease associations in the canary islands. Mol Biol Evol (2018) 35:3010–26. doi: 10.1093/molbev/msy190
62. Bygum A. Hereditary angio-oedema in Denmark: a nationwide survey. Br J Dermatol (2009) 161:1153–8. doi: 10.1111/j.1365-2133.2009.09366.x
63. Faiyaz-Ul-Haque M, Al-Gazlan S, Abalkhail HA, Al-Abdulatif A, Toulimat M, Peltekova I, et al. Novel and recurrent mutations in the C1NH gene of Arab patients affected with hereditary angioedema. Int Arch Allergy Immunol (2010) 151:149–54. doi: 10.1159/000236005
64. Kesim B, Uyguner ZO, Gelincik A, Mete Gökmen N, Sin AZ, Karakaya G, et al. The Turkish hereditary angioedema pilot study (TURHAPS): the first Turkish series of hereditary angioedema. Int Arch Allergy Immunol (2011) 156:443–50. doi: 10.1159/000323915
65. Iwamoto K, Tanaka A, Hiragun M, Kawai M, Mihara S, Takenaka M, et al. Novel and recurrent C1 inhibitor gene mutations in nine Japanese patients with hereditary angioedema. J Dermatol Sci (2012) 68:68–70. doi: 10.1016/j.jdermsci.2012.06.012
66. Martinho A, Mendes J, Simões O, Nunes R, Gomes J, Dias Castro E, et al. Mutations analysis of C1 inhibitor coding sequence gene among Portuguese patients with hereditary angioedema. Mol Immunol (2013) 53:431–4. doi: 10.1016/j.molimm.2012.09.003
67. Psarros F, Koutsostathis N, Farmaki E, Speletas MG, Germenis AE. Hereditary angioedema in Greece: the first results of the greek hereditary angioedema registry. Int Arch Allergy Immunol (2014) 164:326–32. doi: 10.1159/000366276
68. Johnsrud I, Kulseth MA, Rødningen OK, Landrø L, Helsing P, Waage Nielsen E, et al. A nationwide study of Norwegian patients with hereditary angioedema with C1 inhibitor deficiency identified six novel mutations in SERPING1. PloS One (2015) 10:e0131637. doi: 10.1371/journal.pone.0131637
69. Steiner UC, Keller M, Schmid P, Cichon S, Wuillemin WA. Mutational spectrum of the SERPING1 gene in Swiss patients with hereditary angioedema. Clin Exp Immunol (2017) 188:430–6. doi: 10.1111/cei.12941
70. Karadža-Lapić L, Barešić M, Vrsalović R, Ivković-Jureković I, Sršen S, Prkačin I, et al. Hereditary angioedema due to C1-inhibitor deficiency in pediatric patients in croatia. first national study, diagnostic and prophylactic challenges. Acta Clin Croat. (2019) 58:139–46. doi: 10.20471/acc.2019.58.01.18
71. Liu S, Xu Y, Liu Y, Zhi Y. Hereditary angioedema: a Chinese perspective. Eur J Dermatol (2019) 29:14–20. doi: 10.1684/ejd.2018.3487
72. Gábos G, Moldovan D, Dobru D, Mihály E, Bara N, Nădăşan V, et al. Mutational spectrum and genotype-phenotype relationships in a cohort of Romanian hereditary angioedema patients caused by C1 inhibitor deficiency. Rev Română Medicină Laborator (2019) 27:255–68. doi: 10.2478/rrlm-2019-0029
73. Schöffl C, Wiednig M, Koch L, Blagojevic D, Duschet P, Hawranek T, et al. Hereditary angioedema in Austria: prevalence and regional peculiarities. J Dtsch Dermatol Ges (2019) 17:416–23. doi: 10.1111/ddg.13815
74. Arce-Ayala YM, Diaz-Algorri Y, Craig T, Ramos-Romey C. Clinical profile and quality of life of Puerto ricans with hereditary angioedema. Allergy Asthma Proc (2019) 40:103–10. doi: 10.2500/aap.2019.40.4200
75. Pagani F, Raponi M, Baralle FE. Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc Natl Acad Sci U S A (2005) 102:6368–72. doi: 10.1073/pnas.0502288102
76. Stergachis AB, Haugen E, Shafer A, Fu W, Vernot B, Reynolds A, et al. Exonic transcription factor binding directs codon choice and affects protein evolution. Science (2013) 342:1367–72. doi: 10.1126/science.1243490
77. Duan J, Shi J, Ge X, Dölken L, Moy W, He D, et al. Genome-wide survey of interindividual differences of RNA stability in human lymphoblastoid cell lines. Sci Rep (2013) 3:1318. doi: 10.1038/srep01318
78. Zeng Z, Aptekmann AA, Bromberg Y. Decoding the effects of synonymous variants. Nucleic Acids Res (2021) 49:12673–91. doi: 10.1093/nar/gkab1159
79. Lord J, Baralle D. Splicing in the diagnosis of rare disease: Advances and challenges. Front Genet (2021) 12:689892. doi: 10.3389/fgene.2021.689892
80. Tomar S, Sethi R, Lai PS. Specific phenotype semantics facilitate gene prioritization in clinical exome sequencing. Eur J Hum Genet (2019) 27:1389–97. doi: 10.1038/s41431-019-0412-7
Keywords: hereditary angioedema, genetic cause, rare disease, variant interpretation, precision medicine
Citation: Mendoza-Alvarez A, Tosco-Herrera E, Muñoz-Barrera A, Rubio-Rodríguez LA, Alonso-Gonzalez A, Corrales A, Iñigo-Campos A, Almeida-Quintana L, Martin-Fernandez E, Martinez-Beltran D, Perez-Rodriguez E, Callero A, Garcia-Robaina JC, González-Montelongo R, Marcelino-Rodriguez I, Lorenzo-Salazar JM and Flores C (2022) A catalog of the genetic causes of hereditary angioedema in the Canary Islands (Spain). Front. Immunol. 13:997148. doi: 10.3389/fimmu.2022.997148
Received: 18 July 2022; Accepted: 23 August 2022;
Published: 20 September 2022.
Edited by:
Erik J. M. Toonen, Hycult Biotech, NetherlandsReviewed by:
Yang Cao, Peking Union Medical College Hospital (CAMS), ChinaChiara Suffritti, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, Italy
Copyright © 2022 Mendoza-Alvarez, Tosco-Herrera, Muñoz-Barrera, Rubio-Rodríguez, Alonso-Gonzalez, Corrales, Iñigo-Campos, Almeida-Quintana, Martin-Fernandez, Martinez-Beltran, Perez-Rodriguez, Callero, Garcia-Robaina, González-Montelongo, Marcelino-Rodriguez, Lorenzo-Salazar and Flores. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Flores, Y2Zsb3Jlc0B1bGwuZWR1LmVz