 Julia Scheiermann1,2
Julia Scheiermann1,2 Annette Künkele1,2,3,4Arend von Stackelberg1
Annette Künkele1,2,3,4Arend von Stackelberg1 Angelika Eggert1,2,3,4
Angelika Eggert1,2,3,4 Peter Lang1,5
Peter Lang1,5 Felix Zirngibl1Luise Martin6
Felix Zirngibl1Luise Martin6 Johannes Hubertus Schulte1,2,3,4†
Johannes Hubertus Schulte1,2,3,4† Horst von Bernuth2,6,7,8*†
Horst von Bernuth2,6,7,8*†- 1Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, Department of Pediatric Hematology, Oncology and Stem Cell Transplantation, University Hospital Center, Berlin, Germany
- 2Berlin Institute of Health at Charité – Universitätsmedizin Berlin, Berlin, Germany
- 3German Cancer Consortium [Deutsches Konsortium für Transnationale Krebsforschung (DKTK)], Berlin, Germany
- 4German Cancer Research Center [Deutsches Krebsforschungszentrum (DKFZ)], Heidelberg, Germany
- 5Department of Pediatric Hematology and Oncology, University Hospital, Tübingen, Germany
- 6Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, Department of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine, University Hospital Center, Berlin, Germany
- 7Department of Immunology, Labor Berlin GmbH, Berlin, Germany
- 8Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Berlin-Brandenburg Center for Regenerative Therapies (BCRT), Berlin, Germany
Chronic granulomatous disease is an inborn error of immunity due to disrupted function of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. This results in impaired respiratory burst of phagocytes and insufficient killing of bacteria and fungi. Patients with chronic granulomatous disease are at increased risk for infections, autoinflammation and autoimmunity. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only widely available curative therapy. While HSCT from human leukocyte antigen (HLA) matched siblings or unrelated donors are standard of care, transplantation from HLA-haploidentical donors or gene therapy are considered alternative options. We describe a 14-month-old male with X-linked chronic granulomatous disease who underwent a paternal HLA-haploidentical HSCT using T-cell receptor (TCR) alpha/beta+/CD19+ depleted peripheral blood stem cells followed by mycophenolate graft versus host disease prophylaxis. Decreasing donor fraction of CD3+ T cells was overcome by repeated infusions of donor lymphocytes from the paternal HLA-haploidentical donor. The patient achieved normalized respiratory burst and full donor chimerism. He remained disease-free off any antibiotic prophylaxis for more than three years after HLA-haploidentical HSCT. In patients with x-linked chronic granulomatous disease without a matched donor paternal HLA-haploidentical HSCT is a treatment option worth to consider. Administration of donor lymphocytes can prevent imminent graft failure.
Introduction
Chronic granulomatous disease (CGD) is an inborn inborn error of immunity (IEI) characterized by a severely impaired or absent respiratory burst in all phagocytes. Pathogenic variants in one of five genes coding for subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex have been identified. Biallelic defects in CYBA, NCF1, NCF2 and CYBC1 (encoding for p22phox, p47phox, p67phox and Eros) cause autosomal recessive CGD (AR-CGD), while mutations in CYBB (encoding for the catalytic domain gp91phox) cause XL-CGD (1). Of note, biallelic pathogenic mutations in NCF4 cause a related, but distinct disease (2). Malfunctional NADPH oxidase predisposes patients to infections with catalase positive bacteria such as Staphylococcus aureus, Burkholderia cepatia, Serratia marcescens, and fungi such as Aspergillus spp. and other molds (3). Residual superoxide production correlates with longer overall survival (OS) (4). Additionally, autoinflammation and autoimmunity with or without granuloma formation can occur potentially in any organ, most often in the bladder and gastrointestinal tract. Although the frequency of infections can be significantly reduced with the prophylactic use of antimicrobials (e.g. trimethoprim-sulfamethoxazole, TMP-SMX), antifungals (e.g. itraconazole or posaconazole) and interferon-γ (INF-γ), overall survival begins to decline in the second decade of life (3). Consequently, the use of allogeneic HSCT in younger patients, preferentially those younger than 8 years of age, improves overall survival (3). In addition, improving medical care and safer conditioning protocols with reduced toxicity conditioning (RTC) were established enabling HSCT for adult CGD patients who have been suffering from fungal infections and/or autoinflammation prior to HSCT (5). If a HLA-matched donor is not available for XL-CGD patients, paternal haplo-HSCT can be used.
However, non-engraftment occurs in a considerable portion of patients with CGD (3). Administration of donor lymphocyte infusions (DLI) is an established immunotherapeutic treatment in patients, who underwent haploidentical-HSCT (haplo-HSCT) and present with mixed donor chimerism (6). However, experience of DLI application for children transplanted for the treatment of CGD is still scarce. Pediatric IEI patients receiving DLI after HSCT responded to the treatment by increasing donor chimerism more effectively in matched unrelated donors. In approximately 80% of patients DLI could prevent graft rejection. Moreover, there is concern, that administration of DLI may induce or aggravate GVHD. Published literature reports acute and chronic GVHD occurrence in 10% to 60%, yet GVHD was mostly rather mild (grade I-II) and effected mostly the skin (7–11). Here we report on a 14-month-old boy with XL-CGD who was treated with paternal haplo-HSCT followed by eight DLI administrations.
Materials and methods
Informed consent for haplo-HSCT was obtained from the patient’s parents in accordance with local and European Society for Blood and Marrow Transplantation (EBMT) guidelines. Before submission of the manuscript, additional informed consent for publication was obtained from both parents. The day of engraftment was defined as the first of three consecutive days without transfusion and with absolute neutrophil counts (ANC) > 500/μl. The patient underwent weekly polymerase chain reaction (PCR) analysis of blood to detect adenovirus (ADV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV). Acute GVHD was assessed using the modified Seattle Glucksberg criteria (12). Chronic GVHD was scored according to the National Institutes of Health (NIH) criteria (13). Chimerism was monitored in bone marrow and peripheral blood specimens to assess donor cell engraftment. Donor chimerism was measured by labelling blood with anti-CD3 and anti-CD34 micro beads and cell lines were separated using an autoMACS automated benchtop magnetic cell sorter (Miltenyi Biotec). Separated cells were assayed using short tandem repeats via multiplex PCR and capillary array electrophoresis (ABI Prism 3100, ThermoFischer). The neutrophil respiratory burst was performed on fresh whole blood samples with Phagoburst kit (DB biosciences) according to manufacturer’s instructions.
Case description
We describe a 14-month-old male patient diagnosed with XL-CGD (mutation CYBB c.175 T>C), who was successfully transplanted with peripheral blood stem cells (PBMC) from his HLA-haploidentical father. The patient was diagnosed with XL- CGD after birth upon a history of CGD in his maternal uncle and his mother being a carrier for the same pathogenic mutation in CYBB as identified in her brother.
He received oral itraconazole (4 - 6 mg/kg/d) and cefuroxime (20 mg/kg/d) in the first 3 months of life. The latter was switched to TMP-SMX (4 - 6 mg/kg/d TMP component) thereafter. By the end of the first year of life the patient had developed mild CGD colitis. On pre-HSCT examination, ultrasound showed increased bowel wall thickness. Histology revealed follicular lymphatic hyperplasia and mild eosinophilia in tissue biopsies from caecum, colon, sigma and rectum in line with CGD colitis.
The patient underwent a myeloablative conditioning (MAC) regimen at the age of 14 months after a course of antibiotic treatment using cefotaxime and metronidazole from day - 12 to day - 9. Due to CGD colitis, the patient received additional methylprednisolone treatment (20 mg/kg/d) from day - 11 to day - 9. Levetiracetam (20 mg/kg/d) was administered form day - 9 through day + 5 for seizure prophylaxis. The conditioning regimen consisted of rabbit anti-thymocyte globulin (ATG, Grafalon) 20 mg/kg/d (day - 11 to day - 9), fludarabine 30 mg/m2/d (day - 8 to day - 4), and thiotepa 10 mg/kg/d on day - 2. Busulfan was given from day - 7 to day - 4 with therapeutic drug monitoring and a final myeloablative cumulative dosage of 174 mg (18.9 mg/kg) and total area under the curve (AUC) of 93553 ng x h/ml. Peripheral blood stem cell graft infusion from the patient’s father was administered on day 0 after TCR alpha/beta+/CD19+ depletion (Figure 1). The cell product contained total nucleated cells 3.35 x 108 cells/kg, with CD34+, 53.96 x 106 cells/kg; CD3+ 53.28 x 106 cells/kg; TCR alpha/beta+ 12.31 x 103 cells/kg; and TCR gamma/delta+ 52.27 x 106 cells/kg.
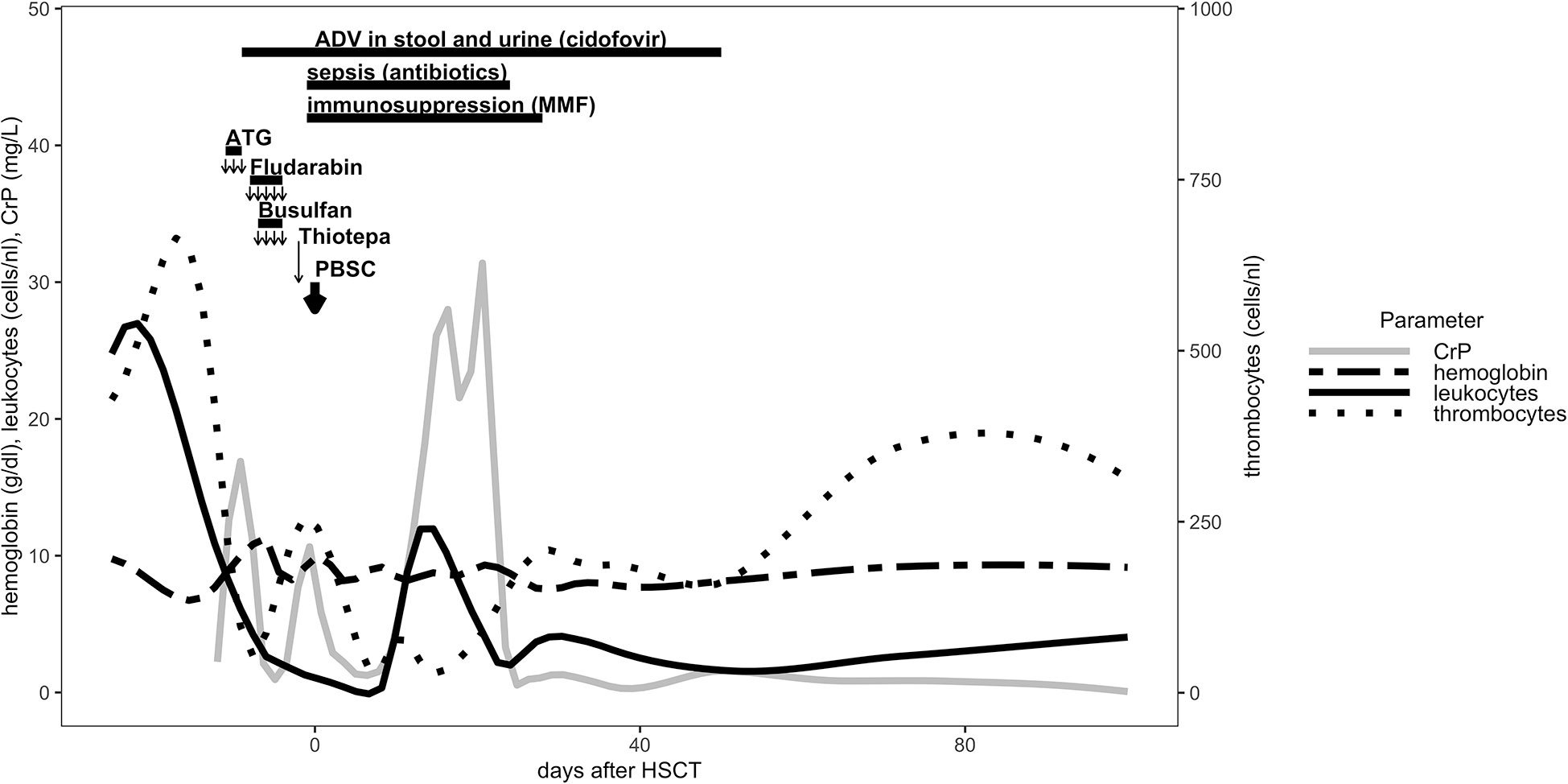
Figure 1 Conditioning regiment, immunosuppression and cellular reconstitution are shown for the first 100 days post HLA-haploidentical HSCT.
Granulocyte colony stimulating factor (GCSF) (5 μg/kg/d) was administered from day + 5 for 6 consecutive days. Full engraftment occurred on day + 13. The patient received immunosuppressive therapy with mocophenolate mofetil (MMF) at the dosage of 2 x 600 mg/m2/d as GVHD prophylaxis from day + 1 until day + 28 (Figure 1). During the course of conditioning regimen, the patient developed fever and sepsis with Serratia marcescens on day - 1 and received antimicrobial therapy through day + 24. Adenovirus was isolated day – 9 in the stool and urine samples and was treated with cidofovir until day + 50 (Figure 1). The prophylactic antifungal treatment with liposomal amphotericin B was administered intravenously starting on day + 1 and was replaced by posaconazol p.o. at day + 30. At day + 14 the patient developed a veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) under prophylactic intravenous heparin according to European society for blood and marrow transplantation (EBMT) diagnostic criteria (14), which was treated with defibrotide (starting at 40 mg/kg/d and escalating to 60 mg/kg/d) from day + 15 until day + 33. Aciclovir and posaconazol were continued prophylactically until day + 251 after HSCT.
The patient developed mixed donor chimerism beginning at day + 52 with the lowest values for CD3+ chimerism of 20% on day + 80 and total chimerism of 89% on day + 95 after HSCT. In parallel counts of CD3+ T-cell in peripheral blood also declined, whereas the respiratory burst of patient’s granulocytes, measured by Dihydrorhodamine (DHR) assays upon stimulation with E. coli PMA continuously exhibited a normal production of H2O2 (Figures 2, 3). In total, eight DLI were administered for rescue of the decreasing donor chimerism starting with a cell number of 2.5 x 104 CD3+ cells/kg and increasing up to 30 x 104 CD3+ cells/kg of patient’s body weight between days + 66 and + 329 (Figures 2, 3; Table 1). Peripheral T cell numbers as well as donor chimerism rose post DLI. This was particularly observed after the first four DLI, whereas the following four DLI had a less obvious effect. The patient`s ADV infection neither effected the chimerism nor the number of peripheral T cells. No signs of GVHD occurred after the administration of the DLI. 14 months post transplantation the patient achieved complete donor chimerism which remained stable thereafter. No acute or chronic GVHD developed. Four years after haplo-HSCT our patient is cured from CGD and off any treatment.
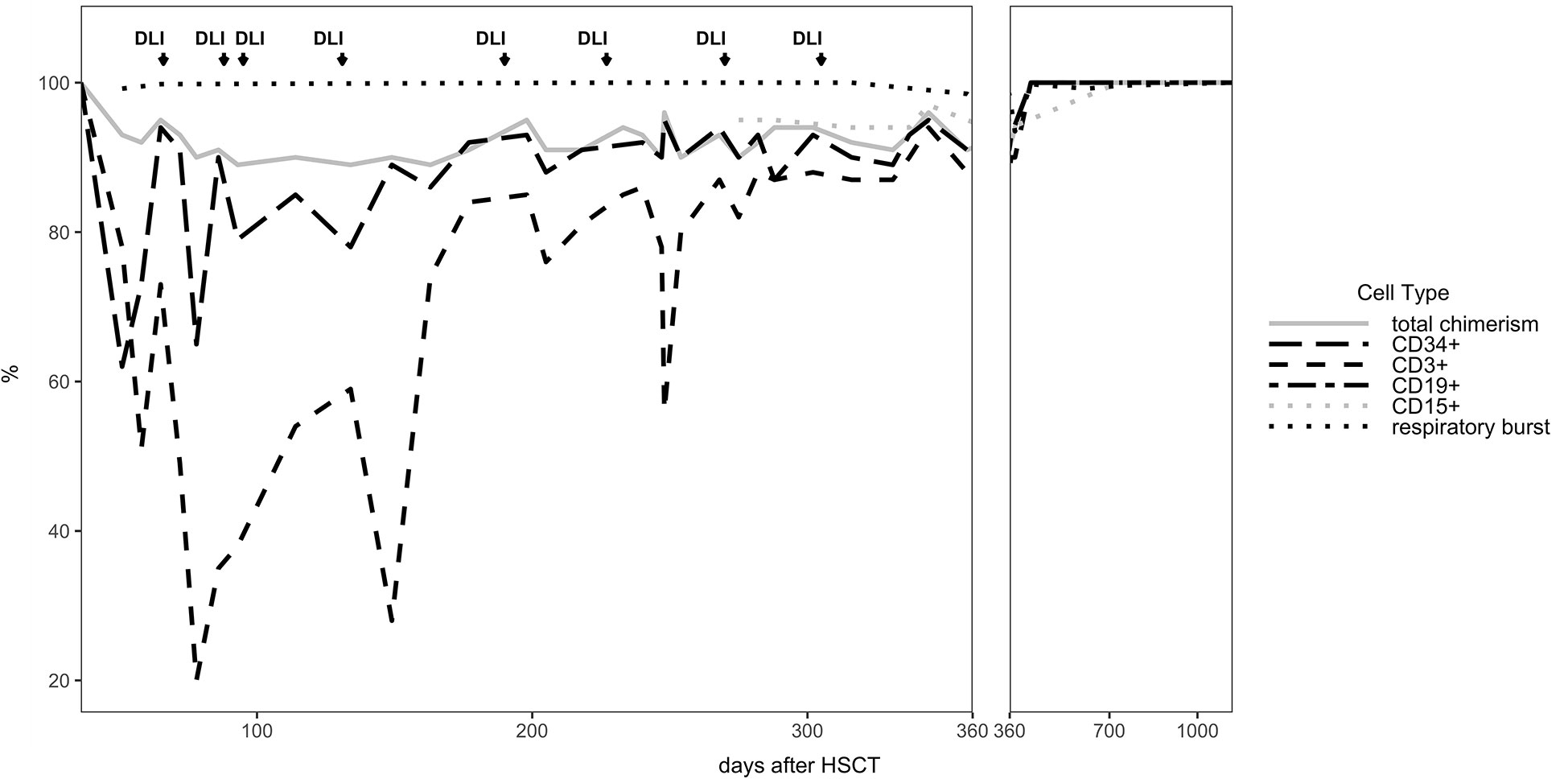
Figure 2 Donor Chimerism in peripheral blood for CD34+, CD3+, total cells (total chimerism) and respiratory burst in patient’s neutrophils measured by DHR test are shown for the entire follow up time of 3 years.
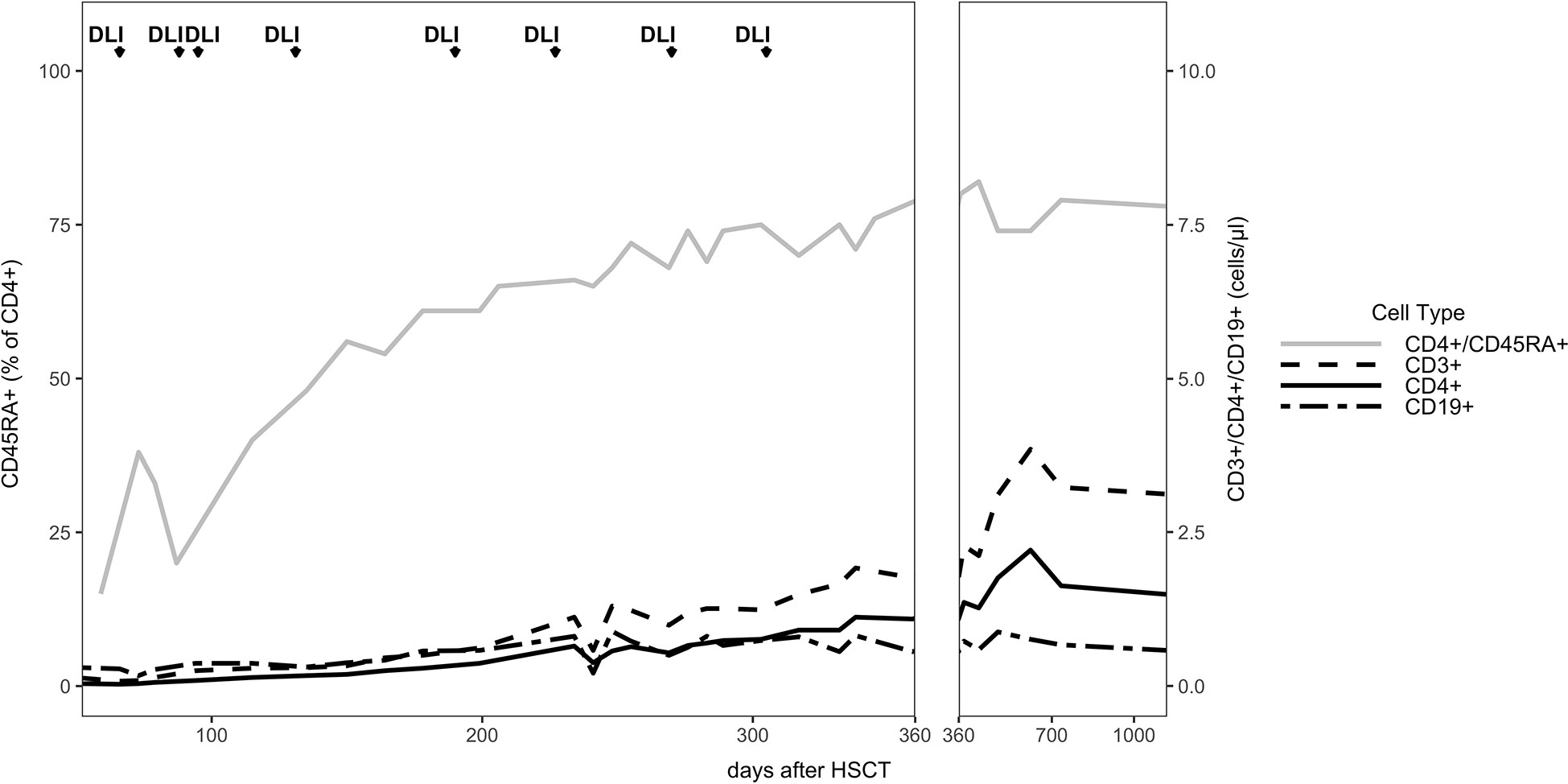
Figure 3 Immune reconstitution: The absolute cell numbers per μl are shown for the entire follow up time of 3 years. The CD45RA+ (CCR7+) percentage of the CD4+ cells are shown.
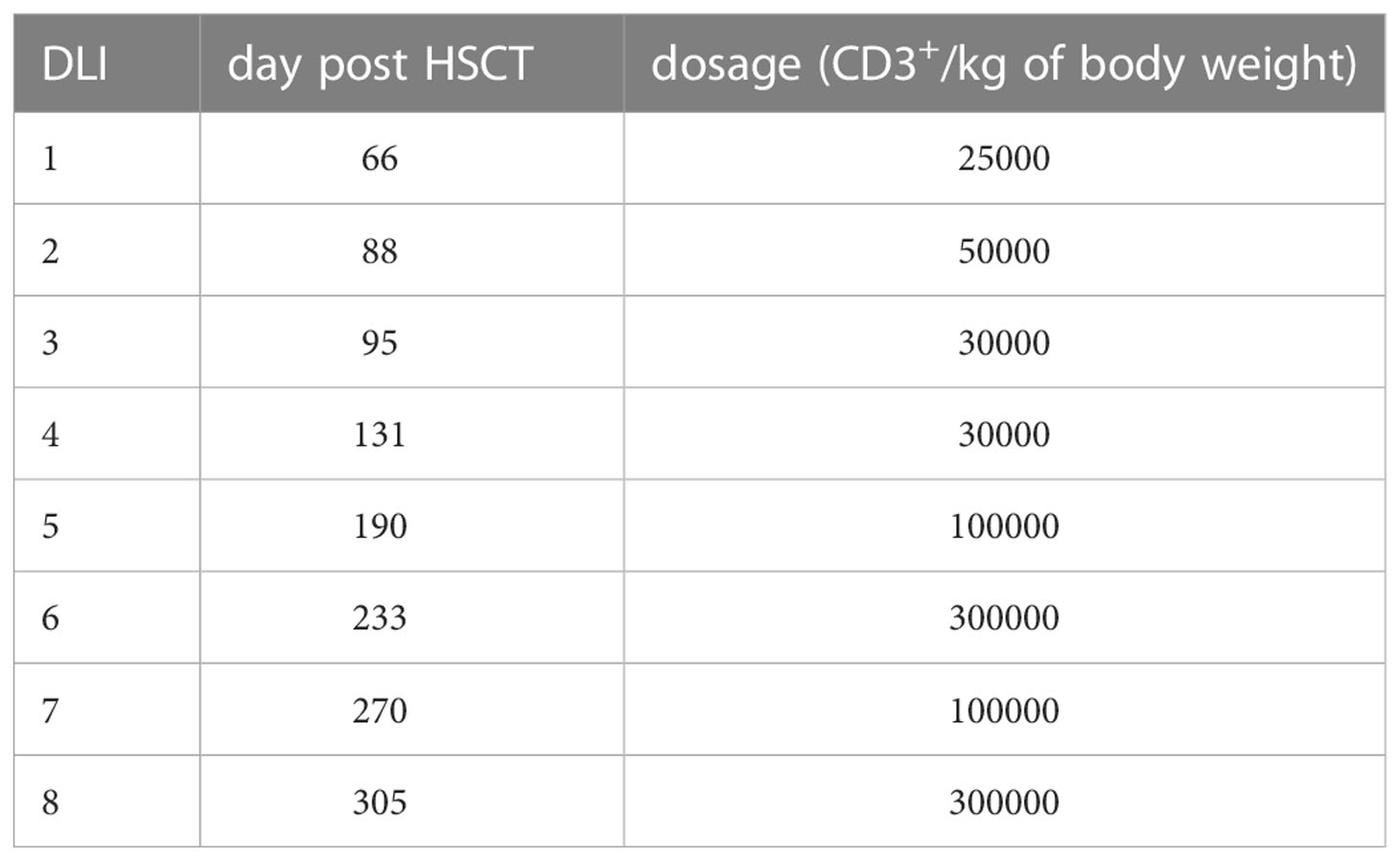
Table 1 DLI characteristics: dates and dosages of administered CD3+ cells are shown.
Discussion
The introduction of safer protocols and effective GVHD prophylaxis has improved the overall survival of CGD patients receiving allogeneic HSCT (5). This seems particularly the case, if CGD patients are transplanted at young age, ≤ 8 years while organ functions are fully retained and severe infectious and autoinflammatory complications have not yet occurred (3). Yet, matched related or unrelated donors are not available for all patients with CGD. Gene therapy is yet not widely available and must still be regarded as rather experimental and prone to serious complications (15). Along this line, haplo-HSCT seems the more evident option for children with CGD in the absence of a matched unrelated donor. Moreover, haplo-HSCT became an attractive option because most patients with CGD still have living parents who can perform as a possible donor. In XL-CGD patients’ mothers are often carriers for a pathogenic mutation in CYBB. As this heterozygous state may exhibit skewed lyonisation later in life, only paternal haplo-HSCT is a reasonable option in XL-CGD.
We here report the 22nd published successful haplo-HSCT in CGD (16–29) and the 11th published patient transplanted using a TCR alpha/beta+/CD19+ depleted PBSC graft (19, 22, 24, 25, 29) (Supplementary Table 1). The first haplo-HSCT described by Kikuta et al. in 2006 was performed in a 2-year-old using RIC conditioning, however, that patient developed graft failure on day + 77 and needed a second transplant from a MUD. The use of HLA-haploidentical donors historically entailed an increased risk of graft rejection. To our knowledge, we report the first successful HLA-haploidentical HSCT, who showed mixed chimerism with CD3+ T cell donor chimerism as low as 20% at day + 80, as sign of imminent graft failure, in which the graft could be rescued with repeated DLI infusions from his paternal donor. By the time of 14 months after haplo-HSCT stable donor chimerism was observed in this patient (Table 1).
Until today only few pediatric case series could show beneficial use of DLI cellular immunotherapy in settings of graft rejection after HSCT in IEI (7–11). Our case shows how important it is to monitor the lineage specific chimerism after haplo-HSCT in CGD to initiate DLI administrations. Similarly, in patients with hemoglobinopathies mixed donor chimerism and graft rejection post HSCT, regardless of the donor source, are frequently observed and may also be rescued by DLI (9, 30, 31). Of note, until this day no definite recommendation from the EBMT working party exists regarding the use of DLI after haplo-HSCT (6). Our report enforces the notion that if a HLA-matched donor is not available for XL-CGD patients, paternal haplo-HSCT using TCR alpha/beta+/CD19+ depletion is an option worth to consider. In case of mixed donor chimerism and imminent graft failure, DLI administerd early in the course post HSCT, may allow to rescue a graft.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Informed consent for haploHSCT was obtained from the patient’s parents in accordance with local and EBMT guidelines. Before submission of the manuscript, additional informed consent for publication was obtained from both parents.
Author contributions
HB diagnosed CGD. AS, JHS and HB planed the HSCT. JS, JHS and HB planned the manuscript. JS, AK, AS, FZ, LM, JHS and HB cared “hands on” for the patient, prior and post HSCT. AE and PL supervised HSCT. JS collected and analyzed the data, wrote the initial version of the manuscript and made figures and tables. HB finalized the manuscript. All authors read and approved the manuscript. All authors agree to be accountable for the content of this work.
Funding
JS is participant in the BIH Charité Junior Clinician Scientist Program partly funded by the Charité – Universitätsmedizin Berlin, and the Berlin Institute of Health at Charité (BIH). We acknowledge financial support from the Open Access Publication Fund of Charité – Universitätsmedizin Berlin and the German Research Foundation (DFG).
Conflict of interest
Author HB was employed by Labor Berlin GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1042650/full#supplementary-material
Glossary
References
1. Yu HH, Yang YH, Chiang BL. Chronic granulomatous disease: a comprehensive review. Clin Rev Allergy Immunol (2021) 61(2):101–13. doi: 10.1007/s12016-020-08800-x
2. van de Geer A, Nieto-Patlan A, Kuhns DB, Tool AT, Arias AA, Bouaziz M, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest (2018) 128(9):3957–75. doi: 10.1172/JCI97116
3. Dedieu C, Albert MH, Mahlaoui N, Hauck F, Hedrich C, Baumann U, et al. Outcome of chronic granulomatous disease - conventional treatment vs stem cell transplantation. Pediatr Allergy Immunol (2021) 32(3):576–85. doi: 10.1111/pai.13402
4. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med (2010) 363(27):2600–10. doi: 10.1056/NEJMoa1007097
5. Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet (2014) 383(9915):436–48. doi: 10.1016/S0140-6736(13)62069-3
6. Dholaria B, Savani BN, Labopin M, Luznik L, Ruggeri A, Mielke S, et al. Clinical applications of donor lymphocyte infusion from an HLA-haploidentical donor: consensus recommendations from the acute leukemia working party of the EBMT. Haematologica. (2020) 105(1):47–58. doi: 10.3324/haematol.2019.219790
7. Haines HL, Bleesing JJ, Davies SM, Hornung L, Jordan MB, Marsh RA, et al. Outcomes of donor lymphocyte infusion for treatment of mixed donor chimerism after a reduced-intensity preparative regimen for pediatric patients with nonmalignant diseases. Biol Blood Marrow Transpl (2015) 21(2):288–92. doi: 10.1016/j.bbmt.2014.10.010
8. Umeda K, Adachi S, Tanaka S, Miki M, Okada K, Hashii Y, et al. Comparison of second transplantation and donor lymphocyte infusion for donor mixed chimerism after allogeneic stem cell transplantation for nonmalignant diseases. Pediatr Blood Cancer (2016) 63(12):2221–9. doi: 10.1002/pbc.26141
9. Swaminathan VV, Uppuluri R, Patel S, Sivashankaran M, Ravichandran N, Ramanan KM, et al. Safety and efficacy of fresh whole blood donor lymphocyte infusion in children. Bone Marrow Transpl (2019) 54(11):1892–7. doi: 10.1038/s41409-019-0580-7
10. Ali T, Behfar M, Mohseni R, Salajegheh P, Kheder M, Abou-Fakher F, et al. Escalated dose donor lymphocyte infusion treatment in patients with primary immune deficiencies after HSCT with reduced-intensity conditioning regimen. Hematol Oncol Stem Cell Ther (2021) 15(4):189–95. doi: 10.1016/j.hemonc.2021.06.002
11. Gabelli M, Stepensky P, Ottaviano G, Mullanfiroze K, Lazareva A, Zaidman I, et al. Incremental donor lymphocyte infusion to treat mixed chimerism after allogeneic stem cell transplantation in children with non-malignant diseases. Bone Marrow Transpl (2022) 58(1):109–11. doi: 10.1038/s41409-022-01844-x
12. Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, et al. 1994 Consensus conference on acute GVHD grading. Bone Marrow Transpl (1995) 15(6):825–8.
13. Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-Host disease: I. the 2014 diagnosis and staging working group report. Biol Blood Marrow Transpl (2015) 21(3):389–401.e1. doi: 10.1016/j.bbmt.2014.12.001
14. Mohty M, Malard F, Abecassis M, Aerts E, Alaskar AS, Aljurf M, et al. Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a new classification from the European society for blood and marrow transplantation. Bone Marrow Transpl (2016) 51(7):906–12. doi: 10.1038/bmt.2016.130
15. Kohn DB, Booth C, Kang EM, Pai SY, Shaw KL, Santilli G, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med (2020) 26(2):200–6. doi: 10.1038/s41591-019-0735-5
16. Kikuta A, Ito M, Mochizuki K, Akaihata M, Nemoto K, Sano H, et al. Nonmyeloablative stem cell transplantation for nonmalignant diseases in children with severe organ dysfunction. Bone Marrow Transpl (2006) 38(10):665–9. doi: 10.1038/sj.bmt.1705511
17. Hoenig M, Niehues T, Siepermann K, Jacobsen EM, Schutz C, Furlan I, et al. Successful HLA haploidentical hematopoietic SCT in chronic granulomatous disease. Bone Marrow Transpl (2014) 49(10):1337–8. doi: 10.1038/bmt.2014.125
18. Parta M, Hilligoss D, Kelly C, Kwatemaa N, Theobald N, Malech H, et al. Haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in a patient with chronic granulomatous disease and active infection: A first report. J Clin Immunol (2015) 35(7):675–80. doi: 10.1007/s10875-015-0204-y
19. Morillo-Gutierrez B, Beier R, Rao K, Burroughs L, Schulz A, Ewins AM, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. (2016) 128(3):440–8. doi: 10.1182/blood-2016-03-704015
20. Klein OR, Chen AR, Gamper C, Loeb D, Zambidis E, Llosa N, et al. Alternative-donor hematopoietic stem cell transplantation with post-transplantation cyclophosphamide for nonmalignant disorders. Biol Blood Marrow Transpl (2016) 22(5):895–901. doi: 10.1016/j.bbmt.2016.02.001
21. Zhou L, Dong LJ, Gao ZY, Yu XJ, Lu DP. Haploidentical hematopoietic stem cell transplantation for a case with X-linked chronic granulomatous disease. Pediatr Transpl (2017) 21(1). doi: 10.1111/petr.12861
22. Shah RM, Elfeky R, Nademi Z, Qasim W, Amrolia P, Chiesa R, et al. T-Cell receptor alphabeta(+) and CD19(+) cell-depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J Allergy Clin Immunol (2018) 141(4):1417–26.e1. doi: 10.1016/j.jaci.2017.07.008
23. Regueiro-Garcia A, Farina-Nogueira S, Porto-Arceo JA, Couselo-Sanchez JM. Haploidentical stem cell transplantation in a boy with chronic granulomatous disease. Allergol Immunopathol (2018) 46(4):385–8. doi: 10.1016/j.aller.2017.09.020
24. Lum SH, Flood T, Hambleton S, McNaughton P, Watson H, Abinun M, et al. Two decades of excellent transplant survival for chronic granulomatous disease: a supraregional immunology transplant center report. Blood. (2019) 133(23):2546–9. doi: 10.1182/blood.2019000021
25. Brettig T, Smart J, Choo S, Mechinaud F, Mitchell R, Raj TS, et al. Use of TCR alpha(+)beta(+)/CD19(+)-depleted haploidentical hematopoietic stem cell transplant is a viable option in patients with primary immune deficiency without matched sibling donor. J Clin Immunol (2019) 39(5):505–11. doi: 10.1007/s10875-019-00648-x
26. Neven B, Diana JS, Castelle M, Magnani A, Rosain J, Touzot F, et al. Haploidentical hematopoietic stem cell transplantation with post-transplant cyclophosphamide for primary immunodeficiencies and inherited disorders in children. Biol Blood Marrow Transpl (2019) 25(7):1363–73. doi: 10.1016/j.bbmt.2019.03.009
27. Parta M, Hilligoss D, Kelly C, Kwatemaa N, Theobald N, CS Z, et al. Failure to prevent severe graft-Versus-Host disease in haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in chronic granulomatous disease. J Clin Immunol (2020) 40(4):619–24. doi: 10.1007/s10875-020-00772-z
28. Holzer U, Doring M, Eichholz T, Ebinger M, Queudeville M, Turkiewicz D, et al. Matched versus haploidentical hematopoietic stem cell transplantation as treatment options for primary immunodeficiencies in children. Transplant Cell Ther (2021) 27(1):71.e1– e12. doi: 10.1016/j.bbmt.2020.09.010
29. Merli P, Pagliara D, Galaverna F, Li Pira G, Andreani M, Leone G, et al. TCRalphabeta/CD19 depleted HSCT from an HLA-haploidentical relative to treat children with different nonmalignant disorders. Blood Adv (2022) 6(1):281–92. doi: 10.1182/bloodadvances.2021005628
30. Fouzia NA, Edison ES, Lakshmi KM, Korula A, Velayudhan SR, Balasubramanian P, et al. Long-term outcome of mixed chimerism after stem cell transplantation for thalassemia major conditioned with busulfan and cyclophosphamide. Bone Marrow Transpl (2018) 53(2):169–74. doi: 10.1038/bmt.2017.231
Keywords: chronic granulomatous disease, HLA-haploidentical hematopoietic stem cell transplantation, TCR alpha/beta+/CD19+ depleted peripheral blood HSCT, donor lymphocyte infusion (DLI), graft verses host disease
Citation: Scheiermann J, Künkele A, von Stackelberg A, Eggert A, Lang P, Zirngibl F, Martin L, Schulte JH and von Bernuth H (2023) Case report: HLA-haploidentical HSCT rescued with donor lymphocytes infusions in a patient with X-linked chronic granulomatous disease. Front. Immunol. 14:1042650. doi: 10.3389/fimmu.2023.1042650
Received: 12 September 2022; Accepted: 02 February 2023;
Published: 16 February 2023.
Edited by:
Mary Slatter, Newcastle University, United KingdomReviewed by:
Revathi Raj, Apollo Speciality Hospitals, Chennai, IndiaTheresa Cole, Royal Children’s Hospital, Australia
Copyright © 2023 Scheiermann, Künkele, von Stackelberg, Eggert, Lang, Zirngibl, Martin, Schulte and von Bernuth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Horst von Bernuth, aG9yc3Qudm9uLWJlcm51dGhAY2hhcml0ZS5kZQ==
†These authors have contributed equally to this work and share senior authorship