 Marc Burian
Marc Burian Morna F. Schmidt
Morna F. Schmidt Amir S. Yazdi
Amir S. Yazdi- Department of Dermatology and Allergology, RWTH University Hospital Aachen, Aachen, Germany
Healthy human skin is constantly exposed to sterile and microbial agents. The skin immune system plays an important role in immune surveillance between tolerance and immune activation. This is mainly mediated by neutrophils, macrophages and most importantly lymphocytes. Keratinocytes, which form the outer skin barrier (epidermis) are also critical for cutaneous homeostasis. Being a non-professional immune cell, recognition of danger signals in keratinocytes is mediated by innate immune receptors (pattern recognition receptors, PRR). While Toll-like receptors are located on the cell membrane or the endosomes, nucleotide-binding domain and leucine-rich repeat containing gene family receptors (NLR) are intracellular PRRs. Some of these, once activated, trigger the formation of inflammasomes. Inflammasomes are multiprotein complexes and serve as platforms that mediate the release of innate cytokines after successful recognition, thereby attracting immune cells. Moreover, they mediate the pro-inflammatory cell death pyroptosis. Best characterized is the NLRP3 inflammasome. The function of inflammasomes differs significantly between different cell types (keratinocytes versus immune cells) and between different species (human versus mouse). In recent years, great progress has been made in deciphering the activation mechanisms. Dysregulation of inflammasomes can lead to diseases with varying degrees of severity. Here we focus on the structure, function, and associated pathologies of the NLRP1 inflammasome, which is the most relevant inflammasome in keratinocytes.
From the scratch
Keratinocytes not only form the outer structural barrier of human skin but also express molecules that are actively involved in immune responses, such as the secretion of proinflammatory cytokines (1). The recognition of molecules frequently found in pathogens (pathogen-associated molecular patterns [PAMPs]) or non-microbial danger-associated molecular patterns (DAMPs) is sensed by pattern recognition receptors (PRRs) on the surface or intracellularly (2–4). While Toll-like receptors are located on the cell surface or the endosome, intracellular recognition of danger signals triggers the activation of nucleotide-binding domain and leucine-rich repeat containing gene family receptors (NLR) (formerly referred to NOD-like receptors) (5, 6). Some extracellular stimuli such as ATP or Flagellin can activate NLRs via specific receptors, while NLRs, such as NLRP1 and NLRP3 can form intracellular protein complexes, so-called inflammasomes. Inflammasomes activate caspase-1, which cleaves cytokines of the innate immune system (mainly IL-1β and IL-18). Once being unconventionally secreted, these cytokines induce a cascade which attracts immune cells (7, 8). Additionally, inflammasome activation can initiate a lytic form of cell death called pyroptosis (9, 10).
All known inflammasomes usually consist of an intracellular sensor (NLRP1, NLRP3, NLRC4, AIM2, Pyrin or CARD8) and can recruit a proinflammatory caspase (cysteine protease). Some require an additional adaptor protein to be able to bind caspases via CARD-CARD interactions, and can therefore be viewed as a cytosolic hub that recognizes a signal and subsequently triggers an inflammatory response (8, 11–16).
After activation by stress or danger signals, oligomerization with the adaptor molecule ASC (apoptosis-associated speck-like protein containing a CARD) occurs. As the ASC specks can be visualized in the cytosol, this is used as a read-out for inflammasome activation (17–19). ASC consists of one caspase recruitment domain (CARD) and one pyrin domain (PYD). Through its CARD domain, ASC interacts with pro-caspase-1, which in turn leads to dimerization of the caspase with subsequent self-activation (12, 20, 21). In humans, 12 inflammatory caspases have been identified, distinguishing initiator and effector caspases (the latter involved in apoptosis) (22). Caspase-1, the most well characterized inflammatory caspase, is expressed as an inactive pro-caspase-1 consisting of a CARD domain and subunits p20 and p10 (catalytically active domains). Dimerization and autoproteolytic processes induce its activation and the cleavage of the N-terminal CARD domain (23–26). Substrates of active caspase-1 are pro-IL-1β and pro-IL-18, which are cleaved into their biologically active forms (27). Furthermore, caspase-1 processes Gasdermin D (GSDMD), which is incorporated into the cell membrane leading to pyroptosis via pore formation. Secreted and activated proinflammatory cytokines can enter the extracellular component, bind to their receptors, and attract immune cells. The loss of membrane integrity, possibly due to the formation of GSDMD pores in organellar membranes, contributes to pyroptotic cell death (28, 29).
In the past years, inflammasomes have been studied mainly in immune cells (macrophages or dendritic cells) where NLRP3 (nucleotide-binding domain and leucine-rich repeat pyrin domain containing protein 3) was the focus of interest due to its involvement in neurodegenerative or cardiovascular diseases and cancer (30–32). Additionally, other NLRP3-associated hereditary autoinflammatory syndromes from the group of cryopyrin-associated periodic syndromes including Muckle-Wells-Syndrome, familial cold autoinflammatory syndrome and chronic infantile neurological cutaneous articular syndrome have been classified (33). However, inflammasomes are also expressed in epithelial cells such as keratinocytes. Here, NLRP1 (nucleotide-binding domain and leucine-rich repeat pyrin domain containing protein 1) appears to play a central role (34–36). Innate cytokines (IL-1α, IL-1β and IL-18) are constitutively expressed in keratinocytes, unlike in myeloid cells. For this reason, keratinocytes do not need a priming step by PAMPs (LPS) or cytokines (TNF-α, IFN-γ or IL-1) to induce pro-IL1 cytokines or the NLRs (37–40). In this review, we present the basic features of the structure and function of the NLRP1 inflammasome to understand the pathogenesis of cutaneous diseases in particular.
From form to function
Human NLRP1 (hNLRP1) consists of an N-terminal pyrin domain (PYD) followed by a NACHT domain and leucine-rich repeats (LRRs). At the C-terminal end of hNLRP1 there is a function-to-find domain (FIIND) and a CARD (Figure 1A) that are autoproteolyzed between the ZU5 and UPA subdomains (43, 44). Constitutive autoproteolysis between the two subdomains is required for NLRP1 activation, but is not sufficient on its own (45, 46). Despite constitutive autoproteolysis, the amino-terminal fragment of NLRP1 (N-NLRP1) remains associated with the C-terminal effector fragment (C-NLRP1), promoted by interaction with dipeptidyl peptidase (DPP) 9 (47, 48). Activation signals such as UVB (38) induce the proteosomal degradation of N-NLRP1, which thus can no longer inhibit the C-NLRP1. The CARD-domain of C-NLRP1 recruits ASC and caspase-1 to form the NLRP1 inflammasome (49).
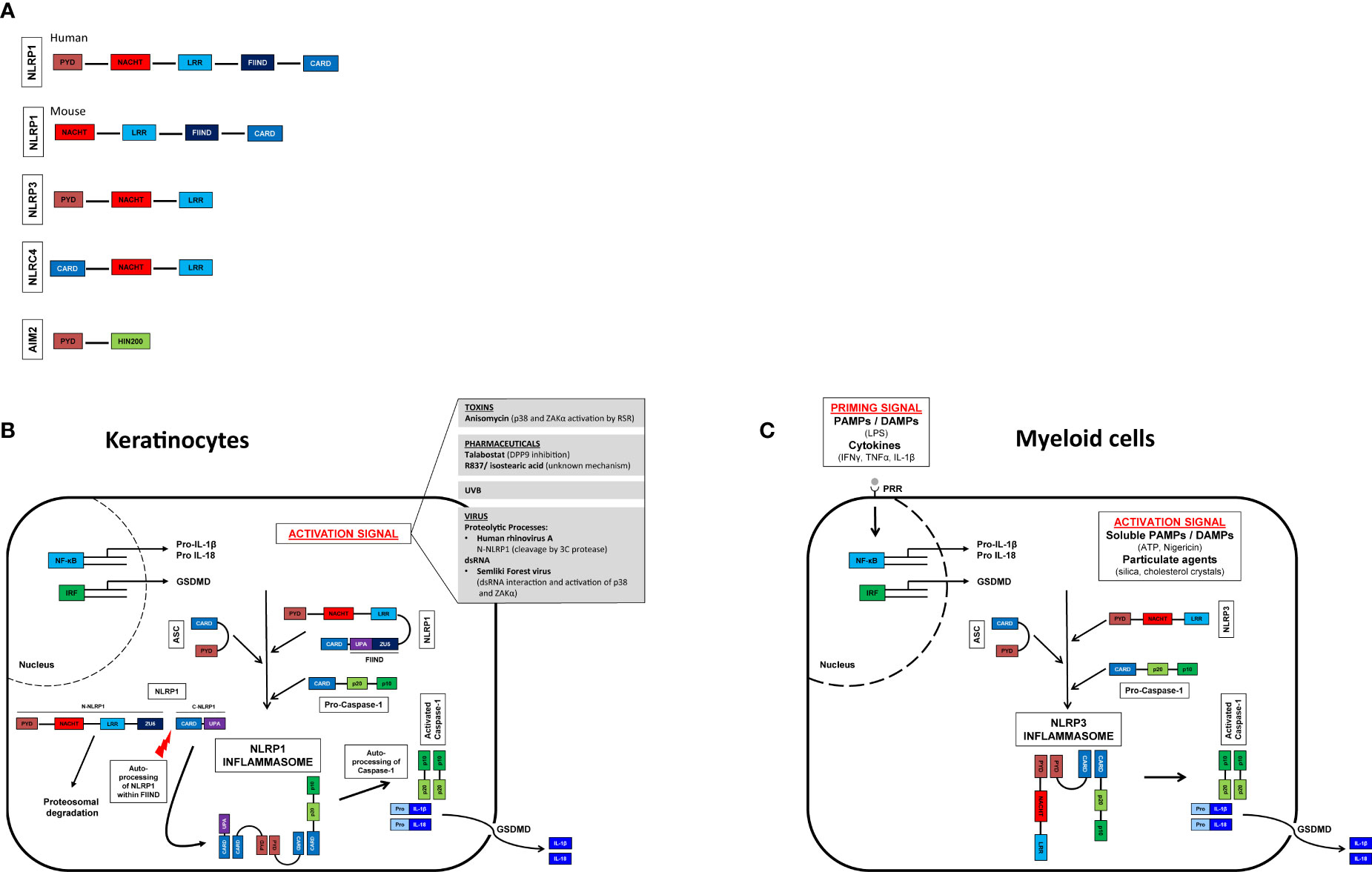
Figure 1 Inflammasome activation in myeloid cells and keratinocytes and their domain structure. Domain structure of human and murine NLRP1 (nucleotide-binding domain and leucine-rich repeat [NLR], pyrin domain [PYD] containing protein 1), NLRP3 (NLR family PYD domain containing protein 3), NLRC4 (NLR family caspase CARD [Caspase recruitment domain] domain containing protein 4), AIM2 (Absent in melanoma 2). GSDMD, gasdermin D (A). Activation signals lead to oligomerization of the sensor (NLRP1/NLRP3) with ASC and caspase 1. Self-activation of caspase 1 and subsequent activation of pro-IL-1β and pro-IL-18. Pore formation in the cell membrane allows secretion of mature IL-1β and IL-18. Myeloid cells (C), unlike keratinocytes (B), do not constitutively express pro-IL-1β and pro-IL-18 and therefore require a priming step (41, 42).
In contrast to humans, mice express two NLRP1 paralogs (NLRP1a and NLRP1b) and one pseudogene (NLRP1c) all of which lack the N-terminal PYD domain (Figure 1A) (50, 51). Interestingly, UVB irradiation of murine keratinocytes does not lead to inflammasome activation; expression of pro-IL-1ß and NLRP1 is only marginal (52). Unlike human keratinocytes, which secrete IL-1ß upon UVB irradiation, this appears to be mediated by a yet unidentified cell type. Therefore, murine models investigating NLRP1-activation might not be transferable to the human system. Murine NLRP1b is activated by bacterial enzymes, as for example the E3 ubiquitin ligase IpaH7.8 of Shigella flexneri or the protease lethal factor of Bacillus anthracis (53, 54).
In contrast to murine keratinocytes and human myeloid cells, human keratinocytes do not require an initial “priming” signal but can be activated directly by one stimulus. Among the best studied activation mechanisms are (i.) proteolytic processes, (ii.) DPP9 inhibition, (iii.) double-stranded RNA, and (iv.) phosphorylation (35). Briefly, the human rhinovirus (HRV) 3C protease cleaves hNLRP1 between the PYD and NACHT domains and subsequently the newly formed amino terminus of NLRP1 is ubiquitinated and degraded by the proteasome (55). NLRP1 is also activated by the anticancer drug talabostat (Val-boroPro, PT-100) (56). At steady state DPP9 binds to one fragment of N-NLRP1 and two fragments of C-NLRP1. Talabostat releases C-NLRP1 from DPP9, leading to proteasomal degradation of N-NLRP1 (47, 48, 57, 58). Double-stranded RNA (dsRNA) from alphaviruses can bind to the LRR domain, most likely leading to a conformational change of N-NLRP1 and proteasomal degradation and consequently activation of C-NLRP1 (58). UVB and certain bacterial toxins activate the ribotoxic stress response (RSR) and in turn induce NLRP1 phosphorylation between PYD and NACHT domains via ZAKα (a MAPK kinase activated by the RSR) and downstream p38. Phosphorylation results in ubiquitination and proteasomal degradation of N-NLRP1 (55, 59).
In summary, similarly to the NLPR3 inflammasome in myeloid cells, the NLRP1 inflammasome in keratinocytes is activated by a variety of microbial or sterile danger signals, leading to de-stabilization of the N-terminal fragment of NLRP1 and, in turn, driving NLRP1 activation (Figure 1B). As inflammasome activation leads to the secretion of highly potent pro-inflammatory innate cytokines, all inflammasomes are strictly regulated. Dysregulation of inflammasomes of their downstream receptors or their antagonists sometimes leads to severe diseases. While a putative involvement of NLRP3 in numerous pathologies have been described for NLRP3 in recent years (Figure 1C), the exact role of NLRP1 or inflammasomes in the human skin immune system in vivo is poorly understood and needs further investigation.
From health to disease
The intrinsic inborn function of inflammasomes is to maintain the balance of the organism in the defense against pathogens or non-microbial danger signals (60). Impairment of inflammasome regulation is known in many common and less common diseases in the human system. There are diseases which are associated with certain inflammasome subtypes. NLRP3 inflammasome for example is associated with frequent diseases such as artherosclerosis and Alzheimer’s disease, most probably activated during the frustrated phagocytosis of cholesterin or fibrillar β-amyloid proteins (31, 61). Once activated, the secretion of IL-1β perpetuates inflammation.
When looking at inflammasome-associated diseases, it becomes obvious that there are above mentioned tissue-specific differences in function, as organ-specific symptoms can occur without a systemic impairment (62). As stated above, NLRP1 is the most prominent inflammasome sensor in the skin (63). Its function is not strictly linked to the innate immune system, as common inflammatory skin diseases (e.g. psoriasis and atopic dermatitis) are associated with the NLRP1 inflammasome and more importantly proinflammatory cytokines from IL-1 family (36, 60, 64–66). Atopic dermatitis is a prototypical disease that is shaped by genetic and environmental influences. A single nucleotide polymorphism (SNP) in the NLRP1 gene from Swedish patients with atopic dermatitis was classified as a possible susceptibility for the occurrence of a disease manifestation, although not the only contributing factor (67). Similarly, SNPs in the NLRP1 gene have been studied in patients with psoriasis vulgaris and have shown a strong association with disease susceptibility (68). In addition, the impaired endogenous production of IL-1 by keratinocytes also seems to play a role in the pathogenesis of psoriasis. Here, the dysbalance of the skin microbiome could be identified as a main cause (36, 69). A closer look at autoimmune diseases reveals the double-edged role of inflammasomes. On the one hand, activation is important for pathogen and sterile defense, especially at border surfaces, such as the epithelia of the skin and the gastrointestinal tract; on the other hand, overactivation and dysbalance can lead to disease manifestation in almost any organ [reviewed in (60)]. With regard to the skin, a classic inflammasome-associated disease is vitiligo, a multifactorial disease that is based on abnormal melanocyte function. Several SNPs in the NLRP1 gene were reported to be associated with vitiligo (70) and patient-derived monocytes were shown to secrete increased amounts of IL-1β upon activation (71). A putative role of IL-1 cytokines is also described in systemic lupus erythematosus (72, 73), leprosy (74, 75), oral pemphigus vulgaris (76) and Addison’s disease (hyperpigmentation) (77, 78). However, some of these studies lack the clear demonstration of the functional pathogenetic role of NLRP1, as they only describe genetic findings in the way of SNPs.
A functional connection between diseases and inflammasomes was shown in autoinflammatory diseases, where the term inflammasomopathies was shaped. It is an emerging group of multisystemic diseases characterized by elevated levels of proinflammatory cytokines in the serum, some of which present with cutaneous symptoms (79–82). Several of those autoinflammatory diseases with skin manifestations have been described in which germline-activating mutations in the inflammasome sensor NLRP1 are present. Depending on the site in which the mutations occur, clinical pictures vary from localized cutaneous symptoms to accompanying systemic reactions with fever (63). In multiple self-healing palmoplantar carcinoma (MSPC), gain-of-functions are present in the pyrin domain of NLRP1 (63). Clinically, ulcerated, hyperkeratotic nodules termed keratoacanthomas are seen predominantly on the palmoplantar skin as well as on the conjunctiva and corneal epithelium. These lesions normally regress spontaneously but can predispose to the development of squamous cell carcinoma (SCC), demonstrating a link between inflammasome and carcinogenesis. Another disease with gain-of function mutations in the LRR domain of NLRP1 is familial keratosis lichenoides chronica (FKLC), in which affected individuals show disseminated lichenoid papules predominantly on the extremities and trunk. In both cases, the mutations lead to a loss of the autoinhibitory effect in NLRP1 with a consecutive increase in proinflammatory cytokines (63). Secretion of IL-1 cytokines implies the release of further proinflammatory cytokines and growth factors creating a proinflammatory milieu. This manifests in focal epidermal hyperplasia and hyperkeratosis, one of the morphological hallmarks of psoriasis (63, 68).
A further disease called NLRP1-associated autoinflammation with arthritis and dyskeratosis (NAIAD) is coined by a mutation between the NACHT and the LRR region or in the FIIND domain. It is accompanied by systemic reactions, especially fever, and high levels of proinflammatory cytokines in the blood (83).
A promising therapeutic approach in treatment of systemic autoinflammatory syndromes used in clinical practice is the blockade of the IL-1 pathway. Here, three modalities are available: the IL-1 receptor antagonist anakinra, the fusion protein rilonacept or canakinumab, a human monoclonal antibody directed against IL-1β (84–86).
The last two decades elucidated several diseases associated with mainly NLRP3 inflammasome dysregulation or IL-1 production and it remains to be found out why certain inflammasome subtypes lead to a distinct clinical picture. Although NLRP1 is also highly expressed in myeloid cells, the germline mutations have a more severe phenotype in the skin compared to systemic functions (63). It is still currently under investigation if this might be due to unknown, non-specific mutations, e.g. in non-coding areas or through post-translational modifications.
There is also growing evidence of overlapping autoimmune- autoinflammatory mechanisms. Innate and adaptive immune processes cannot be strictly separated from each other, precisely because they show similarities in the pathophysiology of clinically different diseases (87).
Gain-of function mutations in NLRP1 inflammasome demonstrate the link between inflammasome dysfunction and carcinogenesis (63). Inflammation and recognition of tumor cells by the immune system is crucial to control tumor growth. In clinical practice, inducing inflammation is used to treat superficial non-melanoma skin cancer (88). However, proinflammatory processes also drive tumorigenesis, for instance in lichen planus or chronic wounds (89). UVB radiation is the major risk factor for nonmelanoma skin cancer (90) as well as a known activator of the NLRP1 inflammasome in human keratinocytes leading to secretion of proinflammatory cytokines (38). Additionally, it results in decreased levels of anti-apoptotic proteins (Bcl-2 and Bcl-XL) (91). Other recent reports present downregulation of inflammasome components in SCC, which may contribute to tumor progression, especially at later time points (92). One might postulate different functions of innate immunity or inflammasomes at different stages of disease progression. In tumorigenesis, inflammation might favor tumor progression, while once the tumor is established, innate immunity might control tumor growth or even induces tumor regression. The proinflammatory anti-carcinogenic effect of inflammasomes is also exploited in the topical therapy of early SCC or its precursor, actinic keratosis. Imiquimod which is used as a topical treatment is both an activator of TLR7 and of NLRP3 (93, 94). In primary malignant melanoma, a pro-tumorigenic role of the NLRP3 inflammasome was shown (95), while anti-apoptotic influence of the NLRP1 inflammasome seems to play a role particularly in metastatic malignant melanoma (96).
Besides the previously discussed cancers, rare mutations associated with DPP9 deficiency should also be mentioned. Here, not only immunological and neurological deficits, but skin changes can also occur in the setting of DPP9 deficiency (97). In addition, a mutation leading to impaired binding of DPP9 to the inflammasome, resulting in loss of autoinhibition with enhanced activation was recently described (16). The clinical relevance and the question of why NLRP1 deficiencies or functional disorders lead to isolated skin manifestations is still the subject of current research.
For an overview of the diseases mentioned, see Figure 2.
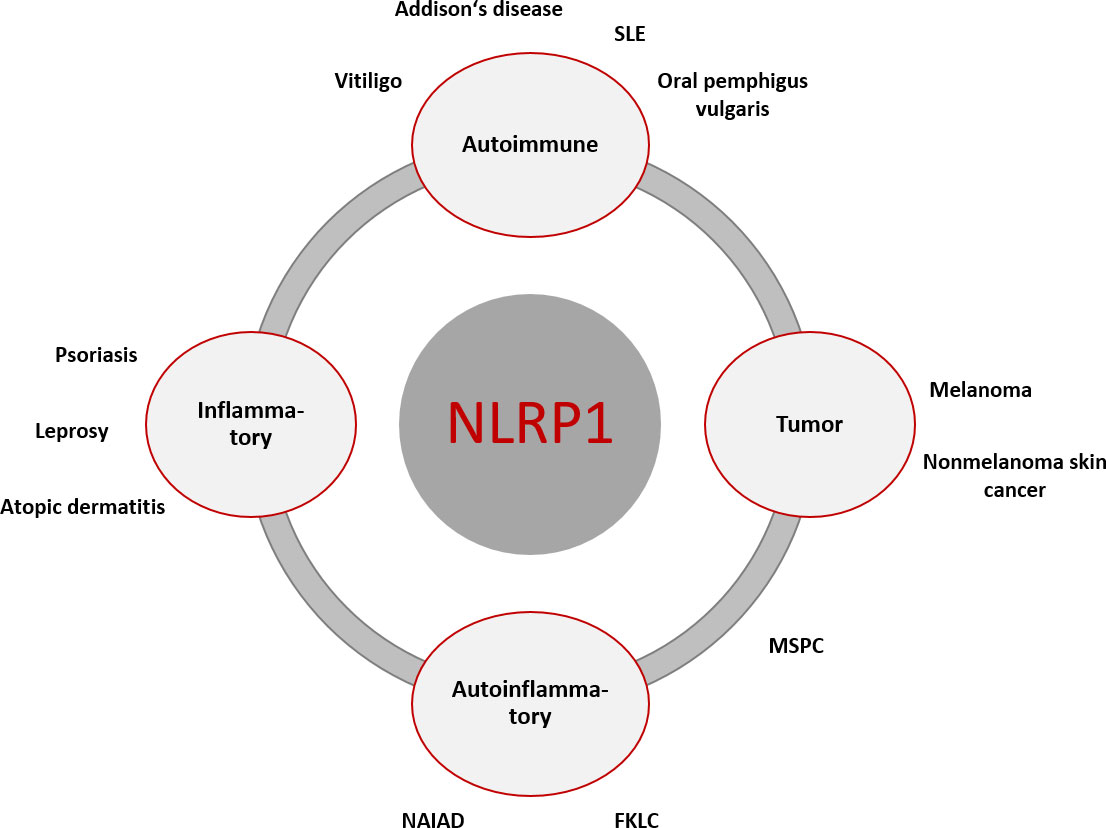
Figure 2 Overview of NLRP1-associated diseases with primary cutaneous manifestation or associated cutaneous clinical symptoms. The primary genesis may be inflammatory, autoimmune, autoinflammatory, and cancerogenic. There is overlap between the different entities, which can lead to symptom complexes that are not clearly assignable diagnostically and therapeutically. [FKLC, familial keratosis lichenoides chronica; MSPC, Multiple Self-Healing Palmoplantar Carcinoma; NAIAD, NLRP1- associated autoinflammation with arthritis and dyskeratosis; SLE, systemic lupus erythematodes].
From now to future
Since the discovery of inflammasomes in 2002 (8) there have been major advances in understanding how PAMPs and DAMPs are recognized by the immune system and how it triggers both innate and adaptive inflammation. Likewise, during the past two decades, the impact of IL-1, inflammatory caspases or inflammasomes on various human diseases, such as gout, arteriosclerosis, pyoderma gangrenosum became more apparent, with a still growing list of diseases (62). The investigation of the activation and regulation of inflammasomes at molecular levels is most advanced in monogenic hereditary syndromes described above, where a targeted treatment is now possible (98). The effect of anti-IL-1 treatments in other non-monogenetic disorders is still under investigation (99). However, some essential questions have not yet been answered. With regard to the skin, no physiologically relevant activator of NLRP1 inflammasomes has been identified so far, apart from UVB (38). In the future, it will be important not to overlook the role of commensal microorganism on the skin and their secreted products (such as metabolites but also secreted colonization/virulence factors). A challenge will be to differentiate the effect of commensals or pathogens on the NLRP1 inflammasome in the skin. Microbe-specific virulence factors could have distinct effects on the fine regulation of inflammasome activity. It is yet cryptic if NLRP1 activation in keratinocytes itself, the subsequent activation of inflammatory caspases or the secretion of innate cytokines impair the skin barrier or induce the recruitment of inflammatory cells to the epidermis. As the skin is involved in most of the “inflammasopathies” and inflammasome activation was reported in various diseases, NLRP1 inflammasome might be a promising pathway to develop targeted therapies. Future inflammasome studies should primarily be investigated in the human model to depict physiological conditions.
Author contributions
MB, MS and AY contributed to the manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the START-Program of the Faculty of Medicine of the RWTH Aachen University and by the Deutsche Forschungsgemeinschaft (TRR 156, 246807620 and 264 FOR2497, 289113135 to AY).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol (2014) 14(5):289–301. doi: 10.1038/nri3646
2. Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol (2004) 4(6):469–78. doi: 10.1038/nri1372
3. Lai Y, Gallo RL. Toll-like receptors in skin infections and inflammatory diseases. Infect Disord Drug Targets (2008) 8(3):144–55. doi: 10.2174/1871526510808030144
4. Janeway CA Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
5. Wicherska-Pawlowska K, Wrobel T, Rybka J. Toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-Like receptors (RLRs) in innate immunity. TLRs, NLRs, and RLRs ligands as immunotherapeutic agents for hematopoietic diseases. Int J Mol Sci (2021) 22(24):13397. doi: 10.3390/ijms222413397
6. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity. (2008) 28(3):285–7. doi: 10.1016/j.immuni.2008.02.005
7. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
8. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/S1097-2765(02)00599-3
9. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe (2010) 8(6):471–83. doi: 10.1016/j.chom.2010.11.007
10. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9(3):113–4. doi: 10.1016/S0966-842X(00)01936-3
11. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440(7081):237–41. doi: 10.1038/nature04516
12. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and ipaf. Nature. (2004) 430(6996):213–8. doi: 10.1038/nature02664
13. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. (2009) 458(7237):509–13. doi: 10.1038/nature07710
14. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458(7237):514–8. doi: 10.1038/nature07725
15. Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med (2018) 24(8):1151–6. doi: 10.1038/s41591-018-0082-y
16. Zhong FL, Robinson K, Teo DET, Tan KY, Lim C, Harapas CR, et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem (2018) 293(49):18864–78. doi: 10.1074/jbc.RA118.004350
17. Franklin BS, Latz E, Schmidt FI. The intra- and extracellular functions of ASC specks. Immunol Rev (2018) 281(1):74–87. doi: 10.1111/imr.12611
18. Smatlik N, Drexler SK, Burian M, Rocken M, Yazdi AS. ASC speck formation after inflammasome activation in primary human keratinocytes. Oxid Med Cell Longev (2021) 2021:7914829. doi: 10.1155/2021/7914829
19. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
20. Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem (2002) 277(24):21119–22. doi: 10.1074/jbc.C200179200
21. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med (2018) 215(3):827–40. doi: 10.1084/jem.20172222
22. Julien O, Wells JA. Caspases and their substrates. Cell Death Differ (2017) 24(8):1380–9. doi: 10.1038/cdd.2017.44
23. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
24. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. (1992) 356(6372):768–74. doi: 10.1038/356768a0
25. Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA, et al. Structure and mechanism of interleukin-1 beta converting enzyme. Nature. (1994) 370(6487):270–5. doi: 10.1038/370270a0
26. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. (1992) 256(5053):97–100. doi: 10.1126/science.1373520
27. Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol (2009) 10(3):241–7. doi: 10.1038/ni.1703
28. Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
29. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526(7575):660–5. doi: 10.1038/nature15514
30. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. (2013) 493(7434):674–8. doi: 10.1038/nature11729
31. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464(7293):1357–61. doi: 10.1038/nature08938
32. Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol (2021) 22(5):550–9. doi: 10.1038/s41590-021-00886-5
33. Meier-Schiesser B, French LE. Autoinflammatory syndromes. J Dtsch Dermatol Ges (2021) 19(3):400–26. doi: 10.1111/ddg.14332
34. Fenini G, Karakaya T, Hennig P, Di Filippo M, Beer HD. The NLRP1 inflammasome in human skin and beyond. Int J Mol Sci (2020) 21(13):4788. doi: 10.3390/ijms21134788
35. Fenini G, Karakaya T, Hennig P, Di Filippo M, Slaufova M, Beer HD. NLRP1 inflammasome activation in keratinocytes: Increasing evidence of important roles in inflammatory skin diseases and immunity. J Invest Dermatol (2022) 142(9):2313–22. doi: 10.1016/j.jid.2022.04.004
36. Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Goss C, Anz D, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med (2011) 3(82):82ra38. doi: 10.1126/scitranslmed.3002001
37. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
38. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol (2007) 17(13):1140–5. doi: 10.1016/j.cub.2007.05.074
39. Naik SM, Cannon G, Burbach GJ, Singh SR, Swerlick RA, Wilcox JN, et al. Human keratinocytes constitutively express interleukin-18 and secrete biologically active interleukin-18 after treatment with pro-inflammatory mediators and dinitrochlorobenzene. J Invest Dermatol (1999) 113(5):766–72. doi: 10.1046/j.1523-1747.1999.00750.x
40. Companjen AR, van der Velden VH, Vooys A, Debets R, Benner R, Prens EP. Human keratinocytes are major producers of IL-18: Predominant expression of the unprocessed form. Eur Cytokine Netw (2000) 11(3):383–90.
41. Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res (2020) 30(4):315–27. doi: 10.1038/s41422-020-0295-8
42. Patel MN, Carroll RG, Galvan-Pena S, Mills EL, Olden R, Triantafilou M, et al. Inflammasome priming in sterile inflammatory disease. Trends Mol Med (2017) 23(2):165–80. doi: 10.1016/j.molmed.2016.12.007
43. Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, et al. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem (2012) 287(30):25030–7. doi: 10.1074/jbc.M112.378323
44. Frew BC, Joag VR, Mogridge J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PloS Pathog (2012) 8(4):e1002659. doi: 10.1371/journal.ppat.1002659
45. D'Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, Reed JC. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PloS One (2011) 6(11):e27396. doi: 10.1371/journal.pone.0027396
46. Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, Johnson DC, et al. Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem Biol (2018) 25(3):262–7 e5. doi: 10.1016/j.chembiol.2017.12.013
47. Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, Dagbay KB, et al. DPP9 sequesters the c terminus of NLRP1 to repress inflammasome activation. Nature. (2021) 592(7856):778–83. doi: 10.1038/s41586-021-03350-4
48. Huang M, Zhang X, Toh GA, Gong Q, Wang J, Han Z, et al. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature. (2021) 592(7856):773–7. doi: 10.1038/s41586-021-03320-w
49. Robert Hollingsworth L, David L, Li Y, Griswold AR, Ruan J, Sharif H, et al. Mechanism of filament formation in UPA-promoted CARD8 and NLRP1 inflammasomes. Nat Commun (2021) 12(1):189. doi: 10.1038/s41467-020-20320-y
50. Yu CH, Moecking J, Geyer M, Masters SL. Mechanisms of NLRP1-mediated autoinflammatory disease in humans and mice. J Mol Biol (2018) 430(2):142–52. doi: 10.1016/j.jmb.2017.07.012
51. Sastalla I, Crown D, Masters SL, McKenzie A, Leppla SH, Moayeri M. Transcriptional analysis of the three Nlrp1 paralogs in mice. BMC Genomics (2013) 14:188. doi: 10.1186/1471-2164-14-188
52. Sand J, Haertel E, Biedermann T, Contassot E, Reichmann E, French LE, et al. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis (2018) 9(2):24. doi: 10.1038/s41419-017-0009-4
53. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science. (2019) 364(6435):eaau1330. doi: 10.1126/science.aau1330
54. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet (2006) 38(2):240–4. doi: 10.1038/ng1724
55. Robinson KS, Teo DET, Tan KS, Toh GA, Ong HH, Lim CK, et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science (2020) 370(6521):eaay2002. doi: 10.1126/science.aay2002
56. Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res (2004) 64(15):5471–80. doi: 10.1158/0008-5472.CAN-04-0447
57. Gai K, Okondo MC, Rao SD, Chui AJ, Ball DP, Johnson DC, et al. DPP8/9 inhibitors are universal activators of functional NLRP1 alleles. Cell Death Dis (2019) 10(8):587. doi: 10.1038/s41419-019-1817-5
58. Bauernfried S, Hornung V. DPP9 restrains NLRP1 activation. Nat Struct Mol Biol (2021) 28(4):333–6. doi: 10.1038/s41594-021-00580-y
59. Jenster LM, Lange KE, Normann S, vom Hemdt A, Wuerth JD, Schiffelers LDJ, et al. P38 kinases mediate NLRP1 inflammasome activation after ribotoxic stress response and virus infection. J Exp Med (2023) 220(1):e20220837. doi: 10.1084/jem.20220837
60. Tupik JD, Nagai-Singer MA, Allen IC. To protect or adversely affect? the dichotomous role of the NLRP1 inflammasome in human disease. Mol Aspects Med (2020) 76:100858. doi: 10.1016/j.mam.2020.100858
61. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
62. Alehashemi S, Goldbach-Mansky R. Human autoinflammatory diseases mediated by NLRP3-, pyrin-, NLRP1-, and NLRC4-inflammasome dysregulation updates on diagnosis, treatment, and the respective roles of IL-1 and IL-18. Front Immunol (2020) 11:1840. doi: 10.3389/fimmu.2020.01840
63. Zhong FL, Mamai O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. (2016) 167(1):187–202 e17. doi: 10.1016/j.cell.2016.09.001
64. Iznardo H, Puig L. IL-1 family cytokines in inflammatory dermatoses: Pathogenetic role and potential therapeutic implications. Int J Mol Sci (2022) 23(16):9479. doi: 10.3390/ijms23169479
65. Migliorini P, Italiani P, Pratesi F, Puxeddu I, Boraschi D. The IL-1 family cytokines and receptors in autoimmune diseases. Autoimmun Rev (2020) 19(9):102617. doi: 10.1016/j.autrev.2020.102617
66. Tang L, Zhou F. Inflammasomes in common immune-related skin diseases. Front Immunol (2020) 11:882. doi: 10.3389/fimmu.2020.00882
67. Bivik C, Verma D, Winge MC, Lieden A, Bradley M, Rosdahl I, et al. Genetic variation in the inflammasome and atopic dermatitis susceptibility. J Invest Dermatol (2013) 133(10):2486–9. doi: 10.1038/jid.2013.168
68. Ekman AK, Verma D, Fredrikson M, Bivik C, Enerback C. Genetic variations of NLRP1: susceptibility in psoriasis. Br J Dermatol (2014) 171(6):1517–20. doi: 10.1111/bjd.13178
69. Cai Y, Xue F, Quan C, Qu M, Liu N, Zhang Y, et al. A critical role of the IL-1beta-IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol (2019) 139(1):146–56. doi: 10.1016/j.jid.2018.07.025
70. Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med (2007) 356(12):1216–25. doi: 10.1056/NEJMoa061592
71. Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proc Natl Acad Sci U S A (2013) 110(8):2952–6. doi: 10.1073/pnas.1222808110
72. Pontillo A, Reis EC, Liphaus BL, Silva CA, Carneiro-Sampaio M. Inflammasome polymorphisms in juvenile systemic lupus erythematosus. Autoimmunity. (2015) 48(7):434–7. doi: 10.3109/08916934.2015.1064399
73. Perez-Alamino R, Cuchacovich R, Espinoza LR, Porretta CP, Zea AH. Role of inflammasome activation in systemic lupus erythematosus: Are innate immune cells activated? Reumatol Clin (Engl Ed) (2021) 17(4):187–91. doi: 10.1016/j.reuma.2019.10.007
74. Silva LM, de Sousa JR, Hirai KE, Dias LB Jr., Furlaneto IP, Carneiro FRO, et al. The inflammasome in leprosy skin lesions: an immunohistochemical evaluation. Infect Drug Resist (2018) 11:2231–40. doi: 10.2147/IDR.S172806
75. Pontillo A, Laurentino W, Crovella S, Pereira AC. NLRP1 haplotypes associated with leprosy in Brazilian patients. Infect Genet Evol (2013) 19:274–9. doi: 10.1016/j.meegid.2013.06.006
76. Shamsabadi RM, Basafa S, Yarahmadi R, Goorani S, Khani M, Kamarehei M, et al. Elevated expression of NLRP1 and IPAF are related to oral pemphigus vulgaris pathogenesis. Inflammation. (2015) 38(1):205–8. doi: 10.1007/s10753-014-0023-y
77. Magitta NF, Boe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, et al. A coding polymorphism in NALP1 confers risk for autoimmune addison's disease and type 1 diabetes. Genes Immun (2009) 10(2):120–4. doi: 10.1038/gene.2008.85
78. Zurawek M, Fichna M, Januszkiewicz-Lewandowska D, Gryczynska M, Fichna P, Nowak J. A coding variant in NLRP1 is associated with autoimmune addison's disease. Hum Immunol (2010) 71(5):530–4. doi: 10.1016/j.humimm.2010.02.004
79. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and muckle-wells syndrome. Nat Genet (2001) 29(3):301–5. doi: 10.1038/ng756
80. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46(10):1140–6. doi: 10.1038/ng.3089
81. Ozdogan H, Ugurlu S. Familial Mediterranean fever. Presse Med (2019) 48(1 Pt 2):e61–76. doi: 10.1016/j.lpm.2018.08.014
82. Borst C, Symmank D, Drach M, Weninger W. Cutaneous signs and mechanisms of inflammasomopathies. Ann Rheum Dis (2022) 81(4):454–65. doi: 10.1136/annrheumdis-2021-220977
83. Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis (2017) 76(7):1191–8. doi: 10.1136/annrheumdis-2016-210021
84. Hoffman HM. Therapy of autoinflammatory syndromes. J Allergy Clin Immunol (2009) 124(6):1129–38. doi: 10.1016/j.jaci.2009.11.001
85. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med (2009) 360(23):2416–25. doi: 10.1056/NEJMoa0810787
86. Goldbach-Mansky R, Shroff SD, Wilson M, Snyder C, Plehn S, Barham B, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheumatol (2008) 58(8):2432–42. doi: 10.1002/art.23620
87. Doria A, Zen M, Bettio S, Gatto M, Bassi N, Nalotto L, et al. Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev (2012) 12(1):22–30. doi: 10.1016/j.autrev.2012.07.018
88. Voiculescu VM, Lisievici CV, Lupu M, Vajaitu C, Draghici CC, Popa AV, et al. Mediators of inflammation in topical therapy of skin cancers. Mediators Inflamm (2019) 2019:8369690. doi: 10.1155/2019/8369690
89. Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol (2012) 13(4):343–51. doi: 10.1038/ni.2224
90. Green AC, Olsen CM. Cutaneous squamous cell carcinoma: an epidemiological review. Br J Dermatol (2017) 177(2):373–81. doi: 10.1111/bjd.15324
91. Reagan-Shaw S, Breur J, Ahmad N. Enhancement of UVB radiation-mediated apoptosis by sanguinarine in HaCaT human immortalized keratinocytes. Mol Cancer Ther (2006) 5(2):418–29. doi: 10.1158/1535-7163.MCT-05-0250
92. Sand J, Fenini G, Grossi S, Hennig P, Di Filippo M, Levesque M, et al. The NLRP1 inflammasome pathway is silenced in cutaneous squamous cell carcinoma. J Invest Dermatol (2019) 139(8):1788–97 e6. doi: 10.1016/j.jid.2019.01.025
93. Walter A, Schafer M, Cecconi V, Matter C, Urosevic-Maiwald M, Belloni B, et al. Aldara activates TLR7-independent immune defence. Nat Commun (2013) 4:1560. doi: 10.1038/ncomms2566
94. Navi D, Huntley A. Imiquimod 5 percent cream and the treatment of cutaneous malignancy. Dermatol Online J (2004) 10(1):4. doi: 10.5070/D34VW339W4
95. Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem (2010) 285(9):6477–88. doi: 10.1074/jbc.M109.064907
96. Zhai Z, Liu W, Kaur M, Luo Y, Domenico J, Samson JM, et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene. (2017) 36(27):3820–30. doi: 10.1038/onc.2017.26
97. Harapas CR, Robinson KS, Lay K, Wong J, Moreno Traspas R, Nabavizadeh N, et al. DPP9 deficiency: An inflammasomopathy that can be rescued by lowering NLRP1/IL-1 signaling. Sci Immunol (2022), 7(75):eabi4611. doi: 10.1126/sciimmunol.abi4611
98. Nigrovic PA, Lee PY, Hoffman HM. Monogenic autoinflammatory disorders: Conceptual overview, phenotype, and clinical approach. J Allergy Clin Immunol (2020) 146(5):925–37. doi: 10.1016/j.jaci.2020.08.017
Keywords: inflammasomes, nucleotide-binding domain and leucine-rich repeat containing gene family, interleukin-1β, caspase-1, keratinocytes, skin, innate immunity, therapeutic target
Citation: Burian M, Schmidt MF and Yazdi AS (2023) The NLRP1 inflammasome in skin diseases. Front. Immunol. 14:1111611. doi: 10.3389/fimmu.2023.1111611
Received: 29 November 2022; Accepted: 13 February 2023;
Published: 23 February 2023.
Edited by:
Ralf J. Ludwig, University of Lübeck, GermanyReviewed by:
Knut Schäkel, Heidelberg University, GermanyChristian David Sadik, University of Lübeck, Germany
Copyright © 2023 Burian, Schmidt and Yazdi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amir S. Yazdi, YXlhemRpQHVrYWFjaGVuLmRl
† These authors share first authorship