 Hao Zhou1,2
Hao Zhou1,2 Jun Li1
Jun Li1 Dewei Zhou1Yingjie Wu1Xingliang Wang3
Dewei Zhou1Yingjie Wu1Xingliang Wang3 Jiang Zhou3
Jiang Zhou3 Qingqing Ma1
Qingqing Ma1 Xinsheng Yao1*
Xinsheng Yao1* Long Ma1*
Long Ma1*- 1Department of Immunology, Center of Immunomolecular Engineering, Innovation & Practice Base for Graduate Students Education, Zunyi Medical University, Zunyi, China
- 2Department of Genome Informatics, Research Institute for Microbial Diseases, Osaka University, Suita, Japan
- 3School of Life Sciences, Guizhou Normal University, Guiyang, China
Introduction: Bats are recognized as natural reservoirs for many viruses, and their unique immune system enables them to coexist with these viruses without frequently exhibiting disease symptoms. However, the current understanding of the bat adaptive immune system is limited due to the lack of a database or tool capable of processing T-cell receptor (TCR) sequences for bats.
Methods: We performed germline gene annotation in three bat species using homologous genes and RSSs (Recombinational Signal Sequences) scanning method. Then we used the conserved C gene to construct the TCRβ chain receptor library of the Intermediate Horseshoe Bat. Bats' TCRβ data will be analyzed using MiXCR and constructed reference library.
Results: Regarding the annotation results, we found that the Pale Spear-nosed Bat has 37 members in the TRBV12 family, which is more than the total number of TRBV genes in the Greater Horseshoe Bat. The average number of unique TCRβ chain receptor sequences in each Intermediate Horseshoe Bat sample reached 24,904.
Discussion: The distinct variations in the distribution of TRBV genes among the three types of bats could have a direct impact on the diversity of the TCR repertoire, as evidenced by the presence of conserved amino acids that indicate the T-cell recognition of antigens in bats is MHC-restricted. The bats’ TCRβ repertoire is formed through the rearrangement of the V-D-J-C genes, with D-J/V-D deletions and insertions resulting in high diversity.
1 Introduction
Bats belong to the order Chiroptera, are the second largest order of mammals in the world (1). Despite carrying numerous virulent zoonotic viruses, bats do not often show serious clinical symptoms (2–5). This has led to increased interest in the differences between the bat immune system and those of other mammals.
Studies have identified several differences between the innate immune systems of bats and those of humans or mice. For instance, in humans or mice, activation of pattern recognition receptors (PRRs) by RNA viruses, danger signals, or intracellular double-stranded DNA triggers the activation of NLR-family pyrin domain containing 3 (NLRP3) or Interleukin-1β (IL-1β), resulting in inflammation. In contrast, bats inhibit the transcriptional initiation of NLRP3, leading to reduced functionality of interferon-inducible proteins AIM2 and IFI-16 (6–9), This, in turn, leads to lower caspase-1 activity and IL-1β cleavage, resulting in overall reduced inflammation. A recent study also found that deletion of the 358th serine site of the bat STING protein inhibits interferon secretion (10). Moreover, Pavlovich et al. reported a “high amplification and non-classical distribution of genes” at MHC-I loci in bats, suggesting that the combination of these abundant non-classically distributed MHC-I type genes with highly expressed NKG2, which contain inhibitory interaction motifs, increases the activation threshold of NK cells and reduces their response (11, 12).
The lack of suitable reagents and models for cellular biology studies, the challenge of isolating viruses, and the diverse nature of bat species (belonging to 2 orders, 21 families, and over 1350 species) hinder the understanding of the adaptive immune system of bats (13). Next Generation Sequencing (NGS) technology has facilitated high-throughput sequencing (HTS) for genome analysis of various species. The Bat1k Project and Vertebrate Genome Project (VGP) have produced high-quality bat genomic data, which has improved our knowledge of the bat immune system, particularly their response to viral infections (14, 15). However, the annotation of T/B cell receptor germline genes in bats remains limited. The TCR/BCR repertoire comprises all functional T or B cells in an individual’s circulatory system, each with its own antigen-specific receptor. Deep sequencing of the repertoire is a commonly used method for studying the adaptive immune system, assessing an individual’s health status, developing antibodies, and detecting and treating targeted diseases (16). Various tools, such as IMGT/HighV-QUEST, TRUST, MiXCR, have been used to process TCR/BCR sequences (17–19). Our previous work involved annotating R. ferrumequinum TRB loci, indicating that the bat’s adaptive immune response is similar to that of humans and mice, and that its TCR is rearranged from germline genes with high diversity (20). In this study, we expanded the annotation to include the TRB loci of P. discolor and P. pipistrellus, and compared the TRB loci of all three bat species. The TRBC Exon1 sequences of various bats were aligned, and primers were developed to amplify the TCR β-chain CDR3 repertoire of R. affinis using the 5’ Rapid Amplification of cDNA Ends (5’RACE) technique. Our study provides a crucial theoretical foundation, a novel research method, and a comparable database for studying genetic evolution and adaptive immune response in Chiroptera.
2 Materials and methods
2.1 TRB locus location and annotation
Figure 1A presents the experimental flow of the study, which involved identifying the TRB loci in the genome by detecting two genes, MOXD2 and EPHB6, located at the boundary of the TRB loci. Two gene annotation methods were used to annotate the regions between the two genes, including IMGT-LIGMotif (21) and 12/23 RSS(Recombination Signal Sequence) scanning (22). For the IMGT-LIGMotif method, representative animals from three different subjects, namely human(primate), mouse(rodents), and pig(artiodactyla), were selected, and TRBV genes of all known species in IMGT were obtained to ensure that no information was missed due to evolutionary divergence. The mapping of these genes to the TRB loci of the three bat species was performed using Geneious Prime (Version 2022.2.1). The 12/23 RSS scanning method involved screening all RSS motifs within the three bat TRB loci and searching for V/D/J genes upstream and downstream of the RSS. The annotation result, including the number of genes and locus composition information, is summarized in Table 1. Homology of the TRB loci among the three annotated bats was compared using Easyfig (Version 2.2.5), with a threshold of 70% identity (Figure 1C). Following the IMGT guidelines (23), the key components of each germline gene within the locus were labeled after annotation with Geneious Prime, and the annotation file was exported in GenBank format (Supplementary Data 1 and Supplement Figure 1).
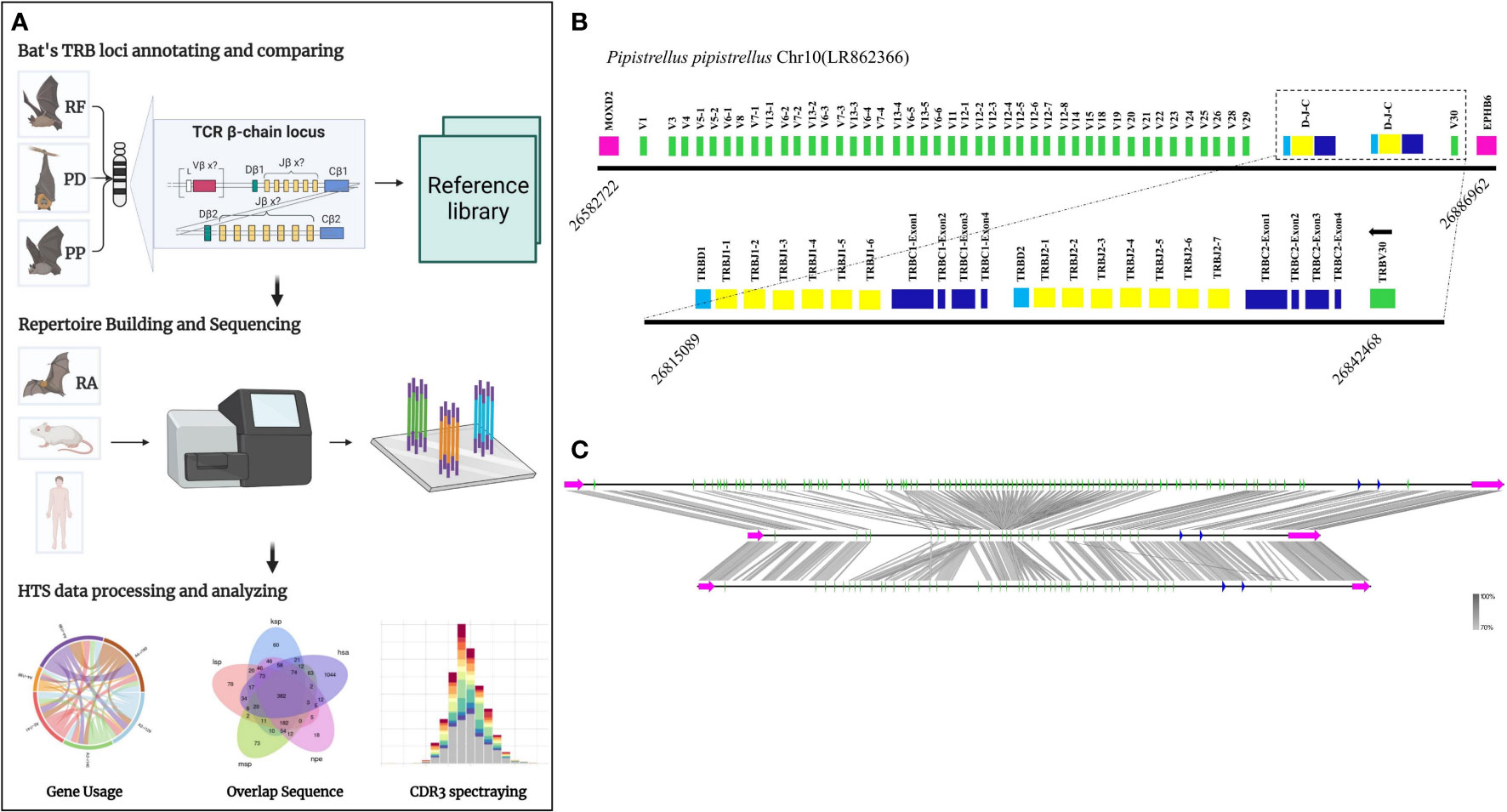
Figure 1 The TRB loci annotation and correlation analysis of three bat species. (A) Overview of this study design. (B) P. pipistrellus TRB loci on Chromosome 10, with colored boxes representing each annotated gene, including 44 TRBV genes, 2 TRBD genes, 13 TRBJ genes, and 2 TRBC genes. (C) A genome homology comparison map of the TRB loci for the three annotated bat species.

Table 1 Number of germline genes retrived by homologous genes and RSSs method.
2.2 Naming of germline genes and clustering of TRBV families
The germline gene names were assigned following the guidelines provided by IMGT. In order to classify V genes into families, a threshold of 75% nucleotide identity was set. To verify familial homology, a phylogenetic tree of TRBV nucleotide sequences was constructed using the Neighbor-Joining method in MEGA7 (version 7.0.26). The TRBV genes from human, mouse, and pig were retrieved from IMGT-GeneDB (https://www.imgt.org/genedb/), and only functional genes and ORFs were selected, with one gene per family. Supplementary Table 1 lists the accession numbers used in the analysis. The distribution of TRBV gene families in twelve mammalian species belonging to four different subjects (Artiodactyla, Carnivores, and Primates) was compared to understand the evolutionary direction of the V gene (Figure 2A). The D, J, and C genes were named based on their position within the cluster they belong to in the locus.
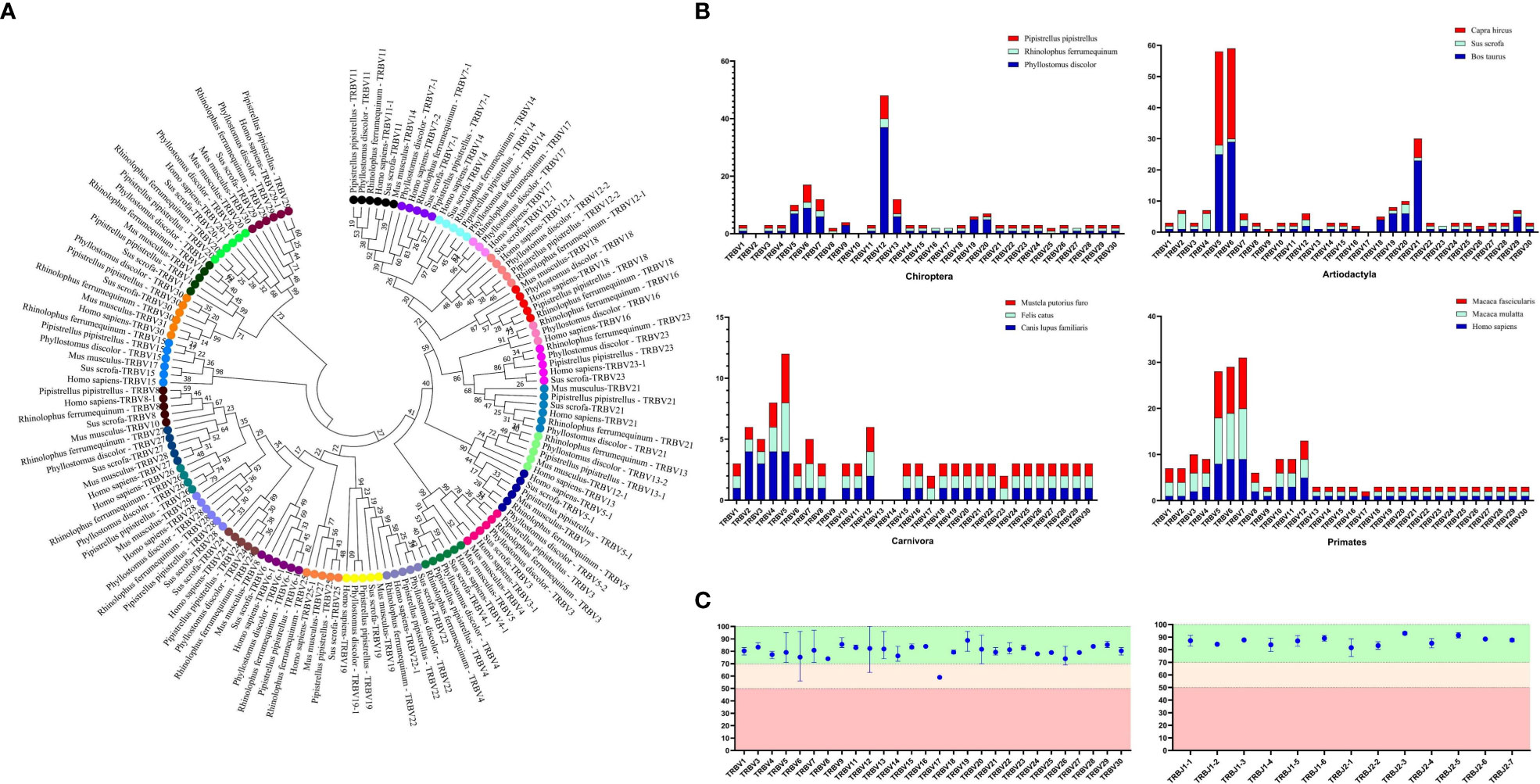
Figure 2 Analysis of TRBV gene homology in different bat species, as well as in human, mice, and pigs. (A) An NJ-phylogenetic tree was constructed from functional TRBV gene nucleotide sequences, with each color representing one family and bootstrap is 1000. (B) A comparison of TRBV gene family distribution in three mammalian orders (Carnivora, Artiodactyla, and Primates) with the three bat species is shown, with data from https://www.imgt.org/IMGTrepertoire/. (C) A comparison of the nucleotide sequence indentity of TRBV and TRBJ genes is presented for the nature annotated bat species.
2.3 Establishment of reference library
To establish a reference library, we utilized Geneious Prime (Clustal Omega) to align the amino acid sequences of the germline V and J genes. Priority was given to aligning three conserved sites, 23CYS, 41TRP, and 104CYS, based on their known structure and specific positions in mammalian TRBV (23). Structural domains were classified based on alignment results, with V genes including FR1 (1–26), CDR1 (27–38), FR2 (39–55), CDR2 (56–65), FR3 (66-104), and CDR3 (105-117), and J genes including FR4 (phenylalanine to the end of the FGNG motif sequence). Functional descriptions of germline genes were based on IMGT, incorporating stop codons, RSS, conserved amino acids, and splice sites. Supplementary Table 2 provides information on pseudogenes and ORFs. The germline gene nucleic sequences and structural domain information were exported as reference library files identifiable by MiXCR, enabling processing of bat TRB data.
2.4 Sample collection
Bat samples were collected with the assistance of Zhou’s group in Xishui County, Zunyi City, Guizhou Province. Muscle tissues were used to extract genetic information by amplifying the Cytb gene with designed primers (Supplementary Table 5), and PCR products were sequenced to determine bats’ genotypes based on E-value and Percent Identity. Although we did not find any annotated bat species based on BLAST results, we selected three R. affinis as our experimental model due to their expected high homology with the target species, R. ferrumequinum. Spleen tissues from three 5-month-old BALB/c mice were used for RNA extraction and TCRβ chain library construction. Moreover, we collected molecular cells from 5 ml of peripheral blood from three individuals at the Affiliated Hospital of Zunyi Medical University. The study was approved by the Animal Protection and Ethics Committee of Zunyi Medical University, with work involving humans approved under permit number (2021)1-022 and the bat and mouse project approved under permit number (2018)2-261.
2.5 TCR-β chain repertoire sequencing and analysis
To construct and amplify the TCR-β library for R. affinis, we collected and aligned fifteen known bats’ TRBC Exon1 nucleotide sequences(accession number in Supplementary Table 1), designed primers in conserved regions, and used the 5’RACE method to prepare repertoires of R. affinis. Library construction and sequencing were performed by ImmuQuad Company in Hangzhou, China. We used samples from humans and mice for species repertoire comparison. Spleen tissue was used for TCR-β repertoire construction in mice using the 5’RACE method, while peripheral blood mononuclear cells (PBMC) were collected from three individuals for humans, and the multiplex PCR method was used for TCR-β repertoire construction.
We analyzed the sequencing results of bats, humans, and mice using the MiXCR software (Version 3.0.13). MiXCR was used to assemble and present the sequencing results of bats based on the library constructed from annotated genes. The output data of the nine samples are shown in Table 2 and Supplementary Table 6. To ensure the quality of the repertoire, we examined the sequences of bat1 sample, which contained 25 TRBV genes and 14 TRBJ genes (Figure 4E). The evolutionary relationship between the sequenced samples and the three annotated bats was determined by sequence attribution, Cytb, and TRBC gene alignment. To assess the homogeneity and heterogeneity of TCR-β repertoires among the three species, we used Productive-Clonetype sequences for subsequent analyses to exclude the effects of repertoire construction and sequencing, as well as physiological health status. Our analyses included: (1) statistics on repertoire overlap index among the three species; (2) usage of TRBV gene and TRBJ gene, V-J gene pairing, and tracking of conserved motifs; and (3) evaluation of CDR3 length and amino acid usage, as well as nucleotide deletion and insertion.
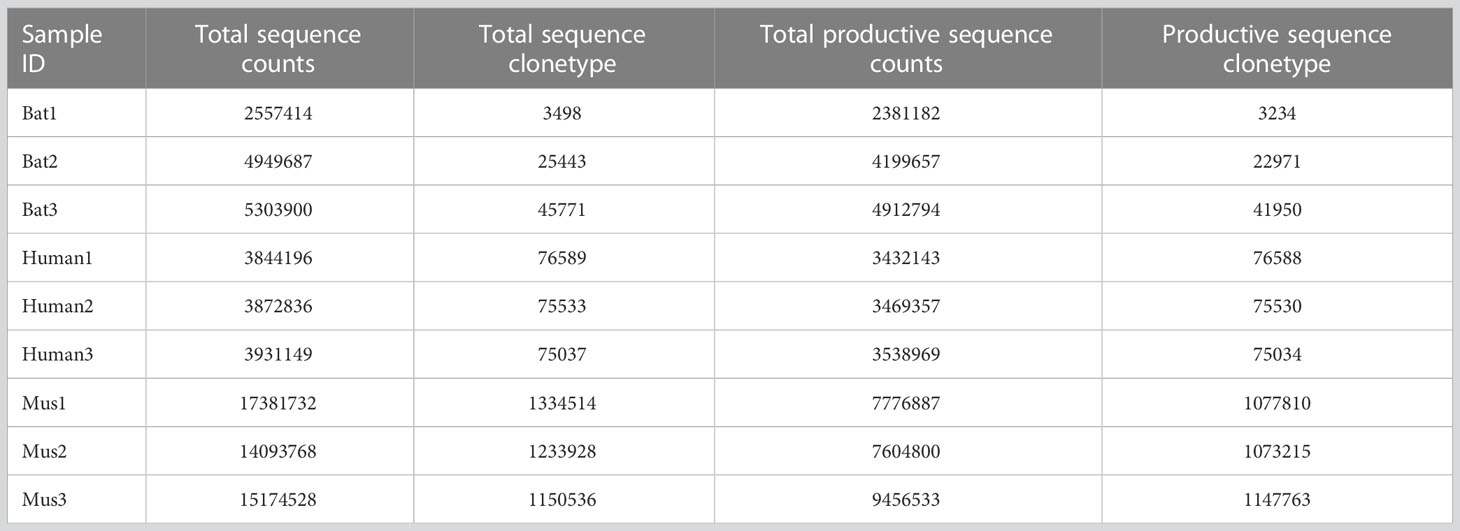
Table 2 MiXCR outputted sequence statistics from a total of nine samples, including bats, humans, and mice.
2.6 Statistical analysis
The figures were draw with GraphPad Prism (Version 8.0.2), R package “ggplot2”, and R package “Immunarch”. Data analysis was performed by R studio (v3.3.3) and GraphPad Prism. P-values were calculated with the aid of the t test. P<0.05 was considered statistically significant.
3 Results
3.1 TR loci annotation and Reference directory establishment
Table 1 records the summary of TRB loci using homologous genes and RSS methods. The TRB location in P. discolor was found on chromosome 10(NC_040912: 85517683-85943353), R. ferrumequinum on chromosome 26(NC_046309: 7911840-8171343), and P. pipstrellus on chromosome10(LR862366.1: 26582722-26886962). The TRB loci in all three species followed the classical mammalian structure, but with differences in length distribution and orientation as shown in Figure 1B and Supplementary Figure 1.
We identified 100 TRBV genes in P. discolor (including 2 pseudogenes), 30 in R. ferrumequinum, and 45 in P. pipistrellus. A phylogenetic tree was constructed using the nucleotide sequences of TRBV genes, which showed that all genes could be homologous to each other or to those found in pigs and humans. Functional V gene families did not cluster separately in bats (Figure 2A). The main differences in the number of germline genes were due to internal duplications and deletions of the V gene family. The evolutionary direction of Chiroptera and Artiodactyla was more similar, as shown in Figure 2B. Most of the TRBV and TRBJ gene families of the three bat species had more than 70% identity, except for the TRBV17 family (Figure 2C). We identified a specific TRBV12 family of P. discolor containing 37 members, which was close to almost all TRBV genes of P. pipistrellus and even exceeded the total TRBV genes of R. ferrumequinum.
In the Multiple Sequence Alignment (MSA) of TRBV genes, we identified several conserved sites, including 23Cys, 41Trp, 89Leu, and 104Cys, as well as Gln at position 6 and Tyr at position 42. Gln at position 44 was also highly conserved in P. pipistrellus. Additionally, most TRBV genes contained CASS motifs at the end (Figure 3A and Supplementary Figure 2A). P. discolor had a higher number of germline genes and more gene duplications, resulting in a greater number of pseudogenes (17%) compared to R. ferrumequinum (7%) and P. pipistrellus (11%). However, the number of functional genes (V and J genes) in P. discolor, which exceeded that of R. ferrumequinum and P. pipistrellus, was 97 (Supplementary Table 2). Like primates and carnivores, Chiroptera contains two D-J-C clusters named according to their chromosomal location. The only difference between the three bat species regarding the number of genes in each cluster was the number of J genes in the second cluster. All TRBJ genes in bats contained conserved FGNG motifs except for TRBJ2-3 in R. ferrumequinum (Figure 3B and Supplementary Figure 2B). In addition, all bats’ TRBD genes were guanine-rich sequences, with 23RSS and 12RSS downstream and upstream (Figure 3C). There were no significant differences in the RSS between bats and other mammals, and the most conserved site was the first four sites (CACA) of the heptamer, with three continuous adenosines in the middle position of the nonamer (Figure 3D and Supplementary Figure 3). We created a MiXCR reference library for HTS data analysis using nucleotide sequences and amino acid sequence MSA results. To organize the germline genes family of three bat species, we combined them into a single family with allelic nomenclature methods. (Supplementary Table 3).
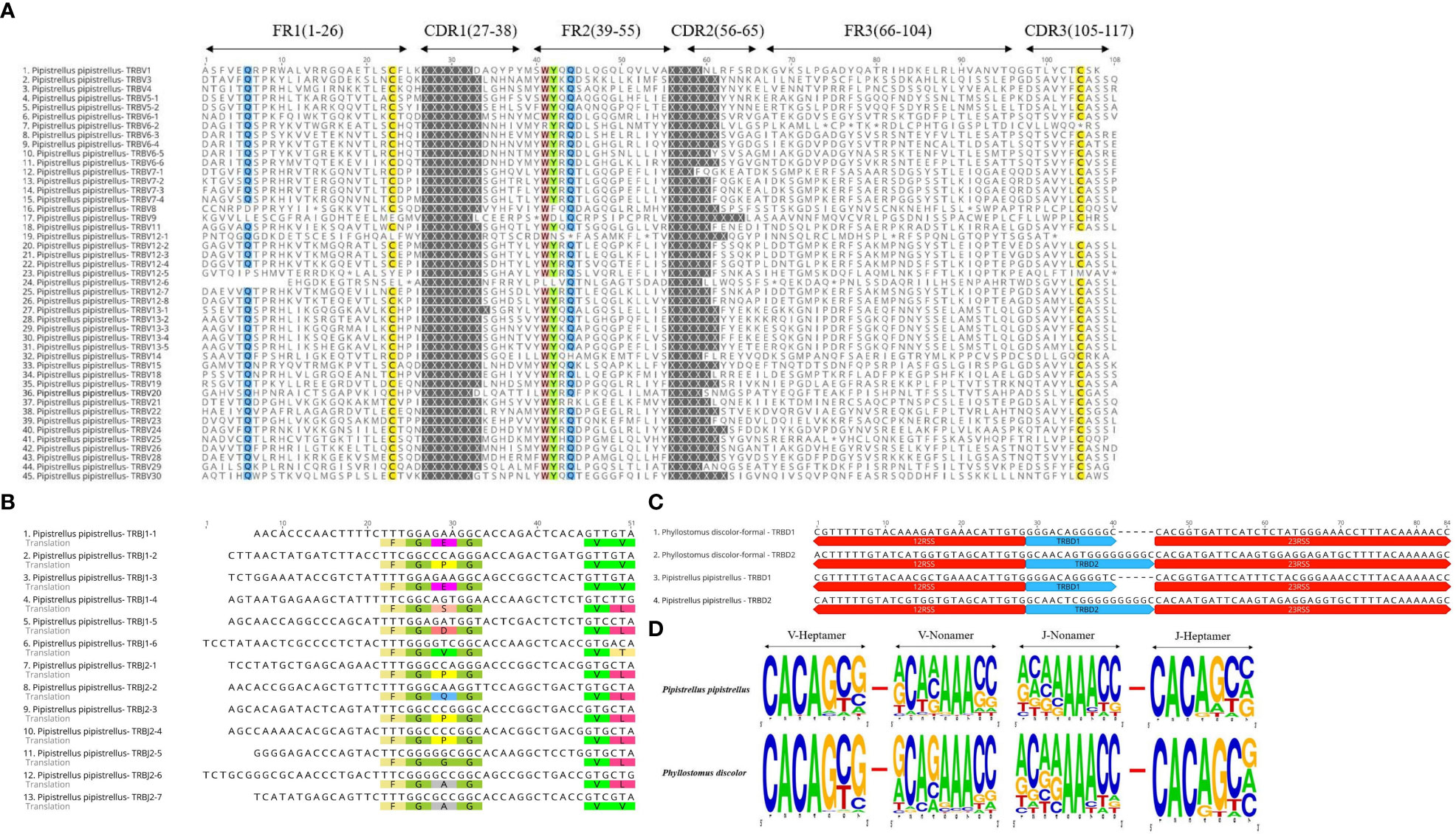
Figure 3 Annotated Germline Genes Display. (A) Displays 45 TRBV amino acid sequences with a consistency threshold of 90%. (B) Displays the nucleotide and amino acid sequences of 13 TRBJ genes with translated conserved FGNG motifs. (C) Shows four TRBD genes along with their 23RSS and 12RSS nucleotide sequences. (D) Compares the conserved sites of TRBV’23RSS and TRBJ’12RSS in P. discolor and P. pipistrellus.
3.2 TCRβ repertoire constructing, sequencing, analyzing
The Cytb sequencing results of the three experimental bats were analyzed, and an average sequence length of 488 base pairs and 98.5% sequence identity was observed. The bats were identified as R. affinis through a BLAST analysis, as shown in Supplementary Table 4. Furthermore, we constructed the TCRβ chain repertoire using the 5’RACE method, which involved comparing the conserved regions of the TRBC Exon1 nucleotide sequences of fifteen different types of bats and designing primers accordingly. The nucleotide sequence identity of 19 TRBC Exon1 varied from 77.66% to 98.9%, and several fully conserved regions were suitable for repertoire construction and sequencing, as illustrated in Figure 4A.

Figure 4 Homology analysis of R. affinis samples and annotated bat species. (A) Alignment of TRBC Exon1 nucleotide sequences of 15 Chiroptera species used for primer design in the 5’RACE method of R. affinis. (B) Comparison of Cytb nucleotide sequence results of R. affinis samples with P. discolor (PD), R. ferrumequinum (RF), and P. pipistrellus (PP). (C) Comparison of a partial C gene fragment from the bat1 sample with those of the three annotated bat species. (D) Attribution statistics of the sequencing results of the three R. affinis samples in the three annotated bat species. (E) Output of 25 TRBV genes and 14 TRBJ genes in the bat1 sample.
ImmuQuad Company conducted the sequencing, and the library was confirmed by a peak between 600 and 700 base pairs in the R. affinis samples, as shown in Supplementary Figure 4. MiXCR was used to process raw data from nine samples, which were analyzed and can be accessed at NCBI_PRJNA877449 (SRX17468175, SRX17468176, and SRX17468177). The 5’race method yielded an average of 4.27 million TCR reads and 24,904 clonotypes per bat sample, while the mouse samples had 15 million TCR reads and 1.23 million clonotypes per sample. The multiplex-PCR method resulted in 3.8 million TCR reads and 75,000 clonotypes per human sample. Despite variations in sequencing depth across bat samples, all samples met the CDR3 sequence depth analysis requirements for HTS sequencing, as indicated in Table 2.
To explore the degree of bat species divergence, we compared the Cytb nucleotide sequences of the three experimental bats (R. affinis) with those of R. ferrumequinum and P. pipistrellus (Figure 4B). The results showed that R. ferrumequinum had a higher identity (87.9%) to R. affinis than P. pipistrellus (74.6%). Additionally, the TRBC nucleotide fragment exhibited identity between R. affinis and P. discolor, R. ferrumequinum, and P. pipistrellus, with respective identities of 77.1%, 93.8%, and 75.0% (Figure 4C). After categorizing the sequencing results of the experimental samples, we found that most of the output sequences of R. affinis belonged to R. ferrumequinum (Figure 4D). These comparison results further indicated that the sequences of R. affinis were more similar to those of R. ferrumequinum. We then demonstrated the completeness of the repertoire by exporting 25 V and 14 J gene sequences from the Bat1 samples (Figure 4E).
3.3 Comparison of CDR3 repertoire in bats, humans, and mice
Four indices (overlap coefficient, Jaccard, Morisita, and Tversky) were used to assess the CDR3 repertoire in bats, humans, and mice (Figure 5A). The analysis showed that the bat CDR3 repertoires were variable and significantly different from each other, with some overlap coefficients slightly higher than humans but significantly lower than mice.
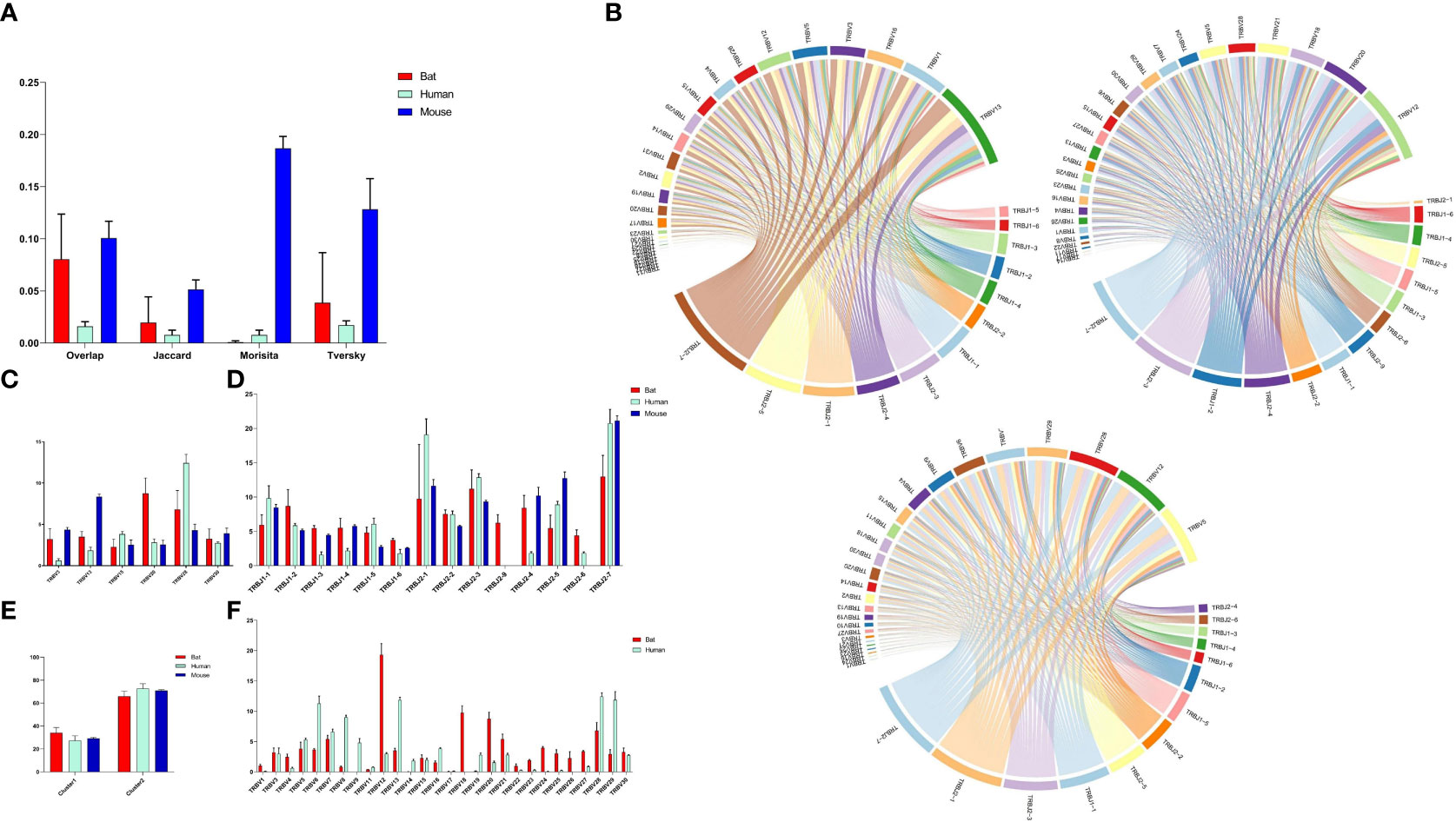
Figure 5 Comparative Analysis of TCR β Chain Repertoire Genes and Motif Usage in Bats, Humans, and Mice. (A) Overlap index of CDR3 sequences in the three species. (B) V-J pairing analysis of the three species, with the order being Bat1, Human1, and Mouse1. (C) Homologous TRBV gene usage analysis of the three species. (D) TRBJ gene usage analysis. (E) J gene usage in cluster. (F) TRBV gene usage analysis of bats and humans.
Based on the phylogenetic tree result, we analyzed the homology TRBV gene usage and V-J pairing (Figure 5B and Supplementary Figure 5) in bats, humans, and mice. Six homology TRBV genes were expressed, with TRBV15 and TRBV30 exhibiting similar usage (Figure 5C). Furthermore, we compared the usage of all TRBV genes in bats and humans (Figure 5F). The most commonly used TRBV genes in bats were TRBV12, TRBV18, and TRBV20, whereas in humans, they were TRBV13, TRBV28, and TRBV29. Notably, TRBV9 and TRBV19 were either not expressed or exhibited low expression in the bat samples. Additionally, the TRBV9 group was exclusive to P. discolor, with all four TRBV9 members being pseudogenes. Among the three species, all functional TRBJ genes, with the exception of TRBJ2-8 (pseudogene) in bats and TRBJ2-6 (pseudogene) in mice, were expressed (Figure 5D). Through comparative TRBJ analysis, we observed that high usage of TRBJ2-1, TRBJ2-3, and TRBJ2-7 occurred concurrently in bats, humans, and mice. Additionally, most of the J genes used in the three species were derived from the second D-J-C cluster (Figure 5E). This finding suggests a correlation between gene usage during rearrangement and distance.
We analyzed the CDR3 region motifs (5 amino acids) and found that four of the top ten high-frequency motifs were shared among bats, humans, and mice, with most being CASSN motifs (Figures 6A, B). This suggests that the cell populations involved in the immune response in the three species may be similar. The CDR3 region length was also characterized, with a bell-shaped distribution of 14 amino acids for bats and mice, and 15 amino acids for humans (Figure 6C). We assessed the length effects caused by insertion and deletion of nucleotides in the CDR3 region and found that humans and mice had similar deletions at the V’3 end, while bat and mouse had similar insertions at the V 3’ end and the J gene 5’ end (Figures 6D, E). Finally, we observed that the amino acids in the CDR3 region were consistent, with high frequencies observed for S, G, and A in all three species (Figure 6F).
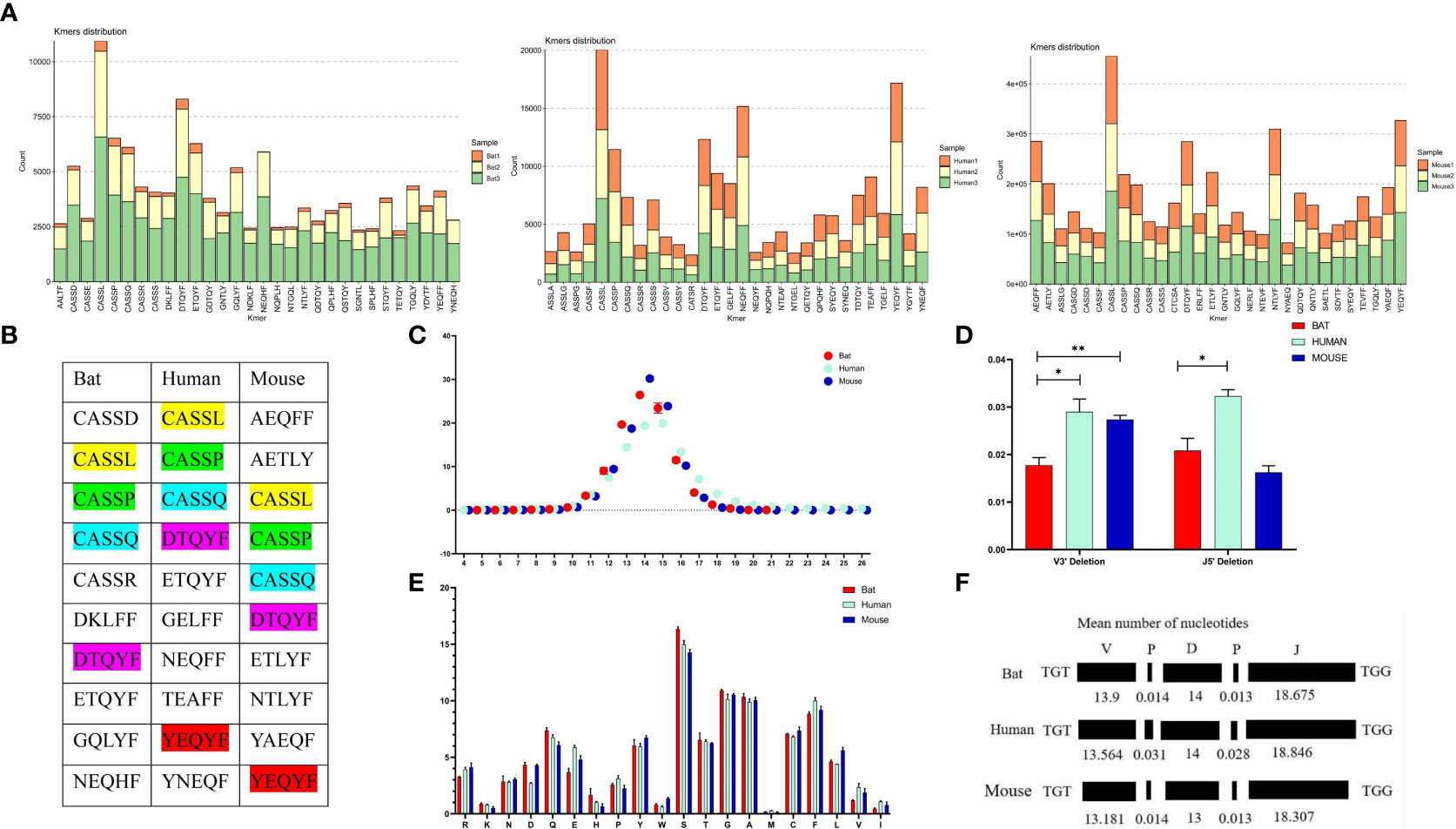
Figure 6 Comparative analysis of the CDR3 region features of the TCRβ chain repertoire in bat, human, and mouse is presented in this figure. (A) The top 30 5 AA length motifs used in the CDR3 region are analyzed. (B) The top 10 shared motifs are labeled in the same color. (C) The CDR3 region length is analyzed. (D) Deletions at the 3’ end of the V gene and 5’end of the J gene are compared. (E) AA usage in the CDR3 region is analyzed. (F) The composition of the CDR3 region is presented.
4 Discussion
The study of bat immunity has been hindered by the lack of basic genetic information, despite their importance in virus transmission and possession of unique physiological traits. Previous studies have identified T, B, and macrophage populations in Pteropus giganteus, with a higher T/B cell ratio in the spleen and lymph nodes when compared to mice (24). Further investigations have revealed that CD4+ T cells dominate the blood lymph, whereas CD8+ T cells dominate the spleen in fruit-eating bat Pteropus alecto (25). These findings suggest that T/B cell immunity plays a role in bats, but the understanding of TCR remains limited.
In 2021, the TR/IG loci of the Greater horseshoe bat and the Egyptian rousette bats were annotated separately (20, 26). However, no public database or tool has been used to analyze TCR/BCR sequences for bats, and the TCR/BCR repertoire characterization of bats has not been reported. To create a bat germline gene database, a complete annotation of TR/IG loci was a priority. It is important to note that the TR/IG loci annotation is different from normal genome annotation, requiring a high-quality genome due to the short genes (such as D and J genes) and the loci’s large span on the genome. For our research, we selected three bat species with high-quality genomes from the database: R. ferrumequinum, P. discolor, and P. pipistrellus. Creating germline gene databases involves identifying all potential genes based on specific structural features, which is the central issue. Due to the short sequence of D (10-15 bp) and J (30-50 bp) genes, we used the RSS sequence search method simultaneously to avoid losing sequences from the homology search. However, non-classical RSS (12/23+/-1 nt) may impact the final result, although such genes are rare in humans and mice. Moreover, pseudogenes lacking RSS cannot be involved in the rearrangement process, implying that they have no impact on the productive repertoire and pathogenic response (27).
In Chiroptera, previous studies have found varying degrees of replication of immune-related genes, but for TCR, gene duplication events were mostly reported in Artiodactyla such as cattle (28, 29) and goats (30), which have diverse T cell receptor pools. Surprisingly, massive gene duplication events of the TRBV12 family in P. discolor and considerable variation in TRBV gene numbers among Chiroptera have never been reported in mammals. In comparison, studies of primates (31–33), carnivores (34–36), and camels (37) have shown minimal differences in TRB loci structure and the number of germline genes among species of the same orders. The distribution and evolutionary direction of TRBV gene families in the three bat species are similar to Artiodactyla, with random and massive replications. The lack of gut-associated lymphoid tissue (GALT) in R. Hildebrandti and P. pipistrellus makes it unlikely that they possess a gene conversion-related mechanism to generate receptor diversity, as observed in the chicken immunoglobulin system (38–41). Instead, T cells use gene rearrangement as the predominant mechanism for generating TCR diversity in bats. This difference in rearrangement likelihood suggests that TCRβ chain repertoire diversity might be present in at least the three annotated bat species. The anchor residues in TRBV genes are important for MHC peptide binding, and while MHC genes in bats are more diverse than in other mammals, previous research has shown high sequence similarity between bat and human, mouse, dog, and cow MHC genes (42–44). Moreover, the anchor residues in the annotated TRBV and TRBJ genes of bats are highly conserved, indicating that TCR recognition in bats is MHC-restricted.
We have established a bat TCR β chain reference library and analyzed the TCR β chain repertoire using three R. affinis samples. From a total of 4.2 million reads on average, we found that the number of TCR clonotypes differed among the bat samples, with bat1 having 3498, while bat2 and bat3 had 25443 and 45771, respectively. There are several possible explanations for this variation. Firstly, it may be due to differences in the expression of TCRs between individual bats. This suggests that the diversity of individual bats may vary. Second, since this is the first time we have performed repertoire building and sequencing of bats, the sequencing depth may have an impact, and increasing the number of sequenced samples would help to address potential issues of individual bat diversity and low TCR expression due to limited sample collection. After analyzing the sequence output of each bat sample, we discovered that 25 TRBV genes and 14 TRBJ gene families were present in every experimental bat. This finding indicates that we have, for the first time, achieved a high level of completeness in identifying the TCR β chain CDR3 in bats. Our results provide valuable technical resources and data analysis tools to assist in the design and optimization of primers for high-throughput sequencing analysis of TCR repertoire from bats belonging to different families using 5’ RACE. The TCR β chain sequences of R. affinis were highly similar to the annotated genes of R. ferrumequinum. Specifically, 99% of the C genes belonged to R. ferrumequinum, and the TRBV and TRBJ genes were nearly identical to the annotated V and J genes of R. ferrumequinum. This suggests that certain immune response genes are genetically conserved among bat species of the same family.
We conducted an analysis on the usage and pairing patterns of TRBV and TRBJ genes in bats, humans, and mice. Despite humans having over 50 and mice over 30 germline V genes, their specific immune response may rely on only one or a few of them (45). The low expression levels of several gene families may be explained by a variety of factors. Firstly, it’s possible that the low expression levels are due to R. affinis itself. Additionally, the TRBV17 family has very low homology in the three annotated bats, which suggests that it may undergo significant mutations in the R. affinis genome and therefore not be detected. These factors could contribute to the overall low expression levels of certain gene families. A kind of mutation might lead to functional genes becoming pseudogenes, a situation that is very common across species. For example, the TRBV1 gene in bats is functional gene, while TRBV1 in humans is pseudogene. There are also cases where functional genes are not detected as expressed, for example, TRBV18 is barely expressed in humans but relatively highly expressed in bats; similar to TRBV23, TRBV24, TRBV26, etc. TRBV30, which is located in a specific position downstream of the second C gene and in a position opposite to transcription in bats, humans, and mice (TRBV31), was observed to be common in all three species. Several studies have reported that V and J gene usage during rearrangement correlates with position in the locus (46–48). Interestingly, in all three species, over 60% of the J gene is from the second D-J-C cluster. TRBV12-5 in humans and TRBV13-2 in mice are often linked to autoimmune diseases and account for over 50% of NK-T cells. Additionally, mouse MAIT cells express a TCR-α chain with TRAV1 and TRAJ33 and paired β chains of TRBV13 and TRBV19 (49–53). Notably, both TRBV12 in bats and TRBV13 in mice are highly utilized V genes. Bats exhibit significant differences from humans in their usage of multiple TRBV genes, indicating a specific biased amplification in the evolution of bat TCR immune response genes. This may be linked to evolutionary pressure or coevolution with viruses.
The usage of TRB CDR3 region amino acids is highly similar in bats, humans, and mice, with a consistent pattern of high-frequency motifs. However, there are significant differences in motif usage throughout the CDR3 region, indicating that the V-terminal and J-front ends of the CDR3 regions in these mammals are more conserved. This suggests that typical antigenic selection encountered in evolution may play a role. The length of the TRB CDR3 region is shorter in bats and mice compared to humans, which we attribute to differences in insertion and deletion at the V3’ and J5’ ends across the three species, as well as differences in the evolutionary length of the D gene and V/J involvement in the CDR3 region genes. While the overlap between individual bat CDR3 regions is higher than that of humans, it is significantly lower than that of mice. This finding suggests a higher concordance of CDR3 regions between different individual BALB/c mice with the same genetic background.
We annotated the complete TRB loci in three species of bats and constructed a database for analyzing the bat TCR repertoire. This allowed us to study the TCR evolution and specific immune response mechanism in bats and provide a new theory, technical tools, and data for comparative analysis of the mechanism of virus tolerance in bats.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA877449.
Ethics statement
The studies involving human participants were reviewed and approved by Zunyi Medical University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Zunyi Medical University.
Author contributions
XY, LM and HZ designed the experiment and wrote the paper, while LM and HZ conducted the experiments, analyzed data, and created graphs. Other contributors, including JL, LL, DZ, YW, XW, JZ and QM, assisted with sample collection, data analysis, manuscript revision and library construction for humans and mice. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (31860257&81860300) and the Guizhou Provincial Hundred Talent Fund [No. (2018) 5637].
Acknowledgments
We would like to express our gratitude to Jiang Zhou and Xingliang Wang from Guizhou Normal University for assisting in collecting the bat samples. We also thank ImmuQuad Company for their help in constructing the libraries and conducting high-throughput sequencing of the bat samples. Additionally, we would like to thank the Vertebrate Genome Project workers for their contribution in sequencing the bat genomes and sharing their data. Lastly, we extend our thanks to Dianita S. Saputri for revising the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1147859/full#supplementary-material
Supplementary Data Sheet 1 | GenBank files for Phyllostomus discolor, Rhinolophus ferrumequinum, and Pipistrellus pipistrellus.
References
1. Burgin CJ, Colella JP, Kahn PL, Upham NS. How many species of mammals are there? J Mammalogy (2018) 99:1–14. doi: 10.1093/jmammal/gyx147
2. Letko M, Seifert SN, Olival KJ, Plowright RK, Munster VJ. Bat-borne virus diversity, spillover and emergence. Nat Rev Microbiol (2020) 18:461–71. doi: 10.1038/s41579-020-0394-z
3. Ruiz-Aravena M, McKee C, Gamble A, Lunn T, Morris A, Snedden CE, et al. Ecology, evolution and spillover of coronaviruses from bats. Nat Rev Microbiol (2022) 20:299–314. doi: 10.1038/s41579-021-00652-2
4. Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
5. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med (2020) 26:450–2. doi: 10.1038/s41591-020-0820-9
6. Goh G, Ahn M, Zhu F, Lee LB, Luo D, Irving AT, et al. Complementary regulation of caspase-1 and IL-1beta reveals additional mechanisms of dampened inflammation in bats. Proc Natl Acad Sci U.S.A. (2020) 117:28939–49. doi: 10.1073/pnas.2003352117
7. Ahn M, Anderson DE, Zhang Q, Tan CW, Lim BL, Luko K, et al. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat Microbiol (2019) 4:789–99. doi: 10.1038/s41564-019-0371-3
8. Irving AT, Ahn M, Goh G, Anderson DE, Wang LF. Lessons from the host defences of bats, a unique viral reservoir. Nature (2021) 589:363–70. doi: 10.1038/s41586-020-03128-0
9. Ahn M, Cui J, Irving AT, Wang LF. Unique loss of the PYHIN gene family in bats amongst mammals: Implications for inflammasome sensing. Sci Rep (2016) 6:21722. doi: 10.1038/srep21722
10. Xie J, Li Y, Shen X, Goh G, Zhu Y, Cui J, et al. Dampened STING-dependent interferon activation in bats. Cell Host Microbe (2018) 23:297–301.e294. doi: 10.1016/j.chom.2018.01.006
11. Pavlovich SS, Lovett SP, Koroleva G, Guito JC, Arnold CE, Nagle ER, et al. The Egyptian rousette genome reveals unexpected features of bat antiviral immunity. Cell (2018) 173:1098–1110.e1018. doi: 10.1016/j.cell.2018.03.070
12. Randolph HE, Barreiro LB. Holy immune tolerance, batman! Immunity (2018) 48:1074–6. doi: 10.1016/j.immuni.2018.05.016
13. Sotero-Caio CG, Baker RJ, Volleth M. Chromosomal evolution in chiroptera. Genes (Basel) (2017) 8:272. doi: 10.3390/genes8100272
14. Jebb D, Huang Z, Pippel M, Hughes GM, Lavrichenko K, Devanna P, et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature (2020) 583:578–84. doi: 10.1038/s41586-020-2486-3
15. Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature (2021) 592:737–46. doi: 10.1038/s41586-021-03451-0
16. Six A, Mariotti-Ferrandiz ME, Chaara W, Magadan S, Pham H-P, Lefranc M-P, et al. The past, present, and future of immune repertoire biology - the rise of next-generation repertoire analysis. Front Immunol (2013) 4:413. doi: 10.3389/fimmu.2013.00413
17. Li S, Lefranc M-P, Miles JJ, Alamyar E, Giudicelli V, Duroux P, et al. IMGT/HighV QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat Commun (2013) 4:2333. doi: 10.1038/ncomms3333
18. Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: Software for comprehensive adaptive immunity profiling. Nat Methods (2015) 12:380–1. doi: 10.1038/nmeth.3364
19. Song L, Cohen D, Ouyang Z, Cao Y, Hu X, Liu XS. TRUST4: Immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat Methods (2021) 18:627–30. doi: 10.1038/s41592-021-01142-2
20. Zhou H, Ma L, Liu L, Yao X. TR locus annotation and characteristics of rhinolophus ferrumequinum. Front Immunol (2021) 12:741408. doi: 10.3389/fimmu.2021.741408
21. Lane J, Duroux P, Lefranc MP. From IMGT-ONTOLOGY to IMGT/LIGMotif: the IMGT® standardized approach for immunoglobulin and T cell receptor gene identification and description in large genomic sequences. BMC Bioinf (2010) 11:1–16. doi: 10.1186/1471-2105-11-223
22. Merelli I, Guffanti A, Fabbri M, Cocito A, Furia L, Grazini U, et al. RSSsite: A reference database and prediction tool for the identification of cryptic recombination signal sequences in human and murine genomes. Nucleic Acids Res (2010) 38:W262–7. doi: 10.1093/nar/gkq391
23. Giudicelli V, Lefranc MP. Imgt-ontology 2012. Front Genet (2012) 3:79. doi: 10.3389/fgene.2012.00079
24. Sarkar SK CA. Analysis of immunocompetent cells in the bat, pteropus giganteus: Isolation and scanning electron microscopic characterization. Dev Comp Immunol (1991) 15:423–30. doi: 10.1016/0145-305X(91)90034-V
25. Gómez JMM, Periasamy P, Dutertre C-A, Irving AT, Ng JHJ, Crameri G, et al. Phenotypic and functional characterization of the major lymphocyte populations in the fruit-eating bat pteropus alecto. Sci Rep (2016) 6:37796. doi: 10.1038/srep37796
26. Larson PA, Bartlett ML, Garcia K, Chitty J, Balkema-Buschmann A, Towner J, et al. Genomic features of humoral immunity support tolerance model in Egyptian rousette bats. Cell Rep (2021) 35:109140. doi: 10.1016/j.celrep.2021.109140
27. Ramsden DA, Baetz K, Wu GE. Conservation of sequence in recombination signal sequence spacers. Nucleic Acids Res (1994) 22:1785–96. doi: 10.1093/nar/22.10.1785
28. Connelley T, Aerts J, Law A, Morrison WI. Genomic analysis reveals extensive gene duplication within the bovine TRB locus. BMC Genomics (2009) 10:192. doi: 10.1186/1471-2164-10-192
29. Connelley TK, Degnan K, Longhi CW, Morrison WI. Genomic analysis offers insights into the evolution of the bovine TRA/TRD locus. BMC Genomics (2014) 15:1–17. doi: 10.1186/1471-2164-15-994
30. Giannico F, Massari S, Jambrenghi AC, Soriano A, Pala A, Linguiti G, et al. The expansion of the TRB and TRG genes in domestic goats (Capra hircus) is characteristic of the ruminant species. BMC Genomics (2020) 21:623. doi: 10.1186/s12864-020-07022-x
31. Jaiswal S, Nyquist SK, Boyce S, Jivanjee T, Ibrahim S, Bromley JD, et al. Identification and characterization of the T cell receptor (TCR) repertoire of the cynomolgus macaque (Macaca fascicularis). BMC Genomics (2022) 23:647. doi: 10.1186/s12864-022-08867-0
32. Pégorier P, Bertignac M, Chentli I, Ngoune VN, Folch G, Jabado-Michaloud J, et al. IMGT(R) biocuration and comparative study of the T cell receptor beta locus of veterinary species based on homo sapiens TRB. Front Immunol (2020) 11:821. doi: 10.3389/fimmu.2020.00821
33. Greenaway HY, Kurniawan M, Price DA, Douek DC, Davenport MP, Venturi V, et al. Extraction and characterization of the rhesus macaque T-cell receptor beta-chain genes. Immunol Cell Biol (2009) 87:546–53. doi: 10.1038/icb.2009.38
34. Mineccia M, Massari S, Linguiti G, Ceci L, Ciccarese S, Antonacci R, et al. New insight into the genomic structure of dog T cell receptor beta (TRB) locus inferred from expression analysis. Dev Comp Immunol (2012) 37:279–93. doi: 10.1016/j.dci.2012.03.010
35. Radtanakatikanon A, Keller SM, Darzentas N, Moore PF, Folch G, Ngoune VN, et al. Topology and expressed repertoire of the felis catus T cell receptor loci. BMC Genomics (2020) 21:20. doi: 10.1186/s12864-019-6431-5
36. Gerritsen B, Pandit A, Zaaraoui-Boutahar F, van den Hout MCGN, van IJcken WFJ, de Boer RJ, et al. Characterization of the ferret TRB locus guided by V, d, J, and c gene expression analysis. Immunogenetics (2020) 72:101–8. doi: 10.1007/s00251-019-01142-9
37. Antonacci R, Bellini M, Linguiti G, Ciccarese S, Massari S. Comparative analysis of the TRB locus in the camelus genus. Front Genet (2019) 10:482. doi: 10.3389/fgene.2019.00482
38. Strobel S, Encarnacao JA, Becker NI, Trenczek TE. Histological and histochemical analysis of the gastrointestinal tract of the common pipistrelle bat (Pipistrellus pipistrellus). Eur J Histochem (2015) 59:2477. doi: 10.4081/ejh.2015.2477
39. Makanya AN, Maina JN. The morphology of the intestine of the insectivorous horseshoe bat (Rhinolophus hildebvandti, peters): A scanning electron and light microscopic study. Afr. J Ecol (1994) 32:158–68. doi: 10.1111/j.1365-2028.1994.tb00566.x
40. Claude-Agnès Reynaud VA, Grimal Hélène, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell (1987) 48:379–88. doi: 10.1016/0092-8674(87)90189-9
41. RG. M. Diversification of rabbit VH genes by gene-conversion-like and hypermutation mechanisms. Immunol Rev (1998) 162:49–54. doi: 10.1111/j.1600-065X.1998.tb01428.x
42. Qu Z, Li Z, Ma L, Wei X, Zhang L, Liang R, et al. Structure and peptidome of the bat MHC class I molecule reveal a novel mechanism leading to high-affinity peptide binding. J Immunol (2019) 202:3493–506. doi: 10.4049/jimmunol.1900001
43. Qurkhuli T, Schwensow N, Brandel SD, Tschapka M, Sommer S. Can extreme MHC class I diversity be a feature of a wide geographic range? The example of seba's short-tailed bat (Carollia perspicillata). Immunogenetics (2019) 71:575–87. doi: 10.1007/s00251-019-01128-7
44. Ng JHJ, Tachedjian M, Deakin J, Wynne JW, Cui J, Haring V, et al. Evolution and comparative analysis of the bat MHC-I region. Sci Rep (2016) 6:21256. doi: 10.1038/srep21256
45. Turner SJ, Doherty PC, McCluskey J, Rossjohn J. Structural determinants of T-cell receptor bias in immunity. Nat Rev Immunol (2006) 6:883–94. doi: 10.1038/nri1977
46. Hansen TO, Lange AB, Barington T. Sterile DJH rearrangements reveal that distance between gene segments on the human ig h chain locus influences their ability to rearrange. J Immunol (2015) 194:973–82. doi: 10.4049/jimmunol.1401443
47. Fu L, Li X, Zhang W, Wang C, Wu J, Yang H, et al. A comprehensive profiling of T- and b-lymphocyte receptor repertoires from a Chinese-origin rhesus macaque by high-throughput sequencing. PLoS One (2017) 12:e0182733. doi: 10.1371/journal.pone.0182733
48. Park J-E, Botting RA, Domínguez Conde C, Popescu D-M, Lavaert M, Kunz DJ, et al. A cell atlas of human thymic development defines T cell repertoire formation. Science (2020) 367(6480):eaay3224. doi: 10.1126/science.aay3224
49. Alli R, Zhang ZM, Nguyen P, Zheng JJ, Geiger TL. Rational design of T cell receptors with enhanced sensitivity for antigen. PLoS One (2011) 6:e18027. doi: 10.1371/journal.pone.0018027
50. Cibotti R CJ, Pannetier C, Delarbre C, Vergnon I, Kanellopoulos JM, Kourilsky P. Public and private V beta T cell receptor repertoires against hen egg white lysozyme (HEL) in nontransgenic versus HEL transgenic mice. J Exp Med (1994) 180:861–72. doi: 10.1084/jem.180.3.861
51. Haqqi TM, Gary D. Anderson, Subhashis banerjee, and chella s. David, restricted heterogeneity in T-cell antigen receptor vβ gene usage in the lymph nodes and arthritic joints of mice. Proc Natl Acad Sci U.S.A. (1992) 89:1253–5. doi: 10.1073/pnas.89.4.1253
52. Masuda YMK, Cui J, Ito T, Tokuhisa T, Takahama Y, Koseki H, et al. Phenotypes and invariant alpha beta TCR expression of peripheral V alpha 14+ NK T cells. J Immunol (1997) 158:2076–82. doi: 10.4049/jimmunol.158.5.2076
Keywords: bat, TR loci annotation, high-throughput sequencing, TCRβ chain repertoire, germline genes
Citation: Zhou H, Li J, Zhou D, Wu Y, Wang X, Zhou J, Ma Q, Yao X and Ma L (2023) New insights into the germline genes and CDR3 repertoire of the TCRβ chain in Chiroptera. Front. Immunol. 14:1147859. doi: 10.3389/fimmu.2023.1147859
Received: 19 January 2023; Accepted: 07 March 2023;
Published: 27 March 2023.
Edited by:
Michelle Baker, Australian Centre for Disease Preparedness (CSIRO), AustraliaReviewed by:
Robert David Miller, University of New Mexico, United StatesIram Maqsood, Shaheed Benazir Bhutto Women University, Pakistan
Copyright © 2023 Zhou, Li, Zhou, Wu, Wang, Zhou, Ma, Yao and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinsheng Yao, aW1tdW5vbG9neUAxMjYuY29t; Long Ma, MTY0ODQ5NjcxQHFxLmNvbQ==