 Beatriz Escudero-Pérez
Beatriz Escudero-Pérez Philip Lawrence3
Philip Lawrence3 Javier Castillo-Olivares
Javier Castillo-Olivares- 1WHO Collaborating Centre for Arbovirus and Haemorrhagic Fever Reference and Research, Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany
- 2German Center for Infection Research (DZIF), Partner Site Hamburg-Luebeck-Borstel-Reims, Braunschweig, Germany
- 3CONFLUENCE: Sciences et Humanités (EA 1598), Université Catholique de Lyon (UCLy), Lyon, France
- 4Laboratory of Viral Zoonotics, University of Cambridge, Cambridge, United Kingdom
Correlates of protection (CoP) are biological parameters that predict a certain level of protection against an infectious disease. Well-established correlates of protection facilitate the development and licensing of vaccines by assessing protective efficacy without the need to expose clinical trial participants to the infectious agent against which the vaccine aims to protect. Despite the fact that viruses have many features in common, correlates of protection can vary considerably amongst the same virus family and even amongst a same virus depending on the infection phase that is under consideration. Moreover, the complex interplay between the various immune cell populations that interact during infection and the high degree of genetic variation of certain pathogens, renders the identification of immune correlates of protection difficult. Some emerging and re-emerging viruses of high consequence for public health such as SARS-CoV-2, Nipah virus (NiV) and Ebola virus (EBOV) are especially challenging with regards to the identification of CoP since these pathogens have been shown to dysregulate the immune response during infection. Whereas, virus neutralising antibodies and polyfunctional T-cell responses have been shown to correlate with certain levels of protection against SARS-CoV-2, EBOV and NiV, other effector mechanisms of immunity play important roles in shaping the immune response against these pathogens, which in turn might serve as alternative correlates of protection. This review describes the different components of the adaptive and innate immune system that are activated during SARS-CoV-2, EBOV and NiV infections and that may contribute to protection and virus clearance. Overall, we highlight the immune signatures that are associated with protection against these pathogens in humans and could be used as CoP.
1 Introduction
The human immune system responds through multiple interactive mechanisms to any invading pathogen, ultimately controlling the virus infection or clearing it from our system and enabling a more rapid response on subsequent encounters with such a pathogen. Correlates of protection (CoP) may be defined as those immunological parameters, characteristic of a specific immune mechanism, that are associated with protection against infection or disease (1). Understanding these specific immune mechanisms can help to identify specific immune CoP, which can then be used as surrogate measurements of vaccine protective efficacy and to assess the susceptibility of individuals and populations to a specific pathogen. It is important to note that whilst protection against different viral infections can be mediated by similar immune effector mechanisms, CoP are specific for a viral disease or infection (or even a specific manifestation of a disease), for a specific population group (elderly vs children) and even for a specific type of vaccine (CoP for an inactivated Influenza A vaccine may not necessarily coincide with that of an intra-nasal live Influenza vaccine, for example) (2–4).
The immune system is classically divided into two branches: the innate and the adaptive immune response. While innate immunity consists of a rapid but less specific inflammatory response, adaptive immunity develops more slowly, but is long-lasting and highly specific (5). However, this is a non-strict dichotomy since both arms of the immune response are strongly inter-connected. The interplay between the different immune cell populations and the complexity of immune reactions renders the rational design of effective vaccines against a specific pathogen difficult. For instance, while protection mediated through antibodies is prominent during the acute phase of an infection, cell‐mediated responses normally play an important role in virus infection clearance and/or during the chronic phase of infection. However, this is not the case for all pathogens (6).
The precise protective role of the different effector mechanisms of the immune system have only been fully characterized for a small number of pathogens. However, this has not prevented the statistical association of specific immune mechanism signatures with protection against a disease manifestation. These statistical correlations are built from data (immunological, virological and clinical readouts), collected from field infections and vaccine clinical trials. Once a statistical association has been made between protection and an immunological biomarker it is difficult to improve, modify or introduce a novel CoP. Therefore, in order to derive reliable CoP, it is necessary first to analyze and review in detail how the immune responses develop in experimental and natural infections. This process would in turn lead to the selection of relevant immunological bio-markers with which to evaluate vaccine efficacy in clinical trials, which ultimately will result in establishing a CoP.
2 Types of protective immune responses
There is a wide range of cell populations and soluble factors involved in the development of a protective immune reponse against a particular pathogen. Each of these immunological parametres is measurable and constitutes the basis from which to define a CoP. Hereinafter, we describe the main players of the immune responses that can lead to pathogen clearance and protection against disease.
2.1 Innate immunity
Innate immunity is the first line of defense against invading pathogens. There are several cell types involved in the innate immune response: monocytes, dendritic cells (DC), macrophages, mast cells, basophils, eosinophils, natural killer (NK) cells and innate lymphoid effector cells. Other than the anatomic and physiologic barriers, innate immune responses comprise endocytic or phagocytic and inflammatory processes as defense mechanisms (7). Both phagocytic and inflammatory immune processes promote the clearance of pathogens and activation of the adaptive immune response (7–9).
For instance, the innate immune response may lead to the activation of the complement cascade which will induce the opsonization of certain pathogens thus rendering them susceptible to phagocytosis, enzymatic degradation or lysis in the case of bacterial pathogens. Another mechanism used during the innate immune response is the production of cytokines and chemokines. This process can lead to an inflammatory state with local activation of cellular responses and recruitment of additional immune cells to sites of infection. The presence of foreign nucleic acid molecules, such as double-stranded RNA (dsRNA) inside the cell, precedes the secretion of interferons (IFNs), the major soluble factors of the innate immune response.
There are several types of IFN but the most notable in virus clearance are type I IFN, which includes IFN-α and IFN-β, IFN type II (IFN-γ) and IFN type III (IFN-λ) (10). The main role of IFN consists in inhibiting viral replication in cells that are already infected but it can also contribute to the protection of neighboring, uninfected cells. Interferon activates signaling pathways that lead to the degradation of the invading pathogen and the activation of some kinase proteins that will shut down the embattled host cell, thus inhibiting viral replication without killing the cell. In some cases, however, the infected cell can also die to prevent viral replication. Hematopoietic cells are the main producers of IFN-α and amongst them, plasmacytoid DCs (pDCs) are the major source (11). On the other hand, most infected cells are capable of producing IFN-β (12). However, the function of innate immune cell populations can be significantly affected during certain infections.
Early innate immune responses do not represent an isolated compartment of the immune system since their activation influences the type of adaptive immune response that develops during the course of the infection (9).
Thus, since soluble factors, molecules and cells involved in the innate immune response influence the development of specific effector mechanisms of the adaptive immune response (antibodies, T cells, immunological memory), which typically define CoP, the measurement of specific innate immune response signatures could potentially be used during an early phase of a vaccination trial to determine early on the protective capacity of a specific vaccine. In other words, innate immune response signatures can serve as alternative CoP. This is an area that has not been sufficiently exploited so far and deserves further investigation.
2.1.1 Innate effector cells
Innate effector T cells can act without previous exposure to antigens. Examples of innate effector T cells include invariant natural killer T (iNKT) cells and γδ T cell receptor expressing cells. Despite their limited T cell receptor (TCR) diversity and low capacity for proliferation, these cells can rapidly execute effector functions, such as the release of various cytokines, chemokines and growth factors. These mechanisms can initially control infection, interact with the adaptative immune response and even promote the development of effector and memory T cells (13). An example of a robust γδ T cell and effector memory T cell response is that observed after smallpox vaccination (14).
2.2 Adaptive immunity
The adaptive immune system, by virtue of one of its most defining features, immunological memory, enables a fast and effective response against an invading pathogen upon a second exposure to that pathogen and, in many cases, confers long lasting protection. However, for some diseases, it has been shown that protection declines over time. Whilst the level of circulating antibodies is commonly used as a CoP to assess the protective efficacy against many viral diseases, there are instances where protection has been observed in the face of very low virus-specific antibody titers. In these instances, measurements of virus-specific memory B-cell frequency might serve as more realistic CoP than simply measuring serum antibody levels. In other cases, cellular immunity might play a more important protective role than previously recognized. Indeed, during Influenza virus infections it has been clearly demonstrated that virus-specific T-cell responses limit the severity of disease (15).
As a general rule, antibodies tend to prevent cell infection whereas cellular immune responses rather act once replication of the pathogen takes place (16, 17).
2.2.1 Humoral immunity
Humoral immunity results in the production of antibodies that target specific pathogens. There are 5 isotypes or classes of antibodies, IgA, IgD, IgE, IgG and IgM, that have different biological functions. This classification is made according to their heavy chain, namely alpha, delta, epsilon, gamma or mu respectively (7). Additionally, antibodies can be subdivided into several subclasses (IgG1, IgG2, IgG3, IgG4, IgA1, IgA2) that are structurally and functionally different. While the Fab region of an antibody performs mostly a recognition and/or neutralization role, the Fc region is rather used in cell-mediated immune functions (18, 19).
After a first encounter with a pathogen, B cells will differentiate into effector B cells or plasma cells, which will then secrete antibodies specific to the pathogen encountered. A fraction of these cells will then become memory B cells which are long-lived and can respond quickly after a second exposure to the pathogen. IgG antibody production against a first time encountered pathogen can take up to two weeks to develop, however, if re-infection occurs, antibodies are produced after only a day or two thanks to these antigen-specific memory cells (20).
Antibody production is generated by B lymphocytes, however, CD4+ T cells are required in this process. When antigen specific CD4+ helper T cells interact with activated B cells, they produce IL-4 and IL-5 that will then induce B cell proliferation and antibody production. These antibodies can bind to pathogens and prevent their proliferation through different mechanisms such as neutralization, opsonization and complement activation. Neutralization consists of the binding of antibodies to the surface of the pathogen, thus blocking the pathogen’s attachment to the cell or interfering with virus uncoating within the cell. Opsonization on the other hand, requires the pathogen to be ‘marked’ by opsonins (such as IgG antibody, C3b or C1q molecules of the complement) for the subsequent phagocytic removal of the pathogen (21). Complement-dependent cytotoxicity (CDC) takes place when complement protein C1q (in the classical complement pathway), C3b (in the alternative complement pathway) or Mannose binding lectin (MBL), in the Lectin complement pathway, bind to the Fc region of IgG or IgM, coupled to a pathogen antigen expressed on the surface of an infected cell. This activates the complement pathway that will lead to the formation of a membrane attack complex (MAC) that will then cause cell lysis (22, 23).
In some cases, B cells can direct other immune cells to eliminate the pathogen via Fc-Fc receptor (FcR) interactions, thus combining the strong antiviral functions of innate immune effector cells with the specificity of the adaptive humoral activity. These mechanisms comprise Antibody Dependent Cellular Cytotoxicity (ADCC) and Antibody Dependent Cellular Phagocytosis (ADCP) (24). During these cell-mediated immune mechanisms, antibodies are produced that will bind the pathogen and these will then be recognized by effector cells that have FcR, namely NK cells, neutrophils, macrophages and dendritic cells. In the case of ADCC, the effector cell will lyse the targeted cell containing the pathogen on the surface coated with IgG1 or IgG3 containing the bound Fc. Pathogen infected cells can be eliminated through the action of cytokines, reactive oxygen species (ROS), perforin and/or granzymes. In contrast, during ADCP, the targeted cell will be engulfed and processed for phagolysosomal degradation. The main leukocytes involved in ADCP include monocytes, macrophages, neutrophils, and eosinophils (25, 26). In addition, B cells can activate and present antigens directly to effector T cells. B cells can directly recognize certain antigens via their surface IgG. These specifically bound antigens will be endocytosed, processed and their peptides presented to specific antigen matching T helper cells. As a result of this interaction, B cells express costimulatory molecules that can activate the T helper cells that will then coordinate effector functions.
It is also important to mention the role of immune memory, which during certain infections can be highly correlated with protection. B cell memory is generated by two different cell subsets: memory B cells and long-lived plasma cells or memory plasma cells. Thus, upon a second antigen exposure, memory plasma-cells can rapidly produce antibodies and memory B cells can differentiate faster into plasma cells and start a quick and robust response producing antibodies, isotype switching, effector functions and affinity maturation besides rapid proliferation (27). These processes can play an important role when low, pre-existing antibody levels are present or if the existing antibodies are overcome by the infectious agent (8, 16).
The ability to induce a strong humoral response is the hallmark of an effective host defense against certain infections (7). There are several factors that may affect the efficiency of the antibody response such as the titer, location, subclass of antibody, time of appearance and durability. However, the specific threshold levels of antibody titers conferring protection against many specific pathogens are either currently undetermined or variable amongst pathogens (28). Nevertheless, due to the ease of measuring antigen-specific antibody levels in various clinical specimens and bodily fluids, CoP based on antibody level measurement (e.g. virus neutralization, antibody binding assays, hemagglutination inhibition assays) have been used extensively to assess the immunity of populations against a specific pathogen and to evaluate vaccine efficacy. Furthermore, collection and processing of clinical material for antibody analysis is relatively simple in comparison to collecting, storing and processing PBMC for the assessment of cell-mediated immune responses.
2.2.2 Cell-mediated immunity
Most infectious pathogens are susceptible to the action of antibodies during the extracellular phase of their infection cycle. However, humoral immune responses are not completely effective at clearing pathogens when they are inside cells and cell-mediated immune effector mechanisms are called upon to clear viral infection. These mechanisms are mediated typically by CD8+ cytotoxic T-lymphocytes, which bind in a specific manner via their T-cell receptors, to the MHC-I molecules of infected cells that display viral antigen-derived peptides. However, this is not the only cell-mediated effector mechanism of T lymphocytes. Indeed, upon encountering infected cells, T cells secrete pro-inflammatory cytokines, co-stimulatory soluble factors and other regulatory signals. Thus, cell-mediated immunity (CMI) is relevant for intracellular pathogens and this protective mechanism can also synergize with an antibody production strategy in order to achieve a protective response. For instance, it has been shown that certain antibodies can activate Th1 cells through FcR, thus facilitating the rapid processing of antigens (29). Likewise, as alluded to earlier, ADCC and ADCP can also be considered as hybrid effector mechanisms of immunity involving the synergistic action of antibodies and innate immune effector cells.
T cells are considered as the main mediators of cellular adaptive immune responses. They have a crucial role in immunosurveillance since they can discriminate pathogen-derived peptides from native “self” proteins. In order to mount an efficient immune response, after recognition of foreign peptides these cells undergo activation.
During infection, antigen-presenting cells (APCs) recognize and process invading pathogens thus presenting these foreign epitopes to T cells through major histocompatibility complex (MHC) molecules. Such peptides can only be recognized by T cells when they are presented by MHC molecules. There are two types of MHC: class I, which is expressed on the surface of all nucleated cells, and class II which is located on surfaces of specialized APCs. CD8+ and CD4+ T cells will bind MHC I and MHC II respectively.
CD4+ T cells play a central role in the development of the adaptative immune response since they direct downstream effector mechanisms of other immune cells through the secretion of different types of cytokines and chemokines. Through their MHC-II molecules, APCs can present pathogen peptides to naïve CD4+ T cells. If activated, APCs then provide specific co‐stimulatory signals resulting in T cell proliferation and differentiation of naïve CD4+ T cells into specific functional T helper (Th) cell subsets, namely: Th1, Th2, Th9, Th17, Th22, T follicular helper (Tfh) and regulatory T (Treg) cells, amongst others (Figure 1). These Th cells contribute to immunoregulation of inflammatory, humoral or CMI responses through the release of effector molecules. A Th1 response for instance, involves the release of tumor necrosis factor-alpha (TNF-α), IFN-γ, and interleukin-2 (IL- 2) amongst others, that will mainly help to clear intracellular pathogens. Th2 responses will release IL-4, IL-5, IL-6 and IL-13, that are mainly involved in the clearance of extracellular pathogens. Th17 effector cells will secrete IL-17, IL-21 and IL-22 and are responsible for the clearance of some extracellular pathogens, however they are also involved in auto immune processes. On the other hand, Treg cells are involved in tolerance and secrete mainly TGF-β and IL-10. It is important to take into consideration that the release of the mentioned cytokines is not exclusive to T cells and that some other immune cells are also an important source of cytokines and chemokines that play crucial roles during infection. In addition, CD4+ T cells also release soluble factors that contribute to the generation and maintenance of CD8+ T cells (30).
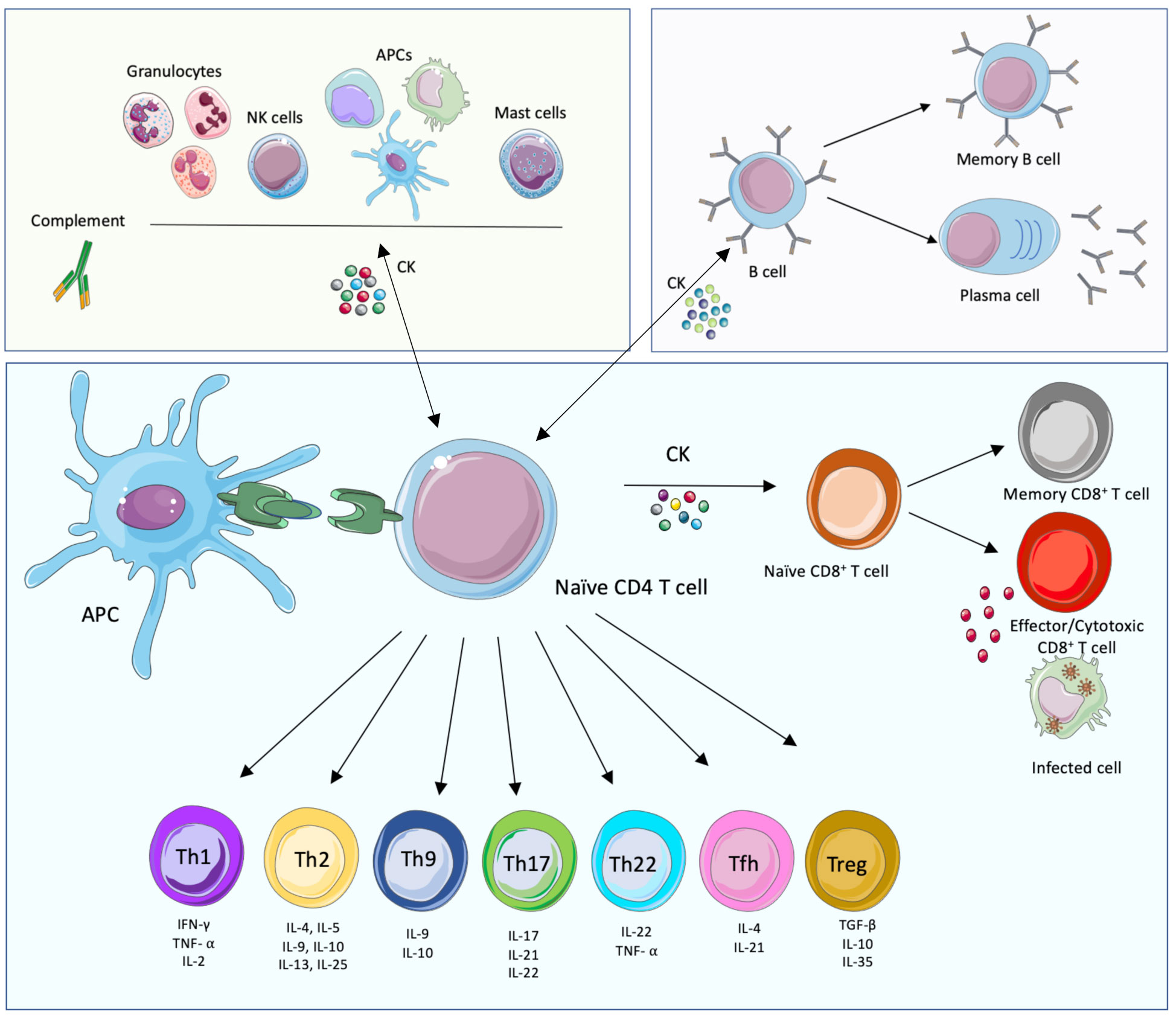
Figure 1 Innate, humoral and cellular-mediated immune responses. The main cellular immune players of the cell-mediated innate (green background, top left), humoral (purple, top right) and adaptative cell-mediated (blue, bottom) immune responses and their interconnections are displayed. The components of the innate immune system provide, together with their effector functions and soluble mediators, an immediate response to pathogens. This response triggers in turn the adaptive immune system, mostly T cell-mediated immune responses that lead to the activation of effector T cells and the activation of B cell functions. This branch of immunity provides specific, long-lasting immune responses. The adaptive and innate immune systems are connected; importantly, while soluble mediators are important to link both arms of immunity, the presentation of foreign peptides (in green) by Antigen Presenting Cells (APCs) is also necessary, together with immune mediators such as cytokines (CK). This figure was created with smart.servier.com.
As mentioned earlier, cytotoxic CD8+ T lymphocytes (CTL) also play an important role during CMI responses. When activated through MHC I presentation of certain intracellular antigens, these cells release cytotoxic proteins such as perforin, granzyme and cytokines including IFN-γ, TNF-α, IL-2, IL-4, and IL-10, that trigger the killing of specific target cells (8, 31).
Once infection is cleared, antigen‐specific effector T cell (CD4+ and CD8+ T) populations decline and a small cellular subset is maintained as antigen‐specific effector and long-lived memory T cells (CD4+ and CD8+ T cells) (32). Hence, in a secondary immune response the numbers and the activation status of T cells rapidly increase, the stimulatory antigen requirement to induce a response is reduced and as a consequence, a faster response of the effector functions takes place compared to a first contact with the pathogen (32). There are two main subpopulations of memory cells: effector-memory T cells and central-memory T cells. Effector memory T cells circulate through non-lymphoid tissues and provide an immediate response at pathogen sites of entry, but they have a poor proliferative capacity. Central memory T cells, on the other hand, are located in secondary lymphoid tissues, have a long-life span and a high proliferative capacity. Together, effector and central memory T cells have been shown to protect and reduce infection levels in several vaccine studies (32, 33).
Factors such as T-cell phenotype, which antigen the cells are specific to and their function can influence the potential of the CMI response to be an immune CoP during infection. Thus, T cell proliferation and the specific cytokine profile secreted by immune cells in response to specific antigens could be used as CoP for certain infections.
3 Protective immune responses during SARS-CoV-2, Nipah virus and Ebola virus infection
3.1 SARS-CoV-2
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is an enveloped virus with a positive single-stranded RNA genome that belongs to the Coronaviridae family and the ß-Coronavirus genus (34).
SARS-CoV-2 is transmitted mainly via respiratory droplets and can cause the syndrome known as coronavirus disease 2019 (COVID-19). While most patients are asymptomatic or mildly symptomatic with flu-like symptoms, 13.9% of patients can experience complications that may lead to acute respiratory distress syndrome (ARDS), disseminated intravascular coagulation (DIC) or organ-failure, amongst others (35, 36). This, together with its high transmissibility, makes this virus a public health threat for humans.
Upon entry into a host’s cells, SARS-CoV-2 causes cell damage and triggers a host immune response. There are several molecular mechanisms by which the human immune system can be hijacked by SARS-CoV-2. In this way, innate immune responses are affected, adaptive immune responses are delayed and as a consequence, viral clearance is inefficient and the virus can spread systemically.
3.1.1 The role of cytokines in SARS-CoV-2 infection
While inflammation is crucial for the development of an efficient and coordinated antiviral immune response, an exacerbated inflammatory response can become detrimental for the host. This is the case for SARS-CoV-2 infection where a ‘cytokine storm’ signature has been shown to be a common denominator in severe cases of COVID-19 (37–40). These high levels of proinflammatory cytokines have been associated with injury and loss of lung function, increased levels of SARS-CoV-2 load and severe or fatal outcomes (41). Some proinflammatory markers that are elevated during severe COVID-19 include IL-6, IL-8, IL-1β, TNF-α, MCP-3, TGF-β, CXCL10 and IL-17, amongst others (42, 43). Severe cases of SARS-CoV-2 also correlate with the release of ROS. It is believed that ROS, in turn, increases the expression of proinflammatory cytokines, which further contribute to disease severity (44, 45). The elevated expression of multiple chemokines during COVID-19 also leads to high numbers of neutrophils and monocytes, which in severe cases, can infiltrate the alveolar spaces and are believed to contribute to lung injury and increased disease severity (46, 47). Moreover, this cytokine storm also has an impact on the adaptive immune response, since the low expression of HLA-DR induced by high concentrations of IL-6 and TNF-α leads to a pronounced lymphopenia in severe COVID-19 cases (48).
Amongst cytokines, it is also important to mention IFN. Despite its importance in viral clearance, the precise role of IFN during COVID-19 has still not been elucidated; IFN-I production can be partially impaired by SARS-CoV-2 proteins such as the M protein or non-structural proteins nsp1, nsp6, nsp13-15 and orf6 (49–51). The efficacy of IFN during COVID-19 is however controversial, while some studies have shown a potential protective role during SARS-CoV-2 infection (52, 53), others have suggested that it may be detrimental during infection (54). For instance, it has been shown that high levels of IFN-α are associated with high viral loads and severity, thus indicating that in some severe cases, IFN signatures fail to clear the viral load (55). However, this could be explained by a delayed production of IFN when SARS-CoV-2 titers are already too high and thus IFN cannot clear the virus.
On the contrary, it seems that an early production of IFN and an efficient adaptive immune response correlate with control of SARS-CoV-2 infection, whilst both absence or prolonged presence of IFN can lead to cellular hyperactivation with high inflammation levels that may cause a detrimental clinical outcome (56). Some other studies have suggested a link between severe human cases of COVID-19 and defective IFN responses (52, 57, 58). For instance, it has shown that the impairment of IFN responses, caused by insufficient production of IFN or the presence of autoantibodies against interferons in the host, is correlated with COVID-19 severity (58, 59). In agreement with an IFN protective role, several treatments have been shown to alleviate the severity of COVID-19 by accelerating viral clearance and decreasing levels of certain pro-inflammatory cytokines (60–63). Interestingly, a recent study showed that treatment with a single subcutaneous injection of pegylated interferon lambda in (majoritarily vaccinated) patients with acute COVID-19 decreased disease severity by 51% when compared with placebo-treated patients (64). While not all aspects of IFN effects during COVID-19 are clear, it would appear that IFN treatment during COVID-19 is more efficient in the early phases of the disease (65–67).
In addition to the timing of IFN production or its presence or absence during COVID-19, the anatomical location of IFN also seems to be relevant to determine disease severity. Currently, complete data regarding the circulating levels of IFN-I in association with the severity of COVID-19 disease are lacking. Some studies have shown that circulating IFN-α levels were not significantly different between severe and mild cases when measured in plasma (68) and that prolonged IFN production during SARS-CoV-2 infection can be detrimental for the host and cause negative clinical outcomes (54, 69). However, with regards to local IFN lung production, high IFN-III levels in the upper respiratory tract have been seen to be protective during COVID-19 resulting in mild cases, while high IFN-I and IFN-II levels in the lower respiratory tract have been associated with severe cases of COVID-19 (70). Other factors, such as the age of the patients, seem to be relevant for the severity of the disease. This could be related to the fact that IFN production is impaired by age through the decrease in RIG-I signaling efficiency and pDC IFN production capacity (71). For instance, in a recent study on SARS-CoV-2 infected macaques, it was shown that, in aged macaques, there was higher expression of pro-inflammatory cytokines, lower IFN-I and increased lung pathology (72).
Therefore, the role of IFN during COVID-19 may be strongly influenced, in addition to its presence or absence, by its anatomical location, the moment at which it is produced and the pre-existing cytokine host environment and pre-immune status. Thus, depending on the host and disease context, IFN kinetics can result in a protective or detrimental outcome.
3.1.2 The role of T cell immunity in SARS-CoV-2 infection
Adaptive immune responses are essential in controling and clearing SARS-CoV-2 infection, thus cellular and humoral immunity can confer protection during COVID-19. However, adaptative immune responses are highly influenced during infection by multiple factors, including the immune status of the host (genetic and acquired factors), the efficacy of the innate immune response and the initial virus load, amongst others (73–75).
T cell responses seem to correlate with protection during SARS-CoV-2 infection, however, they are partially impaired in severe cases of COVID-19 thus leading to exacerbated activation and lymphopenia (56). Although the mechanisms responsible for this phenomenon are not fully understood; it would appear that lymphocyte hyperactivation, exhaustion and impaired lymphocyte proliferation can contribute to disease, especially in severe COVID-19 cases (76, 77). The type of T cell response seems to also be relevant. While a biased Th1 phenotype seems to be associated with milder COVID-19 cases and good clinical outcome, Th2 and Th17 responses have been shown to be more prominent and detrimental in severe cases (76, 78). However, it is difficult to draw a general conclusion from such studies, as the observations are often based on relatively low numbers of patients.
In terms of cellular immunity, it is important to identify SARS-CoV-2 specific epitopes that elicit efficient responses in humans. The immunodominance and immunoprevalence of a peptide correspond respectively to how strongly and how frequently a given peptide sequence is recognized by T cells. In the context of SARS-CoV-2, it is possible that an optimized immunodominance and immunoprevalence could improve the efficiency of host immune responses. Therefore, by knowing which specific epitopes can elicit an efficient T cell response, it is possible to modulate the immune responses, thus possibly improving the outcome of the disease by providing immunological memory.
It has previously been shown that convalescent COVID-19 patients harbor an efficient CD4+ T cell response against the SARS-CoV-2 spike (S) glycoprotein. This response also seems to correlate with the presence of specific IgG and IgA titers. Interestingly, several studies showed that some individuals unexposed to SARS-CoV-2 had S specific CD4+ T cells and, at a low level, specific CD8+ T cells (79–81). The presence of these specific responses in non-previously infected individuals could be explained by cross-reactivity responses from previous coronavirus infection. In support of this, an additional study showed that some human samples obtained before the SARS-CoV-2 virus pandemic harbor preexisting memory CD4+ T cells that are cross-reactive to specific SARS-CoV-2 epitopes but also to other common cold coronaviruses (82). Of note, while CD4+ T cells from healthy donors mostly target the C terminal part of the S glycoprotein, CD4+ T cells from COVID-19 patients target almost equally both the N- and C-terminal parts of the SARS-CoV-2 S glycoprotein. This is probably due to the fact that the C-terminal part of the S glycoprotein of many betacoronaviruses has high homology (83). Another difference is that CD4+ T cells from convalescent patients from mild to severe COVID-19 are in an activated state (81). In addition, S glycoprotein-specific T CD4+ cell responses are considered to support antibody generation, thus correlating cellular with humoral immunity in the memory phase (84). Whether S specific T cell responses provide a potential protective role or modulate the severity of the disease in healthy individuals when exposed to SARS-CoV-2 remains to be determined. Another mechanism of protective immunity that has been asasociated with protection against COVID-19 is the presence of resident memory T cells in the lungs, which can last up to 10 months post-infection regardless of the severity of COVID-19 (85, 86).
CD8+ T cells responses seem to be highly heterogeneous between COVID-19 patients. A correlation has been shown between a high expression level of effector molecules by CD8+ T cells and a positive clinical outcome (78). This is supported by non-human primate (NHP) models, where, in SARS-CoV-2 infection in macaques, CD8+ T cell responses have an important role in protection even when neutralizing antibody levels are low (87). The relevance of CD8+ T cells was also supported in recovered COVID-19 patients, where they were shown to harbor not only SARS-CoV-2-specific CD8+ T cells, but also CD8+ T cell memory cells (88, 89). Likewise, clonal expansion of CD8+ T cells has been suggested to be present in mild COVID-19 cases (56). Several studies have identified the importance of respiratory CD8+ T cell responses and the importance of the interaction between cytotoxic CD8+ T cell and epithelial cells in the upper respiratory tract (90).
In conclusion, severe COVID-19 cases correlate with a delayed and excessive adaptive immune response, whilst in milder and convalescent cases, it appears of importance to have an early robust T cell response that leads to SARS-2 clearance. Moreover, T cell responses are probably not redundant thus cellular and humoral responses can be simultaneously considered as CoP.
3.1.3 The role of B cells in SARS-CoV-2 infection
In general, a specific level of circulating anti-viral antibodies is necessary to confer humoral protection against infection. Most patients with COVID-19 develop IgM and IgG within days to weeks after the onset of symptoms (91). However, the relevance of SARS-CoV-2 specific antibodies during infection is not yet clear.
The protective effect of humoral immune responses against SARS-CoV-2 re-infection depends on how long the humoral response lasts and the antigenic characteristics of the re-infecting virus. After natural, SARS-CoV-2 infection, virus-specific T cells, memory B cells and protective neutralizing antibodies can be detected more than 1 year after infection (92–94). In general, memory B cells and specific protective antibodies are still present at 12-18 months post-infection (at least as long as the studies lasted). Indeed, in one study, 20 months after initial SARS-CoV-2 infection, natural antibody and cellular immunity were still shown to confer protection against infection and hospitalization in 95% and 87% of cases respectively compared to patients that presented no immunity (95). In comparison, vaccine-induced immunity decays faster than natural immunity. Thus, after vaccination a hook effect is observed, while protection is very efficient in the first months, it has been shown to decline more rapidly, nearly disappearing five months after the second dose (96, 97). Moreover, after vaccination the immunogenic reaction takes place against the spike S protein only and IgA is minimally elicited.
With regards to the natural humoral response, it was shown that 300 days after natural infection, IgG antibodies against SARS-CoV-2 S and N proteins were present in 68% and 87% of subjects respectively (98). In fact, many studies have shown the presence of SARS-CoV-2 IgG neutralizing antibodies months after contracting COVID-19 (88, 99–103). Importantly, recent studies have detected the presence of neutralizing IgAs on the surface of the upper nasopharyngeal airway mucosa, lasting for several months (104, 105).
When hybrid immunity takes place (natural + vaccination immunity) the data is slightly contradictory. Some studies show that vaccination in recovered COVID-19 patients improves the disease outcome or increases antibody titers (106–110). For instance, in the previous mentioned (95) study, hybrid immunity induced by either one or two doses of a COVID-19 vaccine was associated with an additional risk reduction of SARS-CoV-2 reinfection compared with natural immunity for up to 9 months, although with small absolute differences. An additional study has shown that infection probability after vaccination is significantly lower than the possibility of reinfection after natural infection (111). For instance, it has also been shown that hybrid immunity is 95.3% and 97.4% effective in preventing hospital admission and severe disease respectively at 6 and 12 months, with the first vaccination dose after the most recent infection. With regards to reinfection, hybrid immunity effectiveness after primary vaccination decreased to 46.5% and 41.8% at 6 and 12 months respectively (112). On the contrary, several studies have shown no statistically significant differences between natural or hybrid immunity effectiveness in terms of increase in neutralizing antibodies, cellular immunity, or specific memory B cells in recovered COVID-19 patients after the second vaccine dose (113–116).
Age is also an important factor with regards to antibody titers against SARS-CoV-2. It has been shown that adaptive immune humoral responses wane with age not only after COVID-19 illness but also after vaccination (117–119).
Overall, there are many studies determining the potential protective role conferred by previous infection and/or vaccination and while there is conflicting evidence, it has been shown that in most cases the presence of high antibody titers decreases the risk of infection by SARS-CoV-2, albeit this risk is not completely eliminated. These studies are summarized in (120). Importantly, when reinfection takes place, previously SARS-CoV-2-exposed patients or recently vaccinated individuals seem to be protected from relevant clinical repercussions and against infection by certain variants (121–123). In these studies, neutralizing antibody titers correlated with the level of protection and thus this parameter is often used as a CoP for COVID-19. In contrast, with regards to the potential protection of SARS-CoV-2 pre-existing humoral immunity towards new molecular variants of the virus, some studies in rhesus macaques have shown that neutralizing antibodies developed during a first SARS-CoV2 infection confer clinical protection against some of the new variants (124–126). Moreover, there are indications that existing humoral cross-reactivity does occur between SARS-CoV-2 and other Beta-coronaviruses. For instance, it has been shown that neutralizing antibodies from the 2003 SARS-1 outbreak can neutralize SARS-CoV-2 (82).
It is also important to consider that one-fifth of SARS-CoV-2 infections result in long-term COVID-19, where despite viral clearance, certain symptoms persist and can lead to post-acute sequelae of COVID-19 (PASC). The dysregulation of the host immune response and virus persistence are believed to account for the development of PASC. Among the immune responses noted in PASC, a distinct humoral immune response was observed, with more avid IgM, weaker Fcγ receptor binding anti-SARS-CoV-2 antibodies and an expanded inflammatory antibody response recognizing the human Betacoronavirus OC43 that can cross-react across SARS-CoV-2 and other coronaviruses. In some cases, CD8+ T cells against Cytomegalovirus (CMV) and Epstein-Barr virus (EBV) reactivation have also been detected (127, 128). The mechanism by which these markers lead to PASC are still not well known and may involve different pathophysiological mechanisms that translate into PASC being an heterogenous syndrome with specific endotypes.
In summary, protection against SARS-CoV-2 depends on a coordinated immune response involving various effector mechanisms of the adaptive immune system. The information acquired to date on SARS-CoV-2 immunity is enabling the development of effective treatments and vaccines to reverse the detrimental immune responses sometimes associated with infection. In conclusion, protection against SARS-CoV-2 depends on: (i) eliciting an early non-exacerbated, innate immune response with limited early IFN production, (ii) inducing a robust cellular response without hyperactivation of T cells, (iii) inducing an effective humoral response with neutralizing antibody production, (iv) generating immunological memory and (v) producing cross-reactive, non-specific innate and adaptive immune responses to generate heterologous protection against COVID-19. The main COVID-19 immune CoP candidates are displayed in Supplementary Table 1.
3.1.4 SARS-CoV-2 variants and their interplay with immune responses
The increasing SARS-CoV-2 genomic diversity poses a potential threat to vaccination efficiency since antigenic changes can lead to the appearance of variants of concern (VOCs) with improved viral fitness that can jeopardize a host’s immunity in comparison to previous circulating strains. VOCs sometimes have significant mutations that give them unique properties with a functional impact affecting virus-host interactions and infection capacity, transmission and/or replication, amongst others. As of March 2023, the following major VOCs have been indentified: Alpha (B.1.1.7), Beta (B.1.315), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529. */BA.*) (129).
Interestingly, SARS-CoV-2 variants have been shown to differ in their capacity to bind to the SARS-CoV-2 receptor Angiotensin-converting enzyme 2 (ACE 2), in their antibody escape capacity or in triggering different host immune responses. For instance, while the first SARS-CoV-2 variants tended to induce a stronger innate immune response, SARS-CoV-2 Delta has integrated multiple improved mechanisms to evade an IFN response by suppressing the host innate immune response (130).
SARS-CoV-2 Omicron has recently been shown to include many concerning mutations that affect several viral proteins. SARS-CoV-2 proteins can be classified into three categories: structural, non-structural and accessory proteins. Structural SARS-CoV-2 proteins notably play a role in virion assembly and formation. There are four major SARS-CoV-2 structural proteins: the spike protein S, the envelope protein E, the membrane protein M and the nucleocapsid protein N. The Omicron variant has been shown to contain unique mutations mainly in the receptor-binding domain (RBD) and the N-terminal domain of the S1 spike subunit (131). Omicron spike mutations increase binding to ACE 2 and enable antibody escape, thus adding an increased immune evasion capacity to an already higher transmission and replication fitness. While the most explored mutations are found in the spike protein, further mutations in Omicron have also been detected in the N-terminus region of the structural E protein which is known to interact with NSP3 for ubiquitination and glycosylation (132), and also in the M protein that promotes the assembly of new viral particles, affecting membrane integrity and post-translational modifications (133). Unique mutations are also found in the N-terminal region of N which translates into a more significant inhibition of RNA-induced IFN expression (134). Overall, while the functional effect of these mutations has not fully been studied, it is believed that they can modulate host-virus interactions and thus, increase SARS-CoV-2 Omicron replication, pathogenicity, and fitness (135).
Mutations in non-structural proteins for Omicron may also have a crucial effect on immune regulation, transcriptional regulation and viral pathogenesis. For instance, some mutations take place in NSP1, that binds to ribosomal subunits to stop host protein translation (136), or in NSP3, NSP4, and NSP6 that are responsible for viral budding by modifying the endoplasmic reticulum (ER) into double-membrane vesicles (137, 138). In addition, mutations in NSP14 can cause post-transcriptional modifications (139). Mutations have also been observed in NSP5, the main viral protease which also harbors the binding site for this enzyme, and that might which affect viral enzymatic processing activity. Additionaly, mutations detected in NSP6 could help virus survival through the avoidance of autophagosome fusion with lysosomes (140).
Accessory proteins normally act as virulence factors mainly through immune evasion mechanisms that increase viral survival in the host. In SARS-CoV-2, there are eleven accessory proteins of which some are known to be potent interferon antagonists. For instance, in the Omicron variant some mutations have appeared in ORF3a and ORF7b that inhibit STAT signaling phosphorylation and ISGs expression (141, 142). Other examples include ORF3b, ORF6, ORF7a, ORF8 and ORF9b, that are also known to have IFN-antagonistic activity (143–145). ORF9b and ORF9c are also known to interact with cellular organelles, reducing antiviral responses (129, 144–146).
Altogether, it has been shown that Omicron can evade the host immune response more efficiently than previous VOCs. This is credited to decreased recognition by neutralizing antibodies but also to new acquired mutations that lead to increased viral fitness, higher transmission rates and better host immune evasion amongst others.
3.2 Nipah virus
Nipah virus (NiV) is an enveloped virus with an 18 kb negative-sense single-stranded RNA genome that belongs to the Paramyxoviridae family (147). There are two different strains: NiV Malaysia (NiV-M) and NiV Bangladesh (NiV-B) (148).
NiV outbreaks are reported almost yearly, the most recent occurring in India in 2021, notably with one of the highest fatality rates (92%) observed in the last few years (149). NiV infection in humans is generally associated with an acute respiratory and neurological syndrome resulting in a high fatality rate of between 40% and 92%, depending on the local capacity for epidemiological surveillance and clinical management (150–152).
The NiV reservoir has been identified as fruit bats of the Pteropus genus (153). It is known that NiV can also cause severe disease in domestic animals such as pigs, resulting in significant economic losses for farmers (154). Currently, there are no approved treatments or vaccines available for either humans or swine infected with NiV.
Here we aim to summarize the main features of the innate and the adaptive immune response to NiV and discuss the identification of potential immune CoP.
3.2.1 The role of cytokines in NiV infection
NiV infection triggers a robust inflammatory and IFN-I response involving the expression of various IFN-induced antiviral genes. However, to counteract this, NiV expresses several structural and non-structural proteins that can efficiently antagonize a host immune response (155). For instance, the structural matrix M protein and non-structural accessory proteins C, V and W play important roles in preventing IFN-I activation and production at many stages of the signaling pathways involved [summarized in (156)]. In addition, the disproportionate production of pro-inflammatory cytokines at the very early steps of NiV infection in humans considerably contributes to its pathogenicity by causing vasculitis and encephalitis characterized by inflammatory cell infiltration (150).
In this regard, neutrophils are very important during the early steps of the innate immune response since they are involved in several defense mechanisms, including the production of antimicrobial peptides or ROS-induced neutrophil extracellular traps (NETs). However, while in general, NETs can trap and act upon viral particles, in some respiratory virus infections, such as with respiratory syncytial virus (RSV) and influenza A virus (IAV), an exacerbated release of cytokines can lead to high levels of neutrophil activation and excessive NET formation leading to airway occlusion and increased lung inflammation that is detrimental for the host (157). While this has not yet been specifically shown for NiV infection, it is very likely that, similar to what occurs in IAV and RSV infection, a strong release of proinflammatory cytokines leads to hyper-activation of neutrophils which can result in tissue damage. Moreover, it has previously been shown that during NiV infection, while neutrophils do not seem to be infected, they play a prominent role in disseminating NiV (158).
Some of the pro-inflammatory mediators released during NiV infection in humans include TNF-α, CXCL10 and interleukin-1β (IL-1β). The three have been shown to have an important role in disrupting the blood-brain barrier (BBB) and contribute to the neurological symptoms observed in severe cases of NiV disease (NiVD) (150, 157, 159). In addition, other pro-inflammatory mediators such as IL-6, IL-8, MCP-1, GM-CSF and G-CSF have been shown to be released at high levels in severe cases of NiV infection, particularly in the lungs. It has also been shown that this increase in inflammatory chemokines correlates with increased monocyte and T lymphocyte chemotaxis (155).
As a result, it is believed that IFN-I impairment and the release of high levels of pro-inflammatory mediators can contribute to the worsening of clinical symptoms (157). Altogether, data indicates that NiV employs many strategies to counteract the innate immune response and that specific levels of IFN-I and pro-inflammatory cytokines could be used to determine the outcome of the infection. Further study is needed in order to establish these correlations more specifically.
3.2.2 The role of T cell immunity in NiV infection
To date, very little information is available on human cellular immune responses to NiV infection. During the 2018 NiV outbreak in Kerala, India, 18 patients were confirmed to be infected with NiV, of which 2 survived the disease. Cell mediated and humoral immune responses were studied during the acute and convalescent phases of the disease (160). Throughout these periods, surviving patients presented stable T lymphocyte absolute numbers and CD4+ T cells were not more activated than in healthy individuals. However, this was not the case for CD8+ T cells that were more activated and indicated active proliferation and effector functions during the acute phase of the illness and returned to basal levels during the convalescence phase.
Interestingly, the clearance of NiV from the blood seemed to happen before the humoral response (NiV-specific IgG antibodies) took place and rather coincided with the aforementioned activation of CD8+ T cells. In a study where African green monkeys (AGM) were infected with NiV, analysis of the peripheral immune response also showed high levels and activation of T CD8 effector memory cells in surviving AGMs, correlating with an increased release of cytokines and associated cell-mediated immunity (161). This is interesting since CMI was not shown to be relevant in animals that succumbed to NiV infection, thus suggesting that effector memory cells were only relevant in survivors. Interestingly, the activation and proliferation of CD8+ T cells was also observed in the only two survivors of the NiV outbreak in Kerala (160). In contrast, in another study performed in a porcine model, a reduction of CD4+ T cell populations was shown in individuals with a poor clinical outcome (162).
There are several limitations that make it difficult to draw conclusions about cellular immune responses during NiV infection. In human studies, small sample sizes and the lack of samples from disease victims to compare with survivor samples often limit the robustness of the conclusions drawn. Moreover, a complete overview of the relevance of cellular immune responses during NiV infection is lacking, in part due to an absence of CMI response studies in NiV animal models.
During infection, robust T cell responses would enable the development of a faster transition between innate and adaptative immune responses and thus accelerate the production of antibodies and protective immunity. While more data is required, preliminary studies in humans and non-human primates indicate that cellular immune responses, specifically CD8+ T cell activation, seem to be important for protection and therefore CoP for NiVD can be derived from CD8+ T cell measurements.
3.2.3 The role of B cells in NiV infection
Similarly, humoral immune response studies of NiV infection in humans are very limited. However, in the previously mentioned study on the two Kerala NiV survivors in 2018, both patients showed an increased number of B lymphocytes that correlated with the presence of NiV-specific IgG and IgM antibodies within a week after exposure. Moreover, an increased level of activated B cells and plasmablasts was present in both survivor patients (160). However, the specific NiV antigens targeted by the NiV-specific humoral response are yet to be identified.
The correlation between protection against NiV and the presence of antibodies has also been demonstrated in several animal models of infection. For instance, in NiV-infected swine, neutralizing antibodies were detected a week post-infection, with considerably increased titers observed two weeks post-infection. However, NiV RNA could still be detected several months after the initial infection (163). In an AGM model, B cell numbers decreased at twelve days post-infection, a fact that correlated with disease progression and a detrimental outcome (161). In contrast, the only surviving animal in the study showed robust IgM and IgG responses which correlated with an increase in B cell lymphocytes, suggesting that humoral immune responses are relevant during NiV infection and may afford protection against the virus. Moreover, humoral immunity relevance during NiV infection has also been shown in several models including ferrets, hamsters and again in AGM. In these models, the administration of sera or NiV-specific monoclonal antibodies was shown to protect from NiV challenge (164–168).
With regards to fatal cases, there is almost no data for the acute phases of NiVD in humans, and the existing data derives mostly from histopathological analysis of post-mortem samples. However, it has been shown in AGM that lymphopenia takes place in fatal cases of infection with both NiV-M and NiV-B strains (169).
Despite the limitations in human and animal model studies for NiV infection, humoral immune responses seem to play an active role in protection and it is likely that CoP could be derived from humoral immunity parameters such as numbers of plasmablasts and activated B-cells and specific titers of IgM and IgG antibodies.
The main NiVD immune CoP candidates are summarized in Supplementary Table 1.
3.2.4 NiV interplay with immune responses
NiV has several proteins that modulate the host immune response. For instance, NiV viral proteins P, C, V and W can antagonize the IFN signaling response (170). While NiV-W protein sequesters STAT1 in the nucleus to inhibit subsequent ISG activation, NiV-V protein antagonizes IFN by binding STAT1 and STAT2 thus preventing their dimerization and transport to the nucleus for transcriptional activation of ISG genes. NiV-P protein is also able to bind and sequester STAT-1 in the nucleus (171, 172). NiV-C protein prevents IFN production in the cytoplasm, but the details of this process are still not well known (156, 173). Further IFN antagonistic mechanisms of P gene products are produced through interactions with TANK-binding kinase 1 (TBK1), Inhibitor of κB kinase ϵ (IKKϵ) and IRF-3 by the NiV-W protein (174, 175) or through inhibition of STAT2 (176), LGP2, RIG-I (177), and MDA5 (178) by NiV-V, thus preventing downstream signaling.
Besides P gene products, NiV matrix protein (NiV-M), can also inhibit IFN-I. When NiV-M interacts with TRIM6, it promotes its degradation and reduces IKKϵ polyubiquitination thus reducing IFN-mediated responses (179). Moreover, NiV nucleoprotein N can either directly prevent STAT nuclear import or hamper STAT-complex formation, thus also reducing STAT nuclear accumulation and inhibiting type I and II IFN responses (180).
3.3 EBOV virus
The genus Ebolavirus contains six virus species, namely Zaire ebolavirus (EBOV), Sudan ebolavirus (SUDV), Taï Forest ebolavirus (TAFV), Bundibugyo ebolavirus (BDBV), Reston ebolavirus (RESTV) and Bombali virus (BOMV). Out of the six, EBOV is the most prominent member having caused many highly lethal outbreaks in the past. EBOV is a single-negative stranded RNA virus from the Filoviridae family (181) and is highly pathogenic for humans and non-human primates. There have been many EBOV outbreaks with high morbidity and mortality since 1976, the 2014 outbreak in West Africa being the deadliest, with more than 28000 recorded cases and 13000 fatalities (182). Due to the multiple transmission mechanisms of EBOV, the broad cellular tropism of the virus and the multiple mechanisms used by EBOV to evade human immune responses, EBOV is considered a highly infectious, category A pathogen. EBOV can cause a highly pathogenic disease, known as Ebola Virus Disease (EVD), with a fatality rate of up to 90% in humans. Cases of EVD are often associated with a septic-shock-like syndrome characterized by an exacerbated inflammatory immune response and coagulopathy, that when combined, lead in many cases to multiple organ failure and death (183).
Whilst many treatments that have been tested in animal models and several vaccines have been shown to induce a very promising immune response against EBOV infection in animal models and humans (some of which are licensed for human use), it is not yet completely clear what are the main protective mechanisms of a successful immune response against EBOV. Although virus neutralizing antibodies and enumeration of polyfunctional T cells (IFN-γ, TNF-α, IL-2) have been associated with protection against EBOV, more data is required to validate these as reliable CoP (184–187). Determining more accurate CoP could facilitate the development of novel, better targeted treatments and vaccines.
3.3.1 The role of cytokines in EBOV infection
EBOV has a broad cellular tropism, with monocytes, dendritic cells and macrophages all being primary cellular targets of the virus. After becoming infected by EBOV, these cells have a pivotal role in the systemic dissemination of the virus through the blood and the lymphatic system. Moreover, their infection also triggers the release of inflammatory mediators such as IL-16, TNF-α, MIP-1α, IL-1β, IL-6, IL-10, amongst others (188, 189). Specifically, high levels of IL-10 and TNF-α are believed to correlate with fatal outcomes from EVD (190, 191).
The virus glycoprotein (GP) and soluble viral proteins such as shed GP are released from infected cells into the extracellular medium, where they have been shown to contribute to the release of proinflammatory cytokines, however, the exact mechanisms responsible for the early cytokine storm are yet to be determined (192, 193).
While monocytes, macrophages and dendritic cells are the main producers of proinflammatory products, other cells such as T cells and endothelial cells are also involved in the release of multiple inflammatory mediators. This results in an immunological disbalance that is believed to, in part, contribute to the severity of EVD.
Overall, the immune disbalance observed during EVD has been shown to be a crucial factor in determining disease severity, since fatal cases often present an exacerbated immune response while survivors, in contrast, mostly display a well-regulated inflammatory response (194).
Other soluble mediators that appear to be extremely relevant during EVD include ROS and IFN I. In the case of IFN I, EBOV VP35 and VP24 proteins act as IFN I transcription and signaling antagonists, respectively (195, 196). Thus, while an early and short-lived production of Type I IFN has been associated with a survival outcome, the absence of IFN is believed to contribute to EBOV dissemination (197). In contrast, an early IFN-γ response followed by lymphopenia is believed to correlate with fatal cases of EVD (188, 190).
ROS has also been shown to have a relevant role in EBOV pathogenesis. For instance, high levels of nitric oxide (NO) are associated with mortality in infected patients (198). Abnormal NO levels are believed to contribute to several pathological disorders such as tissue damage, lymphocyte apoptosis and the disruption of vascular integrity.
There are several coagulopathies associated with EVD such as thrombocytopenia or the presence of high levels of fibrin degradation products. In some cases, this leads to DIC, which frequently contributes to multiorgan failure (199). While not all of the mechanisms responsible for triggering EBOV-related coagulopathy are fully understood, the results of several studies strongly suggest that the exacerbated release of proinflammatory mediators considerably contributes to these characteristic EVD coagulopathies. For instance, the hyperproduction of proinflammatory cytokines activates coagulation factors such as procoagulant protein tissue factor (TF), fibrin fragment E and thrombin, which in turn, upregulate the production of proinflammatory cytokines (190, 200).
It has also been observed that endothelial cells are severely affected in late stages of EVD. Due to exceedingly high levels of proinflammatory cytokines (ROS and TF amongst other soluble mediators) endothelial cells are activated and endothelial leakage occurs (201).
Therefore, upon EBOV infection, a chain reaction initiated by an exacerbated inflammation response leads to a disbalanced immune response, systemic virus spread, vascular damage and coagulopathies that altogether will lead to a septic-shock like syndrome and multiorgan failure.
3.3.2 The role of T cell immunity in EBOV infection
Although EBOV does not infect lymphocytes, it can interact with T cells, affecting the development of immune responses. T-cell mediated immune responses during EVD involve a robust activation of T cells followed by their proliferation in both fatal cases and survivor patients (202). The magnitude and diversity of T-cell mediated immune responses in survivors during EVD are more robust when compared to fatal cases. In fatal cases there is an early T cell activation followed by a T cell population collapse, probably due to T-cell exhaustion (203, 204). Moreover, oligoclonal T-cell responses and higher expression of T cell inhibitory molecules CTLA-4 and PD-1 in CD8+ and CD4+ T cells are believed to contribute to an inefficient T cell response in fatal cases that is associated with higher viral loads when compared to survivors (205, 206). In this regard, the early cytokine storm observed in fatal cases correlates with high later expression levels of CTLA-4 and PD-1 in T cells (206). In contrast, survivors would appear to develop a very diverse T cell response with low levels of CTLA-4 and PD-1 T cell inhibitors, thus contributing to viral clearance. However, a more recent study has shown that West African EVD survivors from 2013-2016, presented an increase in activation and proliferation markers in CD4+ and CD8+ T cell populations by 30% and 50% respectively, when compared with healthy individuals (202). This increased activation and proliferation suggests that survivor patients can develop a robust immune response. This activation was shown to last more than one month after recovery.
Interestingly, during the EVD convalescent phase, CD4+ and CD8+ T lymphocytes from survivor patients were able to respond to EBOV nucleoprotein (NP) thus indicating that EBOV NP can stimulate virus-specific-T cell responses in humans after resolution of the disease. Similarly, in another study in EVD patients from the 2013-2016 outbreak, it was shown that survivor memory CD8+ T cells can secrete IFN-γ and TNF-α and mainly responded to viral NP and to a lesser degree to VP24, VP40, VP35 and GP. This data would appear to corroborate the immunodominance of the EBOV NP-specific T cell responses described in previous studies (205). Studies in mice, guinea pigs and NHP models have also highlighted the importance of T cell responses during EVD and the involvement of the viral NP in generating T-cell immunity (207–209).
In cases of EVD, lymphocytes are severely affected and undergo apoptosis thus making lymphoid depletion a prominent feature of the disease (208, 210). In fatal cases, there is approximatively one fourth less lymphocytes when compared with levels found in survivors (190). This loss of lymphocytes is believed to be due to several factors, including the combined impairment of DC associated with the previously mentioned abnormal release of inflammatory cytokines, including TNF-related apoptosis-inducing ligand (TRAIL) and Fas death receptor, upon EBOV infection (194, 211). Abnormal levels of NO and direct interactions between EBOV and lymphocytes are also believed to contribute to the loss of bystander lymphocytes during infection (183, 212). In addition, it has also been shown that phosphatidylserine associated with EBOV GP can bind and stimulate CD4+ T cells through T-cell immunoglobulin mucin receptor 1 (TIM-1). These cells then release proinflammatory mediators believed to contribute to the cytokine storm and the lymphopenia observed during EVD (213). Other studies have determined that abortive infection of T lymphocytes causes ER-stress in these cells thus contributing to their own apoptosis (214). Importantly, lymphopenia was shown to correlate with fatal cases during the 2000 Ebola Sudan outbreak in Uganda (198).
To summarize, the proliferation of lymphocytes is observed in both survivors and fatal human cases, however in the latter, T lymphocytes display less immune response diversity and frequently show lymphopenia in the later stages of EVD. While it has been determined that robust T cell mediated immune responses can be a CoP during EVD, it is clear that immune responses are not independent compartments and the appreciating the interplay between innate and adaptive immunity may be crucial in understanding the complexity required to produce an efficient protective immune response during EVD.
3.3.3 The role of B cell immunity in EBOV infection
Even though T cell mediated immune responses are crucial during EVD, humoral immune responses also play a very relevant role in EBOV clearance. It has previously been shown that survivors tend to produce early and sustained levels of IgG, while fatalities have rather an impaired humoral response characterized by the absence of EBOV-specific IgG, low levels of IgM and lymphopenia (215, 216). Upon EBOV infection, IgM can be detected as early as day 2 after symptoms appear and in the case of IgG, antibodies are normally present between days 5 to 18 days after symptom onset (202, 217, 218). After one year of symptom onset, an IgM repertoire against VP40 and GP was observed in survivors despite undetectable virus levels. This however could also be an indicator of hidden viral persistence (194). Interestingly, serological surveys of IgG levels in rural villages in Gabon showed EBOV antibody seroprevalence, suggesting either prior exposure to EBOV or the presence of cross-reactive antibodies (219).
It is however unclear how long the immunity in EBOV survivors lasts; in some cases, it has been shown that EBOV-specific antibodies are present for forty years after symptomatic infection (220). These antibodies have been shown to have pan-neutralizing capacity against EBOV in vitro and were associated with protective roles in several animal models such as mice, guinea pigs and ferrets (221–225). However, whether these antibodies have a protective potential against EBOV reinfection in survivors remains undetermined. It should be considered that EBOV neutralization may not always translate into protection in humans and frequently, other antibody functions (complement, opsonization…) have been shown to be important in surviving EVD (226, 227).
In order to assess serological immune profiles of EVD survivors, antibody isotypes were analyzed and showed changes in the antibody repertoire over time. While neutralizing EBOV-specific IgG1 persisted over time, IgG3 decreased in early phases and IgG4 appeared later on. Moreover, IgA with innate immune effector functions and long-lasting IgG/IgM/IgA epitope diversity were described in EVD survivors (228, 229).
Not all of the antibodies detected can recognize EBOV GP. Survivors from the 1976 Yambuku outbreak for example have been shown to harbor antibodies with reactivity to GP, NP and to a lesser extent VP40. However, all identified antibodies with neutralizing capacity were GP-specific in humans and animal models (220, 230, 231). It is for this reason that most vaccines are based on EBOV GP.
Antibodies can target nearly any region on the surface of EBOV GP. Conserved GP regions include the receptor binding site (RBS), the base, the internal fusion loop (IFL) and the heptad repeat 2 (HR2). Other regions such as the glycan cap region and mucin-like domain (MLD) are less conserved (232). While conserved regions are normally targeted by cross-reactive antibodies, most of the antibody responses found in survivors target less conserved regions since they are structurally more exposed. However, these antibodies are frequently non-neutralizing, show weak affinity and are non-cross-reactive (233). There are nevertheless some antibodies that target the glycan cap and are pan-protective, neutralizing several EBOV species (234) Recently, a conserved site named the MLD cradle that connects the MLD to the glycan cap has been identified as an antibody target region that destabilizes the GP quaternary structure, blocking the receptor binding required for effective EBOV infection (235). This is an important step forward in determining the molecular basis of EBOV neutralization by targeting conserved exposed epitopes and could be used to design universal antibody therapeutics.
There are currently two approved vaccines against EBOV, namely rVSV-ZEBOV (Ervebo) and ChAd3-MVA (Ad26) (236, 237). They have both been shown to induce EBOV-specific humoral and cellular responses, however these immune responses are not identical to the ones observed in survivors. In order to generate efficient vaccines, it is thus important to compare immunogenicity and protection between vaccinees and survivors. For instance, there are serological studies of immune memory responses showing that EVD survivors (2-6 months after infection) from the 2013-2016 EBOV outbreak have higher antibody levels and stronger antibody affinity when compared to ChAd3-MVA vaccinees at 2-12 months. Moreover, while this cohort of vaccinees had a predominant IgM response, survivors displayed a higher level of IgG with a more diverse antibody repertoire than the vaccinees (230, 238). Interestingly, survivor antibodies were shown to preferentially target the fusion peptide and HR2 domains of the viral GP2 protein and provide neutralization (238).
In the case of rVSV-ZEBOV, a serological study comparing survivors versus rVSV-ZEBOV vaccinees showed that survivor IgM and IgG do not bind the same EBOV GP epitopes when compared to rVSV-ZEBOV vaccinees (239). Additionally, another study in a similar cohort showed no significant differences in circulating antibody subclass levels. However, survivor antibodies had a higher neutralization capacity and a higher capacity to induce cellular responses than those from vaccinee samples. Importantly, IgG1 levels in survivors correlated with EBOV neutralization capacity, which was not the case in vaccinees (240).
These studies provide a good overview of the potential differences between survivor and vaccinee immune responses and will surely contribute to the development of more efficient next-generation vaccines. The main EVD immune CoP candidates are shown in Supplementary Table 1.
3.3.4 EBOV interplay with immune responses
EBOV has two main strategies to interfere with the host immune response. First, EBOV blocks IFN signaling and production through VP24 and VP35, respectively. This way, both proteins together ensure that IFN production is hampered and in the case that IFN is produced, the infected cell is unable to respond (241). In the case of VP24, this protein can either directly bind to STAT-1 thus blocking its transport to the nucleus or it can bind to karyopherin α1 and consequently block the IFN antiviral response (242, 243). EBOV VP35 on the other hand, antagonizes IFN mainly through blocking Interferon regulatory factor 3 (IRF-3) phosphorylation and protein kinase R (196, 244).
The second mechanism used by EBOV for immune diversion involves several glycoproteins. The EBOV fourth gene encodes for three different glycoproteins depending on the number of uracyls (Us) added at a so-called editing site. The viral structural surface glycoprotein (EBOV GP) is transcribed when 8 Us are found the editing site (245, 246). When 7Us and 6U/9Us are present, different soluble glycoproteins are produced, namely secreted glycoprotein (sGP) and small soluble GP (ssGP) respectively. Moreover, an additional soluble glycoprotein is generated when a percentage of the EBOV GP expressed on the surface of infected cells is cleaved by proteases releasing it in a soluble form with no transmembrane domain, known as shed GP. Due to its structural similarity to EBOV GP, it has been suggested that shed GP has a role in recruiting new primary targets and also binds antibodies directed to the virus (192). While not all functions have clearly been elucidated, it is suggested that soluble glycoprotein sGP may also play a role in immune evasion by binding antibodies initially directed against the viral surface glycoprotein EBOV GP. However, since not all amino acids are identical to the surface glycoprotein, it is believed that sGP also acts as decoy antigen and reduces specific antibody production against surface GP, possibly resulting in antigenic subversion (247). Importantly, sGP also exhibits anti-inflammatory activities in the endothelium and by reducing the amount of CD16b receptor on human neutrophils thus preventing their activation and consequently stunting an innate immune response (248). Regarding ssGP, while it is believed that it could share some of the functions described for sGP (249), its specific role during EBOV pathogenesis has not yet been clearly elucidated.
4 Comparison of immune profiling in SARS-CoV-2, NiV and EBOV infection
To effectively tackle emerging viruses, it is essential to understand the potential similarities and differences between virus families, the viruses themselves and the immune responses that they elicit upon infection in order to establish realiable CoP. SARS-CoV-2, NiV and EBOV share some immune signatures, yet currently not all of the molecular mechanisms involved in fighting infection with these viruses have been elucidated. These emerging viruses all have in common that they dysregulate host immune responses, including both early and late events, and this dysregulation is associated with viral progression during COVID-19, NiVD and EVD. Below we provide a comparison of the key features of immune responses to SARS-CoV-2, NiV and EBOV infection in humans (Table 1), to better understand differences and the potential pathways to derive CoP (or pathology) for the three diseases.
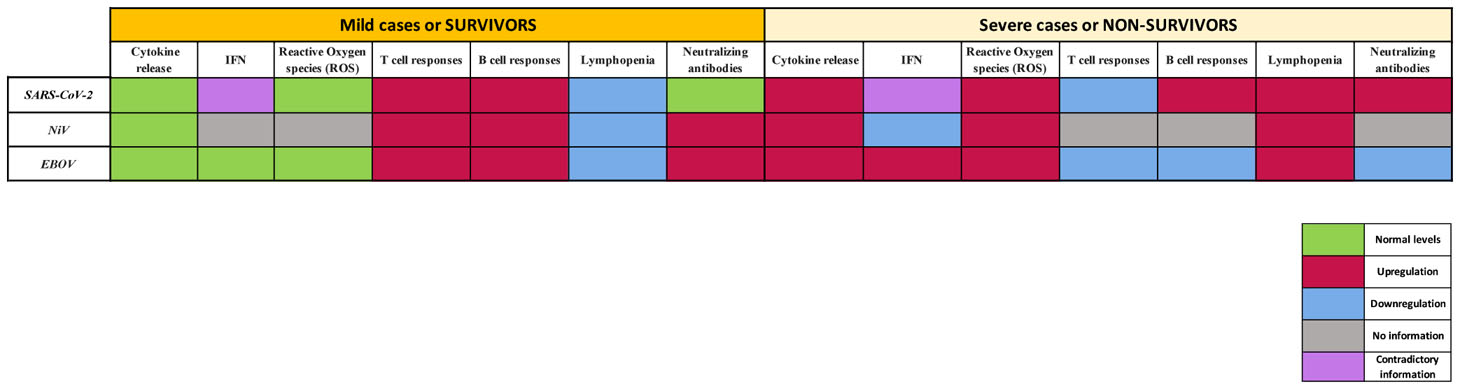
Table 1 Key features of immune responses to SARS-CoV-2, NiV and EBOV infecCon in humans.
The exacerbated release of cytokines and chemokines plays a major role in SARS-CoV-2, NiV and EBOV immunopathology, as this can eventually lead to severe complications and in some cases death during all three diseases. Moreover, abnormal inflammation levels have a big influence in late-stage cellular and humoral immune responses. In this regard, the level of activation, proliferation, phenotype and kinetics of T lymphocyte populations can direct specific T cell mediated and humoral responses and influence the severity of the aforementioned emerging diseases.
The presence of neutralizing IgG in serum is in general used as a CoP, however the titer and the type of antibodies that are needed to reach protection for each disease (or disease outcome) are not clear and need to be defined more precisely and more specifically for each manifestation of infection (protection against infection, protection against death or severe disease, chronic infection, etc…). The kinetics of humoral responses and additional antibody functions such as ADCC may also play a crucial role in protection. Moreover, currently, most vaccines are focused on the glycoproteins of the virus envelope as the immunogen, however, as described above, the viral nucleoproteins and other non-envelope proteins should also be considered in future vaccine designs, and consequently the definition of CoP be updated.
Taken in combination, all of these factors highlight the importance of the different immune compartments and their interactions in achieving viral clearance and highlight the need to further understand innate, cellular and humoral responses and their interplay in order to identify more specific CoP. It is also important to consider the relevance of the potential differences amongst host immune responses during disease progression and to appreciate the role of host diversity in determining the ability to survive infection. The evaluation of common immune signatures that lead to the transition from a mild disease state to a severe one will help in finding novel preventive measures and treatments that could reduce mortality rates.
5 Conclusions
In summary, defining which immune effector mechanisms play a role in protective immunity is crucial for the rational design of vaccines and therapeutics and also for deriving CoP. The latter could be used as surrogates of protection so that evaluation of the protective efficacy of new medical countermeasures can be facilitated. This will in turn guide the pathway for the acceleration of the licensing of these products by regulatory agencies.
For certain pathogens, when survival rates are low, it is not easy to define the underlying immune mechanisms that correlate with protection. The lack of a full understanding of natural immune responses and the potential of certain pathogens to evade them complicates the derivation of CoP. The immune response components described above are an example of the variety of the different and non-exclusive mechanisms that the human immune system uses to evoke the desired protective immunity. Moreover, these mechanisms are not always systemic and for instance, there are often organ-specific mechanisms of immunity (mucosal immunity). More research towards the definition of organ-specific protective mechanisms would help in determining more reliable CoP for certain diseases and vaccines.
The nature and complexity of the interactions of the different cells and soluble effectors of the immune system that are involved in protection is remarkable. Indeed, humoral and cell-mediated immune responses do not act in isolation and the innate immune response strongly influences both T-cell mediated and humoral responses. The best protection against most pathogens is achieved when both arms of the immune system act cooperatively in synergy. While pre-existing antibodies and natural immunity mechanisms may provide the first line of adaptive immune defense, when it is breached, memory T and B cell responses come into play. Thus, an ideal vaccine should offer an integrative approach that triggers both protective antibody levels with robust immunological memory and rapid and efficient effector functions. The high degree of variability of surface antigens in certain pathogens and the complex and dynamic nature of host-pathogen interactions, render the development of vaccines against intracellular infections a challenging process. Notably, such infections often also require cell‐mediated immunity.
A limitation of current vaccine development strategies however, is the reductionist approach of measuring vaccine efficacy as a function of measurable antibody responses, which are often used as CoP, mainly for reasons related to ease of detection, quantification and ease of standardization. Whilst this strategy has proven useful for certain vaccines, it has also shown limitations. This is exemplified by the case of using a serum antibody titer of 1/40 in hemagglutination inhibition tests as a CoP for Influenza vaccines. This way of assessing protective immunity is becoming obsolete as further studies have revealed that different population sub-groups (i.e. the elderly and children) require different titers for predicting protection, particularly as new vaccine strategies for flu based on viral vectors (including other antigens in addition to HA) and mucosal delivery routes (which induce different type of immune effector mechanisms) become available. In the case of EBOV, NiV and SARS-CoV-2, virus neutralizing antibodies have been used as CoP, but again as explained above, this parameter has its own limitations as it is clear that other arms of the immune system do play a role in protection that may not necessarily correlate with neutralizing Ab levels. Furthermore, it is important to emphasize that CoP need to be defined for a specific set of conditions that are related to host, population group, specific disease manifestation that the vaccine intends to protect against and dose, amongst other factors. The large variation in immune responses of the host and the heterogeneity in terms of genetics, age, sex, individual variation and environmental factors, including previous infection status, adds to the challenge of obtaining efficient vaccines.
For all the reasons described above, gaining a deeper understanding of the underlaying immune mechanisms and requirements for successful outcomes during infection is essential in order to derive CoP that are accurate and reliable. This would translate to the development of more effective vaccines and provide more confidence in the ways in which these vaccines are assessed. Vaccine efficacy could be dramatically improved by targeting specific immune CoP such as the generation and maintenance of distinct memory T cell subsets, the specific release of cytokines or facilitating the production of neutralizing antibodies.
The increasing focus on characterizing immune responses to viral infections has led to the development of novel approaches to detect common immune features conferring protection. This has resulted in the development of in silico prediction targets that ultimately may result in the definition of CoP against prominent current pathogens but also for future emerging ones. From a long-term perspective, understanding immune CoP that are specific for certain pathogens could help to promote long-term immunological health. Hence, at a time when emerging infections seem to be more and more frequent, the speed of the efficient establishment of immune CoP appears to be a critical factor in the fight against present and future health threatening diseases.
Author contributions
Conceptualization was done by BE-P and JC-O; writing—original draft preparation, BE-P, PL and JC-O; writing—review and editing, BE-P, PL and JC-O. All authors contributed to the article and approved the submitted version.
Funding
JC-O was funded by the NIHR grant HICC: 'Humoral Immune Correlates for COVID19: Defining protective responses and critical readouts for Clinical Trials of Vaccines and Therapeutics' (NIHR COV0170) and the MRC grant 'Investigation of proven vaccine breakthrough by SARS-CoV-2 variants in established UK healthcare worker cohorts: SIREN consortium & PITCH Plus Pathway' (MRC MR/W02067X/1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1156758/full#supplementary-material
References
1. Sadoff JC, Wittes J. Correlates, surrogates, and vaccines. J Infect Dis (2007) 196(9):1279–81. doi: 10.1086/522432
2. Fiore AE, Bridges CB, Cox NJ. Seasonal influenza vaccines. Curr Top Microbiol Immunol (2009) 333:43–82. doi: 10.1007/978-3-540-92165-3_3
3. Feng S, Phillips DJ, White T, Sayal H, Aley PK, Bibi S, et al. Correlates of protection against symptomatic and asymptomatic SARS-CoV-2 infection. Nat Med (2021) 27(11):2032–40. doi: 10.1038/s41591-021-01540-1
4. Plotkin SA. Complex correlates of protection after vaccination. Clin Infect Dis (2013) 56(10):1458–65. doi: 10.1093/cid/cit048
5. Schenten D, Medzhitov R. The control of adaptive immune responses by the innate immune system. Adv Immunol (2011) 109:87–124. doi: 10.1016/B978-0-12-387664-5.00003-0
6. Meyer M, Malherbe DC, Bukreyev A. Can Ebola virus vaccines have universal immune correlates of protection? Trends Microbiol (2019) 27(1):8–16. doi: 10.1016/j.tim.2018.08.008
7. Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol (2018) 14(Suppl 2):49. doi: 10.1186/s13223-018-0278-1
8. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S33–40. doi: 10.1016/j.jaci.2009.09.017
9. Turvey SE, Broide DH. Innate immunity. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S24–32. doi: 10.1016/j.jaci.2009.07.016
10. Huang Y, Dai H, Ke R. Principles of effective and robust innate immune response to viral infections: A multiplex network analysis. Front Immunol (2019) 10:1736. doi: 10.3389/fimmu.2019.01736
11. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med (1999) 5(8):919–23. doi: 10.1038/11360
12. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol (2015) 15(4):231–42. doi: 10.1038/nri3806
13. Dai R, Huang X, Yang Y. gammadeltaT cells are required for CD8(+) T cell response to vaccinia viral infection. Front Immunol (2021) 12:727046. doi: 10.3389/fimmu.2021.727046
14. Shao L, Huang D, Wei H, Wang RC, Chen CY, Shen L, et al. Expansion, reexpansion, and recall-like expansion of Vgamma2Vdelta2 T cells in smallpox vaccination and monkeypox virus infection. J Virol (2009) 83(22):11959–65. doi: 10.1128/JVI.00689-09
15. La Gruta NL, Turner SJ. T Cell mediated immunity to influenza: Mechanisms of viral control. Trends Immunol (2014) 35(8):396–402. doi: 10.1016/j.it.2014.06.004
16. Castellino F, Galli G, Del Giudice G, Rappuoli R. Generating memory with vaccination. Eur J Immunol (2009) 39(8):2100–5. doi: 10.1002/eji.200939550
17. Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol (2010) 17(7):1055–65. doi: 10.1128/CVI.00131-10
18. Lu LL, Suscovich TJ, Fortune SM, Alter G. Beyond binding: Antibody effector functions in infectious diseases. Nat Rev Immunol (2018) 18(1):46–61. doi: 10.1038/nri.2017.106
19. Crowe JE Jr. Principles of broad and potent antiviral human antibodies: Insights for vaccine design. Cell Host Microbe (2017) 22(2):193–206. doi: 10.1016/j.chom.2017.07.013
21. Burton DR. Antibodies, viruses and vaccines. Nat Rev Immunol (2002) 2(9):706–13. doi: 10.1038/nri891
22. Wang B, Yang C, Jin X, Du Q, Wu H, Dall'Acqua W, et al. Regulation of antibody-mediated complement-dependent cytotoxicity by modulating the intrinsic affinity and binding valency of IgG for target antigen. MAbs (2020) 12(1):1690959. doi: 10.1080/19420862.2019.1690959
23. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
24. Kemp SA, Collier DA, Datir RP, Ferreira I, Gayed S, Jahun A, et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature (2021) 592(7853):277–82. doi: 10.1038/s41586-021-03291-y
25. Stewart WE 2nd, Scott WD, Sulkin SE. Relative sensitivities of viruses to different species of interferon. J Virol (1969) 4(2):147–53. doi: 10.1128/jvi.4.2.147-153.1969
26. Tay MZ, Wiehe K, Pollara J. Antibody-dependent cellular phagocytosis in antiviral immune responses. Front Immunol (2019) 10:332. doi: 10.3389/fimmu.2019.00332
27. Akkaya M, Kwak K, Pierce SK. B cell memory: Building two walls of protection against pathogens. Nat Rev Immunol (2020) 20(4):229–38. doi: 10.1038/s41577-019-0244-2
28. Thakur A, Pedersen LE, Jungersen G. Immune markers and correlates of protection for vaccine induced immune responses. Vaccine (2012) 30(33):4907–20. doi: 10.1016/j.vaccine.2012.05.049
29. Lee HG, Cho MZ, Choi JM. Bystander CD4(+) T cells: crossroads between innate and adaptive immunity. Exp Mol Med (2020) 52(8):1255–63. doi: 10.1038/s12276-020-00486-7
30. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol (2012) 12(2):136–48. doi: 10.1038/nri3152
31. Schijns V, Majhen D, van der Ley P, Thakur A, Summerfield A, Berisio R, et al. Rational vaccine design in times of emerging diseases: The critical choices of immunological correlates of protection, vaccine antigen and immunomodulation. Pharmaceutics (2021) 13(4). doi: 10.3390/pharmaceutics13040501
32. Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol (2003) 4(9):835–42. doi: 10.1038/ni969
33. Harris NL, Watt V, Ronchese F, Le Gros G. Differential T cell function and fate in lymph node and nonlymphoid tissues. J Exp Med (2002) 195(3):317–26. doi: 10.1084/jem.20011558
34. Zumla A, Chan JF, Azhar EI, Hui DS, Yuen KY. Coronaviruses - drug discovery and therapeutic options. Nat Rev Drug Discovery (2016) 15(5):327–47. doi: 10.1038/nrd.2015.37
35. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol (2020) 20(6):363–74. doi: 10.1038/s41577-020-0311-8
36. Jelinek HF, Mousa M, Alefishat E, Osman W, Spence I, Bu D, et al. Evolution, ecology, and zoonotic transmission of betacoronaviruses: A review. Front Vet Sci (2021) 8:644414. doi: 10.3389/fvets.2021.644414
37. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
38. Pedersen SF, Ho Y-C. SARS-CoV-2: a storm is raging. J Clin Invest (2020) 130(5):2202–5. doi: 10.1172/JCI137647
39. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
40. Khalil BA, Shakartalla SB, Goel S, Madkhana B, Halwani R, Maghazachi AA, et al. Immune profiling of COVID-19 in correlation with SARS and MERS. Viruses (2022) 14(1). doi: 10.3390/v14010164
41. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol (2020) 146(1):119–127 e4. doi: 10.1016/j.jaci.2020.04.027
42. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in wuhan, China. Clin Infect Dis (2020) 71(15):762–8. doi: 10.1093/cid/ciaa248
43. Arunachalam PS, Wimmers F, Mok CKP, Perera R, Scott M, Hagan T, et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science (2020) 369(6508):1210–20. doi: 10.1126/science.abc6261
44. Sarzi-Puttini P, Giorgi V, Sirotti S, Marotto D, Ardizzone S, Rizzardini G, et al. COVID-19, cytokines and immunosuppression: What can we learn from severe acute respiratory syndrome? Clin Exp Rheumatol (2020) 38(2):337–42. doi: 10.55563/clinexprheumatol/xcdary
45. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-Mediated inflammatory responses: From mechanisms to potential therapeutic tools. Virol Sin (2020) 35(3):266–71. doi: 10.1007/s12250-020-00207-4
46. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol (2008) 295(3):L379–99. doi: 10.1152/ajplung.00010.2008
47. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe (2020) 27(6):883–890 e2. doi: 10.1016/j.chom.2020.04.017
48. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe (2020) 27(6):992–1000 e3. doi: 10.1016/j.chom.2020.04.009
49. Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep (2020) 33(1):108234. doi: 10.1016/j.celrep.2020.108234
50. Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerging Microbes infections (2020) 9(1):1418–28. doi: 10.1080/22221751.2020.1780953
51. Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther (2020) 5(1):299. doi: 10.1038/s41392-020-00438-7
52. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
53. Trouillet-Assant S, Viel S, Gaymard A, Pons S, Richard JC, Perret M, et al. Type I IFN immunoprofiling in COVID-19 patients. J Allergy Clin Immunol (2020) 146(1):206–208 e2. doi: 10.1016/j.jaci.2020.04.029
54. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol (2020) 5(49). doi: 10.1126/sciimmunol.abd1554
55. Wei L, Ming S, Zou B, Wu Y, Hong Z, Li Z, et al. Viral invasion and type i interferon response characterize the immunophenotypes during COVID-19 infection. (2020).
56. Moss P. The T cell immune response against SARS-CoV-2. Nat Immunol (2022) 23(2):186–93. doi: 10.1038/s41590-021-01122-w
57. Combes AJ, Courau T, Kuhn NF, Hu KH, Ray A, Chen WS, et al. Global absence and targeting of protective immune states in severe COVID-19. Nature (2021) 591(7848):124–30. doi: 10.1038/s41586-021-03234-7
58. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
59. Beck DB, Aksentijevich I. Susceptibility to severe COVID-19. Science (2020) 370(6515):404–5. doi: 10.1126/science.abe7591
60. Rahmani H, Davoudi-Monfared E, Nourian A, Khalili H, Hajizadeh N, Jalalabadi NZ, et al. Interferon β-1b in treatment of severe COVID-19: a randomized clinical trial. Int Immunopharmacol (2020) 88:106903. doi: 10.1016/j.intimp.2020.106903
61. Pereda R, Gonzalez D, Rivero HB, Rivero JC, Perez A, Lopez LDR, et al. Therapeutic effectiveness of interferon-alpha2b against COVID-19: The Cuban experience. J Interferon Cytokine Res (2020) 40(9):438–42. doi: 10.1089/jir.2020.0124
62. Zhou Q, Chen V, Shannon CP, Wei XS, Xiang X, Wang X, et al. Interferon-alpha2b treatment for COVID-19. Front Immunol (2020) 11:1061. doi: 10.3389/fimmu.2020.01061
63. Walz L, Cohen AJ, Rebaza AP, Vanchieri J, Slade MD, Dela Cruz CS, et al. JAK-inhibitor and type I interferon ability to produce favorable clinical outcomes in COVID-19 patients: A systematic review and meta-analysis. BMC Infect Dis (2021) 21(1):47. doi: 10.1186/s12879-020-05730-z
64. Reis G, Moreira Silva EAS, Medeiros Silva DC, Thabane L, Campos VHS, Ferreira TS, et al. Early treatment with pegylated interferon lambda for covid-19. N Engl J Med (2023) 388(6):518–28. doi: 10.1056/NEJMoa2209760
65. King C, Sprent J. Dual nature of type I interferons in SARS-CoV-2-Induced inflammation. Trends Immunol (2021) 42(4):312–22. doi: 10.1016/j.it.2021.02.003
66. Haji Abdolvahab M, Moradi-Kalbolandi S, Zarei M, Bose D, Majidzadeh AK, Farahmand L. Potential role of interferons in treating COVID-19 patients. Int Immunopharmacol (2021) 90:107171. doi: 10.1016/j.intimp.2020.107171
67. Park A, Iwasaki A. Type I and type III interferons - induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe (2020) 27(6):870–8. doi: 10.1016/j.chom.2020.05.008
68. da Silva RP, Goncalves JIB, Zanin RF, Schuch FB, de Souza APD. Circulating type I interferon levels and COVID-19 severity: A systematic review and meta-analysis. Front Immunol (2021) 12:657363. doi: 10.3389/fimmu.2021.657363
69. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
70. Sposito B, Broggi A, Pandolfi L, Crotta S, Clementi N, Ferrarese R, et al. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell (2021) 184(19):4953–4968 e16. doi: 10.1016/j.cell.2021.08.016
71. Bencze D, Fekete T, Pazmandi K. Correlation between type I interferon associated factors and COVID-19 severity. Int J Mol Sci (2022) 23(18). doi: 10.3390/ijms231810968
72. Smits SL, de Lang A, van den Brand JM, Leijten LM, van IWF, Eijkemans MJ, et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PloS Pathog (2010) 6(2):e1000756. doi: 10.1371/journal.ppat.1000756
73. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
74. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell (2020) 181(5):1036–1045 e9. doi: 10.1016/j.cell.2020.04.026
75. Pujadas E, Chaudhry F, McBride R, Richter F, Zhao S, Wajnberg A, et al. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir Med (2020) 8(9):e70. doi: 10.1016/S2213-2600(20)30354-4
76. Notarbartolo S, Ranzani V, Bandera A, Gruarin P, Bevilacqua V, Putignano AR, et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci Immunol (2021) 6(62). doi: 10.1126/sciimmunol.abg5021
77. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
78. Su Y, Chen D, Yuan D, Lausted C, Choi J, Dai CL, et al. Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell (2020) 183(6):1479–95:e20. doi: 10.1016/j.cell.2020.10.037
79. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell (2020) 181(7):1489–1501 e15. doi: 10.1016/j.cell.2020.05.015
80. Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, et al. Presence of SARS-CoV-2-reactive T cells in COVID-19 patients and healthy donors. MedRxiv (2020).
81. Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature (2020) 584(7821):457–62. doi: 10.1038/s41586-020-2550-z
82. Mateus J, Grifoni A, Tarke A, Sidney J, Ramirez SI, Dan JM, et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science (2020) 370(6512):89–94. doi: 10.1126/science.abd3871
83. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe (2020) 27(3):325–8. doi: 10.1016/j.chom.2020.02.001
84. Juno JA, Tan HX, Lee WS, Reynaldi A, Kelly HG, Wragg K, et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat Med (2020) 26(9):1428–34. doi: 10.1038/s41591-020-0995-0
85. Jung JH, Rha M-S, Sa M, Choi HK, Jeon JH, Seok H, et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat Commun (2021) 12(1):1–12. doi: 10.1038/s41467-021-24377-1
86. Cohen KW, Linderman SL, Moodie Z, Czartoski J, Lai L, Mantus G, et al. Longitudinal analysis shows durable and broad immune memory after SARS-CoV-2 infection with persisting antibody responses and memory b and T cells. Cell Rep Med (2021) 2(7):100354. doi: 10.1016/j.xcrm.2021.100354
87. McMahan K, Yu J, Mercado NB, Loos C, Tostanoski LH, Chandrashekar A, et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature (2021) 590(7847):630–4. doi: 10.1038/s41586-020-03041-6
88. Ni L, Ye F, Cheng ML, Feng Y, Deng YQ, Zhao H, et al. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity (2020) 52(6):971–977. e3. doi: 10.1016/j.immuni.2020.04.023
89. Peng Y, Mentzer AJ, Liu G, Yao X, Yin Z, Dong D, et al. Broad and strong memory CD4 (+) and CD8 (+) T cells induced by SARS-CoV-2 in UK convalescent COVID-19 patients. bioRxiv (2020).
90. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
91. Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clin Infect Dis (2020) 71(16):2027–34. doi: 10.1093/cid/ciaa344
92. Haveri A, Ekstrom N, Solastie A, Virta C, Osterlund P, Isosaari E, et al. Persistence of neutralizing antibodies a year after SARS-CoV-2 infection in humans. Eur J Immunol (2021) 51(12):3202–13. doi: 10.1002/eji.202149535
93. Gussarow D, Bonifacius A, Cossmann A, Stankov MV, Mausberg P, Tischer-Zimmermann S, et al. Long-lasting immunity against SARS-CoV-2: Dream or reality? Front Med (Lausanne) (2021) 8:770381. doi: 10.3389/fmed.2021.770381
94. Petersen MS, Hansen CB, Kristiansen MF, Fjallsbak JP, Larsen S, Hansen JL, et al. SARS-CoV-2 natural antibody response persists for at least 12 months in a nationwide study from the faroe islands. Open Forum Infect Dis (2021) 8(8):ofab378. doi: 10.1093/ofid/ofab378
95. Nordstrom P, Ballin M, Nordstrom A. Risk of SARS-CoV-2 reinfection and COVID-19 hospitalisation in individuals with natural and hybrid immunity: A retrospective, total population cohort study in Sweden. Lancet Infect Dis (2022) 22(6):781–90. doi: 10.1016/S1473-3099(22)00143-8
96. Shenai MB, Rahme R, Noorchashm H. Equivalency of protection from natural immunity in COVID-19 recovered versus fully vaccinated persons: A systematic review and pooled analysis. Cureus (2021) 13(10):e19102. doi: 10.7759/cureus.19102
97. Hammerman A, Sergienko R, Friger M, Beckenstein T, Peretz A, Netzer D, et al. Effectiveness of the BNT162b2 vaccine after recovery from covid-19. N Engl J Med (2022) 386(13):1221–9. doi: 10.1056/NEJMoa2119497
98. Alfego D, Sullivan A, Poirier B, Williams J, Adcock D, Letovsky S. A population-based analysis of the longevity of SARS-CoV-2 antibody seropositivity in the united states. EClinicalMedicine (2021) 36:100902. doi: 10.1016/j.eclinm.2021.100902
99. Alejo JL, Mitchell J, Chang A, Chiang TPY, Massie AB, Segev DL, et al. Prevalence and durability of SARS-CoV-2 antibodies among unvaccinated US adults by history of COVID-19. JAMA (2022) 327(11):1085–7. doi: 10.1001/jama.2022.1393
100. Yang Y, Yang M, Peng Y, Liang Y, Wei J, Xing L, et al. Longitudinal analysis of antibody dynamics in COVID-19 convalescents reveals neutralizing responses up to 16 months after infection. Nat Microbiol (2022) 7(3):423–33. doi: 10.1038/s41564-021-01051-2
101. Ng OW, Chia A, Tan AT, Jadi RS, Leong HN, Bertoletti A, et al. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine (2016) 34(17):2008–14. doi: 10.1016/j.vaccine.2016.02.063
102. Ripperger TJ, Uhrlaub JL, Watanabe M, Wong R, Castaneda Y, Pizzato HA, et al. Orthogonal SARS-CoV-2 serological assays enable surveillance of low-prevalence communities and reveal durable humoral immunity. Immunity (2020) 53(5):925–933 e4. doi: 10.1016/j.immuni.2020.10.004
103. Yao L, Wang GL, Shen Y, Wang ZY, Zhan BD, Duan LJ, et al. Persistence of antibody and cellular immune responses in coronavirus disease 2019 patients over nine months after infection. J Infect Dis (2021) 224(4):586–94. doi: 10.1093/infdis/jiab255
104. Sterlin D, Mathian A, Miyara M, Mohr A, Anna F, Claer L, et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci Transl Med (2021) 13(577). doi: 10.1126/scitranslmed.abd2223
105. Wang Z, Muecksch F, Schaefer-Babajew D, Finkin S, Viant C, Gaebler C, et al. Naturally enhanced neutralizing breadth against SARS-CoV-2 one year after infection. Nature (2021) 595(7867):426–31. doi: 10.1038/s41586-021-03696-9
106. van Gils MJ, van Willigen HDG, Wynberg E, Han AX, van der Straten K, Burger JA, et al. A single mRNA vaccine dose in COVID-19 patients boosts neutralizing antibodies against SARS-CoV-2 and variants of concern. Cell Rep Med (2022) 3(1):100486.
107. Chen X, Chen Z, Azman AS, Sun R, Lu W, Zheng N, et al. Neutralizing antibodies against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants induced by natural infection or vaccination: A systematic review and pooled analysis. Clin Infect Dis (2022) 74(4):734–42. doi: 10.1093/cid/ciab646
108. Anichini G, Terrosi C, Gandolfo C, Gori Savellini G, Fabrizi S, Miceli GB, et al. SARS-CoV-2 antibody response in persons with past natural infection. N Engl J Med (2021) 385(1):90–2. doi: 10.1056/NEJMc2103825
109. Prunas O, Warren JL, Crawford FW, Gazit S, Patalon T, Weinberger DM, et al. Vaccination with BNT162b2 reduces transmission of SARS-CoV-2 to household contacts in Israel. Science (2022) 375(6585):1151–4. doi: 10.1126/science.abl4292
110. Pajon R, Paila YD, Girard B, Dixon G, Kacena K, Baden LR, et al. Initial analysis of viral dynamics and circulating viral variants during the mRNA-1273 phase 3 COVE trial. Nat Med (2022) 28(4):823–30. doi: 10.1038/s41591-022-01679-5
111. Ronchini C, Gandini S, Pasqualato S, Mazzarella L, Facciotti F, Mapelli M, et al. Lower probability and shorter duration of infections after COVID-19 vaccine correlate with anti-SARS-CoV-2 circulating IgGs. PloS One (2022) 17(1):e0263014. doi: 10.1371/journal.pone.0263014
112. Bobrovitz N, Ware H, Ma X, Li Z, Hosseini R, Cao C, et al. Protective effectiveness of previous SARS-CoV-2 infection and hybrid immunity against the omicron variant and severe disease: A systematic review and meta-regression. Lancet Infect Dis (2023). doi: 10.1016/S1473-3099(22)00801-5
113. Goel RR, Apostolidis SA, Painter MM, Mathew D, Pattekar A, Kuthuru O, et al. Distinct antibody and memory b cell responses in SARS-CoV-2 naive and recovered individuals following mRNA vaccination. Sci Immunol (2021) 6(58). doi: 10.1126/sciimmunol.abi6950
114. Krammer F, Srivastava K, Alshammary H, Amoako AA, Awawda MH, Beach KF, et al. Antibody responses in seropositive persons after a single dose of SARS-CoV-2 mRNA vaccine. N Engl J Med (2021) 384(14):1372–4. doi: 10.1056/NEJMc2101667
115. Lozano-Ojalvo D, Camara C, Lopez-Granados E, Nozal P, Del Pino-Molina L, Bravo-Gallego LY, et al. Differential effects of the second SARS-CoV-2 mRNA vaccine dose on T cell immunity in naive and COVID-19 recovered individuals. Cell Rep (2021) 36(8):109570.
116. Hwang JY, Kim Y, Lee KM, Jang EJ, Woo CH, Hong CU, et al. Humoral and cellular responses to COVID-19 vaccines in SARS-CoV-2 infection-naive and -recovered Korean individuals. Vaccines (Basel) (2022) 10(2). doi: 10.3390/vaccines10020332
117. Anastassopoulou C, Antoni D, Manoussopoulos Y, Stefanou P, Argyropoulou S, Vrioni G, et al. Age and sex associations of SARS-CoV-2 antibody responses post BNT162b2 vaccination in healthcare workers: A mixed effects model across two vaccination periods. PloS One (2022) 17(4):e0266958. doi: 10.1371/journal.pone.0266958
118. Yang HS, Costa V, Racine-Brzostek SE, Acker KP, Yee J, Chen Z, et al. Association of age with SARS-CoV-2 antibody response. JAMA Netw Open (2021) 4(3):e214302. doi: 10.1001/jamanetworkopen.2021.4302
119. Muller L, Andree M, Moskorz W, Drexler I, Walotka L, Grothmann R, et al. Age-dependent immune response to the Biontech/Pfizer BNT162b2 coronavirus disease 2019 vaccination. Clin Infect Dis (2021) 73(11):2065–72. doi: 10.1093/cid/ciab381
120. Perry J, Osman S, Wright J, Richard-Greenblatt M, Buchan SA, Sadarangani M, et al. Does a humoral correlate of protection exist for SARS-CoV-2? a systematic review. PloS One (2022) 17(4):e0266852. doi: 10.1371/journal.pone.0266852
121. Mercado NB, Zahn R, Wegmann F, Loos C, Chandrashekar A, Yu J, et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature (2020) 586(7830):583–8. doi: 10.1038/s41586-020-2607-z
122. Feng L, Wang Q, Shan C, Yang C, Feng Y, Wu J, et al. An adenovirus-vectored COVID-19 vaccine confers protection from SARS-COV-2 challenge in rhesus macaques. Nat Commun (2020) 11(1):4207. doi: 10.1038/s41467-020-18077-5
123. Yu J, Tostanoski LH, Peter L, Mercado NB, McMahan K, Mahrokhian SH, et al. DNA Vaccine protection against SARS-CoV-2 in rhesus macaques. Science (2020) 369(6505):806–11. doi: 10.1126/science.abc6284
124. Deng W, Lv Q, Li F, Liu J, Song Z, Qi F, et al. Sequential immunizations confer cross-protection against variants of SARS-CoV-2, including omicron in rhesus macaques. Signal Transduct Target Ther (2022) 7(1):124. doi: 10.1038/s41392-022-00979-z
125. Chandrashekar A, Liu J, Martinot AJ, McMahan K, Mercado NB, Peter L, et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science (2020) 369(6505):812–7. doi: 10.1126/science.abc4776
126. Wu J, Liang B, Chen C, Wang H, Fang Y, Shen S, et al. SARS-CoV-2 infection induces sustained humoral immune responses in convalescent patients following symptomatic COVID-19. Nat Commun (2021) 12(1):1813. doi: 10.1038/s41467-021-22034-1
127. Herman JD, Atyeo C, Zur Y, Cook CE, Patel NJ, Vanni KM, et al. Impact of cross-coronavirus immunity in post-acute sequelae of COVID-19. medRxiv (2022). doi: 10.1101/2022.09.25.22280335
128. Su Y, Yuan D, Chen DG, Ng RH, Wang K, Choi J, et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell (2022) 185(5):881–895 e20. doi: 10.1016/j.cell.2022.01.014
129. Hossain A, Akter S, Rashid AA, Khair S, Alam A. Unique mutations in SARS-CoV-2 omicron subvariants' non-spike proteins: Potential impacts on viral pathogenesis and host immune evasion. Microb Pathog (2022) 170:105699. doi: 10.1016/j.micpath.2022.105699
130. Tandel D, Sah V, Singh NK, Potharaju PS, Gupta D, Shrivastava S, et al. SARS-CoV-2 variant delta potently suppresses innate immune response and evades interferon-activated antiviral responses in human colon epithelial cells. Microbiol Spectr (2022) 10(5):e0160422. doi: 10.1128/spectrum.01604-22
131. Ke H, Chang MR, Marasco WA. Immune evasion of SARS-CoV-2 omicron subvariants. Vaccines (Basel) (2022) 10(9). doi: 10.3390/vaccines10091545
132. Schoeman D, Fielding BC. Coronavirus envelope protein: Current knowledge. Virol J (2019) 16(1):69. doi: 10.1186/s12985-019-1182-0
133. Kannan SR, Spratt AN, Sharma K, Chand HS, Byrareddy SN, Singh K. Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J Autoimmun (2022) 126:102779. doi: 10.1016/j.jaut.2021.102779
134. Bai Z, Cao Y, Liu W, Li J. The SARS-CoV-2 nucleocapsid protein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses (2021) 13(6). doi: 10.3390/v13061115
135. Johnson BA, Zhou Y, Lokugamage KG, Vu MN, Bopp N, Crocquet-Valdes PA, et al. Nucleocapsid mutations in SARS-CoV-2 augment replication and pathogenesis. PloS Pathog (2022) 18(6):e1010627. doi: 10.1371/journal.ppat.1010627
136. Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler LA, et al. Author correction: SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol (2020) 27(11):1094. doi: 10.1038/s41594-020-00533-x
137. Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res (2018) 149:58–74. doi: 10.1016/j.antiviral.2017.11.001
138. Gorkhali R, Koirala P, Rijal S, Mainali A, Baral A, Bhattarai HK. Structure and function of major SARS-CoV-2 and SARS-CoV proteins. Bioinform Biol Insights (2021) 15:11779322211025876. doi: 10.1177/11779322211025876
139. Saramago M, Barria C, Costa VG, Souza CS, Viegas SC, Domingues S, et al. New targets for drug design: Importance of nsp14/nsp10 complex formation for the 3'-5' exoribonucleolytic activity on SARS-CoV-2. FEBS J (2021) 288(17):5130–47. doi: 10.1111/febs.15815
140. Sun X, Liu Y, Huang Z, Xu W, Hu W, Yi L, et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ (2022) 29(6):1240–54. doi: 10.1038/s41418-021-00916-7
141. Yang R, Zhao Q, Rao J, Zeng F, Yuan S, Ji M, et al. SARS-CoV-2 accessory protein ORF7b mediates tumor necrosis factor-alpha-Induced apoptosis in cells. Front Microbiol (2021) 12:654709. doi: 10.3389/fmicb.2021.654709
142. Beyer DK, Forero A. Mechanisms of antiviral immune evasion of SARS-CoV-2. J Mol Biol (2022) 434(6):167265. doi: 10.1016/j.jmb.2021.167265
143. Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci U.S.A. (2020) 117(45):28344–54. doi: 10.1073/pnas.2016650117
144. Wu J, Shi Y, Pan X, Wu S, Hou R, Zhang Y, et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep (2021) 34(7):108761. doi: 10.1016/j.celrep.2021.108761
145. Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J Med Virol (2021) 93(9):5376–89. doi: 10.1002/jmv.27050
146. Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol (2020) 17(9):998–1000. doi: 10.1038/s41423-020-0514-8
147. Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, et al. Nipah virus: a recently emergent deadly paramyxovirus. Science (2000) 288(5470):1432–5. doi: 10.1126/science.288.5470.1432
148. Yadav PD, Shete AM, Kumar GA, Sarkale P, Sahay RR, Radhakrishnan C, et al. Nipah virus sequences from humans and bats during nipah outbreak, kerala, India, 2018. Emerg Infect Dis (2019) 25(5):1003–6. doi: 10.3201/eid2505.181076
149. Shete AM, Radhakrishnan C, Pardeshi PG, Yadav PD, Jain R, Sahay RR, et al. Antibody response in symptomatic & asymptomatic nipah virus cases from kerala, India. Indian J Med Res (2021) 154(3):533–5.
150. Ang BSP, Lim TCC, Wang L. Nipah virus infection. J Clin Microbiol (2018) 56(6). doi: 10.1128/JCM.01875-17
151. Chadha MS, Comer JA, Lowe L, Rota PA, Rollin PE, Bellini WJ, et al. Nipah virus-associated encephalitis outbreak, siliguri, India. Emerg Infect Dis (2006) 12(2):235–40. doi: 10.3201/eid1202.051247
152. Escudero-Pérez B, Lalande A, Mathieu C, Lawrence P. Host–Pathogen interactions influencing zoonotic spillover potential and transmission in humans. Viruses (2023) 15(3):599.
153. Chua KB, Koh CL, Hooi PS, Wee KF, Khong JH, Chua BH, et al. Isolation of nipah virus from Malaysian island flying-foxes. Microbes Infect (2002) 4(2):145–51. doi: 10.1016/S1286-4579(01)01522-2
154. Mohd Nor MN, Gan CH, Ong BL. Nipah virus infection of pigs in peninsular Malaysia. Rev Sci Tech (2000) 19(1):160–5.
155. Lo MK, Miller D, Aljofan M, Mungall BA, Rollin PE, Bellini WJ, et al. Characterization of the antiviral and inflammatory responses against nipah virus in endothelial cells and neurons. Virology (2010) 404(1):78–88. doi: 10.1016/j.virol.2010.05.005
156. Lawrence P, Escudero-Perez B. Henipavirus immune evasion and pathogenesis mechanisms: Lessons learnt from natural infection and animal models. Viruses (2022) 14(5). doi: 10.3390/v14050936
157. Liew YJM, Ibrahim PAS, Ong HM, Chong CN, Tan CT, Schee JP, et al. The immunobiology of nipah virus. Microorganisms (2022) 10(6). doi: 10.3390/microorganisms10061162
158. Mathieu C, Pohl C, Szecsi J, Trajkovic-Bodennec S, Devergnas S, Raoul H, et al. Nipah virus uses leukocytes for efficient dissemination within a host. J Virol (2011) 85(15):7863–71. doi: 10.1128/JVI.00549-11
159. Mathieu C, Guillaume V, Sabine A, Ong KC, Wong KT, Legras-Lachuer C, et al. Lethal nipah virus infection induces rapid overexpression of CXCL10. PloS One (2012) 7(2):e32157. doi: 10.1371/journal.pone.0032157
160. Arunkumar G, Devadiga S, McElroy AK, Prabhu S, Sheik S, Abdulmajeed J, et al. Adaptive immune responses in humans during nipah virus acute and convalescent phases of infection. Clin Infect Dis (2019) 69(10):1752–6. doi: 10.1093/cid/ciz010
161. Lara A, Cong Y, Jahrling PB, Mednikov M, Postnikova E, Yu S, et al. Peripheral immune response in the African green monkey model following nipah-Malaysia virus exposure by intermediate-size particle aerosol. PloS Negl Trop Dis (2019) 13(6):e0007454. doi: 10.1371/journal.pntd.0007454
162. Stachowiak B, Weingartl HM. Nipah virus infects specific subsets of porcine peripheral blood mononuclear cells. PloS One (2012) 7(1):e30855. doi: 10.1371/journal.pone.0030855
163. Berhane Y, Weingartl HM, Lopez J, Neufeld J, Czub S, Embury-Hyatt C, et al. Bacterial infections in pigs experimentally infected with nipah virus. Transbound Emerg Dis (2008) 55(3-4):165–74. doi: 10.1111/j.1865-1682.2008.01021.x
164. Geisbert TW, Mire CE, Geisbert JB, Chan YP, Agans KN, Feldmann F, et al. Therapeutic treatment of nipah virus infection in nonhuman primates with a neutralizing human monoclonal antibody. Sci Transl Med (2014) 6(242):242ra82. doi: 10.1126/scitranslmed.3008929
165. Mire CE, Chan YP, Borisevich V, Cross RW, Yan L, Agans KN, et al. A cross-reactive humanized monoclonal antibody targeting fusion glycoprotein function protects ferrets against lethal nipah virus and hendra virus infection. J Infect Dis (2020) 221(Suppl 4):S471–9. doi: 10.1093/infdis/jiz515
166. Bossart KN, Zhu Z, Middleton D, Klippel J, Crameri G, Bingham J, et al. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute nipah virus infection. PloS Pathog (2009) 5(10):e1000642. doi: 10.1371/journal.ppat.1000642
167. Guillaume V, Contamin H, Loth P, Grosjean I, Courbot MC, Deubel V, et al. Antibody prophylaxis and therapy against nipah virus infection in hamsters. J Virol (2006) 80(4):1972–8. doi: 10.1128/JVI.80.4.1972-1978.2006
168. Guillaume V, Contamin H, Loth P, Georges-Courbot MC, Lefeuvre A, Marianneau P, et al. Nipah virus: Vaccination and passive protection studies in a hamster model. J Virol (2004) 78(2):834–40. doi: 10.1128/JVI.78.2.834-840.2004
169. Mire CE, Satterfield BA, Geisbert JB, Agans KN, Borisevich V, Yan L, et al. Pathogenic differences between nipah virus Bangladesh and Malaysia strains in primates: Implications for antibody therapy. Sci Rep (2016) 6:30916. doi: 10.1038/srep30916
170. Basler CF. Nipah and hendra virus interactions with the innate immune system. Curr Top Microbiol Immunol (2012) 359:123–52. doi: 10.1007/82_2012_209
171. Shaw ML, Garcia-Sastre A, Palese P, Basler CF. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol (2004) 78(11):5633–41. doi: 10.1128/JVI.78.11.5633-5641.2004
172. Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF. Nipah virus sequesters inactive STAT1 in the nucleus via a p gene-encoded mechanism. J Virol (2009) 83(16):7828–41. doi: 10.1128/JVI.02610-08
173. Horie R, Yoneda M, Uchida S, Sato H, Kai C. Region of nipah virus c protein responsible for shuttling between the cytoplasm and nucleus. Virology (2016) 497:294–304. doi: 10.1016/j.virol.2016.07.013
174. Shaw ML, Cardenas WB, Zamarin D, Palese P, Basler CF. Nuclear localization of the nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J Virol (2005) 79(10):6078–88. doi: 10.1128/JVI.79.10.6078-6088.2005
175. Yoneda M, Guillaume V, Sato H, Fujita K, Georges-Courbot MC, Ikeda F, et al. The nonstructural proteins of nipah virus play a key role in pathogenicity in experimentally infected animals. PloS One (2010) 5(9):e12709. doi: 10.1371/journal.pone.0012709
176. Rodriguez JJ, Parisien JP, Horvath CM. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol (2002) 76(22):11476–83. doi: 10.1128/JVI.76.22.11476-11483.2002
177. Childs K, Randall R, Goodbourn S. Paramyxovirus V proteins interact with the RNA helicase LGP2 to inhibit RIG-i-dependent interferon induction. J Virol (2012) 86(7):3411–21. doi: 10.1128/JVI.06405-11
178. Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, et al. Mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology (2007) 359(1):190–200. doi: 10.1016/j.virol.2006.09.023
179. Bharaj P, Wang YE, Dawes BE, Yun TE, Park A, Yen B, et al. The matrix protein of nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the IKKepsilon kinase-mediated type-I IFN antiviral response. PloS Pathog (2016) 12(9):e1005880.
180. Sugai A, Sato H, Takayama I, Yoneda M, Kai C. Nipah and hendra virus nucleoproteins inhibit nuclear accumulation of signal transducer and activator of transcription 1 (STAT1) and STAT2 by interfering with their complex formation. J Virol (2017) 91(21). doi: 10.1128/JVI.01136-17
181. Ascenzi P, Bocedi A, Heptonstall J, Capobianchi MR, Di Caro A, Mastrangelo E, et al. Ebolavirus and marburgvirus: Insight the filoviridae family. Mol Aspects Med (2008) 29(3):151–85. doi: 10.1016/j.mam.2007.09.005
182. Bausch DG. West Africa 2013 Ebola: From virus outbreak to humanitarian crisis. Curr Top Microbiol Immunol (2017) 411:63–92. doi: 10.1007/82_2017_69
183. Feldmann H, Geisbert TW. Ebola Haemorrhagic fever. Lancet (2011) 377(9768):849–62. doi: 10.1016/S0140-6736(10)60667-8
184. Sarwar UN, Costner P, Enama ME, Berkowitz N, Hu Z, Hendel CS, et al. Safety and immunogenicity of DNA vaccines encoding ebolavirus and marburgvirus wild-type glycoproteins in a phase I clinical trial. J Infect Dis (2015) 211(4):549–57. doi: 10.1093/infdis/jiu511
185. De Santis O, Audran R, Pothin E, Warpelin-Decrausaz L, Vallotton L, Wuerzner G, et al. Safety and immunogenicity of a chimpanzee adenovirus-vectored Ebola vaccine in healthy adults: A randomised, double-blind, placebo-controlled, dose-finding, phase 1/2a study. Lancet Infect Dis (2016) 16(3):311–20. doi: 10.1016/S1473-3099(15)00486-7
186. Marzi A, Robertson SJ, Haddock E, Feldmann F, Hanley PW, Scott DP, et al. EBOLA VACCINE. VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science (2015) 349(6249):739–42.
187. Marzi A, Reynolds P, Mercado-Hernandez R, Callison J, Feldmann F, Rosenke R, et al. Single low-dose VSV-EBOV vaccination protects cynomolgus macaques from lethal Ebola challenge. EBioMedicine (2019) 49:223–31. doi: 10.1016/j.ebiom.2019.09.055
188. Wauquier N, Becquart P, Padilla C, Baize S, Leroy EM. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PloS Negl Trop Dis (2010) 4(10). doi: 10.1371/journal.pntd.0000837
189. McElroy AK, Harmon JR, Flietstra TD, Campbell S, Mehta AK, Kraft CS, et al. Kinetic analysis of biomarkers in a cohort of US patients with Ebola virus disease. Clin Infect Dis (2016) 63(4):460–7. doi: 10.1093/cid/ciw334
190. Falasca L, Agrati C, Petrosillo N, Di Caro A, Capobianchi MR, Ippolito G, et al. Molecular mechanisms of Ebola virus pathogenesis: Focus on cell death. Cell Death Differ (2015) 22(8):1250–9. doi: 10.1038/cdd.2015.67
191. Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, et al. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J Infect Dis (1999) 179(Suppl 1):S188–91. doi: 10.1086/514283
192. Escudero-Perez B, Volchkova VA, Dolnik O, Lawrence P, Volchkov VE. Shed GP of Ebola virus triggers immune activation and increased vascular permeability. PloS Pathog (2014) 10(11):e1004509. doi: 10.1371/journal.ppat.1004509
193. Wahl-Jensen V, Kurz S, Feldmann F, Buehler LK, Kindrachuk J, DeFilippis V, et al. Ebola Virion attachment and entry into human macrophages profoundly effects early cellular gene expression. PloS Negl Trop Dis (2011) 5(10):e1359. doi: 10.1371/journal.pntd.0001359
194. Baize S, Leroy EM, Georges AJ, Georges-Courbot MC, Capron M, Bedjabaga I, et al. Inflammatory responses in Ebola virus-infected patients. Clin Exp Immunol (2002) 128(1):163–8. doi: 10.1046/j.1365-2249.2002.01800.x
196. Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol (2003) 77(14):7945–56. doi: 10.1128/JVI.77.14.7945-7956.2003
197. Hutchinson KL, Rollin PE. Cytokine and chemokine expression in humans infected with Sudan Ebola virus. J Infect Dis (2007) 196(Suppl 2):S357–63. doi: 10.1086/520611
198. Sanchez A, Lukwiya M, Bausch D, Mahanty S, Sanchez AJ, Wagoner KD, et al. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: Cellular responses, virus load, and nitric oxide levels. J Virol (2004) 78(19):10370–7. doi: 10.1128/JVI.78.19.10370-10377.2004
199. Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: Overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis (2003) 188(11):1618–29. doi: 10.1086/379724
200. Jain S, Khaiboullina SF, Baranwal M. Immunological perspective for Ebola virus infection and various treatment measures taken to fight the disease. Pathogens (2020) 9(10). doi: 10.3390/pathogens9100850
201. Feldmann H, Bugany H, Mahner F, Klenk HD, Drenckhahn D, Schnittler HJ, et al. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J Virol (1996) 70(4):2208–14. doi: 10.1128/jvi.70.4.2208-2214.1996
202. McElroy AK, Akondy RS, Davis CW, Ellebedy AH, Mehta AK, Kraft CS, et al. Human Ebola virus infection results in substantial immune activation. Proc Natl Acad Sci U.S.A. (2015) 112(15):4719–24. doi: 10.1073/pnas.1502619112
203. Speranza E, Ruibal P, Port JR, Feng F, Burkhardt L, Grundhoff A, et al. T-Cell receptor diversity and the control of T-cell homeostasis mark Ebola virus disease survival in humans. J Infect Dis (2018) 218(suppl_5):S508–18. doi: 10.1093/infdis/jiy352
205. Sakabe S, Sullivan BM, Hartnett JN, Robles-Sikisaka R, Gangavarapu K, Cubitt B, et al. Analysis of CD8(+) T cell response during the 2013-2016 Ebola epidemic in West Africa. Proc Natl Acad Sci U.S.A. (2018) 115(32):E7578–86. doi: 10.1073/pnas.1806200115
206. Ruibal P, Oestereich L, Ludtke A, Becker-Ziaja B, Wozniak DM, Kerber R, et al. Unique human immune signature of Ebola virus disease in Guinea. Nature (2016) 533(7601):100–4. doi: 10.1038/nature17949
207. Xu L, Sanchez A, Yang Z, Zaki SR, Nabel EG, Nichol ST, et al. Immunization for Ebola virus infection. Nat Med (1998) 4(1):37–42. doi: 10.1038/nm0198-037
208. Geisbert TW, Hensley LE, Gibb TR, Steele KE, Jaax NK, Jahrling PB. Apoptosis induced in vitro and in vivo during infection by Ebola and marburg viruses. Lab Invest (2000) 80(2):171–86. doi: 10.1038/labinvest.3780021
209. Ludtke A, Ruibal P, Wozniak DM, Pallasch E, Wurr S, Bockholt S, et al. Ebola Virus infection kinetics in chimeric mice reveal a key role of T cells as barriers for virus dissemination. Sci Rep (2017) 7:43776. doi: 10.1038/srep43776
210. Zaki SR, Goldsmith CS. Pathologic features of filovirus infections in humans. Curr Top Microbiol Immunol (1999) 235:97–116. doi: 10.1007/978-3-642-59949-1_7
211. Hensley LE, Young HA, Jahrling PB, Geisbert TW. Proinflammatory response during Ebola virus infection of primate models: Possible involvement of the tumor necrosis factor receptor superfamily. Immunol Lett (2002) 80(3):169–79. doi: 10.1016/S0165-2478(01)00327-3
212. Chepurnov AA, Tuzova MN, Ternovoy VA, Chernukhin IV. Suppressive effect of Ebola virus on T cell proliferation in vitro is provided by a 125-kDa GP viral protein. Immunol Lett (1999) 68(2-3):257–61. doi: 10.1016/S0165-2478(99)00058-9
213. Younan P, Iampietro M, Nishida A, Ramanathan P, Santos RI, Dutta M, et al. Ebola Virus binding to Tim-1 on T lymphocytes induces a cytokine storm. mBio (2017) 8(5). doi: 10.1128/mBio.00845-17
214. Younan P, Santos RI, Ramanathan P, Iampietro M, Nishida A, Dutta M, et al. Ebola Virus-mediated T-lymphocyte depletion is the result of an abortive infection. PloS Pathog (2019) 15(10):e1008068. doi: 10.1371/journal.ppat.1008068
215. Baize S, Leroy EM, Georges-Courbot MC, Capron M, Lansoud-Soukate J, Debre P, et al. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat Med (1999) 5(4):423–6. doi: 10.1038/7422
216. Baize S, Leroy EM, Mavoungou E, Fisher-Hoch SP. Apoptosis in fatal Ebola infection. does the virus toll the bell for immune system? Apoptosis (2000) 5(1):5–7.
217. Wauquier N, Becquart P, Gasquet C, Leroy EM. Immunoglobulin G in Ebola outbreak survivors, Gabon. Emerg Infect Dis (2009) 15(7):1136–7. doi: 10.3201/eid1507.090402
218. Ksiazek TG, Rollin PE, Williams AJ, Bressler DS, Martin ML, Swanepoel R, et al. Clinical virology of Ebola hemorrhagic fever (EHF): Virus, virus antigen, and IgG and IgM antibody findings among EHF patients in kikwit, democratic republic of the Congo, 1995. J Infect Dis (1999) 179(Suppl 1):S177–87. doi: 10.1086/514321
219. Becquart P, Wauquier N, Mahlakoiv T, Nkoghe D, Padilla C, Souris M, et al. High prevalence of both humoral and cellular immunity to Zaire ebolavirus among rural populations in Gabon. PloS One (2010) 5(2):e9126.
220. Rimoin AW, Lu K, Bramble MS, Steffen I, Doshi RH, Hoff NA, et al. Ebola Virus neutralizing antibodies detectable in survivors of theYambuku, Zaire outbreak 40 years after infection. J Infect Dis (2018) 217(2):223–31. doi: 10.1093/infdis/jix584
221. Dowall SD, Kempster S, Findlay-Wilson S, Mattiuzzo G, Graham VA, Page M, et al. Towards quantification of protective antibody responses by passive transfer of the 1st WHO international standard for Ebola virus antibody in a guinea pig model. Vaccine (2020) 38(2):345–9. doi: 10.1016/j.vaccine.2019.10.009
222. Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science (2016) 351(6277):1078–83. doi: 10.1126/science.aad5788
223. Flyak AI, Shen X, Murin CD, Turner HL, David JA, Fusco ML, et al. Cross-reactive and potent neutralizing antibody responses in human survivors of natural ebolavirus infection. Cell (2016) 164(3):392–405. doi: 10.1016/j.cell.2015.12.022
224. Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science (2016) 351(6279):1339–42. doi: 10.1126/science.aad5224
225. Bornholdt ZA, Herbert AS, Mire CE, He S, Cross RW, Wec AZ, et al. A two-antibody pan-ebolavirus cocktail confers broad therapeutic protection in ferrets and nonhuman primates. Cell Host Microbe (2019) 25(1):49–58 e5. doi: 10.1016/j.chom.2018.12.005
226. Oswald WB, Geisbert TW, Davis KJ, Geisbert JB, Sullivan NJ, Jahrling PB, et al. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PloS Pathog (2007) 3(1):e9. doi: 10.1371/journal.ppat.0030009
227. Radinsky O, Edri A, Brusilovsky M, Fedida-Metula S, Sobarzo A, Gershoni-Yahalom O, et al. Sudan Ebolavirus long recovered survivors produce GP-specific abs that are of the IgG1 subclass and preferentially bind FcgammaRI. Sci Rep (2017) 7(1):6054.
228. Davis CW, Jackson KJL, McElroy AK, Halfmann P, Huang J, Chennareddy C, et al. Longitudinal analysis of the human b cell response to Ebola virus infection. Cell (2019) 177(6):1566–1582 e17. doi: 10.1016/j.cell.2019.04.036
229. Gunn BM, Roy V, Karim MM, Hartnett JN, Suscovich TJ, Goba A, et al. Survivors of Ebola virus disease develop polyfunctional antibody responses. J Infect Dis (2020) 221(1):156–61. doi: 10.1093/infdis/jiz364
230. Longet S, Mellors J, Carroll MW, Tipton T. Ebolavirus: Comparison of survivor immunology and animal models in the search for a correlate of protection. Front Immunol (2020) 11:599568. doi: 10.3389/fimmu.2020.599568
231. Marzi A, Engelmann F, Feldmann F, Haberthur K, Shupert WL, Brining D, et al. Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc Natl Acad Sci U.S.A. (2013) 110(5):1893–8. doi: 10.1073/pnas.1209591110
232. Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature (2008) 454(7201):177–82. doi: 10.1038/nature07082
233. Murin CD, Fusco ML, Bornholdt ZA, Qiu X, Olinger GG, Zeitlin L, et al. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc Natl Acad Sci U.S.A. (2014) 111(48):17182–7. doi: 10.1073/pnas.1414164111
234. Gilchuk P, Murin CD, Milligan JC, Cross RW, Mire CE, Ilinykh PA, et al. Analysis of a therapeutic antibody cocktail reveals determinants for cooperative and broad ebolavirus neutralization. Immunity (2020) 52(2):388–403 e12. doi: 10.1016/j.immuni.2020.01.001
235. Murin CD, Gilchuk P, Ilinykh PA, Huang K, Kuzmina N, Shen X, et al. Convergence of a common solution for broad ebolavirus neutralization by glycan cap-directed human antibodies. Cell Rep (2021) 35(2):108984. doi: 10.1016/j.celrep.2021.108984
236. Tomori O, Kolawole MO. Ebola Virus disease: current vaccine solutions. Curr Opin Immunol (2021) 71:27–33. doi: 10.1016/j.coi.2021.03.008
237. Woolsey C, Geisbert TW. Current state of Ebola virus vaccines: A snapshot. PloS Pathog (2021) 17(12):e1010078. doi: 10.1371/journal.ppat.1010078
238. Fuentes S, Ravichandran S, Coyle EM, Klenow L, Khurana S. Human antibody repertoire following Ebola virus infection and vaccination. iScience (2020) 23(3):100920. doi: 10.1016/j.isci.2020.100920
239. Heidepriem J, Krahling V, Dahlke C, Wolf T, Klein F, Addo MM, et al. Epitopes of naturally acquired and vaccine-induced anti-Ebola virus glycoprotein antibodies in single amino acid resolution. Biotechnol J (2020) 15(9):e2000069. doi: 10.1002/biot.202000069
240. Koch T, Rottstegge M, Ruibal P, Gomez-Medina S, Nelson EV, Escudero-Pérez B, et al. Ebola Virus disease survivors show more efficient antibody immunity than vaccinees despite similar levels of circulating immunoglobulins. Viruses (2020) 12(9):915. doi: 10.3390/v12090915
241. Audet J, Kobinger GP. Immune evasion in ebolavirus infections. Viral Immunol (2015) 28(1):10–8. doi: 10.1089/vim.2014.0066
242. Zhang AP, Bornholdt ZA, Liu T, Abelson DM, Lee DE, Li S, et al. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PloS Pathog (2012) 8(2):e1002550. doi: 10.1371/journal.ppat.1002550
243. Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, et al. Ebola Virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol (2006) 80(11):5156–67. doi: 10.1128/JVI.02349-05
244. Feng Z, Cerveny M, Yan Z, He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol (2007) 81(1):182–92. doi: 10.1128/JVI.01006-06
245. Cook JD, Lee JE. The secret life of viral entry glycoproteins: moonlighting in immune evasion. PloS Pathog (2013) 9(5):e1003258. doi: 10.1371/journal.ppat.1003258
246. Sanchez A, Trappier SG, Mahy BW, Peters CJ, Nichol ST. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc Natl Acad Sci U.S.A. (1996) 93(8):3602–7. doi: 10.1073/pnas.93.8.3602
247. Mohan GS, Li W, Ye L, Compans RW, Yang C. Antigenic subversion: a novel mechanism of host immune evasion by Ebola virus. PloS Pathog (2012) 8(12):e1003065. doi: 10.1371/journal.ppat.1003065
248. Wahl-Jensen VM, Afanasieva TA, Seebach J, Stroher U, Feldmann H, Schnittler HJ. Effects of Ebola virus glycoproteins on endothelial cell activation and barrier function. J Virol (2005) 79(16):10442–50. doi: 10.1128/JVI.79.16.10442-10450.2005
Keywords: immune correlates of protection, emerging viruses, humoral immunity, cell-mediated immunity, SARS-CoV-2, Nipah virus, Ebola virus
Citation: Escudero-Pérez B, Lawrence P and Castillo-Olivares J (2023) Immune correlates of protection for SARS-CoV-2, Ebola and Nipah virus infection. Front. Immunol. 14:1156758. doi: 10.3389/fimmu.2023.1156758
Received: 01 February 2023; Accepted: 20 March 2023;
Published: 17 April 2023.
Edited by:
Amol Suryawanshi, Auburn University, United StatesReviewed by:
Rahul K. Suryawanshi, Gladstone Institutes, United StatesPawan Singh, University of Missouri System, United States
Copyright © 2023 Escudero-Pérez, Lawrence and Castillo-Olivares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz Escudero-Pérez, YmVhdHJpei5lc2N1ZGVyb0Bibml0bS5kZQ==; Javier Castillo-Olivares, ZmpjMzdAY2FtLmFjLnVr