 Hongdian Li
Hongdian Li Mingxuan Li
Mingxuan Li Cong Liu1
Cong Liu1- 1Department of Nephrology, Dongfang Hospital, Beijing University of Chinese Medicine, Beijing, China
- 2Department of Cardiology, Beijing Hospital of Traditional Chinese Medicine, Capital Medical University, Beijing, China
- 3Department of Nephrology, Tianjin Academy of Traditional Chinese Medicine Affiliated Hospital, Tianjin, China
Background: While targeted systemic inflammatory modulators show promise in preventing chronic kidney disease (CKD) progression, the causal link between specific inflammatory factors and CKD remains uncertain.
Methods: Using a genome-wide association study of 41 serum cytokines from 8,293 Finnish individuals, we conducted a bidirectional two-sample Mendelian randomization (MR) analysis. In addition, we genetically predicted causal associations between inflammatory factors and 5 phenotypes, including CKD, estimated glomerular filtration rate (eGFR), dialysis, rapid progression of CKD, and rapid decline in eGFR. Inverse variance weighting (IVW) served as the primary MR method, while MR-Egger, weighted median, and MR-pleiotropy residual sum and outlier (MR-PRESSO) were utilized for sensitivity analysis. Cochrane’s Q test for heterogeneity. Leave-one-out method ensured stability of MR results, and Bonferroni correction assessed causal relationship strength.
Results: Seventeen cytokines were associated with diverse renal outcomes. Among them, after Bonferroni correction test, higher tumor necrosis factor alpha levels were associated with a rapid decrease in eGFR (OR = 1.064, 95% CI 1.028 – 1.103, P = 0.001), higher interleukin-4 levels were associated with an increase in eGFR (β = 0.003, 95% CI 0.001 – 0.005, P = 0.002), and higher growth regulated oncogene alpha (GROα) levels were associated with an increased risk of CKD (OR=1.035, 95% CI 1.012 - 1.058, P = 0.003). In contrast, genetic susceptibility to CKD was associated with an increase in GROa, and a decrease in eGFR may lead to an increase in stem cell factor. We did not find the presence of horizontal pleiotropy during the analysis.
Conclusion: We discovered causally related inflammatory factors that contribute to the initiation and progression of CKD at the genetic prediction level.
1 Introduction
Chronic kidney disease (CKD) is a growing global health burden with increasing prevalence and incidence. According to a systematic analysis of the Global Burden of Disease Study, the global mortality rate from CKD has increased by 41.5% between 1990 and 2017, resulting in approximately 1.23 million deaths in 2017 (1). Moreover, CKD imposes a substantial economic burden, with the cost of CKD and ESRD representing 24% of total annual Medicare expenditures in the United States (2). In low- and middle-income countries, more than half of ESDR patients are unable to continue dialysis due to the high cost of treatment (3–5). Despite the high prevalence and economic burden of CKD, there is a lack of effective treatments to slow the progressive loss of renal function and the development of ESRD (6). Therefore, a better understanding of the pathogenesis of CKD is essential to identify new treatment options.
Inflammation may be a promising target for intervention in CKD (7). Regardless of the etiology of CKD, chronic inflammation may be present as a cause and consequence of glomerular and tubulointerstitial pathology (8–11), as a microinflammatory state significantly different from that of the normal renal function population is observed in patients with many forms of CKD with asymptomatic proteinuria (12, 13). Although there are several mechanisms that contribute to the pathological alterations of glomeruli and tubules, inflammation is the key link between them. Transcription factors induce chronic hypoxia in the tubular mesenchyme, leading to peripheral capillary sparing, which triggers adverse phenotypes such as apoptosis. Consequently, mediators mediate inflammatory cell infiltration and fibrosis, impairing local oxygenation and causing aseptic inflammation (14–16). Persistent microinflammation aggravates reactive oxygen species (ROS) loss, exacerbating progressive renal function decline in a mutually reinforcing manner throughout CKD progression (17–19). Oxidative stress induces inflammation through the activation of nuclear factor kappa-B (NF-κB) (20), and the subsequent production of inflammatory factors is associated with a progressive decrease in estimated glomerular filtration rate (eGFR) (21, 22). Observational studies have shown that inflammation is one of the most important pathways for the decline in renal function in European patients with nine different types of CKD (23). These evidences suggest that inflammation is directly associated with CKD and its complications and that inflammation is both the initiator and the outcome of a vicious cycle. Current population-based clinical studies have not yet demonstrated a direct causal association between inflammation and CKD.
Mendelian randomization (MR) uses genetic variants to establish causal links, overcoming biases, resembling nature’s own large-scale randomized controlled trial (RCT) (24). A recent MR analysis has shed new light on the potential therapeutic role of inflammatory modulators in CKD by identifying a causal association between elevated C-reactive protein levels and diabetic nephropathy (25). Bidirectional MR analysis, an extension of conventional MR, has been instrumental in untangling intricate relationships in biological systems, including feedback loops between exposure and outcome variables (26).
To comprehensively evaluate the causal association between systemic inflammatory regulators and CKD, we conducted a bidirectional MR study. Given the prolonged progression of CKD, we included multiple endpoints in our analysis, including eGFR, rapid decline in renal function (Rapid3, defined as a decline in eGFR exceeding 3 mL/min/1.73 m2 per year), rapid progression to CKD (CKDi25, defined as a decline in eGFR ≥25% of baseline while progressing from no CKD to CKD), and dialysis. By utilizing a bidirectional MR approach, we aimed to provide a more comprehensive understanding of the complex relationships between inflammatory factors and dynamic changes in renal function.
2 Method
2.1 Data source of inflammatory markers
We obtained genome-wide association analysis (GWAS) data for circulating concentrations of 41 inflammatory factors from meta-analyses involving 8293 individuals from three independent population cohorts: the Cardiovascular Risk in Young Finns Study (YFS), FINRISK1997, and FINRISK2002 (27). A total of 48 cytokines were measured in YFS and FINRISK2002, following the instructions of the Pro Human Cytokine assay kit (Bio-Rad, Hercules, California, USA). Seven cytokines with missing values exceeding 90% were removed, and 17 cytokines that overlapped with those in FINRISK2002 and YFS were searched for in FINRISK1997. Cytokines were quantified from EDTA plasma in FINRISK1997, from heparin plasma in FINRISK2002, and from serum in YFS. In the original study, a series of rigorous interventions were employed to standardize the expression of cytokine effect sizes, ensuring robust and reliable results. To begin with, cytokine distributions were meticulously normalized through an inverse transformation process. Subsequently, the transformed phenotypes underwent meticulous adjustments for significant genetic principal components, such as age, sex, and body mass index. To ensure the adherence to normal distribution assumptions, another round of meticulous inverse transformation was performed on the model residuals. Subsequently, genome-wide association testing was conducted with the Snptest2 software v.2.5beta. Meta-analyses were performed using METAL software (v.2011-03-25). We have placed these cytokine source details in the Supplementary Table.
2.2 Data source of CKD and kidney function
We utilized data from the CKDGen Consortium for instrumental variables associated with CKD and eGFR, which were the primary outcomes. CKD was defined as eGFR < 60 ml/min/1.73m2, and the GWAS data for CKD were obtained from a meta-analysis involving 23 cohorts of European origin comprising 41,395 patients and 439,303 controls (28). eGFR GWAS data were obtained from meta-analyses conducted in the UK Biobank (n = 436,581, European origin) and the CKDGen consortium (n = 765,348, predominantly European origin) (29). In the UK Biobank, serum creatinine was measured using a Beckman Coulter AU5800 analysis and substituted into the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula to calculate eGFR (30, 31). For individuals aged less than 18 years, the Schwartz formula was used instead (32). Additionally, to assess the relationship between inflammatory factors and dynamic changes in renal function, we included three cohort studies as endpoints. Rapid3 (34,874 cases and 107,090 controls) and CKDi25 (19,901 cases and 175,244 controls) data were obtained from the CKDGen Consortium and a GWAS meta-analysis of 42 studies primarily conducted in UK Biobank with European ancestry (33). Dialysis data were obtained from the Finngen database (r8) of Finns, including 954 cases and 330,300 controls (34).
2.3 Filtering of single nucleotide polymorphisms (SNPs)
Selection of appropriate SNPs is critical for the success of MR analysis. The fundamental assumption of MR requires that all SNPs strongly and independently predict exposure at the genome-wide significance level. In our study, we used 41 inflammatory factor-associated SNPs as instrumental variables for exposure. However, a strict threshold of 5 × 10-8 would have excluded the majority of SNPs. Therefore, we set a relatively lenient but still strongly significant threshold of 5 × 10-6, based on previous studies (35), to include most inflammatory modulator-associated SNPs with an R2 < 0.001 and kb = 10,000 to eliminate chain imbalance. For CKD and renal function-related phenotypes, we used a threshold of 5 × 10-8 for all instrumental variables except for Rapid3, for which we used a threshold of 5 × 10-6 due to fewer eligible SNPs, and we similarly set R2 < 0.001 and kb = 10,000 to eliminate linkage disequilibrium. Additionally, we removed weakly validated SNPs with F values less than 10 (equation: F = β2 exposure/SE2 exposure) to ensure the strength of association between instrumental variables and exposure factors. These screening conditions ensure the credibility of the results of our study.
2.4 Two-sample MR
We used two-sample MR analysis to assess the causal effect of systemic inflammatory modifiers on CKD and renal function. Instrumental estimates of MR for individual SNPs were derived using instrumental variable ratios. Assuming valid instruments without pleiotropy, we performed inverse variance-weighted fixed effects (IVW-FE) MR between instrumental estimates and standard errors (36), following the three main assumptions of MR (see Figure 1). To address horizontal pleiotropy, caused by genetic variation influencing outcomes through pathways other than the exposure, we used multiple methods: inverse variance-weighted random effects (IVW-RE), weighted median (WM), and MR-Egger approaches (37, 38). Moreover, we performed a comprehensive sensitivity analysis employing multiple methods to ensure the robustness of our findings. These methods included the heterogeneity test, horizontal pleiotropy assessment, funnel plot analysis, and leave-one-out analysis of IVW-RE. In the leave-one-out analysis, we systematically excluded one variant at a time to examine its impact on the results. To assess the individual instrumental variables, we employed the instrumental variable ratio (Wald) estimator. This estimator allowed us to evaluate the strength and validity of each instrument used in our instrumental variable analysis. To assess heterogeneity in individual causal effects, we calculated Q-statistics, with p-values < 0.05 indicating heterogeneity (39). In addition to the aforementioned approaches, we also employed a complementary WM method. This method ensures a reliable MR estimate by validating at least 50% of the inverse-variance and ranking its weighted variance. To tackle horizontal pleiotropy, we employed MR pleiotropy residual sum and outlier (MR-PRESSO) test for correction (40). When multiple sets of data are processed and compared simultaneously, there is a risk of encountering false positive results due to random effects. To mitigate this potential issue, we employed the Bonferroni correction test to assess the strength of the causal relationship between the exposure and outcome variables. A significance level of P < 0.05 was considered as suggestive evidence for a causal relationship. We used a significance threshold of 0.0045 for chemokines (11 factors), 0.0056 for growth factors (9 factors), 0.003125 for interleukins (16 factors), and 0.01 for other types (5 factors).
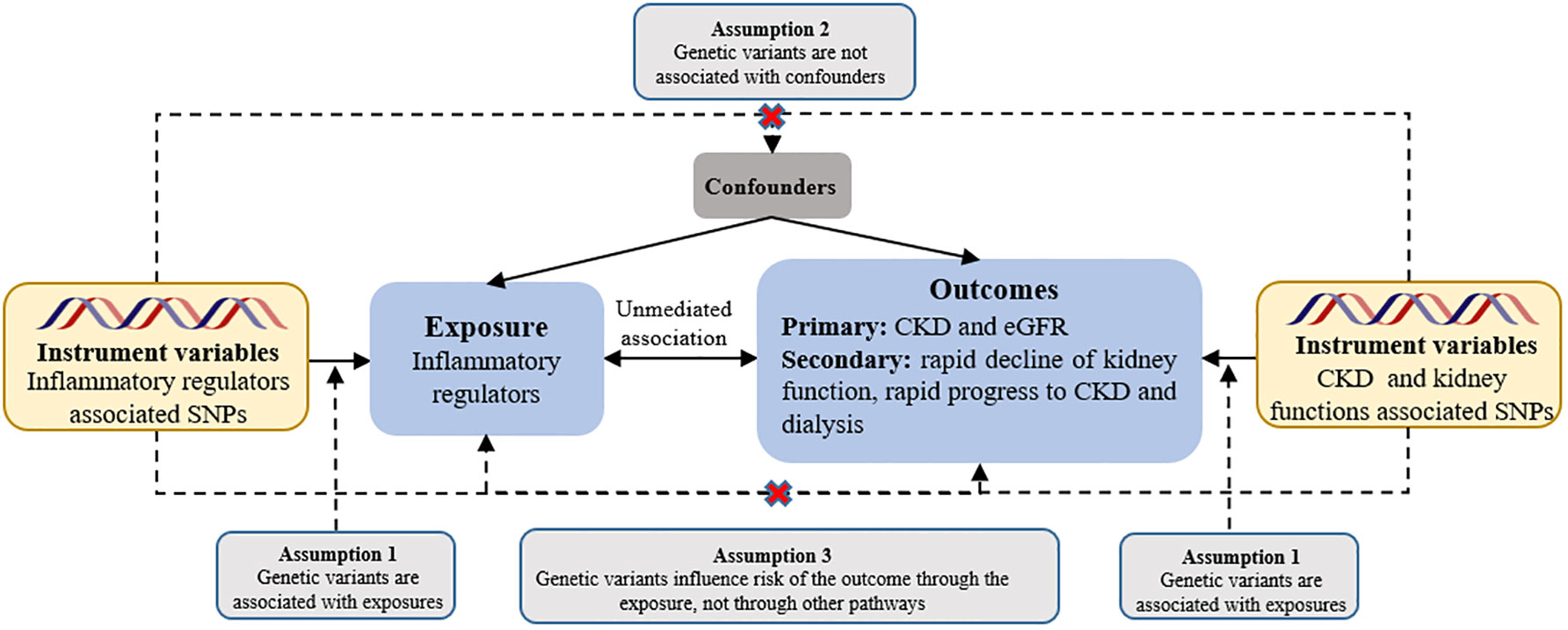
Figure 1 Assumptions of a mendelian randomization analysis for inflammatory regulators and risk of CKD. Broken lines represent potential pleiotropic or direct causal effects between variables that would violate Mendelian randomization assumptions. eGFR, estimated glomerular filtration rate.
3 Results
3.1 Selection of instrumental variables
In our study, we initially screened 41 inflammatory factors for instrumental variables separately, resulting in a total of 452 SNPs that met our set screening criteria, all of which exhibited strong association strength (F-statistics range of 11-789). In the reverse MR analysis, we screened 5 instrumental variables related to CKD, resulting in 60390 SNPs meeting the significance range. After removing chain imbalance, we retained 514 SNPs. Further calculation of the F-statistics of these SNPs revealed that only 1 SNP, namely rs13329952, was strongly correlated with the exposure factor for Rapid3 with an F-statistic of 14, and rs12922822 was identified as an instrumental variable for CKDi25 with an F-statistic of 27. Additionally, we identified 23, 340, and 5 instrumental variables that exhibited strong association with CKD, eGFR, and dialysis, with F-statistics ranging from 25-693. The details of these SNPs are shown in the Supplementary Table.
3.2 Causal link between inflammatory factors and CKD
The study found evidence of a causal link between 10 inflammatory factors and an increased risk of developing CKD (as shown in Figure 2). The IVW-RE method for genetic prediction revealed that higher levels of cutaneous T-cell attracting (CRACK) (OR=1.128, 95% CI 1.014 - 1.255, P = 0.027), growth regulated oncogene alpha (GROα) (OR=1.035, 95% CI 1.012 - 1.058, P = 0.003), beta-nerve growth factor (NGF-β) (OR=1.074, 95% CI 1.005 - 1.148, P = 0.036), stem cell growth factor beta (SCGF-β) (OR=1.053, 95% CI 1.008 - 1.100, P = 0.021), interleukin-8 (IL-8) (OR=1.025, 95% CI 1.006 - 1.046, P = 0.011), and interleukin-7 (IL-7) (OR=1.044, 95% CI 1.011 - 1.077, P = 0.008) were associated with an increased risk of CKD, and the results were similar with the IVW-FE, MR-Egger and weighted median analyses. The results from IVW-FE showed that higher levels of IL-13 were associated with a higher risk of CKD (OR=1.035, 95% CI 1.001 - 1.071, P = 0.049). We identified 2 of 5 SNPs for TNF-β and 2 of 10 SNPs for stromal-cell-derived factor 1 alpha (SDF-1α) associated with CKD. The study found that higher levels of SDF-1α (OR=1.211, 95% CI 1.018 - 1.442, P = 0.033) and TNF-β (OR=1.114, 95% CI 1.022 - 1.215, P = 0.011) were associated with an increased risk of CKD using Wald analysis. However, the IVW-FE analysis revealed that lower levels of TNF-related apoptosis inducing ligand (TRAIL) were associated with a higher risk of CKD (OR=0.968, 95% CI 0.938 - 0.999, P = 0.047), which was observed in the IVW-RE, MR-Egger and weighted median analyses that were consistent. Although there was some evidence of heterogeneity based on Q-statistics in the IL-13 analysis, no evidence of heterogeneity was found in the analysis of other inflammatory factors. Supplementary eFigure 1 displays a scatter plot depicting the association between genetically predicted inflammatory regulators and CKD. Horizontal pleiotropy was not detected through MR-Egger and MR-PRESSO tests (P >0.05). Additionally, the leave-one-out test did not identify any variants that significantly influenced the overall outcome (Supplementary eFigure 2). The funnel plot displayed a symmetrical distribution (Supplementary eFigure 3). Results of the Bonferroni correction test demonstrated that higher levels of GROα remained significantly associated with an increased risk of CKD.
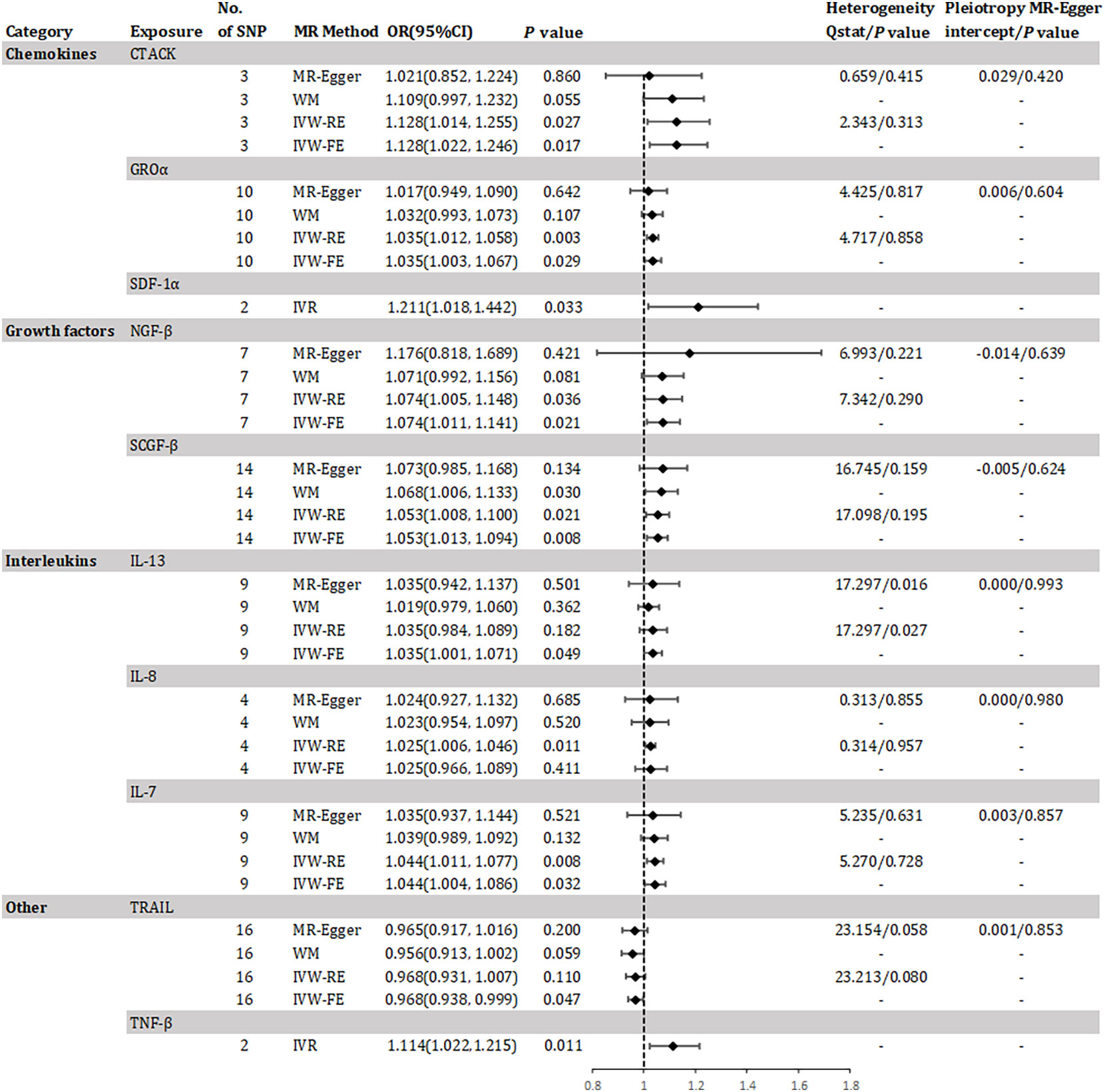
Figure 2 Odds ratio for association of genetically predicted systemic inflammatory regulators with chronic kidney disease. CTACK, cutaneous T-cell attracting; GROa, growth regulated oncogene alpha; SDF-1a, stromal-cell-derived factor 1 alpha; NGF-B, beta-nerve growth factor; SCGF-B, stem cell growth factor beta; IL, interleukin; TNF-β, tumor necrosis factor beta; TRAIL, TNF-related apoptosis inducing ligand; MR, mendelian randomization; CI, confidence internal; OR, odds ratio; IVW-FE, inverse-variance weighted fixed-effects MR; IVW-RE, inverse-variance weighted random-effects MR; WM, weighted median; IVR, instrumental variable ratio (Wald) estimator; SNP, single nucleotide polymorphism. P value for heterogeneity based on Cochran's Q statistic for IVW, and Rücker's Q for MR-Egger.
3.3 Causal link between inflammatory factors and eGFR
The study found evidence of a causal link between 10 inflammatory factors and an increased risk of declined eGFR (as shown in Figure 3). The IVW method for genetic prediction revealed that 1 SD-increase in SDF-1α(β = -0.005, 95% CI -0.007 - -0.003, P = 0.000), NGF-β (β = -0.003, 95% CI -0.005 - -0.001, P = 0.001), SCGF-β (β = -0.002, 95% CI -0.003 - -0.001, P = 0.001) and stem cell factor (SCF)(β = -0.004, 95% CI -0.005 - -0.002, P = 0.000) by the allele were associated with decreased eGFR. The difference is that a genetically predicted 1 SD-increase in platelet-derived growth factor BB (PDGF) (β = 0.001, 95% CI 0.000 – 0.002, P = 0.019), IL-4 (β = 0.003, 95% CI 0.001 – 0.005, P = 0.002) and IL-2ra (β = 0.001, 95% CI 0.000 - 0.002, P = 0.032) by the allele were associated with increased eGFR. The analysis of SDF-1α, NGF-β, SCGF-β and SCF showed some evidence of heterogeneity based on Q-statistics, while no evidence of heterogeneity was found in the analysis of other inflammatory factors. Supplementary eFigure 4 provided scatter plots showing the relationship between these inflammatory regulators and eGFR. MR-Egger and MR-PRESSO tests did not show evidence of horizontal pleiotropy (P >0.05). The leave-one-out test did not identify any associated variants that strongly influenced the overall results (Supplementary eFigure 5), and the funnel plot showed overall symmetry (Supplementary eFigure 6). According to the Bonferroni correction test, higher levels of IL-4 maintained a strong causal relationship with elevated eGFR.
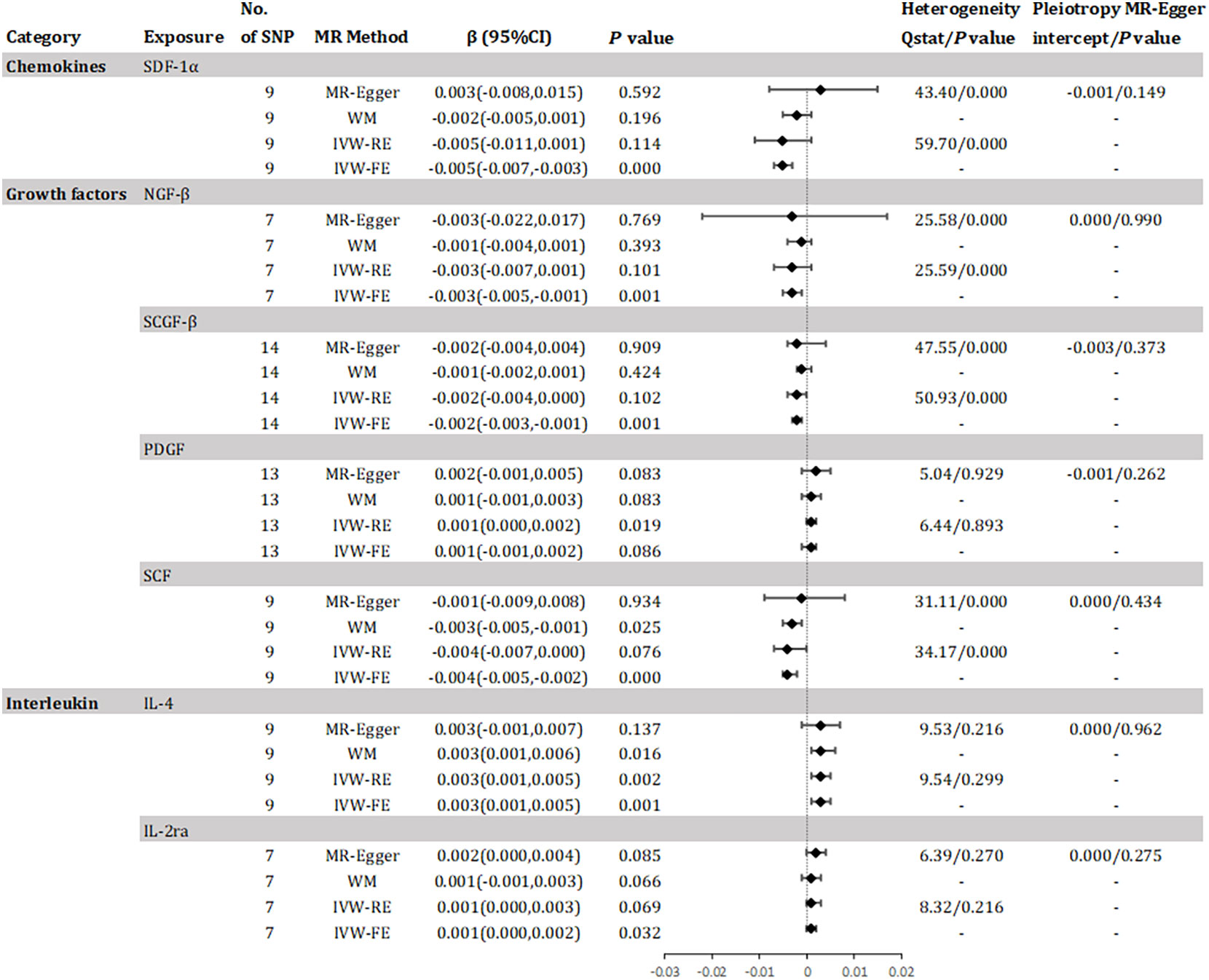
Figure 3 Effect for association of genetically predicted systemic inflammatory regulators with estimated glomerular filtration rate. SCGF-B, stem cell growth factor beta; NGF-β, beta-nerve growth factor; IL, interleukin; SDF-1a, stromal-cell-derived factor 1 alpha; SCF, stem cell factor; PDGF, platelet-derived growth factor BB; MR, mendelian randomization; CI, confidence internal; OR, odds ratio; IVW-FE, inverse-variance weighted fixed-effects MR; IVW-RE, inverse-variance weighted random-effects MR; WM, weighted median; SNP, single nucleotide polymorphism. P value for heterogeneity based on Cochran's Q statistic for IVW, and Rücker's Q for MR-Egger.
3.4 Causal link between inflammatory factors and dynamic changes in renal function
Results has revealed a causal relationship between seven inflammatory factors and changes in renal function, with five factors associated with Rapid3 risk, two with CKDi25 risk, and two with dialysis risk (see Figure 4). A genetically predicted 1 SD-increase in IL-13 (OR = 1.011, 95% CI 1.001 - 1.021, P = 0.029), IL-8 (OR = 1.030, 95% CI 1.006 - 1.053, P = 0.013) and IL-7 (OR = 1.017, 95% CI 1.002 - 1.032, P = 0.031) by the allele was associated with a higher risk of Rapid3. Macrophage inflammatory protein 1 beta (MIP1β) was associated with an elevated risk of CKDi25 (OR = 1.029, 95% CI 1.002 - 1.056, P = 0.036) and SCF was associated with an elevated risk of dialysis (OR = 1.596, 95% CI 1.076 – 2.368, P = 0.020). TNF-α was associated with an elevated risk of CKDi25 (OR = 1.034, 95% CI 1.006 - 1.063, P = 0.018) and Rapid3 (OR = 1.064, 95% CI 1.028 – 1.103, P = 0.001). Interferon gamma (IFN-γ) was associated with an increased risk of dialysis (OR = 1.692, 95% CI 1.124 – 2.458, P = 0.012) and Rapid3 (OR = 1.029, 95% CI 1.002 – 1.056, P = 0.036). We found no evidence of heterogeneity in the course of our analysis. Scatter plots of the relationships between these inflammatory regulators and Rapid3, CKDi25, and dialysis are provided in Supplementary eFigures 7, 10, 13, respectively. MR-Egger and MR-PRESSO tests did not show evidence of horizontal pleiotropy (P >0.05). The leave-one-out test did not identify any associated variants that strongly influenced the overall results (Supplementary eFigures 8, 11, 14), and the funnel plot showed overall symmetry (Supplementary eFigures 9, 12, 15). The results of the Bonferroni correction test showed that higher levels of TNF-α maintained a strong causal relationship with higher risk of Rapid3.
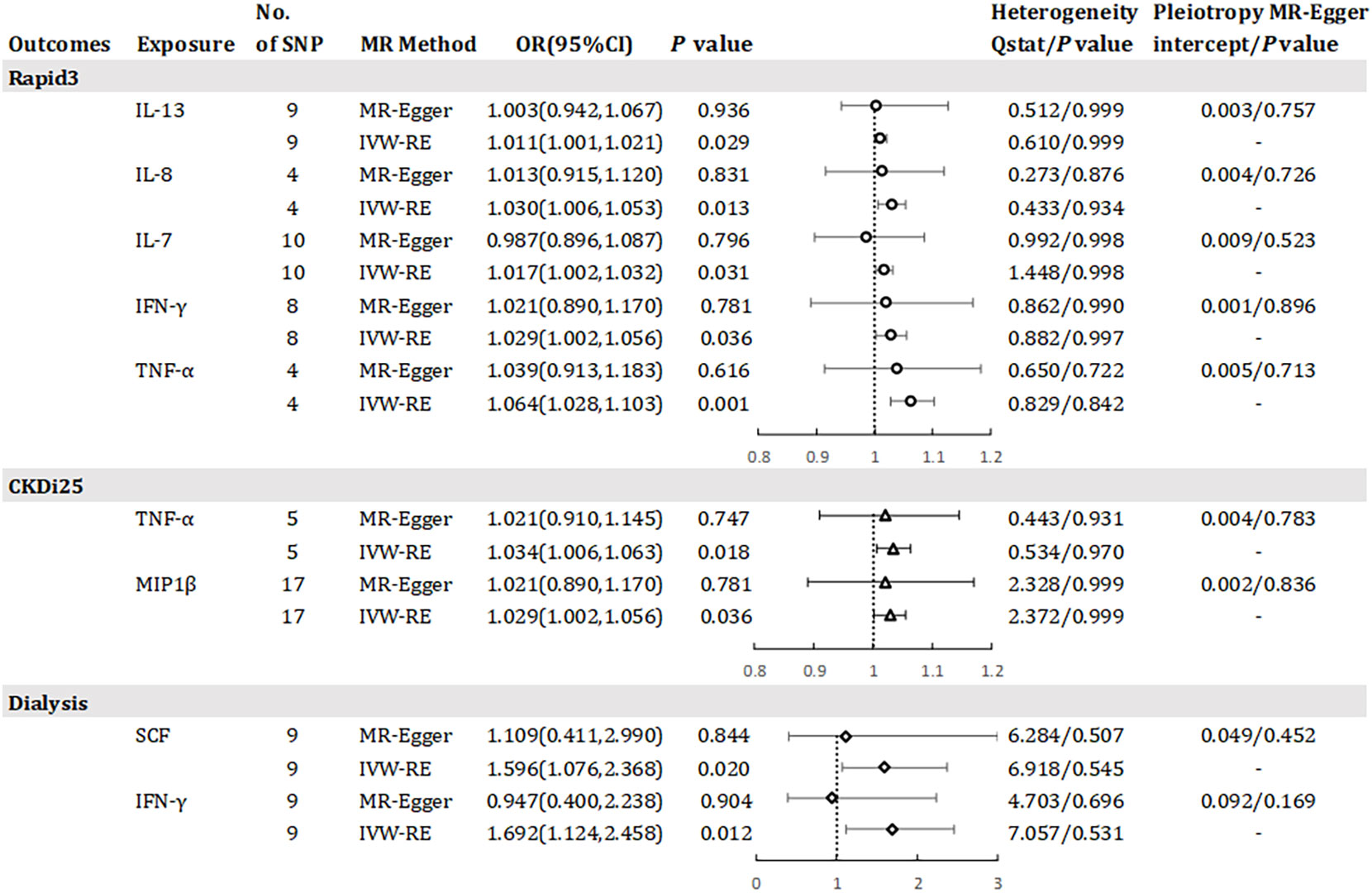
Figure 4 Odds ratio for association of genetically predicted systemic inflammatory regulators with Rapid3, CKDI25 and dialysis. IL, interleukin; TNF-a, tumor necrosis factor alpha; IFN-y, interferon gamma; MIP1bβ, macrophage inflammatory protein 1 beta; SCF, stem cell factor; MR, mendelian randomization; CI, confidence internal; OR, odds ratio; IVW-RE, inverse-variance weighted random- effects MR; SNP, single nucleotide polymorphism; Rapid3, rapid decline in renal function, i.e., a decline in eGFR of more than 3 mL/min/1.73 m2 per year; CKDI25, rapid progression to chronic kidney disease (CKD), i.e., a decrease in eGFR ≥ 25% of baseline, along with progression from no CKD to CKD. P value for heterogeneity based on Cochran's Q statistic for IVW, and Rücker's Q for MR- Egger.
3.5 Reverse analysis
In this study, we identified causal associations between 17 inflammatory factors and various CKD outcomes through forward analyses. We also conducted a reverse study to determine the genetic association between CKD and these 17 inflammatory factors. The ivw-RE analysis revealed that CKD may lead to higher levels of GROα (β = 0.144, 95% CI 0.010 - 0.278, P = 0.036), and reduced eGFR may lead to elevated levels of SCF (β = -1.998, 95% CI -2.773 - -1.223, P = 0.000). However, we did not observe clear associations of other inflammatory factors with Rapid3, CKDi25, and dialysis risk (Supplementary eFigures 16–18). Figure 5 illustrates a bidirectional causal relationship between inflammatory factors and CKD. During the analysis, we found no evidence of horizontal pleiotropy (P >0.05).
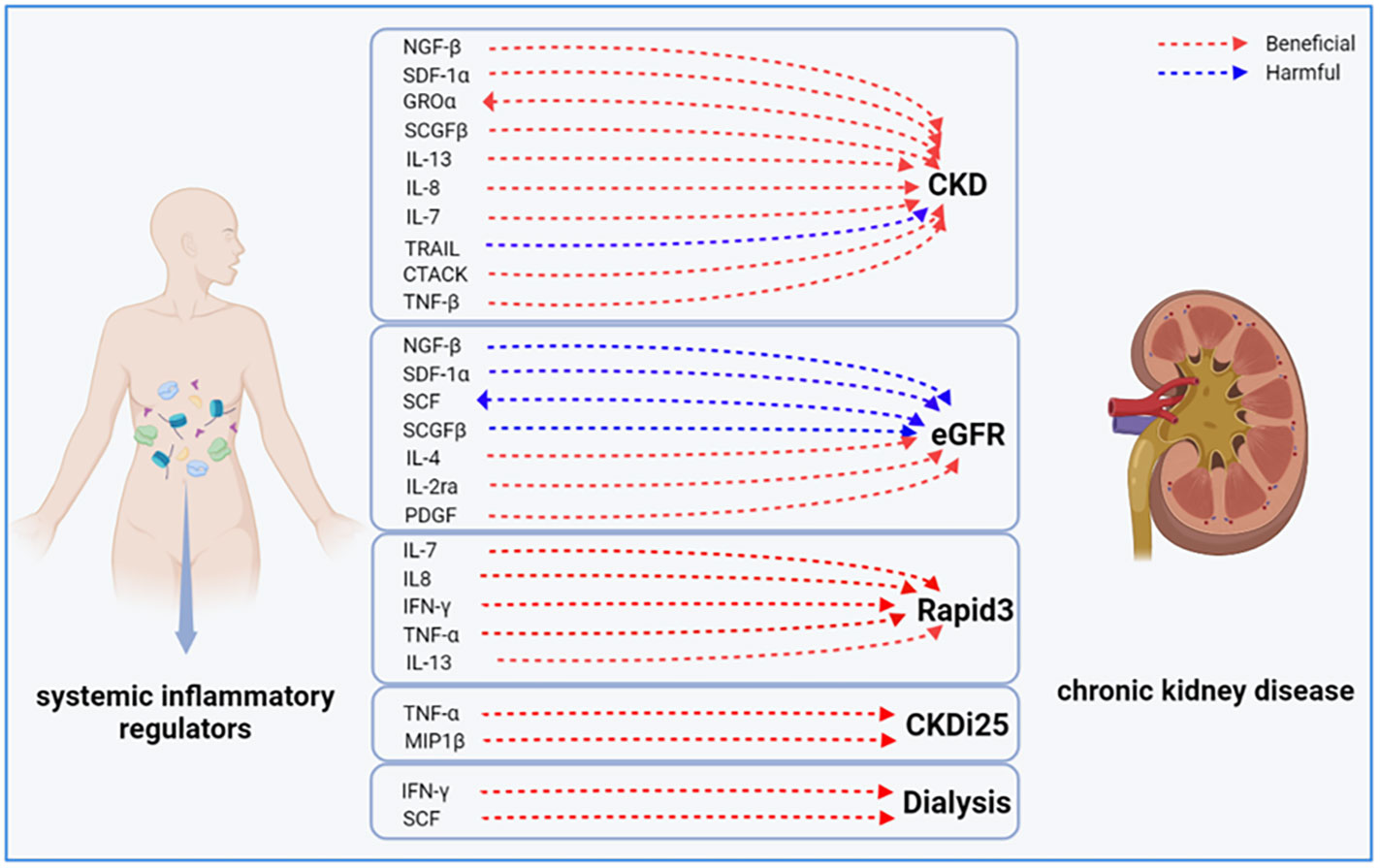
Figure 5 Bidirectional causal link between systemic inflammatory factors and chronic kidney disease. CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; CKDI25, rapid progression to CKD, i.e., a decrease in eGFR ≥ 25% of baseline, along with progression from no CKD to CKD; Rapid3, rapid decline in renal function, i.e., a decline in eGFR of more than 3 mL/min/1.73 m2 per year.
4 Discussion
To our knowledge, our study represents the first large-scale and comprehensive MR investigation to explore the genetic causal relationship between systemic inflammatory regulators and CKD. Previous studies have mainly focused on the cellular or animal level, examining local inflammation in renal tissues or cells rather than the systemic inflammatory response of the organism (41, 42). Observational studies based on clinical settings are often limited by confounding factors and reverse causation bias, leading to distorted causal relationships between variables. By integrating GWAS data from multiple large populations, our study identified 17 inflammatory factors that are genetically associated with various renal outcomes. Specifically, we found strong causal associations between GROα and CKD, IL4 and eGFR, and TNF-α and Rapid3. Subsequent analysis revealed that genetic susceptibility to CKD resulted in an increase in GROa and SCF. These findings highlight the genetic regulation of systemic inflammatory factors by chronic kidney injury.
In this study, we established links between inflammatory factors and multiple outcomes related to CKD, with some inflammatory factors playing important regulatory roles in various outcomes, consistent with previous research. For instance, elevated NGF-β, SDF-1α, and SCGFβ were not only associated with a higher risk of CKD, but also had the potential to decrease eGFR; increased IL7, IL8, and IL13 not only contributed to the likelihood of CKD, but also may have contributed to Rapid3; TNF was associated with both CKD and CKDi25; increased IFN-γ not only contributed to a higher risk of Rapid3, but also may be causally associated with dialysis. There is ample evidence to suggest that unresolved inflammatory processes can lead to renal fibrosis and eventual ESKD, as seen in conditions such as Alport syndrome, autosomal dominant polycystic kidney disease (ADPKD), IgA nephropathy, and focal segmental glomerulosclerosis (43). Our results further validate the direct causal association between systemic inflammation and renal function.
IL are the most diverse and extensively studied family of cytokines with a complex impact on renal function (44). Within this family, IL-8 is a multifunctional factor that specifically attracts neutrophils to inflamed tissues, triggering degranulation, superoxide anion production, respiratory burst, and promoting the release of inflammatory mediators. IL-8 has been established as a critical factor in inducing CKD (45). Studies have demonstrated that not only is IL-8 elevated in adult CKD patients (46), but also in pre-dialysis CKD children compared to healthy controls (47, 48). Meanwhile, IL8 has emerged as an independent risk predictor of CKD and its associated vascular damage. For instance, in patients with type 2 diabetes mellitus, abnormally elevated IL8 levels were found to be associated with a 1.41-fold increased risk of urinary protein (49). Moreover, in patients on hemodialysis, proteomics-detected IL8 was found to predict all-cause mortality risk ratios of up to 1.17, with an elevated cardiovascular all-cause mortality risk ratio of up to 1.34 (50). These predicted risk values align closely with the findings from our study. Previous investigations have demonstrated that chemokines with a sequence similar to IL-8 were highly expressed in rodent models with renal cysts (51). In two different ADPKD cell lines (WT9-7 and WT9-12), IL-8 secretion and expression were found to be significantly increased compared to normal human renal cortical epithelial cell lines. Cell proliferation, mediated by IL-8 signaling, was inhibited by antagonists or siRNAs targeting the IL-8 receptor, and in vitro cystogenesis was attenuated after blocking IL-8 receptor signaling, thus confirming the promoting effect of IL-8 on CKD development (52). However, a recent study further elucidated the effects of different types of IL-8 release on renal structure. In glomerulonephritis specimens, the expression of 72-AA type IL-8 was found to increase in podocytes, whereas 77-A type IL-8 was predominantly expressed in podocytes and interstitial vascular endothelial cells from healthy kidneys and may be associated with preserving glomerular structure (53). Thymocyte differentiation antigen 1 (Thy-1) regulates several fundamental fibroblast functions associated with fibrogenesis and is detectable in serum and urine as soluble Thy-1 (sThy-1). Serum creatinine is a significant and independent predictor of sThy-1 levels. Pro-fibrotic cytokines, including IL-7, IL-13, and IL-8, promote the expression of Thy-1 genes and sThy-1 release from renal interstitial fibroblasts, leading to renal fibrosis and ultimately the development of CKD (54). Our findings highlight the detrimental effects of elevated levels of IL-13, IL-8, and IL-7 on renal function, further supporting the causal link between pro-fibrotic mediators and CKD.
While NGF is traditionally known for its involvement in neuronal injury, recent studies have identified its potential association with kidney function. For instance, single-cell RNA sequencing in a mouse model of renal fibrosis demonstrated an unexpected upregulation of NGF (55). SDF-1α, a CXC-type chemokine, is a known biomarker for angiogenesis, and the SDF-1/CXCR4 pathway plays a crucial role in the development of renal vasculature. Prior research has demonstrated increased renal expression of SDF-1 in rats with subtotal nephrectomy treated with angiotensin-converting enzyme inhibitors (56). Additionally, in a CKD rat model, therapy with peripheral blood-derived endothelial progenitor cells elevated SDF-1α expression at the protein level, suggesting a role for local SDF-1/CXCR4 signaling in preserving microvascular integrity and preventing renal fibrosis (57). However, systemic SDF-1 expression in CKD patients differs from in vivo models, with one study showing higher serum and SDF-1 levels in dialysis patients with chronic renal failure than in healthy controls (58), which aligns with our findings. Activation of CD56bright natural killer cells produced by IFN-γ can play a critical role in the fibrotic process and progression to CKD (59). Clinical evidence suggests that serum IFN-γ levels are often elevated in individuals with CKD and uremia compared to healthy individuals (60, 61). Control of inflammatory factors may decrease body IFN-γ levels and thus regulate renal depletion and CKD-related plaque vulnerability (62, 63). Our findings are consistent with this, suggesting that controlling the body’s inflammatory response is not only protective against chronic renal impairment but also has additional benefits for other pathological responses such as vascular damage and nerve damage associated with CKD (64).
Our study has provided a noteworthy contribution to the understanding of the bidirectional association between systemic inflammation and CKD. The systemic inflammatory response is not only the initiating factor of CKD but is also a consequence of renal impairment. Previous research has primarily focused on the causes of CKD formation, neglecting the contribution of renal disease to the systemic inflammatory state (65–67). Renal disease is a potent modulator of normal gut microbiota composition and metabolism, which can lead to the production of toxins and inflammatory factors. In CKD, factors like gut dysbiosis, slow intestinal transit, low fiber intake, metabolic acidosis, intestinal ischemic edema, iron therapy, and frequent antibiotics exposure can promote the leakage of gut-derived factors (e.g., bacterial components, endotoxins, and intestinal metabolites) into circulation. This triggers immune activation and pro-inflammatory signaling (68, 69). CKD-associated impaired intestinal integrity can promote the leakage of intestinal metabolites, such as trimethylamine N-oxide, p-cresol, and indolol sulfate, as well as lipid peroxidation products, which can directly disrupt cholesterol metabolism, increase scavenger receptor expression, and promote foam cell formation. Moreover, renal disease leads to increased production of these potentially harmful compounds, which can pose a threat to systemic homeostasis, resulting in a range of cardiovascular complications closely linked to the inflammatory response (70). Oxidative stress and metabolic acidosis, which develop as glomerular filtration rate decreases, contribute to the proinflammatory state associated with CKD (71). The impaired nuclear factor erythroid 2-related factor 2 (Nrf2) system plays a pivotal role in this intricate process. Its activity is influenced by factors such as the etiology of renal disease, comorbidities, CKD stage, and the severity of uremic toxin accumulation and inflammation. Notably, early stages of CKD and rapid disease progression are associated with heightened Nrf2 system activation, while in later stages, a stronger inhibition of the Nrf2 system is observed (72). Therefore, systemic inflammation in CKD is not only a well-established risk factor for mortality, but also a potent catalyst for other complications. In the context of renal function decline, coupled with systemic inflammation, there exists a notable escalation in the susceptibility to disturbances in homeostasis. This heightened risk stems from the dual impact of reducing both the functional and structural tissue reserves within the body, while concurrently hampering the intricate interplay between organs that aids in recovery from both internal and external stressors (73).
Currently, there are ongoing drug investigations targeting inflammatory factors in CKD. An earlier proof-of-concept trial in lupus nephritis patients failed to establish the effectiveness of IL-6 inhibition alongside conventional therapy (74). Nevertheless, Pergola and colleagues conducted a RCT assessing the impact of ziltivekimab, a novel anti-IL-6 ligand antibody, in hemodialysis patients harboring polymorphisms predisposing them to IL-6-induced inflammation. Their findings revealed that treated patients exhibited not only improvements in inflammatory markers but also elevated serum albumin levels (75). Additionally, a case report demonstrated the protective effects of tocilizumab, utilized in patients with rheumatoid arthritis accompanied by AA amyloidosis and CKD, manifesting as diminishing proteinuria (76). Ongoing investigations are delving into the role of IL-1 inhibition in renal function. Buckley et al. observed a reduction in serum inflammatory markers following administration of recombinant human IL-1 receptor antagonists in individuals with cardiorenal syndrome (77). Conversely, Nowak et al. did not find enhancements in CKD-associated mineral and bone disorders or physical function after 12 weeks of rilonacept treatment, an IL-1 inhibitor (78). However, their subsequent study addressing vascular function demonstrated that the use of lixonacept in CKD was linked to improved brachial artery flow-mediated dilation and diminished systemic inflammation (79). Moreover, in the CANTOS trial, a human monoclonal antibody targeting IL-1β was employed in CKD patients, resulting in a reduction of major cardiovascular events (80). IL-8 exhibits a heightened specificity within renal pathophysiology, particularly in the context of diabetic nephropathy, and its expression inhibition could hold promise as a potential therapeutic target (81). IL-17A, a constituent of the IL-17 family, contributes to countering bacterial and fungal infections of the skin. It is also implicated in autoimmune and inflammatory disorders, along with its involvement in the pathogenesis of CKD (82, 83). A murine study demonstrated that blocking TNF-α in diabetic nephropathy led to reductions in albuminuria, serum creatinine, histopathological changes, and macrophage infiltration into the kidneys (84). Interferon, often employed as a non-renal therapeutic modality, has recently shown an emerging link to renal pathology. Notably, a case report highlighted a patient with hypereosinophilic syndrome who developed progressive renal failure and nephrotic-range proteinuria after a year of recombinant IFN-α-2b therapy. Renal injury reversed upon cessation of cytokine treatment (85). A similar nephrotic syndrome induced by IFN-β treatment was observed in a multiple sclerosis patient (86). These insights find partial clarification in Migliorini et al.’s investigation, which delineated distinct yet synergistic effects of INF-α and IFN-β on podocytes and parietal epithelial cells, ultimately culminating in glomerulosclerosis (87).
It is worth noting that the Bonferroni correction test may yield false negatives (88). Our study revealed that several systemic inflammatory factors were correlated with various phenotypes, but many of these correlations did not survive the Bonferroni correction test. This could be attributed to the intricate interplay between inflammatory mediators and the kidney, which is usually orchestrated by multiple factors. The pathogenesis of a single inflammatory factor may not be as significant as previously believed, and clusters of inflammatory factors may collaborate with each other to induce disease. By comprehending the pathophysiology of the combined response of these clusters of inflammatory factors in the body and their interaction with renal disease, we may gain a better understanding of the intricate mechanisms involved and develop targeted multi-inflammatory modulators in the future.
This pioneering MR study, utilizing recent pooled data, is the first to examine the causal connection between CKD and systemic inflammatory regulators. Unlike traditional observational studies, which are vulnerable to reverse causality bias due to the involvement of non-renal metabolic pathways in the systemic inflammatory response, this MR analysis minimizes confounding factors and reverse causality, providing a robust estimate of causality. Secondly, our study not only includes a GWAS cohort of CKD events alone but also incorporates multiple dynamic renal function indicators and phenotypes to synthesize the association between renal disease and inflammatory factors, which is more clinically relevant and informative. Finally, serum is one of the most readily accessible biological specimens in clinical practice, and the evidence from this study has broad implications for conducting future relevant clinical research.
It is equally important to acknowledge certain limitations in our study. First and foremost, the genetic data used for this analysis primarily came from individuals of European descent. As a result, the findings may not be applicable or generalizable to other ethnic groups, and caution should be exercised when extrapolating the results to diverse populations. Secondly, despite our best efforts to exclude SNPs that could be associated with potential confounders and conducting various sensitivity analyses under different assumptions, it is still possible that there are complex and multidirectional effects that might not be fully detected. Additionally, the use of instrumental variables from the GWAS meta-analysis prevented us from exploring potential stratification effects and nonlinear relationships, leaving room for further investigation into these aspects. We employed strict Bonferroni correction thresholds and other criteria, such as checking for evidence of pleiotropy, to identify the most reliable MR results. However, this strategy might lead to some false-negative results, although it minimizes the likelihood of identifying false-positive associations. Lastly, while MR analysis is a robust method for estimating causality, it should not be considered a substitute for RCTs. Therefore, the causality inferred from this study may not necessarily align with the results observed in RCTs. It is essential to conduct individual-based genetic observations and potentially incorporate RCTs in future research to further validate the causal relationships identified here.
In summary, our study using publicly available GWAS summary data and MR analysis has identified causal associations between 17 systemic inflammatory modulators and CKD outcomes, including bidirectional associations between GROα and SCF levels and renal function impairment. These findings offer promising genetic evidence for the development of targeted treatments for CKD across different stages.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
All the data utilized in this study have been sourced from the public domain, ensuring their accessibility and transparency. It is imperative to note that all participants involved in this research provided informed consent, thus upholding ethical standards. Furthermore, the study protocols underwent rigorous scrutiny and received the necessary approvals from the local ethical committees associated with each participant. These meticulous steps taken to adhere to ethical guidelines underscore the integrity and reliability of the findings presented in this study.
Author contributions
HL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, and Writing - original draft. ML: Conceptualization, Formal analysis, Investigation, and Writing - original draft. CL: Data curation, Formal analysis; PH: Visualization, and Validation. SD: Funding acquisition and Supervision. AD: Investigation. MZ: Funding acquisition, Conceptualization, and Writing - review & editing. All authors contributed to this article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (No. 82074242) and Research Project of Tianjin Academy of Traditional Chinese Medicine Affiliated Hospital (No. 2022008).
Acknowledgments
We thank all researchers and research platforms that provided raw data. Special thanks to the National Natural Science Foundation of China and Tianjin Academy of Traditional Chinese Medicine Affiliated Hospital for providing us with funding and support for this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1229636/full#supplementary-material
References
1. GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (2020) 395(10225):709–33. doi: 10.1016/S0140-6736(20)30045-3
2. Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, Johansen K, et al. U.S. Renal Data System, USRDS 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (2013).
3. Modi GK, Jha V. The incidence of end-stage renal disease in India: a population-based study. Kidney Int (2006) 70(12):2131–3. doi: 10.1038/sj.ki.5001958
4. Robinson BM, Akizawa T, Jager KJ, Kerr PG, Saran R, Pisoni RL, et al. Factors affecting outcomes in patients reaching end-stage kidney disease worldwide: differences in access to renal replacement therapy, modality use, and haemodialysis practices. Lancet (2016) 388(10041):294–306. doi: 10.1016/S0140-6736(16)30448-2
5. Van Biesen W, Jha V, Abu-Alfa AK, Andreoli SP, Ashuntantang G, Bernieh B, et al. Considerations on equity in management of end-stage kidney disease in low- and middle-income countries. Kidney Int Suppl (2020) 10(1):e63–71. doi: 10.1016/j.kisu.2019.11.004
6. National Kidney Foundation. KDOQI clinical practice guideline for diabetes and CKD: 2012 update. Am J Kidney Dis (2012) 60(5):850–86. doi: 10.1053/j.ajkd.2012.07.005
7. Shacham Y. Inflammation in chronic kidney disease - Something old, something new. Int J Cardiol (2023) 370:407–8. doi: 10.1016/j.ijcard.2022.10.022
8. Akchurin OM, Kaskel F. Update on inflammation in chronic kidney disease. Blood Purif (2015) 39(1-3):84–92. doi: 10.1159/000368940
9. Imig JD, Ryan MJ. Immune and inflammatory role in renal disease. Compr Physiol (2013) 3(2):957–76. doi: 10.1002/cphy.c120028
10. Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol (2010) 298(3):F662–71. doi: 10.1152/ajprenal.00421.2009
11. Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol (2014) 10(9):493–503. doi: 10.1038/nrneph.2014.114
12. Ucha I, Mamven M, Adejumo O, Nwankwo EA. Malnutrition inflammation complex syndrome in pre-dialysis chronic kidney disease patients in a Nigerian tertiary hospital. West Afr J Med (2022) 39(12):1253–9.
13. Das A, Barman B, Bhattacharya P, Lynrah KG, Tiewsoh I, Phukan P. Inflammation and its determinants in patients with chronic kidney disease: A study from North Eastern region of India. Cureus (2022) 14(1):e20917. doi: 10.7759/cureus.20917
14. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell (2012) 148(3):399–408. doi: 10.1016/j.cell.2012.01.021
15. Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int Suppl (2000) 75:S22–6. doi: 10.1046/j.1523-1755.57.s75.12.x
16. Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol (2006) 17(1):17–25. doi: 10.1681/ASN.2005070757
17. Cobo G, Lindholm B, Stenvinkel P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol Dial Transplant (2018) 33(suppl_3):iii35–40. doi: 10.1093/ndt/gfy175
18. Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int (2002) 62(5):1524–38. doi: 10.1046/j.1523-1755.2002.00600.x
19. Zhang K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int J Clin Exp Med (2010) 3(1):33–40.
20. Queisser N, Schupp N. Aldosterone, oxidative stress, and NF-κB activation in hypertension-related cardiovascular and renal diseases. Free Radic Biol Med (2012) 53(2):314–27. doi: 10.1016/j.freeradbiomed.2012.05.011
21. Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int (2013) 83(6):1029–41. doi: 10.1038/ki.2012.439
22. Grande MT, Pérez-Barriocanal F, López-Novoa JM. Role of inflammation in túbulo-interstitial damage associated to obstructive nephropathy. J Inflammation (Lond) (2010) 7:19. doi: 10.1186/1476-9255-7-19
23. Martini S, Nair V, Keller BJ, Eichinger F, Hawkins JJ, Randolph A, et al. Integrative biology identifies shared transcriptional networks in CKD. J Am Soc Nephrol (2014) 25(11):2559–72. doi: 10.1681/ASN.2013080906
24. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med (2008) 27(8):1133–63. doi: 10.1002/sim.3034
25. Lin CC, Li CI, Liu CS, Liao LN, Yang CW, Lin CH, et al. Association of high-sensitivity C-reactive protein and diabetic nephropathy in patients with type 2 diabetes: a Mendelian randomization study. BMJ Open Diabetes Res Care (2023) 11(1):e003197. doi: 10.1136/bmjdrc-2022-003197
26. Richmond RC, Davey Smith G, Ness AR, den Hoed M, McMahon G, Timpson NJ. Assessing causality in the association between child adiposity and physical activity levels: a Mendelian randomization analysis. PloS Med (2014) 11(3):e1001618. doi: 10.1371/journal.pmed.1001618
27. Ahola-Olli AV, Würtz P, Havulinna AS, Aalto K, Pitkänen N, Lehtimäki T, et al. Genome-wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet (2017) 100(1):40–50. doi: 10.1016/j.ajhg.2016.11.007
28. Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet (2019) 51(6):957–72. doi: 10.1038/s41588-019-0407-x
29. Stanzick KJ, Li Y, Schlosser P, Gorski M, Wuttke M, Thomas LF, et al. Discovery and prioritization of variants and genes for kidney function in >1.2 million individuals. Nat Commun (2021) 12(1):4350. doi: 10.1038/s41467-021-24491-0
30. Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med (2012) 367(1):20–9. doi: 10.1056/NEJMoa1114248
31. Pattaro C, Riegler P, Stifter G, Modenese M, Minelli C, Pramstaller PP. Estimating the glomerular filtration rate in the general population using different equations: effects on classification and association. Nephron Clin Pract (2013) 123(1-2):102–11. doi: 10.1159/000351043
32. Schwartz GJ, Schneider MF, Maier PS, Moxey-Mims M, Dharnidharka VR, Warady BA, et al. Improved equations estimating GFR in children with chronic kidney disease using an immunonephelometric determination of cystatin C. Kidney Int (2012) 82(4):445–53. doi: 10.1038/ki.2012.169
33. Gorski M, Jung B, Li Y, Matias-Garcia PR, Wuttke M, Coassin S, et al. Meta-analysis uncovers genome-wide significant variants for rapid kidney function decline. Kidney Int (2021) 99(4):926–39. doi: 10.1016/j.kint.2020.09.030
34. Kurki MI, Karjalainen J, Palta P, Sipila TP, Kristiansson K, Donner K, et al. FinnGen: Unique genetic insights from combining isolated population and national health register data. medRxiv (2022), 22271360. doi: 10.1101/2022.03.03.22271360
35. Shi Q, Wang Q, Wang Z, Lu J, Wang R. Systemic inflammatory regulators and proliferative diabetic retinopathy: A bidirectional Mendelian randomization study. Front Immunol (2023) 14:1088778. doi: 10.3389/fimmu.2023.1088778
36. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol (2016) 40(4):304–14. doi: 10.1002/gepi.21965
37. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol (2013) 37(7):658–65. doi: 10.1002/gepi.21758
38. Tan JS, Liu NN, Guo TT, Hu S, Hua L. Genetically predicted obesity and risk of deep vein thrombosis. Thromb Res (2021) 207:16–24. doi: 10.1016/j.thromres.2021.08.026
39. Burgess S, Bowden J, Dudbridge F, Thompson SG. Robust instrumental variable methods using multiple candidate instruments with application to mendelian randomization. (2016). doi: 10.48550/arXiv.1606.03729
40. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet (2018) 50(5):693–8. doi: 10.1038/s41588-018-0099-7
41. Widowati W, Prahastuti S, Tjokropranoto R, Onggowidjaja P, Kusuma HSW, Afifah E, et al. Quercetin prevents chronic kidney disease on mesangial cells model by regulating inflammation, oxidative stress, and TGF-β1/SMADs pathway. PeerJ (2022) 10:e13257. doi: 10.7717/peerj.13257
42. Olivari V, Di Modica SM, Lidonnici MR, Aghajan M, Cordero-Sanchez C, Tanzi E, et al. A single approach to targeting transferrin receptor 2 corrects iron and erythropoietic defects in murine models of anemia of inflammation and chronic kidney disease. Kidney Int (2023) 104(1):61–73. doi: 10.1016/j.kint.2023.03.012
43. Stenvinkel P, Chertow GM, Devarajan P, Levin A, Andreoli SP, Bangalore S, et al. Chronic inflammation in chronic kidney disease progression: role of Nrf2. Kidney Int Rep (2021) 6(7):1775–87. doi: 10.1016/j.ekir.2021.04.023
44. Matsushima K, Yang D, Oppenheim JJ. Interleukin-8: An evolving chemokine. Cytokine (2022) 153:155828. doi: 10.1016/j.cyto.2022.155828
45. Kalantar-Zadeh K. The latest addition to the inflammatory homeboys in chronic kidney disease: interleukin-8. Nephron Clin Pract (2006) 102(2):c59–60. doi: 10.1159/000088924
46. Romanova Y, Laikov A, Markelova M, Khadiullina R, Makseev A, Hasanova M, et al. Proteomic analysis of human serum from patients with chronic kidney disease. Biomolecules (2020) 10(2):257. doi: 10.3390/biom10020257
47. Mazaheri M. Serum interleukin-6 and interleukin-8 are sensitive markers for early detection of pyelonephritis and its prevention to progression to chronic kidney disease. Int J Prev Med (2021) 12:2. doi: 10.4103/ijpvm.IJPVM_50_19
48. Tunçay SC, Doğan E, Hakverdi G, Tutar ZÜ, Mir S. Interleukin-8 is increased in chronic kidney disease in children, but not related to cardiovascular disease. J Bras Nefrol (2021) 43(3):359–64. doi: 10.1590/2175-8239-JBN-2020-0225
49. Liu SY, Chen J, Li YF. Clinical significance of serum interleukin-8 and soluble tumor necrosis factor-like weak inducer of apoptosis levels in patients with diabetic nephropathy. J Diabetes Investig (2018) 9(5):1182–8. doi: 10.1111/jdi.12828
50. Wu PH, Glerup RI, Svensson MHS, Eriksson N, Christensen JH, Laval P, et al. Novel biomarkers detected by proteomics predict death and cardiovascular events in hemodialysis patients. Biomedicines (2022) 10(4):740. doi: 10.3390/biomedicines10040740
51. Yoo KH, Sung YH, Yang MH, Jeon JO, Yook YJ, Woo YM, et al. Inactivation of Mxi1 induces Il-8 secretion activation in polycystic kidney. Biochem Biophys Res Commun (2007) 356(1):85–90. doi: 10.1016/j.bbrc.2007.02.103
52. Lee EJ, Song SA, Mun HW, Yoo KH, Choi SY, Park EY, et al. Blockade of interleukin-8 receptor signalling inhibits cyst development in vitro, via suppression of cell proliferation in autosomal polycystic kidney disease. Nephrol (Carlton) (2014) 19(8):471–8. doi: 10.1111/nep.12261
53. Niemir ZI, Stein H, Ciechanowicz A, Olejniczak P, Dworacki G, Ritz E, et al. The in situ expression of interleukin-8 in the normal human kidney and in different morphological forms of glomerulonephritis. Am J Kidney Dis (2004) 43(6):983–98. doi: 10.1053/j.ajkd.2004.02.011
54. Deng YJ, Lin XP, Li XQ, Lu PF, Cai Y, Liu LL, et al. Interleukin-7 is associated with clinical and pathological activities in immunoglobulin A nephropathy and protects the renal proximal tubule epithelium from cellular fibrosis. Curr Med Sci (2021) 41(5):880–7. doi: 10.1007/s11596-021-2409-z
55. Ó hAinmhire E, Wu H, Muto Y, Donnelly EL, MaChado FG, Fan LX, et al. A conditionally immortalized Gli1-positive kidney mesenchymal cell line models myofibroblast transition. Am J Physiol Renal Physiol (2019) 316(1):F63–75. doi: 10.1152/ajprenal.00460.2018
56. Huang TH, Chen YT, Sung PH, Chiang HJ, Chen YL, Chai HT, et al. Peripheral blood-derived endothelial progenitor cell therapy prevented deterioration of chronic kidney disease in rats. Am J Transl Res (2015) 7(5):804–24.
57. Chen LH, Advani SL, Thai K, Kabir MG, Sood MM, Gibson IW, et al. SDF-1/CXCR4 signaling preserves microvascular integrity and renal function in chronic kidney disease. PloS One (2014) 9(3):e92227. doi: 10.1371/journal.pone.0092227
58. Chen YT, Cheng BC, Ko SF, Chen CH, Tsai TH, Leu S, et al. Value and level of circulating endothelial progenitor cells, angiogenesis factors and mononuclear cell apoptosis in patients with chronic kidney disease. Clin Exp Nephrol (2013) 17(1):83–91. doi: 10.1007/s10157-012-0664-9
59. Law BMP, Wilkinson R, Wang X, Kildey K, Lindner M, Rist MJ, et al. Interferon-γ production by tubulointerstitial human CD56bright natural killer cells contributes to renal fibrosis and chronic kidney disease progression. Kidney Int (2017) 92(1):79–88. doi: 10.1016/j.kint.2017.02.006
60. Park J, Cai H, Zhang X, Wang XH. The impact of senescence on muscle wasting in chronic kidney disease. J Cachexia Sarcopenia Muscle (2023) 14(1):126–41. doi: 10.1002/jcsm.13112
61. He T, Xia Y, Yang J. Systemic inflammation and chronic kidney disease in a patient due to the RNASEH2B defect. Pediatr Rheumatol Online J (2021) 19(1):9. doi: 10.1186/s12969-021-00497-2
62. Mason DL, Godugu K, Nnani D, Mousa SA. Effects of sevelamer carbonate versus calcium acetate on vascular calcification, inflammation, and endothelial dysfunction in chronic kidney disease. Clin Transl Sci (2022) 15(2):353–60. doi: 10.1111/cts.13151
63. Dei Cas M, Vischi M, Cozzolino MG, Fogagnolo P, Riva A, Petrangolini G, et al. Curcumin supplementation (Meriva®) modulates inflammation, lipid peroxidation and gut microbiota composition in chronic kidney disease. Nutrients (2022) 14(1):231. doi: 10.3390/nu14010231
64. Bi X, Du C, Wang X, Wang XY, Han W, Wang Y, et al. Mitochondrial damage-induced innate immune activation in vascular smooth muscle cells promotes chronic kidney disease-associated plaque vulnerability. Adv Sci (Weinh) (2021) 8(5):2002738. doi: 10.1002/advs.202002738
65. Graterol Torres F, Molina M, Soler-Majoral J, Romero-González G, Rodríguez Chitiva N, Troya-Saborido M, et al. Evolving concepts on inflammatory biomarkers and malnutrition in chronic kidney disease. Nutrients (2022) 14(20):4297. doi: 10.3390/nu14204297
66. Chao CT, Kuo FC, Lin SH. Epigenetically regulated inflammation in vascular senescence and renal progression of chronic kidney disease. Semin Cell Dev Biol (2022). doi: 10.1016/j.semcdb.2022.09.012
67. Magliocca G, Mone P, Di Iorio BR, Heidland A, Marzocco S. Short-chain fatty acids in chronic kidney disease: focus on inflammation and oxidative stress regulation. Int J Mol Sci (2022) 23(10):5354. doi: 10.3390/ijms23105354
68. Kon V, Shelton EL, Pitzer A, Yang HC, Kirabo A. Inflammation, lymphatics, and cardiovascular disease: amplification by chronic kidney disease. Curr Hypertens Rep (2022) 24(10):455–63. doi: 10.1007/s11906-022-01206-4
69. Andersen K, Kesper MS, Marschner JA, Konrad L, Ryu M, Kumar Vr S, et al. Intestinal dysbiosis, barrier dysfunction, and bacterial translocation account for CKD-related systemic inflammation. J Am Soc Nephrol (2017) 28(1):76–83. doi: 10.1681/ASN.2015111285
70. Zhong J, Yang HC, YerMalitsky V, Shelton EL, Otsuka T, Wiese CB, et al. Kidney injury-mediated disruption of intestinal lymphatics involves dicarbonyl-modified lipoproteins. Kidney Int (2021) 100(3):585–96. doi: 10.1016/j.kint.2021.05.028
71. Tinti F, Lai S, Noce A, Rotondi S, Marrone G, Mazzaferro S, et al. Chronic kidney disease as a systemic inflammatory syndrome: update on mechanisms involved and potential treatment. Life (Basel) (2021) 11(5):419. doi: 10.3390/life11050419
72. Aranda-Rivera AK, Cruz-Gregorio A, Pedraza-Chaverri J, Scholze A. Nrf2 activation in chronic kidney disease: promises and pitfalls. Antioxid (Basel) (2022) 11(6):1112. doi: 10.3390/antiox11061112
73. Kooman JP, Dekker MJ, Usvyat LA, Kotanko P, van der Sande FM, Schalkwijk CG, et al. Inflammation and premature aging in advanced chronic kidney disease. Am J Physiol Renal Physiol (2017) 313(4):F938–50. doi: 10.1152/ajprenal.00256.2017
74. Rovin BH, van Vollenhoven RF, Aranow C, Wagner C, Gordon R, Zhuang Y, et al. A multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of treatment with Sirukumab (CNTO 136) in patients with active lupus nephritis. Arthritis Rheumatol (2016) 68(9):2174–83. doi: 10.1002/art.39722
75. Pergola PE, Devalaraja M, Fishbane S, Chonchol M, Mathur VS, Smith MT, et al. Ziltivekimab for treatment of anemia of inflammation in patients on hemodialysis: results from a phase 1/2 multicenter, randomized, double-blind, placebo-controlled trial. J Am Soc Nephrol (2021) 32(1):211–22. doi: 10.1681/ASN.2020050595
76. Fukuda M, Sawa N, Hoshino J, Ohashi K, Motoaki M, Ubara Y. Tocilizumab preserves renal function in rheumatoid arthritis with AA amyloidosis and end-stage kidney disease: Two case reports. Clin Nephrol (2021) 95(1):54–61. doi: 10.5414/CN109971
77. Buckley LF, Canada JM, Carbone S, Trankle CR, Kadariya D, Billingsley H, et al. Potential role for interleukin-1 in the cardio-renal syndrome. Eur J Heart Fail (2019) 21:385–6. doi: 10.1002/ejhf.1403
78. Nowak KL, Hung A, Ikizler TA, Farmer-Bailey H, Salas-Cruz N, Sarkar S, et al. Interleukin-1 inhibition, chronic kidney disease-mineral and bone disorder, and physical function. Clin Nephrol (2017) 88:132–43. doi: 10.5414/CN109122
79. Nowak KL, Chonchol M, Ikizler TA, Farmer-Bailey H, Salas N, Chaudhry R, et al. IL-1 inhibition and vascular function in CKD. J Am Soc Nephrol (2017) 28:971–80. doi: 10.1681/ASN.2016040453
80. Ridker PM, MacFadyen JG, Glynn RJ, Koenig W, Libby P, Everett BM, et al. Inhibition of interleukin-1β by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J Am Coll Cardiol (2018) 71:2405–14. doi: 10.1016/j.jacc.2018.03.490
81. Elsherbiny NM, Al-Gayyar MM. The role of IL-18 in type 1 diabetic nephropathy: The problem and future treatment. Cytokine (2016) 81:15–22. doi: 10.1016/j.cyto.2016.01.014
82. Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol (2018) 10:a028522. doi: 10.1101/cshperspect.a028522
83. Cortvrindt C, Speeckaert R, Moerman A, Delanghe JR, Speeckaert MM. The role of interleukin-17A in the pathogenesis of kidney diseases. Pathology (2017) 49:247–58. doi: 10.1016/j.pathol.2017.01.003
84. Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, et al. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int (2015) 88:722–33. doi: 10.1038/ki.2015.162
85. Nassar GM, Pedro P, Remmers RE, Mohanty LB, Smith W. Reversible renal failure in a patient with the hypereosinophilia syndrome during therapy with alpha interferon. Am J Kidney Dis (1998) 31:121–6. doi: 10.1053/ajkd.1998.v31.pm9428462
86. Ozturk M, Basoglu F, Yilmaz M, Ozagari AA, Baybas S. Interferon β associated nephropathy in a Multiple Sclerosis patient: A case and review. Mult Scler Relat Disord (2016) 9:50–3. doi: 10.1016/j.msard.2016.06.012
87. Migliorini A, Angelotti ML, Mulay SR, Kulkarni OO, Demleitner J, Dietrich A, et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: Implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am J Pathol (2013) 183:431–40. doi: 10.1016/j.ajpath.2013.04.017
Keywords: chronic kidney disease, systemic inflammation, inflammatory modulators, Mendelian randomization, genetic causal association
Citation: Li H, Li M, Liu C, He P, Dong A, Dong S and Zhang M (2023) Causal effects of systemic inflammatory regulators on chronic kidney diseases and renal function: a bidirectional Mendelian randomization study. Front. Immunol. 14:1229636. doi: 10.3389/fimmu.2023.1229636
Received: 26 May 2023; Accepted: 14 August 2023;
Published: 30 August 2023.
Edited by:
Giuseppe Murdaca, University of Genoa, ItalyCopyright © 2023 Li, Li, Liu, He, Dong, Dong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mianzhi Zhang, emhhbmdtaWFuemhpQHZpcC5zaW5hLmNvbQ==
†These authors share first authorship