 Silvia Ricci1,2†
Silvia Ricci1,2† Walter Maria Sarli1,2*†
Walter Maria Sarli1,2*† Lorenzo Lodi1,2Clementina Canessa2Francesca Lippi2Donata Dini3Marta Ferrari3Laura Pisano3
Lorenzo Lodi1,2Clementina Canessa2Francesca Lippi2Donata Dini3Marta Ferrari3Laura Pisano3 Elena Sieni4Giuseppe Indolfi3,5Massimo Resti3Chiara Azzari1,2
Elena Sieni4Giuseppe Indolfi3,5Massimo Resti3Chiara Azzari1,2- 1Department of Health Sciences, University of Florence, Florence, Italy
- 2Immunology Division, Section of Pediatrics, Meyer Children’s Hospital IRCCS, Florence, Italy
- 3Department of Pediatrics, Meyer Children’s Hospital IRCCS, Florence, Italy
- 4Pediatric Hematology-Oncology Department, Meyer Children’s Hospital IRCCS, Florence, Italy
- 5Department Neurofarba, University of Florence, Florence, Italy
Background: Hemophagocytic Lymphohistiocytosis (HLH) is a rare and life-threatening condition characterized by a severe impairment of the immune homeostasis. While Familial-HLH (FHL) is a known cause, the involvement of other Inborn Errors of Immunity (IEI) in pediatric-HLH remains understudied.
Objective: This systematic review aimed to assess the clinical features, triggers, laboratory data, treatment, and outcomes of pediatric HLH patients with IEI other than FHL (IEInotFHL), emphasizing the importance of accurate identification and management.
Methods: A systematic search for studies meeting inclusion criteria was conducted in PubMed, EMBASE, MEDLINE, and Cochrane Central. Quality assessment was performed through JBI criteria.
Results: A comprehensive search yielded 108 records meeting inclusion criteria, involving 178 patients. We identified 46 different IEI according to IUIS 2022 Classification. Combined immunodeficiencies, immune dysregulation disorders, and phagocyte defects were the IEI most frequently associated with HLH. In 75% of cases, HLH preceded the IEI diagnosis, often with an unrecognized history of severe infections. Triggers reflected the specific infection susceptibilities within IEI groups. Liver and central nervous system involvement were less common than in FHL cases. Treatment approaches and outcomes varied, with limited long-term follow-up data, limiting the assessment of therapeutic efficacy across IEI groups.
Conclusion: A comprehensive evaluation encompassing immunological, infectious, and genetic aspects is essential in pediatric-HLH. Relying solely on FHL or EBV susceptibility disorders tests is insufficient, as diverse other IEI can contribute to HLH. Early recognition of HLH as a potential warning sign can guide timely diagnostic investigations and facilitate tailored therapeutic interventions for improved outcomes.
Systematic review registration: https://www.crd.york.ac.uk/prospero/display_record.php?RecordID=371425, PROSPERO, CRD42022371425.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare, hyper-acute and potentially life-threatening clinical entity caused by a severe impairment of the immune homeostasis. Previously seen and managed as a single disease, HLH represents a potential clinical expression of several diseases. HLH occurring in patients bearing known mutations in genes related to granule-dependent cytotoxicity are termed “primary” or familial HLH (FHL) (1). According to the International Union of Immunological Societies (IUIS) (2), FHL are Inborn Errors of Immunity (IEI) presenting with HLH as their predominant clinical feature. On the contrary, when HLH is triggered by infections, autoimmune manifestations or malignancy in the absence of specific mutations in FHL-related genes, it is termed “secondary” or “acquired” (1). However, this classification may be considered overly simplistic, since primary HLH are often triggered by infections or other events that activate the immune system as well as secondary HLH might hide unrecognized or unknown genetic causes. Moreover, since HLH both primary and secondary stems from a loss of immune homeostasis, several IEI other than FHL (IEInotFHL) could predispose to HLH. This systematic review aims to characterize HLH in patients with IEInotFHL, especially those in pediatric age. In fact, since the first diagnostic and therapeutic steps for HLH are often taken in the general pediatric setting, pediatricians should be aware of the existence of possible underlying IEI beyond FHL to minimize the potentially fatal risks associated with a missed diagnosis.
Methods
This systematic review followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) recommendations (3). The study protocol was registered as PROSPERO CRD42022371425.
Definitions
IEI: 485 inherited disorders, often due to mutations in a single gene, involving specific impairment of normal development and immune function. IUIS2022 Classification groups IEI into 9 major categories and multiple subgroups based on which part of immune system is impaired (see Supplemental Data for complete classification) (2). IUIS2022 Classification also considers an additional group for phenocopies of IEI.
FHL: IEI which share HLH as their predominant clinical feature. According to IUIS 2022 Classification FHL can be distinguished in two subgroups based on the presence of hypopigmentation. The first subgroup includes pathogenic variants in PRF1, STX1, UNC13D, STXBP2, FAAP24, SLC7A7, and RHOG, while the second subgroup consists of gene variants associated with Chediak-Higashi syndrome, Griscelli type 2, and, less commonly, Hermansky-Pudlak syndrome types 2 and 10, in addition to the neofunction of CEBPE.
IEInotFHL: IEI not belonging to FHL subgroups according to IUIS 2022 Classification.
Study design and search strategy
On 30th September 2022 an extensive search for publications on HLH associated to IEInotFHL was conducted on PubMed, MEDLINE, EMBASE, and Cochrane Central databases (see Supplementary Data for strings). The search string included IEInotFHL previously described as possibly associated to HLH (4–6) and Epstein-Barr Virus (EBV) susceptibility disorders that are not comprised in FHL subgroups according to IUIS 2022 Classification (2). Reference lists were hand-searched for further relevant studies.
Study eligibility and quality assessment
After duplicates were removed through Rayyan online database (7), screening on title and abstract was conducted by three independent reviewers (W.M.S, S.R and M.F). Discrepancies were discussed until a common decision was reached.
A study was considered eligible when the following criteria were met: at least 5 diagnostic criteria of HLH (8), confirmed diagnosis or high suspicion of IEInotFHL according to IUIS Classification 2022, clinical data report. Records were excluded if they did not describe IEInotFHL, described animal experiments, contained no clinical data, or were not peer reviewed. Phenocopies of IEI were not considered. Non-English records were finally excluded for practical purposes. Quality assessment was performed with Critical Appraisal Tools of the Johanna Briggs Institute (JBI) by two independent reviewers (W.M.S and S.R) (9, 10). No records were excluded because of methodological quality.
Data extraction, synthesis of results and analysis
All data were extracted by a reviewer (W.M.S) using a Microsoft® Excel® spreadsheet developed by the author team and verified by a second reviewer (S.R.). Common decision was to extract the worst laboratory values. The main characteristics of the included studies have been analyzed and summarized in tables. Quantitative variables are expressed as mean and standard deviations (DS) or median and interquartile range (IQR). GraphPad® Prism 9 was used for statistical analysis. Mann-Whitney test was used to compare non-normal values. Fisher and χ2 tests were used to evaluate differences between groups. The level of significance was set to p<0.05.
Results
Study selection
This review included 108 studies (Figure 1) for a total of 178 patients (4, 5, 11–116). Through database search and reference lists screening, 4570 records were identified. After 1304 duplicates were removed, 3263 titles/abstracts were screened and subsequently, 261 full-text articles were assessed for eligibility. All details are shown in Figure 1.
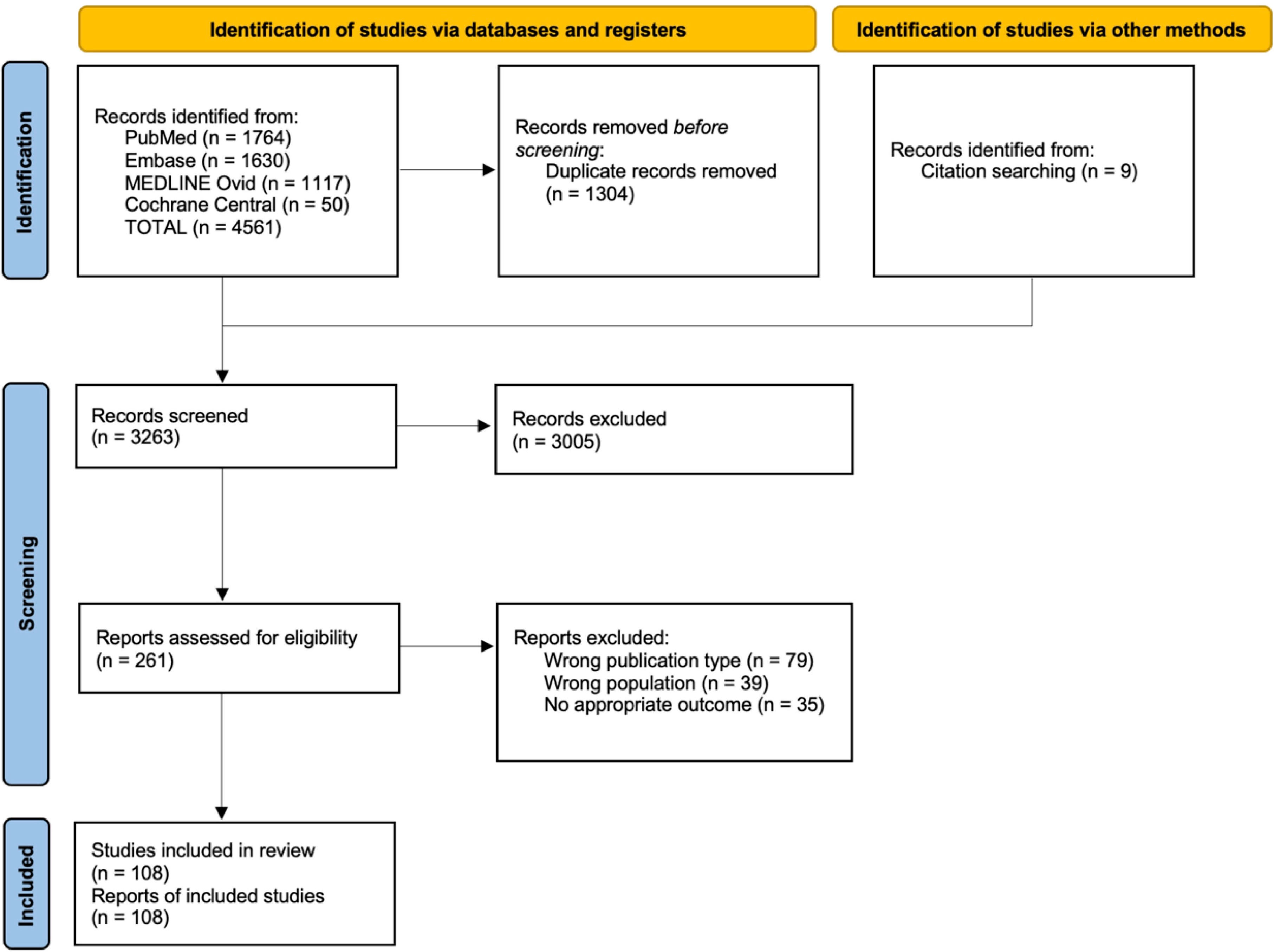
Figure 1 Selected reports flow-chart according to PRISMA Guidelines.
Demographics
At HLH onset, 159/178 patients (89%) were aged 0-18 years, 7/159 pediatric patients (4%) experienced HLH within first 30 days of life (IQR 9-23.5 days) (19, 41, 61, 63, 92, 95, 110) and one of them during fetal life (55). The median age at HLH diagnosis, considering only pediatric patients, was 17.5 months (IQR 4-60 months).
Gender was declared or deductible from genetic diagnosis for 149/178 patients (84%) with a M:F ratio 3:1. As expected, gender ratio varied across distinct IEI groups, according with the inheritance pattern. When excluding all X-linked defects, the M:F ratio was established to 1.5:1. Consanguinity between parents was reported in 28/178 patients (16%) while familiarity for IEI or sudden infant death was reported in 30/178 patients (17%).
HLH in IEInotFHL
The frequencies of HLH diagnostic criteria in selected patients are shown in Table 1.
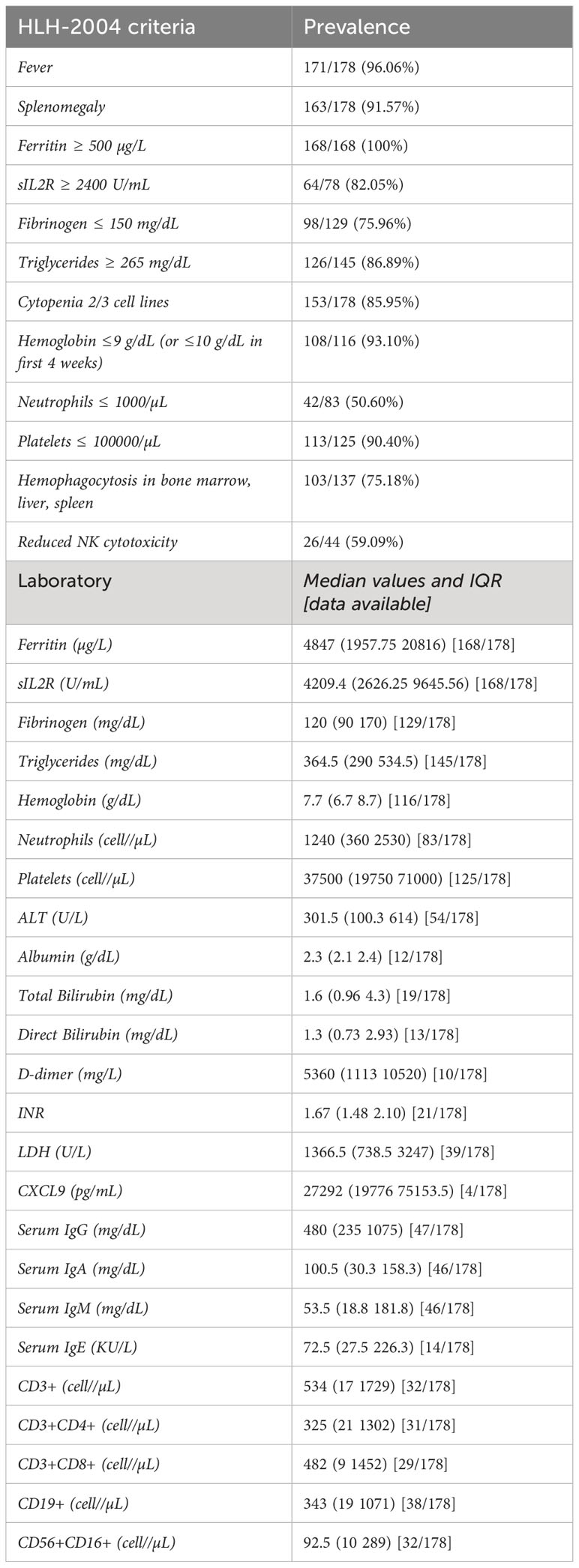
Table 1 Clinical and Laboratory data of patients with HLH and IEInotFHL.
Genetic diagnosis of IEI was available for 149/178 patients (84%) and causative mutations were reported for 115/149 patients (77%). Precise characterization of the genetic investigations has been challenging due to incomplete data and a lack of information regarding the specific timing of these analyses. For the remaining 16% of cases, the diagnosis of IEInotFHL was established without genetic analysis but relied on criteria defined by ESID (European Society for Immunodeficiencies) based on medical history, clinical presentation, and functional laboratory parameters (117). However, for other IEInotFHL, such as those related to innate immunity (12/12 patients, 100%) and autoinflammatory disorders (23/24 patients, 96%), specific diagnosis was only possible through genetic analysis. Otherwise, these conditions would have remained undiagnosed due to the absence of decisive functional tests.
Data collection identified 46 different IEI complicated by HLH at the onset or during disease course (See Supplementary Data for complete list). All IUIS 2022 IEI major groups were represented, apart from complement defects (Figure 2). Most HLH cases identified belonged to groups of IEI affecting both cellular and humoral immunity (45/178, 25%), immune dysregulation disorders (39/178, 22%), defects of phagocyte number or function (30/178, 17%), and defects of intrinsic and innate immunity (24/178, 13%). Recurrence of HLH was reported in 21/178 patients (12%), mainly in intrinsic and innate immunity defects (7/24 vs 14/154, p=0.0048). The distribution of Groups and Subgroups of IEInotFHL is shown in Figure 2, according to IUIS 2022 classification (2). (See Supplementary Data for complete list of identified IEInotFHL).
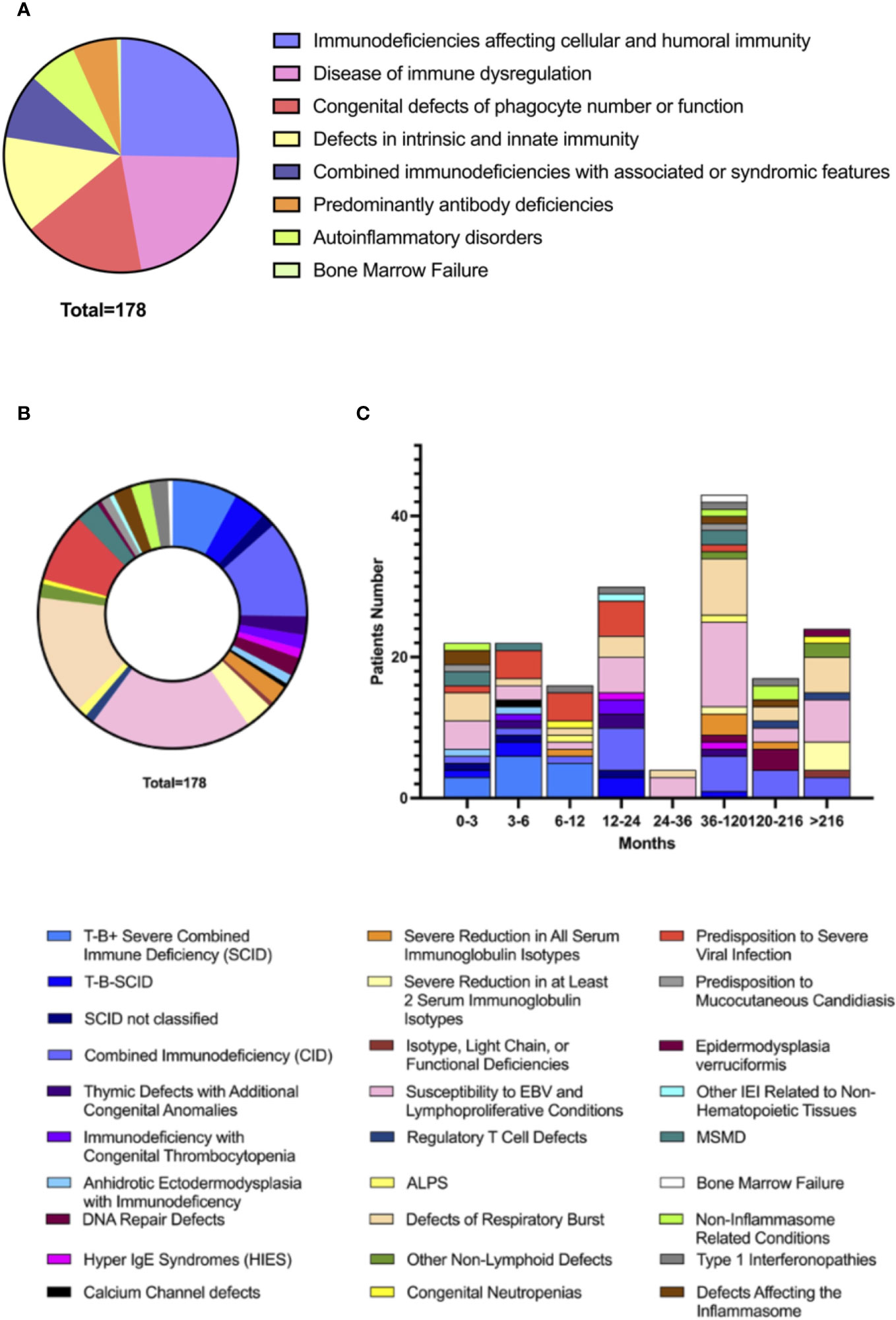
Figure 2 Distribution of HLH in Groups (A) of IEInotFHL and Subgroups (B) of IEInotFHL according with IUIS 2022 Classification of IEI and distribution of subgroup according to age (C).
HLH as first sign of IEInotFHL
The age of HLH onset in the different immune defects are detailed in Figure 2. Predominantly antibody deficiencies presented with HLH at an older age than all the other groups of IEI (mean values 207.9 ± 203.42 vs 67.64 ± 101.04 months; p=0.0039) while patients with SCID showed the earliest onset of HLH compared to all the other subgroups of IEI (mean values 8.22 ± 12.16 vs 87.04 ± 119.46 months; p<0.0001).
Temporal relation between HLH and IEI diagnosis was available for 172/178 patients (97%). In 127/172 patients (74%) HLH preceded IEI diagnosis. However, when evaluating the medical history prior to the HLH event (available for 98/172 patients; 57%), a history of infections could be identified in 38/127 patients (30%), and in 15/127 (12%) at multiple sites. The most frequent infections were pneumonia 17/127 (13%), recurrent in more than half of the cases (59%), upper respiratory tract infections 11/127 (9%), acute otitis media 7/127 (6%), chronic or recurrent sinusitis 4/127 (3%), sepsis 4/127 (3%), chronic/recurrent muco-cutaneous candidiasis 4/127 (3%), skin or visceral abscesses 3/127 (2%), severe skin infections 2/127 (2%), live strain vaccine viral infections 2/127 (2%), meningoencephalitis and osteomyelitis 1/127 each (1%).
Furthermore, other non-infectious signs or symptoms suggestive of IEI were reported prior to the first episodes of HLH. This included failure to thrive in 10/127 patients (8%) or chronic/recurrent bloody or watery diarrhea in 8/127 patients (6%). One patient was previously suspected for Inflammatory Bowel Disease (IBD) (73). Facial dysmorphisms were described in 5/127 patients (4%) while hematologic anomalies such as persistent/recurrent cytopenia or splenomegaly in 13/127 patients (10%). Additionally, a history of hypogammaglobulinemia was reported in 3/127 patients (2%), although in one case it was likely due to nephrotic syndrome (40, 96, 98). Despite available data are limited, 20/127 patients (16%) had silent clinical history before HLH (See Supplementary Data for detailed clinical data).
HLH laboratory data
Laboratory data referred to each group of IEI according to IUIS 2022 Classification are extensively detailed in the Supplement. No significant differences were found among groups of IEI neither in ferritin values, nor in triglycerides, fibrinogen, hemoglobin, and platelets values (Table 1). Conversely, patients with IEI affecting both cellular and humoral immunity had significantly lower sIL2R values than all the other IEInotFHL (mean values 2407.25 ± 1467.54 U/mL vs 8015.04 ± 7049.91 U/mL; p=0.0012). Moreover, patients with defects of intrinsic or innate immunity had significantly higher values of neutrophils (mean values 8448 ± 7818 cell/µL vs 1960± 3266cell/µL; p<0.0001) and aspartate aminotransferase (mean values 3831 ± 4855 U/L vs 1188 ± 2015 U/L; p=0.0273) than all other IEInotFHL. Additional laboratory data are listed in Table 1.
Triggers and clinical signs at onset of HLH
Infectious triggers were reported in 121/178 patients (68%). Multiple concomitant triggers were identified in 21/178 patients (12%). EBV, alone or combined with other pathogens, was found in 48/178 patients (27%), and it was the most frequent trigger of HLH in most groups of IEI (Figure 3). However, in 4 patients included in the “susceptibility to EBV and lymphoproliferative disorders” subgroup, HLH was triggered by different viruses (Parvovirus, HHV8, and CMV) (20, 61, 68). Additionally, in 12 patients within the same subgroup a trigger could not be clearly identified after EBV infection was ruled out.
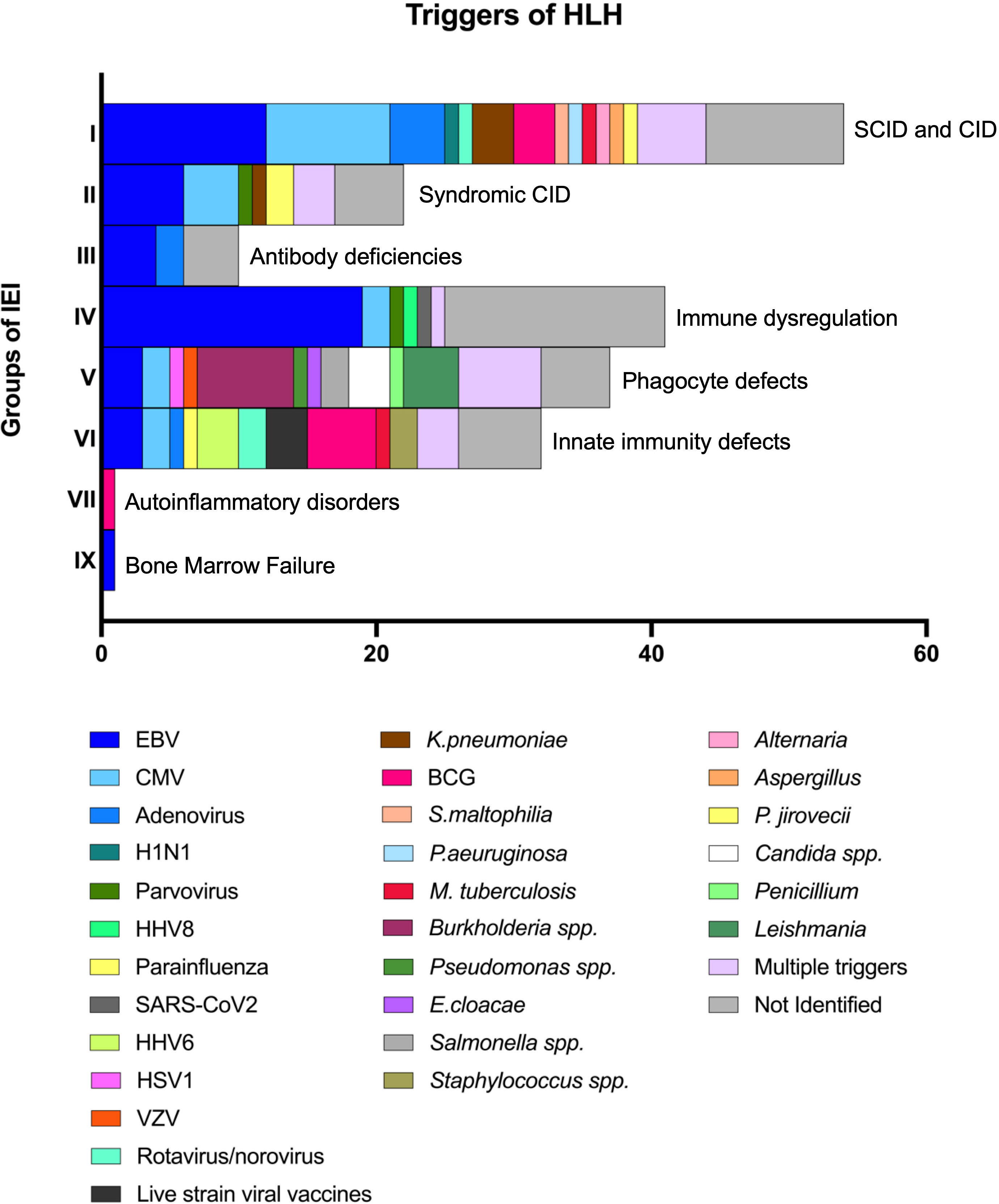
Figure 3 Triggers of HLH in patients with underlying IEInotFHL.
HLH was infrequently triggered by infections in patients with IEI classified as autoinflammatory disorders (e.g., type 1 interferonopathies or defects affecting the inflammasome) when compared to all the others (1/12 vs 120/166, p<0.0001) (25). Selective triggers were identified for specific IEI: fungal infections were more frequent in patients with defects of phagocyte number or function (5/25 vs 5/96, p=0.0167) while BCG was more commonly found in patients with intrinsic or innate immunity disorders (5/19 vs 3/102, p=0.0024). Leishmania was identified only in patients with CGD (3 X-linked and 1 AR-CGD) (4, 62) while live-strain viral vaccine were only in patients with intrinsic or innate immunity disorders (21, 53, 103).
Regardless of infectious trigger, airways were the most frequent site of infection with symptomatic presentation such as cough or dyspnea. Respiratory signs were followed by skin rash (30/178, 17%) and gastrointestinal signs (25/178, 14%). Neurologic signs as consciousness impairment, seizures, ataxia, or focal signs were reported in 21/178 patients (12%) at onset of HLH. Nevertheless, Central Nervous System (CNS) involvement was confirmed by lumbar puncture or brain Magnetic Resonance Imaging (MRI) in 5/21 patients (24%) and 7/21 patients (33%), respectively. Overall, liver involvement, inclusively considering hepatomegaly and transaminases above 100 U/L, was reported in 97/178 patients (54%), while jaundice and liver failure were reported in 5/178 (3%) (30, 38, 63, 70, 80) and 3/178 patients (2%) (45, 61, 80), respectively.
Treatment and outcome of HLH in IEInotFHL
Treatment data were available for 157/178 patients (88%) and are listed in Table 2. HLH-94/04 protocol was set up in 63/157 patients (40%), whereas 21/157 patients (13%) received corticosteroids only, which alone provided resolution in 12/21 patients (57%).
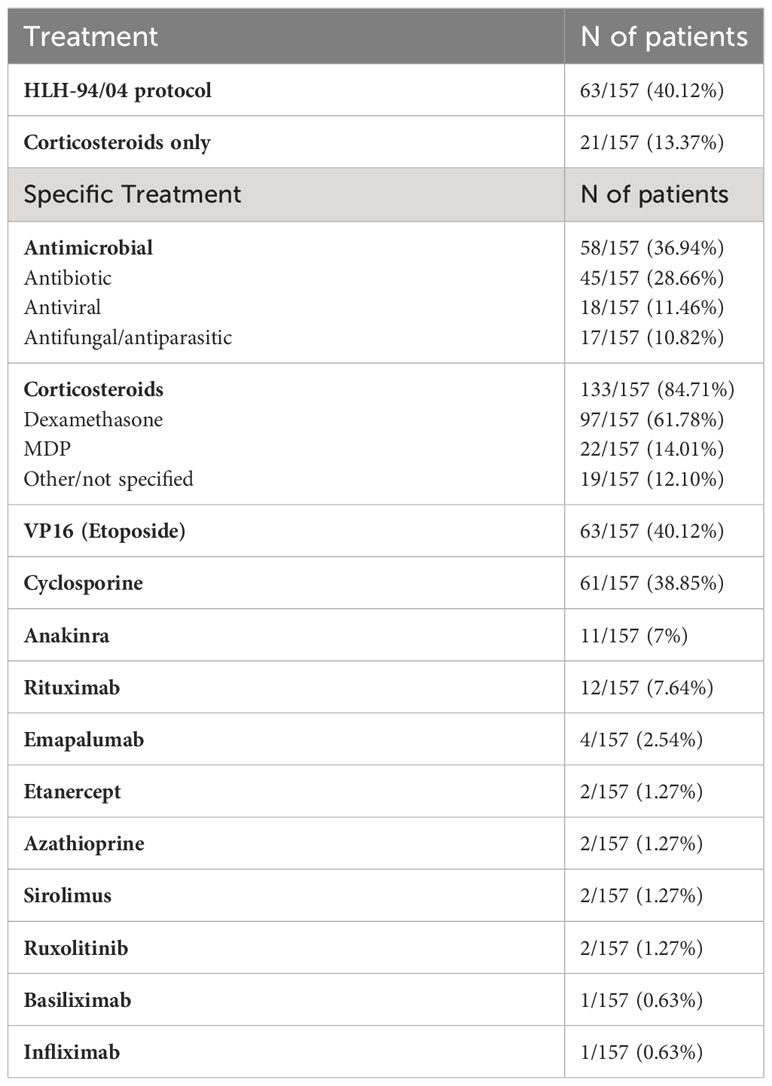
Table 2 Treatment of HLH in patients with IEInotFHL.
Biologic agents or immunosuppressive therapies, as shown in Table 2, were used in addition to complete HLH treatment protocol in 8/157 patients (5%) and used as an alternative approach to delay chemotherapy with VP16 in 22/157 patients (14%). Finally, at the time of reports submission, 32/178 patients (18%) underwent Hematopoietic Stem Cell Transplantation (HSCT). More specific details regarding transplantation could not be provided because data were incomplete. Information about type of donor and conditioning regimen were reported for only 14/32 patients (44%) and 13/32 patients (41%), respectively.
Outcome was clearly described for 171/178 patients (96%). At the time of reports submission 84/171 patients (49%) were alive. Age at death was clearly described for 43/171 patients (25%). The leading cause of death were multiorgan failure (22/47, 47%), respiratory failure (16/47, 34%), and sepsis (7/47, 15%). Outcome was significantly worse when the onset of HLH was in the first 12 months (36/57 vs 51/114 patients dead, p=0.0231). Overall mortality was significantly higher among patients with IEI affecting cellular and humoral immunity (CID and SCID) compared to all the other IEI (38/60 vs 49/111 patients dead, p=0.0166). After HSCT, 13/32 patients (41%) died (mean of 57.9 ± 95.8 days). No differences in survival outcome were found between transplanted and not transplanted patients (13/32 vs 74/139 patients dead, p=0.1982). No significative differences were also found between patients with or without CNS involvement (15/25 vs 72/146 patients dead, p=0.3234), or between patients who received etoposide and the ones who did not (35/60 vs 52/111 patients dead, p=0.1516).
Discussion
Characterization of HLH as a possible clinical manifestation of an underlying IEI is critical to properly manage this life-threatening condition. Moreover, raising awareness among pediatricians and neonatal/pediatric intensivists, who often deal with HLH at onset, is crucial to ensure rapid recognition, appropriate treatment of any underlying IEI, and improved outcomes both during HLH and, even more so, after its remission.
Chinn et al. in 2018 highlighted significant genetic diversity within patients meeting HLH criteria (118). Among 122 subjects enrolled over a 17-year period, genetic testing was conducted on 101 subjects. Notably, whole-exome sequencing (WES) analysis identified IEInotFHL in 14 cases and dysregulated immune activation or proliferation disorders in 8 cases. However, it has long been known that IEInotFHL can predispose to the development of HLH. In 2015 a large survey and literature review on patients with HLH and IEI other than FHL or XLP performed by Bode et al. found that combined immunodeficiencies (CID and SCID) and chronic granulomatous disease (CGD) were the most frequent underlying IEInotFHL (4). Similar conclusions were obtained by Cetinkaya et al. in another single-center study enrolling 28 patients with HLH, between the years 2013 and 2017, in which combined immunodeficiencies represented the most frequent IEInotFHL (5).
In accordance with previous reports (4, 5), we noticed that most cases of HLH occurred in the groups of SCID and CID, in the group of immune dysregulation disorders such as XLP-1 and XLP-2, and in CGD patients. However, all groups in the IUIS classification except complement deficiencies were found to underlie HLH development. Specifically, 46 different IEIs were identified in patients with HLH in this review.
Recognizing the methodology limitation of a systematic review, we acknowledge its non-quantitative nature, impeding the determination of individual IEInotFHL prevalence. Notably, the exclusion of numerous studies exploring the association between EBV susceptibility disorders (e.g., XLP1 or XLP2) and HLH (119–121) was guided by our specific criteria. This exclusion may have potentially led to an underestimation of the significance of each IEInotFHL, especially the well-established EBV susceptibility disorders. Indeed, Gadoury-Levesque et al. proposed that SAP and XIAP deficiency contribute to approximately 15% of genetically confirmed HLH disorders (122). This prevalence aligns with the combination of HLH susceptibility with pigmentary defects and is slightly lower than each of the three common FHL disorders, as indicated by the same study.
Nevertheless, we believe that focusing only on FHL or EBV susceptibility disorders in patients with HLH might be reductive and lead to missed diagnoses of other IEI resulting in increased numbers of complications, sequelae, or death in undiagnosed patients. Therefore, these results suggest that specific functional tests beyond flow cytometric assessment of perforin and NK degranulation activity should be included in first diagnostic steps in all patients, followed by extensive genetic analysis not limited to FHL solely. Genetic investigations, ranging from basic to more in-depth testing, were performed in over 80% of cases, being decisive especially in cohorts of IEInotFHL such as autoinflammatory disorders or innate immunity defects, wherein non-genetic testing was inconclusive. These observations underscore the pivotal role of genetics in discerning intricate immune-related conditions when suspicion is elevated, and conventional immunological assessments remain inconclusive. Additionally, in line with the suggestions of Chinn et al. (118), these findings emphasize the constraints of targeted FHL gene sequencing for the majority of HLH patients, while accentuating the potential of WES to precisely identify other IEI and pinpoint specific therapeutic approaches.
Bode et al. demonstrated that HLH was the initial presentation of IEI in 57% of patients (4). Similar conclusions were obtained by Cetinkaya et al (5). In the present review HLH episodes preceded the diagnosis of IEI in three quarters of patients, often as the first manifestation of the underlying disease. This was also true for SCID, which are usually suspected in first months of life because of severe life-threatening infections. Therefore, it can be speculated that widespread implementation of newborn screening for SCID may also serve as a preventive measure against potentially life-threatening HLH episodes triggered by infections (123–125). On the other hand, a previous history of susceptibility to recurrent or severe infections was reported in one-third of cases diagnosed with IEInotFHL after HLH, but these infections were probably not considered relevant enough to prompt a suspicion of IEI.
The application of clinical screening based on the 10 Jeffrey Modell Foundations warning signs based on infectious disease susceptibility, has greatly promoted knowledge of immunodeficiencies in the last decades. Nevertheless the International Immunology Network emphasized the importance of inflammatory and autoimmune manifestations among the warning signs to reduce the missed diagnoses of IEI (126). In this regard it could be useful to consider HLH as a possible warning sign for IEI and include it among the many outlined by the Jeffrey Modell Foundation.
The present review showed that HLH triggers usually reflect the specific susceptibility to infections of different groups of IEI. Fungal triggers indeed, were more represented among patients with underlying defects of phagocyte number and function while live strain vaccines like BCG were among patients with specific disorders of innate immunity like mendelian susceptibilities to mycobacterial disease. These data reinforce the theory that HLH is often a state of “immune frustration” due to the inability of the immune system to efficiently fight infection and achieve complete clearance or control of the pathogen resulting in a self-sustaining cycle of antigenic stimulation and hyperinflammation. The identification of specific pathogens in the context of an episode of HLH could help direct clinical suspicion to a specific underlying IEInotFHL. For instance, CGD should always be investigated after the identification of Leishmania spp., Candida spp. or Burkholderia spp. in HLH patients, even more so if they have recurrent or persistent HLH. Anyway, in accordance with the literature, viruses were the primary triggers of HLH in almost all groups of IEInotFHL (127). Other viruses were also identified as triggers in patients with EBV-related disorders, as well as EBV was the trigger even in non-EBV-related disorders. Therefore, infectious workup should be as wide as possible and not limited to the detection of EBV.
Regardless of the infectious trigger, children with IEInotFHL may develop HLH in the context of respiratory or gastrointestinal infections more frequently (25%) than those with FHL who rarely show signs of infection at the onset of HLH (128). In contrast to the inflammatory phenotype, liver and CNS involvement was reported more rarely in patients with HLH and underlying IEInotFHL than in FHL, where CNS involvement ranges from 30% to 73% of cases and implies a worse outcome, as confirmed by Amirifar et al (129, 130).
Comparison of data from this review with those in the literature found no significant differences in routine diagnostic markers of HLH compared with secondary HLH or FHL.
Regarding treatment, the data obtained from this review are uneven and not based on long follow-up, making them inconsistent for speculations on therapeutic efficacy in the different IEI groups. However, as expected in view of the high infectious risk, less than half of patients received HLH-1994 or HLH-2004 treatment with chemotherapy. This may partly be attributed to the identification of specific pathogens as trigger of HLH which prompted clinicians to immediately start targeted antimicrobial treatment and delay the application of chemotherapy. In two cases, this approach resolved HLH without additional treatments (32, 47).
Acknowledging the partial limitations of the data, it is critical to point out that the results of chemotherapy were not significantly better. While FHL is a more uniform category, HLH due to IEInotFHL requires subcategorization for tailored therapy, as it does not always involve T-cell activation. In fact, in the North American Consortium for Histiocytosis (NACHO) recommendations, Jordan et al. introduced the term “HLH disease mimics” to refer to all those conditions that, while meeting HLH criteria, would not benefit from immunosuppression (131). Based on the observations from this systematic review, some HLH due to IEInotFHL might actually meet this definition. Thus, although data should be interpreted with caution due to potential sources of bias, our suggestion is that patients with IEInonFHL may not necessarily require comprehensive treatment protocols for HLH.
Therefore, cautious evaluation and monitoring, along with individualized treatment strategies based on underlying IEInotFHL or identified triggers, remain mandatory.
In addition, pathway-specific target therapies are progressively gaining more space in the treatment of HLH at the expense of broad-spectrum etoposide-based chemotherapy, especially for patients with IEI, both because of a better understanding of individual pathogenetic defects (e.g., defects in interferon pathways) and because of greater availability of new molecules that necessarily exert a less global immunosuppressive effect.
Although the overall outcomes did not appear to be significantly influenced by HSCT, it is important to note that these findings should be interpreted with caution due to the lack of long-term follow-up data and insufficient details on conditioning regimens and prophylaxis of graft-versus-host disease (GVHD) in the selected studies. Further research and comprehensive analysis are needed to provide a more conclusive understanding of the impact of treatments and outcomes of HLH in the context of IEI.
The main limitation of this work is the retrospective design of the study which does not allow conclusions about significant differences in laboratory features for specific IEIs or treatment approaches that are often not well described.
Conclusions
HLH represents a significant and unpredictable clinical challenge for pediatricians and pediatric intensivists who often manage this clinical emergency at its onset. To the best of our knowledge this is the first systematic review about HLH and IEI other than FHL. The data presented within this study suggest that HLH could potentially emerge as a clinical hallmark across various forms of IEInotFHL, often serving as their initial recognizable indicator. Proficiency in discerning HLH indicators and, notably, uncovering the etiology of this potentially fatal condition can notably enhance patient prognoses, extending benefits even beyond HLH remission. The recognition that early detection of underlying genetic origins can reshape patient management in next future is evident. As the genetic landscape unfolds, an appealing transition to personalized approaches emerges, enriching therapeutic options and directing us toward precision interventions, ultimately leading to improved patient outcomes.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
SR: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. WS: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. LL: Writing – review & editing. CC: Writing – review & editing. FL: Writing – review & editing. DD: Writing – review & editing. MF: Writing – review & editing. LP: Writing – review & editing. ES: Supervision, Validation, Writing – review & editing. GI: Supervision, Validation, Writing – review & editing. MR: Supervision, Validation, Writing – review & editing. CA: Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported in part by funds from the ‘Current Research Annual Funding’ of the Italian Ministry of Health.
Acknowledgments
We would like to express our gratitude to all those who contributed to this article. Your support and contributions are greatly appreciated.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1282804/full#supplementary-material
Abbreviations
HLH, Hemophagocytic Lymphohistiocytosis; FHL, Familial Hemophagocytic Lymphohistiocytosis; IEI, Inborn Errors of Immunity; ESID, European Society for Immunodeficiencies.
References
1. Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Childhood. (1952) 27:519. doi: 10.1136/ADC.27.136.519
2. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2022) 42(7):1473–1507. doi: 10.1007/S10875-022-01289-3
3. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ (2021) 372:n71. doi: 10.1136/BMJ.N71
4. Bode SFN, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. (2015) 100:978–88. doi: 10.3324/HAEMATOL.2014.121608
5. Cetinkaya PG, Cagdas D, Gumruk F, Tezcan I. Hemophagocytic lymphohistiocytosis in patients with primary immunodeficiency. J Pediatr Hematology/Oncology. (2020) 42:E434–9. doi: 10.1097/MPH.0000000000001803
6. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. (2020) 135:1332–43. doi: 10.1182/BLOOD.2019000936
7. Ouzzani M, Hammady H, Fedorowicz Z, Elmagarmid A. Rayyan—a web and mobile app for systematic reviews. Systematic Rev. (2016) 5:210. doi: 10.1186/s13643-016-0384-4
8. Henter JI, Horne AC, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood cancer. (2007) 48:124–31. doi: 10.1002/PBC.21039
9. Munn Z, MClinSc SM, Lisy K, Riitano D, Tufanaru C. Methodological guidance for systematic reviews of observational epidemiological studies reporting prevalence and cumulative incidence data. Int J Evidence-Based healthcare. (2015) 13:147–53. doi: 10.1097/XEB.0000000000000054
10. Ma LL, Wang YY, Yang ZH, Huang D, Weng H, Zeng XT. Methodological quality (risk of bias) assessment tools for primary and secondary medical studies: what are they and which is better? Military Med Res. (2020) 7:7. doi: 10.1186/s40779-020-00238-8
11. Agarwal A, Sharma S, Airun M. Symptomatic primary selective IgM immunodeficiency - B lymphoid cell defect in adult man with secondary HLH syndrome. J Assoc Physicians India. (2016) 64(7):91–93.
12. Al-Hammadi S, Yahya AM, Al-Amri A, Shibli A, Balhaj GB, Tawil MI, et al. Case report: BCG-triggered hemophagocytic lymphohistiocytosis in an infant with X-linked recessive mendelian susceptibility to mycobacterial disease due to a variant of chronic granulomatous disease. Front pediatrics. (2021) 9:687538. doi: 10.3389/fped.2021.687538
13. Alawbathani S, Westenberger A, Ordonez-Herrera N, Al-Hilali M, Al Hebby H, Alabbas F, et al. Biallelic ZNFX1 variants are associated with a spectrum of immuno-hematological abnormalities. Clin Genet. (2022) 101(2):247–254. doi: 10.1111/cge.14081
14. Alsalamah M, Roifman CM. Hemophagocytic ymphohistiocytosis associated with ataxia telangiectasia. LymphoSign J. (2017) 4(3):113–116. doi: 10.14785/lymphosign-2017-0007
15. Alsalamah M, Sarpal A, Siu VM, Gibson P, Rupar CA, Barton M, et al. Hemophagocytic lymphohistiocytosis in a patient with CD3δ deficiency. LymphoSign J. (2015) 2(4):201–206. doi: 10.14785/lpsn-2015-0006
16. Arico M, Bettinelli A, Maccario R, Clementi R, Bossi G, Danesino C. Hemophagocytic lymphohistiocytosis in a patient with deletion of 22q11.2. Am J Med Genet. (1999) 87(4):329–30. doi: 10.1002/(SICI)1096-8628(19991203)87:4<329::AID-AJMG9>3.3.CO;2-D
17. Aytekin ES, Cagdas; D, Tan C, Cavdarlj B, Bilgic I, Tezcan I. Hematopoietic stem cell transplantation complicated with EBV associated hemophagocytic lymphohistiocytosis in a patient with DOCK2 deficiency. Turkish J Pediatrics. (2021) 63:1072–7. doi: 10.24953/turkjped.2021.06.016
18. Bajaj P, Clement J, Bayerl MG, Kalra N, Craig TJ, Ishmael FT. High-grade fever and pancytopenia in an adult patient with common variable immune deficiency. Allergy Asthma Proc. (2014) 35(1):78–82. doi: 10.2500/aap.2014.35.3704
19. Barsalou J, Blincoe A, Fernandez I, Dal-Soglio D, Marchitto L, Selleri S, et al. Rapamycin as an adjunctive therapy for NLRC4 associated macrophage activation syndrome. Front Immunol. (2018) 9:2162. doi: 10.3389/fimmu.2018.02162
20. Bird JA, McClain KL, Rosenblatt HM, Abramson SL, Hanson IC. Hemophagocytic lymphohistiocytosis in a patient with x-linked lymphoproliferative disease. Allergy Asthma proceedings : Off J regional state Allergy societies. (2009) 30(4):458–62. doi: 10.2500/aap.2009.30.3259
21. Boehmer DFR, Koehler LM, Magg T, Metzger P, Rohlfs M, Ahlfeld J, et al. A novel complete autosomal-recessive STAT1 LOF variant causes immunodeficiency with hemophagocytic lymphohistiocytosislike hyperinflammation. J Allergy Clin Immunology: In Practice. (2020) 8(9):3102–3111. doi: 10.1016/j.jaip.2020.06.034
22. Burak N, Jan N, Kessler J, Oei E, Patel P, Feldman S. Diagnosis of GATA2 deficiency in a young woman with hemophagocytic lymphohistiocytosis triggered by acute systemic cytomegalovirus infection. Am J Case Rep. (2021) 22:e927087. doi: 10.12659/AJCR.927087
23. Burns C, Cheung A, Stark Z, Choo S, Downie L, White S, et al. A novel presentation of homozygous loss-of-function STAT-1 mutation in an infant with hyperinflammation—A case report and review of the literature. J Allergy Clin Immunology: In Practice. (2016) 4(4):777–9. doi: 10.1016/j.jaip.2016.02.015
24. Butt FF, Mir FF, Madasu A, Humad H, Rana AN. Recurrent infection and immune dysfunction: A case of NCF-2 gene mutation with secondary hemophagocytic lymphohistiocytosis. Dubai Med J. (2022) 5(2):129–132. doi: 10.1159/000521700
25. Castro CN, Rosenzwajg M, Carapito R, Shahrooei M, Konantz M, Khan A, et al. NCKAP1L defects lead to a novel syndrome combining immunodeficiency, lymphoproliferation, and hyperinflammation. J Exp Med. (2020) 217(12):e20192275. doi: 10.1084/JEM.20192275
26. Celiksoy MH, Ozyavuz Cubuk P, Guner SN, Yildiran A. A case of ataxia-telangiectasia presented with hemophagocytic syndrome. J Pediatr Hematology/Oncology. (2018) 40(8):e547–e549. doi: 10.1097/MPH.0000000000001134
27. Cesaro S, Messina C, Sainati L, Danesino C, Arico M. Del 22Q11.2 and Hemophagocytic lymphohistiocytosis: A non-random association [3]. Am J Med Genet. (2003) 116:208–9. doi: 10.1002/ajmg.a.10122
28. Chidambaram AC, Maulik K, Ramamoorthy JG, Parameswaran N. A novel mutation of adenosine deaminase causing SCID presenting as hemophagocytic lymphohistiocytosis with acute kidney injury. Br J Haematology. (2020) 191:509–12. doi: 10.1111/bjh.17058
29. Cui T, Wang Y, Wang J, Zhang J, Gao Z, Wang Z. The role of allogeneic hematopoietic stem cell transplantation and Epstein-Barr virus infection on the treatment for child primary hemophagocytic lymphohistiocytosis patients with X-linked lymphoproliferative disease: A rare case report and family survey study. Pediatr Transplantation. (2020) 24:e13635. doi: 10.1111/petr.13635
30. De La Varga-Martinez R, Mora-Lopez F, Garcia-Cuesta D, Garrastazul-Sánchez MP, Quintero S, Rodríguez C, et al. X-linked lymphoproliferative disease type 1 in a patient with the p.Gly93Asp SH2D1A gene mutation and hemophagocytic lymphohistiocytosis. J Pediatr Hematology/Oncology. (2017) 39(8):e483–e485. doi: 10.1097/MPH.0000000000000938
31. Dvorak CC, Sandford A, Fong A, Cowan MJ, George TI, Lewis DB. Maternal T-cell engraftment associated with severe hemophagocytosis of the bone marrow in untreated X-linked severe combined immunodeficiency. J Pediatr Hematology/Oncology. (2008) 30(5):396–400. doi: 10.1097/MPH.0b013e318168e7a0
32. Eng V, Zomorodian TJ, Samant SA, Maarup TJ, Sheikh J. Signal transducer and activator of transcription 1 gain-of-function with refractory hemophagocytic lymphohistiocytosis. Ann allergy Asthma immunology : Off Publ Am Coll Allergy Asthma Immunol. (2020) 125:605–607.e1. doi: 10.1016/J.ANAI.2020.06.042
33. Escaron C, Ralph E, Bibi S, Visser J, Aricò M, Rao K, et al. Diagnosis of HLH: two siblings, two distinct genetic causes. Clin Exp Immunol. (2022) 207:205–7. doi: 10.1093/cei/uxab019
34. Gera A, Misra A, Tiwari A, Singh A, Mehndiratta S. A hungry Histiocyte, altered immunity and myriad of problems: Diagnostic challenges for Pediatric HLH. Int J Lab Hematology. (2021) 43. doi: 10.1111/ijlh.13626
35. Gothe F, Hatton CF, Truong L, Klimova Z, Kanderova V, Fejtkova M, et al. A novel case of homozygous interferon alpha/beta receptor alpha chain (IFNAR1) deficiency with hemophagocytic lymphohistiocytosis. Clin Infect Diseases. (2022) 74(1):136–139. doi: 10.1093/cid/ciaa1790
36. Greil J, Verga-Falzacappa MV, Echner NE, Behnisch W, Bandapalli OR, Pechanska P, et al. Mutating heme oxygenase-1 into a peroxidase causes a defect in bilirubin synthesis associated with microcytic anemia and severe hyperinflammation. Haematologica. (2016) 101(11):e436–e439. doi: 10.3324/haematol.2016.147090
37. Grunebaum E, Zhang J, Dadi H, Roifman CM. Haemophagocytic lymphohistiocytosis in X-linked severe combined immunodeficiency. Br J Haematology. (2000) 108:834–7. doi: 10.1046/j.1365-2141.2000.01923.x
38. Halasa NB, Whitlock JA, McCurley TL, Smith JA, Zhu Q, Ochs H, et al. Fatal hemophagocytic lymphohistiocytosis associated with epstein-barr virus infection in a patient with a novel mutation in the signaling lymphocytic activation moleculeassociated protein. Clin Infect Diseases. (2003) 37(10):e136–41. doi: 10.1086/379126
39. Han SP, Lin YF, Weng HY, Tsai SF, Fu LS. A novel BTK gene mutation in a child with atypical X-linked agammaglobulinemia and recurrent hemophagocytosis: A case report. Front Immunol. (2019) 10:1953. doi: 10.3389/fimmu.2019.01953
40. Harnisch E, Buddingh EP, Thijssen PE, Brooks AS, Driessen GJ, Kersseboom R, et al. Hematopoietic stem cell transplantation in a patient with ICF2 syndrome presenting with EBV-induced hemophagocytic lymphohystiocytosis. Transplantation. (2016) 100(7):e35–6. doi: 10.1097/TP.0000000000001210
41. Higuchi T, Izawa K, Miyamoto T, Honda Y, Nishiyama A, Shimizu M, et al. An efficient diagnosis: A patient with X-linked inhibitor of apoptosis protein (XIAP) deficiency in the setting of infantile hemophagocytic lymphohistiocytosis was diagnosed using high serum interleukin-18 combined with common laboratory parameters. Pediatr Blood Cancer. (2022) 69(8):e29606. doi: 10.1002/pbc.29606
42. Honda K, Kanegane H, Eguchi M, Kimura H, Morishima T, Masaki K, et al. Large deletion of the X-linked lymphoproliferative disease gene detected by fluorescence. Situ hybridization. Am J Hematology. (2000) 64(2):128–32. doi: 10.1002/(sici)1096-8652(200006)64:2<128::aid-ajh11>3.0.co;2-%23
43. Horneff G, Rhouma A, Weber C, Lohse P. Macrophage activation syndrome as the initial manifestation of tumour necrosis factor receptor 1-associated periodic syndrome (TRAPS). Clin Exp Rheumatol. (2013) 31(3 Suppl 77):99–102.
44. Hoshino T, Kanegane H, Doki N, Irisawa H, Sakura T, Nojima Y, et al. X-linked lymphoproliferative disease in an adult. Int J Hematology. (2005) 82(1):55–8. doi: 10.1532/IJH97.05020
45. Hugle B, Astigarraga I, Henter JI, Porwit-Macdonald A, Meindl A, Schuster V. Simultaneous manifestation of fulminant infectious mononucleosis with haemophagocytic syndrome and B-cell lymphoma in X-linked lymphoproliferative disease. Eur J Pediatrics. (2007) 166(6):589–93. doi: 10.1007/s00431-006-0290-1
46. Imashuku S, Miyagawa A, Chiyonobu T, Ishida H, Yoshihara T, Teramura T, et al. Epstein-Barr virus-associated T-lymphoproliferative disease with hemophagocytic syndrome, followed by fatal intestinal B lymphoma in a young adult female with WHIM syndrome. Ann Hematology. (2002) 81(8):470–3. doi: 10.1007/s00277-002-0489-9
47. Jain G, Kalra S, Sharma S, Kumar Vasnik G, Gupta R. Hemophagocytic lymphohistiocytosis in a child with chronic granulomatous disease: A rare complication of a rare disorder. Med journal Armed Forces India. (2022) 78:99–102. doi: 10.1016/J.MJAFI.2018.11.012
48. Jiang MY, Guo X, Sun SW, Li Q, Zhu YP. Successful allogeneic hematopoietic stem cell transplantation in a boy with X-linked inhibitor of apoptosis deficiency presenting with hemophagocytic lymphohistiocytosis: A case report. Exp Ther Med. (2016) 12(3):1341–1344. doi: 10.3892/etm.2016.3498
49. Kashiwagi Y, Kawashima H, Sato S, Ioi H, Amaha M, Takekuma K, et al. Virological and immunological characteristics of fatal virus-associated haemophagocytic syndrome (VAHS). Microbiol Immunol. (2007) 51(1):53–62. doi: 10.1111/j.1348-0421.2007.tb03890.x
50. Klemann C, Ammann S, Heizmann M, Fuchs S, Bode SF, Heeg M, et al. Hemophagocytic lymphohistiocytosis as presenting manifestation of profound combined immunodeficiency due to an ORAI1 mutation. J Allergy Clin Immunol. (2017) 140(6):1721–1724. doi: 10.1016/j.jaci.2017.05.039
51. Kuijpers TW, Baars PA, Aan De Kerk DJ, Jansen MH, Dors N, van Lier RA, et al. Common variable immunodeficiency and hemophagocytic features associated with a FAS gene mutation. J Allergy Clin Immunol. (2011) 127(6):1411–4.e2. doi: 10.1016/j.jaci.2011.01.046
52. Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. (2019) 216(12):2778–2799. doi: 10.1084/jem.20190147
53. Le Voyer T, Sakata S, Tsumura M, Khan T, Esteve-Sole A, Al-Saud BK, et al. Genetic, immunological, and clinical features of 32 patients with autosomal recessive STAT1 deficiency. J Immunol. (2021) 207(1):133–152. doi: 10.4049/jimmunol.2001451
54. Lekbua A, Ouahed J, OConnell AE, Kahn SA, Goldsmith JD, Imamura T, et al. Risk-factors associated with poor outcomes in VEO-IBD secondary to XIAP deficiency: A case report and literature review. J Pediatr Gastroenterol Nutr. (2019) 69(1):e13–e18. doi: 10.1097/MPG.0000000000002297
55. Liang J, Alfano DN, Squires JE, Riley MM, Parks WT, Kofler J, et al. Novel NLRC4 mutation causes a syndrome of perinatal autoinflammation with hemophagocytic lymphohistiocytosis, hepatosplenomegaly, fetal thrombotic vasculopathy, and congenital anemia and ascites. Pediatr Dev pathology : Off J Soc Pediatr Pathol Paediatric Pathol Society. (2017) 20:498–505. doi: 10.1177/1093526616686890
56. Liang JH, Zhu HY, Xu DM, Wang L, Wang Y, Qiao C, et al. A new SH2D1A mutation in a female adult XLP disease with hemophagocytic lymphohistiocytosis and NK-cell leukemia. Ann Hematology. (2019) 98(12):2829–2831. doi: 10.1007/s00277-019-03810-y
57. Loganathan A, Munirathnam D, Sundaram B. X-linked lymphoproliferative disease (XLP1) presenting as non-epstein barr virus (EBV) — Related hemophagocytic lymphohistiocytosis (HLH). Indian Pediatrics. (2020) 57(11):1077–1078. doi: 10.1007/s13312-020-2043-z
58. Lougaris V, Baronio M, Castagna A, Tessarin G, Rossi S, Gazzurelli L, et al. Paediatric MAS/HLH caused by a novel monoallelic activating mutation in p110δ. Clin Immunol. (2020) 219:108543. doi: 10.1016/j.clim.2020.108543
59. Maignan M, Verdant C, Bouvet GF, Van Spall M, Berthiaume Y. Undiagnosed Chronic Granulomatous Disease, Burkholderia cepacia complex Pneumonia, and Acquired Hemophagocytic Lymphohistiocytosis: A Deadly Association. Case Rep Pulmonology. (2013) 2013:874197. doi: 10.1155/2013/874197
60. Malkan UY, Gunes G, Aslan T, Etgul S, Aydin S, Buyukasik Y. Common variable immune deficiency associated hodgkins lymphoma complicated with EBV-linked hemophagocytic lymphohistiocytosis: A case report. Int J Clin Exp Med. (2015) 8(8):14203–6.
61. Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. (2010) 116:1079–82. doi: 10.1182/BLOOD-2010-01-256099
62. Martin A, Marques L, Soler-Palacin P, Caragol I, Hernandez M, Figueras C, et al. Visceral leishmaniasis associated hemophagocytic syndrome in patients with chronic granulomatous disease. Pediatr Infect Dis J. (2009) 28(8):753–4. doi: 10.1097/INF.0b013e31819c6f3a
63. Marzollo A, Conti F, Rossini L, Rivalta B, Leonardi L, Tretti C, et al. Neonatal manifestations of chronic granulomatous disease: MAS/HLH and necrotizing pneumonia as unusual phenotypes and review of the literature. J Clin Immunol. (2022) 42:299–311. doi: 10.1007/s10875-021-01159-4
64. Mischler M, Fleming GM, Shanley TP, Madden L, Levine J, Castle V, et al. Epstein-Barr virus-induced hemophagocytic lymphohistiocytosis and X-linked lymphoproliferative disease: A mimicker of sepsis in the Pediatric Intensive Care Unit. Pediatrics. (2007) 119(5):e1212–8. doi: 10.1542/peds.2006-1534
65. Ozturk C, Sutcuoglu S, Atabay B, Berdeli A. X-linked agammaglobulinemia presenting with secondary hemophagocytic syndrome: A case report. Case Rep Med. (2013) 2013:742795. doi: 10.1155/2013/742795
66. Pachlopnik Schmid JM, Junge SA, Hossle JP, Schneider EM, Roosnek E, Seger RA, et al. Transient hemophagocytosis with deficient cellular cytotoxicity, monoclonal immunoglobulin M gammopathy, increased T-cell numbers, and hypomorphic NEMO mutation. Pediatrics. (2006) 117(5):e1049–56. doi: 10.1542/peds.2005-2062
67. Parekh C, Hofstra T, Church JA, Coates TD. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr Blood cancer. (2011) 56:460–2. doi: 10.1002/PBC.22830
68. Pasic S, Cupic M, Lazarevic I. HHV-8-related hemophagocytic lymphohistiocytosis in a boy with XLP phenotype. J Pediatr Hematology/Oncology. (2012) 34(6):467–71. doi: 10.1097/MPH.0b013e3182375372
69. Pasic S, Micic D, Kuzmanovic M. Epstein-Barr virus-associated haemophagocytic lymphohistiocytosis in Wiskott-Aldrich syndrome. Acta Paediatrica Int J Paediatrics. (2003) 92(7):859–61. doi: 10.1080/08035250310003631
70. Patiroglu T, Haluk Akar H, van den Burg M, Unal E, Akyildiz BN, Tekerek NU, et al. X-linked severe combined immunodeficiency due to a novel mutation complicated with hemophagocytic lymphohistiocytosis and presented with invagination: A case report. Eur J Microbiol Immunol. (2014) 4(3):174–6. doi: 10.1556/eujmi-d-14-00019
71. Prader S, Felber M, Volkmer B, Trück J, Schwieger-Briel A, Theiler M, et al. Life-threatening primary varicella zoster virus infection with hemophagocytic lymphohistiocytosis-like disease in GATA2 haploinsufficiency accompanied by expansion of double negative T-lymphocytes. Front Immunol. (2018) 9:2766. doi: 10.3389/fimmu.2018.02766
72. Prader S, Ritz N, Baleydier F, Andre MC, Stähli N, Schmid K, et al. X-linked lymphoproliferative disease mimicking multisystem inflammatory syndrome in children—A case report. Front Pediatrics. (2021) 9:691024. doi: 10.3389/fped.2021.691024
73. Qiu KY, Liao XY, Wu RH, Huang K, Fang JP, Zhou DH. X-linked hyper-igM syndrome: A phenotype of crohns disease with hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol. (2017) 34(8):428–434. doi: 10.1080/08880018.2017.1409301
74. Razaghian A, Parvaneh L, Delkhah M, Abbasi A, Sadeghirad P, Shahrooei M, et al. Bacillus CalmetteGuerin (BCG)-associated hemophagocytic lymphohistiocytosis in the setting of IFN-γR1 deficiency: A diagnostic dilemma. eJHaem. (2020) 1(1):334–337. doi: 10.1002/jha2.5
75. Ren Y, Xiao F, Cheng F, Huang X, Li J, Wang X, et al. Whole exome sequencing reveals a novel LRBA mutation and clonal hematopoiesis in a common variable immunodeficiency patient presented with hemophagocytic lymphohistiocytosis. Exp Hematol Oncol. (2021) 10:38. doi: 10.1186/s40164-021-00229-y
76. Ricci S, Romano F, Nieddu F, Picard C, Azzari C. OL-EDA-ID syndrome: A novel hypomorphic NEMO mutation associated with a severe clinical presentation and transient HLH. J Clin Immunol. (2017) 37(1):7–11. doi: 10.1007/s10875-016-0350-x
77. Rossi-Semerano L, Hermeziu B, Fabre M, Kone-Paut I. Macrophage activation syndrome revealing familial mediterranean fever. Arthritis Care Res. (2011) 63(5):780–3. doi: 10.1002/acr.20418
78. Spergel AR, Walkovich K, Price S, Niemela JE, Wright D, Fleisher TA, et al. Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis. Pediatrics. (2013) 132(5):e1440-4. doi: 10.1542/peds.2012-2748
79. Salzer E, Daschkey S, Choo S, Gombert M, Santos-Valente E, Ginzel S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica. (2013) 98(3):473–8. doi: 10.3324/haematol.2012.068791
80. Schaballie H, Renard M, Vermylen C, Scheers I, Revencu N, Regal L, et al. Misdiagnosis as asphyxiating thoracic dystrophy and CMV-associated haemophagocytic lymphohistiocytosis in Shwachman-Diamond syndrome. Eur J Pediatrics. (2013) 172(5):613–22. doi: 10.1007/s00431-012-1908-0
81. Scheffler-Mendoza SC, Yamazaki-Nakashimada MA, Olaya-Vargas A, Morin-Contreras A, Juárez-Echenique JC, Alcántara-Ortigoza MA, et al. Successful stem cell transplantation in a child with chronic granulomatous disease associated with contiguous gene deletion syndrome and complicated by macrophage activation syndrome. Clin Immunol. (2014) 154(2):112–5. doi: 10.1016/j.clim.2014.07.004
82. Schmid I, Reiter K, Schuster F, Wintergerst U, Meilbeck R, Nicolai T, et al. Allogeneic bone marrow transplantation for active Epstein-Barr virus-related lymphoproliferative disease and hemophagocytic lymphohistiocytosis in an infant with severe combined immunodeficiency syndrome. Bone Marrow Transplantation. (2002) 29(6):519–21. doi: 10.1038/sj.bmt.1703396
83. Schultz KAP, Neglia JP, Smith AR, Ochs HD, Torgerson TR, Kumar A. Familial hemophagocytic lymphohistiocytosis in two brothers with X-linked agammaglobulinemia. Pediatr Blood Cancer. (2008) 51(2):293–5. doi: 10.1002/pbc.21573
84. Shadur B, Abuzaitoun O, NaserEddin A, Even-Or E, Zaidman I, Stepensky P. Management of XLP-1 and ITK deficiency: The challenges posed by PID with an unpredictable spectrum of disease manifestations. Clin Immunol. (2019) 198:39–45. doi: 10.1016/j.clim.2018.12.016
85. Shahin T, Mayr D, Shoeb MR, Kuehn HS, Hoeger B, Giuliani S, et al. Identification of germline monoallelic mutations in IKZF2 in patients with immune dysregulation. Blood Advances. (2022) 6(7):2444–2451. doi: 10.1182/bloodadvances.2021006367
86. Sheth J, Patel A, Shah R, Bhavsar R, Trivedi S, Sheth F. Rare cause of Hemophagocytic Lymphohistiocytosis due to mutation in PRF1 and SH2D1A genes in two children - A case report with a review. BMC Pediatrics. (2019) 19(1):73. doi: 10.1186/s12887-019-1444-4
87. Shi B, Chen M, Xia Z, Xiao S, Tang W, Qin C, et al. Hemophagocytic syndrome associated with Mycobacterium bovis in a patient with X-SCID: A case report. BMC Infect Diseases. (2020) 20(1):711. doi: 10.1186/s12879-020-05421-9
88. Seidel MG. CD27: A new player in the field of common variable immunodeficiency and EBV-associated lymphoproliferative disorder? J Allergy Clin Immunol. (2012) 129(4):1175. doi: 10.1016/j.jaci.2012.01.053
89. Sieni E, Cetica V, Piccin A, Gherlinzoni F, Sasso FC, Rabusin M, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: Clinical and genetic features of a small series. PloS One. (2012) 7(9):e44649. doi: 10.1371/journal.pone.0044649
90. Sirinavin S, Techasaensiri C, Pakakasama S, Vorachit M, Pornkul R, Wacharasin R. Hemophagocytic syndrome and Burkholderia cepacia splenic microabscesses in a child with chronic granulomatous disease. Pediatr Infect Dis J. (2004) 23(9):882–4. doi: 10.1097/01.inf.0000137565.23501.03
91. Spinner MA, Ker JP, Stoudenmire CJ, Fadare O, Mace EM, Orange JS, et al. GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. (2016) 137(2):638–40. doi: 10.1016/j.jaci.2015.07.043
92. Squire JD, Vazquez SN, Chan A, Smith ME, Chellapandian D, Vose L, et al. Case report: Secondary hemophagocytic lymphohistiocytosis with disseminated infection in chronic granulomatous disease—A serious cause of mortality. Front Immunol. (2020) 11:581475. doi: 10.3389/fimmu.2020.581475
93. Staines-Boone AT, Deswarte C, Montoya EV, Sánchez-Sánchez LM, García Campos JA, Muñiz-Ronquillo T, et al. Multifocal recurrent osteomyelitis and hemophagocytic lymphohistiocytosis in a boy with partial dominant IFN-γR1 deficiency: Case report and review of the literature. Front Pediatrics. (2017) 5:75. doi: 10.3389/fped.2017.00075
94. Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, et al. IL-2-inducible T-cell kinase deficiency: Clinical presentation and therapeutic approach. Haematologica. (2011) 96(3):472–6. doi: 10.3324/haematol.2010.033910
95. Suzuki N, Morimoto A, Ohga S, Kudo K, Ishida Y, Ishii E. Characteristics of hemophagocytic lymphohistiocytosis in neonates: A nationwide survey in Japan. J Pediatrics. (2009) 155(2):235–8.e1. doi: 10.1016/j.jpeds.2009.02.050
96. Szczawinska-Poplonyk A, Ploski R, Bernatowska E, Pac M. A novel CDC42 mutation in an 11-year old child manifesting as syndromic immunodeficiency, autoinflammation, hemophagocytic lymphohistiocytosis, and Malignancy: A case report. Front Immunol. (2020) 11:318. doi: 10.3389/fimmu.2020.00318
97. Tesi B, Sieni E, Neves C, Romano F, Cetica V, Cordeiro AI, et al. Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN-γ receptor deficiency. J Allergy Clin Immunol. (2015) 135(6):1638–41. doi: 10.1016/j.jaci.2014.11.030
98. Triebwasser MP, Barrett DM, Bassiri H, Bunin N, Elgarten C, Freedman J, et al. Combined use of emapalumab and ruxolitinib in a patient with refractory hemophagocytic lymphohistiocytosis was safe and effective. Pediatr Blood Cancer. (2021) 68(7):e29026. doi: 10.1002/pbc.29026
99. Tucci F, Gallo V, Barzaghi F, Ferrua F, Migliavacca M, Calbi V, et al. Emapalumab treatment in an ADA-SCID patient with refractory hemophagocytic lymphohistiocytosis-related graft failure and disseminated bacillus Calmette-Guerin infection. Haematologica. (2021) 106:641–6. doi: 10.3324/HAEMATOL.2020.255620
100. Uslu N, Demir H, Balta G, Saltik-Temizel IN, Ozen H, Gürakan F, et al. Hemophagocytic syndrome in a child with severe Crohns disease and familial Mediterranean fever. J Crohns Colitis. (2010) 4(3):341–4. doi: 10.1016/j.crohns.2009.12.005
101. Valentine G, Thomas TA, Nguyen T, Lai YC. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: A case report. Pediatrics. (2014) 134(6):e1727–30. doi: 10.1542/peds.2014-2175
102. Van Montfrans JM, Rudd E, Van Corput LD, Henter JI, Nikkels P, Wulffraat N, et al. Fatal hemophagocytic lymphohistiocytosis in X-linked chronic granulomatous disease associated with a perforin gene variant. Pediatr Blood cancer. (2009) 52:527–9. doi: 10.1002/PBC.21851
103. Vavassori S, Chou J, Faletti LE, Haunerdinger V, Opitz L, Joset P, et al. Multisystem inflammation and susceptibility to viral infections in human ZNFX1 deficiency. J Allergy Clin Immunol. (2021) 148(2):381–393. doi: 10.1016/j.jaci.2021.03.045
104. Vieth S, Ammann S, Schwarz K, Härtel C, Schultz C, Lehmberg K, et al. Clinical phenotype and functional analysis of a rare XIAP/BIRC4 mutation. Klinische Padiatrie. (2013) 225(6):343–6. doi: 10.1055/s-0033-1355393
105. Vignesh P, Anjani G, Kumrah R, Singh A, Mondal S, Nameirakpam J, et al. Features of hemophagocytic lymphohistiocytosis in infants with severe combined immunodeficiency: Our experience from chandigarh, North India. Front Immunol. (2022) 13:867753. doi: 10.3389/FIMMU.2022.867753
106. Voeten M, Maes P, Wojciechowski M, Vandenbossche L, Meyts I, Ceulemans B. Extremely elevated cerebrospinal fluid protein levels in a child with neurologic symptoms: Beware of haemophagocytic lymphohistiocytosis. Eur J Paediatric Neurology. (2014) 18(3):427–9. doi: 10.1016/j.ejpn.2013.11.012
107. Wegehaupt O, Groß M, Wehr C, Marks R, Schmitt-Graeff A, Uhl M, et al. TIM-3 deficiency presenting with two clonally unrelated episodes of mesenteric and subcutaneous panniculitis-like T-cell lymphoma and hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2020) 67(6):e28302. doi: 10.1002/pbc.28302
108. Wei A, Ma H, Zhang L, Li Z, Zhang Q, Wang D, et al. Hemophagocytic lymphohistiocytosis resulting from a cytokine storm triggered by septicemia in a child with chronic granuloma disease: A case report and literature review. BMC Pediatrics. (2020) 20(1):100. doi: 10.1186/s12887-020-1996-3
109. White S, Mowrer MC, Jesudas R, Keles ES, Roberts JC. Hemophagocytic lymphohistiocytosis in a patient with hyper IgE syndrome. Pediatr Blood Cancer. (2019) 66(10):e27894. doi: 10.1002/pbc.27894
110. Yang X, Hoshino A, Taga T, Kunitsu T, Ikeda Y, Yasumi T, et al. A female patient with incomplete hemophagocytic lymphohistiocytosis caused by a heterozygous XIAP mutation associated with non-random X-chromosome inactivation skewed towards the wild-type XIAP allele. J Clin Immunol. (2015) 35(3):244–8. doi: 10.1007/s10875-015-0144-6
111. Yang X, Wada T, Imadome KI, Nishida N, Mukai T, Fujiwara M, et al. Characterization of Epstein-Barr virus (EBV)-infected cells in EBV-associated hemophagocytic lymphohistiocytosis in two patients with X-linked lymphoproliferative syndrome type 1 and type 2. Herpesviridae. (2012) 3(1):1. doi: 10.1186/2042-4280-3-1
112. Yao J, Gu H, Mou W, Chen Z, Ma J, Ma H, et al. Various phenotypes of LRBA gene with compound heterozygous variation: A case series report of pediatric cytopenia patients. Int J immunopathology Pharmacol. (2022) 36:3946320221125591. doi: 10.1177/03946320221125591
113. Zhang Q, Ma H, Ma J, Wang D, Zhao Y, Wang T, et al. Clinical and genetic analysis of immunodeficiency-related diseases associated with PIK3CD mutations. Pediatr Invest. (2018) 2(4):257–262. doi: 10.1002/ped4.12101
114. Zheng F, Li J, Zha H, Zhang J, Zhang Z, Cheng F. ITK gene mutation: Effect on survival of children with severe hemophagocytic lymphohistiocytosis. Indian J Pediatrics. (2016) 83(11):1349–1352. doi: 10.1007/s12098-016-2079-1
115. Zhou S, Ma H, Gao B, Fang G, Zeng Y, Zhang Q, et al. Characterization of a novel disease-causing mutation in exon 1 of SH2D1A gene through amplicon sequencing: A case report on HLH. BMC Med Genet. (2017) 18(1):15. doi: 10.1186/s12881-017-0376-9
116. Zhou Z, Zondag T, Hermans M, van Hagen PM, van Laar JAM. Hemophagocytic lymphohistiocytosis in activated PI3K delta syndrome: An illustrative case report. J Clin Immunol. (2021) 41(7):1656–1659. doi: 10.1007/s10875-021-01080-w
117. ESID. European Society for Immunodeficiencies . Available online at: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria (Accessed March 7, 2023).
118. Chinn IK, Eckstein OS, Peckham-Gregory EC, Goldberg BR, Forbes LR, Nicholas SK, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. (2018) 132:89–100. doi: 10.1182/blood-2017-11-814244
119. Booth C, Gilmour KC, Veys P, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. (2011) 117:53–62. doi: 10.1182/blood-2010-06-284935
120. Yang L, Booth C, Speckmann C, Seidel MG, Worth AJJ, Kindle G, et al. Phenotype, genotype, treatment, and survival outcomes in patients with X-linked inhibitor of apoptosis deficiency. J Allergy Clin Immunol. (2022) 150:456–66. doi: 10.1016/j.jaci.2021.10.037
121. Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. (2011) 117:1522–9. doi: 10.1182/blood-2010-07-298372
122. Gadoury-Levesque V, Dong L, Su R, Chen J, Zhang K, Risma KA, et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. (2020) 4:2578–94. doi: 10.1182/bloodadvances.2020001605
123. Borte S, von Dobeln U, Hammarstrom L. Guidelines for newborn screening of primary immunodeficiency diseases. Curr Opin Hematol. (2013) 20:48–54. doi: 10.1097/MOH.0b013e32835a9130
124. Lodi L, Ricci S, Romano F, Ghiori F, Canessa C, Lippi F, et al. Newborn screening for PIDs using both TREC and KREC identifies late occurrence of B cells. Pediatr Allergy Immunol. (2017) 28:498–500. doi: 10.1111/pai.12733
125. Fazi C, Lodi L, Magi L, Canessa C, Giovannini M, Pelosi C, et al. Case report: Zellweger syndrome and humoral immunodeficiency: The relevance of newborn screening for primary immunodeficiency. Front Pediatr. (2022) 10:852943. doi: 10.3389/fped.2022.852943
126. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, members of the CEREDIH French PID study group. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. (2017) 140:1388–1393.e8. doi: 10.1016/J.JACI.2016.12.978
127. Brisse E, Wouters CH, Andrei G, Matthys P. How viruses contribute to the pathogenesis of hemophagocytic lymphohistiocytosis. Front Immunol. (2017) 8:1102. doi: 10.3389/fimmu.2017.01102
128. Amirifar P, Ranjouri MR, Abolhassani H, Moeini Shad T, Almasi-Hashiani A, Azizi G, et al. Clinical, immunological and genetic findings in patients with UNC13D deficiency (FHL3): A systematic review. Pediatr Allergy Immunol. (2021) 32:186–97. doi: 10.1111/PAI.13323
129. Horne AC, Wickstrom R, Jordan MB, Yeh EA, Naqvi A, Henter JI, et al. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis? Curr Treat options neurology. (2017) 19(1):3. doi: 10.1007/S11940-017-0439-4
130. Henter JI, Nennesmo I. Neuropathologic findings and neurologic symptoms in twenty-three children with hemophagocytic lymphohistiocytosis. J pediatrics. (1997) 130:358–65. doi: 10.1016/S0022-3476(97)70196-3
Keywords: hemophagocytic lymphohistiocytosis, inborn errors of immunity, macrophage activation syndrome, immune deficiency, familial hemophagocytic lymphohistiocytosis, hemophagocytic syndrome
Citation: Ricci S, Sarli WM, Lodi L, Canessa C, Lippi F, Dini D, Ferrari M, Pisano L, Sieni E, Indolfi G, Resti M and Azzari C (2024) HLH as an additional warning sign of inborn errors of immunity beyond familial-HLH in children: a systematic review. Front. Immunol. 15:1282804. doi: 10.3389/fimmu.2024.1282804
Received: 25 August 2023; Accepted: 29 January 2024;
Published: 13 February 2024.
Edited by:
Austen Worth, Great Ormond Street Hospital for Children NHS Foundation Trust, United KingdomReviewed by:
Eduardo Lopez-Granados, University Hospital La Paz, SpainSujal Ghosh, Heinrich Heine University of Dusseldorf, Germany
Copyright © 2024 Ricci, Sarli, Lodi, Canessa, Lippi, Dini, Ferrari, Pisano, Sieni, Indolfi, Resti and Azzari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter Maria Sarli, d2FsdGVybWFyaWEuc2FybGlAdW5pZmkuaXQ=
†These authors have contributed equally to this work and share first authorship