 Jiayu Zhang
Jiayu Zhang Kexuan Li
Kexuan Li Xiuyue Qiu
Xiuyue Qiu- Nursing School, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China
Objectives: Knee osteoarthritis (KOA) and certain inflammatory cytokines (such as interleukin 1 [IL-1] and tumor necrosis factor alpha [TNF-a]) are related; however, the causal relationship remains unclear. Here, we aimed to assess the causal relationship between 41 inflammatory cytokines and KOA using Mendelian randomization (MR).
Methods: Two-sample bidirectional MR was performed using genetic variation data for 41 inflammatory cytokines that were obtained from European Genome-Wide Association Study (GWAS) data (n=8293). KOA-related genetic association data were also obtained from European GWAS data (n=40,3124). Inverse variance weighting (IVW), MR, heterogeneity, sensitivity, and multiple validation analyses were performed.
Results: Granulocyte colony-stimulating factor (G-CSF) or colony-stimulating factor 3 (CSF-3) levels were negatively associated with the risk of developing KOA (OR: 0.93, 95%CI:0.89–0.99, P=0.015). Additionally, macrophage inflammatory protein-1 alpha (MIP-1A/CCL3) was a consequence of KOA (OR: 0.72, 95%CI:0.54–0.97, P=0.032). No causal relationship was evident between other inflammatory cytokines and KOA development.
Conclusion: This study suggests that certain inflammatory cytokines may be associated with KOA etiology. G-CSF exerts an upstream influence on KOA development, whereas MIP-1A (CCL-3) acts as a downstream factor.
1 Introduction
Knee osteoarthritis (KOA) is a chronic degenerative disease that profoundly affects the joints and associated tissues (1). The pathological features of KOA encompass degenerative changes in the articular cartilage, secondary osteophyte formation, subchondral bone sclerosis, and synovial inflammation (2, 3). Epidemiological surveys have revealed that the prevalence of osteoarthritis (OA) is as high as 29% in the middle-aged population (4). Furthermore, the prevalence of primary KOA is as high as 46.3% in people aged > 40 years in China (5). The number of patients with KOA increases with an aging population (6). The primary clinical manifestations of KOA include pain, limited mobility, and joint deformity (7), which can affect joint stability and may lead to disability in severe cases (8). Approximately 100 million people worldwide are disabled due to KOA. Moreover, KOA accounts for 2.2% of the global disease burden and is the fourth most disabling disease worldwide (9, 10). Beyond its physical toll, KOA further affects the mental well-being of afflicted individuals and profoundly diminishes their overall quality of life (11). Simultaneously, the economic ramifications impose a substantial burden on patients, their families, and society (12). The annual healthcare costs associated with arthritis exceed $ 300 billion (13). Given the staggering incidence and disability associated with KOA, identifying modifiable risk factors is imperative for developing strategies to control the disease and alleviate its societal burden.
Cytokines are small-molecule proteins with a wide range of biological activities that are synthesized and secreted by immune and certain non-immune cells in response to various stimuli (14). Cytokines have various functions, including immunomodulation, cell growth, and tissue repair (15). Cytokines can be categorized into interleukins, chemokines, and growth factors (16). The exact etiology and pathogenesis of KOA remain unclear. However, it is now generally accepted that inflammation is involved in the development of the disease (17), and it has been suggested that KOA is triggered by multiple complex factors mediated by inflammatory cytokines and their associated signaling pathways (18, 19). Inflammatory cytokines are closely associated with functional alterations in the synovium, cartilage, and subchondral bone and can impair the synthetic pathways required to repair the integrity of degenerating chondrocytes (20). The association between inflammation and KOA has been demonstrated in animal model studies (21). Furthermore, certain inflammatory cytokines, such as IL-1 and tumor necrosis factor (TNF-ɑ), can be detected during early lesions of KOA, a finding that suggests that inflammatory cytokines are associated with KOA (22–24). The expression levels of inflammatory cytokines in joint fluids influence KOA to some extent (25). However, relevant studies have mainly focused on the effects of IL-1 and TNF-a on KOA (26–28), and few studies regarding the association between other inflammatory cytokines and KOA exist. In addition, owing to the limitations of the traditional study design and the inability of the current study to completely exclude confounding factors, the causal relationship between inflammatory cytokines and KOA remains unclear and should be confirmed by additional studies.
Mendelian randomization (MR) uses genetic variants associated with modifiable exposures (or risk factors) to assess their possible causal relationship with outcomes (28–31). Single nucleotide polymorphisms (SNPs) are commonly used as IVs to assess potential causal associations with outcomes (32, 33). Because genetic variants are randomly assigned at the time of conception, potential sources of error are eliminated, and a clear causal chain is established (34). This minimizes potential bias due to confounding factors and reverse causation, thus increasing the reliability of the results (35). Current MR in KOA mostly focuses on MR between two diseases or influencing factors, such as OA with cardiovascular disease and osteoporosis (36, 37). However, there is a notable gap between the studies addressing the potential causal relationship between KOA and inflammatory cytokine levels. To address this gap, we aimed to assess the potential causal association between inflammatory cytokines and KOA using two-sample bidirectional MR analysis. We first extracted validated genetic IVs for 41 inflammatory cytokines from the genome-wide association study (GWAS) data and analyzed their association with KOA. The direction of causality was further explored using reverse exposure and outcomes. Our study findings potentially provide new strategies for KOA prevention.
2 Materials and methods
2.1 MR assumptions
MR was used to analyze the relationship between inflammatory cytokines and KOA. Figure 1 shows the two-sample bidirectional MR study design. There are three core assumptions of MR analysis: relevance, independence, and exclusion. (1) Relevance: the variables selected as genetic instruments are closely associated with exposures; (2) independence: genetic variation is not associated with confounding factors; and (3) exclusion restrictions: genetic variation affects outcomes only through exposures instead of other pathways (32).
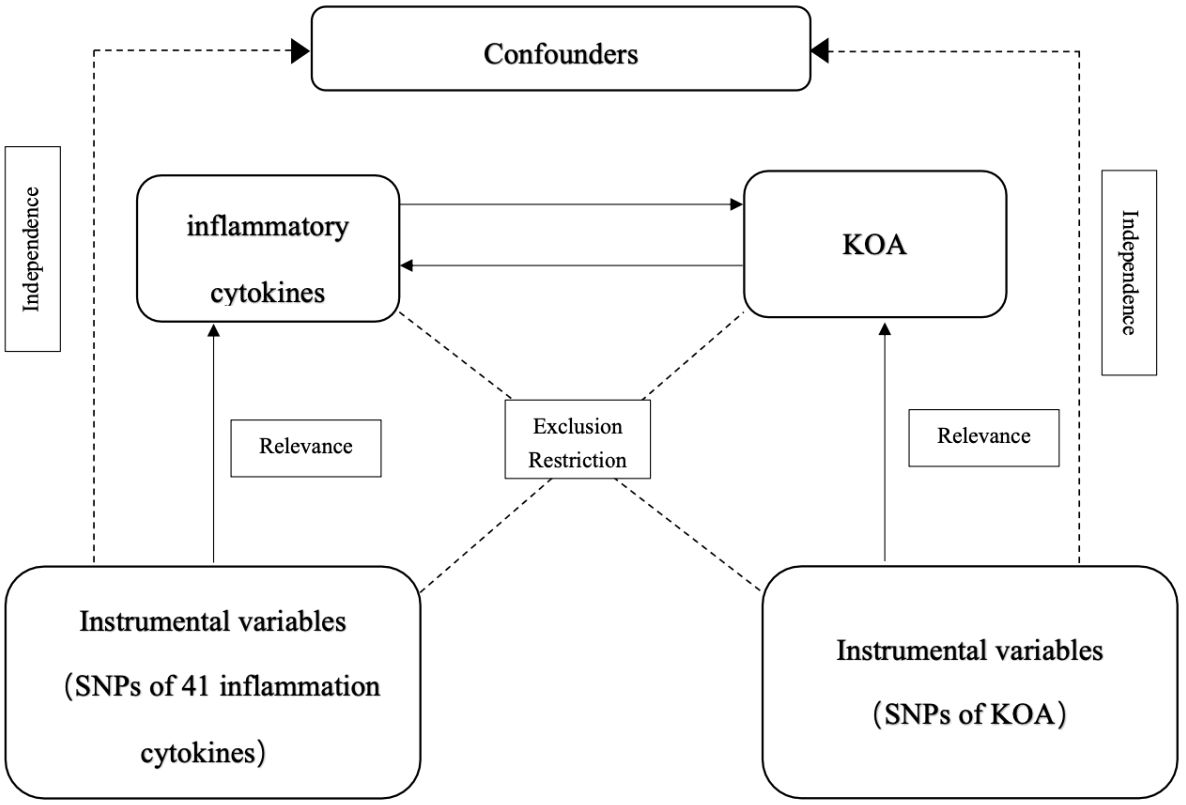
Figure 1 Schematic representation of the study design in this bidirectional Mendelian randomization (MR) analysis.
2.2 Data source
The two datasets used in the MR analysis were derived from publicly available GWAS summary data. The KOA data for this study were obtained from the UK Biobank and Arthritis Research UK Osteoarthritis Genetics (arcOGEN). This study included 24,955 patients and 378,169 controls of European ancestry (38) (https://was.mrcieu.ac.uk/datasets/ebi-a-GCST007090/). The UK Biobank recruited participants aged 40–69 between 2006–2010. ArcOGEN is a collection of patients of European ancestry with KOA. Diagnosis of KOA was based on clinical signs of the disease and the need for arthroplasty or imaging evidence (Kelgren-Lawrence≥2) to determine the diagnosis. For 41 inflammatory cytokines, data were obtained from a meta-analysis combining the results of the Finnish Young Adult Cardiovascular Risk Study (YFS) and the FINRISK survey between 1980–2011, which included 8,293 Finnish individuals (39) (https://data.bris.ac.uk/data/dataset/3g3i5smgghp0s2uvm1doflkx9x). The mean age of the participants in the YFS an FINRISK studies were 37 and 60 years, respectively.
2.3 Instrumental variable selection
First, we set P<5×10-8 as the genome-wide significance threshold to select SNPs that were strongly associated with KOA and inflammatory cytokines. Because very few SNPs were identified for some of the cytokines upon exposure, a higher cutoff value (P<5×10-6) to ensure that there were enough SNPs for further MR analysis (40, 41). Secondly, to avoid linkage disequilibrium, instrumental variables were removed to ensure mutual independence of these instrumental variables (r2 = 0.001, kb=10,000). Third, the F statistic was used to determine the IV exposure correlations. The F value of the SNP was calculated to determine the presence of a weak IV bias. If F>10, the correlation is considered strong enough to avoid weak IV bias.
2.4 Statistical analysis
In this study, we explored the potential causal relationship between inflammatory cytokines and KOA using inflammatory cytokines and KOA as exposure and outcome variables, respectively. Analysis was performed using R4.3.2 software and the R package “Two sample MR”. Causality assessments were conducted using diverse methods including inverse variance weighting (IVW), MR-Egger, weighted median, weighted modal, and simple modal analyses. The IVW method served as the primary approach for the MR analysis, and Cochran’s Q value was computed for both the IVW and MR-Egger estimates to gauge heterogeneity. Horizontal pleiotropy was evaluated using the MR-Egger intercept test (42) and anomalous SNPs were identified using MR-PRESSO (43). The stability of the results was examined using the leave-one-out method, wherein each SNP was systematically excluded to assess its impact on supporting a causal association (44). In cases where IVW analysis yielded a significant result (P<0.05) and no evidence of horizontal pleiotropy or heterogeneity was found, the result could be considered positive, even if other methods did not yield a significant result, as long as the direction of the beta remained consistent (42). In cases where horizontal pleiotropy was present but no heterogeneity was detected, the MR-Egger method was used. In cases where heterogeneity was observed, but multidirectionality was not present, the analysis was performed using the IVW method.
3 Results
To ensure an adequate number of SNPs for further MR analysis, a significance threshold of P<5×10-6 was selected when screening for SNPs related to each inflammatory cytokine and KOA. The F-statistics for the SNPs of the 41 inflammatory cytokines were 20.77–782.26. The F-statistic for all instrumental variables was >10, indicating no weak instrumental variable bias (Supplementary Tables 1-3).
To determine the primary analytical tool, hypothesis checks were performed for all 41 inflammatory cytokines. The IVW method was used as the primary analytical method for all cytokines, except IL-1RA, for which there was no evidence of heterogeneity or weak interference. For IL-1RA, MR-Egger was chosen as the primary method and the reason for this was because the P-values of the Q-tests for both IVW and MR-Egger were <0.05 (P=0.032, P=0.040).
3.1 Influence of 41 inflammatory cytokines on KOA
The main results of the MR analysis using 41 inflammatory cytokines as exposures and KOA as the outcome are shown in Tables 1 and Supplementary Table 1. The IVW results showed that genetically predicted elevated levels of Granulocyte colony-stimulating factor (G-CSF) were negatively correlated with the risk of developing KOA (OR:0.93, 95%CI:0.89–0.99, P=0.015). There was no evidence of potential horizontal pleiotropy (P=0.173) or heterogeneity (P=0.597) (Supplementary Table 2). MR-PRESSO analyses did not identify any SNP abnormalities in the G-CSF. The leave-one-out method demonstrates the stability of the results (Supplementary Figure 1). The beta values of the MR-Egger, simple model, weighted median, and weighted model analyses are consistent with the direction of the results. Figures 2A, C shows a scatter plot and funnel plot of the causal relationship between G-CSF and the occurrence of KOA. There is no evidence suggesting that other circulating inflammatory cytokines are associated with KOA.

Table 1 Causality of 41 circulating inflammatory cytokines on KOA.
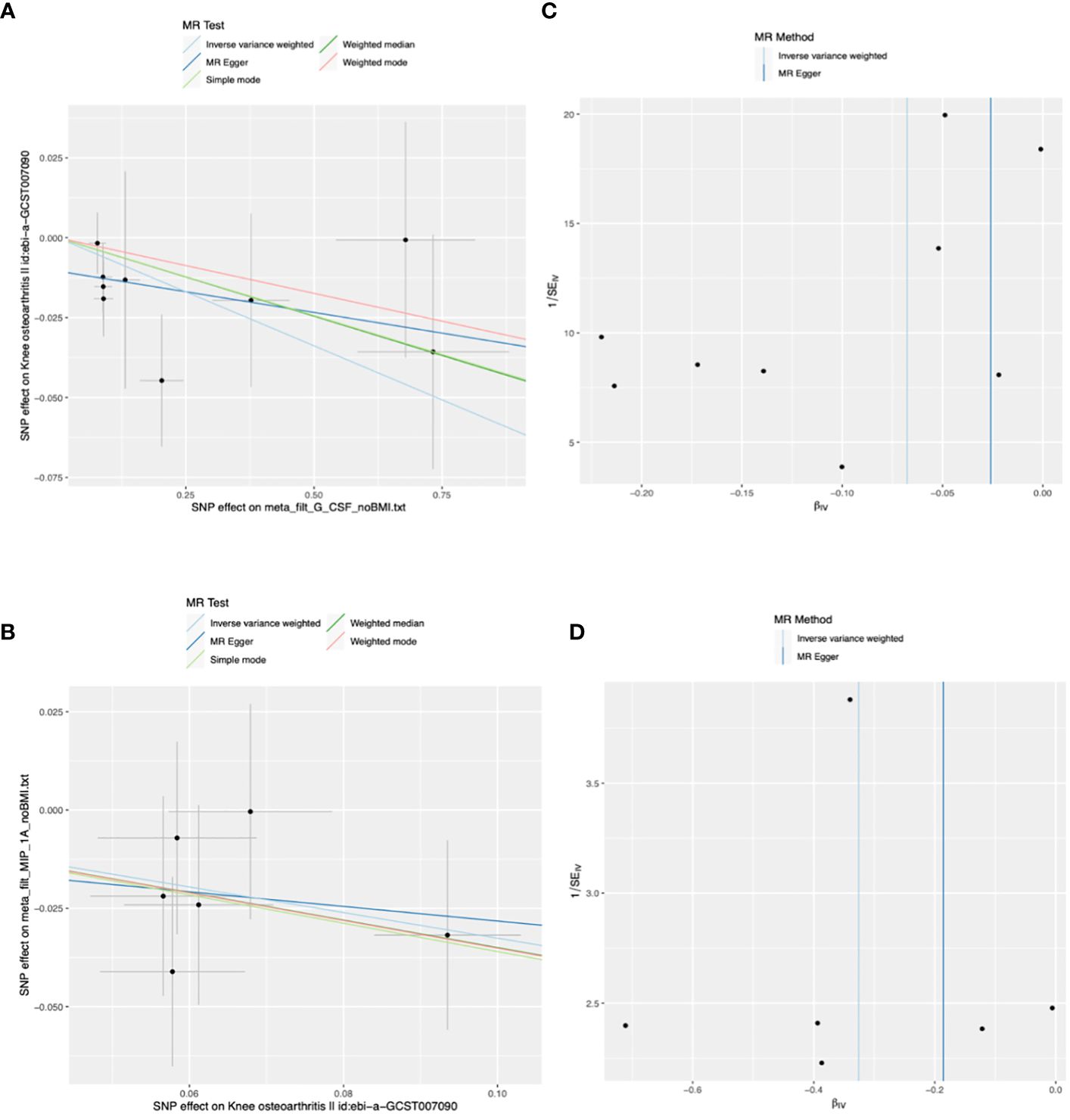
Figure 2 Scatter and funnel plots of Mendelian randomization (MR) analyses for granulocyte colony-stimulating factor (G-CSF) and macrophage inflammatory protein-1 alpha (MIP-1A) in patients with knee osteoarthritis (KOA). (A) The exposure is G-CSF and outcome is KOA. (B) The exposure is KOA and outcome is MIP-1A. (C, D) Funnel plots show the inverse variance weighted MR estimate of each cytokine single-nucleotide polymorphism (SNP) with KOA versus 1/standard error (1/SEIV).
3.2 Influence of KOA on 41 inflammatory cytokines
The main results of the MR analysis when KOA was used as the exposure and the 41 inflammatory cytokines were used as outcomes are shown in Tables 2 and Supplementary Table 4. Six significant SNPs were extracted as instrumental variables for KOA, and the F-statistics for SNPs of KOA were 32.15–96.87(Supplementary Tables 4-6). As no heterogeneity or weak instruments were observed, the IVW method was used for the primary evaluation of SNP in KOA. The IVW results showed that genetically predicted increases in KOA occurrence were negatively associated with MIP-1A levels (OR: 0.72, 95%CI:0.54–0.97, P=0.032). MR-Egger analysis did not reveal any potential horizontal pleiotropy (P=0.862) or heterogeneity (P=0.880) of SNPs for exposure factors. MR-PRESSO analyses did not reveal outliers in the SNPs for the exposure factors (Supplementary Table 5). Leave-one-out analysis demonstrated the stability of the results (Supplementary Figure 2). The directions of the beta of the MR-Egger, simple model, weighted median, and weighted model analysis results were consistent. Scatter and funnel plots of the causality of MIP-1A in KOA are shown in Figures 2B, D. There is no evidence suggesting that KOA development is associated with other circulating inflammatory cytokines.

Table 2 Causality of KOA and 41 circulating inflammatory cytokines.
4 Discussion
This study used a two-sample MR analysis to explore the potential causal relationship between inflammatory cytokines and KOA. First, we identified causal relationships between 41 inflammatory cytokines and KOA and found that lower levels of G-CSF were associated with a high risk of developing KOA, suggesting that G-CSF may be a potential upstream cause of KOA. In addition, when KOA is used as an exposure variable in MR, it may lead to lower MIP-1A levels via pathogenic pathways; therefore, MIP-1A may be located downstream of disease progression. In summary, our analysis suggests that G-CSF is involved in KOA pathogenesis. MIP-1A has a protective effect against KOA, as evidenced by the reduction in MIP-1A levels when KOA was considered an exposure factor.
Our findings suggested that reduced G-CSF levels were associated with an increased risk of KOA. G-CSF (also known as CSF-3) is a hematopoietic growth factor produced primarily by macrophages, vascular endothelial cells, fibroblasts, and bone marrow mesenchymal stromal cells (45). G-CSF is widely found in the body and the circulatory system (46) and is involved in chronic inflammatory autoimmune diseases such as rheumatoid arthritis (RA) (47). RA contributes directly to joint deterioration by mediating the release of a variety of cells, including macrophages and lymphocytes, as well as inflammatory factors: TNF-α, IL-1 and matrix metalloproteinases (48), which in turn manifests as joint deformities and dysfunction. This finding implies that G-CSF may have similar effects in patients with KOA. G-CSF primarily acts on hematopoietic progenitor cells to promote proliferation and differentiation, and stimulates granulocyte maturation and release (49–51). Reduced levels of G-CSF lead to decreased chemotaxis of granulocytes to damaged tissues (52), leading to decreased granulocyte numbers and activity (53, 54), thereby affecting immune cell function (55) and leading to reduced repair capacity of tissues, such as articular cartilage. Damage to articular cartilage and subchondral bone is an underlying pathological process of OA (56). G-CSF affects the survival and function of chondrocytes in the joint fluid (57), and reduced levels alter the state of these cells, affecting the stability of joint tissues (58), ultimately leading to KOA.
MIP-1A (also known as CCL3), belongs to the chemokine CC subfamily. MIP-1A is produced by a variety of cell types, including monocytes, fibroblasts, vascular endothelial cells, and smooth muscle cells (59), has chemotactic effects on monocytes, T cells, B cells (60, 61), and is involved in the pathogenesis of several inflammatory diseases such as RA and asthma (62–64). In patients with RA, MIP-1A triggers signaling pathways by binding to receptors on the surface of chondrocytes, leading to altered activation of intracellular molecules and inhibition of proteoglycan synthesis, ultimately affecting chondrocyte function (65, 66). In patients with RA, decreased MIP-1A expression correlates with disease severity, and there is a trend toward decreased MIP-1A expression with decreased macrophage infiltration (67). RA and KOA are both inflammatory diseases (68); therefore, it is hypothesized that MIP-1A may have a similar effect on KOA. A study on the synovial fluid also reported that MIP-1A levels decreased with OA progression (69).
Lowering MIP-1A levels may exert a protective effect against KOA through multiple pathways, including influencing immune cell activity, slowing inflammatory responses, and maintaining articular cartilage stability. First, lowering MIP-1A levels decreases immune cell activity (70). MIP-1A reduces the destructive effects of macrophages on joint tissues by attenuating their activation, thereby protecting joint structures, maintaining joint tissues (71), and slowing the progression of arthritis. Secondly, as an important chemokine (72), reduced levels of MIP-1A may lead to reduced immune cell chemotaxis in the affected joint region (73). This in turn slows the inflammatory response (74), thereby reducing inflammation and attenuating joint tissue degradation. Finally, MIP-1A affects chondrocyte function (75), leading to limited cellular synthesis of proteoglycans, which reduces proteoglycan synthesis and exacerbates articular cartilage damage (76). Therefore, a reduction in MIP-1A levels promotes proteoglycan synthesis, which reduces the risk of articular cartilage damage and protects joints.
Here, we found that MIP-1A levels were decreased in patients with KOA, whereas the study by Guo et al. (77) reported that MIP-1A levels were increased in patients with KOA, leading to a discrepancy, which may be due to the difference in the sample sizes of the studies, as well as the racial differences between the European and Asian populations. Therefore, future studies should explore the potential mechanism of the negative correlation between KOA and MIP-1A to reveal the specific role of MIP-1A in the pathogenesis of KOA, which will provide new perspectives on the treatment strategy of KOA and provides new ideas for discovering new ways to treat KOA.
The previous studies investigated the potential causal relationship between cytokines and OA, suggesting a correlation between CX3CL1, MCP4, and CCL25 and OA (78). Another study noted a causal relationship between the inflammatory cytokines MCSF and vascular endothelial growth factor (VEGF) and OA (79), but these results were not validated in our analysis. Potential explanations include differences in study focus, with previous research encompassing various types of OA, while our study specifically examined KOA. Variation in the selection of inflammatory cytokines across studies, each with distinct biological functions and expression patterns, may also contribute to differing results. Additionally, the heterogeneity of study populations could impact cytokine expression levels and patterns, influencing study outcomes. Notably, our study had a large sample size and assessed numerous cytokines. Furthermore, we identified previously unexplored causal relationships, such as the association between G-CSF and MIP-1A with KOA. These novel findings warrant further validation in future research to ascertain the potential of G-CSF and MIP-1A as biomarkers for KOA prevention and treatment.
This study has several limitations. First, our survey data were derived from two large-scale GWAS datasets, which make this study less statistically valid. However, owing to the lack of data, we were unable to perform subgroup analyses of the variables to refine the results of this study. Therefore, they must be validated in future large-sample clinical trials. Second, the GWAS data used in this study were derived from individuals of European origin, and there may be racial bias. Thus, the generalizability of our findings to other populations should be validated using local data. Third, a significance threshold with a P<5×10-6 was used in the exposed GWAS data because the number of genome-wide significant SNP at a threshold of P<5×10-8 was too small to support this study. Fourth, the scope of this study was limited to examining the correlation between the 41 inflammatory cytokines and KOA. Given the diversity of inflammatory cytokines, future studies should be extended to the latest 91 inflammatory cytokines to deepen our understanding of the causal relationships between inflammatory cytokines and KOA.
5 Conclusion
The results of this study suggest a causal relationship between inflammation and KOA. G-CSF (CSF-3) may be an upstream factor in KOA, whereas MIP-1A (CCL3) may be a downstream effect of KOA. However, further studies are required to determine whether these cytokines can be used to predict or prevent KOA.
Author contributions
JZ: Writing – original draft. KL: Writing – review & editing. XQ: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank all participants who contributed to the GWAS data used in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1362012/full#supplementary-material
References
3. Cross M, Smith E, Hoy D, Nolte S, Ackerman I, Fransen M, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. (2014) 73:1323–30. doi: 10.1136/annrheumdis-2013-204763
4. Li Y, Xie W, Xiao W, Xiao W, Dou D. Progress in osteoarthritis research by the National Natural Science Foundation of China. Bone Res. (2022) 10:41. doi: 10.1038/s41413-022-00207-y
5. The Joint Surgery Branch of the Chinese Orthopaedic Association, The Subspecialty Group of Osteoarthritis, Chinese Association of Orthopaedic Surgeons, The National Clinical Research Center for Geriatric Disorders (Xiangya Hospital), Editorial Office of Chinese Journal of Orthopaedics, Wang K, Lei G, et al. Chinese guidelines for the diagnosis and treatment of osteoarthritis (2021 edition). Chin J Orthopedics. (2021) 18):1291–314. doi: 10.3760/cma.j.cn121113-20210624-00424
6. Qu B, Ma Y, Yan M, Wu HH, Fan L, Liao DF, et al. The economic burden of fracture patients with osteoporosis in western China. Osteoporos Int. (2014) 25:1853–60. doi: 10.1007/s00198-014-2699-0
7. Zhang R, Zhang X, Zhao M. Pathogenesis and treatment of osteoarthritis of the knee. Rheum Arthritis. (2019) 8:68–72.
8. Kolasinski SL, Neogi T, Hochberg MC, Oatis C, Guyatt G, Block J, et al. 2019 American College of Rheumatology/Arthritis Foundation guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis Rheumatol. (2020) 72:220–33. doi: 10.1002/art.41142
9. Safiri S, Kolahi AA, Smith E, Hill C, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of osteoarthritis 1990-2017: a systematic analysis of the Global Burden of Disease Study 2017. Ann Rheum Dis. (2020) 79:819–28. doi: 10.1136/annrheumdis-2019-216515
10. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nat Rev Dis Primers. (2016) 2:16072. doi: 10.1038/nrdp.2016.72
11. Palazzo C, Nguyen C, Lefevre-Colau MM, Rannou F, Poiraudeau S. Risk factors and burden of osteoarthritis. Ann Phys Rehabil Med. (2016) 59:134–8. doi: 10.1016/j.rehab.2016.01.006
12. Hunter DJ, Schofield D, Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol. (2014) 10:437–41. doi: 10.1038/nrrheum.2014.44
13. Murphy LB, Cisternas MG, Pasta DJ, Helmick CG, Yelin EH. Medical expenditures and earnings losses among US adults with arthritis in 2013. Arthritis Care Res (Hoboken). (2018) 70:869–76. doi: 10.1002/acr.23425
14. Floege J, Lüscher B, Müller-Newen G. Cytokines and inflammation. Eur J Cell Biol. (2012) 91:427. doi: 10.1016/j.ejcb.2012.01.003
15. Akdis M, Burgler S, Crameri R, Eiwegger T, Fujita H, Gomez E, et al. Interleukins, from 1 to 37, and interferon-γ: receptors, functions, and roles in diseases. J Allergy Clin Immunol. (2011) 127:701–21.e1-70. doi: 10.1016/j.jaci.2010.11.050
16. Cavaillon JM. Exotoxins and endotoxins: Inducers of inflammatory cytokines. Toxicon. (2018) 149:45–53. doi: 10.1016/j.toxicon.2017.10.016
17. Shen J, Abu-Amer Y, O’Keefe RJ, McAlinden A. Inflammation and epigenetic regulation in osteoarthritis. Connect Tissue Res. (2017) 58:49–63. doi: 10.1080/03008207.2016.1208655
18. Mabey T, Honsawek S, Tanavalee A, Yuktanandana P, Wilairatana V, Poovorawan Y. Plasma and synovial fluid inflammatory cytokine profiles in primary knee osteoarthritis. Biomarkers. (2016) 21:639–44. doi: 10.3109/1354750X.2016.1171907
19. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. (2011) 7:33–42. doi: 10.1038/nrrheum.2010.196
20. Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. (2002) 39:237–46.
21. Pincus T. Clinical evidence for osteoarthritis as an inflammatory disease. Curr Rheumatol Rep. (2001) 3:524–34. doi: 10.1007/s11926-001-0068-4
22. Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. (2013) 5:77–94. doi: 10.1177/1759720X12467868
23. Attur M, Belitskaya-Lévy I, Oh C, Krasnokutsky S, Greenberg J, Samuels J, et al. Increased interleukin-1β gene expression in peripheral blood leukocytes is associated with increased pain and predicts risk for progression of symptomatic knee osteoarthritis. Arthritis Rheum. (2011) 63:1908–17. doi: 10.1002/art.v63.7
24. Cao R, Yang HL, He RD. Clinical observation on 45 cases of knee osteoarthritis treated by acupuncture. J Tradit Chin Med. (2016) 57:1133–6. doi: 10.13288/j.11-2166/r.2016.13.014
25. Huang P-C, Ma Y. Exploring the medication pattern of external treatment of knee osteoarthritis in traditional Chinese medicine. Chin J Exp Formulary. (2016) 22:195–9. doi: 10.13422/j.cnki.syfjx.2016020195
26. Wang T, He C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. (2018) 44:38–50. doi: 10.1016/j.cytogfr.2018.10.002
27. Molnar V, Matišić V, Kodvanj I, Bjelica R, Jeleč Ž, Hudetz D, et al. Cytokines and chemokines involved in osteoarthritis pathogenesis. Int J Mol Sci. (2021) 22(17):9208. doi: 10.3390/ijms22179208
28. Van Den Bosch MHJ, Van Lent P, van der Kraan PM. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthritis Cartilage. (2020) 28:532–43. doi: 10.1016/j.joca.2020.01.016
29. Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. Jama. (2021) 326:1614–21. doi: 10.1001/jama.2021.18236
30. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomization studies: a guide, glossary, and checklist for clinicians. Bmj. (2018) 362:k601. doi: 10.1136/bmj.k601
31. Richmond RC, Davey Smith G. Mendelian randomization: concepts and scope. Cold Spring Harb Perspect Med. (2022) 12. doi: 10.1101/cshperspect.a040501
32. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. Jama. (2017) 318:1925–6. doi: 10.1001/jama.2017.17219
33. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Smith GD. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. (2008) 27:1133–63. doi: 10.1002/sim.3034
34. Wang B, Liu Y, Zhang YC, Han ZY, Hou JL, Chen S, et al. Assessment of causal effects of physical activity on the risk of osteoarthritis: a two-sample Mendelian randomization study. BMC Med Genomics. (2023) 16:237. doi: 10.1186/s12920-023-01681-x
35. Burgess S, Scott RA, Timpson NJ, Smith GD, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. (2015) 30:543–52. doi: 10.1007/s10654-015-0011-z
36. Lin L, Luo P, Yang M, Wang J, Hou W, Xu P. Causal relationship between osteoporosis and osteoarthritis: A two-sample Mendelian randomized study. Front Endocrinol (Lausanne). (2022) 13:1011246. doi: 10.3389/fendo.2022.1011246
37. Wang Z, Kang C, Xu P, Zhang S, Song J, Wang D, et al. Osteoarthritis and cardiovascular disease: A Mendelian randomization study. Front Cardiovasc Med. (2022) 9:1025063. doi: 10.3389/fcvm.2022.1025063
38. Tachmazidou I, Hatzikotoulas K, Southam L, Esparza-Gordillo J, Haberland V, Zheng J, et al. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat Genet. (2019) 51:230–6. doi: 10.1038/s41588-018-0327-1
39. Ahola-Olli AV, Würtz P, Havulinna AS, Aalto K, Pitkänen N, Lehtimäki T, et al. Genome-wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet. (2017) 100:40–50. doi: 10.1016/j.ajhg.2016.11.007
40. Fang P, Liu X, Qiu Y, Wang Y, Wang D, Zhao J, et al. Exploring causal correlations between inflammatory cytokines and ankylosing spondylitis: a bidirectional mendelian-randomization study. Front Immunol. (2023) 14:1285106. doi: 10.3389/fimmu.2023.1285106
41. Guan S, Bai X, Ding J, Zhuang R. Circulating inflammatory cytokines and hypertensive disorders of pregnancy: a two-sample Mendelian randomization study. Front Immunol. (2023) 14:1297929. doi: 10.3389/fimmu.2023.1297929
42. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. (2015) 44:512–25. doi: 10.1093/ije/dyv080
43. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. (2018) 50:693–8. doi: 10.1038/s41588-018-0099-7
44. Xiang M, Wang Y, Gao Z, Wang J, Chen Q, Sun Z, et al. Exploring causal correlations between inflammatory cytokines and systemic lupus erythematosus: A Mendelian randomization. Front Immunol. (2022) 13:985729. doi: 10.3389/fimmu.2022.985729
45. Fan Z, Xing D, Dong Z, Zheng X, Gao B, Feng L, et al. The mechanism of granulocyte colony-stimulating factor-induced bone pain and its therapeutic progress. Chin J New Drugs Clinics. (2020) 39:390–3. doi: 10.14109/j.cnki.xyylc.2020.07.02
46. Adachi Y, Imagawa J, Suzuki Y, Yogo K, Fukazawa M, Kuromaru O, et al. G-CSF treatment increases side population cell infiltration after myocardial infarction in mice. J Mol Cell Cardiol. (2004) 36:707–10. doi: 10.1016/j.yjmcc.2004.03.005
47. Campbell IK, Rich MJ, Bischof RJ, Hamilton JA. The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol. (2000) 68:144–50. doi: 10.1189/jlb.68.1.144
48. Adlan AM, Panoulas VF, Smith JP, Fisher JP, Kitas GD. Association between corrected QT interval and inflammatory cytokines in rheumatoid arthritis. J Rheumatol. (2015) 42:421–8. doi: 10.3899/jrheum.140861
49. Zhang B, Lu S, Tao S, Rong J. Role of granulocyte colony-stimulating factor in inducing osteogenesis. Chin Tissue Eng Res. (2014) 18:5276–81.
50. Haas R, Murea S. The role of granulocyte colony-stimulating factor in mobilization and transplantation of peripheral blood progenitor and stem cells. Cytokines Mol Ther. (1995) 1:249–70.
51. Rahi V, Jamwal S, Kumar P. Neuroprotection through G-CSF: recent advances and future viewpoints. Pharmacol Rep. (2021) 73:372–85. doi: 10.1007/s43440-020-00201-3
52. He Z-Y, He P-Q, Ying X-X, Lin Y, Wang F. Study on the correlation between NF-κB and GM-CSF expression and breast cancer bone metastasis. J Pract Cancer. (2011) 26:250–4.
53. Roseren F, Pithioux M, Robert S, Balasse L, Guillet B, Lamy E, et al. Systemic administration of G-CSF accelerates bone regeneration and modulates mobilization of progenitor cells in a rat model of distraction osteogenesis. Int J Mol Sci. (2021) 22(7):3505. doi: 10.3390/ijms22073505
54. Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, et al. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood. (2008) 112:5193–201. doi: 10.1182/blood-2008-02-139535
55. Martin KR, Wong HL, Witko-Sarsat V, Wicks IP. G-CSF - A double edge sword in neutrophil mediated immunity. Semin Immunol. (2021) 54:101516. doi: 10.1016/j.smim.2021.101516
56. Blaney Davidson EN, Vitters EL, Mooren FM, van den Berg WB, van der Kraan PM. Connective tissue growth factor/CCN2 overexpression in mouse synovial lining results in transient fibrosis and cartilage damage. Arthritis Rheum. (2006) 54:1653–61. doi: 10.1002/art.21795
57. Tang S, Zhang R, Bai H, Shu R, Chen D, He L, et al. Endogenus chondrocytes immobilized by G-CSF in nanoporous gels enable repair of critical-size osteochondral defects. Mater Today Bio. (2024) 24:100933. doi: 10.1016/j.mtbio.2023.100933
58. Cameron ML, Fu FH, Paessler HH, Schneider M, Evans CH. Synovial fluid cytokine concentrations as possible prognostic indicators in the ACL-deficient knee. Knee Surg Sports Traumatol Arthrosc. (1994) 2:38–44. doi: 10.1007/BF01552652
59. Wu Y, Zhang Y. Progress of macrophage inflammatory protein 1α. Jiangsu Med. (2000) 10):811–3. doi: 10.19460/j.cnki.0253-3685.2000.10.034
60. Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J. (2008) 275:4448–55. doi: 10.1111/j.1742-4658.2008.06580.x
61. Sato T, Terai M, Yasuda R, Watanabe R, Berd D, Mastrangelo MJ, et al. Combination of monocyte-derived dendritic cells and activated T cells which express CD40 ligand: a new approach to cancer immunotherapy. Cancer Immunol Immunother. (2004) 53:53–61. doi: 10.1007/s00262-003-0419-2
62. Yang J, Kuang M, Wang S, Jiang Z, Wen G. Relationship between peripheral blood macrophage inflammatory protein 1α levels and plasma fibrinogen in ACS patients. J Clin Cardiovasc Dis. (2014) 30:43–5. doi: 10.13201/j.issn.1001-1439.2014.01.013
63. Feng Z, Niu Y, Wen H, Yin N, Zhang J, Pang Y, et al. Correlation between rheumatoid arthritis and macrophage inflammatory protein-1α. Hebei Med. (2011) 33:1619–22.
64. Chen M, Hou X, Yan B, Wu J. Changes in serum macrophage inflammatory protein-1α levels and its significance in children with cough variant asthma. Chin Family Med. (2009) 12:275–6.
65. Li S, Cao J, Caterson B, Hughes CE. Proteoglycan metabolism, cell death and Kashin-Beck disease. Glycoconj J. (2012) 29:241–8. doi: 10.1007/s10719-012-9421-2
66. Zhang GQ, Liu JX. An experimental animal model of Kashin-Beck disease. Ann Rheum Dis. (1989) 48:149–52. doi: 10.1136/ard.48.2.149
67. Katrib A, Smith MD, Ahern MJ, Slavotinek J, Stafford L, Cuello C, et al. Reduced chemokine and matrix metalloproteinase expression in patients with rheumatoid arthritis achieving remission. J Rheumatol. (2003) 30:10–21.
68. Chen Z, Bozec A, Ramming A, Schett G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat Rev Rheumatol. (2019) 15:9–17. doi: 10.1038/s41584-018-0109-2
69. Rübenhagen R, Schüttrumpf JP, Stürmer KM, Frosch KH. Interleukin-7 levels in synovial fluid increase with age and MMP-1 levels decrease with progression of osteoarthritis. Acta Orthop. (2012) 83:59–64. doi: 10.3109/17453674.2011.645195
70. Schaller TH, Batich KA, Suryadevara CM, Desai R, Sampson JH. Chemokines as adjuvants for immunotherapy: implications for immune activation with CCL3. Expert Rev Clin Immunol. (2017) 13:1049–60. doi: 10.1080/1744666X.2017.1384313
71. Nedunchezhiyan U, Varughese I, Sun AR, Crawford R, Prasadam I. Obesity, inflammation, and immune system in osteoarthritis. Front Immunol. (2022) 13:907750. doi: 10.3389/fimmu.2022.907750
72. Lu J, Shen H, Xiao W, Zhao L. Levels and significance of macrophage inflammatory protein-1α in synovial fluid of RA and OA patients. Chin J Modern Med. (2005) 19):2926–8.
73. González-Amaro R, Sánchez-Madrid F. [Intercellular adhesion molecules and chemotaxic factors in the pathogenesis of multiple sclerosis]. Rev Neurol. (2002) 35:985–93.
74. Ntanasis-Stathopoulos I, Fotiou D, Terpos E. CCL3 signaling in the tumor microenvironment. Adv Exp Med Biol. (2020) 1231:13–21. doi: 10.1007/978-3-030-36667-4_2
75. Pulsatelli L, Dolzani P, Piacentini A, Silvestri T, Ruggeri R, Gualtieri G, et al. Chemokine production by human chondrocytes. J Rheumatol. (1999) 26:1992–2001.
76. Borzì RM, Mazzetti I, Marcu KB, Facchini A. Chemokines in cartilage degradation. Clin Orthop Relat Res. (2004) 427 Suppl):S53–61. doi: 10.1097/01.blo.0000143805.64755.4f
77. Guo Q, Liu Z, Wang M, Guo S, Cong H, Liu L. Analysis on the expression and value of CCL2 and CCL3 in patients with osteoarthritis. Exp Mol Pathol. (2021) 118:104576. doi: 10.1016/j.yexmp.2020.104576
78. Jiang Z, Cai X, Yao X, Zhang S, Lan W, Jin Z, et al. The causal effect of cytokine cycling levels on osteoarthritis: a bidirectional Mendelian randomized study. Front Immunol. (2023) 14:1334361. doi: 10.3389/fimmu.2023.1334361
Keywords: knee osteoarthritis, inflammatory cytokines, Mendelian randomization, disease etiology, genetic variation
Citation: Zhang J, Li K and Qiu X (2024) Exploring causal correlations between inflammatory cytokines and knee osteoarthritis: a two-sample Mendelian randomization. Front. Immunol. 15:1362012. doi: 10.3389/fimmu.2024.1362012
Received: 27 December 2023; Accepted: 04 April 2024;
Published: 18 April 2024.
Edited by:
Xiaoqian Dang, The Second Affiliated Hospital of Xi’an Jiaotong University, ChinaReviewed by:
Xiangxuan Wang, Fujian Children’s Hospital (Fujian Branch of Shanghai Children’s Medical Center), ChinaChangyu Huang, Quanzhou Orthopedic-traumatological Hospital, China
Atika Dhar, National Institutes of Health (NIH), United States
Copyright © 2024 Zhang, Li and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuyue Qiu, MzE3MDg4Mjc2QHFxLmNvbQ==