 Yanjia Zhang1,2†
Yanjia Zhang1,2† Dong Li1,3*†
Dong Li1,3*†- 1Tianjin Children’s Hospital (Tianjin University Children’s Hospital), Tianjin, China
- 2Department of Medicine, Tianjin University, Tianjin, China
- 3Department of Neurology, Tianjin Children’s Hospital (Tianjin University Children’s Hospital), Tianjin, China
Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD) is a rare acquired demyelinating syndrome manifesting as optic neuritis (ON), transverse myelitis (TM), acute disseminated encephalomyelitis (ADEM), and brainstem encephalitis. The disease is characterized by serum autoantibodies targeting myelin oligodendrocyte glycoprotein (MOG), which is exclusively expressed on central nervous system (CNS) myelin and oligodendrocyte membranes. Experimental autoimmune encephalomyelitis (EAE) models have been instrumental in elucidating how these antibodies trigger complement-dependent cytotoxicity (CDC) and antibody-dependent cellular responses, leading to inflammatory demyelination. With most patients experiencing relapses and approximately 50% developing permanent disabilities, therapeutic strategies focus on reducing relapse frequency and severity. MOG-EAE models have directly informed acute treatment approaches including corticosteroids, plasma exchange (PLEX), and intravenous immunoglobulin (IVIG). Mechanistic studies in MOG-EAE models have revealed complex treatment responses and identified several translational targets, including complement inhibition, B-cell depletion strategies, and cytokine-directed therapies that are now advancing to clinical trials. Current immunosuppressive therapies include azathioprine (AZA), mycophenolate mofetil (MMF), and rituximab (RTX), with their differential efficacy in MOGAD versus MS and AQP4-NMOSD now explained by EAE model findings on distinct immunopathological mechanisms. Guided by EAE translational insights into MOGAD pathophysiology, ongoing clinical trials are evaluating novel targeted therapies including complement inhibitors, plasma cell-depleting agents, and antigen-specific tolerization approaches. These EAE-derived mechanistic insights are critical for developing personalized treatment strategies that address the unique immunopathology of this challenging condition.
1 Introduction
Myelin oligodendrocyte glycoprotein immunoglobulin G (MOG-IgG) antibody-associated disease (MOGAD) is a rare form of acquired demyelinating syndrome (ADS), distinguished by the presence of serum autoantibodies targeting myelin oligodendrocyte glycoprotein (MOG). This disorder is characterized by central nervous system (CNS) demyelination with mild axonal damage, concurrent with inflammatory cell infiltration, primarily composed of CD4+ T cells and granulocytes (1).MOGAD was not initially acknowledged as a distinct disease entity, but instead was viewed as a subtype or variant of multiple sclerosis (MS) or neuromyelitis optica spectrum disorder (NMOSD) (2, 3).This shift in perception exemplifies the progressively nuanced understanding of autoantibodies and their associated disorders within the realm of neuroimmunology.In 2011, Mader et al. published a seminal study that delineated the clinical characteristics of MOG antibody-positive patients and systematically differentiated them from MS and NMOSD patients (4). Subsequent research in this field progressed rapidly. In 2018, an international expert panel formally proposed MOGAD as a distinct disease entity, established its diagnostic criteria, and emphasized the crucial role of MOG antibody detection in diagnosis (5). The diagnostic differences, clinical aspects, and pathophysiological mechanisms of CNS demyelinating disorders are comprehensively summarized in Table 1.
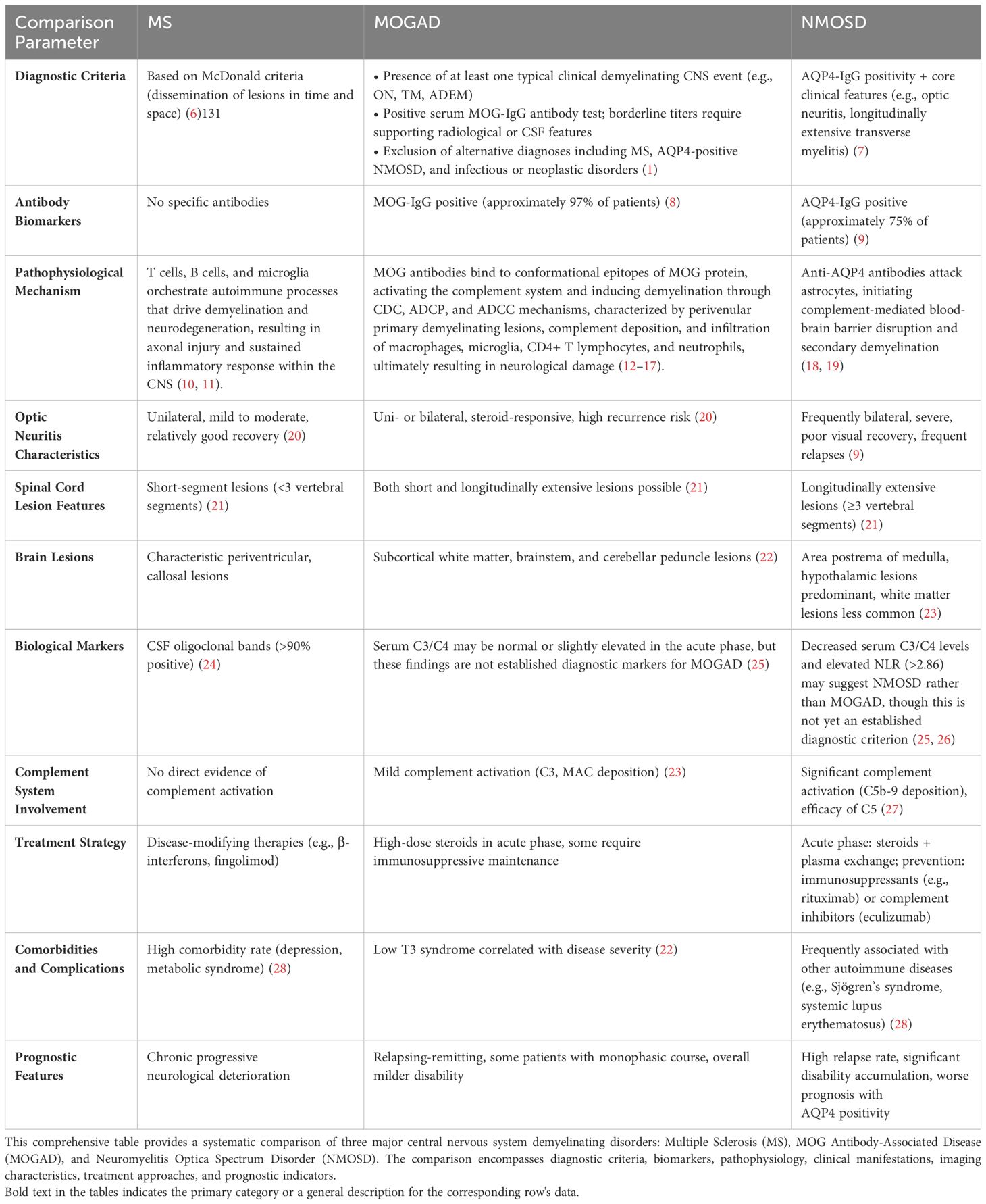
Table 1. CNS demyelinating disorders comparative analysis.
MOG protein, the target antigen of MOGAD, is predominantly expressed on the surface of oligodendrocytes in the outermost layer of central nervous system myelin (29). The molecular basis of its pathogenicity primarily stems from the β-sheet conformation of the MOG protein’s extracelluldomain, which forms a unique immunoglobulin-like structure, exposing multiple potential antigen-binding sites (30). The binding of MOG-IgG autoantibodies (predominantly of the IgG1 subtype (31)) produced by B cells to these exposed epitopes triggers a cascade of immune responses. These responses encompass: [1] complement activation leading to the formation of membrane attack complexes (4), [2] antibody-dependent cell-mediated cytotoxicity (32), and [3] T cell-mediated inflammatory responses (33). Furthermore, activated microglia and infiltrating macrophages secrete pro-inflammatory factors, exacerbating local inflammation (34). These complex pathological mechanisms collectively result in demyelination, oligodendrocyte death, and potential axonal damage (12). Crucially, the persistent inflammatory milieu and the presence of autoantibodies may impede the remyelination process, thereby impacting disease recovery (35). But the underlying pathogenic mechanisms, particularly the interplay between humoral and cellular immunity, remain incompletely understood.
MOGAD exhibits distinctive clinical, radiological, and pathological features compared to other types of ASD (36–39). Firstly, MOGAD manifests with a highly heterogeneous clinical presentation, predominantly encompassing phenotypes such as optic neuritis (ON), transverse myelitis, acute disseminated encephalomyelitis (ADEM), and brainstem encephalitis. These phenotypes can occur in isolation or in combination (40). Pediatric patients predominantly present with ADEM and optic neuritis, necessitating early recognition and prompt initiation of steroid therapy. Conversely, adult patients more frequently manifest with relapsing optic neuritis and myelitis, warranting particular vigilance regarding steroid dependence and relapse risk (41, 42). Secondly, MOGAD typically follows a relapsing-remitting disease course, which is potentially associated with fluctuations in antibody titers and dynamic changes in immune regulation (43). Notably, MOGAD patients often demonstrate a greater capacity for repair and more favorable prognosis compared to those with MS and NMOSD, potentially attributable to the distinct pathophysiological mechanisms underlying MOGAD (36).
In the absence of established MOGAD treatment guidelines, a comprehensive understanding of these mechanisms is imperative for guiding acute attack management, personalized symptom control, and long-term relapse prevention strategies. The current standard of care for acute MOGAD exacerbations involves intravenous high-dose methylprednisolone (IVMP), typically administered at 1g/day for 5 consecutive days (44). However, the risk of disease relapse increases significantly during the steroid tapering and discontinuation phase (45). For patients experiencing further clinical deterioration, plasma exchange or intravenous immunoglobulin (IVIG) may be considered as second-line therapeutic interventions (46). MOGAD therapeutic responses exhibit significant age-dependent variations. Regarding glucocorticoid sensitivity, pediatric patients respond favorably to high-dose corticosteroids during acute phases, although rapid tapering frequently precipitates relapses (41). Research indicates that children with ADEM phenotypes demonstrate steroid sensitivity, yet approximately 20% experience relapse following dose reduction (47). In contrast, while adult patients similarly exhibit marked responses to steroids during acute episodes, they face higher relapse risks, which may be independent of steroid tapering velocity (48). Some adult patients require adjunctive plasma exchange (PLEX) or intravenous immunoglobulin (IVIG) to manage severe exacerbations (49). Age-related distinctions also manifest in maintenance therapy selection, with pediatric patients preferentially receiving IVIG as long-term immunomodulatory treatment due to its superior efficacy compared to other immunosuppressants (such as azathioprine and mycophenolate mofetil) and its more manageable side effect profile (48). Adult patients, however, typically opt for rituximab (B-cell depleting agent) or alternative immunosuppressants, though their relapse prevention efficacy is less pronounced than in NMOSD, necessitating recognition that rituximab offers limited relapse control in certain adult patients (50).
The optimal maintenance therapy for MOGAD patients remains a subject of debate in the medical community. Frequently employed pharmacological interventions include oral corticosteroids, azathioprine (AZA), mycophenolate mofetil (MMF), and B cell-targeted biologics such as rituximab (RTX) and tocilizumab (TCZ). Studies have demonstrated that these agents can significantly mitigate the risk of disease recurrence and improve the annualized relapse rate (ARR) (51, 52).
Regarding relapse risk and treatment duration, approximately 20-34% of pediatric patients progress to relapsing disease courses, particularly when MOG antibodies persist. Oral corticosteroids administered for at least three months (≥0.16 mg/kg/day) reduce relapse risk by 88% (53). Adult patients exhibit substantially higher relapse rates (40-80%), especially within six months following initial presentation, thus requiring more aggressive maintenance therapy. However, standardized protocols remain elusive, and some patients may necessitate extended immunosuppression for several years (42).
Complementing pharmacological approaches, non-pharmacological interventions such as long-term functional rehabilitation, regular MOG antibody serological monitoring, and longitudinal MRI surveillance are equally crucial for pediatric MOGAD patients (54).Furthermore, in pediatric patients, MOG antibody titers typically exceed those observed in adults, potentially indicating more robust immune responses and necessitating more cautious tapering strategies (41). From an age-stratified treatment perspective, pediatric patients often benefit from IVIG and gradual steroid tapering to minimize long-term adverse effects (such as growth suppression and metabolic dysregulation) (48, 50). Adult patients require careful balancing of relapse control against medication toxicity, as rituximab may increase infection susceptibility, thus demanding individualized selection (48).
Experimental autoimmune encephalomyelitis (EAE) has long served as a pivotal animal model in neuroimmunological research, providing fundamental insights into the pathogenesis of various demyelinating disorders (55). The evolution of MOG-specific EAE models, particularly those incorporating human MOG-specific T cells and antibodies, has created unprecedented opportunities for investigating MOGAD-specific immunopathological mechanisms (56). Elucidating the parallels between EAE and MOGAD is crucial for unraveling the underlying pathogenic mechanisms and subsequently developing more effective and targeted therapeutic strategies.
This comprehensive review aims to address critical knowledge gaps by synthesizing experimental and clinical evidence across multiple mechanistic and therapeutic domains. Our primary objectives are threefold: First, to provide a detailed analysis of MOGAD pathogenic mechanisms as elucidated through the lens of EAE models, emphasizing novel insights into disease initiation and progression. Second, to critically evaluate current therapeutic strategies in light of experimental evidence, identifying mechanisms of action and potential areas for optimization. Third, to explore emerging therapeutic approaches based on recent mechanistic discoveries, with particular emphasis on targeted interventions that may offer improved efficacy and safety profiles for diverse patient populations.
In the context of rapidly evolving MOGAD research, this narrative review seeks to provide an integrated perspective on disease mechanisms and therapeutic approaches, bridging preclinical insights with clinical applications. We systematically analyze current mainstream therapeutic strategies and comprehensively summarize ongoing clinical trials, thereby offering valuable insights for both evidence-based clinical practice and translational research directions.
In conclusion, MOGAD’s age-dependent characteristics significantly influence treatment strategies: pediatric patients require focus on achieving complete remission of predominantly monophasic disease courses and minimizing steroid tapering risks, while adult patients necessitate reinforcement of long-term management for more frequently relapsing disease. Given the ongoing advancement of MOGAD-related research, this article provides a critical overview of current treatment strategies, anticipating that additional high-quality clinical studies, particularly randomized controlled trials, will furnish stronger evidence-based guidance for disease management.
2 Pathophysiology of MOGAD
Research on MOG protein has predominantly focused on its role as an autoantigen in EAE and MS (57, 58). However, MOG protein is now recognized as the principal target antigen in MOGAD (5). MOG protein is a crucial encephalitogenic protein, with expression confined to the outermost layer of CNS myelin and the plasma membrane of oligodendrocytes (59). Its extracellular domain exhibits high CNS specificity and can elicit both cellular and humoral immune responses (60, 61). In humans, MOG antibodies (MOG-Ab) exert pathogenicity by recognizing conformational epitopes of the MOG protein and forming bivalent interactions with its extracellular domain (13). This process can activate the complement system, leading to demyelination through complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cellular cytotoxicity (ADCC) mechanisms (14). Notably, while all MOG-IgG subclasses can induce ADCP, the MOG-IgG1 and MOG-IgG3 subclass autoantibodies are particularly potent in inducing CDC (14). Moreover, MOG protein itself can directly activate the classical pathway of the complement system by binding to complement components C1q and C3d, functioning as adhesion molecules, signaling molecules, or activators of the complement cascade (62). This process further amplifies the demyelinating effect.
The current understanding of MOGAD pathogenesis is largely derived from EAE (63, 64), and Table 2 provides a detailed overview of the pathogenic mechanisms and therapeutic insights gained from these models. Additionally, neuropathological and clinical studies have provided corroborating evidence. The CNS pathology in MOGAD patients exhibits complex histological features. Lesions are primarily characterized by confluent primary demyelination surrounding small and medium-sized veins, accompanied by relative axonal preservation in both white and cortical matter, and reactive gliosis. Furthermore, significant complement deposition, along with infiltration and activation of macrophages and microglia, has been observed (12).The inflammatory infiltrate predominantly comprises CD4+ T lymphocytes and neutrophils, reflecting the immune-mediated nature of MOGAD (15). While MOGAD patients exhibit demyelination, the damage to astrocytes and oligodendrocytes is comparatively mild (15). This characteristic suggests fundamental differences in the pathological mechanisms between MOGAD and NMOSD. Moreover, clinical observations have revealed significant peripheral immune activation in MOGAD patients, contrasted with relatively less chronic inflammation within the CNS (12). This feature stands in stark contrast to multiple sclerosis (MS). These findings provide a theoretical foundation for treating MOGAD with plasma exchange (PLEX) or specific immunosuppressants (75), while also paving the way for novel research into targeted therapeutic strategies against specific inflammatory mediators or immune cell subpopulations.
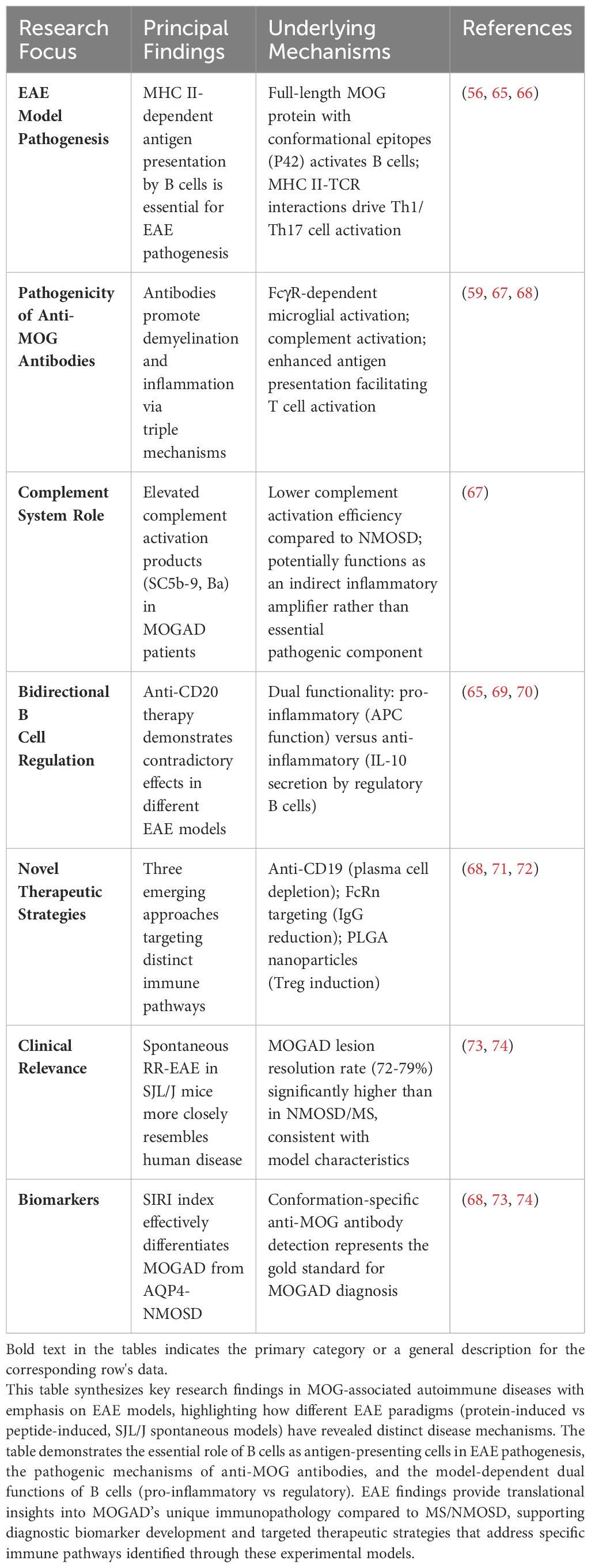
Table 2. Pathogenic mechanisms and therapeutic implications from experimental autoimmune encephalomyelitis models in MOG-associated autoimmune disease.
The EAE model provides a fundamental basis for understanding the immunopathological mechanisms of MOGAD; however, the clinical heterogeneity of MOGAD far exceeds the singular pathological manifestations observed in animal models. EAE is an autoimmune disease animal model induced by immunization with MOG protein, widely used in studying the pathological mechanisms of multiple sclerosis (MS). In comparing EAE with MOGAD, certain similarities exist, such as MOG serving as the target antigen in the EAE model, which aligns with the autoantibody target in MOGAD patients, suggesting that both conditions may share partial immune pathogenic mechanisms (76). Nevertheless, significant differences exist in their pathological mechanisms: MOGAD patients exhibit marked activation of the complement system (e.g., elevated C3 and C4 levels), whereas T cell-mediated inflammation predominates in the EAE model (77). Additionally, MOGAD patients demonstrate a significantly lower positivity rate of oligoclonal bands (OCB) in cerebrospinal fluid compared to MS patients (16.7% versus 94.2%), which differs markedly from the presentation in MS and its EAE model (77). Additionally, MOGAD patients demonstrate a significantly lower positivity rate of oligoclonal bands (OCB) in cerebrospinal fluid compared to MS patients (16.7% versus 94.2%), which differs markedly from the presentation in MS and its EAE model (78, 79). These differences indicate that although the EAE model helps us understand certain pathological aspects of MOGAD, more comprehensive research models are needed to elucidate the disease’s specific immune characteristics and clinical diversity. Future research should further explore the molecular-level similarities and differences between MOGAD and EAE to deepen our understanding of this disease’s heterogeneity.
The 2023 MOGAD expert consensus guidelines emphasize the critical importance of MOG-IgG1 testing in patients presenting with compatible clinical phenotypes (1). MOG-IgG titers exhibit significant correlation with patients’ clinical manifestations and relapse risk. Clinical studies demonstrate that patients with elevated MOG-IgG titers tend to present with more severe clinical symptoms and may exhibit more extensive CNS involvement, including concurrent optic nerve and spinal cord engagement (79). Furthermore, persistently elevated MOG-IgG titers are associated with a less favorable long-term prognosis, characterized by an increased risk of disability and higher frequency of disease relapses (80).
Studies have demonstrated that MOGAD patients exhibit a higher probability of intrathecal MOG-IgG presence compared to other ADS (81). This phenomenon may be attributed to several factors: Firstly, MOGAD patients may exhibit more pronounced blood-brain barrier (BBB) disruption, facilitating antibody penetration into the CNS (17); Secondly, defects in the blood-spinal cord barrier at the central nervous system-peripheral nervous system (CNS-PNS) transition zones may contribute (82); Lastly, intrathecal synthesis (ITS) of MOG-IgG may also be a contributing factor (83). Clinical observations reveal that the degree of disability, cerebrospinal fluid (CSF) leukocyte count, and protein levels in MOGAD patients correlate with CSF MOG-IgG titers, but not significantly with serum MOG-IgG titers (81) Furthermore, studies indicate that patients with MOG-IgG ITS tend to exhibit more severe clinical courses, characterized by more pronounced pyramidal tract involvement and spinal cord lesions, with longitudinally extensive transverse myelitis being particularly prominent (83). The complex immunopathogenesis of MOGAD, leading to oligodendrocyte injury, is schematically depicted in Figure 1, setting the stage for a detailed examination of the contributions from B cells and plasma cells (Section 2.1), T cells (Section 2.2), and innate immunity (Section 2.3).
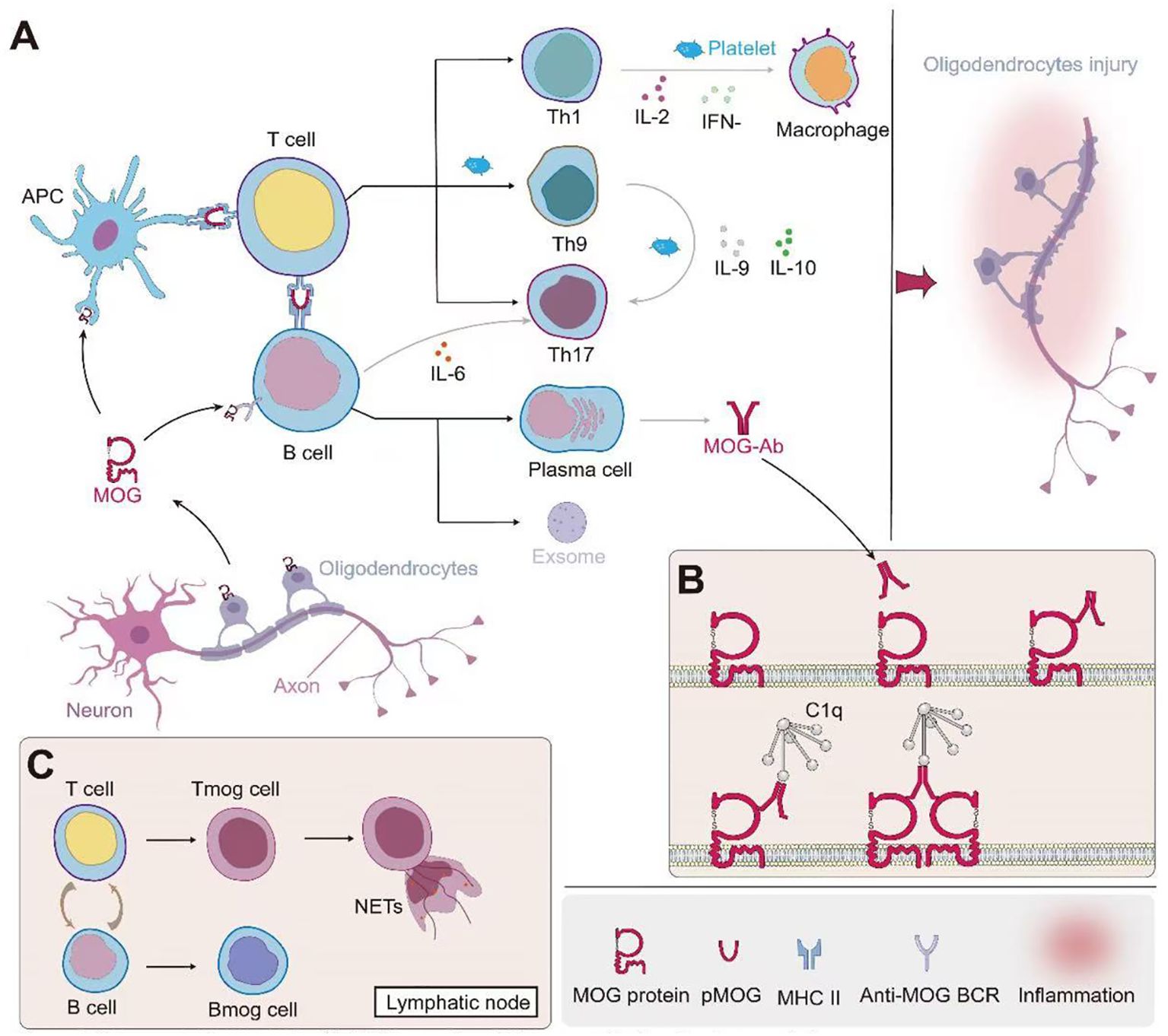
Figure 1. Immunopathogenesis of MOG-associated disease and oligodendrocyte injury. (A) Initiation and propagation of the immune response: MOG from oligodendrocytes is presented by antigen-presenting cells (APCs) to T cells. This leads to the differentiation of various T helper cell subsets (Th1, Th9, Th17) and the activation of B cells. Th1 cells produce IL-2 and IFN-γ, activating macrophages. Th9 and Th17 cells secrete IL-9 and IL-6 respectively, further modulating the immune response. B cells differentiate into plasma cells, producing MOG-specific antibodies (MOG-Ab). Platelets contribute to the inflammatory process. (B) Mechanism of MOG-Ab-mediated oligodendrocyte damage: MOG-Abs bind to MOG on the oligodendrocyte surface. This binding can lead to complement activation through C1q, resulting in the formation of the membrane attack complex (MAC) and subsequent cell lysis. Additionally, MOG-Ab binding may cause crosslinking of MOG proteins, potentially disrupting oligodendrocyte function. (C) Role of lymphoid organs: In lymph nodes, further interactions between T cells and B cells occur. This includes the formation of MOG-specific T cells (Tmog) and B cells (Bmog). Some activated T cells may form neutrophil extracellular traps (NETs), contributing to tissue damage. This cascade of events ultimately leads to oligodendrocyte injury, as depicted on the right side of panel A, potentially resulting in demyelination and axonal damage. The interplay between innate and adaptive immune responses, involving both cellular and humoral immunity, underscores the complexity of MOG-associated autoimmune processes.
2.1 The role of B cells and plasma cells, in MOGAD
B cells and their terminally differentiated form, plasma cells, play a pivotal role in the pathogenesis of MOGAD through the production of MOG-Ab. Analogous to NMOSD, MOG-Ab are predominantly produced by plasmablasts and plasma cells in peripheral tissues (84). Studies have demonstrated that the inflammatory infiltrating B cell population primarily comprises B cells expressing CD79a and CD20, with a subset also expressing CD19 and the activation marker CD27 (85). Within the lesions, approximately 10% of lymphocytes are identified as CD38-expressing plasmablasts and CD138-expressing plasma cells. Of these, more than 90% contain IgG antibodies, potentially mediating disease progression (85). Besides MOG protein, B cell activation and function may be modulated by additional antigens, including butyrophilin (86) and erythrocyte membrane-associated proteins (87, 88).
The role of B cells in MOGAD pathogenesis extends beyond MOG-Ab production, encompassing several other crucial aspects. In MOG protein-induced EAE, B cells can function as APCs (66). The binding of B cell receptors (BCRs) to specific conformational epitopes of MOG protein, primarily involving proline 42, histidine 103, and serine 104 in the CC′ loop (31), triggers a cascade of biological effects: Firstly, it induces natural killer cell-mediated cytotoxicity; Secondly, it activates mitogen-activated protein kinase (MAPK) and protein kinase B (AKT) signaling pathways; Thirdly, it elevates intracellular calcium levels, leading to the activation of stress-related pathways; Lastly, these changes may compromise cellular cytoskeletal integrity (89). Additionally, B cells promote the proliferation of T helper 17 (Th17) cells through interleukin-6 (IL-6) production, thereby exacerbating MOG protein-induced EAE.
MOG-specific B cells (BMOG) exhibit dual functionality: Firstly, they are capable of presenting MOG antigens to MOG-specific T cells (TMOG); Secondly, they interact with TMOG in draining lymph nodes, effectively facilitating the production of autoantibodies (90). Studies demonstrate that BMOG exhibit significantly higher antigen presentation efficiency compared to conventional APCs, approximately 10,000-fold greater (91). However, no correlation has been observed between circulating BMOG and serum anti-MOG-Ab levels (92).
In vitro studies demonstrate that specific MOG-Ab significantly reduce transendothelial electrical resistance (TEER) in blood-brain barrier (BBB) models, directly confirming the capacity of MOG-Ab to compromise BBB integrity (93). Clinical studies have revealed an imbalance in immune cell subsets within the peripheral blood of MOGAD patients: On one hand, there is a reduction in the number of regulatory B cells producing interleukin-10 (IL-10); On the other hand, there is an elevation in levels of pro-inflammatory memory B cells and follicular helper T cells (Tfh), which promote the differentiation of B cells into memory cells and long-lived plasma cells (94).This imbalance in immune cell subsets may be a crucial factor contributing to the persistence of autoimmune responses in MOGAD.
Recent clinical studies have demonstrated significantly elevated levels of multiple immune-related factors in both the CSF and serum of MOGAD patients. These factors primarily fall into two categories: Firstly, activated complement proteins, including C3a, C5a, and Bb (95);Secondly, B cell-associated factors, encompassing α-proliferation-inducing ligand (α-APRIL), B cell activating factor (BAFF), and C-X-C motif chemokine ligand 13 (CXCL13) (96, 97). The elevation of these factors may potentially exacerbate neurological damage in MOGAD patients. This finding not only deepens our understanding of MOGAD pathogenesis but also identifies potential novel targets for future therapeutic strategies.
In conclusion, research indicates that B cells contribute to central nervous system (CNS) damage through multiple mechanisms, including (1): Release of potentially toxic exosomes (2), Secretion of pro-inflammatory cytokines (3), Antigen presentation to T cells, and (4) Production of autoantibodies (98). A significant imbalance in immune cell subsets has been observed in the peripheral blood of MOGAD patients, characterized by a decrease in regulatory B cells and an increase in memory B cells and circulating follicular helper T cells (Tfh). This dysregulation in immune cell proportions may be a critical factor exacerbating the autoimmune response (94). These findings not only enhance our understanding of MOGAD pathogenesis but also provide crucial theoretical foundations and potential targets for developing targeted therapeutic strategies.
2.2 The role of T cells, in MOGAD
Despite the low detection rate of MOG-specific T cells (TMOG) in clinical samples, studies suggest they may play a pivotal role in the autoimmune process of MOGAD (99). Research utilizing EAE models has identified CD4+ T cells as the predominant T cell subpopulation in MOGAD lesions (12). During the initiation phase of the disease, these CD4+ T cells release neutrophil extracellular traps (NETs), providing autocrine co-stimulatory signals for T cells (100). APCs process and present MOG protein (101), exposing epitopes composed of nine amino acid residues from the N-terminus of MOG protein on major histocompatibility complex class II (MHC II) molecules. These epitopes are subsequently recognized by TMOG, leading to T cell activation (102). Notably, MHC II molecules are also expressed on the surface of peripheral APCs. This finding suggests that exogenous MOG peptides may bind to and be presented by MHC II molecules without further antigen processing. This mechanism has significant implications for understanding peripheral nervous system (PNS) involvement in MOGAD patients (103).
Multiple effector T cell subsets (Th1, Th17, and Th9) independently induce EAE through distinct yet partially overlapping mechanisms (104). For instance, Th1 cells, driven by IL-12, secrete IFN-γ, which activates macrophages and promotes EAE progression (105). Upon migration to the CNS, myelin-specific Th1 and Th17 cells can induce demyelination and drive chronic inflammation (106). Th9 cells, under the influence of TGF-β and IL-4, produce IL-9 and IL-10, thereby promoting Th17 differentiation (107). Concurrently, clinical studies have revealed an imbalance in T cell subsets in MOGAD patients, characterized by increased proportions of Th1, Th2, and Th17 cells, as well as upregulation of regulatory T cell (Treg)-related cytokines (97, 108). This imbalance may play a crucial role in the pathogenesis and progression of the disease.
MOG-Ab, upon binding to T cells and activating the complement system, can damage oligodendrocytes (109) and specifically target myelin structures, resulting in extensive structural damage (110). This process further exposes antigens, inducing additional T cell recruitment (90), thereby creating a detrimental positive feedback loop. Simultaneously, B cells activate TMOG through antigen presentation, enhancing its ability to penetrate the BBB and subsequently compromising BBB integrity (90). Furthermore, MOG-Ab triggers the activation and proliferation of peripheral MOG-specific T cells in an Fc receptor-dependent manner, leading to damage to the PNS (68).
In summary, during CNS autoimmunity, the adaptive immune system launches a “double hit” on the brain through independent T cell and B cell effector mechanisms, resulting in severe tissue damage (111). This complex immunopathological mechanism elucidates the pathogenesis of MOGAD, providing crucial insights for understanding disease progression and developing potential therapeutic strategies.
2.3 Innate immunity in MOGAD
The innate immune system plays a crucial role in the progression of MOGAD. Despite the current lack of systematic studies, insights into the underlying mechanisms can be gained through EAE models. The pathological process of MOGAD involves multiple interrelated aspects, including antibody-mediated effects, complement system activation, inflammatory cell infiltration, and complex cytokine network regulation. The core pathogenic mechanism of MOGAD initiates with the specific binding of MOG antibodies to MOG proteins. This interaction triggers a cascade of immune responses, primarily including CDC, ADCP, and ADCC, ultimately resulting in oligodendrocyte damage (14). In the CDC process, complement C9 binds to IgG1 or IgG3 antibody-antigen complexes, initiating the classical pathway and forming the membrane attack complex (MAC), which directly induces cellular damage (14, 112). Notably, although complement activation in MOGAD is relatively minor (113), oligodendrocytes are more susceptible to complement attack due to their lower expression of surface complement regulatory proteins (such as complement receptor 1 (CR1), membrane cofactor protein (MCP), and H factor-related protein (HRF)) (114). Research has shown that disease relapse correlates more strongly with CDC and ADCP activity than with absolute MOG-IgG levels, a finding of significant clinical importance (14) Moreover, MOGAD patients exhibit significantly increased protein levels of activated classical complement pathway (CP) and alternative pathway (AP), which escalate with age (95).These findings have significant practical implications for the clinical management and development of personalized treatment strategies for MOGAD patients.
Biopsy and immunohistochemical analysis of brain tissue from MOGAD patients have revealed perivascular infiltration of various inflammatory cells, including microglia, macrophages, and neutrophils, predominantly concentrated around small and medium-sized veins in the vicinity of demyelinating lesions (12, 15).Notably, microglial infiltration within the cortex often extends beyond the demyelinating lesions (12), potentially elucidating the mechanism of lesion expansion. Clinical observational studies have further confirmed that MOGAD patients exhibit higher neutrophil-to-lymphocyte ratios (NLR) and platelet-to-lymphocyte ratios (PLR) compared to MS patients (115), as well as elevated levels of neutrophil-associated cytokines (such as interleukin-8 [IL-8] and granulocyte colony-stimulating factor [G-CSF]) (97). Moreover, unlike MS and NMOSD, the acute phase of MOGAD is characterized by upregulation of cerebrospinal fluid (CSF) myelin basic protein (MBP) rather than glial fibrillary acidic protein (116).
Neutrophils may play a crucial role in the pathogenesis of MOGAD. Studies on EAE and MS have shown that neutrophils mediate BBB leakage through the secretion of matrix metalloproteinases (MMPs) (117), and their IL-1β secretion may perpetuate inflammatory responses, leading to inflammatory damage in MS (118). As one of the first immune cells recruited from the blood to inflammatory sites, neutrophils exhibit both pro-inflammatory and anti-inflammatory properties, contributing to the balance of immune responses during inflammation (119).
Platelets also play a significant role in CNS inflammatory diseases. Studies have shown that platelets promote the proliferation and differentiation of MOG-specific autoimmune CD4+ T cells into T helper 1 (Th1) and T helper 17 (Th17) cells. Platelets secrete various cytokines, chemokines, and adhesion molecules, becoming key players in CNS inflammatory diseases by influencing leukocyte differentiation, migration, and infiltration (120).
Regarding the cytokine network, MOGAD patients exhibit significantly elevated levels of Th17-related cytokines (such as interleukin-6 [IL-6], IL-8, G-CSF, and granulocyte-macrophage colony-stimulating factor [GM-CSF]) in their CSF, along with altered levels of interferon-γ (IFN-γ), interleukin-10 [IL-10], and interleukin-1 receptor antagonist [IL-1Ra] (97). This distinctive cytokine profile, differing from that of MS, may reflect the unique immunopathological mechanisms of MOGAD.
The EAE model provides valuable insights into the pathogenesis of MOGAD. For instance, neutralization of interleukin-9 (IL-9) reduces mast cell infiltration in the CNS and ameliorates EAE symptoms (121). Dectin-1, a C-type lectin receptor, limits CNS inflammation in EAE and promotes beneficial myeloid cell-astrocyte interactions through oncostatin M-Osm receptor (OsmR) signaling (122). Moreover, IFN-γ plays a complex role in the pathogenesis of EAE. Interactions between IFN-γ and host CNS cells can selectively promote or inhibit neuroinflammation and pathogenesis (123). Interestingly, atypical EAE relies on interleukin-17 (IL-17) signaling, whereas classical EAE depends on GM-CSF and C-X-C chemokine receptor 2 (CXCR2) (124). Atypical EAE is associated with preferential upregulation of C-X-C motif chemokine ligand 2 (CXCL2) in the brainstem and CXCR2-dependent neutrophil recruitment (125).
3 Treatment of MOGAD
Currently, MOGAD treatment approaches are primarily based on clinical experience and extrapolation from other neuroimmunological diseases (particularly MS and AQP4-NMOSD). While large-scale randomized controlled trials (RCTs) specific to this patient population are lacking, observational studies and retrospective analyses have provided relevant evidence for MOGAD treatment.
3.1 Treatments for acute disease phases
Although acute-phase treatment may have limited impact on the long-term progression of MOGAD, timely and effective interventions are crucial for improving prognosis and delaying relapses (126). The primary objectives of acute-phase treatment include suppressing inflammatory responses, limiting central nervous system damage, and ultimately improving long-term neurological function.
Current clinical practice primarily relies on three treatment modalities: corticosteroids, plasma exchange, and intravenous immunoglobulin. Corticosteroids are the first-line treatment for acute MOGAD exacerbations, with the standard regimen typically consisting of intravenous methylprednisolone (IVMP) at a dose of 1g/day for 3–5 days. Research by Ramanathan et al. demonstrated that approximately 80% of patients respond favorably to this regimen (79). However, clinical practice and subsequent studies have revealed limitations of monotherapy with corticosteroids: it may be insufficient for severe attacks (21), and the risk of short-term relapse persists, necessitating consideration of preventive long-term treatment (43, 127–129). Furthermore, timing of treatment is critical. A retrospective study encompassing both AQP4-IgG+ NMOSD and MOGAD suggested that early intervention may lead to better outcomes (130). Notably, timely administration of IVMP in patients with ON can achieve near-complete recovery (21). A clinical study involving 42 patients demonstrated that slow tapering of steroids (up to six months) effectively reduced the risk of relapse (131). These findings underscore the necessity of individualized treatment plans and the importance of combining acute-phase interventions with long-term preventive strategies.Studies in MOG-specific EAE models have revealed complex therapeutic effects of high-dose dexamethasone (DXM, 50 mg/kg), demonstrating improved clinical symptoms but paradoxically enhanced neuroinflammation with cognitive impairment (132).This complexity highlights the critical importance of delivery strategies in glucocorticoid therapy. A novel approach utilizing acetalated dextran microparticles co-encapsulating MOG peptide and DXM demonstrated remarkable efficacy. Subcutaneous administration of these microparticles (MOG 17.6 μg, DXM 8 μg) at three-day intervals reduced clinical scores from 3.4 to 1.6, significantly outperforming conventional delivery methods. This enhanced therapeutic effect was accompanied by substantial suppression of disease-associated cytokines, including IL-17 and GM-CSF (133) Mechanistic investigations revealed that early DXM intervention not only attenuates clinical manifestations but also inhibits myelin and axonal degeneration while suppressing neuroinflammatory processes. Notably, DXM treatment enhanced mesencephalic astrocyte-derived neurotrophic factor (MANF) expression in spinal cord white matter. The therapeutic potential of MANF was further validated through intravenous administration, which improved early-stage EAE symptoms, suggesting its promise as a novel therapeutic target (134).
Plasma exchange (PLEX) is considered a crucial adjunctive therapy for patients with poor response to corticosteroids or severe disease. The standard PLEX regimen typically involves 5–7 sessions, administered every 1–2 days (135), or immunoadsorption (21). As early as 1999, research demonstrated the efficacy of PLEX for patients with severe demyelinating attacks who did not benefit from intravenous corticosteroids (46). An international survey by Whittam et al. further supported this view, indicating that approximately 70% of experts would opt for PLEX after failed steroid treatment (136). In a study of 50 MOG-ab positive patients, PLEX resulted in (near) complete recovery for 40% of patients, benefiting even those who failed IVMP treatment (21). Another study involving 65 pediatric ADS patients revealed that 72% exhibited moderate to complete functional recovery after PLEX, particularly those with ON and TM (137). However, variability in PLEX response may be related to treatment duration, suggesting that in some cases, PLEX might be prematurely discontinued (138). Intravenous immunoglobulin (IVIG), with its immunomodulatory and anti-inflammatory properties, also plays a significant role in the acute treatment of MOGAD. IVIG is particularly suitable for pediatric patients or those with contraindications to PLEX, with a standard regimen of 1–5 days and a total dose of 1-2g/kg (not exceeding 1g/kg per day). Research by Hacohen et al. confirmed the efficacy and safety of IVIG in pediatric MOGAD patients (40). More importantly, multiple studies have shown that IVIG can significantly delay short-term relapses and markedly reduce the annualized relapse rate (ARR) before and after treatment (139–141). For patients with severe attacks, high disability at nadir, or unclear response to IVMP, early escalation to PLEX or IVIG should be considered (21, 89, 142–144). However, the optimal timing for these escalation therapies lacks support from randomized controlled trial (RCT) data. Therefore, clinical decision-making often requires timely adjustment of treatment plans based on individual patient circumstances and disease progression, aiming to maximize acute-phase prognosis and long-term quality of life for MOGAD patients.
3.2 Long-term relapse prevention treatment: overall principles and objectives
The clinical management of MOGAD poses significant challenges, primarily due to its high relapse rate and potential for disability. Studies indicate that approximately 40% of adults and 30% of children experience disease relapse, with recurrent demyelinating episodes leading to varying degrees of neurological damage (44). More concerning is that about half of the patients may develop permanent disabilities, affecting vision, mobility, or sphincter function (52). These data underscore the urgency of developing effective long-term immunosuppressive treatment strategies. Concurrently, an ongoing MOGAD cohort study in China (ClinicalTrials.gov ID NCT06443333) aims to identify expression quantitative trait loci (eQTLs) specific to Chinese MOGAD patients, elucidating pathogenic genes and key mechanisms involved in the onset and progression of neuroimmunological diseases.
Currently, immunosuppressive therapies for MOGAD are largely based on experience with AQP4-NMOSD (40), with commonly used drugs including azathioprine (AZA), mycophenolate mofetil (MMF), and rituximab (RTX). Additionally, intravenous immunoglobulin (IVIG) and tocilizumab (TCZ) have shown potential efficacy (145–147). These treatment regimens have distinct characteristics and require selection and adjustment based on individual patient circumstances. The pharmacological profiles and radiological responses of these immunomodulatory therapies in MOGAD are further detailed in Table 3.
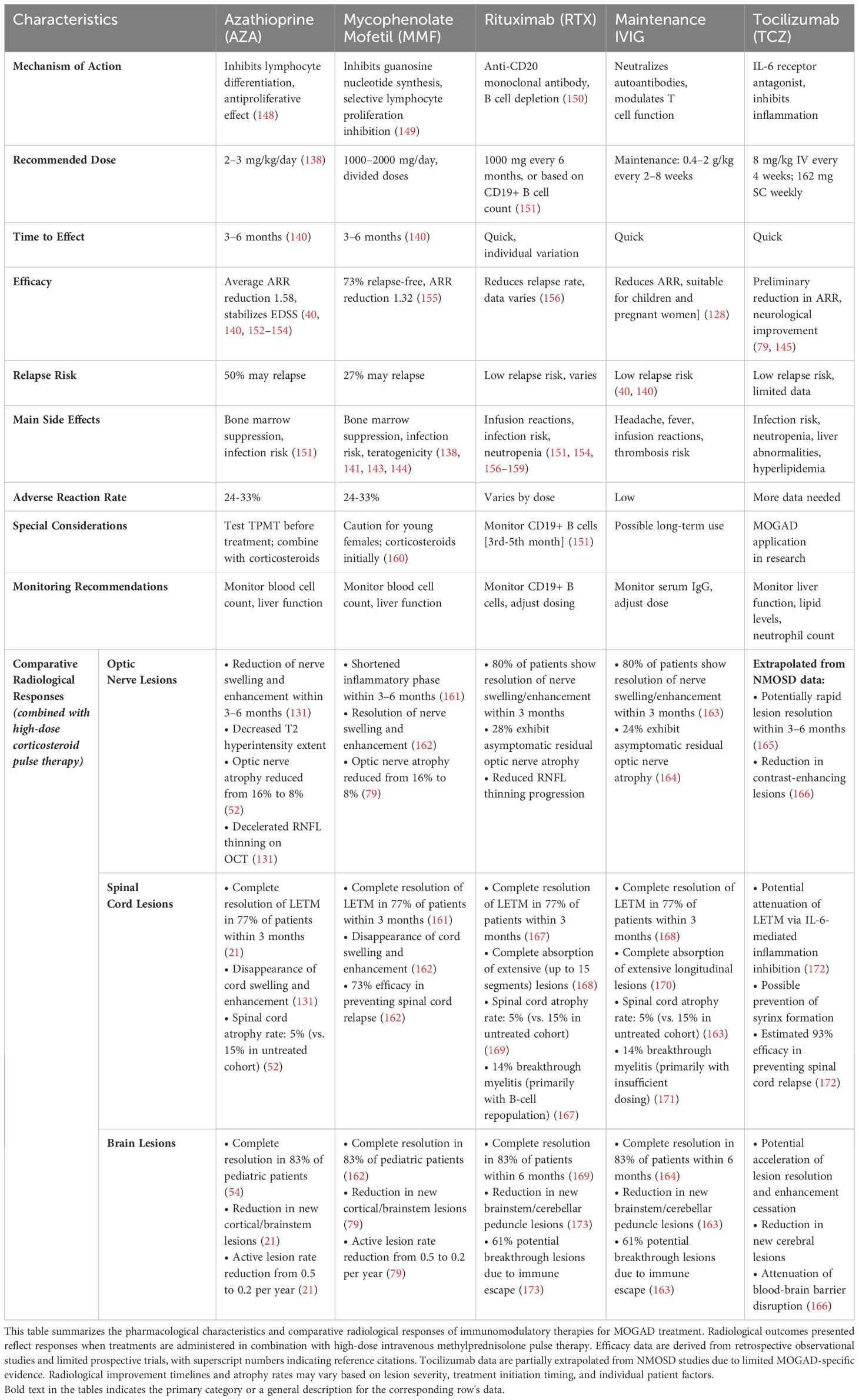
Table 3. Pharmacological profiles and radiological responses of immunomodulatory therapies in MOGAD.
3.3 Long-term relapse prevention treatment: old era
3.3.1 Azathioprine and mycophenolate mofetil
Azathioprine (AZA), a first-line steroid-sparing immunosuppressive therapy, exerts its antiproliferative effect by inhibiting lymphocyte differentiation (148). The recommended dosage is 2–3 mg/kg/day, with full efficacy typically achieved after 3–6 months (138, 140). Multiple retrospective studies have demonstrated that AZA significantly reduces the annualized relapse rate (ARR) by an average of 1.58 and stabilizes EDSS scores (40, 140, 152–154). However, relapses are still observed in approximately 50% of patients, highlighting the need for treatment optimization. Notably, AZA should be initially combined with oral corticosteroids, as most relapses occur in patients not concurrently taking oral prednisone (21, 140) Concurrently, a phase 3 randomized, placebo-controlled trial evaluating AZA for relapse prevention is ongoing (ClinicalTrials.gov ID NCT05349006).
Mycophenolate mofetil (MMF) selectively inhibits B and T lymphocyte proliferation by suppressing de novo guanosine nucleotide synthesis (149). The typical dosage is 1000–2000 mg/day, administered in two divided doses. A meta-analysis revealed that approximately 73% of patients remained relapse-free after MMF treatment, with no significant difference in relapse-free rates between adults and children. MMF treatment reduced the mean ARR by 1.32 (155). Similar to AZA, MMF requires 3–6 months to reach full efficacy and should be initially combined with oral corticosteroids (140).
Although both treatment regimens demonstrate efficacy, they are associated with varying degrees of relapse risk and side effects. The primary side effects of AZA and MMF include bone marrow suppression and increased infection risk, with adverse reactions occurring in approximately 24-33% of patients (58, 133, 136–139, 174). For AZA, testing for TPMT activity prior to treatment initiation is recommended to identify patients at high risk for potentially fatal bone marrow suppression (151). Furthermore, MMF is teratogenic, requiring special consideration when used in young female patients (160).
3.3.2 Rituximab
Rituximab (RTX) is a B-cell depleting monoclonal antibody that targets the CD20 antigen (150). In adults, the typical regimen consists of 1000 mg administered intravenously every 6 months, or individualized dosing based on CD19+ B-cell counts (151). Pediatric dosing protocols differ from those for adults (175). Studies have demonstrated that when employed as a first-line treatment, RTX is associated with a significantly higher reduction in relapse rates compared to alternative therapies (63% vs. 26%) (156). However, the efficacy of RTX in MOGAD appears to be less pronounced than in AQP4-IgG positive NMOSD (167). Recent studies suggest that a treatment regimen based on CD27-positive B-cell repopulation may be more effective. Additionally, FCGR3A gene polymorphism analysis can be employed to assist in evaluating RTX efficacy (176). RTX is associated with a spectrum of adverse effects, including leukopenia, infusion-related reactions, and hypogammaglobulinemia. These side effects lead to treatment discontinuation in approximately 13.71% of patients (151, 154, 156–159).
3.3.3 Maintenance IVIG
Intravenous immunoglobulin (IVIG) may serve as an effective maintenance treatment option for specific patient populations, particularly children and pregnant women (128). However, the widespread application of this therapy is constrained by its high costs and limited availability (144). The typical IVIG treatment protocol consists of an initial loading dose of 0.4 g/kg daily for 5 consecutive days, followed by a maintenance regimen of 0.4–2 g/kg administered every 2–8 weeks. Compared to other conventional immunotherapies, maintenance IVIG therapy has demonstrated a significant reduction in the annualized relapse rate (ARR) (40, 140). Recent studies have demonstrated the safety and efficacy of subcutaneous immunoglobulin (SCIg) in preventing MOGAD relapses (177). SCIg offers several advantages, including better tolerability, the possibility of self-administration, and the option for home-based treatment when infusion services are available, potentially making it a more convenient therapeutic alternative.
3.3.4 Tocilizumab
Tocilizumab (TCZ) has demonstrated notable efficacy in the treatment of refractory MOGAD patients. Small-scale case series studies suggest that TCZ may be highly effective for MOGAD patients who have been refractory to other immunosuppressive treatments (146, 178). The standard dosing regimen for TCZ in adults is 8 mg/kg administered monthly, with a maximum recommended dose of 800 mg/month. Two studies, collectively involving 19 pediatric patients, reported that 93% of patients (95% CI [54%–100%], I2 = 71%, p = 0.06) remained relapse-free during the follow-up period after initiating TCZ treatment. Prior to treatment, the ARRs in these two studies were 1.1 ± 0.4 and 1.78 ± 1.04, respectively. Post-treatment, the ARR in both studies decreased to 0. These results suggest that TCZ treatment significantly reduced the frequency of relapses in MOGAD patients (79, 145). A 2022 study by Ringelstein et al., involving 14 patients, provided data on TCZ’s impact on patient disability. The study utilized the Expanded Disability Status Scale (EDSS) to assess disability. Patients’ EDSS scores decreased from 2.75 ± 1.11 before TCZ treatment to 2.03 ± 1.26 after treatment. These results suggest that TCZ may contribute to improving patients’ functional status (146). To further validate the potential role of TCZ in MOGAD treatment, a randomized, controlled, multicenter study has been initiated (ClinicalTrials.gov Identifier: NCT06452537). This large-scale clinical trial is anticipated to provide more robust and comprehensive data regarding the efficacy of TCZ in MOGAD treatment. Figure 2 offers a comprehensive visual guide to the therapeutic landscape in MOG-associated disease, delineating key intervention points along the immunopathogenic pathway from the peripheral circulation to the central nervous system. It illustrates how diverse strategies—targeting antigen presentation, T and B cell activity, pathogenic antibody clearance, and critical cytokine pathways like IL-6 signaling—aim to disrupt the disease cascade at multiple levels to ultimately reduce CNS inflammation and protect oligodendrocytes.
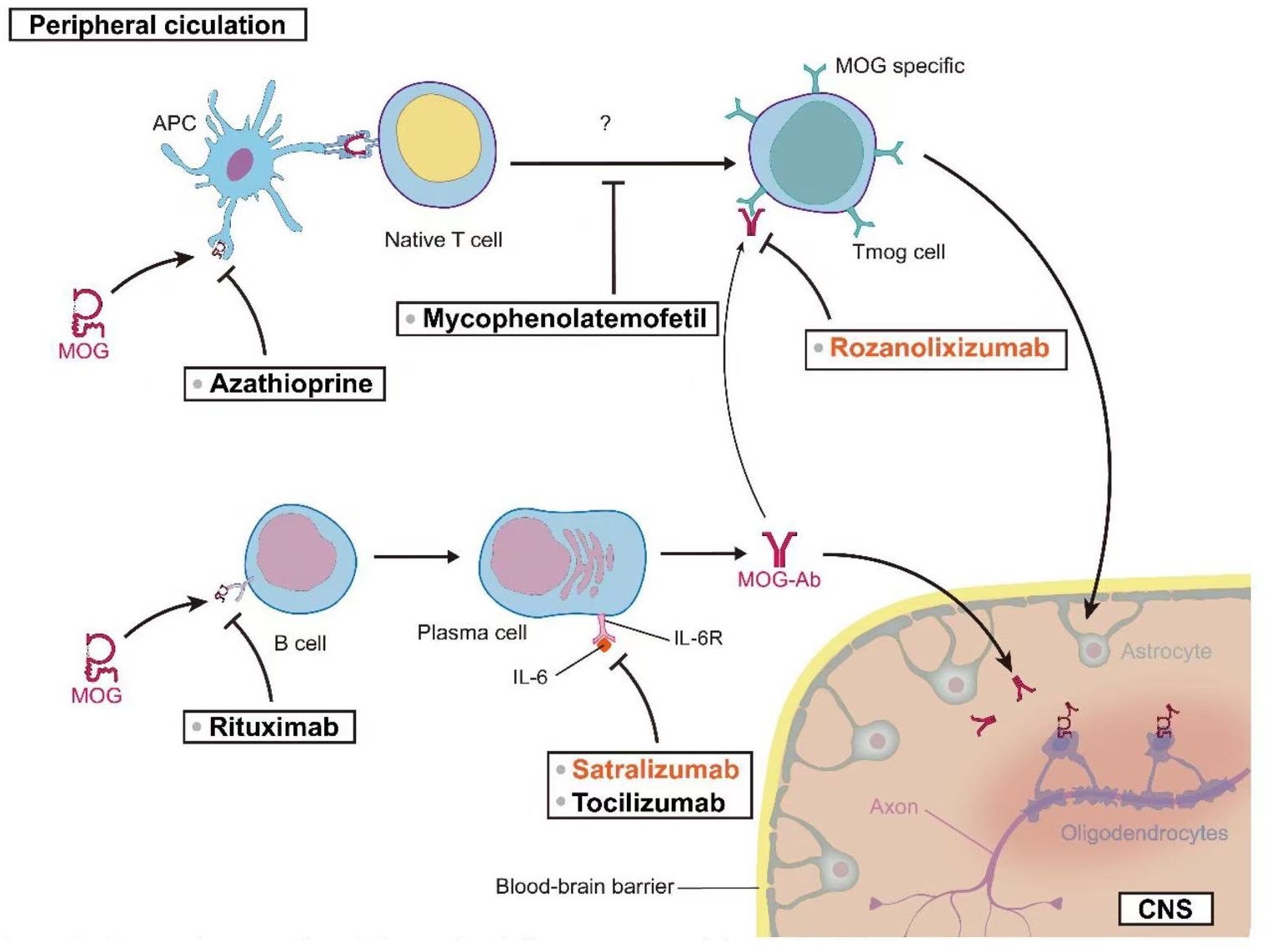
Figure 2. Therapeutic targets in MOG-associated disease: From peripheral circulation to CNS.
3.4 Long-term relapse prevention treatment: future era
3.4.1 Rozanolixizumab
Rozanolixizumab (also known as Rozimab) is a high-affinity humanized immunoglobulin G4 (IgG4) monoclonal antibody that targets the human neonatal Fc receptor (FcRn). This antibody inhibits FcRn activity, resulting in rapid degradation of circulating antibodies, leading to a 70% reduction in antibody levels within 24 hours, an effect comparable to PLEX (179). Currently, rozanolixizumab is primarily indicated for the treatment of myasthenia gravis (180). In February 2022, the first phase 3 placebo-controlled randomized clinical trial for MOGAD was initiated (ClinicalTrials.gov Identifier: NCT05063162). This trial aims to evaluate the efficacy of rozanolixizumab in preventing relapses in MOGAD (181). Results are pending publication.
3.4.2 Satralizumab
Satralizumab is a humanized immunoglobulin G2 (IgG2) monoclonal antibody produced in Chinese hamster ovary cells using recombinant DNA technology. It exerts its therapeutic effect by binding to both membrane-bound and soluble interleukin-6 (IL-6) receptors, thereby inhibiting the IL-6 signaling pathway (182). A phase III, randomized, double-blind, placebo-controlled, multicenter study is currently evaluating the efficacy, safety, pharmacokinetics, and pharmacodynamics of satralizumab (Enspryng®) as monotherapy or as an adjunct to baseline treatment in MOGAD patients (ClinicalTrials.gov Identifier: NCT05271409) (181). Satralizumab received its first global approval in Canada in June 2020 for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in AQP4-IgG seropositive adults and children aged 12 years and older, demonstrating favorable outcomes (183).
3.4.3 CT103A Cells
In recent years, cell-based therapies have garnered widespread attention in the field of autoimmune disease treatment. Chimeric Antigen Receptor T-cell (CAR-T) therapy, an innovative treatment approach, is being explored for various refractory diseases. In this context, a new clinical trial (ClinicalTrials.gov Identifier: NCT04561557) is evaluating the safety and efficacy of a novel CAR-T cell therapy utilizing CT103A cells for the treatment of relapsed/refractory antibody-mediated idiopathic inflammatory diseases. The CT103A CAR-T cell therapy employs genetic engineering techniques to modify T cells, enabling them to specifically recognize and eliminate B cells producing pathological antibodies. This approach aims to fundamentally reduce or eliminate the production of disease-causing autoantibodies, thereby achieving a therapeutic effect (184). While this clinical trial is not specifically targeting MOGAD patients, its outcomes may provide novel insights and approaches for MOGAD treatment.
3.4.4 Calculus Bovis Sativus
Calculus Bovis Sativus (CBS), a traditional Chinese medicine, is documented in historical Chinese medical literature as possessing multiple therapeutic properties, including cardiac relief, phlegm resolution, choleretic effects, and sedative actions. Historically, CBS has been employed to treat a diverse array of conditions, including febrile coma, delirium, epilepsy, infantile convulsions, dental caries, pharyngitis, stomatitis, carbuncles, and furuncles (185). However, these traditional applications are predominantly based on empirical medicine and historical documentation, lacking systematic validation through modern scientific methodologies. To bridge the gap between traditional Chinese medicine theory and modern medical practice, researchers are investigating the potential applications of CBS in specific neurological disorders. Currently, an open-label clinical trial is underway to evaluate the efficacy and safety of CBS in the treatment of idiopathic inflammatory demyelinating diseases (ClinicalTrials.gov Identifier: NCT06474520). This study holds multifaceted significance: it exemplifies methodological innovation by integrating traditional Chinese medicine with modern clinical trial protocols, sets a precedent for the modernization of Chinese medicine research, and potentially offers novel therapeutic options for patients with idiopathic inflammatory demyelinating diseases. Paving the way for future advancements, Table 4 provides a consolidated overview of the dynamic clinical trial landscape in MOGAD and related neuroimmunological disorders.
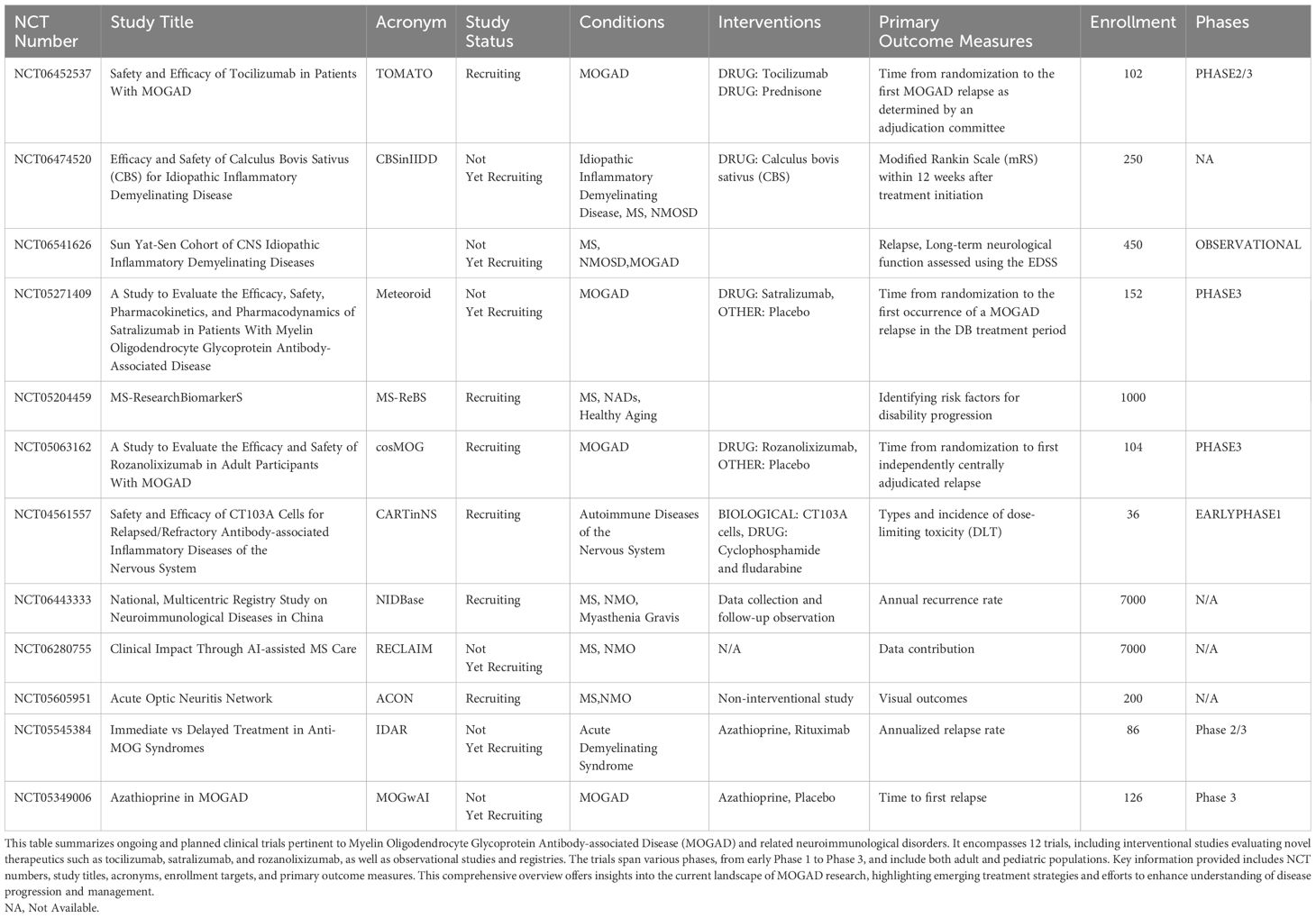
Table 4. Current clinical trials in MOGAD and related neuroimmunological disorders.
4 Conclusions and future perspectives
Translational investigations linking EAE models and MOGAD have significantly progressed our knowledge of disease mechanisms and potential therapeutic goals. In recent years, the pathophysiological mechanisms of MOGAD have been elucidated to a great extend upon lessons learned from EAE models. These experimental results have, in turn, led to lead therapeutic innovations including engineered glucocorticoids and the discovery of novel therapy candidates such as MANF. Systemic evaluation strategies, and borrowing therapeutic approaches from MS and NMOSD have opened up new possibilities for the treatment of MOGAD. The advent of different treatment options, such as monoclonal antibody therapies and cellular-based treatments offers hope for MOGAD patients but needs to be confirmed in future adequately powered clinical trials prior implementing their use widespread.
The integration of EAE models with clinical MOGAD research is of paramount importance for elucidating the pathological mechanisms underlying this disorder. Regarding pathogenesis, EAE models have definitively demonstrated that MOG-specific T cells are essential for disease initiation, while anti-MOG antibodies merely exacerbate demyelination. In contrast, clinical investigations of MOGAD suggest that anti-MOG antibodies may directly participate in the pathogenic process, exemplified by their capacity to disrupt the microtubule cytoskeleton in oligodendrocytes (50, 186). Therefore, when synthesizing findings from both research domains, it is crucial to differentiate between the predominant role of T cells in EAE and the potential direct pathogenic effects of antibodies in MOGAD, while concurrently exploring cooperative mechanisms such as T-B cell interactions in human disease (187, 188). Concerning the controversial role of antibody pathogenicity, although antibodies are not requisite in EAE models, studies have revealed that serum from MOGAD patients can aggravate demyelination in animal models. This observation indicates the necessity for further validation of antibody pathogenicity in human disease through clinical research, such as analyzing correlations between antibody titers and disease activity (50, 189). Notably, transgenic mouse models expressing human MOG have provided valuable platforms for investigating the pathogenicity of human anti-MOG antibodies, further supporting the observation that MOGAD patient serum can exacerbate demyelination in experimental settings (190).
In comparing animal models with clinical phenotypes, EAE models typically manifest as acute monophasic disease courses, whereas approximately 50% of MOGAD patients exhibit relapsing disease trajectories (144). Radiological investigations have revealed that T2 lesions in MOGAD demonstrate greater propensity for complete resolution compared to those in NMOSD and MS, resembling the reversibility of acute inflammation observed in EAE (191). This similarity suggests that EAE models may be valuable for investigating acute-phase mechanisms, though they must be complemented with longitudinal observations that account for the chronic relapsing characteristics of MOGAD. Beyond EAE, toxin-induced demyelination models (such as those utilizing Pseudomonas aeruginosa lipopolysaccharide and lysophosphatidylcholine) have contributed significant insights into myelin repair mechanisms in MOGAD. Particularly, these models demonstrate that remyelination capacity may be more robust in MOGAD compared to MS, which aligns with the clinical observation that MOGAD T2 lesions show greater tendency for complete resolution compared to those in NMOSD and MS (192).Furthermore, MOGAD patients frequently present with multifocal central nervous system involvement, while optic neuritis and myelitis predominate in EAE models (144). This discrepancy indicates the necessity of incorporating multifocal pathology in experimental model design to more accurately recapitulate human disease phenotypes.
Translational research on therapeutic strategies demonstrates that B-cell depletion is ineffective or potentially disease-exacerbating in EAE models, whereas clinical studies of MOGAD indicate that rituximab (anti-CD20) may be efficacious in a subset of patients (144, 193). This discrepancy underscores the necessity for comprehensive analysis of functional differences among B-cell subpopulations (such as plasma cells) in disease pathogenesis, and for exploration of novel B-cell-targeted therapies, including Bruton’s tyrosine kinase inhibitors (194). Regarding antibody-targeted therapeutics, EAE model investigations have demonstrated that anti-FcRn antibodies can reduce IgG circulation, ameliorate neurological dysfunction, and improve visual function, thereby providing a theoretical foundation for clinical MOGAD treatments (such as efgartigimod) (189, 195). However, these findings require further validation through rigorous clinical trials.
In the domain of biomarker development for diagnosis and prognosis, cerebral lesions in MOGAD predominantly involve cortical and subcortical regions, whereas periventricular lesions characterize MS (196, 197). EAE models can be utilized to simulate specific lesion patterns (such as optic neuritis) and, in conjunction with high-resolution MRI, validate human imaging biomarkers (such as “H-type” spinal cord lesions) (144). Concurrently, optimization of anti-MOG antibody detection methodologies (comparing live-cell versus fixed-cell assays) and stratification studies correlating antibody titers with clinical phenotypes (such as monophasic versus relapsing disease) hold significant clinical relevance (144, 189).
Regarding the investigation of cooperative mechanisms and disease model optimization, T-B cell cooperative pathogenesis has been documented in EAE models, and analogous immune interactions may exist in MOGAD patients (188). The establishment of humanized murine models (such as those incorporating transplanted patient T and B lymphocytes) to simulate the human immune microenvironment facilitates comprehensive understanding of these mechanisms (187). Additionally, given that EAE predominantly represents acute disease manifestations while the progression mechanisms of MOGAD remain incompletely elucidated, the development of chronic or relapsing-remitting EAE models, in combination with passive transfer of anti-MOG antibodies, may more accurately recapitulate human disease progression (187, 194).
In conclusion, the integration of EAE and MOGAD research necessitates careful consideration of both similarities and differences between experimental models and clinical manifestations, with particular emphasis on T-B cell interactions, validation of antibody pathogenicity, and translation of therapeutic strategies. Through interdisciplinary collaboration encompassing fundamental immunology, neuroradiology, and clinical trial design, advancements in mechanistic elucidation and precision therapeutics for MOGAD can be substantially accelerated.
There are many challenges on the horizon in MOGAD that need to be addressed, but we anticipate a rapidly changing therapeutic landscape. Given the clinical heterogeneity of MOGAD and limitations in current EAE models to capture all aspects of human disease, continued experimental optimization is required. It is essential that EAE models mimicking MOGAD-specific pathogenic mechanisms be developed, particularly in developing platforms that incorporate human MOG-specific immune responses. Fundamental scientific investigation should push the boundaries of our understanding into molecular processes underlying MOGAD, including production and implications of the MOG IgGs by expansion in B cells as well as functions both regulatory on T-cells and within CNS inflammatory cascades. This knowledge has to be translated into therapeutic targets or biomarkers, and clinical research should combine evaluation of new therapies as well as predictive strategies for differential responsiveness to treatment.
We need to work with researchers, clinicians and patient communities across disciplines. Through enhanced international cooperation and continuous innovation in research methodologies, we anticipate significant improvements in personalized treatment approaches for MOGAD patients. Our ultimate goal extends beyond symptom management to developing comprehensive strategies for preventing disease onset and halting progression. These concerted efforts aim to provide more effective personalized treatment regimens for MOGAD patients, potentially leading to better quality of life and long-term prognosis in the near future.
Author contributions
YZ: Writing – original draft. DL: Funding acquisition, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Public Health and Technology Project of Tianjin [grant number TJWJ2021MS022] and Tianjin Key Medical Discipline (Specialty) Construction Project [grant number TJYXZDXK].
Acknowledgments
The authors are grateful to Mr. Xian Zhang for the design and drawing of the figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. (2023) 22:268–82. doi: 10.1016/S1474-4422(22)00431-8
2. Karni A, Bakimer-Kleiner R, Abramsky O, and Ben-Nun A. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Arch Neurol. (1999) 56:311–5. doi: 10.1001/archneur.56.3.311
3. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, and Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. (2005) 202:473–7. doi: 10.1084/jem.20050304
4. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation. (2011) 8:184. doi: 10.1186/1742-2094-8-184
5. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
6. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
7. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
8. Ciccarelli O, Toosy AT, Thompson A, and Hacohen Y. Navigating through the recent diagnostic criteria for MOGAD: challenges and practicalities. Neurology. (2023) 100:689–90. doi: 10.1212/WNL.0000000000207238
9. Wang X and Bao L. Comparison of ocular changes in multiple sclerosis and neuromyelitis optica spectrum disorder patients. Front Neurol. (2024) 15. doi: 10.3389/fneur.2024.1417814
10. Dendrou CA, Fugger L, and Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
11. Peterson LK and Fujinami RS. Inflammation, demyelination, neurodegeneration and neuroprotection in the pathogenesis of multiple sclerosis. J Neuroimmunol. (2007) 184:37–44. doi: 10.1016/j.jneuroim.2006.11.015
12. Höftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, Hochmeister S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. (2020) 139:875–92. doi: 10.1007/s00401-020-02132-y
13. Macrini C, Gerhards R, Winklmeier S, Bergmann L, Mader S, Spadaro M, et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain. (2021) 144:2375–89. doi: 10.1093/brain/awab105
14. Yandamuri SS, Filipek B, Obaid AH, Lele N, Thurman JM, Makhani N, et al. MOGAD patient autoantibodies induce complement, phagocytosis, and cellular cytotoxicity. JCI Insight. (2023) 8:e165373. doi: 10.1172/jci.insight.165373
15. Takai Y, Misu T, Kaneko K, Chihara N, Narikawa K, Tsuchida S, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. (2020) 143:1431–46. doi: 10.1093/brain/awaa102
16. Reindl M and Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. (2019) 15:89–102. doi: 10.1038/s41582-018-0112-x
17. Corbali O and Chitnis T. Pathophysiology of myelin oligodendrocyte glycoprotein antibody disease. Front Neurol. (2023) 14:1137998. doi: 10.3389/fneur.2023.1137998
18. Papadopoulos MC, Bennett JL, and Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. (2014) 10:493–506. doi: 10.1038/nrneurol.2014.141
19. Saikali P, Cayrol R, and Vincent T. Anti-aquaporin-4 auto-antibodies orchestrate the pathogenesis in neuromyelitis optica. Autoimmun Rev. (2009) 9:132–5. doi: 10.1016/j.autrev.2009.04.004
20. Sechi E, Buciuc M, Pittock SJ, Chen JJ, Fryer JP, Jenkins SM, et al. Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol. (2021) 78:741–6. doi: 10.1001/jamaneurol.2021.0912
21. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
22. Wu Y, Zhou H, Ci X, and Lu J. Low T3 syndrome is associated with the severity of myelin oligodendrocyte glycoprotein antibody-associated disease exacerbation. Front Neurosci. (2024) 18. doi: 10.3389/fnins.2024.1357633
23. Negro-Demontel L, Maleki AF, Reich DS, and Kemper C. The complement system in neurodegenerative and inflammatory diseases of the central nervous system. Front Neurol. (2024) 15. doi: 10.3389/fneur.2024.1396520
24. Dobson R and Giovannoni G. Multiple sclerosis - a review. Eur J Neurol. (2019) 26:27–40. doi: 10.1111/ene.2019.26.issue-1
25. Wang J, Cui C, Lu Y, Chang Y, Wang Y, Li R, et al. Therapeutic response and possible biomarkers in acute attacks of neuromyelitis optica spectrum disorders: A prospective observational study. Front Immunol. (2021) 12:720907. doi: 10.3389/fimmu.2021.720907
26. Gklinos P and Dobson R. Myelin oligodendrocyte glycoprotein-antibody associated disease: an updated review of the clinical spectrum, pathogenetic mechanisms and therapeutic management. Antibodies (Basel). (2024) 13:43. doi: 10.3390/antib13020043
27. Rodin RE and Chitnis T. Soluble biomarkers for Neuromyelitis Optica Spectrum Disorders: a mini review. Front Neurol. (2024) 15:1415535/full. doi: 10.3389/fneur.2024.1415535/full
28. Barzegar M, Drulovic J, and Nociti V. Editorial: Comorbidity in multiple sclerosis and related disorders. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1469614
29. Brunner C, Lassmann H, Waehneldt TV, Matthieu JM, and Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the CNS of adult rats. J Neurochem. (1989) 52:296–304. doi: 10.1111/j.1471-4159.1989.tb10930.x
30. Clements CS, Reid HH, Beddoe T, Tynan FE, Perugini MA, Johns TG, et al. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc Natl Acad Sci U S A. (2003) 100:11059–64. doi: 10.1073/pnas.1833158100
31. Tea F, Lopez JA, Ramanathan S, Merheb V, Lee FXZ, Zou A, et al. Characterization of the human myelin oligodendrocyte glycoprotein antibody response in demyelination. Acta Neuropathol Commun. (2019) 7:145. doi: 10.1186/s40478-019-0786-3
32. Zhou D, Srivastava R, Nessler S, Grummel V, Sommer N, Brück W, et al. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci U S A. (2006) 103:19057–62. doi: 10.1073/pnas.0607242103
33. Chang X, Jiao K, Wang D, Zhou Y, Zhao Z, Xing Y, et al. The immune imbalance between follicular regulatory and helper T cells in myelin oligodendrocyte glycoprotein IgG-associated disease. Clin Immunol. (2023) 255:109734. doi: 10.1016/j.clim.2023.109734
34. Poppell M, Hammel G, and Ren Y. Immune regulatory functions of macrophages and microglia in central nervous system diseases. IJMS. (2024) 24:5925. doi: 10.3390/ijms24065925
35. Sharma G, Gopinath S, and Lakshmi Narasimhan R. Exploring the molecular aspects of glycosylation in MOG antibodyDisease (MOGAD). CPPS. (2022) 23:384–94. doi: 10.2174/1389203723666220815110509
36. Fadda G, Armangue T, Hacohen Y, Chitnis T, and Banwell B. Paediatric multiple sclerosis and antibody-associated demyelination: clinical, imaging, and biological considerations for diagnosis and care. Lancet Neurol. (2021) 20:136–49. doi: 10.1016/S1474-4422(20)30432-4
37. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. (2012) 79:1273–7. doi: 10.1212/WNL.0b013e31826aac4e
38. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. (2014) 71:276–83. doi: 10.1001/jamaneurol.2013.5857
39. Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. (2014) 82:474–81. doi: 10.1212/WNL.0000000000000101
40. Hacohen Y, Wong YY, Lechner C, Jurynczyk M, Wright S, Konuskan B, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. (2018) 75:478–87. doi: 10.1001/jamaneurol.2017.4601
41. Sun X, Qiu W, Wang J, Wang S, Wang Y, Zhong X, et al. Myelin oligodendrocyte glycoprotein-associated disorders are associated with HLA subtypes in a Chinese paediatric-onset cohort. J Neurol Neurosurg Psychiatry. (2020) 91:733–9. doi: 10.1136/jnnp-2019-322115
42. Bartels F, Lu A, Oertel FC, Finke C, Paul F, and Chien C. Clinical and neuroimaging findings in MOGAD-MRI and OCT. Clin Exp Immunol. (2021) 206:266–81. doi: 10.1111/cei.13641
43. Cobo-Calvo A, Ruiz A, Rollot F, Arrambide G, Deschamps R, Maillart E, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. (2021) 89:30–41. doi: 10.1002/ana.25909
44. Marignier R, Hacohen Y, Cobo-Calvo A, Pröbstel AK, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. (2021) 20:762–72. doi: 10.1016/S1474-4422(21)00218-0
45. Ruskamo S, Raasakka A, Pedersen JS, Martel A, Škubník K, Darwish T, et al. Human myelin proteolipid protein structure and lipid bilayer stacking. Cell Mol Life Sci. (2022) 79:419. doi: 10.1007/s00018-022-04428-6
46. Weinshenker BG, O’Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. (1999) 46:878–86. 10.1002/1531-8249(199912)46:6<878::aid-ana10>3.0.co;2-q
47. Yılmaz Ü. The diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) in children. Explor Neuroprot Ther. (2024) 4:38–54. doi: 10.37349/ent
48. Messias K, Marques VD, and Messias A. Myelin oligodendrocyte glycoprotein antibody-associated optic neuritis: an update. Arq Bras Oftalmol. (2023) 86:83–92. doi: 10.5935/0004-2749.20230012
49. Wolf AB, Palace J, and Bennett JL. Emerging principles for treating myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). Curr Treat Options Neurol. (2023) 25:437–53. doi: 10.1007/s11940-023-00776-1
50. Al-Ani A, Chen JJ, and Costello F. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): current understanding and challenges. J Neurol. (2023) 270:4132–50. doi: 10.1007/s00415-023-11737-8
51. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study. Neurology. (2018) 90:e1858–69. doi: 10.1212/WNL.0000000000005560
52. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
53. Trewin BP, Dale RC, Qiu J, Chu M, Jeyakumar N, Dela Cruz F, et al. Oral corticosteroid dosage and taper duration at onset in myelin oligodendrocyte glycoprotein antibody-associated disease influences time to first relapse. J Neurol Neurosurg Psychiatry. (2024) 95:1054–63. doi: 10.1136/jnnp-2024-333463
54. Ambrosius W, Michalak S, Kozubski W, and Kalinowska A. Myelin oligodendrocyte glycoprotein antibody-associated disease: current insights into the disease pathophysiology, diagnosis and management. Int J Mol Sci. (2020) 22:100. doi: 10.3390/ijms22010100
55. Glatigny S and Bettelli E. Experimental autoimmune encephalomyelitis (EAE) as animal models of multiple sclerosis (MS). Cold Spring Harb Perspect Med. (2018) 8:a028977. doi: 10.1101/cshperspect.a028977
56. Lyons JA, San M, Happ MP, and Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol. (1999) 29:3432–9. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2
57. Kwon YN, Kim B, Kim J-S, Mo H, Choi K, and Oh S-I. Myelin Oligodendrocyte Glycoprotein-Immunoglobulin G in the CSF: Clinical Implication of Testing and Association With Disability. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1095. doi: 10.1212/NXI.0000000000001095
58. Kerlero de Rosbo N, Milo R, Lees MB, Burger D, Bernard CC, and Ben-Nun A. Reactivity to myelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly to myelin oligodendrocyte glycoprotein. J Clin Invest. (1993) 92:2602–8. doi: 10.1172/JCI116875
59. Peschl P, Bradl M, Höftberger R, Berger T, and Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00529
60. Mayer MC and Meinl E. Glycoproteins as targets of autoantibodies in CNS inflammation: MOG and more. Ther Adv Neurol Disord. (2012) 5:147–59. doi: 10.1177/1756285611433772
61. Iglesias A, Bauer J, Litzenburger T, Schubart A, and Linington C--. and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. (2001) 36:220–34. doi: 10.1002/glia.v36:2
62. Jégou JF, Chan P, Schouft MT, Griffiths MR, Neal JW, Gasque P, et al. C3d binding to the myelin oligodendrocyte glycoprotein results in an exacerbated experimental autoimmune encephalomyelitis. J Immunol. (2007) 178:3323–31. doi: 10.4049/jimmunol.178.5.3323
63. Dagkonaki A, Avloniti M, Evangelidou M, Papazian I, Kanistras I, Tseveleki V, et al. Mannan-MOG35–55 reverses experimental autoimmune encephalomyelitis, inducing a peripheral type 2 myeloid response, reducing CNS inflammation, and preserving axons in spinal cord lesions. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.575451
64. Tanaka K, Kezuka T, Ishikawa H, Tanaka M, Sakimura K, Abe M, et al. Pathogenesis, clinical features, and treatment of patients with myelin oligodendrocyte glycoprotein (MOG) autoantibody-associated disorders focusing on optic neuritis with consideration of autoantibody-binding sites: A review. Int J Mol Sci. (2023) 24:13368. doi: 10.3390/ijms241713368
65. Geladaris A, Häusser-Kinzel S, Pretzsch R, Nissimov N, Lehmann-Horn K, Häusler D, et al. IL-10-providing B cells govern pro-inflammatory activity of macrophages and microglia in CNS autoimmunity. Acta Neuropathol. (2023) 145:461–77. doi: 10.1007/s00401-023-02552-6
66. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. (2013) 210:2921–37. doi: 10.1084/jem.20130699
67. Mader S, Ho S, Wong HK, Baier S, Winklmeier S, Riemer C, et al. Dissection of complement and Fc-receptor-mediated pathomechanisms of autoantibodies to myelin oligodendrocyte glycoprotein. Proc Natl Acad Sci U S A. (2023) 120:e2300648120. doi: 10.1073/pnas.2300648120
68. Kinzel S, Lehmann-Horn K, Torke S, Häusler D, Winkler A, Stadelmann C, et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. (2016) 132:43–58. doi: 10.1007/s00401-016-1559-8
69. Weber MS, Prod’homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. (2010) 68:369–83. doi: 10.1002/ana.22081
70. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, and Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. (2008) 118:3420–30. doi: 10.1172/JCI36030
71. Galicia G, Boulianne B, Pikor N, Martin A, and Gommerman JL. Secondary B cell receptor diversification is necessary for T cell mediated neuro-inflammation during experimental autoimmune encephalomyelitis. PloS One. (2013) 8:e61478. doi: 10.1371/journal.pone.0061478
72. Chen D, Gallagher S, Monson NL, Herbst R, and Wang Y. Inebilizumab, a B cell-depleting anti-CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med. (2016) 5:107. doi: 10.3390/jcm5120107
73. Pöllinger B, Krishnamoorthy G, Berer K, Lassmann H, Bösl MR, Dunn R, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med. (2009) 206:1303–16. doi: 10.1084/jem.20090299
74. Liu R, Du S, Zhao L, Jain S, Sahay K, Rizvanov A, et al. Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.996469
75. Demuth S, Guillaume M, Bourre B, Ciron J, Zephir H, Sirejacob Y, et al. Treatment regimens for neuromyelitis optica spectrum disorder attacks: a retrospective cohort study. J Neuroinflammation. (2022) 19:62. doi: 10.1186/s12974-022-02420-2
76. Wang Y, Danzeng Q, Jiang W, Han B, Zhu X, Liu Z, et al. A retrospective study of myelin oligodendrocyte glycoprotein antibody-associated disease from a clinical laboratory perspective. Front Neurol. (2023) 14. doi: 10.3389/fneur.2023.1187824
77. Vlad B, Reichen I, Neidhart S, Hilty M, Lekaditi D, Heuer C, et al. Basic CSF parameters and MRZ reaction help in differentiating MOG antibody-associated autoimmune disease versus multiple sclerosis. Front Immunol. (2023) 14:1237149. doi: 10.3389/fimmu.2023.1237149
78. Havla J, Pakeerathan T, Schwake C, Bennett JL, Kleiter I, Felipe-Rucián A, et al. Age-dependent favorable visual recovery despite significant retinal atrophy in pediatric MOGAD: how much retina do you really need to see well? J Neuroinflammation. (2021) 18:121. doi: 10.1186/s12974-021-02160-9
79. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
80. Satukijchai C, Mariano R, Messina S, Sa M, Woodhall MR, Robertson NP, et al. Factors associated with relapse and treatment of myelin oligodendrocyte glycoprotein antibody-associated disease in the United Kingdom. JAMA Netw Open. (2022) 5:e2142780. doi: 10.1001/jamanetworkopen.2021.42780
81. Kwon YN, Kim B, Kim JS, Mo H, Choi K, Oh S, et al. Myelin oligodendrocyte glycoprotein-immunoglobulin G in the CSF. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1095. doi: 10.1212/NXI.0000000000001095
82. Gupta P, Paul P, Redenbaugh V, Guo Y, Lucchinetti C, Abdulrahman Y, et al. Peripheral nervous system manifestations of MOG antibody associated disease. Ann Clin Trans Neurol. (2024) 11:1046–52. doi: 10.1002/acn3.52001
83. Greco G, Risi M, Masciocchi S, Businaro P, Rigoni E, Zardini E, et al. Clinical P, prognostic, Jun 06; pathophysiological implications of MI detection in the C the importance of intrathecal MI synthesis JNNP 2024, PracticeUpdate. [cited 2024 Jul 30]. Clinical, Prognostic, and Pathophysiological Implications of CSF MOG-IgG Detection. Neurol J Neurosurgery. (2024) 95:1176–86. doi: 10.1136/jnnp-2024-333554
84. Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. (2011) 108:3701–6. doi: 10.1073/pnas.1017385108
85. Höftberger R, Leisser M, Bauer J, and Lassmann H. Autoimmune encephalitis in humans: how closely does it reflect multiple sclerosis? Acta Neuropathol Commun. (2015) 3:80. doi: 10.1186/s40478-015-0260-9
86. Stefferl A, Schubart A, Storch2 M, Amini A, Mather I, Lassmann H, et al. Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. J Immunol. (2000) 165:2859–65. doi: 10.4049/jimmunol.165.5.2859
87. Xu H, Foltz L, Sha Y, Madlansacay MR, Cain C, Lindemann G, et al. Cloning and characterization of human erythroid membrane-associated protein, human ERMAP. Genomics. (2001) 76:2–4. doi: 10.1006/geno.2001.6600
88. Litzenburger T, Blüthmann H, Morales P, Pham-Dinh D, Dautigny A, Wekerle H, et al. Development of myelin oligodendrocyte glycoprotein autoreactive transgenic B lymphocytes: receptor editing in vivo after encounter of a self-antigen distinct from myelin oligodendrocyte glycoprotein. J Immunol. (2000) 165:5360–6. doi: 10.4049/jimmunol.165.9.5360
89. Brilot F, Dale RC, Selter RC, Grummel V, Reddy Kalluri S, Aslam M, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol. (2009) 66:833–42. doi: 10.1002/ana.21916
90. Flach AC, Litke T, Strauss J, Haberl M, Gómez CC, Reindl M, et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc Natl Acad Sci. (2016) 113:3323–8. doi: 10.1073/pnas.1519608113
91. Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. (1985) 314:537–9. doi: 10.1038/314537a0
92. Winklmeier S, Schlüter M, Spadaro M, Thaler FS, Vural A, Gerhards R, et al. Identification of circulating MOG-specific B cells in patients with MOG antibodies. Neurol Neuroimmunol Neuroinflammation. (2019) 6:625. doi: 10.1212/NXI.0000000000000625
93. Takeshita Y, Obermeier B, Cotleur AC, Spampinato SF, Shimizu F, Yamamoto E, et al. Effects of neuromyelitis optica-IgG at the blood-brain barrier in vitro.. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e311. doi: 10.1212/NXI.0000000000000311
94. Li X, Wang L, Zhou L, ZhangBao J, Miao MZ, Lu C, et al. The imbalance between regulatory and memory B cells accompanied by an increased number of circulating T-follicular helper cells in MOG-antibody-associated demyelination. Mult Scler Relat Disord. (2019) 36:101397. doi: 10.1016/j.msard.2019.101397
95. Keller CW, Lopez JA, Wendel EM, Ramanathan S, Gross CC, Klotz L, et al. Complement activation is a prominent feature of MOGAD. Ann Neurol. (2021) 90:976–82. doi: 10.1002/ana.26226
96. Kothur K, Wienholt L, Tantsis EM, Earl J, Bandodkar S, Prelog K, et al. B cell, th17, and neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PloS One. (2016) 11:e0149411. doi: 10.1371/journal.pone.0149411
97. Kaneko K, Sato DK, Nakashima I, Ogawa R, Akaishi T, Takai Y, et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry. (2018) 89:927–36. doi: 10.1136/jnnp-2018-317969
98. Jain RW and Yong VW. B cells in central nervous system disease: diversity, locations and pathophysiology. Nat Rev Immunol. (2022) 22:513–24. doi: 10.1038/s41577-021-00652-6
99. Hofer LS, Ramberger M, Gredler V, Pescoller AS, Rostásy K, Sospedra M, et al. Comparative analysis of T-cell responses to aquaporin-4 and myelin oligodendrocyte glycoprotein in inflammatory demyelinating central nervous system diseases. Front Immunol. (2020) 11:1188. doi: 10.3389/fimmu.2020.01188
100. Costanza M, Poliani PL, Portararo P, Cappetti B, Musio S, Pagani F, et al. DNA threads released by activated CD4+ T lymphocytes provide autocrine costimulation. Proc Natl Acad Sci. (2019) 116:8985–94. doi: 10.1073/pnas.1822013116
101. Tompkins SM, Padilla J, Dal Canto MC, Ting JPY, Van Kaer L, and Miller SD. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. J Immunol. (2002) 168:4173–83. doi: 10.4049/jimmunol.168.8.4173
102. Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L, and Rothbard JB. T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature. (1986) 324:258–60. doi: 10.1038/324258a0
103. Slavin AJ, Soos JM, Stuve O, Patarroyo JC, Weiner HL, Fontana A, et al. Requirement for endocytic antigen processing and influence of invariant chain and H-2M deficiencies in CNS autoimmunity. J Clin Invest. (2001) 108:1133–9. doi: 10.1172/JCI13360
104. Jäger A, Dardalhon V, Sobel RA, Bettelli E, and Kuchroo VK. Th1, th17, and th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes1. J Immunol. (2009) 183:7169–77. doi: 10.4049/jimmunol.0901906
105. Merrill JE, Kono DH, Clayton J, Ando DG, Hinton DR, and Hofman FM. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc Natl Acad Sci U S A. (1992) 89:574–8. doi: 10.1073/pnas.89.2.574
106. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
107. Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci U S A. (2009) 106:12885–90. doi: 10.1073/pnas.0812530106
108. Horellou P, de Chalus A, Giorgi L, Leroy C, Chrétien P, Hacein-Bey-Abina S, et al. Regulatory T Cells Increase After rh-MOG Stimulation in Non-Relapsing but Decrease in Relapsing MOG Antibody-Associated Disease at Onset in Children. Front Immunol. (2021) 12:679770. doi: 10.3389/fimmu.2021.679770
109. Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, and Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci U S A. (2005) 102:13992–7. doi: 10.1073/pnas.0504979102
110. Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, and Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol. (1993) 143:555–64.
111. Linington C, Bradl M, Lassmann H, Brunner C, and Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. (1988) 130:443–54.
112. Kohyama K, Nishida H, Kaneko K, Misu T, Nakashima I, and Sakuma H. Complement-dependent cytotoxicity of human autoantibodies against myelin oligodendrocyte glycoprotein. Front Neurosci. (2023) 17:1014071. doi: 10.3389/fnins.2023.1014071
113. Lerch M, Schanda K, Lafon E, Würzner R, Mariotto S, Dinoto A, et al. More efficient complement activation by anti–aquaporin-4 compared with anti–myelin oligodendrocyte glycoprotein antibodies. Neurol Neuroimmunol Neuroinflammation. (2023) 10:e200059. doi: 10.1212/NXI.0000000000200059
114. Scolding NJ, Morgan BP, and Compston D a. S. The expression of complement regulatory proteins by adult human oligodendrocytes. J Neuroimmunol. (1998) 84:69–75. doi: 10.1016/S0165-5728(97)00241-5
115. Lin L, Ji M, Wu Y, Hang H, and Lu J. Neutrophil to lymphocyte ratio may be a useful marker in distinguishing MOGAD and MS and platelet to lymphocyte ratio associated with MOGAD activity. Mult Scler Relat Disord. (2023) 71:104570. doi: 10.1016/j.msard.2023.104570
116. Kaneko K, Sato DK, Nakashima I, Nishiyama S, Tanaka S, Marignier R, et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J Neurol Neurosurg Psychiatry. (2016) 87:1257–9. doi: 10.1136/jnnp-2015-312676
117. Rempe RG, Hartz AMS, and Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J Cereb Blood Flow Metab. (2016) 36:1481–507. doi: 10.1177/0271678X16655551
118. Pierson ER, Wagner CA, and Goverman JM. The contribution of neutrophils to CNS autoimmunity. Clin Immunol. (2018) 189:23–8. doi: 10.1016/j.clim.2016.06.017
119. Harbort CJ, Soeiro-Pereira PV, von Bernuth H, Kaindl AM, Costa-Carvalho BT, Condino-Neto A, et al. Neutrophil oxidative burst activates ATM to regulate cytokine production and apoptosis. Blood. (2015) 126:2842–51. doi: 10.1182/blood-2015-05-645424
120. Liu Y, Shields LBE, Gao Z, Wang Y, Zhang YP, Chu T, et al. Current understanding of platelet-activating factor signaling in central nervous system diseases. Mol Neurobiol. (2017) 54:5563–72. doi: 10.1007/s12035-016-0062-5
121. Yin JJ, Hu XQ, Mao ZF, Bao J, Qiu W, Lu ZQ, et al. Neutralization of interleukin-9 decreasing mast cells infiltration in experimental autoimmune encephalomyelitis. Chin Med J. (2017) 130:964. doi: 10.4103/0366-6999.204110
122. Deerhake ME, Danzaki K, Inoue M, Cardakli ED, Nonaka T, Aggarwal N, et al. Dectin-1 limits autoimmune neuroinflammation and promotes myeloid cell-astrocyte crosstalk via Card9-independent expression of Oncostatin M. Immunity. (2021) 54:484–98. doi: 10.1016/j.immuni.2021.01.004
123. Lees JR, Golumbek PT, Sim J, Dorsey D, and Russell JH. Regional CNS responses to IFN-gamma determine lesion localization patterns during EAE pathogenesis. J Exp Med. (2008) 205:2633–42. doi: 10.1084/jem.20080155
124. Kroenke MA, Chensue SW, and Segal BM. EAE mediated by a non-IFN-γ/non-IL-17 pathway. Eur J Immunol. (2010) 40:2340–8. doi: 10.1002/eji.201040489
125. Stoolman JS, Duncker PC, Huber AK, and Segal BM. Site-specific chemokine expression regulates central nervous system inflammation and determines clinical phenotype in autoimmune encephalomyelitis. J Immunol. (2014) 193:564–70. doi: 10.4049/jimmunol.1400825
126. Wendel EM, Thonke HS, Bertolini A, Baumann M, Blaschek A, Merkenschlager A, et al. Temporal dynamics of MOG antibodies in children with acquired demyelinating syndrome. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e200035. doi: 10.1212/NXI.0000000000200035
127. Ren C, Zhang W, Zhou A, Zhou J, Cheng H, Tang X, et al. Clinical and radiologic features among children with myelin oligodendrocyte glycoprotein antibody-associated myelitis. Pediatr Neurol. (2023) 143:96–9. doi: 10.1016/j.pediatrneurol.2023.02.019
128. ZhangBao J, Huang W, Zhou L, Tan H, Wang L, Wang M, et al. Clinical feature and disease outcome in patients with myelin oligodendrocyte glycoprotein antibody-associated disorder: a Chinese study. J Neurol Neurosurg Psychiatry. (2023) 94:825–34. doi: 10.1136/jnnp-2022-330901
129. Wang X, Zhao R, Yang H, Liu C, Wang W, Liu T, et al. Clinical analysis of myelin oligodendrocyte glycoprotein antibody-associated demyelination in children: A single-center cohort study in China. Mult Scler Relat Disord. (2022) 58:103526. doi: 10.1016/j.msard.2022.103526
130. Stiebel-Kalish H, Hellmann MA, Mimouni M, Paul F, Bialer O, Bach M, et al. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurology(R) neuroimmunol neuroinflammation. (2019) 6:e572. doi: 10.1212/NXI.0000000000000572
131. Pandit L, Mustafa S, Nakashima I, Takahashi T, and Kaneko K. MOG-IgG-associated disease has a stereotypical clinical course, asymptomatic visual impairment and good treatment response. Mult Scler J Exp Transl Clin. (2018) 4:2055217318787829. doi: 10.1177/2055217318787829
132. dos Santos N, Novaes LS, Dragunas G, Rodrigues JR, Brandão W, Camarini R, et al. High dose of dexamethasone protects against EAE-induced motor deficits but impairs learning/memory in C57BL/6 mice. Sci Reports. (2019) 9:6673. doi: 10.1038/s41598-019-43217-3
133. Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, et al. Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated peptide and dexamethasone in acetalated dextran microparticles. Mol Pharmaceutics. (2014) 11:828. doi: 10.1021/mp4005172
134. Nam J, Koppinen TK, and Voutilainen MH. MANF is neuroprotective in early stages of EAE, and elevated in spinal white matter by treatment with dexamethasone. Front Cell Neurosci. (2021) 15. doi: 10.3389/fncel.2021.640084
135. Wynford-Thomas R, Jacob A, and Tomassini V. Neurological update: MOG antibody disease. J Neurol. (2019) 266:1280–6. doi: 10.1007/s00415-018-9122-2
136. Whittam DH, Karthikeayan V, Gibbons E, Kneen R, Chandratre S, Ciccarelli O, et al. Treatment of MOG antibody associated disorders: results of an international survey. J Neurol. (2020) 267:3565–77. doi: 10.1007/s00415-020-10026-y
137. Savransky A, Rubstein A, Rios MH, Vergel SL, Velasquez MC, Sierra SP, et al. Prognostic indicators of improvement with therapeutic plasma exchange in pediatric demyelination. Neurology. (2019) 93:e2065–73. doi: 10.1212/WNL.0000000000008551
138. Bruijstens AL, Wendel EM, Lechner C, Bartels F, Finke C, Breu M, et al. E.U. paediatric MOG consortium consensus: Part 5 – Treatment of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatric Neurol. (2020) 29:41–53. doi: 10.1016/j.ejpn.2020.10.005
139. Baumann M, Hennes EM, Schanda K, Karenfort M, Kornek B, Seidl R, et al. Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): Extending the spectrum of MOG antibody positive diseases. Mult Scler. (2016) 22:1821–9. doi: 10.1177/1352458516631038
140. Chen JJ, Flanagan EP, Bhatti MT, Jitprapaikulsan J, Dubey D, Lopez Chiriboga ASS, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. (2020) 95:e111–20. doi: 10.1212/WNL.0000000000009758
141. Yao T, Zeng Q, Xie Y, Bi F, Zhang L, Xiao B, et al. Clinical analysis of adult MOG antibody-associated cortical encephalitis. Mult Scler Relat Disord. (2022) 60:103727. doi: 10.1016/j.msard.2022.103727
142. Armangue T, Olivé-Cirera G, Martínez-Hernandez E, Sepulveda M, Ruiz-Garcia R, Muñoz-Batista M, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol. (2020) 19:234–46. doi: 10.1016/S1474-4422(19)30488-0
143. Huppke P, Rostasy K, Karenfort M, Huppke B, Seidl R, Leiz S, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler. (2013) 19:941–6. doi: 10.1177/1352458512466317
144. Sechi E, Cacciaguerra L, Chen JJ, Mariotto S, Fadda G, Dinoto A, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): A review of clinical and MRI features, diagnosis, and management. Front Neurol. (2022) 13. doi: 10.3389/fneur.2022.885218
145. Rigal J, Pugnet G, Ciron J, Lépine Z, and Biotti D. Off-label use of tocilizumab in neuromyelitis optica spectrum disorders and MOG-antibody-associated diseases: A case-series. Mult Scler Relat Disord. (2020) 46:102483. doi: 10.1016/j.msard.2020.102483
146. Ringelstein M, Ayzenberg I, Lindenblatt G, Fischer K, Gahlen A, Novi G, et al. Interleukin-6 receptor blockade in treatment-refractory MOG-igG-associated disease and neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1100. doi: 10.1212/NXI.0000000000001100
147. Elsbernd PM, Hoffman WR, Carter JL, and Wingerchuk DM. Interleukin-6 inhibition with tocilizumab for relapsing MOG-IgG associated disorder (MOGAD): A case-series and review. Mult Scler Relat Disord. (2021) 48:102696. doi: 10.1016/j.msard.2020.102696
148. Broen JCA and van Laar JM. Mycophenolate mofetil, azathioprine and tacrolimus: mechanisms in rheumatology. Nat Rev Rheumatol. (2020) 16:167–78. doi: 10.1038/s41584-020-0374-8
149. Lipsky JJ. Mycophenolate mofetil. Lancet. (1996) 348:1357–9. doi: 10.1016/S0140-6736(96)10310-X
150. Kridin K and Ahmed AR. Post-rituximab immunoglobulin M (IgM) hypogammaglobulinemia. Autoimmun Rev. (2020) 19:102466. doi: 10.1016/j.autrev.2020.102466
151. Durozard P, Rico A, Boutiere C, Maarouf A, Lacroix R, Cointe S, et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol. (2020) 87:256–66. doi: 10.1002/ana.25648
152. Wong YYM, Hacohen Y, Armangue T, Wassmer E, Verhelst H, Hemingway C, et al. Paediatric acute disseminated encephalomyelitis followed by optic neuritis: disease course, treatment response and outcome. Eur J Neurol. (2018) 25:782–6. doi: 10.1111/ene.2018.25.issue-5
153. Chen JJ, Huda S, Hacohen Y, Levy M, Lotan I, Wilf-Yarkoni A, et al. Association of maintenance intravenous immunoglobulin with prevention of relapse in adult myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. (2022) 79:518–25. doi: 10.1001/jamaneurol.2022.0489
154. Cobo-Calvo A, Sepúlveda M, Rollot F, Armangué T, Ruiz A, Maillart E, et al. Evaluation of treatment response in adults with relapsing MOG-Ab-associated disease. J Neuroinflammation. (2019) 16:134. doi: 10.1186/s12974-019-1525-1
155. Chang X, Zhang J, Li S, Wu P, Wang R, Zhang C, et al. Meta-analysis of the effectiveness of relapse prevention therapy for myelin-oligodendrocyte glycoprotein antibody-associated disease. Mult Scler Relat Disord. (2023) 72:104571. doi: 10.1016/j.msard.2023.104571
156. Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS, Pardo S, Gornall M, Cicconi S, et al. Treatment of MOG-IgG-associated disorder with rituximab: An international study of 121 patients. Mult Scler Relat Disord. (2020) 44:102251. doi: 10.1016/j.msard.2020.102251
157. Zhou J, Lu X, Zhang Y, Ji T, Jin Y, Xu M, et al. Follow-up study on Chinese children with relapsing MOG-IgG-associated central nervous system demyelination. Mult Scler Relat Disord. (2019) 28:4–10. doi: 10.1016/j.msard.2018.12.001
158. Albassam F, Longoni G, Yea C, Wilbur C, Grover SA, and Yeh EA. Rituximab in children with myelin oligodendrocyte glycoprotein antibody and relapsing neuroinflammatory disease. Dev Med Child Neurol. (2020) 62:390–5. doi: 10.1111/dmcn.14336
159. Mao L, Yang L, Kessi M, He F, Zhang C, Wu L, et al. Myelin oligodendrocyte glycoprotein (MOG) antibody diseases in children in central south China: clinical features, treatments, influencing factors, and outcomes. Front Neurol. (2019) 10:868. doi: 10.3389/fneur.2019.00868
160. Perez-Aytes A, Marin-Reina P, Boso V, Ledo A, Carey JC, and Vento M. Mycophenolate mofetil embryopathy: A newly recognized teratogenic syndrome. Eur J Med Genet. (2017) 60:16–21. doi: 10.1016/j.ejmg.2016.09.014
161. Zheng Y, Cai MT, Li EC, Fang W, Shen CH, and Zhang YX. Case report: myelin oligodendrocyte glycoprotein antibody-associated disorder masquerading as multiple sclerosis: an under-recognized entity? Front Immunol. (2021) 12:671425. doi: 10.3389/fimmu.2021.671425
162. Perez-Giraldo G, Caldito NG, and Grebenciucova E. Transverse myelitis in myelin oligodendrocyte glycoprotein antibody-associated disease. Front Neurol. (2023) 14. doi: 10.3389/fneur.2023.1210972
163. Hacohen Y, Wong YY, Lechner C, Jurynczyk M, Wright S, Konuskan B, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody–associated disease. JAMA Neurol. (2018) 75:478–87. doi: 10.1001/jamaneurol.2017.4601
164. Sun X, Liu M, Luo X, Yuan F, Wang C, Wang S, et al. Clinical characteristics and prognosis of pediatric myelin oligodendrocyte glycoprotein antibody-associated diseases in China. BMC Pediatr. (2022) 22:666. doi: 10.1186/s12887-022-03679-3
165. Salvarani C, Magnani L, Catanoso M, Pipitone N, Versari A, Dardani L, et al. Tocilizumab: a novel therapy for patients with large-vessel vasculitis. Rheumatology. (2012) 51:151–6. doi: 10.1093/rheumatology/ker296
166. Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. (2008) 67:1516–23. doi: 10.1136/ard.2008.092932
167. Barreras P, Vasileiou ES, Filippatou AG, Fitzgerald KC, Levy M, Pardo CA, et al. Long-term effectiveness and safety of rituximab in neuromyelitis optica spectrum disorder and MOG antibody disease. Neurology. (2022) 99:e2504–16. doi: 10.1212/WNL.0000000000201260
168. Okubo S, Kakumoto T, Tsujita M, Muramatsu K, Fujiwara S, Hamada M, et al. Extremely longitudinally extensive transverse myelitis in a patient with myelin oligodendrocyte glycoprotein antibody-associated disease. Cureus. (2024) 16:e59938. doi: 10.7759/cureus.59938
169. Kojita Y, Okada N, Hirakawa M, Fujii K, Satou T, and Ishii K. Extensive brainstem lesions in myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): A case report. Radiol Case Rep. (2024) 19:5589–94. doi: 10.1016/j.radcr.2024.08.032
170. Abbas H, Kumar P, Quamar R, and Mani UA. Unravelling the complexity of myelin oligodendrocyte glycoprotein antibody-associated disease. Cureus. (2024) 16:e59840. doi: 10.7759/cureus.59840
171. Chen JJ, Huda S, Hacohen Y, Levy M, Lotan I, Wilf-Yarkoni A, et al. Association of maintenance intravenous immunoglobulin with prevention of relapse in adult myelin oligodendrocyte glycoprotein antibody–associated disease. JAMA Neurol. (2022) 79:518–25. doi: 10.1001/jamaneurol.2022.0489
172. Nishimoto N, Hashimoto J, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis. (2007) 66:1162–7. doi: 10.1136/ard.2006.068064
173. Fujihara K and Cook LJ. Neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody-associated disease: current topics. Curr Opin Neurol. (2020) 33:300. doi: 10.1097/WCO.0000000000000828
174. Montcuquet A, Collongues N, Papeix C, Zephir H, Audoin B, Laplaud D, et al. Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult Scler. (2017) 23:1377–84. doi: 10.1177/1352458516678474
175. Ghezzi A, Banwell B, Bar-Or A, Chitnis T, Dale RC, Gorman M, et al. Rituximab in patients with pediatric multiple sclerosis and other demyelinating disorders of the CNS: Practical considerations. Mult Scler. (2021) 27:1814–22. doi: 10.1177/1352458520932798
176. Bruschi N, Malentacchi M, Malucchi S, Sperli F, Martire S, Sala A, et al. Tailoring rituximab according to CD27-positive B-cell versus CD19-positive B-cell monitoring in neuromyelitis optica spectrum disorder and MOG-associated disease: results from a single-center study. Neurol Ther. (2023) 12:1375–83. doi: 10.1007/s40120-023-00481-w
177. Sotirchos ES, Vasileiou ES, Salky R, Huda S, Mariotto S, Chen JJ, et al. Treatment of myelin oligodendrocyte glycoprotein antibody associated disease with subcutaneous immune globulin. Mult Scler Relat Disord. (2022) 57:103462. doi: 10.1016/j.msard.2021.103462
178. Graf J, Mares J, Barnett M, Aktas O, Albrecht P, Zamvil SS, et al. Targeting B cells to modify MS, NMOSD, and MOGAD: Part 2. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e919. doi: 10.1212/NXI.0000000000000919
179. Hoy SM. Rozanolixizumab: first approval. Drugs. (2023) 83:1341–7. doi: 10.1007/s40265-023-01933-1
180. Zhu LN, Hou HM, Wang S, Zhang S, Wang GG, Guo ZY, et al. FcRn inhibitors: a novel option for the treatment of myasthenia gravis. Neural Regener Res. (2023) 18:1637–44. doi: 10.4103/1673-5374.363824
181. Hoffmann-La Roche. A study to evaluate the efficacy, safety, pharmacokinetics, and pharmacodynamics of satralizumab in patients with myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). ClinicalTrials.gov (2024). Available at: https://clinicaltrials.gov/study/NCT05271409 (Accessed April 30, 2025).
182. Roche. Pharmaceutical products (2024). Available online at: https://www.rocheCanada.com/solutions/pharma-solutions (Accessed April 30, 2025).
183. Heo YA. Satralizumab: first approval. Drugs. (2020) 80:1477–82. doi: 10.1007/s40265-020-01380-2
184. Xu J and Melenhorst JJ. CT103A, a forward step in multiple myeloma immunotherapies. Blood Sci. (2021) 3:59–61. doi: 10.1097/BS9.0000000000000068
185. Paschos KA and ChatziGeorgiadis A. Pathophysiological and clinical aspects of the diagnosis and treatment of bezoars. Ann Gastroenterol. (2019) 32:224–32. doi: 10.20524/aog.2019.0370
186. Sagan SA, Moinfar Z, Moseley CE, Dandekar R, Spencer CM, Verkman AS, et al. T cell deletional tolerance restricts AQP4 but not MOG CNS autoimmunity. Proc Natl Acad Sci U S A. (2023) 120:e2306572120. doi: 10.1073/pnas.2306572120
187. Remlinger J, Bagnoud M, Meli I, Massy M, Hoepner R, Linington C, et al. Modeling MOG antibody-associated disorder and neuromyelitis optica spectrum disorder in animal models: visual system manifestations. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200141. doi: 10.1212/NXI.0000000000200141
188. Lerch M, Bauer A, and Reindl M. The potential pathogenicity of myelin oligodendrocyte glycoprotein antibodies in the optic pathway. J Neuroophthalmol. (2023) 43:5–16. doi: 10.1097/WNO.0000000000001772
189. Rechtman A, Freidman-Korn T, Zveik O, Shweiki L, Hoichman G, and Vaknin-Dembinsky A. Assessing the applicability of the 2023 international MOGAD panel criteria in real-world clinical settings. J Neurol. (2024) 271:5102–8. doi: 10.1007/s00415-024-12438-6
190. Graber DJ, Levy M, Kerr D, and Wade WF. Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflammation. (2008) 5:22. doi: 10.1186/1742-2094-5-22
191. Sechi E, Krecke KN, Messina SA, Buciuc M, Pittock SJ, Chen JJ, et al. Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology. (2021) 97:e1097–109. doi: 10.1212/WNL.0000000000012467
192. Van Delden C and Iglewski BH. Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerg Infect Dis. (1998) 4:551–60. doi: 10.3201/eid0404.980405
193. Gawde S, Siebert N, Ruprecht K, Kumar G, Ko RM, Massey K, et al. Serum proteomics distinguish subtypes of NMO spectrum disorder and MOG antibody-associated disease and highlight effects of B-cell depletion. Neurol Neuroimmunol Neuroinflamm. (2024) 11:e200268. doi: 10.1212/NXI.0000000000200268
194. Evonuk KS, Wang S, Mattie J, Cracchiolo CJ, Mager R, Ferenčić Ž, et al. Bruton’s tyrosine kinase inhibition reduces disease severity in a model of secondary progressive autoimmune demyelination. Acta Neuropathol Commun. (2023) 11:115. doi: 10.1186/s40478-023-01614-w
195. Remlinger J, Madarasz A, Guse K, Hoepner R, Bagnoud M, Meli I, et al. Antineonatal fc receptor antibody treatment ameliorates MOG-igG–associated experimental autoimmune encephalomyelitis. Neurol Neuroimmunol Neuroinflamm. (2025) 9:e1134. doi: 10.1212/NXI.0000000000001134
196. Duan Y, Zhuo Z, Li H, Tian DC, Li Y, Yang L, et al. Brain structural alterations in MOG antibody diseases: a comparative study with AQP4 seropositive NMOSD and MS. J Neurol Neurosurg Psychiatry. (2021) 92:709–16. doi: 10.1136/jnnp-2020-324826
Keywords: myelin oligodendrocyte glycoprotein antibody-associated disorder (MOGAD), experimental autoimmune encephalomyelitis (EAE), pathophysiology, innate immunity, adaptive immunity, treatment strategies
Citation: Zhang Y and Li D (2025) Translational insights from EAE models : decoding MOGAD pathogenesis and therapeutic innovation. Front. Immunol. 16:1530977. doi: 10.3389/fimmu.2025.1530977
Received: 19 November 2024; Accepted: 21 April 2025;
Published: 20 May 2025.
Edited by:
Simona Bonavita, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Reza Rahmanzadeh, TheUltra.ai, SwitzerlandFabrícia Lima Fontes-Dantas, Rio de Janeiro State University, Brazil
Copyright © 2025 Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Li, dGpla3NqQDE2My5jb20=
†Present addresses: Yanjia Zhang, Tianjin Key Laboratory of Birth Defects for Prevention and Treatment, Tianjin, China Dong Li, Tianjin Key Laboratory of Birth Defects for Prevention and Treatment, Tianjin, China