 Lucas Dada1†
Lucas Dada1† Emiko Nagai1†
Emiko Nagai1† Sashank Agrawal2†
Sashank Agrawal2† Ariel S. Wirchnianski3
Ariel S. Wirchnianski3 Ian A. Wilson2,4Kartik Chandran3
Ian A. Wilson2,4Kartik Chandran3 Seiya Kitamura1*
Seiya Kitamura1*- 1Department of Biochemistry, Albert Einstein College of Medicine, Bronx, NY, United States
- 2Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, United States
- 3Department of Microbiology & Immunology, Albert Einstein College of Medicine, Bronx, NY, United States
- 4Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA, United States
Ebola virus (EBOV) causes severe hemorrhagic fever with a high mortality rate in humans. In acute infection, an abnormal immune response results in excessive inflammatory cytokines and uncontrolled systemic inflammation that can result in organ damage and multi-organ failure. While vaccines and monoclonal antibody therapies are available, there is an urgent need for effective small-molecule antivirals against EBOV. Here, we report on the optimization of tamoxifen, an EBOV-glycoprotein (GP) binder that inhibits viral entry, using our Sulfur-Fluoride Exchange (SuFEx) click chemistry-based high-throughput medicinal chemistry (HTMC) strategy. Using a “Direct-to-Biology” approach, we generated a focused library of 2,496 tamoxifen analogs overnight and screened them in a cell-based pseudo-EBOV infection assay. The HTMC workflow enabled the development of a potent EBOV entry inhibitor with submicromolar EC50 cellular antiviral activity and more than 50-fold improvement in binding affinity against EBOV-GP compared to the parent compound. Our findings underscore the use of SuFEx-enabled HTMC for rapidly generating and assessing potential therapeutic candidates against viral and immune-mediated diseases in a cell-based assay.
1 Introduction
Ebola virus (EBOV) is an enveloped, single-stranded, negative-sense RNA virus in the family Filoviridae (1). EBOV infection in humans can lead to Ebola virus disease (EVD), a clinical syndrome initially characterized by nonspecific symptoms, which later progress to severe gastrointestinal issues and hemorrhagic complications with a lethality rate as high as 90% (2–4). While other orthoebolaviruses, such as Sudan virus and Marburg virus, can also cause human disease with substantial mortality, EBOV has been responsible for the majority of recorded human outbreaks and therefore, remains to be of considerable public health concern (5). The unprecedented 2014–2016 EVD epidemic in West Africa, as well as the 2022 outbreak in the Democratic Republic of the Congo have underscored the potential of EVD to trigger severe health emergencies on a regional scale (6, 7).
EBOV causes an acute and serious viral hemorrhagic fever disease, which is often fatal if left untreated. EBOV primarily targets host macrophages leading to cell activation and systemic cytokine storm. Fatal infection is associated with an inhibited interferon response and lymphopenia. Cytokine storms are a hallmark of EVD and play a central role in its pathogenesis, marked by the induction of both pro- and anti-inflammatory responses (8, 9). Despite the high mortality rate associated with EVD, some patients survive and, in certain cases, develop chronic manifestations that may resemble inflammatory or autoimmune conditions (10). It has been demonstrated that this EBOV-induced autoimmunity is involved in post EVD syndrome (11). These features of EVD highlight the necessity for effective therapeutic approaches against EBOV. Recently, EBOV vaccine and monoclonal antibody-based therapeutics have been approved by the FDA (12, 13). However, there are currently no FDA-approved small-molecule drugs with demonstrated efficacy against filovirus infection and/or disease in humans, despite the increasing frequency with which these viruses are causing outbreaks of global concern (14). Therefore, there is a pressing and unmet need for effective therapies to prevent EBOV entry and subsequent infection.
In the pursuit of drug discovery against EVD, several high-throughput screening campaigns have been conducted that yielded hit compounds (15, 16). However, only limited medicinal chemistry optimization and in vivo follow-up studies were performed previously, partially due to the iterative cycle of medicinal chemistry process being time-consuming and labor-intensive. Our group has developed the first-of-its-kind high-throughput medicinal chemistry (HTMC) platform using click chemistry reactions, in particular Sulfur-Fluoride Exchange (SuFEx) reactions (17–21), to accelerate the medicinal chemistry process. The unanticipated discovery that iminosulfur oxydifluoride (difluoride, RN=S(O)F2)-containing molecules react overnight with amines to yield products with >80% conversion in a biocompatible condition (22) allowed us to perform large-scale structure-activity relationship (SAR) studies directly from reaction mixtures. We have used this new type of click chemistry reaction to rapidly synthesize focused libraries of lead compound analogs in a miniaturized format, directly assess the products with biological assays (also known as “Direct-to-Biology (D2B)” approach) (23), and develop drug-like ligands with improved biological potency. We have previously demonstrated the utility of our SuFEx-based HTMC method to improve the potency and specificity of chemical probes against a bacterial pathogenic protease, a human leukemia-associated transcriptional coactivator, the influenza hemagglutinin stem, and molecular glues (17–21).
In this study, we applied our SuFEx-based HTMC platform to an inhibitor of the EBOV-glycoprotein (GP) for the expedited analysis of structure-activity relationships. Tamoxifen, an EBOV-GP inhibitor (24, 25), was used as a starting scaffold for diversification because of its suitability for the synthetic preparation of a SuFExable analog. With the support of an automated liquid handling robot, a focused library of 2,496 compound analogs was synthesized in a single batch and screened using a cell-based EBOV entry assay. This study provides a large-scale SAR dataset of tamoxifen analogs as EBOV entry inhibitors, which will be valuable for the further development of small molecule therapeutics against EVD. Importantly, the HTMC workflow enabled the discovery of an analog with a 50-fold improved binding affinity compared to tamoxifen. Our study showcases the successful application of the SuFEx-based HTMC platform for the accelerated structure-activity relationship study of small molecule inhibitors, ultimately providing next-generation therapeutic modalities against viral infection and autoimmune diseases.
2 Materials and methods
2.1 Cells and viruses
The Vero African green monkey kidney cells in this study were obtained from American Type Culture Collection (ATCC), Cat# CCL-81. Vero cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Corning, NY) containing 10% fetal bovine serum (FBS, Phoenix Scientific, St. Joseph, MO), 1% penicillin-streptomycin (Corning, NY), and GlutaMax (Thermo Fisher Scientific, Waltham, MA). Cells were maintained at 37°C in 5% CO2. Generation and propagation of recombinant vesicular stomatitis virus (rVSV) encoding enhanced green fluorescent protein (eGFP) in the first position and replacing VSV G with the EBOV-GP (EBOV/H.sap-tc/COD/76/Yambuku-Mayinga) or Lassa virus (LASV)-GP (Josiah) were previously described (26, 27).
2.2 Computational protocol
Docking studies were carried out using Glide-SP (Schrödinger modeling suite, versions 2024-1 to 2024-3) to obtain the binding mode for tamoxifen into the proposed binding site. Standard option joint with the SP algorithm was applied for pose generation and evaluation. The X-ray structure for the Zaire EBOV-GP in complex with toremifene (PDB: 5JQ7) was used as a template for the docking.
2.3 Chemical synthesis
The details of chemical synthesis and characterization are described in Supplementary Material. The SuFEx-based library synthesis was adapted from a method described previously (17) in 384-well plate format. Briefly, to a DMSO solution of the iminosulfur oxydifluoride derivative 1 was added amine library in DMSO and sodium phosphate buffer (pH 9.0, 0.2 M) subsequently. The compounds were synthesized with difluoride concentration at 1 mM and a solvent mixture DMSO:buffer 3:1. The reaction mixtures were shaken at room temperature overnight and then used directly for activity measurement with 5000-fold dilution.
2.4 Screening and EC50 measurement using an EBOV-GP-pseudotyped virus
The virus was titrated to achieve an infection rate of approximately 50%. Vero cells were seeded at a density of 2.0 × 104 cells/well in a 384-well plate and incubated for 24 hours at 37°C in 5% CO2. The cell culture medium was then replaced with 20 μL/well of virus culture medium (DMEM containing 2% FBS, 1% penicillin-streptomycin, and GlutaMax), and 0.2 μL/well of the compound library in DMSO was added using the Bravo Automated Liquid Handling Platform (Agilent Technologies, Santa Clara, CA). Subsequently, 20 μL/well of virus solution was added, and the cells were incubated for 16 hours at 37°C in 5% CO2. Infected cells were stained with Hoechst (Thermo Fisher Scientific, Waltham, MA) to visualize nuclei and fixed with 4% paraformaldehyde. Infectivity of VSV pseudotype was measured by automated enumeration of eGFP+ cells using a Cytation 5 reader (BioTek, Winooski, VT), as previously described (26). Quantification was done using Gen5 data analysis software (BioTek, Winooski, VT). For each plate, DMSO-treated infected and non-infected control wells were included to calculate the inhibition rate as: Inhibition rate (%) = (Infection control - Sample)/(Infection control - Non-infection control) × 100.
Dose-response assays were performed under the same experimental conditions as the library screening. Compounds were prepared in two-fold serial dilutions in DMSO and 0.2 μL/well was added to the cell culture media using the Bravo Automated Liquid Handling Platform. The EC50 values were calculated using GraphPad Prism 10.
2.5 Cytotoxicity measurement
Cells were seeded under the same conditions as the antiviral assay. The cell culture medium was replaced with 40 μL/well of virus infection medium without virus, and compound solutions prepared as two-fold serial dilutions were added at 0.2 μL/well using the Bravo platform. After 16 hours, Promega® CellTiter-Glo® 2.0 (Promega, Madison, WI) was added, and cell viability was assessed following the manufacturer’s instruction. The CC50 values were calculated using GraphPad Prism 10.
2.6 Expression and purification of EBOV-GP trimer protein
The gene fragment encoding the extracellular domain of the Zaire EBOV (strain Mayinga-76) glycoprotein (UniProt ID: KB-Q05320) was synthesized as described previously (28). This gene was inserted into the mammalian expression vector pHCMV3, with an Ig Cκ leader sequence at the N-terminus to enable secretion. A foldon trimerization domain from bacteriophage T4 fibritin was added to the C-terminus to promote trimerization, along with a 6×His tag for affinity purification.
The plasmid was transfected into human Expi293S cells to produce the EBOV-GP trimers. After six days of incubation, the culture medium containing the secreted trimers was collected. The protein was purified using Ni-NTA affinity chromatography with Ni Sepharose Excel resin and dialyzed overnight into 1× PBS. The sample was concentrated and further purified via size-exclusion chromatography on a Superdex 200 HiLoad 16/600 column pre-equilibrated with 1× TBS. The protein peak corresponding to the EBOV-GP trimers was identified, collected, and concentrated to a final concentration of 2 mg/mL.
2.7 Microscale thermophoresis (MST)
Recombinant EBOV-GP protein was labeled using the Monolith Protein Labeling Kit RED-tris-NTA 2nd Generation dye (Cat #MO-L018, NanoTemper Technologies, Germany) following the manufacturer’s instructions. Specifically, 125 nM EBOV-GP was incubated with 25 nM dye in 25 mM HEPES pH 7.5, 100 mM NaCl, 0.005% tween-20 in the dark at room temperature for 30 min. The sample was centrifuged for 10 min at 4°C and 14,000 g and the supernatant was transferred to a fresh tube. To determine the KD of EBOV-GP to tamoxifen and its analogs, 125 nM labeled EBOV-GP was incubated with increasing concentrations of small molecules in the same buffer with 1% DMSO. Samples were loaded into standard glass capillaries (Monolith NT.155 Capillaries) and analyzed by MST using a Monolith NT.115 Blue/Red, LED power and IR laser power of 60%. Samples showed no aggregation according to post-run analysis using the Monolith data collection software (NanoTemper). Fraction bound and error were generated by NanoTemper software (MO.Affinity Analysis) and KD values were determined using GraphPad Prism 10 and nonlinear fit of one-site specific binding.
2.8 Crystallization and structure determination
For crystallization of EBOV-GP trimers, the protein was concentrated to 8 mg/mL in a buffer containing 20 mM Tris (pH 8.0) and 150 mM NaCl. Crystals were grown at 20°C in a solution of 9% PEG 6000 and 100 mM sodium citrate (pH 5.2). Complex structures were obtained by soaking EBOV-GP crystals in a 5 mM solution of the target compound for a few minutes, followed by cryoprotection with 20% glycerol and rapid plunging into liquid nitrogen for storage before data collection. Diffraction data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) on beamline BL12-1. EBOV-GP crystals soaked with compound 4R diffracted to a resolution of 2.59 Å. Data indexing, integration, and scaling were performed using HKL2000 (29). Structures were solved by molecular replacement (MR) with Phaser in Phenix (30) with PDB 6F6I as the MR model, and subsequent model building and refinement were conducted using Coot and Phenix.refine (31, 32). Structural quality was assessed with MolProbity (33), and further validation was performed using the PDB validation server. Data collection and refinement statistics are summarized in Supplementary Table 1.
3 Results
3.1 SuFExable tamoxifen analog design and validation of biological activity
The first step of the SuFEx-enabled HTMC workflow requires installation of a SuFExable difluoride moiety on the lead molecule (Figure 1A). This difluoride analog can be readily synthesized by reacting thionyl tetrafluoride (O=SF4) gas with the lead compound functionalized with a primary amine. A structural analysis of EBOV-GP protein + ligand complex was performed to determine the modification site on the lead molecule. Due to the absence of an experimentally determined crystal structure for the complex of EBOV-GP with tamoxifen, molecular docking was used to predict the binding mode of this ligand. For this purpose, we employed a crystal structure of toremifene (i.e. an analog of tamoxifen) bound to EBOV-GP (PDB entry: 5JQ7) as a template (34). Given the chemical structural similarity between tamoxifen and toremifene, we expected that both molecules would share similar binding conformations.
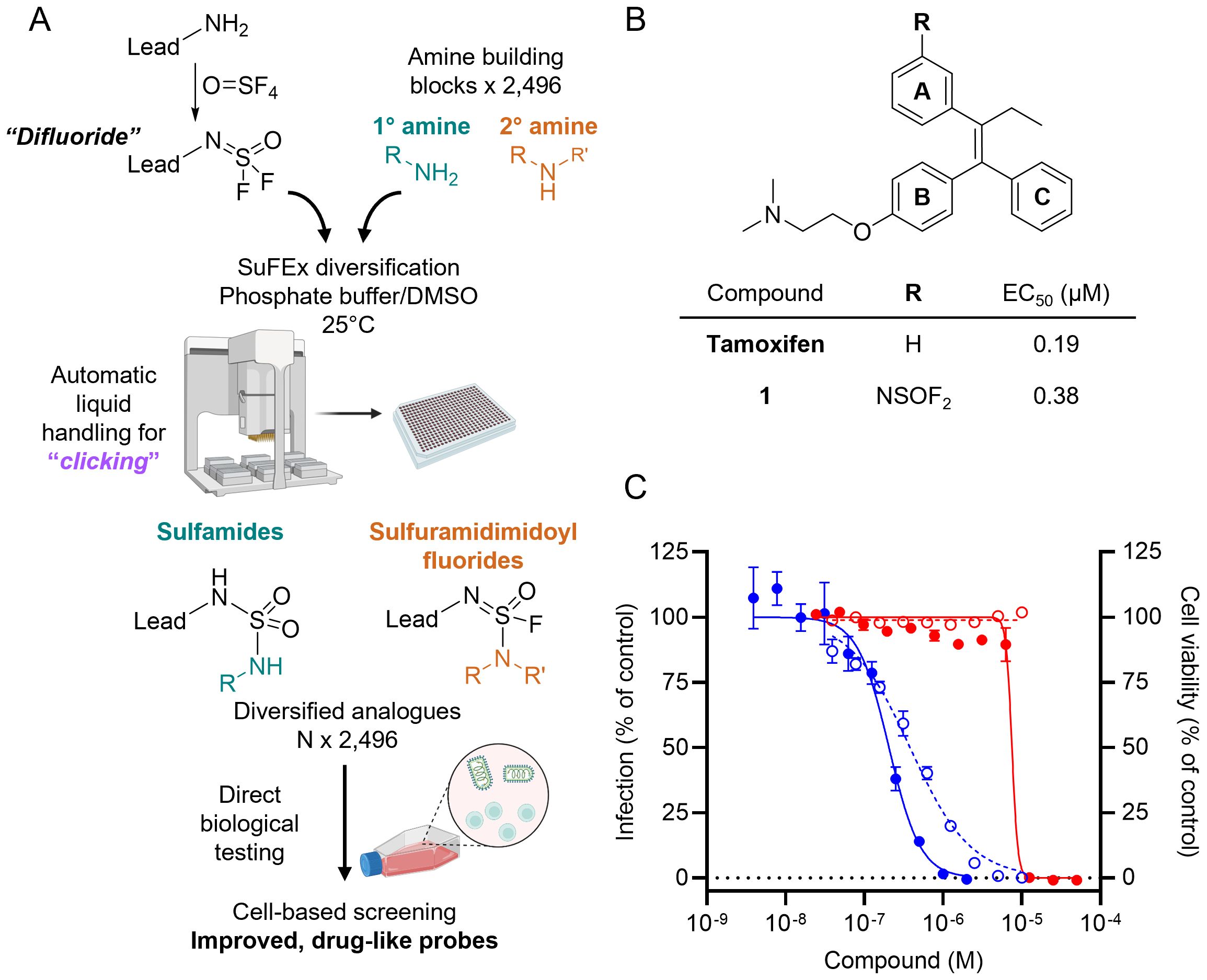
Figure 1. HTMC-based diversification of EBOV entry lead inhibitor tamoxifen. (A) Schematics of SuFEx-based HTMC workflow. (B) Chemical structure and EC50 of tamoxifen and the difluoride analog 1 against VSV-EBOV-GP (Vero cells). (C) Dose-response curves of VSV-EBOV-GP infection (blue) and Vero cell viability (red) for tamoxifen (●, solid lines) and compound 1 (○, dotted lines). Mean ± SD values are shown (n = 3).
The docking suggests that tamoxifen aligns closely with the binding conformation of toremifene, preserving key interactions with crucial residues within the active site (Supplementary Figure 1). The aromatic rings B and C of tamoxifen adopt a similar orientation to the corresponding rings in toremifene, forming a π-stacking between these rings and Tyr517, as well as an ionic interaction between the protonated tertiary amine and the negatively charged side chain of Glu100. Additionally, the docking analysis highlights a solvent-exposed area at the meta position on the aromatic ring A, making it a prime candidate site for further functionalization. By targeting these solvent-exposed positions, we hypothesized that expanding the molecule into unoccupied regions of the binding pocket could form additional interactions with adjacent residues, thus enhancing both binding affinity and cellular anti-infective efficacy.
Guided by the predicted structure, we designed a synthetic route (Supplementary Figure 2) to introduce a difluoride group at the meta position of ring A of tamoxifen based on previous reports for the synthesis of analogs with the triphenylethylene core (25). The difluoride-containing analog 1 was successfully synthesized, and its chemical structure was verified by NMR and LC-MS analyses, as described in the Supplementary Material.
With analog 1 in hand, we assessed its antiviral activity and cytotoxicity, along with that of tamoxifen, using a virus-like particle (VLP) assay (26). The assay employs vesicular stomatitis virus (VSV) particles displaying EBOV-GP (VSV-EBOV-GP), in place of the native glycoprotein G, to infect Vero cells in culture. This approach obviated the need for high-security BSL4 facilities required for working with authentic filoviruses. The VSV-EBOV-GP also encodes an enhanced GFP, which allows for direct quantification of infected cells by fluorescence imaging. The use of a cell-based assay provides advantages for drug discovery, as it more accurately reflects the compounds’ activity in a biologically relevant environment and accelerates the profiling of cell-active compounds. This infection model enabled us to evaluate the inhibitor’s effectiveness in blocking the processing of viral GP, a crucial step for EBOV infection. Additionally, the VSV assay provided insight into whether the proposed structural modifications were tolerated by the lead compound without significantly compromising its activity.
The results showed that the lead compound tamoxifen has an EC50 value of 0.19 μM, while our difluoride-functionalized analog 1 exhibited an EC50 of 0.38 μM, with both values being within the same magnitude (Figure 1B). Importantly, compound 1 did not exhibit cytotoxicity up to 10 µM (Figure 1C). These findings indicate that the structural modifications to introduce the difluoride moiety do not critically reduce the biological activity of the lead compound, consistent with the docking predictions. The results support the effectiveness of the docking-guided design and validate the choice of the modification site on the tamoxifen aromatic ring.
3.2 SuFEx-enabled HTMC
Once an appropriate SuFExable derivative of the lead compound has been identified, the next step in the HTMC workflow (Figure 1A) is the diversification reaction with an amine-fragment library. This strategy allows the preparation of a focused library of lead compound analogs with expanded diversity containing sulfamide or sulfuramidimidoyl fluoride linkages, from primary or secondary amines respectively (Figure 2A).
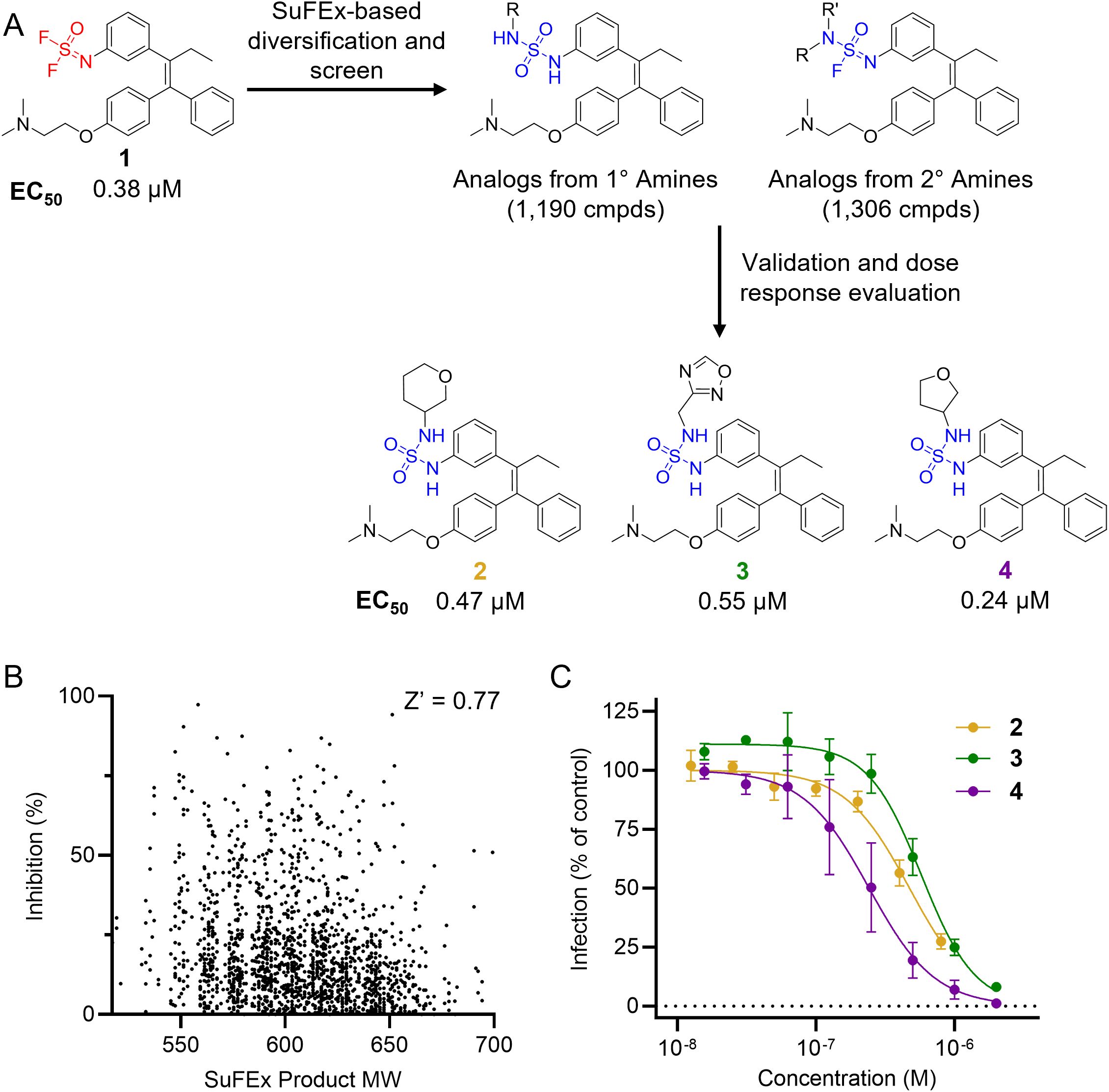
Figure 2. Reaction schematics and screening results of HTMC against EBOV entry. (A) HTMC workflow from a difluoride functionalized lead compound to the improved compounds. Chemical structures and EC50 (VSV-EBOV-GP, Vero cells) of representative hit molecules (compounds 2, 3, and 4) are shown. (B) Scatter plot for the tamoxifen-based HTMC library screening. Screening was performed at a small-molecule concentration of 200 nM. (C) Dose-response curves for representative hits identified from the HTMC library screening. Hit molecules were resynthesized in mg scale, purified, and chemically characterized, and their antiviral potency was measured against VSV-EBOV-GP using Vero cells. Mean ± SD values are shown (n = 3).
A library of 2,496 primary and secondary amine fragments were individually reacted with the difluoride-containing analog in 384-well plates overnight at room temperature using a 1:3 phosphate buffer:DMSO solvent mixture (see Materials and Methods for further details). The reaction conditions employed for diversification were determined in a preliminary assessment of the difluoride reactivity with a small set of representative amines. Reagents concentration, temperature, pH, and solvent composition were evaluated to identify the optimal conditions for generating a high-quality library, allowing us to screen the compound set without additional purification. The synthesis of the library was performed using an automated liquid handling robot Agilent Bravo BenchCel, enabling efficient generation of 2,496 analogs in a single batch. An LC-MS analysis of a randomly selected wells was performed to evaluate the quality of the library. The results indicated a generally high conversion rate, validating the suitability of the library for the D2B screen in the subsequent step.
The synthesized library was screened at 200 nM using the VSV-based EBOV entry assay. The library solutions were dispensed over Vero cells in 384-well plates. Cells treated with the compound were infected with VSV-EBOV-GP, and 16 hours post-infection, their nuclei were stained and then fixed. The infection rate was assessed by measuring eGFP expression levels, normalized to the number of nuclei. This approach allowed us to screen the compound set in singlicate with an assay Z′-factor of 0.77, indicating robust assay performance. The screening results are illustrated in Figure 2B as a scatter plot of % inhibition. After validating the hits by triplicate, the most potent molecules were manually synthesized in milligram quantities for further dose-dependent biochemical and cell-based characterization. This approach allowed us to confirm the efficacy and refine the profile of the most promising candidates. The structure of the representative hits synthesized for validation is shown in Figure 2A (compounds 2-4), and the dose-response inhibition profile in Figure 2C. Compound 4 was identified as one of the top hits in the primary screening, with an EC50 value of 241 nM. Notably, compound 4 demonstrated an almost 2-fold increase in cellular antiviral potency compared to 1.
3.3 Structure-activity relationships and biophysical analysis
To further understand the structure-activity relationships, we synthesized a small library of analogs of the identified molecules and subjected to dose-response antiviral and cytotoxicity studies. Specifically, we focused on modifications to the oxolane ring (Table 1, Supplementary Figure 3). These modifications included altering the ring size (5), investigating different substitution patterns on the oxolane ring (6, 7), and replacing the oxolane with an N-methyl pyrrolidine ring (8) as well as other N-substituted analogs with varying polarity and chain lengths (9 – 15). Despite exploration of these broad structural modifications, no significant improvement in activity was observed with the ring changes. Since compound 4 was identified as a racemic mixture, we synthesized and evaluated both R and S enantiomers (Table 1). Interestingly, the S-configured compound (4S) exhibited a superior EC50 value of 92 nM, compared to its R-counterpart (4R) of 351 nM, indicating the importance of the chirality on the improved potency and specific interaction with GP-protein. The cytotoxicity studies showed variable CC50 values among the synthesized analogs. Compounds 4S and 4R did not show significant cytotoxicity below 10 µM with an excellent selectivity index (SI) of 150 for 4S (Table 1).
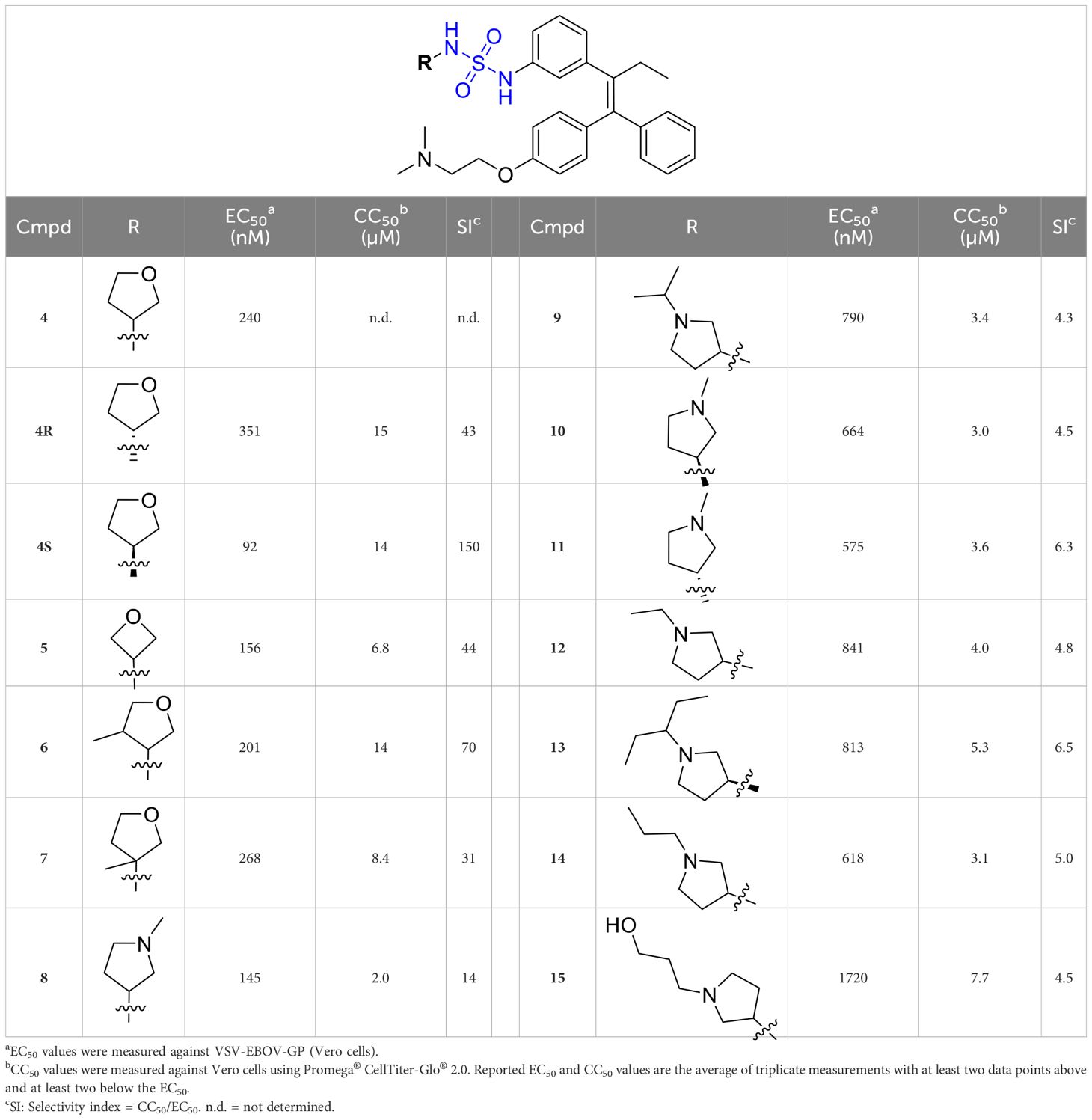
Table 1. Structure-activity relationships of compound 4 analogs against VSV-EBOV-GP.
To confirm the direct interaction of 4R and 4S with EBOV-GP, the equilibrium dissociation constants (KD) for the interaction inhibitor-protein were measured. The recombinant protein was expressed in Expi293S cells and purified using Ni-NTA affinity and size-exclusion chromatography. Then, we evaluated the interaction of both enantiomers as well as the parent tamoxifen with EBOV-GP by microscale thermophoresis (MST). As shown in Figure 3A, 4S displayed a stronger binding affinity against EBOV-GP, with a KD value of 0.63 µM, compared to 4R with a KD value of 6.4 µM. In comparison, the corresponding value for tamoxifen was 32 µM. This trend mirrors the observed differences in cellular potency, where 4S demonstrated enhanced efficacy at low submicromolar concentrations in blocking EBOV entry, while 4R required higher concentrations to achieve a comparable effect (Table 1). It is worth noting the 50-fold affinity enhancement in the biochemical assay between 4S and tamoxifen, providing further validation of improved potency of the inhibitor developed through our HTMC platform.
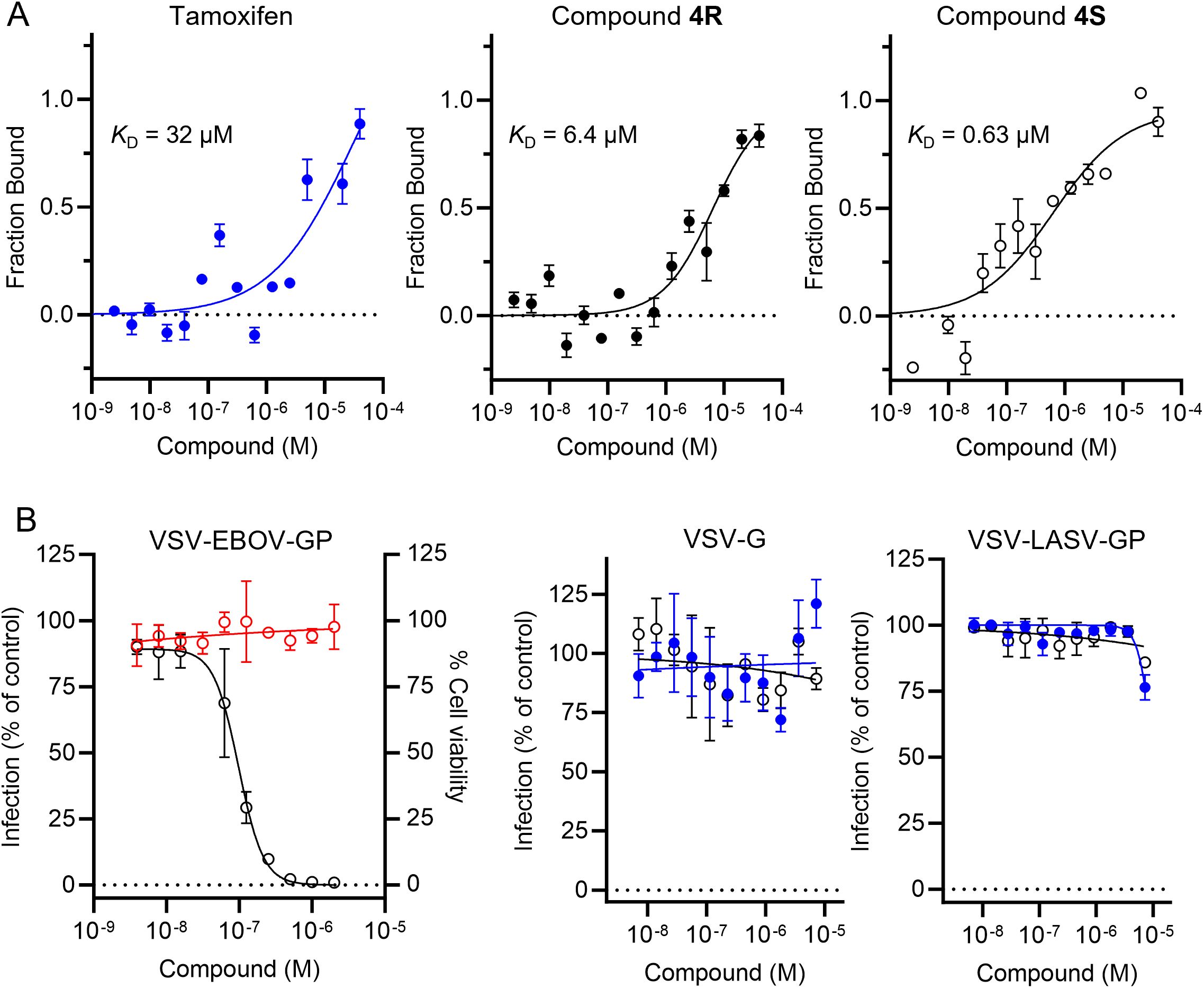
Figure 3. Characterization of the top tamoxifen analog 4S identified from the HTMC campaign. (A) The biophysical affinity of tamoxifen, 4R and 4S towards EBOV-GP as measured by MST. Error bars indicate the standard deviation of three technical replicates (n = 3). (B) Dose-response curves of VSV-EBOV-GP infection (○) and Vero cell viability (○) for compound 4S (left panel). Tamoxifen (●) and compound 4S (○) were tested for their ability to inhibit VLPs displaying the VSV-G glycoprotein (middle panel) and LASV-GP (right panel). Mean ± SD values are shown (n = 3).
We then determined the X-ray structure of 4R in complex with EBOV-GP to 2.59 Å resolution (Figure 4, Supplementary Table 1). The electron density for 4R is well defined (Figure 4B) and the compound binds within the extensive hydrophobic cavity of EBOV-GP, a known target site for other inhibitors. It adopts a similar pose to its analog, toremifene, with conserved interactions involving the three aromatic rings and the dimethylethanamine group of the parent scaffold (34). Compared to toremifene, 4R occupies a larger portion of the available cavity, with its additional moiety engaging a hydrophobic subpocket, potentially contributing to its improved binding. Unfortunately, we were unable to obtain a structure for 4S, as it renders the crystal unstable, possibly due to conformational changes when the compound is soaked into the crystal, and leads to poor diffraction. Nevertheless, the complex structure of EBOV-GP and 4R validates the docking model of our lead molecule and provides structural insights into the improved potency of the developed molecules.
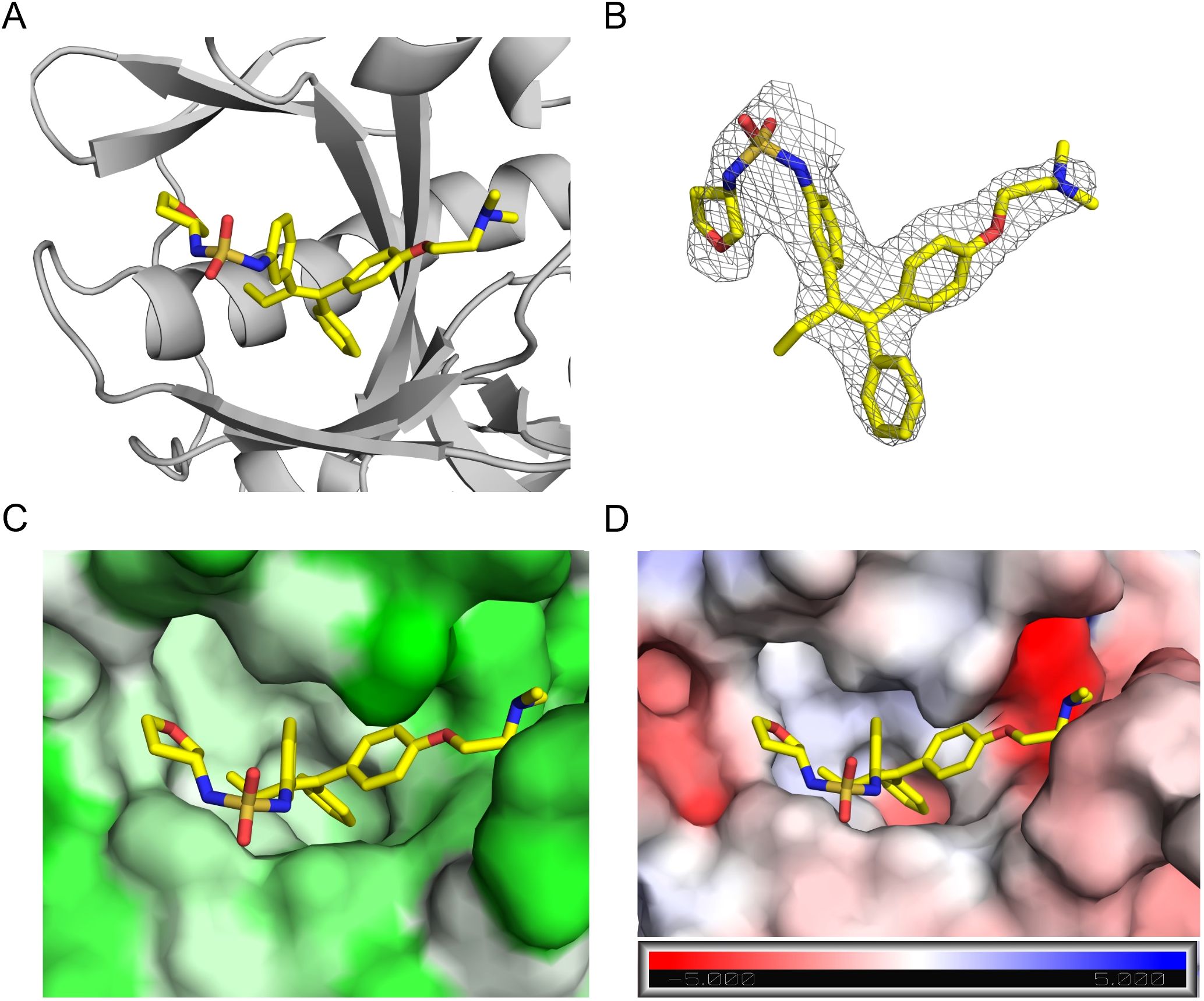
Figure 4. Structure of EBOV-GP in complex with compound 4R. (A) Crystal structure of EBOV-GP trimer bound to 4R. The compound is depicted as sticks, and the protein as a cartoon representation. (B) 2Fo-Fc electron density map contoured at 1 sigma for 4R. (C) 4R bound within the hydrophobic binding pocket of EBOV-GP. The surface is colored according to hydrophobicity, with pale green indicating the most hydrophobic regions and green the least hydrophobic. (D) Electrostatic surface potential of the binding pocket, colored from red (most negative) to blue (most positive), with 4R bound. The color scale for electrostatics is shown at the bottom of the figure.
Finally, we evaluated the viral selectivity and specificity of compound 4S by comparing the inhibitory activity against VSV-EBOV-GP to VSV-G and VSV particles displaying LASV-GP (VSV-LASV-GP, Figure 3B). Tamoxifen and our compound 4S showed selective inhibition of VSV-EBOV-GP but not VSV-G or VSV-LASV-GP, indicating that its antiviral activity is specific to EBOV-GP. This result supports the hypothesis that compound 4S does not induce non-specific effects on viral entry mechanisms shared by multiple viruses or inhibit proteins involved in later stages of the EBOV infection, such as cathepsins, which are known to mediate viral entry through endosomal processing. Furthermore, the lack of activity in VSV-G and VSV-LASV-GP models reduces the likelihood that the compound’s mechanism involves general endosomal disruption or interference with acidic environments. This selectivity, in concert with the chiral preference of 4S over its R-isomer in binding EBOV-GP, suggests that the compound engages specific molecular interactions with EBOV-GP that are crucial for its activity.
4 Discussion
In this study, we leveraged our unique SuFEx-based HTMC platform to generate a large-scale library of tamoxifen analogs with the aim of identifying potent inhibitors of EBOV entry. This approach enabled a rapid SAR analysis that accelerated the identification of novel EBOV inhibitors, highlighting the potential of HTMC workflow as a powerful tool for antiviral drug discovery. Our inhibitor discovery efforts were motivated by the need for effective small-molecule capable of modulating EBOV infection, especially given that current treatment options primarily include vaccines and monoclonal antibodies. While these approaches have shown promise, there remains an unmet need for potent small molecules that can be translated to clinical application. This work also demonstrated a successful application of our SuFEx-based HTMC platform for accelerated optimization of a potential antiviral compound, as we have shown previously for other biological targets (17–21).
To develop this strategy, we employed tamoxifen as a lead scaffold to design an analog with a required SuFExable hub for accelerated diversification. Although other analogs of tamoxifen, such as toremifene and clomiphene, have been extensively characterized as EBOV entry inhibitors (24), tamoxifen was the most suitable scaffold in terms of synthetic design to access to the required functionalized difluoride. The SuFEx-enabled HTMC approach applied here is especially well-suited to drug discovery for infectious diseases such as EVD, where the iterative cycle of lead optimization can be a significant bottleneck. Previous efforts on the optimization of tamoxifen analogs of these compounds have relied on traditional medicinal chemistry. Our SuFEx-based D2B platform eliminates the need for extensive purification. The biocompatible conditions employed for diversification enabled crude products to be tested directly in a cell-based assay. Furthermore, the method facilitated exploration of structural modifications that improve binding affinity and potency, as evidenced by the development of compounds with targeted changes that demonstrated improved antiviral efficacy without compromising cellular viability.
The design basis of our tamoxifen analog functionalized with a difluoride (compound 1) stems from the binding interactions observed in the predicted complex of tamoxifen with EBOV- GP, which suggested sites for functionalization to enhance binding affinity. By targeting the unoccupied pocket, we aimed to improve interaction around the binding pocket. The crystal structure of EBOV-GP with 4R provided further validation to our docking approach for lead design.
This approach led to compound 4S that demonstrated sub-micromolar inhibition of EBOV entry and a 50-fold improvement in binding affinity against EBOV-gp over tamoxifen as measured by MST. The stronger binding of 4S to EBOV-GP, as reflected in its enhanced KD, is consistent with its superior cellular potency, suggesting that improved target affinity is likely to contribute to its increased functional activity. However, it is worth noting that the improvement of cellular potency is around 2-fold compared with tamoxifen. Elucidating the apparent discrepancy between the improvement in biophysical affinity and cellular potency is the subject of further study.
Our structure-activity relationship put in evidence the chiral selectivity of the hit identified, with the analog 4R showing less potency than its corresponding S enantio-counterpart, as measured by biophysical affinity as well as cellular antiviral activity. The significance of stereochemistry in 4S exemplifies the precision achievable with this SAR-guided platform and highlights the potential for designing small molecules for enhanced interactions with viral proteins. These findings underscore the importance of chiral optimization in the development of effective EBOV entry inhibitors and validate 4S as a particularly promising lead compound.
Unfortunately, we could not determine the crystal structure of the most active compound, 4S. However, an analysis of the pose adopted by compound 4R in the binding pocket of EBOV-GP revealed that the newly introduced oxolanyl ring, resulting from the SuFEx reaction, is positioned near the region where the β13-β14 loop (residues 190 to 214) is expected. This region has also been observed to be disordered in previous structures (34, 35). Structural analyses of toremifene bound to EBOV-GP have shown that this scaffold binds at the same site at the entrance of the binding pocket by expelling the DFF lid (residues 192–195) and positioning the A ring of the scaffold (Figure 1B) within this region (34). We propose that this flexible fragment could be modulating the enantioselectivity of the inhibitors identified in this work. However, additional experiments are required to verify this hypothesis.
In conclusion, this study demonstrates the effectiveness of our SuFEx-based HTMC platform in identifying promising EBOV entry inhibitors, contributing to the growing therapeutic modalities available for tackling the challenges associated with viral infections. This work paves the way for future studies to further optimize these lead compounds and assess their in vivo efficacy in EBOV infection. More broadly, the results of our HTMC workflow underscore the capability of this technology to accelerate the design and development of drug candidates in the field of viral infection, immunology, and autoimmune diseases.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. The data presented in the study is deposited in the RCSB PDB repository, accession number 9NNU. Further inquiries can be directed to the corresponding author.
Author contributions
LD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. EN: Formal analysis, Investigation, Writing – review & editing, Data curation, Validation. SA: Formal analysis, Investigation, Data curation, Methodology, Resources, Writing – review & editing. AW: Investigation, Writing – review & editing, Resources, Validation. IW: Writing – review & editing, Supervision, Funding acquisition, Project administration. KC: Funding acquisition, Project administration, Supervision, Writing – review & editing, Investigation, Resources. SK: Funding acquisition, Investigation, Project administration, Resources, Writing – review & editing, Supervision, Conceptualization, Data curation, Formal analysis, Methodology, Validation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported, in part, by the National Institute of General Medical Sciences of the National Institutes of Health (Award Nos. R00GM138758 & R35GM155249 to SK). SK is grateful to Einstein-Montefiore for support to start the lab. The Bruker 600 MHz NMR instrument in the Structural NMR Resource at the Albert Einstein College of Medicine was purchased using funds from NIH award 1S10OD016305 and is supported by a Cancer Center Support Grant (P30 CA013330).
Acknowledgments
We thank Drs. E. Gavathiotis and V.S.K. Venkateshaiah (Albert Einstein College of Medicine) for providing access, training and support for the use of the NanoTemper MST instrument. We would like to thank Dr. R. Stanfield for support in the protein expression experiments and Dr. S. Cahill for providing technical help with the NMR instrument. JChem for Office was used for data management, and we acknowledge Chemaxon (https://www.chemaxon.com) for giving us the academic free license for the software. Additionally, we acknowledge BioRender.com for the creation of a figure in this work. This research used resources of the SSRL, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. To refine English during the revision of the manuscript. The generated sentences were further refined extensively by authors.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1533037/full#supplementary-material
Abbreviations
CC50, 50% cytotoxicity concentration; Direct-to-biology, D2B; DIPEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; EBOV, Ebola virus; EC50, half-maximal effective concentration; eGFP, enhanced green fluorescent protein; EVD, EBOV disease; GP, glycoprotein; HTMC, high-throughput medicinal chemistry; J, NMR coupling constant; KD, dissociation constant; LASV, Lassa virus; MST, microscale thermophoresis; n.d., not determined; NMR, nuclear magnetic resonance; PDB, protein databank; py, pyridine; RT, room temperature; SD, standard deviation; SI, selectivity index; SuFEx; Sulfur(VI)-Fluoride Exchange; THF, tetrahydrofuran; VLP, virus-like particle; VSV, vesicular stomatitis virus.
References
1. Emanuel J, Marzi A, Feldmann H. Filoviruses: ecology, molecular biology, and evolution. Adv Virus Res. (2018) 100:189–221. doi: 10.1016/bs.aivir.2017.12.002
2. Leligdowicz A, Fischer WA, Uyeki TM, Fletcher TE, Adhikari NKJ, Portella G, et al. Ebola virus disease and critical illness. Crit Care. (2016) 20:217. doi: 10.1186/s13054-016-1325-2
3. Kortepeter MG, Bausch DG, Bray M. Basic clinical and laboratory features of filoviral hemorrhagic fever. J Infect Dis. (2011) 204:S810–S6. doi: 10.1093/infdis/jir299
4. Malvy D, McElroy AK, de Clerck H, Günther S, van Griensven J. Ebola virus disease. Lancet. (2019) 393:936–48. doi: 10.1016/S0140-6736(18)33132-5
5. Baseler L, Chertow DS, Johnson KM, Feldmann H, Morens DM. The pathogenesis of ebola virus disease. Annu Rev Pathol. (2017) 12:387–418. doi: 10.1146/annurev-pathol-052016-100506
6. World Health Organization. Situation report: ebola virus disease. (2016) World Health Organization.
7. Spengler J, Ervin E, Towner J, Rollin P, Nichol S. Perspectives on West Africa ebola virus disease outbreak, 2013–2016. Emerging Infect Dis. (2016) 22:956. doi: 10.3201/eid2206.160021
8. Pleet ML, DeMarino C, Stonier SW, Dye JM, Jacobson S, Aman MJ, et al. Extracellular vesicles and ebola virus: A new mechanism of immune evasion. Viruses. (2019) 11:410. doi: 10.3390/v11050410
9. Halajian EA, LeBlanc EV, Gee K, Colpitts CC. Activation of TLR4 by viral glycoproteins: a double-edged sword? Front Microbiol. (2022) 13:1007081. doi: 10.3389/fmicb.2022.1007081
10. The PREVAIL III Study Group. A longitudinal study of ebola sequelae in Liberia. New Engl J Med. (2019) 380:924–34. doi: 10.1056/NEJMoa1805435
11. Fausther-Bovendo H, Qiu X, McCorrister S, Westmacott G, Sandstrom P, Castilletti C, et al. Ebola Virus Infection Induces Autoimmunity against dsDNA and HSP60. Sci Rep. (2017) 7:42147. doi: 10.1038/srep42147
12. Henao-Restrepo AM, Camacho A, Longini IM, Watson CH, Edmunds WJ, Egger M, et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola ça suffit)! Lancet. (2017) 389:505–18. doi: 10.1016/S0140-6736(16)32621-6
13. Mulangu S, Dodd LE, Richard T, Davey J, Mbaya OT, Proschan M, et al. A randomized, controlled trial of ebola virus disease therapeutics. N Engl J Med. (2019) 381:2293–303. doi: 10.1056/NEJMoa1910993
14. El Ayoubi LEW, Mahmoud O, Zakhour J, Kanj SS. Recent advances in the treatment of ebola disease: A brief overview. PloS Pathog. (2024) 20:e1012038. doi: 10.1371/journal.ppat.1012038
15. Picazo E, Giordanetto F. Small molecule inhibitors of ebola virus infection. Drug Discovery Today. (2015) 20:277–86. doi: 10.1016/j.drudis.2014.12.010
16. Liu H-Y, Yang PL. Small-molecule inhibition of viral fusion glycoproteins. Annu Rev Virol. (2021) 8:459–89. doi: 10.1146/annurev-virology-022221-063725
17. Kitamura S, Zheng Q, Woehl JL, Solania A, Chen E, Dillon N, et al. Sulfur(VI) fluoride exchange (SuFEx)-enabled high-throughput medicinal chemistry. J Am Chem Soc. (2020) 142:10899–904. doi: 10.1021/jacs.9b13652
18. Garnar-Wortzel L, Bishop TR, Kitamura S, Milosevich N, Asiaban JN, Zhang X, et al. Chemical inhibition of ENL/AF9 YEATS domains in acute leukemia. ACS Cent Sci. (2021) 7:815–30. doi: 10.1021/acscentsci.0c01550
19. Kitamura S, Lin T-H, Lee C-CD, Takamura A, Kadam RU, Zhang D, et al. Ultrapotent influenza hemagglutinin fusion inhibitors developed through SuFEx-enabled high-throughput medicinal chemistry. Proc Natl Acad Sci United States America. (2024) 121:e2310677121. doi: 10.1073/pnas.2310677121
20. Carter TR, Milosevich N, Dada L, Shaum JB, Barry Sharpless K, Kitamura S, et al. SuFEx-based chemical diversification for the systematic discovery of CRBN molecular glues. Bioorg Med Chem. (2024) 104:117699. doi: 10.1016/j.bmc.2024.117699
21. Shaum JB, Steen EA, Muñoz i Ordoño M, Wenge DV, Cheong H, Hunkeler M, et al. High-throughput diversification of protein-ligand surfaces to discover chemical inducers of proximity. bioRxiv. (2024). doi: 10.1101/2024.09.30.615685
22. Liu F, Wang H, Li S, Bare GAL, Chen X, Wang C, et al. Biocompatible suFEx click chemistry: thionyl tetrafluoride (SOF4)-derived connective hubs for bioconjugation to DNA and proteins. Angew Chem Int Ed. (2019) 58:8029–33. doi: 10.1002/anie.201902489
23. Thomas RP, Heap RE, Zappacosta F, Grant EK, Pogány P, Besley S, et al. A direct-to-biology high-throughput chemistry approach to reactive fragment screening. Chem Sci. (2021) 12:12098–106. doi: 10.1039/D1SC03551G
24. Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, et al. FDA-approved selective estrogen receptor modulators inhibit ebola virus infection. Sci Transl Med. (2013) 5:190ra79. doi: 10.1126/scitranslmed.3005471
25. Cooper L, Schafer A, Li Y, Cheng H, Medegan Fagla B, Shen Z, et al. Screening and reverse-engineering of estrogen receptor ligands as potent pan-filovirus inhibitors. J Med Chem. (2020) 63:11085–99. doi: 10.1021/acs.jmedchem.0c01001
26. Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. A forward genetic strategy reveals destabilizing mutations in the ebolavirus glycoprotein that alter its protease dependence during cell entry. J Virol. (2010) 84:163–75. doi: 10.1128/jvi.01832-09
27. Kleinfelter LM, Jangra RK, Jae LT, Herbert AS, Mittler E, Stiles KM, et al. Haploid genetic screen reveals a profound and direct dependence on cholesterol for hantavirus membrane fusion. mBio. (2015) 6:e00801. doi: 10.1128/mbio.00801-15
28. Ren J, Zhao Y, Fry EE, Stuart DI. Target identification and mode of action of four chemically divergent drugs against ebolavirus infection. J Med Chem. (2018) 61:724–33. doi: 10.1021/acs.jmedchem.7b01249
29. Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymology. (1997) 276:307–26. doi: 10.1016/S0076-6879(97)76066-X
30. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. (2007) 40:658–74. doi: 10.1107/S0021889807021206
31. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr. D. (2010) 66:486–501. doi: 10.1107/S0907444910007493
32. Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr. D. (2012) 68:352–67. doi: 10.1107/S0907444912001308
33. Chen VB, Arendall WBI, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. Molprobity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. (2010) 66:12–21. doi: 10.1107/S0907444909042073
34. Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, et al. Toremifene interacts with and destabilizes the ebola virus glycoprotein. Nature. (2016) 535:169–72. doi: 10.1038/nature18615
Keywords: Ebola, small molecule antiviral drugs, drug discovery, SuFEx, direct-to-biology
Citation: Dada L, Nagai E, Agrawal S, Wirchnianski AS, Wilson IA, Chandran K and Kitamura S (2025) SuFEx-enabled high-throughput medicinal chemistry for developing potent tamoxifen analogs as Ebola virus entry inhibitors. Front. Immunol. 16:1533037. doi: 10.3389/fimmu.2025.1533037
Received: 22 November 2024; Accepted: 25 March 2025;
Published: 28 April 2025.
Edited by:
Feng Dong, AbbVie, United StatesReviewed by:
Hanyong Bae, Sungkyunkwan University, Republic of KoreaJiang Weng, Sun Yat-Sen University, China
Copyright © 2025 Dada, Nagai, Agrawal, Wirchnianski, Wilson, Chandran and Kitamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seiya Kitamura, c2VpeWEua2l0YW11cmFAZWluc3RlaW5tZWQuZWR1
†These authors have contributed equally to this work