 Peter Battisti1Matthew R. Ykema1Darshan N. Kasal1
Peter Battisti1Matthew R. Ykema1Darshan N. Kasal1 Madeleine F. Jennewein1Samuel Beaver1Abbie E. Weight2Derek Hanson3
Madeleine F. Jennewein1Samuel Beaver1Abbie E. Weight2Derek Hanson3 Jasneet Singh1Julie Bakken1Noah Cross1Pauline Fusco1Jacob Archer3Sierra Reed1Alana Gerhardt1
Jasneet Singh1Julie Bakken1Noah Cross1Pauline Fusco1Jacob Archer3Sierra Reed1Alana Gerhardt1 Justin G. Julander2Corey Casper1,4,5,6
Justin G. Julander2Corey Casper1,4,5,6 Emily A. Voigt1*
Emily A. Voigt1*- 1Access to Advanced Health Institute (AAHI), formerly Infectious Disease Research Institute, Seattle, WA, United States
- 2Institute for Antiviral Research, Utah State University, Logan, UT, United States
- 3Infectious Disease Research Institute, Seattle, WA, United States
- 4Department of Medicine, University of Washington, Seattle, WA, United States
- 5Department of Global Health, University of Washington, Seattle, WA, United States
- 6Vaccine and Infectious Disease Division, Fred Hutch Cancer Center, Seattle, WA, United States
Introduction: Yellow fever (YFV) and Zika (ZIKV) viruses cause significant morbidity and mortality, despite the existence of an approved YFV vaccine and the development of multiple ZIKV vaccine candidates to date. New technologies may improve access to vaccines against these pathogens. We previously described a nanostructured lipid carrier (NLC)-delivered self-amplifying RNA (saRNA) vaccine platform with excellent thermostability and immunogenicity, appropriate for prevention of tropical infectious diseases.
Methods: YFV and ZIKV prM-E antigen-expressing saRNA constructs were created using a TC-83 strain Venezuelan equine encephalitis virus-based replicon and complexed with NLC by simple mixing. Monovalent and bivalent vaccine formulations were injected intramuscularly into C57BL/6 mice and Syrian golden hamsters, and the magnitude, durability, and protective efficacy of the resulting immune responses were then characterized.
Results and discussion: Monovalent vaccines established durable neutralizing antibody responses to their respective flaviviral targets, with little evidence of cross-neutralization. Both vaccines additionally elicited robust antigen-reactive CD4+ and CD8+ T cell populations. Notably, humoral responses to YFV saRNA-NLC vaccination were comparable to those in YF-17D-vaccinated animals. Bivalent formulations established humoral and cellular responses against both viral targets, commensurate to those established by monovalent vaccines, without evidence of saRNA interference or immune competition. Finally, both monovalent and bivalent vaccines completely protected mice and hamsters against lethal ZIKV and YFV challenge. We present a bivalent saRNA-NLC vaccine against YFV and ZIKV capable of inducing robust and efficacious neutralizing antibody and cellular immune responses against both viruses. These data support the development of other multivalent saRNA-based vaccines against infectious diseases.
Introduction
Yellow fever virus (YFV) and Zika virus (ZIKV) are mosquito-borne flaviviruses transmitted by Aedes species mosquitos, which cause infectious disease with a significant impact on global health. YFV is found in tropical regions of Africa and South America, and causes fever, jaundice, and liver damage that may become fatal (1). The live-attenuated YF-17D vaccine against YFV has been a gold standard vaccine since its introduction in 1937. The YF-17D vaccine is highly effective in disease prevention, with a single dose inducing likely life-long protection via establishment of non-sterilizing immunity (2, 3). Despite the success of the YF-17D vaccine, production of vaccine stocks has been challenging due to the reliance on cell-based production techniques, limited viral seed stocks due to propagation using an inconsistent process that relies on a limited supply of pathogen-free chicken eggs, and batch-to-batch vaccine production inconsistencies (4–6). Limited production in recent years and increasing outbreaks have led to a near total depletion of global YF-17D vaccine stockpiles, resulting in dose-reducing measures to stretch the supply (7, 8). Additionally, YF-17D is contraindicated in persons with compromised immune systems, and infrequently can cause disseminated disease in the elderly and young infants (9).
ZIKV is found in tropical regions of Africa, Asia, and the Americas, with ZIKV infections potentially leading to fever, rash, joint pain, and conjunctivitis (10). Additionally, ZIKV is one of the few arboviruses to display both horizontal (including sexual contact) and vertical (maternal-fetal) transmission in humans, and can lead to significant birth defects when contracted during pregnancy (11). Both YFV and ZIKV pose a significant public health threat and have caused outbreaks and epidemics since 2000 in multiple areas of the world (12). No approved ZIKV vaccine exists despite the development of vaccine candidates that span a range of traditional and next-generation vaccine technologies (13). Recent Phase 1 clinical trials have demonstrated an excellent safety profile for ZIKV mRNA vaccines, but some of these candidates did not establish a sufficient immune response (14).
A new vaccine platform would be of great use to fully address the global public health issues caused by these flaviviruses (15). Ideal next-generation flavivirus vaccines would provide levels of protection against disease comparable to that induced by YF-17D, allow for adaptation to emerging viral strains, be manufacturable at a low cost with a reliable and consistent process, and be sufficiently thermostable for global distribution. Moreover, given the geographical overlap between viruses spread by Aedes mosquitoes like YFV, ZIKV, dengue, chikungunya, and others, the ability to create multivalent arbovirus vaccine candidates would be of great benefit.
We previously developed a vaccine platform that delivers self-amplifying RNA (saRNA) to target cells via a simple and unique nanostructured lipid carrier (NLC) delivery system (16). These saRNA-NLC vaccine formulations are straightforward to manufacture at scale and readily lyophilizable, increasing shelf life to at least 21+ months when refrigerated and up to 6 months at room temperature (17). This technology has been applied in the development of a clinical-stage SARS-CoV-2 vaccine, among others (16–21). Here, we created saRNA-NLC vaccine candidates against YFV (AAHI-YFV) and ZIKV (AAHI-ZKV) using this established platform, and, in C57BL/6 mice and Syrian golden hamsters, illustrate the ready adaptability of the vaccine platform to support bivalent immunization. The monovalent and bivalent vaccines induced robust humoral and cellular flavivirus-specific immune responses and conferred complete protection against viral challenge, demonstrating the feasibility of multivalent saRNA-NLC vaccines against mosquito-borne diseases.
Methods
Protein modeling
Protein modeling and analysis of the YF-17D (RCSB PDB ID: 6IW4) and ZIKV envelope (E) (RCSB PDB ID: 5JHM) domains were conducted in PyMOL 2.0 (Schrödinger, LLC). Protein alignments and structural similarity evaluation were done using the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) pairwise alignment server under jFATCAT rigid alignment (22–24).
saRNA cloning and production
YFV pre-membrane-envelope (prM-E) (GenBank ID: JN628281.1), ZIKV prM-E (GenBank ID: KJ776791), and secreted alkaline phosphatase protein (SEAP) (GenBank ID: LC380029.1) inserts were subcloned into T7-VEEVRep plasmids (16, 17). Linearized DNA plasmids were used to synthesize YFV prM-E, ZIKV prM-E, and SEAP saRNAs using a previously optimized in vitro transcription and purification protocol (16, 17) (see Supplementary Data Sheet 1).
NLC production
The NLC formulation consisted of an oil core stabilized within an aqueous buffer using appropriate surfactants and was produced by microfluidization as previously described (17) (see Supplementary Data Sheet 1).
Vaccine complexing and characterization
We generated saRNA-NLC vaccine complexes by simple mixing (17, 19) (see Supplementary Data Sheet 1). Dynamic light scattering (DLS) using the Zetasizer Nano ZS (Malvern Panalytical) was used for nanoparticle size determination as previously described (16, 17, 19). The resulting intensity-weighted Z-average diameter was determined for each formulation and averaged from three measurements per formulation.
Transfection, protein harvest, and western blotting
HEK293T cells (American Type Culture Collection (ATCC) #CRL-11268) were transfected with complexed vaccine. YFV and ZIKV E protein expression was then measured by western blot of transfected cell lysates (see Supplementary Data Sheet 1).
Animal studies
Mouse studies
All animal work was done under the oversight of the Bloodworks Northwest Research Institute’s (Seattle, WA) Institutional Animal Care and Use Committee (IACUC), protocol #5389-01. All animal work followed applicable sections of the Final Rules of the Animal Welfare Act regulations (9 CFR Parts 1, 2, and 3) and the Guide for the Care and Use of Laboratory Animals, Eighth Edition.
C57BL/6J mice obtained from The Jackson Laboratory or Charles River Laboratories were used for mouse studies. Mice were 6–8 weeks of age at study onset, and groups were sex balanced. Mice were immunized with saRNA-NLC vaccines by intramuscular injection bilaterally in the rear quadriceps muscle (50 µL/leg, 100 µL total) for a prime (Day 0) and a boost (for some groups) 4 weeks later (Day 28). YF-17D was sourced from the University of Texas Medical Branch World Reference Center for Emerging Viruses and Arboviruses and propagated for >3 passages in Vero cells (ATTC #CCL-81). For mice vaccinated with YF-17D, one 20 µL dose containing 104 PFU was administered via subcutaneous rear footpad injection. Serum and spleen samples were collected in-life and at terminal harvest (see Supplementary Data Sheet 1).
Mice challenged with ZIKV were given 3 mg interferon alpha receptor (IFNAR) blocking monoclonal antibody (MAR1-5A3, BioXCell #BE0241) via intraperitoneal injection on challenge day -1 (D29 or D57), as previously described (16, 25). Additional 1 mg doses were administered on challenge day +1 and +4 (D30 and D33 or D59 and D62). Mice were weighed immediately prior to and daily during the 15-day post-prime and post-boost ZIKV challenge periods (D30–51 and D58-79). Mice were challenged with 106 PFU of ZIKV (strain: MA-Zika Dakar; see Supplementary Data Sheet 1 for virus culture) via bilateral subcutaneous injection (20 µL/rear footpad, 40 µL total) (26). Excess viral stocks used on each challenge were assessed by plaque assay to verify viral titer (actual titer administered = 8.8 × 105 PFU).
Hamster studies
All animal care and husbandry were conducted according to protocols approved by the Utah State University IACUC, protocol #10010 (see Supplementary Data Sheet 1).
Syrian golden hamsters, weighing 80–90 g (approximately 34–40 days old), were purchased from Charles River Laboratories, and randomly divided into sex-balanced groups. Hamsters were immunized with saRNA-NLC vaccines as described above for mice. For YF-17D vaccinated hamsters, a 1-passage stock of human vaccine YF-VAX (Sanofi) was diluted in sterile saline, and 100 µL was administered via subcutaneous injection into the inguinal fold for each immunization. A cohort of hamsters was also left unvaccinated and uninfected as normal health control animals. Serum samples were collected in-life and at terminal harvest (see Supplementary Data Sheet 1).
Hamsters were weighed immediately prior to and daily during the 21-day post-prime and post-boost YFV challenge periods (D30–51 and D58-79). Hamsters were challenged with 200 CCID50 (50% cell culture infectious dose) of hamster-adapted YFV (strain: Jiméńez; see Supplementary Data Sheet 1 for virus culture) via bilateral intraperitoneal injection of 100 µL (200 µL total) (27).
ELISA
Serum YFV and ZIKV E protein-binding IgG antibodies were measured by ELISA (see Supplementary Data Sheet 1) (19). Briefly, plates were coated with 1 µg/mL of recombinant YFV E protein (Meridian Bioscience #R01709) or recombinant ZIKV E protein (Meridian Bioscience #R01635) in PBS and incubated overnight at 4°C. A pan-flavivirus E protein-binding monoclonal antibody (4G2; Novus Biologicals #NBP2-52709) was used as the positive control.
Plaque reduction neutralization test
Serum YFV and ZIKV neutralizing antibody (nAb) titers were determined by a 50% plaque reduction neutralization test (PRNT50)(see Supplementary Data Sheet 1) (17). Briefly, PRNTs were performed with YF-17D or ZIKV FSS13025 viruses.
Flow cytometry
Intracellular cytokine staining and flow cytometry were conducted to measure antigen-reactive splenic T cells (see Supplementary Data Sheet 1 and Supplementary Figure 1) as previously described (19).
Viral titer measurements
See Supplementary Data Sheet 1.
Alanine aminotransferase assay
See Supplementary Data Sheet 1.
Scientific rigor
See Supplementary Data Sheet 1 for discussion of replicates, sample size estimations, randomization, blinding, and inclusion and exclusion criteria.
Statistical analyses
Statistical analysis was conducted using GraphPad Prism 10.3.0. To evaluate immunogenicity data, we examined distribution and variance through Q-Q plot and residual plot analysis. For serum E protein-specific IgG, nAb titers, and viremia, statistics were performed on log10 transformed data using one-way ANOVA with Dunnett’s correction or Tukey’s correction for multiple comparisons, or mixed-effect analysis with Tukey’s correction. For weight change, alanine aminotransferase (ALT), and antigen-reactive T cells, statistics were performed using one-way ANOVA with Dunnett’s correction, Brown-Forsythe and Welch ANOVA with Dunnett’s T3 correction, or Kruskal-Wallis with Dunn’s correction for multiple comparisons. For survival curves, statistics were performed by Mantel-Cox log-rank test.
Results
Design of saRNA-NLC vaccines against YFV and ZIKV
We developed YFV and ZIKV saRNA-NLC vaccines for delivery in monovalent or bivalent formulations. Each saRNA contains two open reading frames (ORFs), a 3’ polyadenylation sequence (42 As encoded into the DNA template), and a 5’ cap-0 structure (Figure 1a). The first ORF encodes the non-structural proteins (nsPs) from the TC-83 strain of Venezuelan equine encephalitis virus (VEEV) (28), and the second ORF encodes codon-optimized prM and E genes from the genomes of YF-17D or ZIKV A/PF/2013, respectively, to produce a fusion protein. The E protein of each virus represents the primary viral antigen targeted by humoral immunity, while the prM domain stabilizes the E protein for proper folding during translation (29, 30). YFV and ZIKV saRNA constructs were complexed as monovalent formulations with AAHI’s unique NLC delivery particle (Figure 1b). The cationic NLC allows for strong electrostatic binding of the negatively-charged saRNA to the outside of the NLC particle, resulting in complexed saRNA-NLC vaccine particles approximately 80 nm in diameter (Supplementary Figure 2) that exhibit a positive charge and robust protection from nuclease-mediated degradation (16, 19). The YFV and ZIKV E domains share significant (40%) protein sequence similarity and 97% structural conservation (Figure 1c), suggesting that they should have similar expression behavior and cellular processing, and may elicit comparable immune responses. The formulated vaccines were then transfected into HEK293T cells, in which YFV or ZIKV E protein expression was detected 24 hours post-transfection by western blot (Figure 1d). Given the similarities in both biophysical properties of the saRNA-NLC complexes and the antigenic proteins expressed, we expected the YFV and ZIKV vaccine candidates to have similar patterns of immunogenicity.
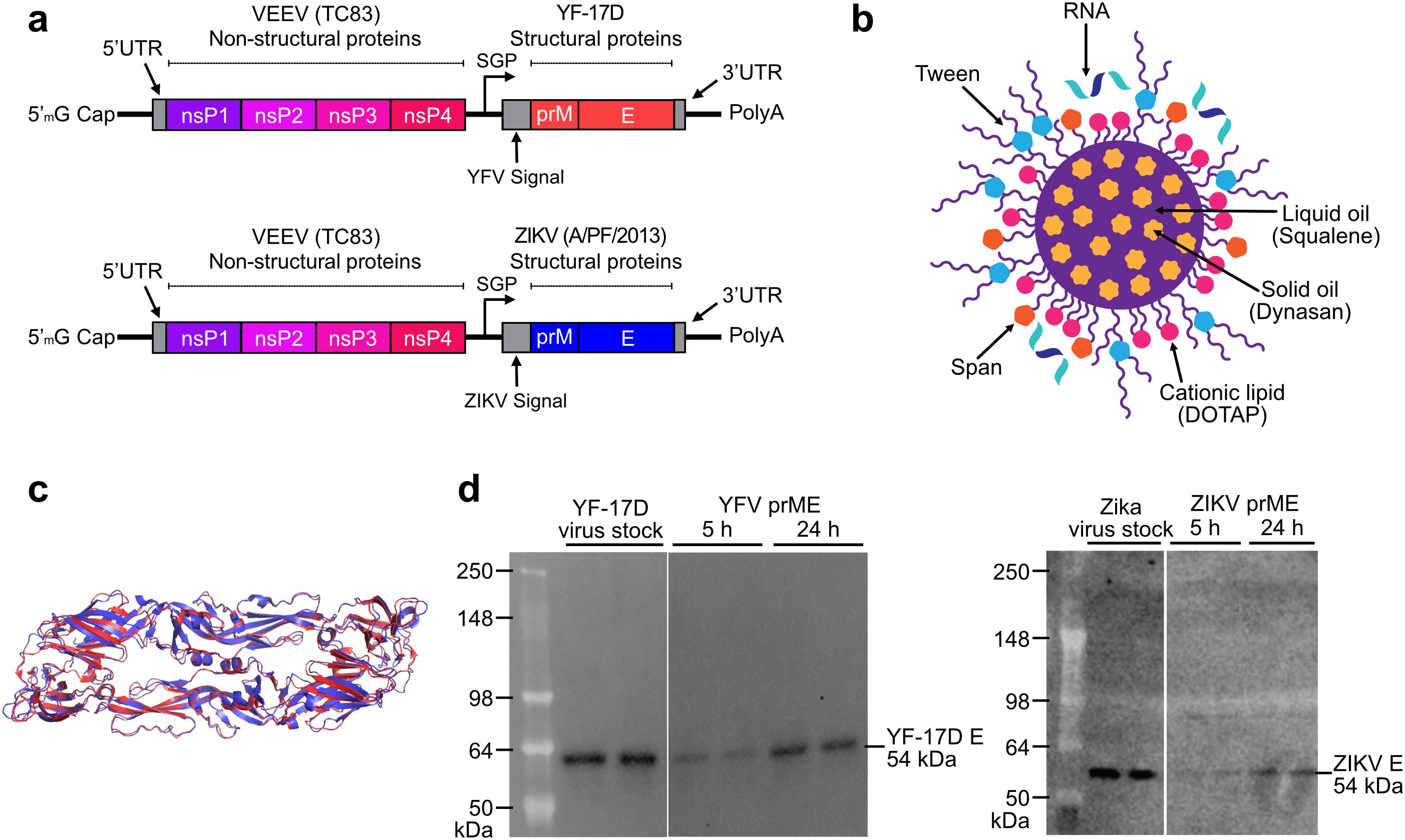
Figure 1. Characterization of the components in AAHI’s bivalent flavivirus saRNA-NLC vaccine. (a) Construct designs for the YFV and ZIKV saRNA vaccine constructs. YFV replicon size is 9.89 kb, and ZIKV replicon size is 9.94 kb. SGP = sub-genomic promoter. (b) Schematic of the NLC RNA delivery particle. Design by Cassandra Baden. (c) Structural alignment of the YF-17D (PDB ID: 6IW4; red) and ZIKV (PDB ID: 5JHM; blue) E protein pre-fusion dimers, indicating significant structural similarity between the two antigens. (d) Western blot verifying in vitro protein expression of 54 kDa YFV and ZIKV E protein in cellular lysates after vaccine HEK293T cell transfection. YF-17D and Zika virus stocks were diluted to 104 PFU/lane and run in duplicate.
A monovalent YFV saRNA vaccine induces robust serum antibody responses in mice
Serum antibody titers are a primary correlate of protection (CoP) against YFV infection (2, 3). Thus, a viable YFV vaccine candidate should establish robust and long-lasting nAb responses, ideally similar to those induced by the approved YF-17D vaccine (2). To study the ability of our monovalent YFV saRNA-NLC vaccine (AAHI-YFV) to induce robust nAb responses, C57BL/6 mice were intramuscularly primed (Day 0) and boosted (Day 28) with various doses (5, 10, 20, and 30 µg) of our monovalent YFV vaccine (AAHI-YFV). Mice immunized with a single dose (104 PFU) of YF-17D were used as a positive vaccine control group. Mice injected with 10 µg of saRNA-NLC expressing the non-immunogenic protein SEAP were used to control for induction of non-specific immune responses.
Serum YFV E protein-specific antibody titers in AAHI-YFV vaccinated mice showed little to no dose-dependency above 10 µg saRNA (Figures 2a, b). While a 5 µg saRNA dose induced more variable serum IgG titers between individual mice, reliably high serum IgG titers were observed in mice immunized with 10 to 30 µg of saRNA, equivalent to those induced by vaccination with 104 PFU of YF-17D.
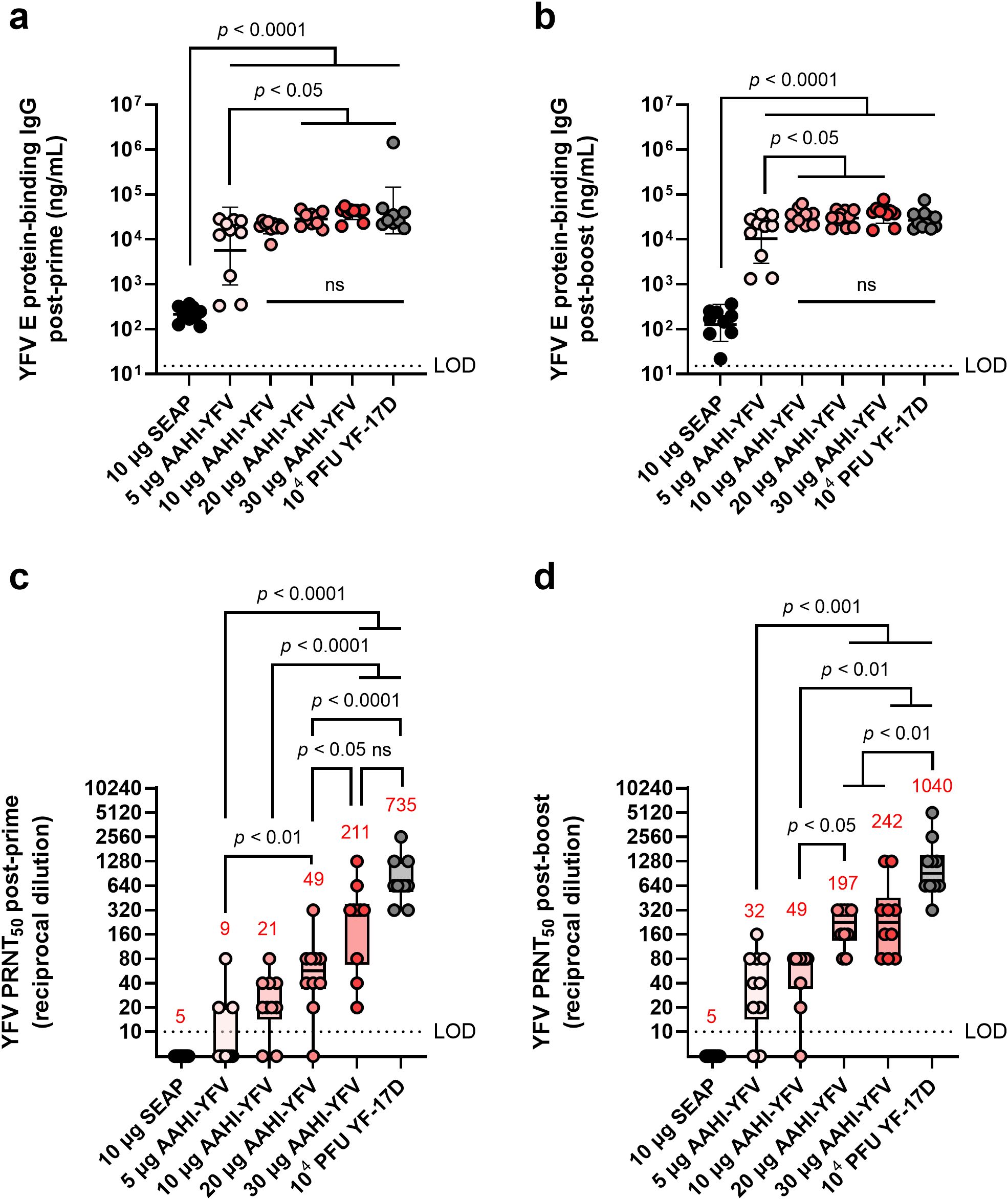
Figure 2. YFV saRNA-NLC vaccination induces serum antigen-binding IgG and neutralizing antibody responses in mice comparable to those induced by the YF-17D vaccine. Mice were vaccinated with a dose range of AAHI-YFV saRNA-NLC and (a, b) serum YFV E protein-binding IgG and (c, d) YFV neutralizing antibody titers were measured (a, c) 28 days post-prime and (b, d) 28 days post-boost. The YF-17D group represents mice that were subcutaneously dosed once with 104 PFU of YF-17D. Statistical analysis was conducted on log10 transformed data using one-way ANOVA with Tukey’s correction for multiple comparisons. ns = non-significant (p > 0.05). Black dotted line shows the limit of detection (LOD) for the assay. Results are from a single independent experiment; n = 10 mice per group, 5 male and 5 female. (a, b) Scatter plots show geometric mean ± geometric SD. (c, d) Box plots show median and IQR ± min/max value. Red numerical values represent group PRNT50 GMT.
We observed a strong dose-dependency in serum YFV nAb titers (Figures 2c, d). All AAHI-YFV dosing groups, apart from the 5 µg group, generated a PRNT50 geometric mean titer (GMT) ≥20 post-prime and ≥40 post-boost, which exceeded the CoP established in other models: PRNT50 ≥10 (humans), PRNT50 ≥40 (hamsters), and PRNT50 ≥20 (non-human primates) (2, 3, 31, 32). Post-boost, both 5 and 10 µg vaccinated groups generated a PRNT50 GMT ≥20, though some individual mouse PRNT50 titers remained below the limit of detection. Conversely, all mice vaccinated with 20 and 30 µg doses developed PRNT50 titers ≥80 post-boost. These high-dose vaccinated groups had a post-boost nAb GMT approaching but not equaling those induced by YF-17D vaccination. The post-boost PRNT50 titers established by all saRNA doses exceeded the CoP for other models, consistent with the post-prime results, and reached levels indicative of protection in other mouse model studies (2, 33, 34). Overall, these data demonstrate that AAHI-YFV induces potent humoral immunity in mice, particularly at 10 to 30 µg doses.
YFV saRNA vaccination establishes durable humoral immune responses at higher doses
The FDA-approved YF-17D vaccine provides life-long protection against disease for most individuals (35). We previously demonstrated that our ZIKV saRNA-NLC vaccine generates a durable serum nAb response up to 7 months post-prime and post-boost immunization (16). Therefore, we investigated the longevity of humoral immunity following AAHI-YFV vaccination. To do this, prime-boost 10 µg, 20 µg, and 30 µg AAHI-YFV and YF-17D vaccinated C57BL/6 mice from the AAHI-YFV dosing study above were maintained for 6 months post-boost to investigate antibody durability.
As expected, serum antigen-binding IgG titers declined significantly over the 6 months post-boost vaccination but remained well in the detectable range for all vaccinated groups (Figure 3a). Notably, mice immunized with all doses of the AAHI-YFV saRNA-NLC vaccine showed serum IgG titers at 6 months post-boost comparable to YF-17D-vaccinated mice (10 µg: p = 0.99, 20 µg: p = 0.99, and 30 µg: p = 0.58) suggesting that the saRNA-NLC vaccine can indeed induce durable humoral immune responses to YFV. Serum nAb titers showed similar results to antigen-binding IgG titers, waning over the 6-month period post-boost but maintaining largely detectable nAb titers in the saRNA-NLC vaccinated mice (Figure 3b). Furthermore, at 6 months post-boost, mice receiving 20 and 30 µg of AAHI-YFV maintained a nAb GMT commensurate to that induced by YF-17D (20 µg: p = 0.1342 and 30 µg: p = 0.9540). Interestingly, while the nAb GMT of the YF-17D group was greater than the 30 µg AAHI-YFV group after the initial vaccinations (Figure 2d), the nAb GMT of the YF-17D group fell more precipitously and was equivalent to the 30 µg AAHI-YFV group at 6 months post-boost (Figure 3b). Critically, both groups maintained nAb titers well above the neutralizing CoP for humans (PRNT50 ≥10) at the final timepoint (3, 36). Together, these data indicate that the potent antibody response induced in mice by our AAHI-YFV vaccine is durable, as serum nAb titers were maintained at levels similar to YF-17D vaccination 6 months after the original vaccination series.
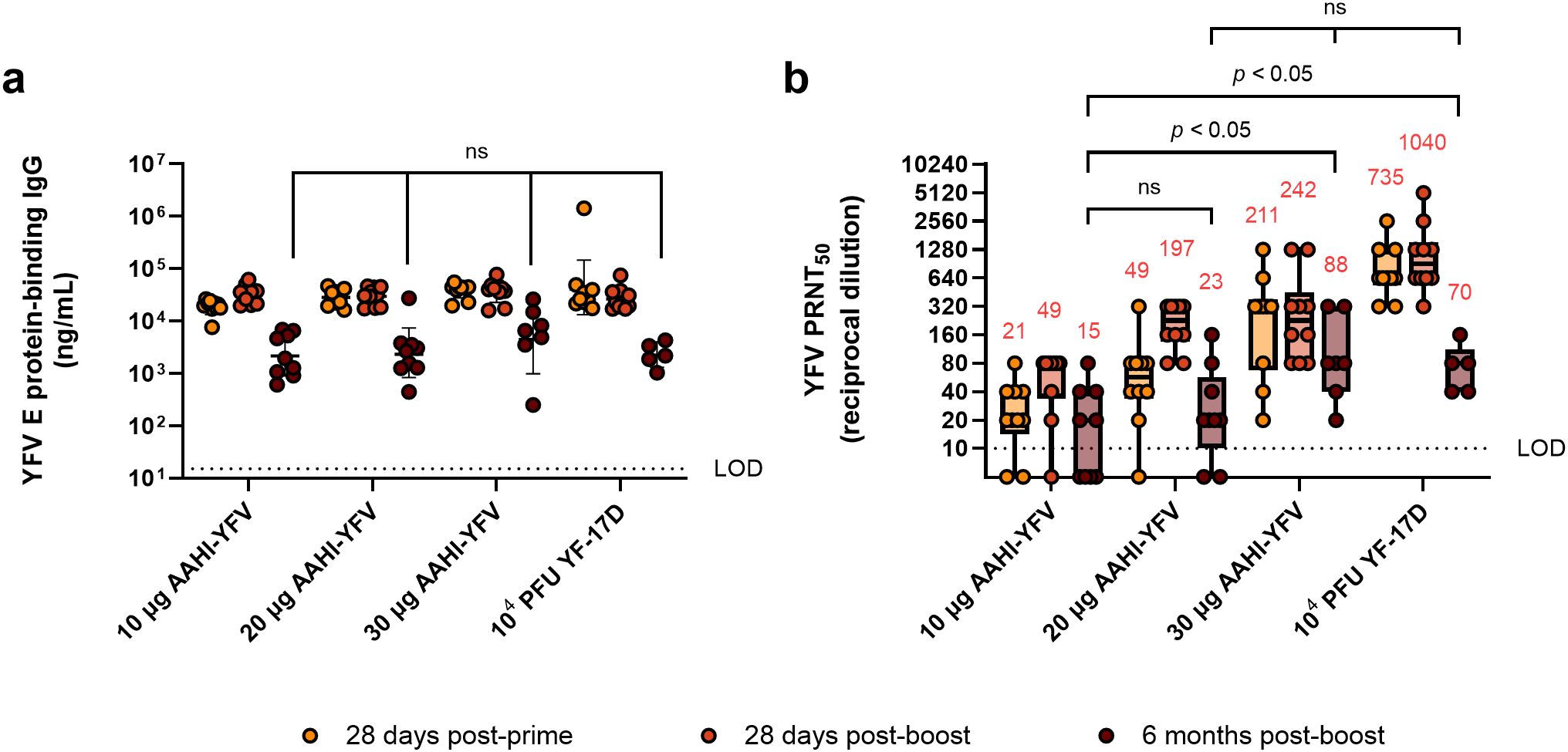
Figure 3. Durability of YFV-specific responses from monovalent saRNA-NLC vaccination. (a) Serum YFV E protein-binding IgG and (b) YFV neutralizing antibody responses were assessed 28 days post-prime, 28 days post-boost, and 6 months post-boost with AAHI-YFV saRNA-NLC vaccines. The YF-17D group represents mice that were subcutaneously dosed once with 104 PFU of YF-17D. Statistical analysis was conducted on log10 transformed data using a mixed-effect analysis with Tukey’s correction for multiple comparisons. Black dotted line shows the limit of detection (LOD) of the assay. ns = non-significant (p > 0.05). Results are from a single independent experiment; n = 10 mice per group, 5 male and 5 female. Animals were removed from the 6-month time point due to length of study: 1 animal from the 10 µg group, 1 animal from the 20 µg group, 3 animals from the 30 µg group, and 5 animals from the YF-17D group. (a) Scatter plots show geometric mean ± geometric SD. (b) Box plots show median and IQR ± min/max value. Red numerical values represent group PRNT50 GMT.
A ZIKV saRNA monovalent vaccine establishes a potent nAb response at various doses
Like other flaviviruses, nAbs to ZIKV are thought to be the best CoP from infection (37). We therefore evaluated the antibody response to our monovalent ZIKV vaccine (AAHI-ZKV) as we did for AAHI-YFV. C57BL/6 mice were prime or prime-boost immunized with escalating doses (0.1, 1, 10, and 30 µg) of AAHI-ZKV, representing a lower focused dose range than tested for the YFV vaccine candidate due to the previously determined high potency of the ZIKV saRNA-NLC vaccine (16).
We observed a dose-dependent increase in ZIKV E protein-specific serum IgG. A dose of 0.1 µg AAHI-ZKV generated responses comparable to background seen in SEAP-expressing vector control saRNA-NLC dosed mice, whereas mice primed and boosted with 10 or 30 µg of saRNA developed the highest serum IgG titers (Figures 4a, b), as was also observed for AAHI-YFV.
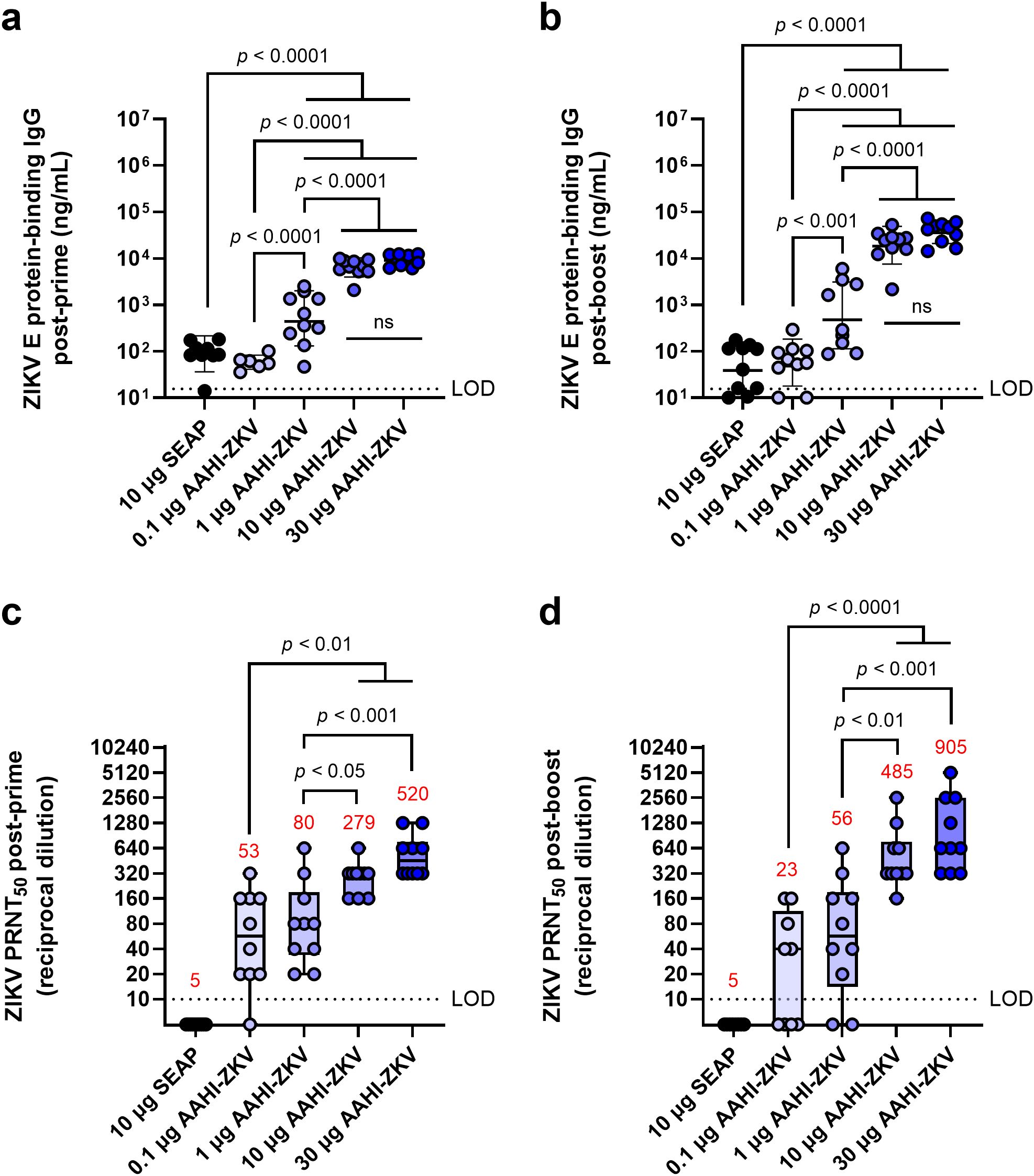
Figure 4. ZIKV-specific serum IgG and neutralizing antibody responses following monovalent saRNA-NLC vaccination. Mice were vaccinated with a dose range of AAHI-ZKV saRNA-NLC and (a, b) serum ZIKV E protein-binding IgG and (c, d) ZIKV neutralizing antibody titers were measured (a, c) 28 days post-prime and (b, d) 28 days post-boost. Statistical analysis was conducted on log10 transformed data using one-way ANOVA with Tukey’s correction for multiple comparisons. Black dotted line shows the limit of detection (LOD) for the assay. ns = non-significant (p > 0.05). Results are from a single independent experiment; n = 10 mice per group, 5 male and 5 female. (a, b) Scatter plots show geometric mean ± geometric SD. (c, d) Box plots show median and IQR ± min/max value. Red numerical values represent group PRNT50 GMT.
We also noted a dose-dependent increase in serum nAb titers. Despite background levels of ZIKV antigen-binding IgG at lower doses of saRNA, 0.1 and 1 µg vaccine doses induced a ZIKV PRNT50 GMT ≥10 (Figures 4c, d). This nAb titer was previously found to confer complete protection against lethal ZIKV challenge in mice (16), despite being lower than the CoP identified through passive transfer studies in mice (PRNT50 GMT ≥100) (38, 39). Whereas low-dose AAHI-ZKV vaccination induced variable PRNT50 titers in individual animals, 10 and 30 µg doses induced PRNT50 titers ≥160 in all mice both post-prime and post-boost. Together, these data demonstrate that both the AAHI-YFV and AAHI-ZKV are highly immunogenic, particularly at the higher saRNA doses tested, with no deleterious outcomes.
Bivalent vaccine formulations induce nAbs to both targets comparable to monovalent formulations
After verification of the monovalent formulations, we next evaluated mixing strategies for bivalent saRNA-NLC vaccine administration using 10 µg doses of each saRNA. Three bivalent vaccine dosing strategies were tested, to determine optimal bivalent vaccine manufacture processes and to evaluate the presence of any immune interference between the two flaviviral antigen-expressing saRNA species. These mixing strategies included (1) delivering individual monovalent saRNA vaccines in anatomically distinct locations represented by separate legs (“Split monovalent”), (2) mixing pre-formed monovalent YFV and ZIKV saRNA-NLC vaccines (“Complex -> Mix”) with each vaccine complex delivering a single saRNA species, or (3) mixing both vaccine saRNAs prior to complexing with NLCs (“Mix -> Complex “), resulting in vaccine complexes delivering both saRNA species (Supplementary Table 1).
Alone, monovalent vaccines induced serum nAb responses against their respective flavivirus as expected, with little to no evidence of cross-neutralization (Figure 5). All mice receiving any of the bivalent vaccines (AAHI-YFV/ZKV), regardless of how the bivalent vaccine was created and injected, induced statistically equivalent nAb titers to those induced by the monovalent formulations. No evidence of any immune interference between the two saRNA-expressed flavivirus antigens was noted, regardless of the bivalent vaccine mixing strategy.
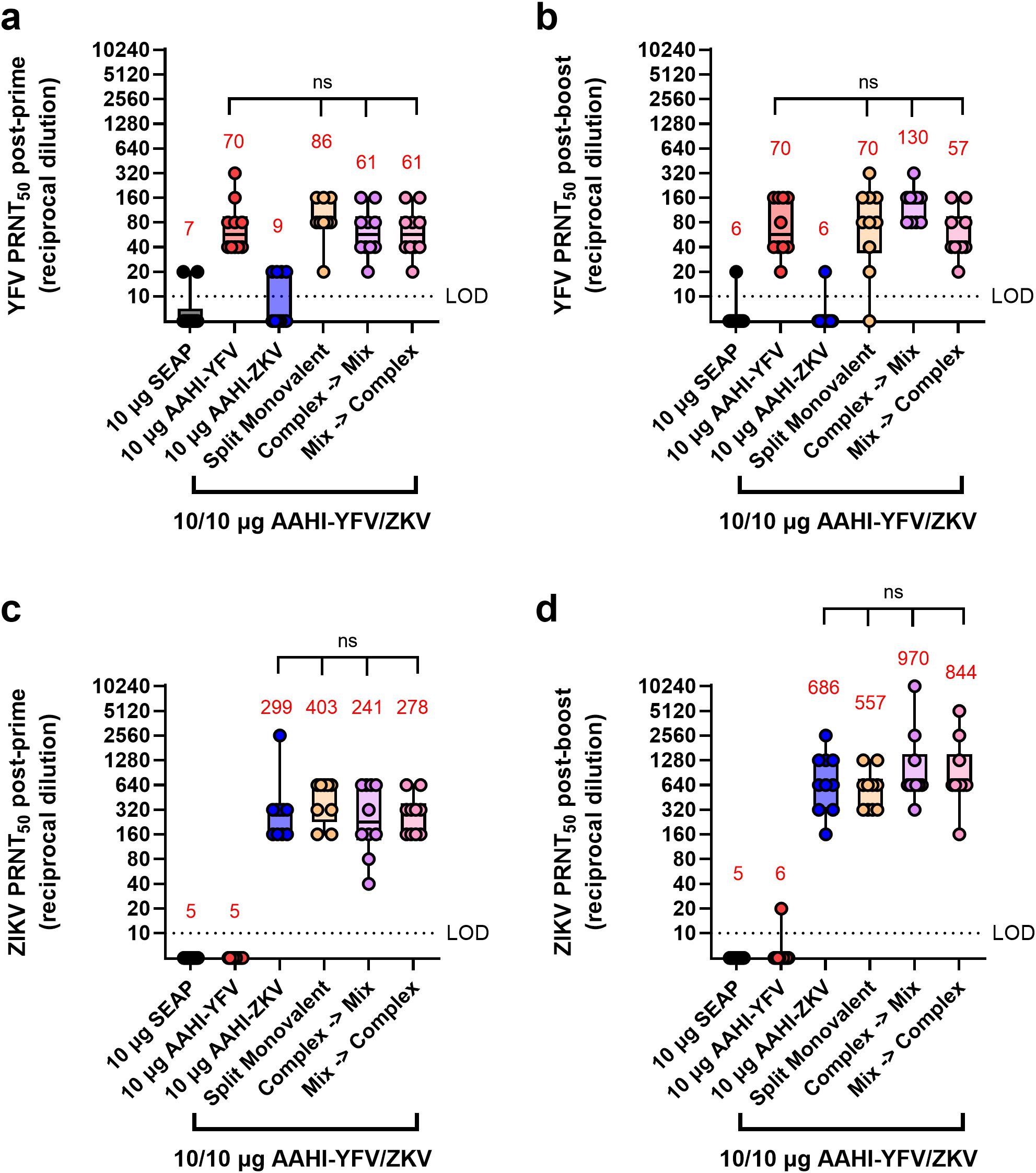
Figure 5. Serum YFV and ZIKV neutralizing antibody titers resulting from different bivalent vaccination strategies. Mice were vaccinated with monovalent or bivalent formulations of AAHI-YFV or AAHI-ZKV saRNA-NLC and serum neutralizing antibody titers against (a, b) YFV and (c, d) ZIKV were measured (a, c) 28 days post-prime and (b, d) 28 days post-boost. Statistical analysis was conducted on log10 transformed data using one-way ANOVA with Tukey’s correction for multiple comparisons. Comparisons were made against monovalent AAHI-YFV or AAHI-ZKV and between bivalent AAHI-YFV/ZKV groups. ns = non-significant (p > 0.05). Black dotted line shows the limit of detection (LOD) for the assay. Results are from a single independent experiment; n = 10 mice per group, 5 male and 5 female. Box plots show median and IQR ± min/max value. Red numerical values represent group PRNT50 GMT.
Bivalent vaccine dosing establishes robust antigen-responsive CD4+ and CD8+ T cell populations
We next studied the cellular responses to the different bivalent dosing strategies investigated in the above mouse study, looking at the antigen-specific responses of splenic CD4+ and CD8+ T cells. In mice, individual antigen-specific T cells are estimated to vary in frequency from 20–200 cells per ~2 x 107 naïve CD4+ or CD8+ T cells (0.0001 – 0.001%) (40, 41). Levels of splenic YFV and ZIKV antigen-reactive activated (CD44+) polyfunctional CD4+ T cells, which are those simultaneously expressing IFN-γ+, IL-2+, and TNF-α+ in response to antigen stimulation, were assessed by flow cytometry. We also examined the antigen-reactive IFN-γ+ CD8+ CD44+ T cell responses in these vaccinated mice.
Flavivirus E protein-reactive polyfunctional CD4+ T cells were readily detectable (>0.5% of total CD4+ CD44+ T cells) post-prime and post-boost in AAHI-YFV or AAHI-ZKV vaccinated mice. Polyfunctional CD4+ T cells were not detected in mice vaccinated with the SEAP-expressing saRNA-NLC negative control (Figures 6a-d). Like the PRNT50 data, splenic CD4+ T cells in groups immunized with monovalent vaccines were only reactive to the corresponding antigen peptide pools, indicating no induction of cross-reactive CD4+ T cells by the two flavivirus vaccines. All bivalent doses, regardless of mixing and dosing strategies, elicited comparable levels of YFV- and ZIKV-reactive polyfunctional CD4+ T cells with respect to each other and corresponding monovalent groups, both post-prime and post-boost.
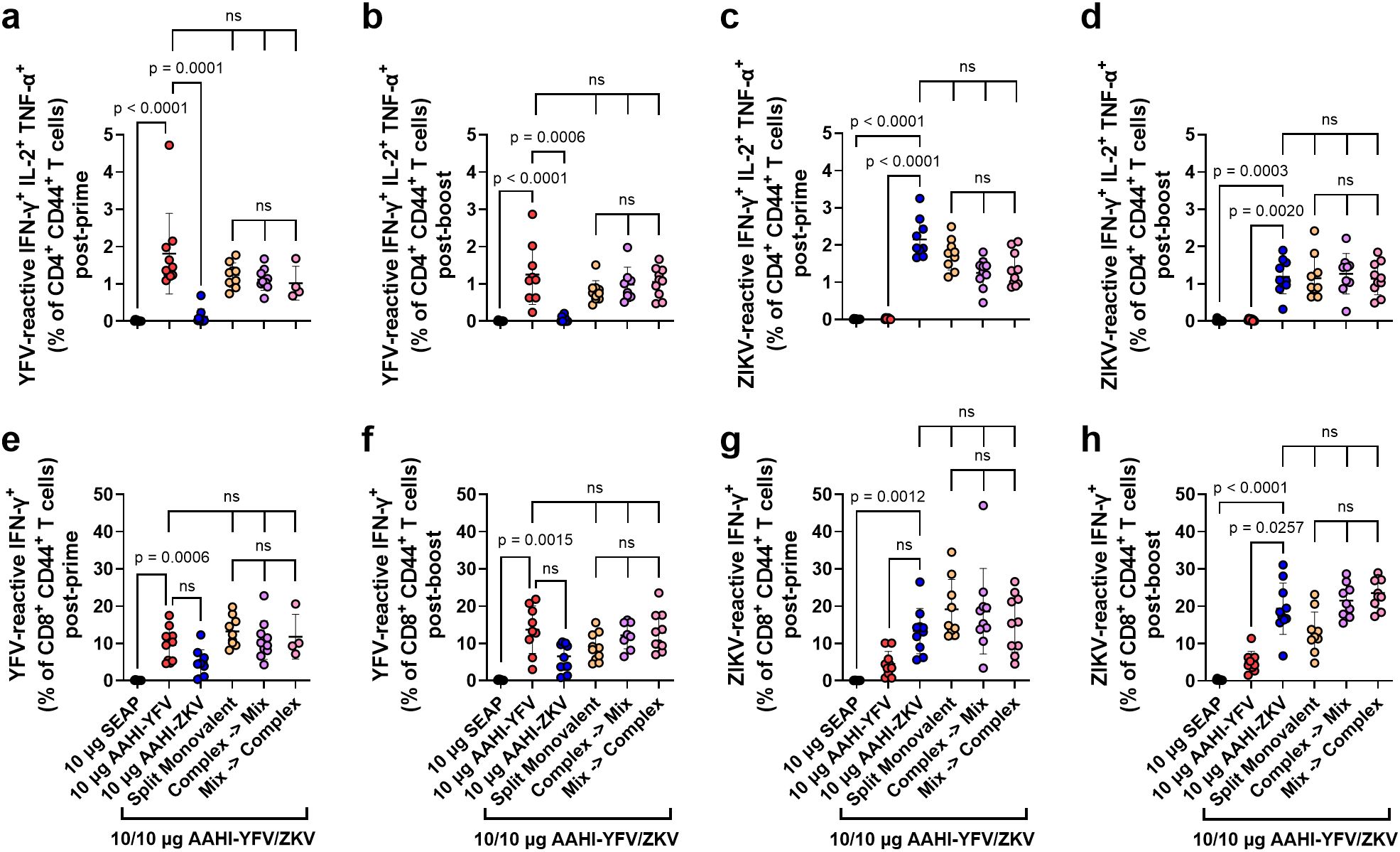
Figure 6. Monovalent and bivalent YFV and ZIKV saRNA-NLC vaccination induces polyfunctional CD4+ and IFN-γ+ CD8+ T cells. Mice were vaccinated with monovalent or bivalent formulations of AAHI-YFV or AAHI-ZKV saRNA-NLC, and splenocytes were isolated (a, c, e, g) 28 days post-prime or (b, d, f, h) 28 days post-boost. The antigen-specific polyfunctional (IFN-γ+ IL-2+ TNFα+) CD4+ T cell response was measured following stimulation with (a, b) YFV or (c, d) ZIKV prM-E peptide pools. The antigen-specific IFN-γ+ CD8+ T cell response was measured following stimulation with (e, f) YFV or (g, h) ZIKV prM-E peptide pools. Statistics were measured using Kruskal-Wallis with Dunn’s correction (a-e, g, h) or Brown-Forsythe and Welch ANOVA with Dunnett’s T3 correction (f) for multiple comparisons. Comparisons were made against monovalent AAHI-YFV or AAHI-ZKV and between bivalent AAHI-YFV/ZKV groups. ns= non-significant (p > 0.05). Results are from a single independent experiment; n = 10 mice per group, 5 male and 5 female. Scatter plots show mean ± SD.
Antigen-reactive IFN-γ+ CD8+ T cells were readily detectable (>1% of total CD8+ CD44+ T cells) in all monovalent AAHI-YFV and AAHI-ZKV vaccinated mice post-prime and post-boost (Figures 6e-h). In contrast with polyfunctional CD4+ T cells, cross-reactive IFN-γ+ CD8+ T cells were detectable at a percentage approximately half or less than that of the matched vaccine antigen. This observation is not surprising as these two related flaviviruses’ prM-E proteins share significant structural and sequence homology, which appear to include some CD8+ T cell epitopes but not CD4+ T cell epitopes. All bivalent AAHI-YFV/ZKV dosing strategies elicited comparable levels of IFN-γ+ CD8+ T cells to those elicited by monovalent vaccines both post-prime and post-boost. Lastly, we saw no consistent differences in YFV- or ZIKV-responsive CD8+ T cells between bivalent AAHI-YFV/ZKV groups regardless of the mixing and dosing strategy. Overall, saRNA-NLC immunization leads to robust multi-fold expansion of antigen-reactive T cells, and the mode of formulation and delivery of bivalent vaccines using this platform appears to have minimal effects on either humoral or cellular vaccine immunogenicity in mice, allowing for flexibility in future bivalent saRNA-NLC vaccine formulation strategies.
Immunization with ZKV saRNA-NLC is protective against lethal ZIKV infection in mice
To evaluate the efficacy of AAHI-ZKV in monovalent and bivalent format, we employed a transiently-immunocompromised mouse ZIKV challenge model (16, 26). C57BL/6 mice were first prime or prime-boost vaccinated with 10 µg monovalent AAHI-YFV or AAHI-ZKV, or 10/10 µg, 5/5 µg, or 1/1 µg bivalent AAHI-YFV/ZKV. A cohort of each study group was evaluated for post-vaccination flavivirus nAb titers and antigen-reactive cellular responses. Thirty days after final vaccination, mice were transiently immunocompromised by injection with α-IFNAR antibody -1, +1, and +4 days post-infection (dpi), and challenged with 105 PFU of MA-Zika Dakar (16, 25). Survival (mortality) and body weight change (morbidity) were assessed as metrics of vaccine efficacy (Figures 7a, e).
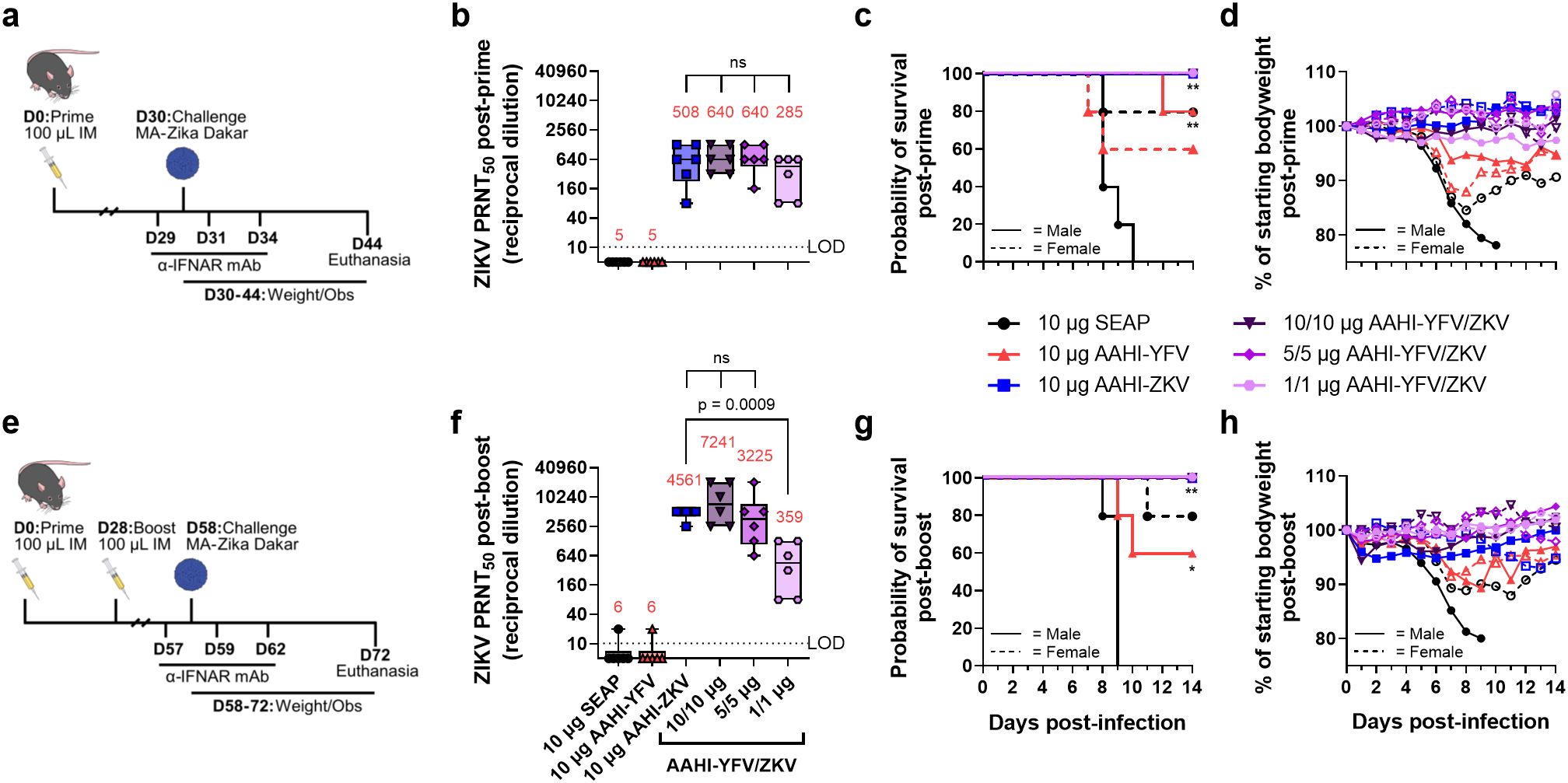
Figure 7. Monovalent and bivalent ZIKV saRNA-NLC vaccination protects mice from lethal ZIKV challenge. (a) Post-prime mouse ZIKV challenge study design (42–44) (b) Serum ZIKV neutralizing antibody titer 28 days post-prime. (c) Post-prime survival curves. (d) Post-prime body weight. (e) Post-boost mouse ZIKV challenge study design (42–44) (f) Serum ZIKV neutralizing antibody titer 28 days post-boost. (g) Post-boost survival curves. (h) Post-boost body weight. (b, d) Statistical analysis was conducted on log10 transformed data using one-way ANOVA with Dunnett’s correction for multiple comparisons. ns = non-significant (p > 0.05). Black dotted line shows the limit of detection (LOD) for the assay. Results are from a single independent experiment; n = 6 mice per group, 3 male and 3 female. Box plots show median and IQR ± min/max value. Red numerical values represent group PRNT50 GMT. (c, g) Statistics were assessed by Mantel-Cox log-rank test against 10 µg SEAP-expressing vector control. *p < 0.05, **p < 0.01. Results are from a single independent experiment; n = 10 mice per vaccination group, 5 male and 5 female.
Consistent with earlier observations (Figures 3, 5), a single prime dose of AAHI-ZKV, in both monovalent and bivalent formulation, induced robust ZIKV nAb titers well above the predicted CoP identified through passive transfer studies in mice (PRNT50 GMT ≥100) (Figure 7b) (38, 39). A boost dose augmented nAb GMT more than 4-fold in 10 µg monovalent AAHI-ZKV and 10/10 µg and 5/5 µg bivalent AAHI-YFV/ZKV dosed groups (Figure 7f). YFV nAb titers were likewise induced by 10 µg AAHI-YFV, and 10/10 µg and 5/5 µg AAHI-YFV/ZKV vaccine doses (Supplementary Figure 3a), with limited improvement with a boost dose (Supplementary Figure 3b). Lastly, indicative of a systemic vaccine-induced cellular response, splenic antigen-reactive polyfunctional CD4+ T cells (>0.2% of total CD4+ CD44+) (Supplementary Figures 4a-d) and CD8+ T cells (>0.2% of total CD8+ CD44+) (Supplementary Figures 4e-h) were generated at similar frequencies by both monovalent and bivalent AAHI-ZKV and AAHI-YFV immunization.
All mice vaccinated with AAHI-ZKV were completely protected against ZIKV-induced mortality, regardless of valency, dose, or dosing regimen (Figures 7c, g and Supplementary Table 2). We observed a strong sex-dependent difference in mortality regardless of dosing regimen, with 0% survival in SEAP-expressing vector control vaccinated males but 80% survival in SEAP-expressing vector control vaccinated females. Changes in weight following challenge mirrored survival (Figures 7d, h and Supplementary Figures 5a, b), with sharp decreases in weight noted in SEAP-expressing vector control vaccinated animals, but maintenance of normal body weight 3–6 dpi in AAHI-ZKV immunized mice regardless of sex or dosing regimen. Notably, mice immunized with 10 µg AAHI-YFV were partially protected from ZIKV challenge, showing 60% - 100% survival depending on both sex and dose regimen, and limited weight loss with survivors regaining normal weight by the end of the study.
Together, these results demonstrate (1) strong protection against ZIKV-induced morbidity and mortality by all AAHI-ZKV doses tested regardless of dosing regimen, (2) no detriment of combining AAHI-YFV and AAHI-ZKV into a bivalent vaccine, and (3) significant cross-protection from ZIKV-induced morbidity and mortality conferred by AAHI-YFV immunization, with no evidence of antibody-dependent enhancement of disease.
Immunization with YFV saRNA-NLC is protective against lethal YFV infection in hamsters
To evaluate the efficacy of AAHI-YFV in monovalent and bivalent format, we employed a hamster YFV challenge model (45). Syrian golden hamsters were first prime or prime-boost vaccinated with 10 µg monovalent AAHI-YFV or AAHI-ZKV, or 10/10 µg, 5/5 µg, or 1/1 µg bivalent AAHI-YFV/ZKV vaccines. Vaccinated hamsters were evaluated for post-vaccination flavivirus serum nAb titers. Thirty days after final vaccination, hamsters were challenged with 200 CCID50 of YFV-Jiménez. Survival (mortality), body weight change and serum ALT (morbidity), and viremia were assessed as metrics of vaccine efficacy (Figures 8a, e).
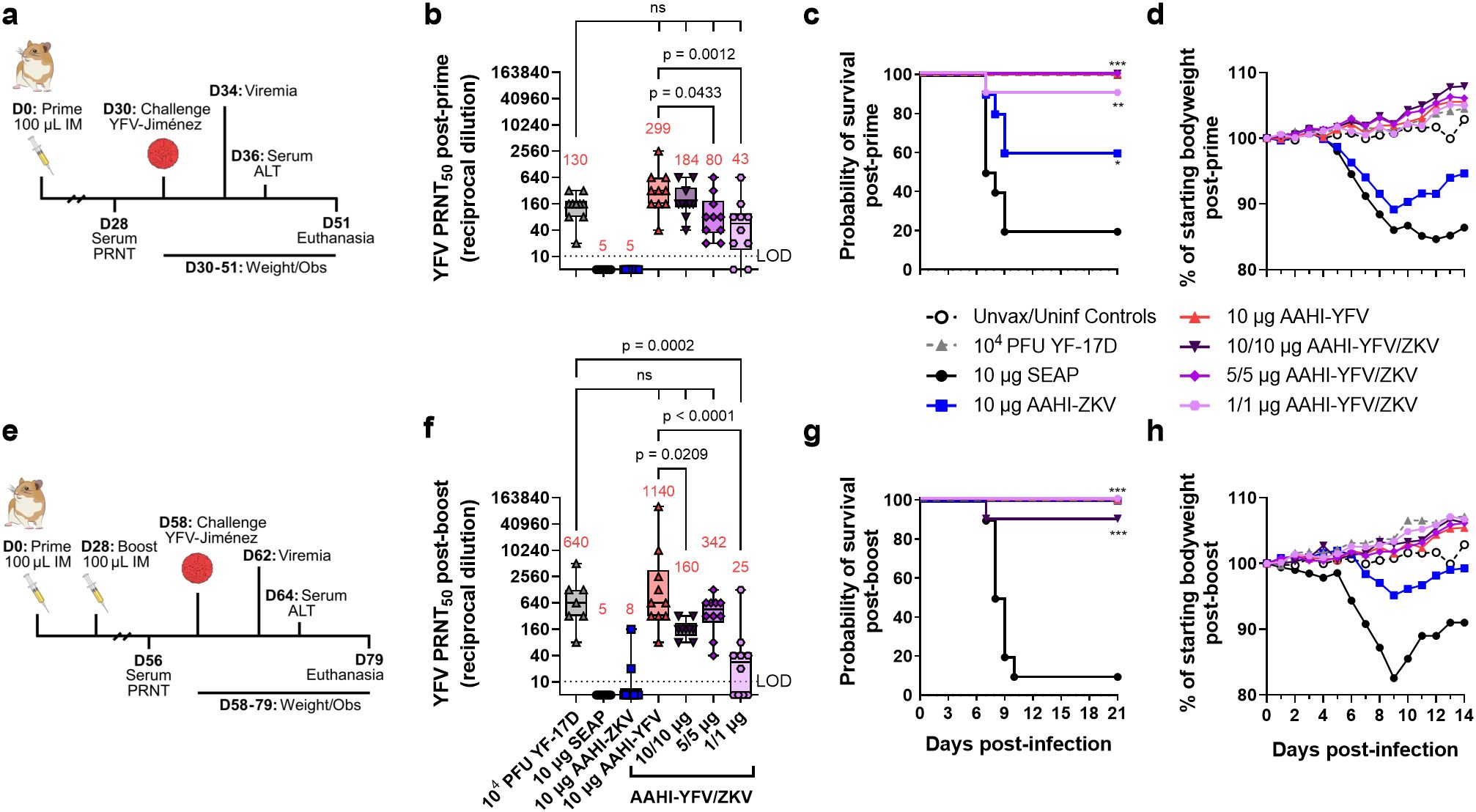
Figure 8. Monovalent and bivalent YFV saRNA-NLC vaccination protects hamsters from lethal YFV challenge. (a) Post-prime hamster YFV challenge study design (42, 44, 46) (b) Pre-challenge serum YFV neutralizing antibody titer 28 days post-prime. (c) Post-prime survival curves. (d) Post-prime body weight. (e) Post-boost hamster YFV challenge study design (42, 44, 46) (f) Pre-challenge serum YFV neutralizing antibody titer 28 days post-boost. (g) Post-boost survival curves. (h) Post-boost body weight. (b, d) Statistical analysis was conducted on log10 transformed data using one-way ANOVA with Dunnett’s correction for multiple comparisons. ns = non-significant (p > 0.05). Black dotted line shows the limit of detection (LOD) for the assay. Box plots show median and IQR ± min/max value. Red numerical values represent group PRNT50 GMT. (c, g) Statistics were assessed by Mantel-Cox log-rank test against 10 µg SEAP-expressing vector control. *p < 0.05, **p < 0.01, ***p < 0.001. Results are from a single independent experiment; n = 10 hamsters per group, 5 male and 5 female, or n = 5 female hamsters for unvaccinated/uninfected (Unvax/Uninf) controls.
Consistent with studies in mice, hamsters prime immunized with AAHI-YFV or AAHI-ZKV in both monovalent and bivalent formulation developed group serum nAb GMTs above the predicted CoP for each flavivirus (YFV: PRNT50 ≥40 in hamsters, ZIKV: PRNT50 ≥100 in mouse passive transfer studies) (Figure 8b and Supplementary Figure 6a) (31, 38, 39, 47). YFV and ZIKV nAb GMTs were generally higher with a prime-boost immunization schedule, though bivalent boosting showed inconsistent improvement for YFV nAb titers in contrast with ZIKV nAb titers (Figure 8f and Supplementary Figure 6b). After prime immunization, groups that received a 10/10 µg dose of AAHI-YFV/ZKV had YFV nAb titers comparable to those vaccinated with 10 µg AAHI-YFV. Lastly, serum YFV nAb titers in 10 µg AAHI-YFV and 10/10 µg and 5/5 µg AAHI-YFV/ZKV vaccinated hamsters were comparable to serum YFV nAb titers in YF-17D vaccinated hamsters.
As expected, a SEAP-expressing vector control immunization did not confer protection (20% survival), and YF-17D vaccination was completely protective (100% survival). Protection against mortality after YFV challenge was demonstrated in all study groups that received a prime-only or prime-boost regimen of AAHI-YFV or AAHI-YFV/ZIKV vaccine (Figures 8c, g and Supplementary Table 3). Prime-only vaccination with AAHI-YFV was highly protective with 100% survival in all doses other than the lowest (1/1 µg) AAHI-YFV/ZKV, which provided significant protection from mortality in 9/10 animals (Figure 8c). Interestingly, partial cross-protection was observed in hamsters dosed with 10 µg AAHI-ZKV, resulting in 60% survival (2/5 females and 4/5 males) after prime vaccination that was significantly improved over the SEAP-expressing vector control, and further improved to 100% survival after boost (Figure 8g).
Morbidity and viremia measurements also revealed a robust protective effect of AAHI-YFV vaccination. Weight loss 3–6 dpi was significantly reduced by prime immunization with AAHI-YFV or YF-17D compared to the SEAP-expressing vector control, whereas AAHI-ZKV vaccination alone did not significantly reduce weight change (Figure 8d and Supplementary Figure 7a). Post-boost, however, all AAHI-YFV and AAHI-ZKV vaccination strategies significantly limited weight change 3–6 dpi (Figure 8h and Supplementary Figure 7d). Average serum ALT levels 6 dpi, reflecting onset of YFV-induced viscerotropic disease, were at or near baseline in post-prime and post-boost YF-17D, AAHI-YFV, AAHI-ZKV, and AAHI-YFV/ZKV vaccinated hamsters (Supplementary Figures 7b, e), and were significantly improved relative to SEAP-expressing vector control-vaccinated hamsters. Lastly, viremia 4 dpi was prevented in animals receiving YF-17D, and any monovalent or bivalent dose of AAHI-YFV via prime-only or prime-boost vaccination, with the sole exception of 3/10 animals in the low 1/1 µg AAHI-YFV/ZKV prime-only vaccine dose group (Supplementary Figure 7c). Prime-only immunization with 10 µg AAHI-ZKV was insufficient to reduce serum viral titers relative to the SEAP-expressing vector control, but prime-boost AAHI-ZKV immunized animals resulted in reduced viremia relative to controls (Supplementary Figure 7f).
In sum, these data demonstrate (1) strong protection against YFV-induced morbidity and mortality provided by AAHI-YFV at all doses tested regardless of dosing regimen, (2) no detriment from bivalent combination of AAHI-YFV and AAHI-ZKV vaccines, and (3) significant cross-protection against YFV infection provided by the AAHI-ZKV vaccine with no evidence of antibody-dependent enhancement of disease.
Discussion
The aim of this study was to demonstrate platform feasibility and evaluate the immunogenicity and efficacy of a bivalent saRNA-NLC vaccine, in this case designed to target the geographically overlapping mosquito-borne flaviviruses YFV and ZIKV. Both monovalent AAHI-YFV and AAHI-ZKV immunization induced strong and durable serum flavivirus E protein-binding IgG and virus-specific nAb responses. Similarly robust nAb titers were elicited following bivalent AAHI-YFV/ZKV vaccination, through various dosing strategies and concentrations, at levels equivalent to those induced by the monovalent vaccines. These results were mirrored in the cellular response, as bivalent AAHI-YFV/ZKV vaccination induced levels of antigen-reactive polyfunctional CD4+ and IFN-γ+ CD8+ T cells comparable to those induced by the monovalent vaccines. Finally, we demonstrated that bivalent AAHI-YFV/ZKV immunization, at various doses, conferred complete protective efficacy in both mouse and hamster models of lethal ZIKV and YFV challenge. Taken together, the potent induction of humoral and cellular responses seen following bivalent vaccination highlights the flexible application of the AAHI saRNA-NLC platform against multiple viruses in a multivalent format.
The presence of serum virus-specific nAbs is critical for protection against flavivirus infection (48, 49). We have previously seen strong and durable induction of nAbs following one- or two-dose immunization with NLC-formulated ZIKV saRNA (16). The nAb GMT induced following monovalent and bivalent vaccination with AAHI-YFV and/or AAHI-ZKV are consistent with prior work, and are above protective PRNT50 values for YFV (PRNT50 ≥10 in humans) and ZIKV (PRNT50 ≥100 in mouse passive transfer studies) observed by other groups (2, 32, 38). Moreover, the magnitude of the nAb responses are comparable to other vaccine strategies, including mRNA-based vaccines, that target YFV (6, 32, 45, 50–56) and ZIKV (38, 39, 57–67). The strong dose-dependent nAb titers induced by AAHI-YFV were notable, which required a high (30 µg) dose or boost vaccination to approach the nAb titers induced in mice by YF-17D immunization. Although levels of YFV E protein-binding IgG and durability of nAb titers were equivalent between AAHI-YFV and YF-17D vaccinated mice, these data suggest a discrepancy in the induction of antigen-binding versus virus-neutralizing antibodies with the current AAHI-YFV saRNA construct. For example, when compared against AAHI-ZKV at comparable saRNA doses, AAHI-YFV induced similar levels of E protein-binding IgG but lower nAb titers. Structural and antigenic differences between prM-E proteins, rather than limitations of the bivalent vaccine strategy, may underpin the differences in antigen-binding versus virus-neutralizing antibodies. Future studies could explore further optimization of the YFV prM-E antigen in an attempt to enhance the ratio of nAbs, for example potentially matching the YFV prM-E structure and function to that of more immunogenic ZIKV prM-E variants (30, 68).
While induction of serum nAb titers is considered the primary CoP for flavivirus vaccination, T cells also play a crucial role in the immune response to viral infections. Vaccination with YF-17D induces a robust and persistent polyfunctional CD4+ and CD8+ T cell response with broad antigen specificity, capable of conferring protection against YFV challenge in conjunction with humoral immunity or through control of early viral loads, respectively (69–76). Various ZIKV vaccination approaches have also been reported to elicit antigen-specific cellular responses (38, 61–65, 77, 78). The induction of antigen-reactive CD4+ and CD8+ T cell responses by our saRNA-NLC formulations paired with strong nAb responses support the value of this vaccine platform. Interestingly, while induction of cross-reactive CD4+ T cells was minimal, cross-reactive CD8+ T cell induction was notable following vaccination with both monovalent flavivirus constructs. This difference may be a consequence of the MHC haplotype in C57BL/6 mice, as other groups have reported CD4+ T cell flavivirus cross-reactivity using MHC transgenic mouse models (79, 80). The consequence of flavivirus cross-reactive cellular responses remains poorly understood but could potentially confer some protection against heterologous flavivirus infections in the absence of robust pre-existing humoral immunity, or shape subsequent T cell responses, as has been proposed for dengue virus and ZIKV (81, 82). A flavivirus cross-reactive cellular response may partially explain the protection conferred by AAHI-YFV and AAHI-ZKV vaccination against ZIKV and YFV challenge, respectively; however, this possibility requires additional confirmation, such as through passive serum transfer and T cell depletion studies. In sum, the ability of this saRNA-NLC vaccine platform to stimulate strong T cell responses is promising, both for these flaviviral-targeted vaccines as well as vaccines targeting other infectious diseases.
To date, only one YFV vaccine has been approved for commercial use, the live attenuated strain YF-17D. Notably, while the safety profile of YF-17D is generally excellent in immunocompetent populations, and manufacturers stockpile 30–80 million doses/year, viscerotropic effects at the extremes of age, dissemination in the immunocompromised, and supply limitations have left public health officials without a complete set of tools for the prevention of YFV despite this durable and highly immunogenic YFV vaccine (6). In contrast with YFV, there are currently no approved ZIKV vaccines, in large part due to difficulties in conducting Phase III efficacy trials following the decline in circulating ZIKV (66). ZIKV still presents pandemic risk in the coming years as carrier mosquito populations continue to expand, and there is a potential risk of mutants escaping existing immunity or current vaccine candidates (83, 84). These unmet medical and public health concerns underscore the need for effective, safe, stable, and rapidly producible YFV and ZIKV vaccines. The potent saRNA-NLC vaccine platform presented here is well suited to address these needs. Intrinsic flexibility in bivalent vaccine mixing and dosing strategies that elicit comparable immune responses permits the efficient co-development of monovalent and bivalent products, and may allow different manufacturing strategies at different phases of product development. Thus, AAHI-YFV/ZKV is a strong candidate for a flavivirus bivalent vaccine.
While the studies conducted here were carefully designed for scientific rigor and reproducibility, some limitations exist. Groups were sex balanced, and while no significant sex-based differences were observed in any of the immunogenicity measures, larger study groups would be necessary to assess more subtle sex-based differences in vaccine-driven immune responses or efficacy against ZIKV challenge. Additionally, no immunogenicity or efficacy data are presented from primate models, which will be a key future direction for these vaccine candidates.
Previous platform development work has demonstrated the inherent flexibility of saRNA synthesis, ease of saRNA and NLC manufacturing, and the stability of lyophilized NLC-formulated saRNA at room temperature for extended periods, allowing for rapid scale-up and stockpiling of vaccine material for emerging viral variants (17, 85). Taken together, our saRNA-NLC system has the potential to improve global access to flavivirus vaccines, particularly in resource-limited areas, and serve as a platform to develop additional vaccines in multivalent format.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by Bloodworks Northwest Research Institute’s (Seattle, WA) Institutional Animal Care and Use Committee (IACUC) and Utah State University IACUC. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
PB: Investigation, Methodology, Validation, Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. MY: Supervision, Investigation, Methodology, Validation, Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. DK: Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. MJ: Supervision, Investigation, Methodology, Validation, Data curation, Formal analysis, Writing – review & editing. SB: Investigation, Validation, Data curation, Formal analysis, Writing – review & editing. AW: Investigation, Writing – review & editing. DH: Investigation, Methodology, Validation, Writing – review & editing. JS: Investigation, Validation, Data curation, Formal analysis, Writing – review & editing. JB: Investigation, Methodology, Validation, Writing – review & editing. NC: Investigation, Writing – review & editing. PF: Investigation, Writing – review & editing. JA: Investigation, Writing – review & editing. SR: Investigation, Validation, Data curation, Writing – review & editing. AG: Supervision, Project administration, Validation, Writing – review & editing. JJ: Supervision, Project administration, Writing – review & editing. CC: Conceptualization, Project administration, Writing – review & editing. EV: Conceptualization, Supervision, Project administration, Funding acquisition, Validation, Formal analysis, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, under Contract No. 75N93019C00059.
Conflict of interest
AG and EV are co-inventors on U.S. patent application nos. PCT/US21/40388, “Co-lyophilized RNA and Nanostructured Lipid Carrier,” and 63/144,169, “A thermostable, flexible RNA vaccine delivery platform for pandemic response.”
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1569454/full#supplementary-material
References
1. Paules CI, Fauci AS. Yellow fever — Once again on the radar screen in the Americas. N Engl J Med. (2017) 376:1397–9. doi: 10.1056/NEJMp1702172
2. Ma J, Boudewijns R, Sanchez-Felipe L, Mishra N, Vercruysse T, Buh Kum D, et al. Comparing immunogenicity and protective efficacy of the yellow fever 17D vaccine in mice. Emerg Microbes Infect. (2021) 10:2279–90. doi: 10.1080/22221751.2021.2008772
3. Staples JE, Barrett ADT, Wilder-Smith A, Hombach J. Review of data and knowledge gaps regarding yellow fever vaccine-induced immunity and duration of protection. NPJ Vaccines. (2020) 5:54. doi: 10.1038/s41541-020-0205-6
4. World Health Organization. WHO expert committee on biological standardization. World Health Organ Tech Rep Ser. (2013) 979:1–366. Available at: https://iris.who.int/handle/10665/89156 (Accessed April 15, 2025).
6. Hansen CA, Barrett ADT. The present and future of yellow fever vaccines. Pharmaceuticals. (2021) 14:891. doi: 10.3390/ph14090891
7. Gardner CL, Ryman KD. Yellow fever: A reemerging threat. Clin Lab Med. (2010) 30:237–60. doi: 10.1016/j.cll.2010.01.001
8. Casey RM, Harris JB, Ahuka-Mundeke S, Dixon MG, Kizito GM, Nsele PM, et al. Immunogenicity of fractional-dose vaccine during a yellow fever outbreak — Final report. N Engl J Med. (2019) 381:444–54. doi: 10.1056/NEJMoa1710430
9. FDA. Package insert - YF-vax(2019). Available online at: https://www.fda.gov/media/76015 (Accessed September 20, 2024).
10. Fauci AS, Morens DM. Zika virus in the americas — Yet another arbovirus threat. N Engl J Med. (2016) 374:601–4. doi: 10.1056/NEJMp1600297
11. Guzeloglu-Kayisli O, Kayisli UA, Schatz F, Lockwood CJ. Vertical zika virus transmission at the maternal-fetal interface. Front Virol. (2022) 2:801778. doi: 10.3389/fviro.2022.801778
12. Nwaiwu AU, Musekiwa A, Tamuzi JL, Sambala EZ, Nyasulu PS. The incidence and mortality of yellow fever in Africa: a systematic review and meta-analysis. BMC Infect Dis. (2021) 21:1089. doi: 10.1186/s12879-021-06728-x
13. Zhou K, Li C, Shi W, Hu X, Nandakumar KS, Jiang S, et al. Current progress in the development of zika virus vaccines. Vaccines. (2021) 9:1004. doi: 10.3390/vaccines9091004
14. Essink B, Chu L, Seger W, Barranco E, Cam NL, Bennett H, et al. The safety and immunogenicity of two Zika virus mRNA vaccine candidates in healthy flavivirus baseline seropositive and seronegative adults: the results of two randomised, placebo-controlled, dose-ranging, phase 1 clinical trials. Lancet Infect Dis. (2023) 23:621–33. doi: 10.1016/S1473-3099(22)00764-2
15. Theiler M, Smith HH. THE USE OF YELLOW FEVER VIRUS MODIFIED BY IN VITRO CULTIVATION FOR HUMAN IMMUNIZATION. J Exp Med. (1937) 65:787–800. doi: 10.1084/jem.65.6.787
16. Erasmus JH, Khandhar AP, Guderian J, Granger B, Archer J, Archer M, et al. A nanostructured lipid carrier for delivery of a replicating viral RNA provides single, low-dose protection against Zika. Mol Ther J Am Soc Gene Ther. (2018) 26:2507–22. doi: 10.1016/j.ymthe.2018.07.010
17. Gerhardt A, Voigt E, Archer M, Reed S, Larson E, Van Hoeven N, et al. A flexible, thermostable nanostructured lipid carrier platform for RNA vaccine delivery. Mol Ther Methods Clin Dev. (2022) 25:205–14. doi: 10.1016/j.omtm.2022.03.009
18. Rice A, Verma M, Voigt E, Battisti P, Beaver S, Reed S, et al. Heterologous saRNA prime, DNA dual-antigen boost SARS-CoV-2 vaccination elicits robust cellular immunogenicity and cross-variant neutralizing antibodies. Front Immunol. (2022) 13:910136. doi: 10.3389/fimmu.2022.910136
19. Voigt EA, Gerhardt A, Hanson D, Jennewein MF, Battisti P, Reed S, et al. A self-amplifying RNA vaccine against COVID-19 with long-term room-temperature stability. NPJ Vaccines. (2022) 7:1–13. doi: 10.1038/s41541-022-00549-y
20. ImmunityBio, Inc. THEMBA II T-CELL Vaccine: A Phase 1/2 Study of the Safety, Reactogenicity, and Immunogenicity of Vaccination With saRNA COVID-19 Vaccines. clinicaltrials.gov. (2022). https://clinicaltrials.gov/ct2/show/NCT05370040 (Accessed April 7, 2025).
21. Jennewein MF, Schultz MD, Beaver S, Battisti P, Bakken J, Hanson D, et al. Intranasal replicon SARS-CoV-2 vaccine produces protective respiratory and systemic immunity and prevents viral transmission. Molecular Therapy. (2025). doi: 10.1016/j.ymthe.2025.04.007
22. Li Z, Jaroszewski L, Iyer M, Sedova M, Godzik A. FATCAT 2.0: towards a better understanding of the structural diversity of proteins. Nucleic Acids Res. (2020) 48:W60–4. doi: 10.1093/nar/gkaa443
23. Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. (2003) 19:ii246–55. doi: 10.1093/bioinformatics/btg1086
24. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
25. Erasmus JH, Archer J, Fuerte-Stone J, Khandhar AP, Voigt E, Granger B, et al. Intramuscular delivery of replicon RNA encoding ZIKV-117 human monoclonal antibody protects against Zika virus infection. Mol Ther Methods Clin Dev. (2020) 18:402–14. doi: 10.1016/j.omtm.2020.06.011
26. Gorman MJ, Caine EA, Zaitsev K, Begley MC, Weger-Lucarelli J, Uccellini MB, et al. An immunocompetent mouse model of Zika virus infection. Cell Host Microbe. (2018) 23:672–685.e6. doi: 10.1016/j.chom.2018.04.003
27. Julander JG, Bantia S, Taubenheim BR, Minning DM, Kotian P, Morrey JD, et al. BCX4430, a novel nucleoside analog, effectively treats yellow fever in a hamster model. Antimicrob Agents Chemother. (2014) 58:6607–14. doi: 10.1128/aac.03368-14
28. Bloom K, van den Berg F, Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. (2021) 28:117–29. doi: 10.1038/s41434-020-00204-y
29. Vratskikh O, Stiasny K, Zlatkovic J, Tsouchnikas G, Jarmer J, Karrer U, et al. Dissection of antibody specificities induced by yellow fever vaccination. PloS Pathog. (2013) 9:e1003458. doi: 10.1371/journal.ppat.1003458
30. Yang C, Zeng F, Gao X, Zhao S, Li X, Liu S, et al. Characterization of two engineered dimeric Zika virus envelope proteins as immunogens for neutralizing antibody selection and vaccine design. J Biol Chem. (2019) 294:10638–48. doi: 10.1074/jbc.RA119.007443
31. Julander JG, Trent DW, Monath TP. Immune correlates of protection against yellow fever determined by passive immunization and challenge in the hamster model. Vaccine. (2011) 29:6008–16. doi: 10.1016/j.vaccine.2011.06.034
32. Amanna IJ, Thomas A, Engelmann F, Hammarlund E, Raué H-P, Bailey AL, et al. Development of a hydrogen peroxide-inactivated vaccine that protects against viscerotropic yellow fever in a non-human primate model. Cell Rep Med. (2024) 5:101655. doi: 10.1016/j.xcrm.2024.101655
33. Yan K, Vet LJ, Tang B, Hobson-Peters J, Rawle DJ, Le TT, et al. A yellow fever virus 17D infection and disease mouse model used to evaluate a chimeric binjari-yellow fever virus vaccine. Vaccines. (2020) 8:368. doi: 10.3390/vaccines8030368
34. Cajaraville AC dos RA, Gomes MP de B, Azamor T, Pereira RC, Neves PC da C, De Luca PM, et al. Evaluation of two adjuvant formulations for an inactivated yellow fever 17DD vaccine candidate in mice. Vaccines. (2023) 11:73. doi: 10.3390/vaccines11010073
35. Gotuzzo E, Yactayo S, Córdova E. Efficacy and duration of immunity after yellow fever vaccination: systematic review on the need for a booster every 10 years. Am J Trop Med Hyg. (2013) 89:434–44. doi: 10.4269/ajtmh.13-0264
36. Mokaya J, Kimathi D, Lambe T, Warimwe GM. What constitutes protective immunity following yellow fever vaccination? Vaccines. (2021) 9:671. doi: 10.3390/vaccines9060671
37. Slon Campos JL, Mongkolsapaya J, Screaton GR. The immune response against flaviviruses. Nat Immunol. (2018) 19:1189–98. doi: 10.1038/s41590-018-0210-3
38. Abbink P, Larocca RA, de la Barrera RA, Bricault CA, Moseley ET, Boyd M, et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science. (2016) 353:1129–32. doi: 10.1126/science.aah6157
39. Baldwin WR, Livengood JA, Giebler HA, Stovall JL, Boroughs KL, Sonnberg S, et al. Purified inactivated zika vaccine candidates afford protection against lethal challenge in mice. Sci Rep. (2018) 8:16509. doi: 10.1038/s41598-018-34735-7
40. Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, et al. Naïve CD4+ T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. (2007) 27:203–13. doi: 10.1016/j.immuni.2007.07.007
41. Blattman JN, Antia R, Sourdive DJD, Wang X, Kaech SM, Murali-Krishna K, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. (2002) 195:657–64. doi: 10.1084/jem.20001021
42. Kissinger R. Syringe. NIH Bioart Source. (2024). https://bioart.niaid.nih.gov/bioart/506 (Accessed April 7, 2025).
43. Kissinger R. Lab mouse. NIH Bioart Source. (2024). https://bioart.niaid.nih.gov/bioart/281 (Accessed April 7, 2025).
44. Kissinger R. Flavivirus. NIH Bioart Source. (2024). https://bioart.niaid.nih.gov/bioart/156 (Accessed April 7, 2025).
45. Julander JG, Testori M, Cheminay C, Volkmann A. Immunogenicity and protection after vaccination with a modified vaccinia virus ankara-vectored yellow fever vaccine in the hamster model. Front Immunol. (2018) 9:1756. doi: 10.3389/fimmu.2018.01756
46. Perry-Gottschalk R. Hamster front view. NIH Bioart Source. (2024). https://bioart.niaid.nih.gov/bioart/188 (Accessed April 7, 2025).
47. Abbink P, Larocca RA, Visitsunthorn K, Boyd M, de la Barrera RA, Gromowski GD, et al. Durability and correlates of vaccine protection against Zika virus in rhesus monkeys. Sci Transl Med. (2017) 9:eaao4163. doi: 10.1126/scitranslmed.aao4163
48. Pierson TC, Fremont DH, Kuhn RJ, Diamond MS. Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: implications for vaccine development. Cell Host Microbe. (2008) 4:229–38. doi: 10.1016/j.chom.2008.08.004
49. Dowd KA, Pierson TC. Antibody-mediated neutralization of flaviviruses: A reductionist view. Virology. (2011) 411:306–15. doi: 10.1016/j.virol.2010.12.020
50. Tretyakova I, Nickols B, Hidajat R, Jokinen J, Lukashevich IS, Pushko P. Plasmid DNA initiates replication of yellow fever vaccine in vitro and elicits virus-specific immune response in mice. Virology. (2014) 468–470:28–35. doi: 10.1016/j.virol.2014.07.050
51. Monath TP, Fowler E, Johnson CT, Balser J, Morin MJ, Sisti M, et al. An inactivated cell-culture vaccine against yellow fever. N Engl J Med. (2011) 364:1326–33. doi: 10.1056/NEJMoa1009303
52. Monath TP, Lee CK, Julander JG, Brown A, Beasley DW, Watts DM, et al. Inactivated yellow fever 17D vaccine: Development and nonclinical safety, immunogenicity and protective activity. Vaccine. (2010) 28:3827–40. doi: 10.1016/j.vaccine.2010.03.023
53. Piras-Douce F, Raynal F, Raquin A, Girerd-Chambaz Y, Gautheron S, Sanchez MEN, et al. Next generation live-attenuated yellow fever vaccine candidate: Safety and immuno-efficacy in small animal models. Vaccine. (2021) 39:1846–56. doi: 10.1016/j.vaccine.2021.02.033
54. Jr MM, Cruz F da SP, Cordeiro MT, Motta MA da, Cassemiro KMS de M, Maia R deCC, et al. A DNA vaccine against yellow fever virus: development and evaluation. PloS Negl Trop Dis. (2015) 9:e0003693. doi: 10.1371/journal.pntd.0003693
55. Schäfer B, Holzer GW, Joachimsthaler A, Coulibaly S, Schwendinger M, Crowe BA, et al. Pre-clinical efficacy and safety of experimental vaccines based on non-replicating vaccinia vectors against yellow fever. PloS One. (2011) 6:e24505. doi: 10.1371/journal.pone.0024505
56. Medina-Magües LG, Mühe J, Jasny E, Medina-Magües ES, Roth N, Lopera-Madrid J, et al. Immunogenicity and protective activity of mRNA vaccine candidates against yellow fever virus in animal models. NPJ Vaccines. (2023) 8:1–13. doi: 10.1038/s41541-023-00629-7
57. Bollman B, Nunna N, Bahl K, Hsiao CJ, Bennett H, Butler S, et al. An optimized messenger RNA vaccine candidate protects non-human primates from Zika virus infection. Immunology. (2022) 8:58. doi: 10.1101/2022.10.11.511814. preprint.
58. Luisi K, Morabito KM, Burgomaster KE, Sharma M, Kong W-P, Foreman BM, et al. Development of a potent Zika virus vaccine using self-amplifying messenger RNA. Sci Adv. (2020) 6:eaba5068. doi: 10.1126/sciadv.aba5068
59. Dowd KA, Ko S-Y, Morabito KM, Yang ES, Pelc RS, DeMaso CR, et al. Rapid development of a DNA vaccine for Zika virus. Science. (2016) 354:237–40. doi: 10.1126/science.aai9137
60. Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell. (2017) 168:1114–1125.e10. doi: 10.1016/j.cell.2017.02.017
61. Van Rompay KKA, Keesler RI, Ardeshir A, Watanabe J, Usachenko J, Singapuri A, et al. DNA vaccination before conception protects Zika virus–exposed pregnant macaques against prolonged viremia and improves fetal outcomes. Sci Transl Med. (2019) 11:eaay2736. doi: 10.1126/scitranslmed.aay2736
62. Gaudinski MR, Houser KV, Morabito KM, Hu Z, Yamshchikov G, Rothwell RS, et al. Safety, tolerability, and immunogenicity of two Zika virus DNA vaccine candidates in healthy adults: randomised, open-label, phase 1 clinical trials. Lancet. (2018) 391:552–62. doi: 10.1016/S0140-6736(17)33105-7
63. Chin W-X, Lee RCH, Kaur P, Lew TS, Yogarajah T, Kong HY, et al. A single-dose live attenuated chimeric vaccine candidate against Zika virus. NPJ Vaccines. (2021) 6:1–12. doi: 10.1038/s41541-021-00282-y
64. Chahal JS, Fang T, Woodham AW, Khan OF, Ling J, Anderson DG, et al. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci Rep. (2017) 7:252. doi: 10.1038/s41598-017-00193-w
65. Tebas P, Roberts CC, Muthumani K, Reuschel EL, Kudchodkar SB, Zaidi FI, et al. Safety and immunogenicity of an anti–zika virus DNA vaccine. N Engl J Med. (2021) 385:e35. doi: 10.1056/NEJMoa1708120
66. Woodson SE, Morabito KM. Continuing development of vaccines and monoclonal antibodies against Zika virus. NPJ Vaccines. (2024) 9:1–8. doi: 10.1038/s41541-024-00889-x
67. Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. (2017) 543:248–51. doi: 10.1038/nature21428
68. Sevvana M, Kuhn RJ. Mapping the diverse structural landscape of the flavivirus antibody repertoire. Curr Opin Virol. (2020) 45:51–64. doi: 10.1016/j.coviro.2020.07.006
69. Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response1. J Immunol. (2009) 183:7919–30. doi: 10.4049/jimmunol.0803903
70. James EA, LaFond RE, Gates TJ, Mai DT, Malhotra U, Kwok WW. Yellow fever vaccination elicits broad functional CD4+ T cell responses that recognize structural and nonstructural proteins. J Virol. (2013) 87:12794–804. doi: 10.1128/JVI.01160-13
71. Watson AM, Lam LKM, Klimstra WB, Ryman KD. The 17D-204 Vaccine Strain-Induced Protection against Virulent Yellow Fever Virus Is Mediated by Humoral Immunity and CD4+ but not CD8+ T Cells. PloS Pathog. (2016) 12:e1005786. doi: 10.1371/journal.ppat.1005786
72. Kongsgaard M, Bassi MR, Rasmussen M, Skjodt K, Thybo S, Gabriel M, et al. Adaptive immune responses to booster vaccination against yellow fever virus are much reduced compared to those after primary vaccination. Sci Rep. (2017) 7:662. doi: 10.1038/s41598-017-00798-1
73. Kohler S, Bethke N, Böthe M, Sommerick S, Frentsch M, Romagnani C, et al. The early cellular signatures of protective immunity induced by live viral vaccination. Eur J Immunol. (2012) 42:2363–73. doi: 10.1002/eji.201142306
74. Wieten RW, Jonker EFF, Leeuwen EMMv, Remmerswaal EBM, Berge IJMt, Visser AW de, et al. A single 17D yellow fever vaccination provides lifelong immunity; characterization of yellow-fever-specific neutralizing antibody and T-cell responses after vaccination. PloS One. (2016) 11:e0149871. doi: 10.1371/journal.pone.0149871
75. Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Abed Maillard S, et al. Long-lasting stem cell-like memory CD8+ T cells with a naive-like profile upon yellow fever vaccination. Sci Transl Med. (2015) 7:282ra48. doi: 10.1126/scitranslmed.aaa3700
76. Bassi MR, Kongsgaard M, Steffensen MA, Fenger C, Rasmussen M, Skjødt K, et al. CD8+ T cells complement antibodies in protecting against yellow fever virus. J Immunol. (2015) 194:1141–53. doi: 10.4049/jimmunol.1402605
77. Larocca RA, Abbink P, Peron JPS, de A, Zanotto PM, Iampietro MJ, et al. Vaccine protection against Zika virus from Brazil. Nature. (2016) 536:474–8. doi: 10.1038/nature18952
78. Wressnigg NV, Hochreiter R, Schneider M, Obersriebnig MJ, Bézay NI, Lingnau K, et al. A randomized, placebo-controlled, blinded phase 1 study investigating a novel inactivated, Vero cell-culture derived Zika virus vaccine. J Travel Med. (2022) 31:taac127. doi: 10.1093/jtm/taac127
79. Wen J, Wang Y-T, Valentine KM, Alves RP dos S, Xu Z, Regla-Nava JA, et al. CD4+ T cells cross-reactive with dengue and zika viruses protect against zika virus infection. Cell Rep. (2020) 31:107566. doi: 10.1016/j.celrep.2020.107566
80. Reynolds CJ, Suleyman OM, Ortega-Prieto AM, Skelton JK, Bonnesoeur P, Blohm A, et al. T cell immunity to Zika virus targets immunodominant epitopes that show cross-reactivity with other Flaviviruses. Sci Rep. (2018) 8:672. doi: 10.1038/s41598-017-18781-1
81. Grifoni A, Pham J, Sidney J, O’Rourke PH, Paul S, Peters B, et al. Prior dengue virus exposure shapes T cell immunity to zika virus in humans. J Virol. (2017) 91:10.1128/jvi.01469-17. doi: 10.1128/jvi.01469-17
82. Schouest B, Grifoni A, Pham J, Mateus J, Sydney J, Brien JD, et al. Pre-existing T cell memory against zika virus. J Virol. (2021) 95:e00132–21. doi: 10.1128/JVI.00132-21
83. Ryan SJ, Carlson CJ, Tesla B, Bonds MH, Ngonghala CN, Mordecai EA, et al. Warming temperatures could expose more than 1.3 billion new people to Zika virus risk by 2050. Glob Change Biol. (2021) 27:84–93. doi: 10.1111/gcb.15384
84. Regla-Nava JA, Wang Y-T, Fontes-Garfias CR, Liu Y, Syed T, Susantono M, et al. A Zika virus mutation enhances transmission potential and confers escape from protective dengue virus immunity. Cell Rep. (2022) 39:110655. doi: 10.1016/j.celrep.2022.110655
Keywords: flavivirus vaccine, self-amplifying RNA, nanostructured lipid carrier, bivalent vaccine, yellow fever virus, Zika virus
Citation: Battisti P, Ykema MR, Kasal DN, Jennewein MF, Beaver S, Weight AE, Hanson D, Singh J, Bakken J, Cross N, Fusco P, Archer J, Reed S, Gerhardt A, Julander JG, Casper C and Voigt EA (2025) A bivalent self-amplifying RNA vaccine against yellow fever and Zika viruses. Front. Immunol. 16:1569454. doi: 10.3389/fimmu.2025.1569454
Received: 31 January 2025; Accepted: 08 April 2025;
Published: 29 April 2025.
Edited by:
Chenming (Mike) Zhang, Virginia Tech, United StatesReviewed by:
Mariah Hassert, The University of Iowa, United StatesHe Hu, ADARx Pharmaceuticals, Inc., United States
Copyright © 2025 Battisti, Ykema, Kasal, Jennewein, Beaver, Weight, Hanson, Singh, Bakken, Cross, Fusco, Archer, Reed, Gerhardt, Julander, Casper and Voigt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emily A. Voigt, ZW1pbHkudm9pZ3RAYWFoaS5vcmc=