 Alexander Biederstädt
Alexander Biederstädt Florian Bassermann
Florian Bassermann Judith S. Hecker
Judith S. Hecker- 1Department of Medicine III, Technical University of Munich (TUM), School of Medicine and Health, Munich, Germany
- 2TranslaTUM, Center for Translational Cancer Research, Technical University of Munich (TUM), Munich, Germany
- 3Deutsches Konsortium für Translationale Krebsforschung (DKTK), Heidelberg, Germany
- 4Bavarian Cancer Research Center (BZKF), Munich, Germany
Introduction: Relapsed/refractory (r/r) large B-cell lymphoma (LBCL) remains a difficult-to-treat disease with limited treatment options and high unmet clinical need, necessitating the development of new therapies with greater potency and broader applicability. While autologous chimeric antigen receptor (CAR)-T cell therapies have transformed the treatment landscape, 60–65% of patients receiving these therapies eventually relapse, underscoring the need for improved approaches. Allogeneic CAR-T and CAR-NK cell therapies have recently emerged as promising alternatives, offering the potential to shorten manufacturing times, reduce costs, and expand access to a broader patient population. This systematic review and meta-analysis compiles the currently available clinical trial data on the efficacy and safety of these novel therapies in adult patients with r/r LBCL.
Methods: A systematic search of MEDLINE, EMBASE, Web of Science, and the Cochrane Central Register of Controlled Trials was conducted for studies published up to January 12, 2025, involving allogeneic CAR-T and CAR-NK cell therapies in R/R LBCL. The primary outcomes assessed were the best overall response rate (bORR) and best complete response rate (bCRR) at any time point. Secondary outcomes included rates of grade 1-2 and grade 3+ cytokine release syndrome (CRS), grade 1-2 and grade 3+ immune effector cell-associated neurotoxicity syndrome (ICANS), grade 1-2 and grade 3+ infections and incidence of graft-versus-host disease (GvHD).
Results: Nineteen studies met the inclusion and exclusion criteria, encompassing 334 patients (155 CAR-NK; 179 CAR-T) evaluable for safety and 235 patients evaluable for response (77 CAR-NK; 158 CAR-T). The pooled estimates for the best overall response rate (bORR) and the best complete response rate (bCRR) were 52.5% [95% CI, 41.0-63.9] and 32.8% [95% CI, 24.2-42.0], respectively. Safety analysis revealed very low incidences of grade 3+ CRS (0.04% [95% CI 0.00-0.49]) or grade 3+ ICANS (0.64% [95% CI 0.01-2.23]) and only one occurrence of a GvH-like reaction across 334 infused patients enrolled in the included studies, highlighting the remarkable safety profile of CAR-engineered “off-the-shelf” allogeneic approaches. The estimated overall incidence of low-grade CRS was 30% [95% CI, 14-48], while the estimated overall incidence of low-grade ICANS was 1% [95% CI, 0%-4%], markedly lower than current-generation autologous CAR-T cell products. The incidence of low-grade and severe infections was 25% [95% CI 14-36%) (n=252) and 7% [95% CI 2-14%] (n=291), respectively.
Discussion: Together, allogeneic CAR-T and CAR-NK cell therapies demonstrate encouraging efficacy in heavily pretreated patients with r/r LBCL. Coupled with their favorable safety profiles and the potential for off-the-shelf availability, allogeneic cell therapies hold great promise to broaden the reach of live cell-based treatments, delivering impactful results to a wider patient population in the coming years.
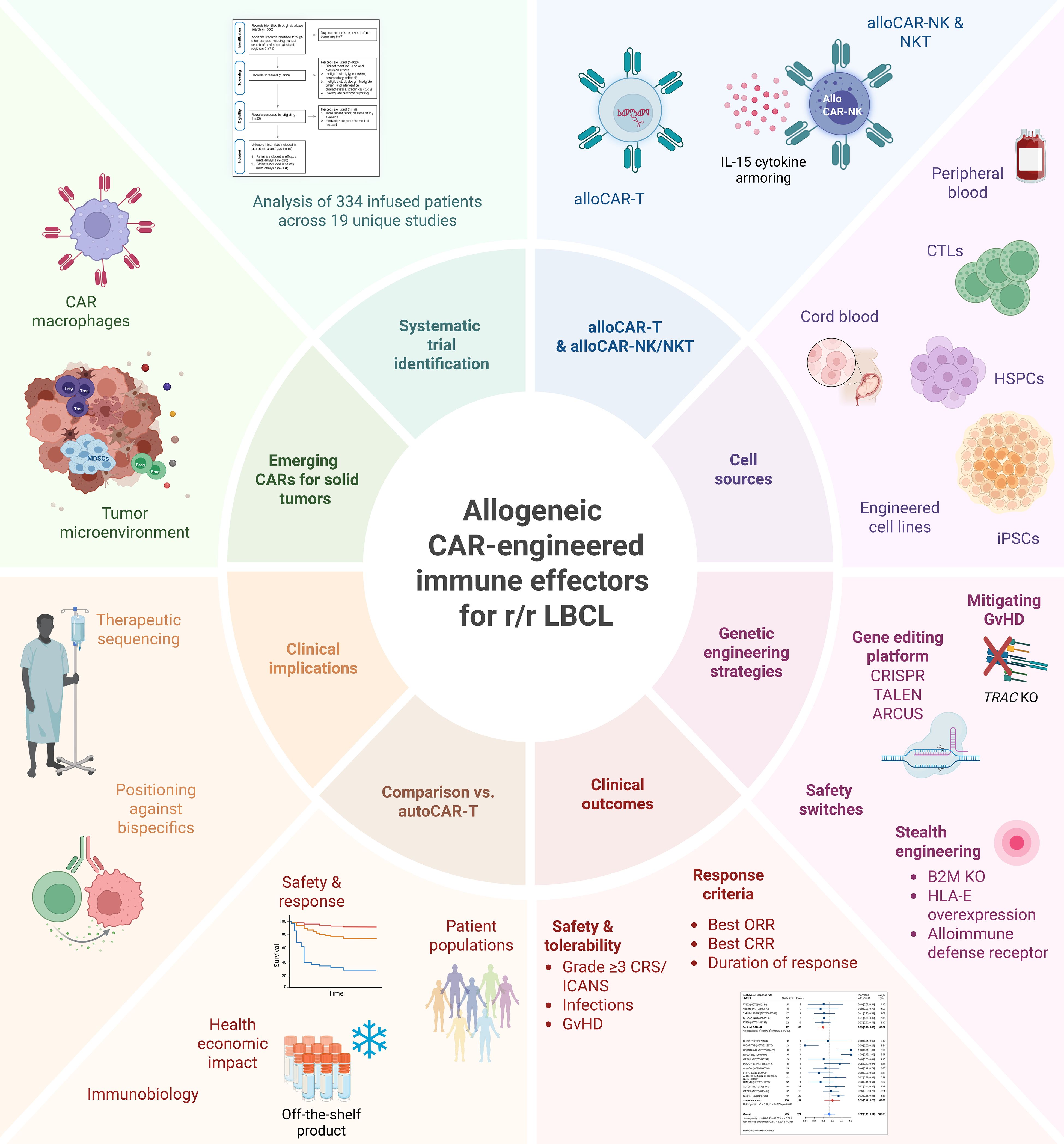
Graphical Abstract. Allogeneic CAR-engineered immune effector cells (CAR-IEC) for relapsed and refractory large B cell lymphoma (r/r LBCL).
Introduction
Chimeric antigen receptor (CAR)-T cell therapy has revolutionized the treatment landscape for relapsed and refractory (r/r) hematologic malignancies, including CD19-positive lymphoid malignancies and multiple myeloma. Over the past decade, seven FDA-approved autologous CAR-T cell products have reached clinical use, each targeting specific indications for patients with r/r disease (1–15). These advances represent a major milestone in oncology, showcasing the potential of cellular immunotherapies.
Long-term and real-world evidence has begun to validate the transformative potential of CAR-T therapy, with durable responses observed in a significant fraction of patients (6, 8, 9, 16). Notably, recent studies have detected CAR-positive T cells persisting up to 10 years post-infusion, underscoring the durability and adaptability of these engineered cellular therapies (17). Despite these remarkable successes, significant challenges remain. Many patients treated with current-generation CAR-T cell products fail to achieve durable responses, and a substantial proportion ultimately relapse (1, 10, 15, 16, 18, 19).
Moreover, the reliance on autologous CAR-T cells introduces logistical and biological challenges. The need for patient-specific lymphocyte apheresis and the complexities of individualized manufacturing extend time to treatment and increase costs. Efforts to decentralize manufacturing of autologous CAR-T cells are being explored to mitigate some of the logistical and cost hurdles (20). However, decentralized manufacturing cannot overcome the fundamental challenge posed by patients with low lymphocyte counts or dysfunctional lymphocytes, which may result from prior chemoimmunotherapy, tumor-induced immunosuppression, or inherent T cell defects, such as those seen in HIV-associated DLBCL. For such patients, allogeneic CAR-T and CAR-NK therapies, which utilize healthy donor-derived or induced pluripotent stem cell (iPSC)-derived cells, may provide a distinct advantage by circumventing the need for patient-derived starting material (21–36).
Recently, the development of bispecific antibodies targeting both CD3 on T cells and tumor antigens such as CD19 and CD20 has provided a potential alternative to autologous CAR-T therapy. These therapies are logistically simpler to deliver, as they are immediately available and typically associated with lower-grade toxicities, particularly in terms of CRS and ICANS, compared to CAR-T cell therapy (37, 38). However, bispecific antibodies are similarly reliant on the fitness of the recipient’s immune system to exert their effects and, unlike CAR-T cells, require extended or indefinite dosing regimens. Furthermore, although direct comparisons between bispecific antibodies and CAR-T cells are limited, observed response rates with bispecific antibodies are generally lower than those achieved with CAR-T cells in similar treatment settings (37, 38). Thus, while bispecific antibodies represent an important addition to the therapeutic armamentarium, they underscore the pressing need for advancements that enhance the efficacy and accessibility of the CAR-T platform.
To address these limitations, the development of novel CAR-equipped allogeneic cell therapies has gained significant momentum. Allogeneic approaches leverage lymphocytes from healthy donors or clonal iPSC master cell lines, enabling the creation of “off-the-shelf” products that bypass the need for patient-specific manufacturing. These cryopreserved therapies are readily available for point-of-care administration, streamlining the treatment process. Furthermore, the use of lymphocytes from healthy donors circumvents issues related to compromised cellular fitness and expands the therapeutic potential. Different lymphocyte sources have been explored for allogeneic CAR engineering, each offering distinct advantages and limitations.
Among the most studied are αβ T cells, which provide robust cytotoxic activity and the potential for immune memory formation, making them highly effective in targeting malignancies. However, the presence of endogenous T-cell receptors (TCRs) in αβ T cells poses a risk of graft-versus-host disease (GvHD), necessitating complex genetic modifications to knock out the TCR (26, 27, 32, 33, 35, 36). Another emerging option are γδ T cells, which blend adaptive and innate immune properties, exhibiting potent anti-tumor activity while demonstrating a reduced risk of GvHD. However, their lower prevalence in peripheral blood and complex biology pose challenges for large-scale clinical application (34).
Natural killer (NK) cells, by contrast, do not express TCRs eliminating the risk of TCR-mediated GvHD, thereby simplifying the engineering process. NK cells also possess innate anti-tumor activity through a set of germline-encoded receptors and function independently of HLA matching, offering broader applicability and heighted anti-tumor immunity (24). NK cells can be sourced from multiple origins including peripheral (23, 25, 28) and cord (21, 22, 24, 31) blood as well as induced pluripotent stem cells (30, 39–41) and immortalized cell lines (42). Advancements in engineering strategies, such as multicistronic constructs incorporating autocrine IL-15 cytokine support, have significantly enhanced their persistence making them attractive contenders for adoptive cell therapy (21–25, 28, 30, 31, 39–41).
Beyond GvHD, a major challenge in the allogeneic setting is host-mediated rejection of infused immune cells, which has spurred the development of ‘stealth’ engineering strategies to help evade immune recognition and promote persistence. These strategies include β2M knockout to disrupt classical HLA class I expression, overexpression of non-classical HLA molecules such as HLA-E or HLA-G, and the incorporation of alloimmune-defense receptors (ADR) to selectively eliminate alloreactive host immune cells.
Accelerated by these advances in cellular engineering technology, allogeneic CAR-T and CAR-NK cell therapies have emerged as clinical candidates for r/r large B cell lymphoma (LBCL) (Figure 1). Early clinical trial data, although limited by small patient cohorts, provide critical insights into the feasibility, safety, and antitumor efficacy of these novel therapeutic modalities.
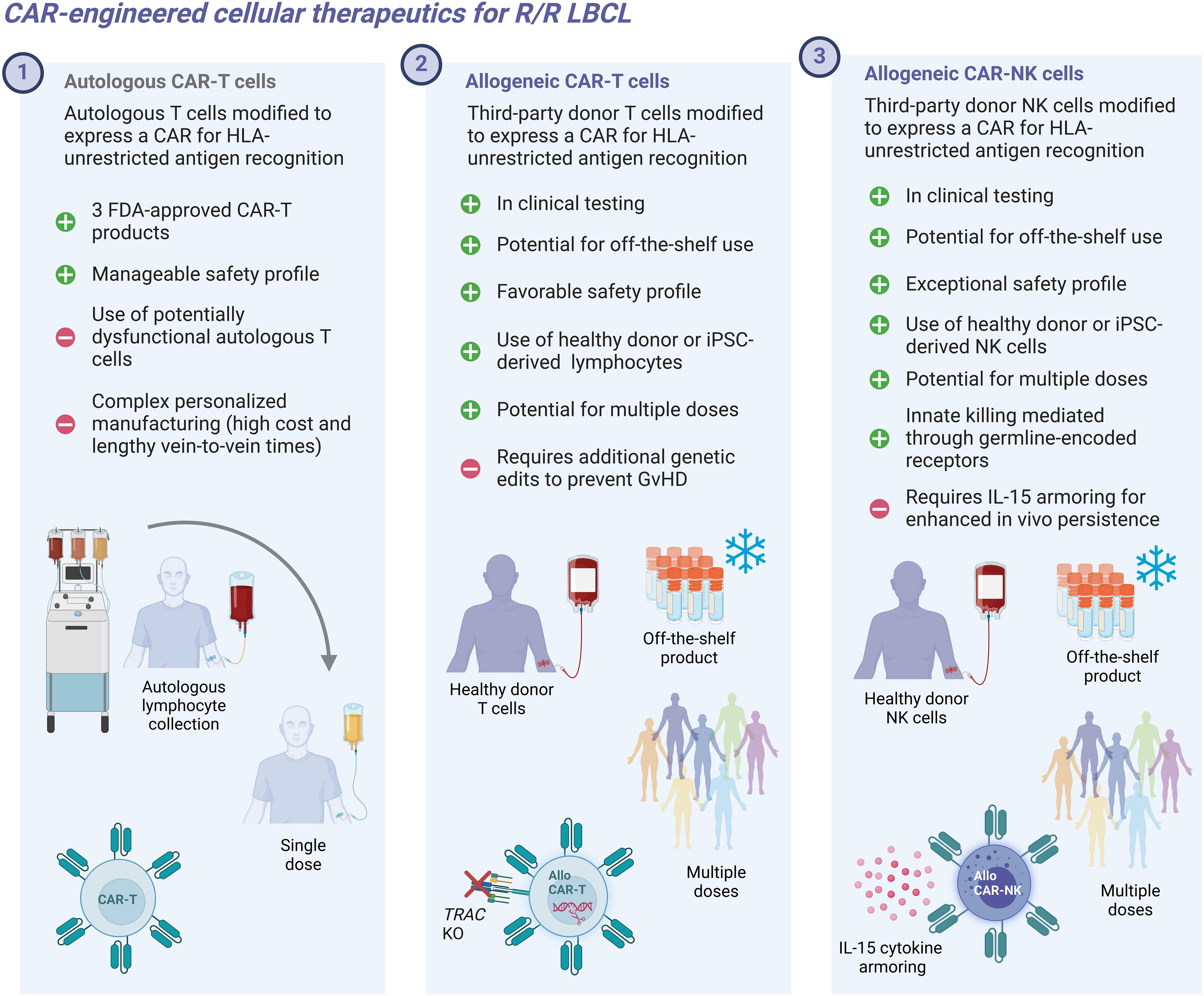
Figure 1. Conceptual depicting characteristics of autologous and allogeneic CAR-engineered cellular therapeutics.
This systematic review and meta-analysis is the first to comprehensively evaluate the emerging clinical evidence on the safety and antitumor efficacy of allogeneic CAR-engineered cell products for the treatment of r/r LBCL. By synthesizing early clinical trial data, we aim to provide an up-to-date and comprehensive evaluation of allogeneic CAR-T and CAR-NK cell therapies. These findings will help assess the feasibility and potential therapeutic impact of these next-generation cell therapies to overcome the limitations of autologous CAR-T cells and transform the therapeutic landscape for r/r LBCL.
Methods
Data sources, eligibility criteria and search strategy
This systematic review and meta-analysis was conducted in accordance with the PRISMA guidelines (43). MEDLINE, EMBASE, Web of Science, and the Cochrane CENTRAL Register of Controlled Trials were searched for interventional clinical trials, with or without comparator, which investigate allogeneic CAR-engineered adoptive cell therapies in patients with large B cell lymphoma from inception to January 12, 2025. Additionally, the conference proceedings of the American Society of Hematology, the American Society of Clinical Oncology, the American Association for Cancer Research, the American Society of Transplantation and Cellular Therapy, the European Hematology Association, International Conference on Malignant Lymphoma were searched manually. Only studies which reported the primary outcomes overall response rate (ORR), and complete response rate (CRR) were eligible for inclusion. Full-length articles, conference abstracts, letters, and case reports were included, whereas reviews, editorials, and commentaries were excluded. Studies without an identifiable associated clinical trial (verified through a clinical trial registration number) were excluded to prevent double counting of participants. In instances where peer-reviewed data or trial results were unavailable, data from conference proceedings or reports issued by trial sponsors were considered. Such data were included only if they provided sufficient methodological detail and could be corroborated through clinical trial registries. In addition to searching study reports, ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform (ICTRP) were consulted to identify and catalog relevant registered clinical trials.
We searched using MeSH terms and keywords for (“Chimeric antigen receptor” OR “T cells” OR Natural Killer Cells” OR “Adoptive Cell Transfer” OR “Adoptive Immunotherapy” OR “Cell- and Tissue-based therapy”) AND “Allogeneic Cells” AND (B-cell Lymphoma OR Non-Hodgkin Lymphoma” OR “Diffuse Large B-Cell Lymphoma”) from the date of inception to January 12, 2025. No filters or publication time limits were applied for the search. A total of 955 unique records were identified using the database search. No language restrictions were applied. All search results were imported to the Endnote reference manager, and duplicates were removed.
Study selection
The search results were imported into Covidence systematic review software (Veritas Health Innovation, Melbourne, Australia). Title and abstract screening, full-text screening, data extraction, and risk of bias assessment were performed independently by two reviewers (AB and JSH). Discrepancies were resolved through discussion or by consultation with a third reviewer (FB). The inclusion criteria were as follows: (1) original studies, specifically early-phase clinical trials; (2) studies including adult participants; (3) studies involving patients with relapsed/refractory large B-cell lymphoma (r/r LBCL), including diffuse large B-cell lymphoma (DLBCL) and high-grade B-cell lymphoma (HGBCL); (4) studies evaluating allogeneic CAR-engineered immune effector cell therapy as the intervention; (5) studies including patients regardless of prior exposure to CD19-targeting agents; and (6) studies including patients with or without previous CAR-T cell therapy. Studies requiring matched or partially HLA matched donors (7) as well as studies utilizing TCR-engineered T cell products (8) were excluded from analysis. Studies evaluating allogeneic CAR-T cells derived from a previous allogeneic hematopoietic stem cell transplantation (allo-HSCT) donor (9) were similarly excluded.
For studies with multiple reports, related records were identified and grouped based on their associated clinical trial number. The most recent full-length article was designated as the primary report for data extraction. If no full-length article was available, the most recent conference abstract or sponsor-issued report was used as the primary report.
Data extraction
Three authors independently extracted data from the 19 selected studies (29, 31–33, 44–58), and the datasheets were cross-checked to resolve any discrepancies. Data collected included available baseline characteristics (e.g., number of patients, diagnosis, prior lines of therapy, prior exposure to CD19-targeting agents, prior CAR-T therapy) as well as primary and secondary outcomes.
The primary outcomes extracted for meta-analysis were the best objective response rate (bORR) and best complete response rate (bCRR), defined as the proportion of patients achieving an objective response (OR) or complete response (CR), respectively, at any time during follow-up.
Response definitions were based on the criteria reported in the respective clinical trials and were not standardized across studies. Cases where response assessments were not explicitly reported were excluded from the analysis.
Secondary efficacy outcomes including overall survival (OS), progression-free survival (PFS), duration of response (DOR), duration of complete remission were extracted when available. Secondary safety outcomes comprised the reported incidence of adverse events, including cytokine release syndrome (CRS), immune-effector cell-associated neurotoxicity syndrome (ICANS), graft-versus-host disease (GVHD), infections, and other reported adverse events. Severity of reported adverse events were recorded and aggregated as low-grade (grade 1-2) or high-grade (grade 3+) toxicities for data analysis purposes.
Data analysis
Meta-analysis was conducted for the primary response criteria bORR and bCRR for r/r LBCL patients. Safety analysis was performed by evaluating all infused r/r B-NHL study patients (including additional histologies) for the incidence of infections, CRS, ICANS and GvHD. Safety analysis for the incidence of infections, CRS and ICANS were performed independently for low-grade (grade 1-2) and high-grade events (grade 3+). Response and safety assessments were performed in aggregate as well as independently by modality (CAR-NK vs. CAR-T). CAR-NKT cells were grouped among CAR-T cells due to their biological similarity to T cells in terms of lineage and TCR expression.
Analyses were performed using STATA/BE statistical software (v.18.0). Binary outcomes were summarized as proportions with 95% confidence intervals (CI). A random-effects model (Restricted Maximum Likelihood) was applied to pool proportions using an arcsine-based transformation, implemented via the meta set function. Due to the prevalence of zero-event observations, particularly in the safety analysis, arcsine transformation was employed for analysis of pooled estimates. Heterogeneity among studies was assessed using several metrics. Between-study variance (τ (2)) was estimated using a random-effects model. The I² statistic was calculated to quantify the proportion of variability in effect sizes due to heterogeneity. Cochran’s Q-test was performed to evaluate the significance of heterogeneity, with the null hypothesis of no heterogeneity tested at a significance level of 0.05. Subgroup analyses were conducted using the subgroup function in STATA/BE, focusing on pre-specified subgroups: CAR-T cells versus CAR-NK cells. Forest plots were created using Graphpad PRISM (Version 10.4.1 Build 532).
Results
Search results
Out of 953 unique references identified, 35 reports (22, 23, 35, 36, 39–41, 44–61) of early-phase clinical trials investigating allogeneic CAR-transduced immune effector cells (CAR-IEC) for the treatment of relapsed and refractory large B cell lymphoma (r/r LBCL) were screened for our review. Among these, 19 unique studies (29, 31–33, 44–58) were included in the meta-analysis, encompassing 235 patients evaluable for efficacy analysis and 334 patients evaluable for safety analysis (Figure 2). Notably, one study (NCT04030195 investigating PBCAR20A) (62) was excluded from both the efficacy and safety meta-analyses due to inconsistent data reporting; however, it is included in the tabular data synthesis. Included studies included phase 1, phase 2 and phase 1/2 early clinical trials, most of which were single-arm, open-label non-randomized trials. Geographically, studies were predominantly sponsored by US-based development programs (11/19) with three trials each sponsored out of Europe and China, respectively, one out of Australia and another out of Japan. Investigational CAR-IEC development programs were mostly industry-sponsored (17/19) with only two investigator-initiated studies sponsored by academic institutions (Supplementary Figure S1). Detailed characteristics of the included studies and interventions are provided in Table 1, while response and safety data are summarized in Tables 2, 3, respectively.
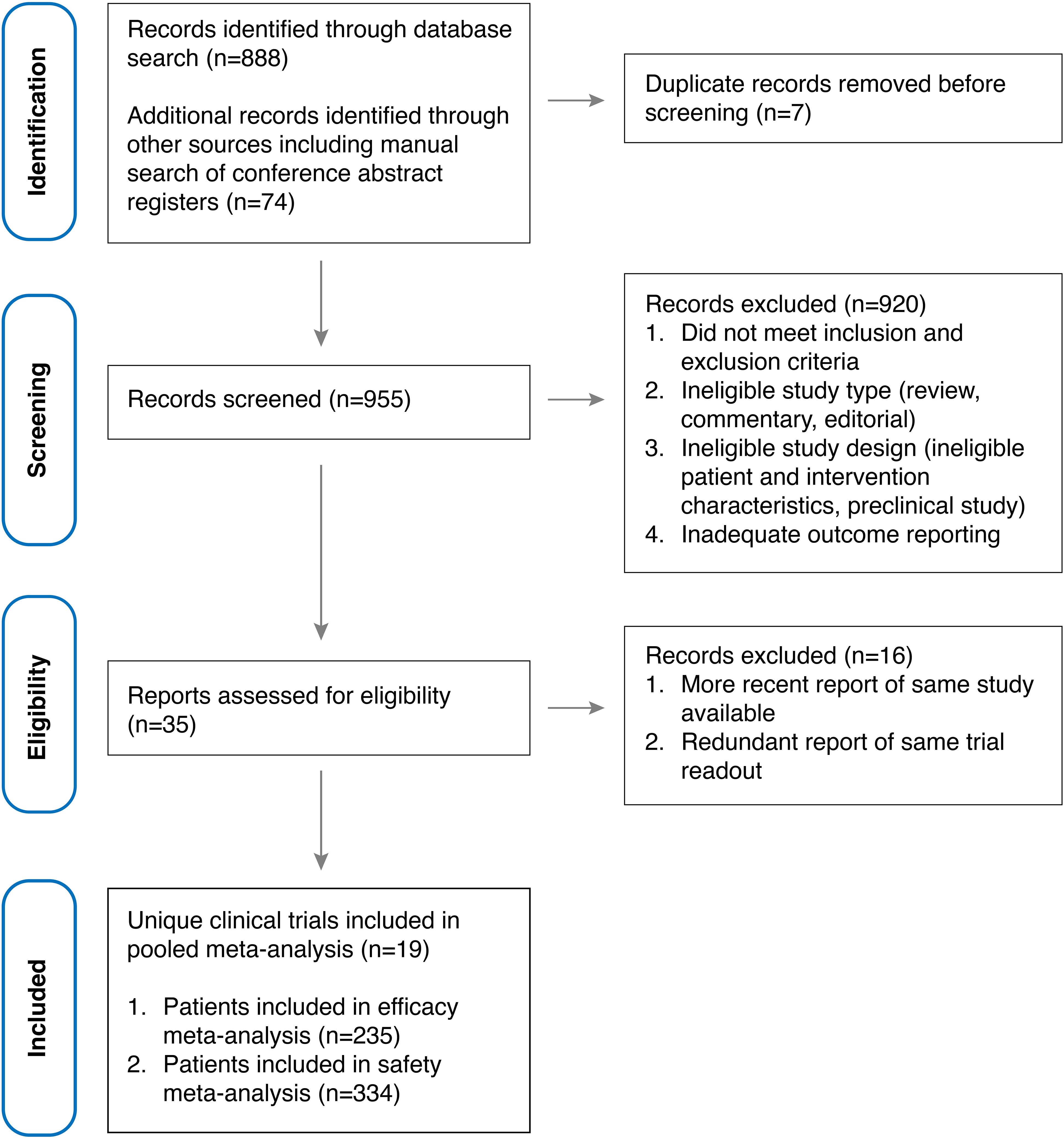
Figure 2. PRISMA flow diagram illustrating the identification, screening, exclusion, and inclusion of clinical trials.
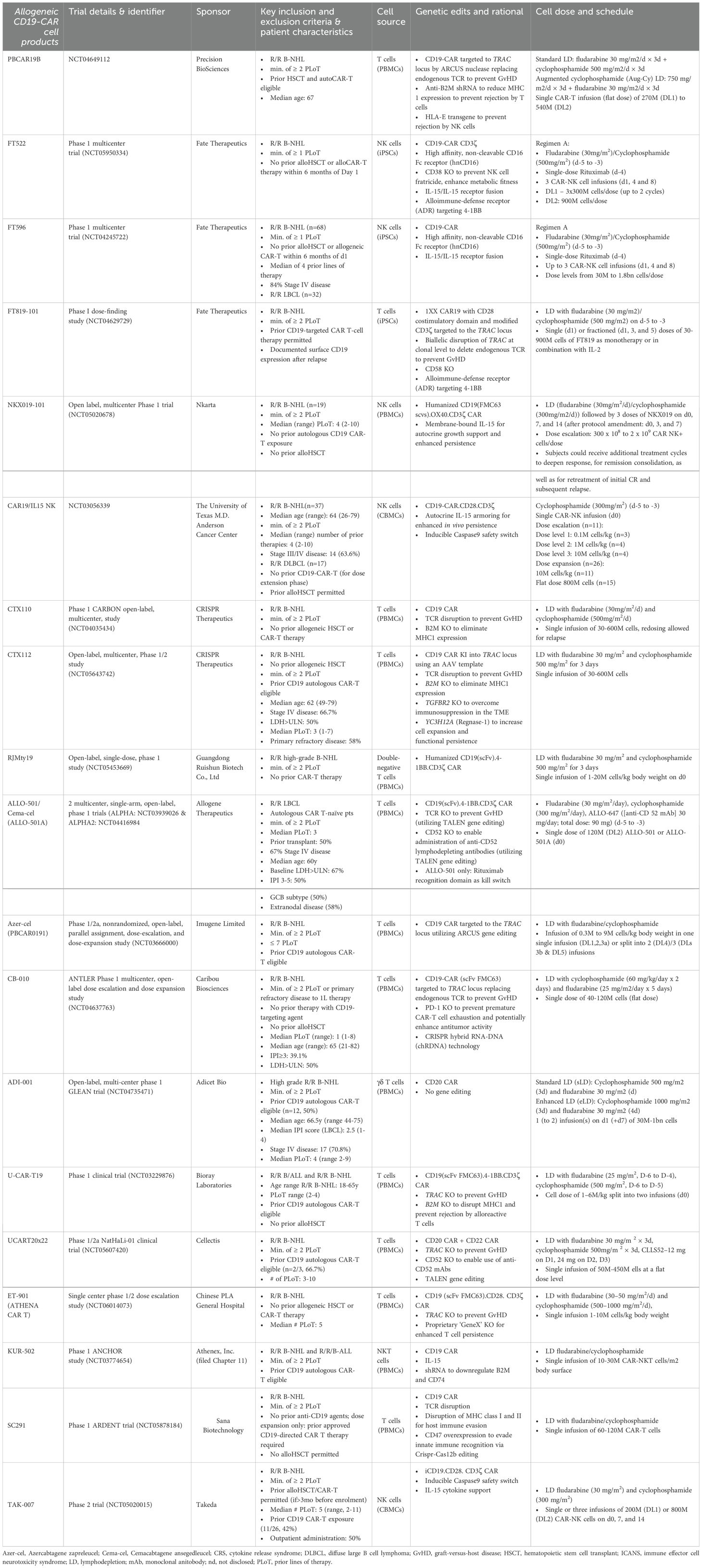
Table 1. Patient and intervention characteristics for studies evaluating allogeneic CD19-CAR-engineered cellular therapeutics in large B cell lymphoma (LBCL).
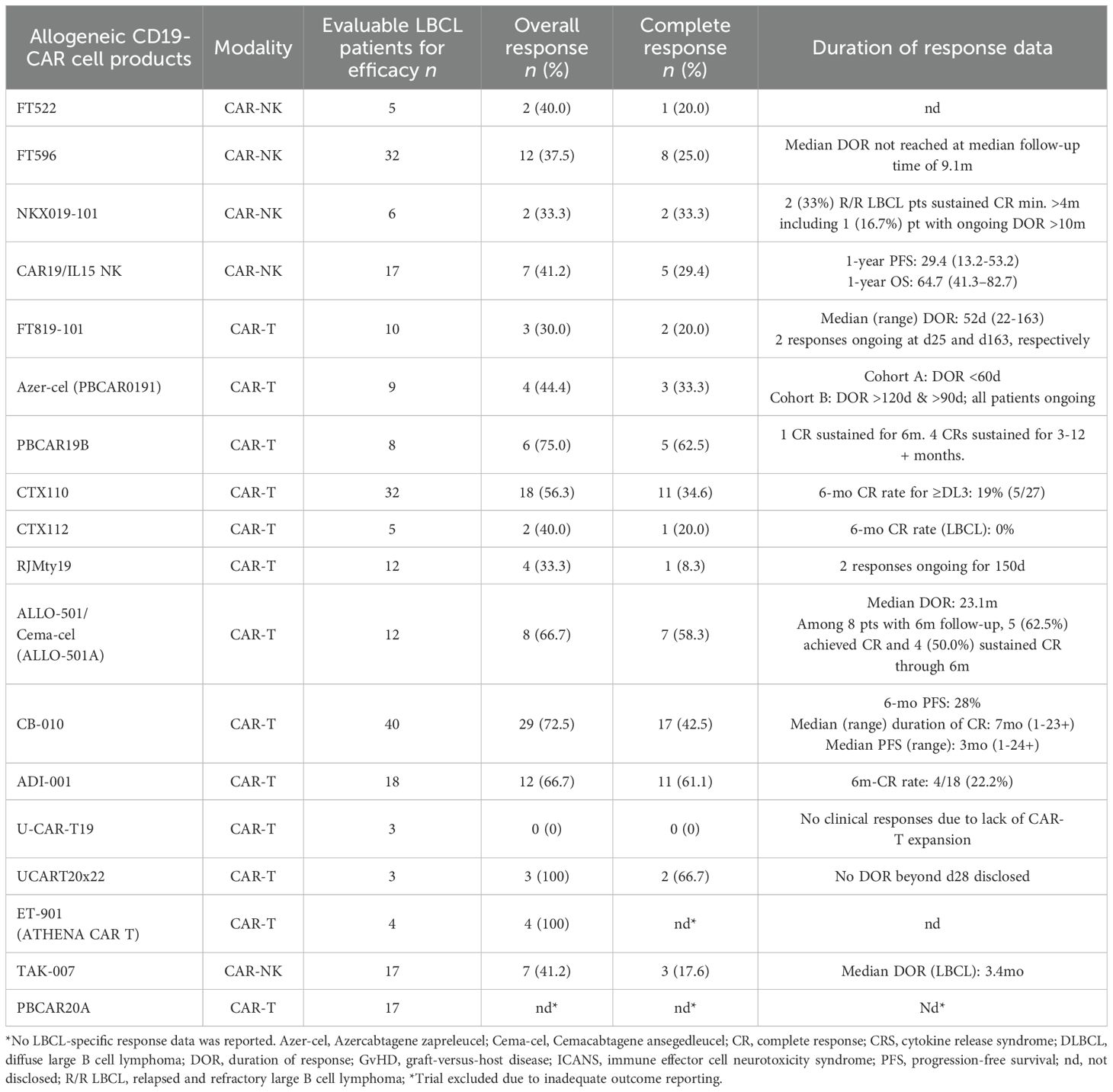
Table 2. Efficacy data of allogeneic CD19-CAR-engineered cellular therapeutics in large B cell lymphoma (LBCL).
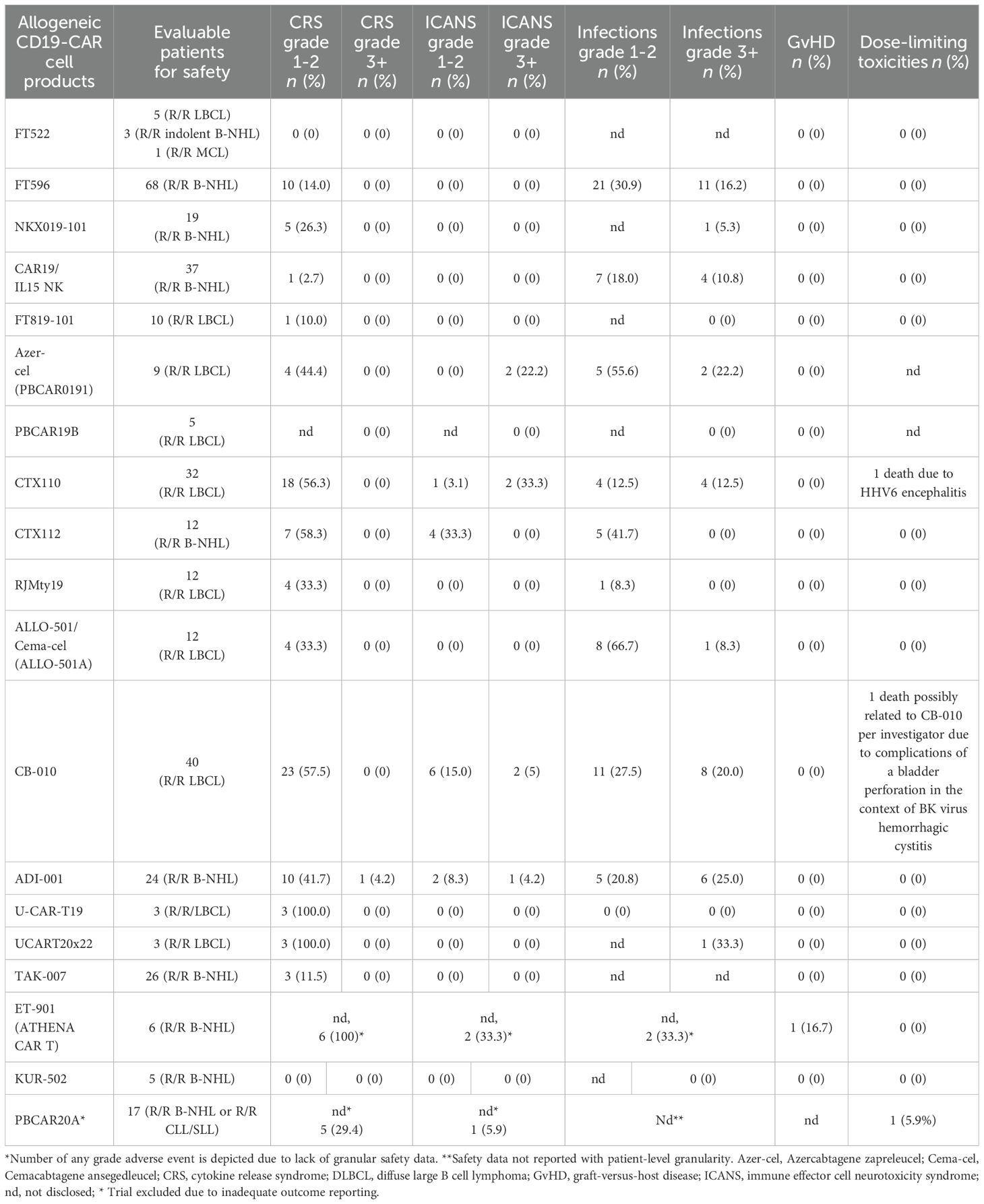
Table 3. Safety data of allogeneic CD19-CAR-engineered cellular therapeutics in large B cell lymphoma (LBCL).
Patient characteristics
All included studies reported response data for r/r LBCL patients, while a significant subset of studies (9/19) also included R/R B-NHL patients with additional histologies encompassing follicular lymphoma (FL), primary mediastinal B cell lymphoma (PMBCL), marginal zone lymphoma (MZL), or mantle cell lymphoma (MCL), which represented the basis for the pooled safety analysis of this review. Of all included studies, 10/19 allowed prior allogeneic hematopoietic cell transplant and 11/19 permitted prior exposure to autologous CD19-CAR-T cell therapy with varying minimum wash-out periods before infusion of investigational allogeneic cell products. Where available, the fraction of trial subjects who previously received autologous CAR-T therapy is specified in Table 1 as well as other information on patient demographics, disease status, and prior lines of therapy. The majority of studies (16/19) investigated patients who had received a minimum of 2 prior lines of therapy (PLoT) while 3 out of 19 studies investigated patients who had received a minimum of 1 prior line of therapy. Included studies encompassed a median of 12 (range 3-68) infused patients.
Intervention characteristics
Included studies investigated CAR-NK (5/19), CAR-T (13/19) and CAR-NKT (1/19) investigational cell products targeting CD19 (17/19), CD20 (1/19) or CD20/CD22 (1/19).
For CAR-NK cell products, cell sources included peripheral (1/5) and cord blood (2/5) as well as induced pluripotent stem cells (2/5). CAR-T cells were manufactured from peripheral blood mononuclear cells (13/14) or induced pluripotent stem cells (1/14). T cell employed for cellular engineering were mostly αβ T cells (13/14) including one study with double-negative CD4-/CD8- αβ T cells (1/14), while one study leveraged γΔ T cells (1/14). Investigational cell products harbored a variety of additional genetic modifications or molecular payloads for enhanced functionality or increased safety leveraging a multitude of gene editing platform including TALEN (2/19), ARCUS (2/19) and CRISPR (7/19) (Table 1). All allogeneic CAR-NK cell candidates (5/5) relied on autocrine IL-15 growth support for enhanced in vivo persistence in form of an IL-15/IL-15 fusion receptor (2/5), membrane-bound IL-15 (1/5) or autocrine secretion of IL-15 (2/5). CAR-NK cell candidates sourced from iPSCs (2/5) were equipped with a high affinity, non-cleavable CD16 Fc receptor (hnCD16) for antibody-dependent cellular toxicity (ADCC). One CAR-NK cell candidate, FT522, included additional gene edits including CD38 disruption to prevent NK cell fratricide as well as a proprietary alloimmune-defense receptor (ADR) targeting 4-1BB to prevent host rejection (1/5). Of note, none of the included CAR-NK cell products required any genetic modifications to prevent GvHD due to the inherent safety profile of NK cells.
CAR-T cell products, on the other hand, mostly relied on TCR disruption to prevent GvHD using CRISPR (7/14), TALEN (2/14) or ARCUS (2/14) gene editing technology with the exception of three studies which employed double-negative αβ T cells, γΔ T cells and NKT cells, respectively, which lack the propensity to induce GvHD. Strategies employed to reduce host rejection of infused cells included anti-B2M (beta 2 microglobulin) shRNA or B2M KO to reduce MHC-1 expression, ectopic expression of HLA-E to prevent host NK cell-mediated rejection, CD47 overexpression to evade innate immune cell rejection, or insertion of an alloimmune-defense receptor (ADR) targeting 4-1BB. Additional genetic edits to enhance functionality, shield from immunosuppressive pressures and augment anti-tumor potency included CD52 KO to enable administration of anti-CD52 lymphodepleting antibodies, PD-1 KO to prevent premature CAR-T cell exhaustion, TGFBR2 KO to overcome TME-induced immunosuppression, Regnase-1 KO for enhanced functional persistence and CD74 KO to prevent formation of immunosuppressive Tregs. Safety switches were present in three out of 19 investigational cell products in form of inducible Caspase9 (2/19) or a rituximab recognition domain (1/19). Of note, there were no reported cases in which safety kill switches were employed to mitigate uncontrollable toxicity.
There was inconsistent reporting on costimulatory domains employed to enhance CAR signaling with only 7/19 studies providing detailed information on CAR design. Costimulatory domains included CD28 (4), OX40 (1) and 4-1BB (2) (Table 1).
Lymphodepletion regimens administered prior to cell infusion consisted of fludarabine (25-30mg/m2) and cyclophosphamide (300-750mg/m2) administration on days d-5 to d-3. Infused cell doses ranged from 2x107 to 2x109 and 8/19 studies allowed infusion of multiple dose, three of which were prespecified split/fractioned doses.
Notably, multiple CAR-T/CAR-NK candidates (CTX110, ALLO-501 and FT596), have already been phased out by their respective sponsors to focus their development programs on next-generation candidates (CTX112, Cema-Cel, previously known as ALLO-501A and FT522), which feature improved functionality through additional genetic modifications. Specifically, CTX112 leverages AAV-template directed site-specific CAR insertion and contains additional genetic edits including KO of Regnase-1 to increase functional persistence, cytokine secretion, KO of TGFBR2 to shield from TME-associated immunosuppression and KO of MHC class 1 for reduced immunogenicity. Cema-Cel (ALLO-501A) lacks the rituximab kill switch present in ALLO-501 to expand the potential patient eligible for treatment including patients with recent exposure to rituximab. Cema-Cel (ALLO-501A) is currently being investigated in the pivotal phase 2 ALPHA3 trial in a first line (1L) LBCL indication to boost first-line cure rates as consolidation after induction therapy. FT522 features an alloimmune-defense receptor (ADR) targeting 4-1BB to reduce alloreactive rejection of infused cells and lacks CD38 to prevent NK cell fratricide and enhance metabolic fitness. Together, these three cell candidate iterations highlight the continuous innovation driven by precision gene editing which allows to custom-build potent cellular therapeutics with advanced functionality for enhanced anti-tumor efficacy.
Response data
This systematic review and meta-analysis included 19 studies (29, 31–33, 44–58) encompassing a total of 235 patients (77 CAR-NK; 158 CAR-T) evaluable for response. The pooled estimate for the best overall response rate (bORR) was 52.5% [95% CI, 41.0-63.9] (n=235) with no significant group-to-group difference (Qb(1)=3.59, p=0.058) between investigated CAR-NK or CAR-T cell products (Figure 3). The random-effects maximum likelihood (REML) model displayed moderate overall heterogeneity (I2 = 63.29%; p<0.001), which was mainly driven by significant heterogeneity among the included CAR-T cell studies (I2 = 74.97%, p<0.001). Included CAR-NK cell trials exhibited considerably less heterogeneity (I2 = 0.00%, p=0.996) in the all-study model.
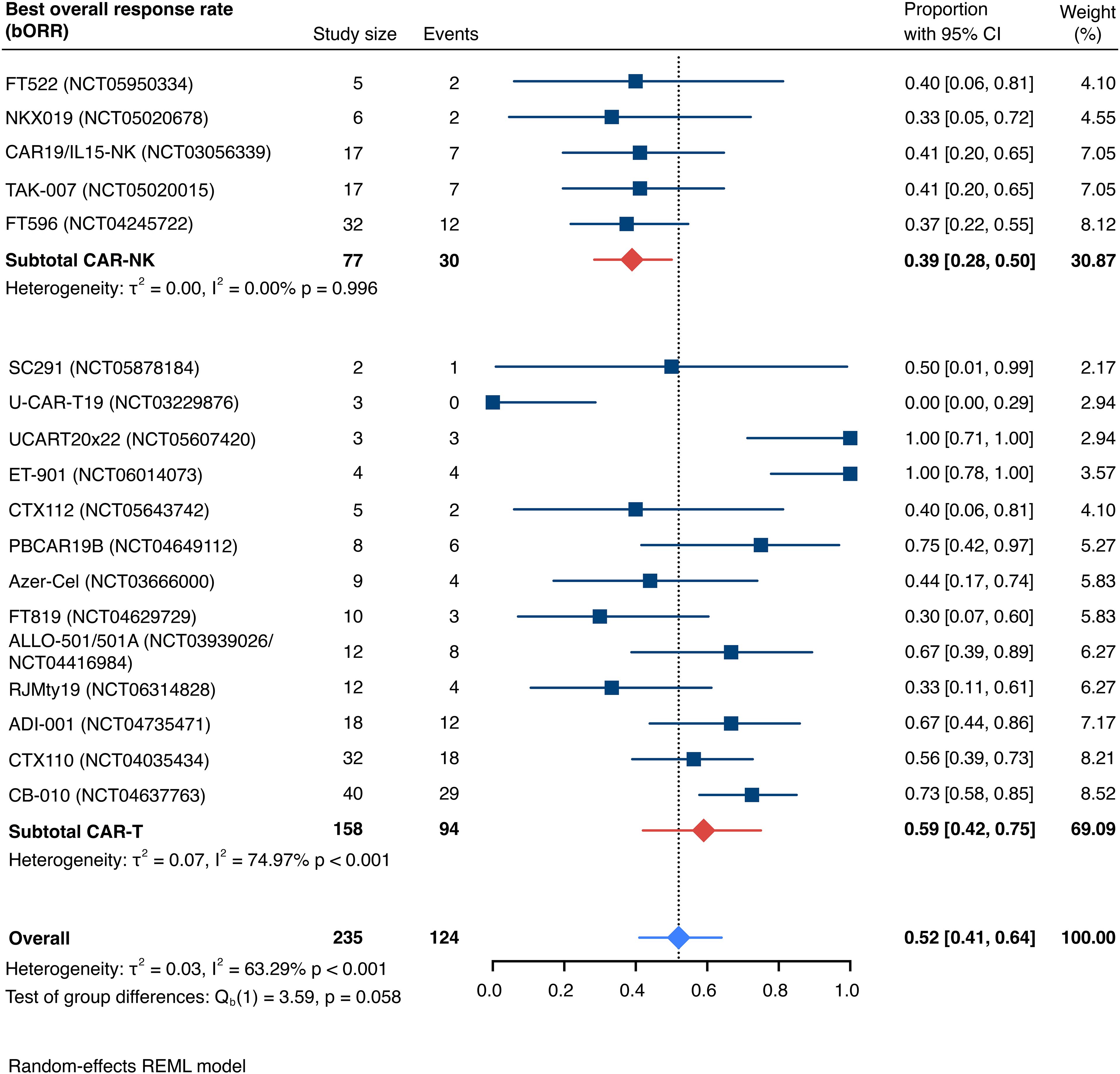
Figure 3. Forest plot illustrating the best overall response rate (bORR), stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
The pooled estimate for the best complete response rate (bCRR) was 32.8% [95% CI, 24.2-42.0] (n=231; Figure 4). Similarly, there was no significant superiority of any cell type (Qb(1)=2.20, p=0.138). We found moderate overall heterogeneity, exclusively driven by moderate intra-group heterogeneity for included CAR-T cell trials (I2 overall=46.07%, p=0.021; I2 CAR-NK<0.01%, p=0.912; I2 CAR-T=57.10%, p=0.013). Patient-level data were insufficient to conduct subgroup analyses based on age, disease stage, international prognostic index (IPI), number of prior lines of therapy, prior hematopoietic stem cell transplantation (HSCT), previous CD19 CAR T-cell therapy, or other prior immunotherapies.
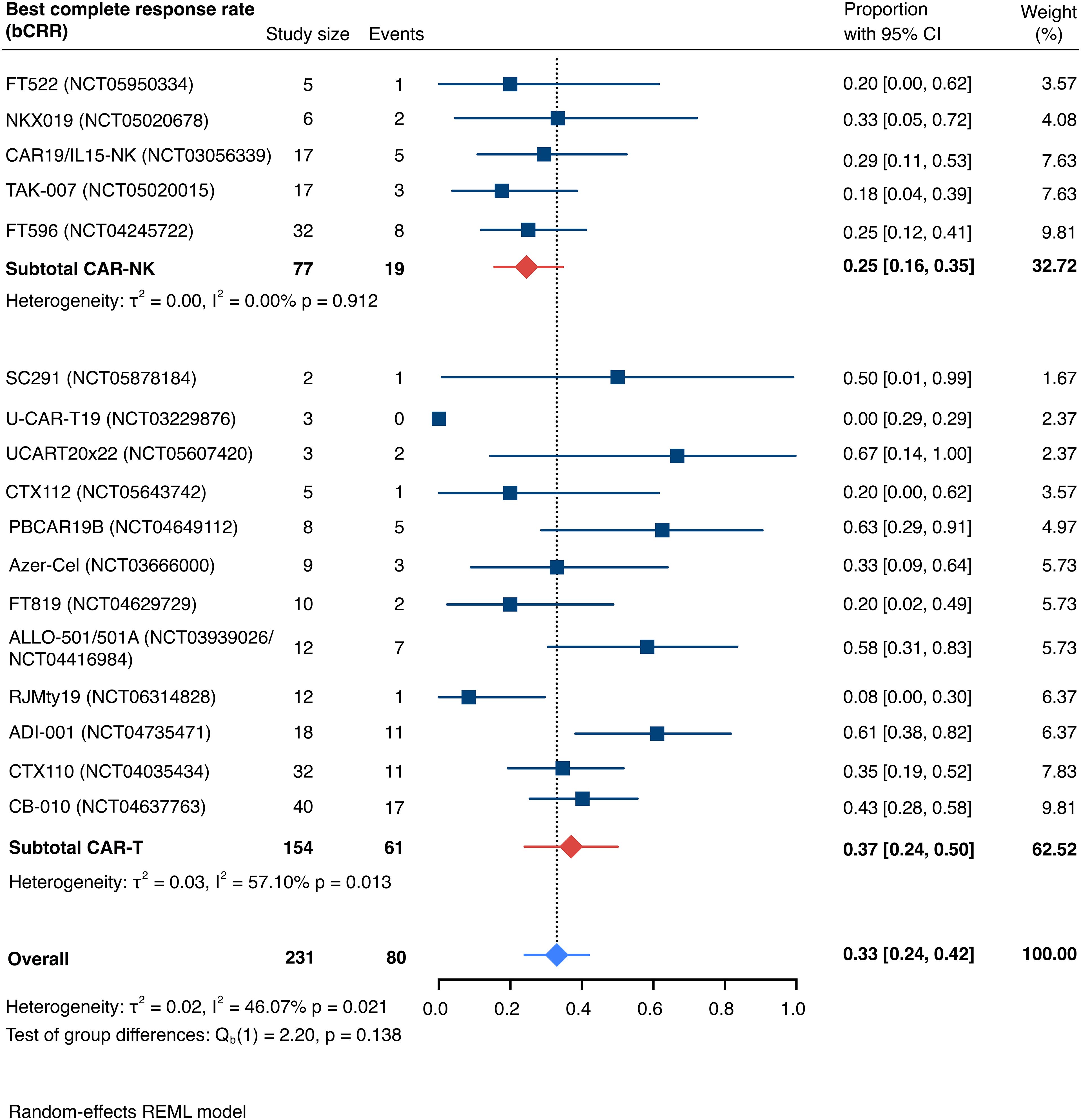
Figure 4. Forest plot illustrating the best complete response rate (bCRR), stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
There was inconsistent reporting on secondary efficacy endpoints including OS, PFS, the duration of response (DOR) and median follow-up across the trials due to the early-stage nature of the clinical data hindering direct comparisons across studies. This variability reflects the limited maturity of outcomes and differing methodologies among early-phase studies. Of note, one trial of CD19-targeted CAR-NK cells reported a 1-year progression free survival rate of 29.4% (13.2-53.2) and a 1-year overall survival rate of 64.7% (41.3-82.7), grossly in line with recent real-world data from autologous CD19-directed CAR-T cell products despite enrolling later-line patients (median # PLoT: 4 [range 3-10] vs. 3 [range 2.6-3.5]) (8). Another study of CD19-targeted CAR-NK cells reported that median duration of response was not reached at a median follow-up of 9.1 months, whereas a study investigating CD19-directed CAR-T cells observed a median duration of response of 23.1 months, indicating the encouraging potential for durable responses with allogeneic CAR-engineered cell products. Table 2 summarizes data on duration of response and follow-up duration, where available.
Safety data
Detailed severity grading for CRS and ICANS incidences was reported in all but two studies. The estimated overall incidence of low-grade CRS was 30% [95% CI, 14%-48%] (Figure 5), while severe CRS (grade 3+) occurred in 0.04% of patients [95% CI, 0.00%-0.49%] (Figure 6). The estimated overall incidence of low-grade ICANS was 1% [95% CI, 0%-4%] (Figure 7), with severe ICANS occurring in another 1% of patients [95% CI, 0%-2%] (Figure 8). No significant differences were observed in the incidence of high-grade CRS between patients infused with CAR-NK vs. CAR-T cell products (p=0.606). On the contrary, low-grade events of both CRS and ICANS as well as high-grade ICANS were significantly more frequent in CAR-T cell-infused patients (CRS grade 1-2: 42% [95%CI 20%-66%] vs. 10% [95%CI 3%-20%], n=323, p=0.007; ICANS grade 1-2: 3% [95%CI 0-8%] vs. 0% [95%CI 0-1%], n=323, p=0.033; ICANS grade 3+: 2% [95% CI 0%-5%] vs. 0% [95% CI 0%-1%], n=328, p=0.016).
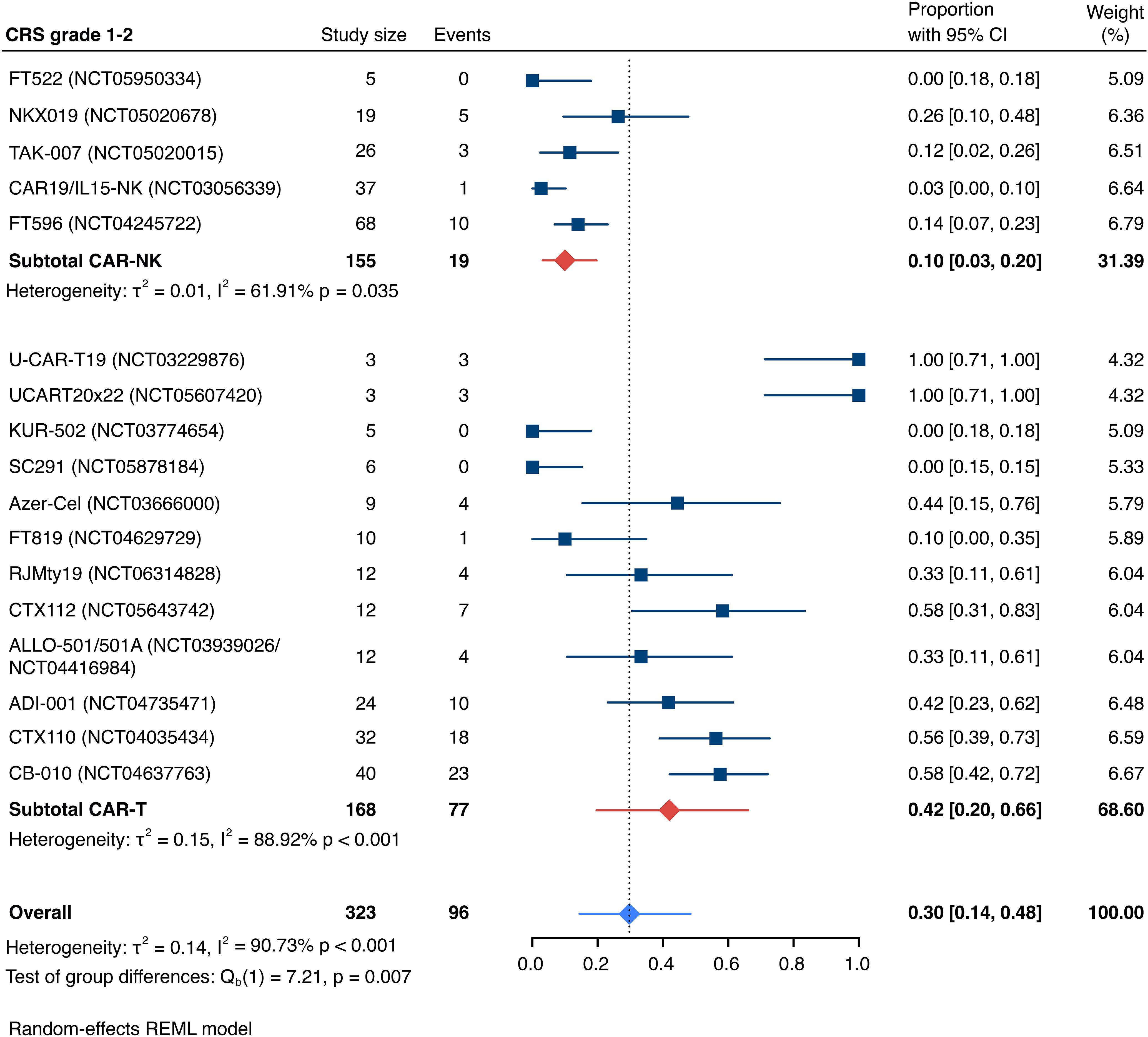
Figure 5. Forest plot illustrating the incidence of grade 1–2 cytokine release syndrome (CRS), stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
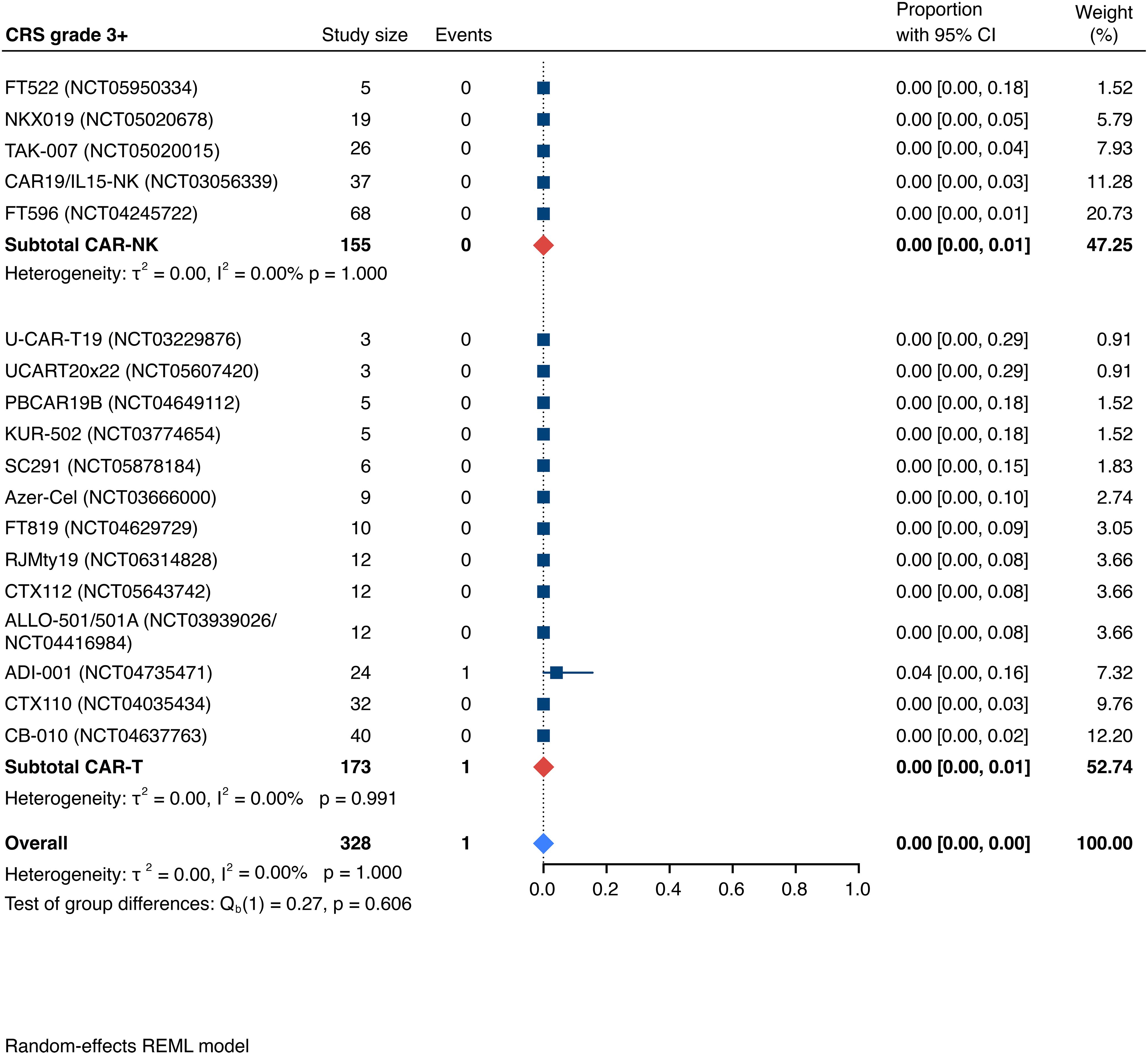
Figure 6. Forest plot illustrating the incidence of grade 3+ cytokine release syndrome (CRS), stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
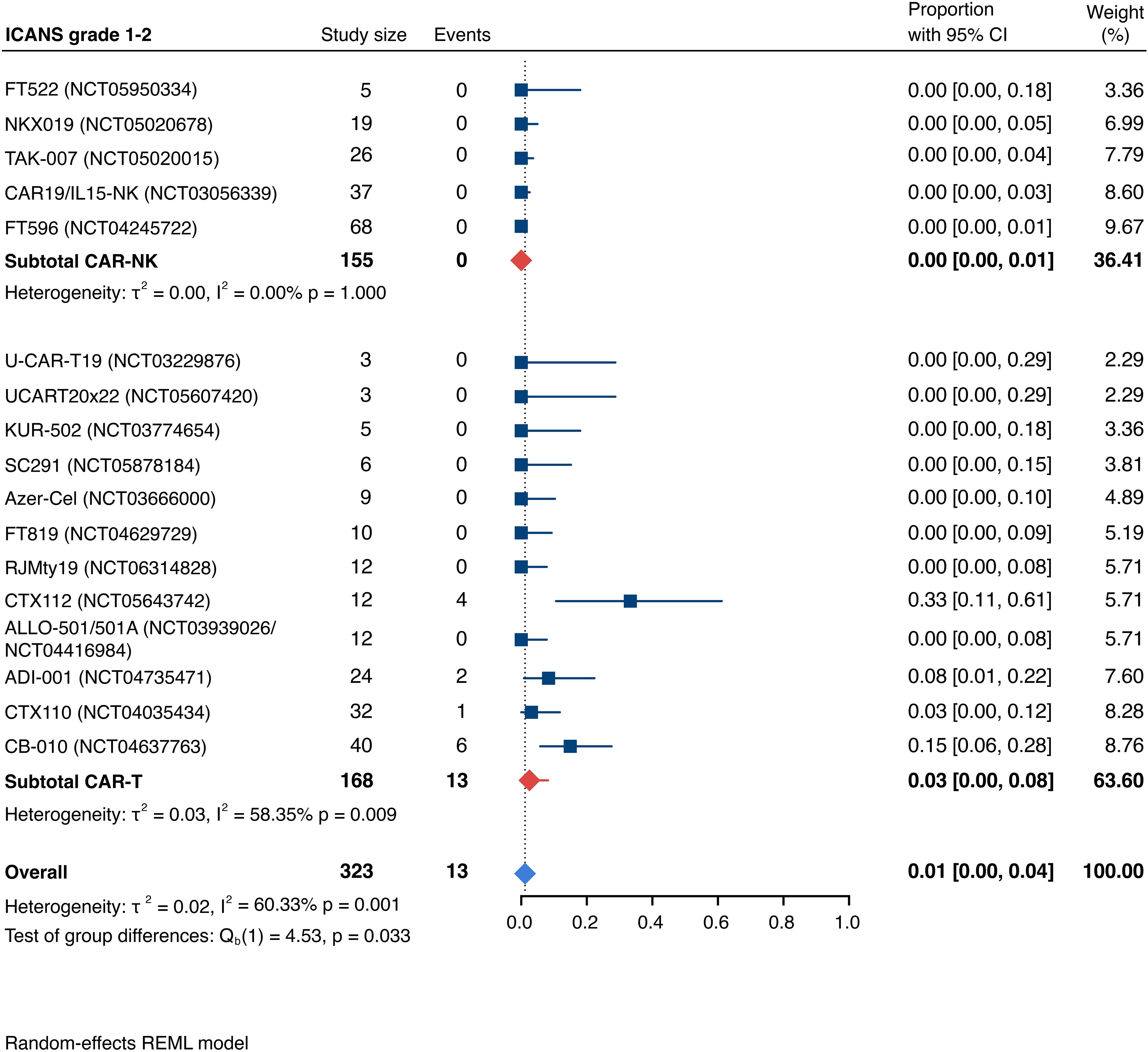
Figure 7. Forest plot illustrating the incidence of grade 1–2 immune effector cell-associated neurotoxicity syndrome (ICANS), stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
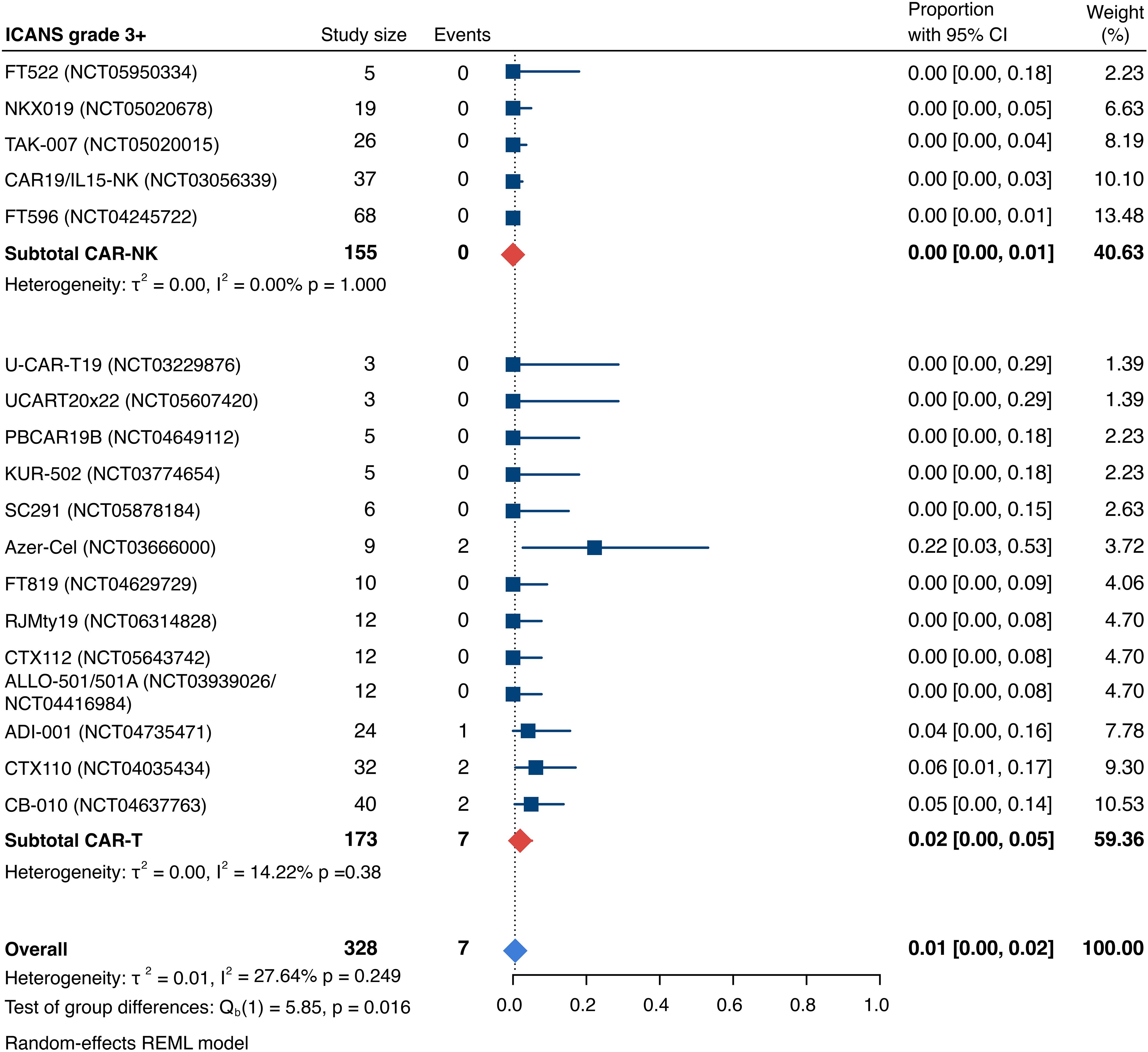
Figure 8. Forest plot illustrating the incidence of grade 3+ immune effector cell-associated neurotoxicity syndrome (ICANS), stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
The incidence of low-grade and severe infections was 25% [95% CI, 14%-36%) (n=252) and 7% [95% CI, 2%-14%] (n=291), respectively, and no significant group-to-group differences were detected across investigated cell types (Figures 9, 10). There was one case of a GvH-like reaction in a patient infused with ET-901 across a total of 334 infused patients, highlighting the impressive safety profile of CAR-engineered allogeneic cell therapies (Figure 11). Notably, there were no occurrences of GvHD in any of the administered HLA-mismatched CAR-NK cell products.
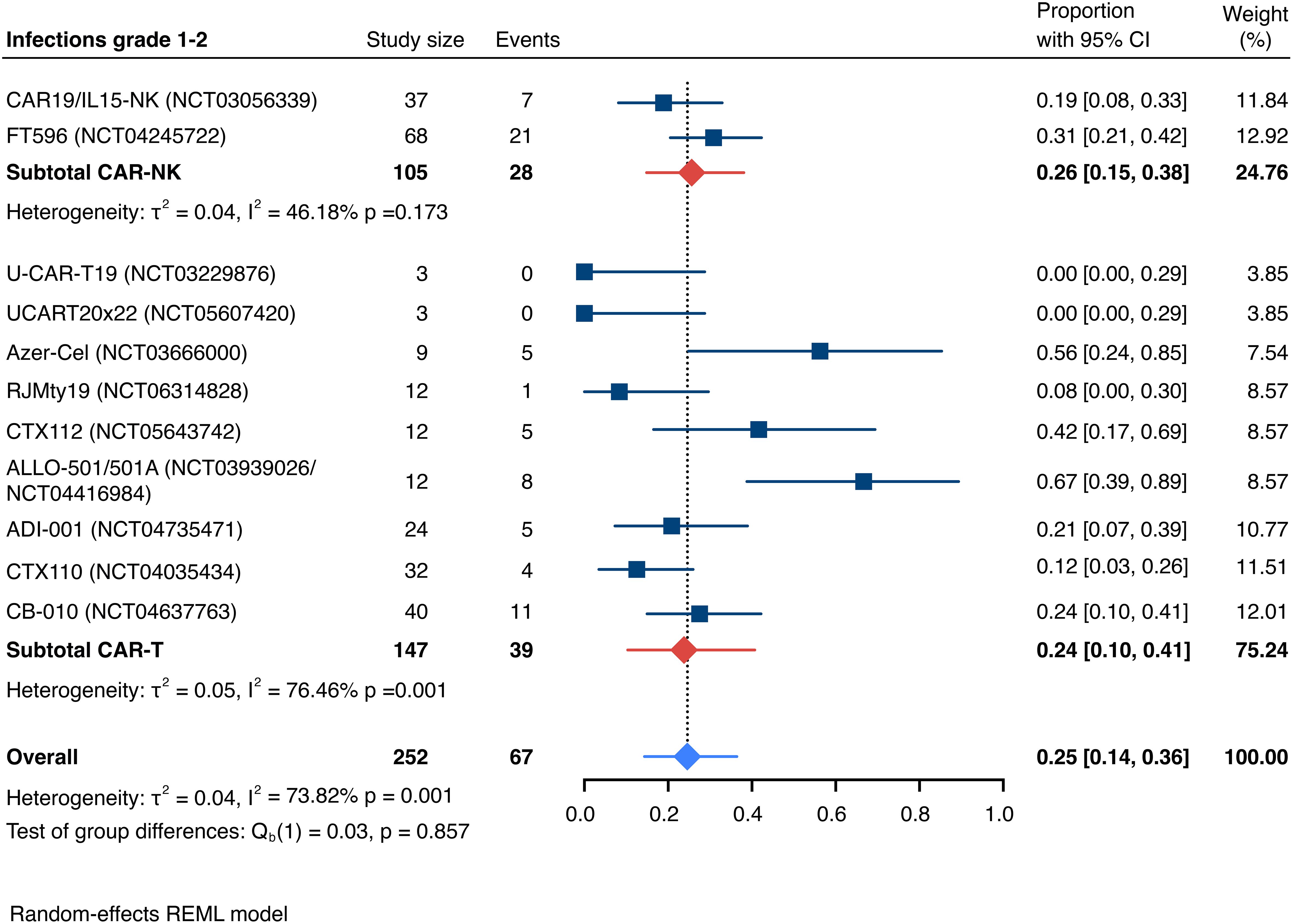
Figure 9. Forest plot illustrating the incidence of grade 1–2 infections, stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
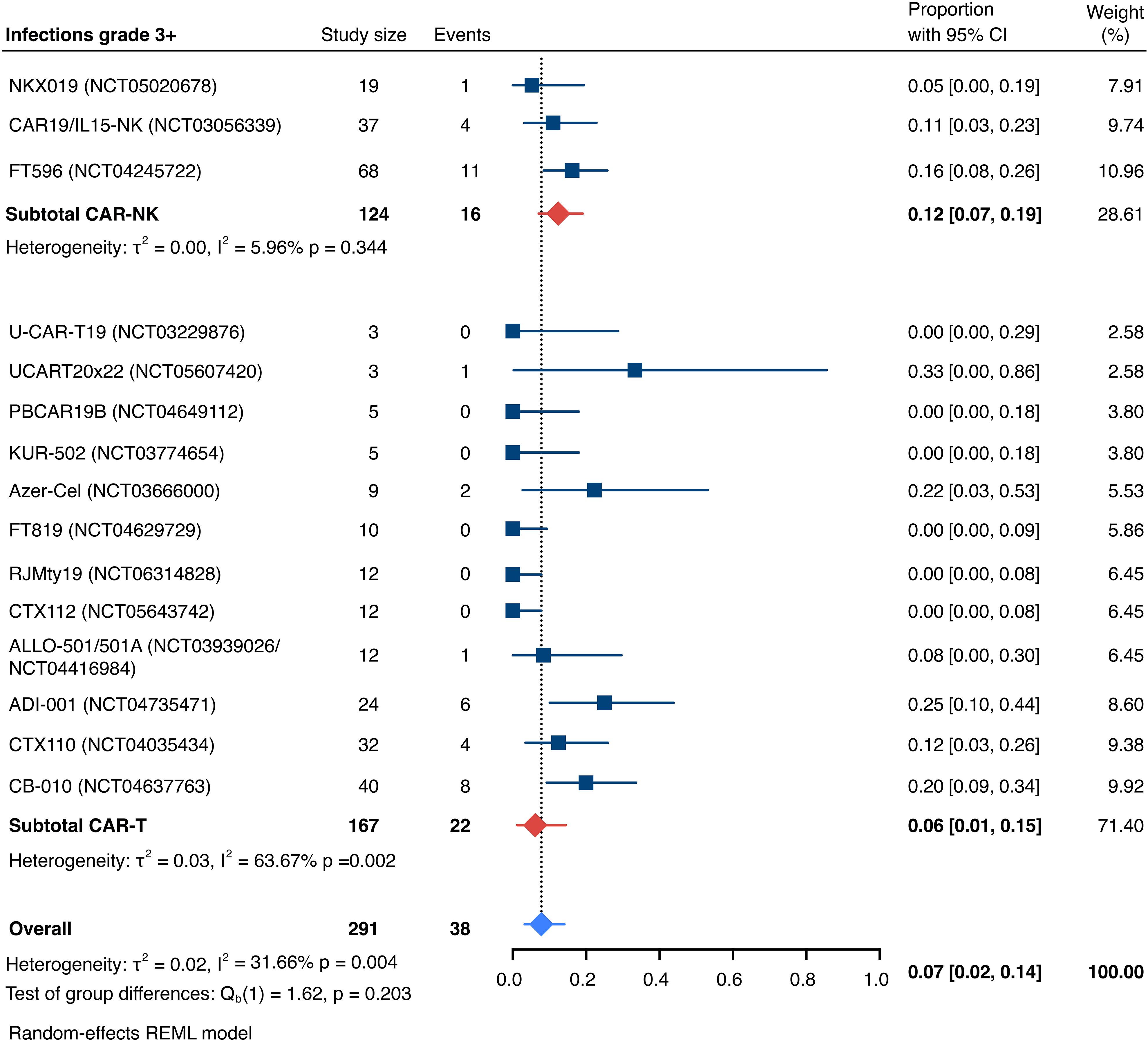
Figure 10. Forest plot illustrating the incidence of grade 3+ infections, stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
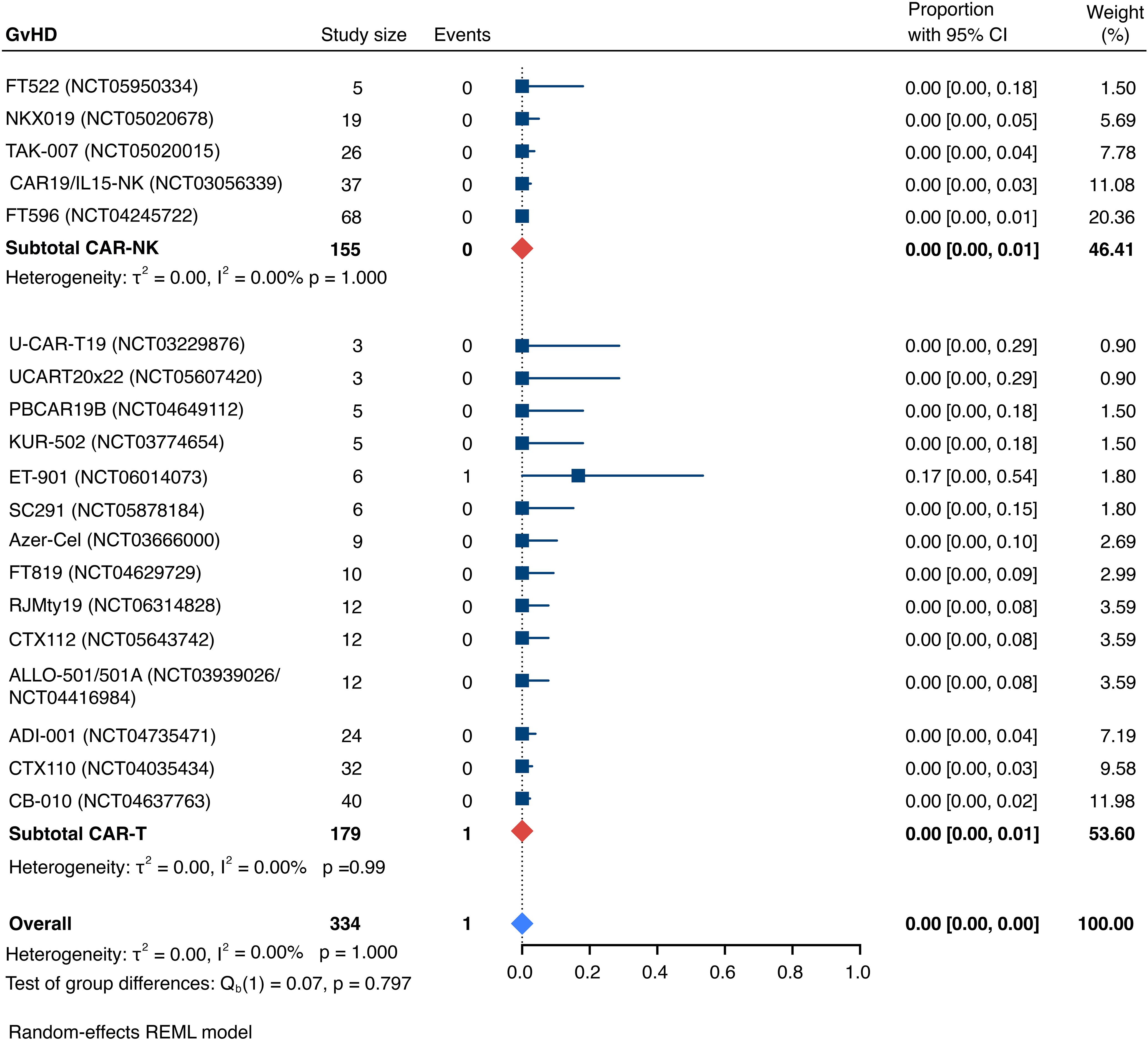
Figure 11. Forest plot illustrating the incidence of graft-versus-host disease (GvHD), stratified by cell source. Pooled estimates were computed for each subgroup and the overall weighted effect.
Dose-limiting toxicities (DLT) were reported for 3 patients including one death to HHV6 encephalitis post CTX110 infusion, one death possibly related to CB-010 infusion in the context of bladder perforation based on BK virus hemorrhagic cystitis and one DLN following PBCAR20A infusion not further classified by the trial sponsor.
Emerging clinical trial landscape
Our search of clinical trial databases identified a total of 47 registered trials (24 recruiting trials; 8 trials active, not recruiting; 2 terminated trials; 3 withdrawn trials; 2 completed trials; 2 trials not yet recruiting; 6 trials with unknown status) investigating allogeneic CAR-modified immune effector cells for the treatment of relapsed/refractory hematologic malignancies, including r/r large B-cell lymphoma (LBCL). Of the identified trials, 32 are investigating investigational cell products targeting CD19, four trials are evaluating cell candidates directed at CD20, two candidates are targeting CD22, 2 candidates are targeting CD70, 3 investigational cell products are targeting CD30, and 4 trials are investigating multi-targeted cell products including CD19/CD20 (2), CD20/CD22 (1) and CD19/CD20/CD22 (1). The majority of trials (36/47) utilize T cells, which include cytotoxic T cells derived from PBMCs, T stem cell memory (Tscm), γδ T cells, double-negative T cells, and T cells derived from iPSCs. NK cells are investigated in 11 trials, derived from sources including PBMCs, CBMCs, the NK-92 cell line, and iPSCs. Overall, cell products are sourced from PBMCs (26), CBMCs (1 trial for NK cells), the NK-92 cell line (2 trials), iPSCs (1 trial each for T cells and NK cells), and EBV-specific T cells (4 trials). Trial sponsors were predominantly based in the US (22) and China (19) with four trials registered by a sponsor based in Switzerland, one based in France and another on in Australia. Table 4 provides detailed information on the employed cell source, cellular engineering and gene editing strategies, sponsor and study status of identified trials.
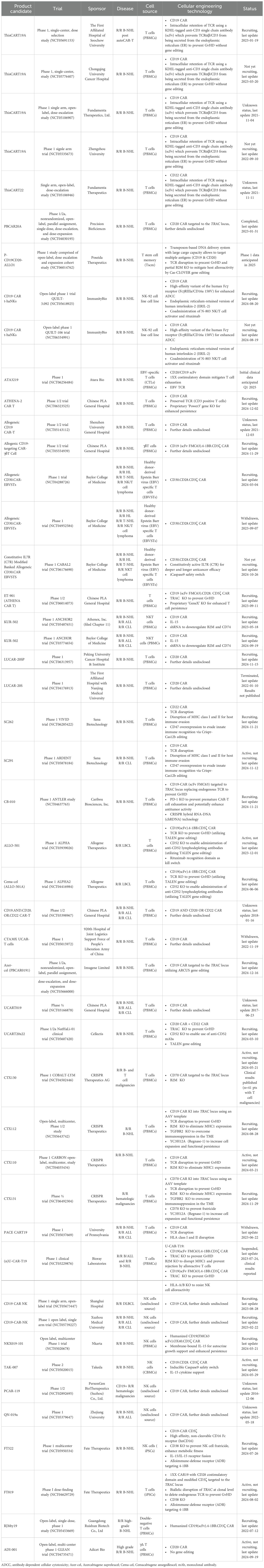
Table 4. Clinical trial landscape of healthy third-party donor allogeneic CAR-engineered cellular therapeutics for relapsed and refractory B cell lymphoma (R/R B-NHL).
Risk of bias
Most of studies the studies included in this systematic review and meta-analysis are early-phase open-label trials, including single center studies which lacked estimates of random variability and blinded outcome assessors. This limitation underscores the need for more robust, multicenter, randomized, and blinded studies to validate these early findings.
Discussion
We conducted, for the first time, a systematic review and meta-analysis to evaluate the emerging clinical evidence on the safety and antitumor efficacy of allogeneic CAR-engineered cell products for the treatment of large B-cell lymphoma (LBCL). Nineteen studies met the inclusion and exclusion criteria, encompassing a total of 334 patients evaluable for safety (155 receiving CAR-NK therapy and 179 receiving CAR-T therapy) and 235 patients evaluable for response (77 receiving CAR-NK therapy and 158 receiving CAR-T therapy). The pooled estimates for the best overall response rate (bORR) and the best complete response rate (bCRR) were 52.5% [95% CI, 41.0-63.9] and 32.8% [95% CI, 24.2-42.0], respectively. This study highlights the exceptional safety profile of allogeneic cell therapies. Across all included studies, only one case of grade 3+ cytokine release syndrome (CRS) was reported, the incidence of grade 3+ immune effector cell-associated neurotoxicity syndrome (ICANS) was 1% [95% CI, 0%-2%], and one occurrence graft-versus-host like reaction (1/334) was reported. Intriguingly, the investigated CAR-NK cell candidates exhibited an even more favorable safety profile compared to allogeneic CAR-T cells with a lower incidence of grade 1–2 CRS (10% [95% CI, 3-20] vs. 42% [95% CI, 20-66]), further underscoring the tolerability of NK cells in the allogeneic setting. Unlike T cells, NK cells naturally tolerate HLA mismatches and have demonstrated excellent safety across numerous clinical studies using unmatched allogeneic NK cell products (63–72). These favorable immunobiological properties are likely due to their germline-encoded repertoire of activating and inhibitory receptors, which finely balance responsiveness and restraint, integrating diverse signals to mediate targeted cytotoxicity without provoking systemic immune overactivation or uncontrolled cytokine release.
Together, these findings highlight the therapeutic potential of allogeneic cell products in r/r LBCL with a significantly reduced toxicity burden compared to their autologous contenders, making them a highly attractive and cost-effective treatment option for this difficult-to-treat patient population, including in the outpatient setting.
Strengths and limitations
The presented study has several notable strengths. First, it is the first study to systematically investigate and compare multiple competing allogeneic cell products aiming to upend the standard-of-care for a patient population with a high unmet medical need. Second, this study incorporates the most current data, including findings presented at recent medical conferences, making it a valuable and up-to-date resource that provides a critical perspective on the emerging clinical trial landscape of allogeneic cell therapies. Third, it aims to harmonize the analyzed patient groups as much as possible despite variability in the underlying patient populations. By stratifying analyses based on cell source, this study provides a meaningful comparison between CAR-NK and CAR-T cell therapies, ensuring nuanced insights into their respective efficacy and safety profiles.
Nevertheless, several limitations must be acknowledged. These are largely inherent in the analyzed primary studies, which often included small cohorts and were conducted in early-phase clinical trial settings. Many of these studies were dose-escalation and dose-expansion trials, where response rates observed during the dose-escalation phase may understate the true effect size due to suboptimal dosing. What is more, limited follow-up times of included trials and variable reporting standards restrict the assessment of therapeutic durability. Additionally, the limited number of studies available and the relatively small overall sample size restrict the generalizability of the findings. Variability in the underlying patient populations further complicates pooled analyses. Differences include the number of prior therapies received (e.g., prior exposure to CD19-targeted therapeutics, prior CAR-T exposure, and prior allogeneic hematopoietic stem cell transplantation), international Prognostic Index (IPI) risk scores, and age. This heterogeneity warrants cautious interpretation of pooled estimates of response and safety particularly across investigated groups.
Lastly, the presented allogeneic CAR-T and CAR-NK cell candidates differ with respect to their respective CAR design including costimulatory domains, employed gene editing platforms and inclusion of additional genetic edits and molecular payloads.
Allogeneic vs. autologous CD19-directed CAR-T: real-world evidence
Recent real-world studies have demonstrated overall and complete response rates in the range of 58-84% and 39-68%, respectively, in patients with r/r LBCL receiving commercially available autologous CAR-T cell products (8, 9, 15, 18, 19). However, these responses rates come at the expense of significant toxicities, with CRS and ICANS occurring in 52-88% and 30-56% of cases, respectively. Higher-grade toxicities (grade ≥3) were observed in 3-23% of cases for CRS and 6-23% for ICANS, underscoring the potential for severe and life-threatening adverse effects. In contrast, our meta-analysis of allogeneic CAR-IEC products showed markedly lower rates of low-grade (grade 1-2) CRS (30%) and ICANS (1%). High-grade CRS and ICANS were rare, with only 1 and 7 cases, respectively, among 328 infused patients. In terms of response, early-phase clinical trial readouts demonstrated encouraging efficacy signals for allogeneic CAR-IEC candidates with pooled estimates for best ORR and best CRR of 52.5% [95% CI, 41.0-63.9] and 32.8% [95% CI, 24.2-42.0], respectively. Beyond their favorable safety profile and promising efficacy, allogeneic CAR-T and CAR-NK cell therapies have the potential to significantly reduce treatment costs and improve accessibility compared to their autologous counterparts.
In principle, gene-editing strategies–while essential for enabling allogeneic use of T cells–may inadvertently compromise cellular efficacy and impinge on clinical response rates. In particular, the requirement for TRAC knockout in alloCAR-T cells, designed to prevent GvHD, eliminates endogenous TCR signaling, which plays a critical role in activation-induced clonal expansion and persistence. Loss of this native signaling axis may reduce the durability and proliferative capacity of the infused product, especially in the absence of supportive cytokine cues.
However, direct comparisons of response rates remain challenging due to fundamental differences in treatment paradigms and differences in examined patient cohorts. Allogeneic cell products eliminate the need for leukapheresis, a process that may fail to yield high-quality starting material in patients with low lymphocyte counts or dysfunctional lymphocytes with impaired fitness due to prior chemoimmunotherapy, tumor-induced immunosuppression, or inherent T cell defects, such as those seen in HIV-associated DLBCL (73).
This distinction makes direct comparison difficult, as most autologous CAR-T cell studies report outcomes only for patients who successfully received the cells (modified intention-to-treat, mITT) rather than from the initial screening population (intention-to-treat, ITT). Furthermore, the time required for CAR-T cell manufacturing often favors patients with less aggressive disease, as those able to wait for production may inherently have a better prognosis (74). Conversely, the “off-the-shelf” nature of allogeneic therapies allows for more rapid treatment initiation, which may better address patients with rapidly progressing disease.
One study directly compared response rates between CAR-NK cells and autologous CAR-T therapies on an ITT basis. When analyzed on an ITT basis, the reported complete response (CR) rate for autologous CAR-T in DLBCL was 34% (95% CI: 27–42%), which was similar to the 27.8% (95% CI: 10–53%) CR rate observed with CAR-NK cells in the same study (31).
This comparison underscores the importance of considering ITT analyses when evaluating the relative efficacy of allogeneic and autologous therapies. Moreover, a number of included studies enrolled patients who relapsed after autologous CD19 CAR-T cell therapies, further complicating any direct comparisons due to inherent differences in the examined patient populations. As allogeneic CAR-engineered cell products, including CAR-NK cells, advance through clinical development and potentially move into earlier lines of therapy, it will be particularly interesting to monitor how their response rates evolve in broader and less heavily pretreated patient populations. Early-line settings may reveal additional benefits of allogeneic products, such as shorter manufacturing times, elimination of leukapheresis, and broader applicability due to reduced cost.
Ongoing trials and expanding targets
In addition to the studies included in this analysis, several clinical trials are currently investigating allogeneic CAR-T and CAR-NK cell therapies in patients with r/r LBCL. These ongoing trials aim to further evaluate the safety, efficacy, and applicability of these therapies across diverse patient populations. Table 4 provides an overview of these ongoing trials, highlighting their design, target populations, and therapeutic approaches.
While the majority of investigational cell products target CD19, other targets for allogeneic CAR therapies are actively being explored. Notably, recent studies of allogeneic CAR-T cells targeting CD70 have demonstrated promising safety and efficacy signals in renal cell carcinoma (RCC) (75, 76) and T-cell non-Hodgkin lymphoma (T-NHL) (77). These findings underscore the expanding potential of allogeneic CAR-based therapies across both hematologic malignancies and solid tumors, paving the way for broader applications of these innovative treatments.
Cell sources and donor selection
The choice of cell sources and donor selection are pivotal in the development of allogeneic CAR-T and CAR-NK cell therapies, influencing scalability, functionality, and clinical outcomes. Peripheral blood mononuclear cells (PBMCs) from healthy donors remain a widely utilized source for allogeneic CAR-T and CAR-NK therapies. These cells offer robust cytotoxic activity and the ability to generate multiple doses from a single donor, enabling rapid treatment delivery and cost-effectiveness. However, reliance on healthy donors introduces variability in cell quality and functionality, prompting exploration of alternative sources.
Cord blood has emerged as a promising source for CAR-NK cell manufacturing and various strategies of IL-15 cytokine armoring enable long-term in vivo persistence of engineered NK cells. Cord blood–derived NK cells demonstrate potent innate cytotoxicity and lack the propensity to induce graft-versus-host disease (GvHD). Recent research has further optimized the use of cord blood by identifying factors that predict enhanced NK cell functionality. A study found that nucleated red blood cell (NRBC) count and time from collection to cryopreservation are key determinants of cord blood unit (CBU) quality. Specifically, CBUs with an NRBC count ≤8×107 and a collection-to-cryopreservation time ≤24 hours correlated with superior NK cell cytotoxicity. These findings support rational donor selection and processing strategies to maximize the therapeutic potential of cord blood–derived NK cells, making them an increasingly attractive option for clinical application (31).
Cell-line–derived platforms, such as high-affinity NK (haNK) cells, have also been explored as alternatives to donor-dependent sources. HaNK cells are engineered with high-affinity CD16 (FcγRIIIa) to enhance antibody-dependent cellular cytotoxicity (ADCC) and are equipped with IL-2 armoring to improve persistence. Due to their origin, haNK cells require irradiation prior to infusion, potentially restricting in vivo persistence (42). Currently, CD19-targeted high affinity NK cells (t-haNKs) are being clinically evaluated in two phase 1 clinical trials (NCT05618925 and QUILT 106).
Induced pluripotent stem cell (iPSC) technology offers another innovative approach to cell sourcing. iPSC-derived NK cells are created from clonal master-engineered iPSC lines. These cells can be produced in large, standardized quantities with consistent quality, enabling off-the-shelf availability and addressing batch variability (30, 39–41). Beyond NK cells, iPSC-derived T cells, such as FT819, are being developed as potential homogeneous CAR-T cell products for clinical use (58, 78). By leveraging genetic engineering, iPSC platforms provide a renewable and versatile option for producing highly customized cell therapies, though their broader clinical applicability remains under investigation.
Lastly, Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes (CTLs) have been investigated as an allogeneic cell platform for CAR T-cell therapeutics due to their established safety profile positioning them as a potentially versatile and immunocompatible cellular backbone for genetically engineered CAR T-cell therapies.
The diversification of cell sources underscores the growing flexibility and innovation in allogeneic cell therapy development. From PBMCs and cord blood to iPSC-derived platforms and cell lines, each source offers unique advantages and challenges. Of note, the use of healthy donor-derived allogeneic cell sources may provide a critical safety benefit as healthy donors presumably are less likely to harbor clonal hematopoiesis or predisposing genetic lesions and have not undergone prior exposure to genotoxic antineoplastic agents. Going forward, rational donor selection could exclude donors with clonal hematopoiesis, thereby reducing the theoretical risk of therapy-related secondary malignancies as have been observed in a limited number of autologous CAR-T products. As the field advances, the optimization of cell sources and donor selection will play a crucial role in improving accessibility, functionality, and therapeutic outcomes for patients with hematologic malignancies and beyond.
Emerging immune cell types for CAR engineering
In addition to CAR-T, CAR-NKT and CAR-NK therapies, CAR-macrophages are emerging as another exciting avenue in cellular immunotherapy (79–84). Macrophages, as innate immune cells with a natural ability to infiltrate solid tumor microenvironments, represent a unique therapeutic platform. Unlike T or NK cells, macrophages can remodel the tumor microenvironment, phagocytose cancer cells, and stimulate secondary immune responses through antigen presentation. CAR-macrophages are engineered to enhance their ability to specifically target tumor cells and sustain immune activation within the immunosuppressive tumor milieu.
Although CAR-macrophage therapies are still in preclinical or early-phase clinical development, their potential to overcome some of the limitations associated with other cell types has garnered significant attention. Preclinical studies have shown promising antitumor activity in both hematologic and solid tumor models. The first clinical data for HER2-directed CAR-macrophages displayed encouraging safety data in patients with recurrent and metastatic solid turmors (81), promising to expand the arsenal of CAR-based treatments and potentially address indications where other immune cell types have shown limited efficacy.
Gene editing platforms and cellular engineering strategies
To enable the safe clinical application of allogeneic CAR-T cell therapies, overcoming graft-versus-host disease (GvHD) and host-versus-graft (HvG) responses remains a central challenge requiring precise gene-editing strategies. To prevent GvHD, most allogeneic CAR-T designs rely on targeted disruption of the TRAC locus, eliminating TCR expression thereby abrogating GvHD risk.
Alloreactive rejection (HvG) of infused CAR-T cell products, driven primarily by αβ T cells targeting mismatched class I molecules, may be circumvented by knocking out β2-microglobulin (B2M), a crucial subunit of HLA class I, thereby, shielding donor cells from cytotoxic CD8+ T cells. However, this approach has the potential to trigger NK cell-mediated CAR-T lysis which in turn has been addressed by HLA-E overexpression, thereby inhibiting NK cell activation via the NKG2A receptor. Another strategy to mitigate NK cell-mediated CAR-T cell rejection is by HLA-A/B deletion. Additional modifications include CD47 overexpression to inhibit macrophage-mediated phagocytosis and CD52 knockout to protect donor cells from depletion by alemtuzumab-based lymphodepletion regimens.
Besides circumventing alloreactivity, cell engineering strategies are similarly being employed to prevent fratricide, for instance in CD70-targeting CAR-T cells (85) or to augment cellular fitness by targeted deletion of functional checkpoints, including PD-1 (46) and other previously unrecognized negative immune regulators which have recently emerged from large-scale CRISPR screening efforts in primary immune cells (86–90).
Lastly, safety switches in form of rituximab recognition domains (29) or inducible Caspase9 (21, 22, 31) are incorporated into some cell products to enable rapid in vivo elimination of engineered cells the event of inadvertent toxicities.
Multiple gene-editing platforms enable these modifications, each with unique strengths and limitations. Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are early-generation technologies that induce double-stranded breaks (DSBs) at specific loci. ZFNs rely on protein-DNA interactions but face challenges related to off-target effects and complex design. TALENs offer greater specificity but require labor-intensive customization.
The CRISPR-Cas system revolutionized gene editing by introducing RNA-guided targeting, enabling efficient multiplex editing. However, concerns persist about off-target effects, chromosomal rearrangements, and translocations following DSB-induced repair. To reduce these risks, base editing has emerged, allowing precise single-nucleotide changes without inducing DSBs. This approach enhances safety while maintaining editing efficiency.
Proprietary ARCUS nucleases, based on engineered homing endonucleases, offer an alternative platform with a single-cut mechanism and enhanced specificity. ARCUS’ small size enables easier delivery into cells, while its reduced off-target profile promises improved genomic stability. As these platforms continue to evolve, balancing precision, scalability, and clinical feasibility will be essential for advancing next-generation allogeneic adoptive cell therapies.
Positioning against bispecific antibodies
Allogeneic CAR-equipped cell products must also strategically position themselves against a growing number of bispecific antibodies, which are rapidly becoming an important therapeutic option in r/r LBCL. Bispecific antibodies, such as epcoritamab (CD3×CD20) (91, 92) and glofitamab (CD3×CD20) (93–95), have shown promise in providing effective, off-the-shelf treatments with lower manufacturing complexity and reduced treatment costs. These agents also tend to be associated with fewer severe adverse events, such as CRS or ICANS, further enhancing their appeal as practical alternatives (91–95).
However, bispecific antibodies may come with a trade-off in terms of long-term efficacy, particularly progression-free survival (PFS). Recent studies suggest that while bispecifics achieve impressive initial response rates, their durability may not match that of CAR-T therapies, which benefit from the ability of engineered cells to persist and provide sustained immune surveillance (37, 38). As allogeneic CAR-T and CAR-NK therapies continue to evolve, demonstrating superior durability of responses and the ability to achieve long-term remissions will be critical for distinguishing themselves in an increasingly competitive treatment landscape.
Impact of therapeutic sequencing
The sequencing of therapeutic agents, particularly bispecific antibodies, autologous CAR-T cells, and allogeneic CAR-T cells, plays a critical role in determining response rates and optimizing outcomes for patients with r/r LBCL. Exposure to previous CD19-targeting agents, including tafasitamab (96), a CD19-targeting monoclonal antibody, and loncastuximab tesirine (97), a CD19-directed antibody-drug conjugate, may exert selective pressure that promotes antigen escape, potentially complicating treatment in later-line patients and warranting the exploration of additional targets or dual-targeting strategies (6, 98). Conversely, preclinical data demonstrated reduced toxicity and improved antitumor efficacy in mice receiving CD19-CAR-T treatment following tafasitamab pretreatment, potentially by modulating CD19 antigen density available for CAR-T binding, thereby ameliorating CAR-T cell activation and pyroptosis of lymphoma cells (99).
It remains to be seen when in the treatment sequence allogeneic CAR-T cells may have their highest therapeutic impact. A recent decision by a clinical trial sponsor to deprioritize and terminate their ongoing trial investigating cemacabtagene ansegedleucel (Cema-cel) in a third-line indication reflects this uncertainty. Instead, the sponsor refocused their trial initiatives to pursue a frontline consolidation indication for patients with LBCL who remain MRD positive after completion of a full course of standard 1L induction therapy. This pivotal ALPHA3 trial (NCT06500273) builds on the growing appreciation that both safety and efficacy are improved in patients with low disease burden at the time of infusion.
Conversely, PBCAR19B is being developed as a potential first-in-class therapy specifically for relapse following autologous CD19-CAR-T therapy. This underscores the expanding potential of CAR-engineered products to target unique indications and refine therapeutic sequencing strategies.
Novel CAR-T cell constructs targeting alternative pathways or antigens are also advancing the therapeutic landscape. Transposon-engineered BAFF ligand–based autologous CAR T cells (LMY-920) have demonstrated encouraging responses with a tolerable safety profile in a phase 1 trial (NCT05312801) among 3 patients with r/r B-NHL (100). Notably, the therapy induced a complete response in 1 patient with diffuse large B-cell lymphoma (DLBCL) without causing higher-grade CRS or ICANS. An allogeneic version of the BAFF CAR-T cell is currently under development, which could further expand therapeutic options and improve accessibility for this patient population.
These developments highlight the importance of optimizing the timing and sequencing of CAR-based therapies within the treatment paradigm for LBCL. Biomarker-driven patient selection and strategic placement of CAR-T and CAR-NK therapies in earlier lines of treatment hold significant potential to enhance outcomes and maximize their therapeutic impact.
Funding of highly innovative cell therapy development programs
Besides ingenuity in cellular engineering, the future of cell therapy programs critically depends on efficient mechanisms of resource allocation to balance risk and reward in a field which has garnered tremendous momentum while it seeks to exploit the promises of precision gene editing.
Of note, the vast majority of included studies in the meta-analysis originated from industry-led cell therapy programs, while academic sponsorship remained limited. While industry funding accelerates commercialization, it also introduces potential bias, which may impinge on scientific independence. US biotech firms in particular benefit from a competitive and mature venture capital ecosystem, while China’s state-backed investments emphasize commercialization. However, venture capital dependence can shift priorities toward short-term gains over high-risk academic innovation.
To balance this, public funding models like the NIH SBIR (Small Business Innovation Research)/STTR (Small Business Technology Transfer) and CPRIT (Cancer Prevention and Research Institute of Texas) emulate venture capital funding mechanics by making high-risk investments and reinvesting financial returns based on revenue-sharing agreements. In contrast, European programs such as IMI (Innovative Medicines Initiative) and EDCTP (European and Developing Countries Clinical Trials Partnership) focus on public-private partnerships but lack the same reinvestment mechanisms that help derisk early-stage clinical assets to enable commercial scale-up. These differences in funding may explain the strong US footprint and present a potential path towards increasing Europe’s presence in the cell therapy space.
Conclusion
The very low incidences of higher-grade CRS, ICANS, and GvHD across the included studies highlights the exceptional safety profile of allogeneic CAR-T and CAR-NK cell therapies, setting them apart as a safer alternative to autologous CAR-T products. Early efficacy signals are very encouraging, especially considering that this is the first generation of allogeneic cell products. Multi-engineered cell products with additional genetic edits for enhanced functionality are constantly being added to the pipelines of cell therapy development programs and are already being evaluated in the clinic (101). Recent studies leveraging large-scale screening approaches have uncovered previously unrecognized regulators of immune cell function, thereby providing a catalogue of actionable targets to further increase the anti-tumor potency of next-generation cellular therapeutics (86–90, 102).
It will be interesting to see the therapeutic efficacy of allogeneic CAR-T and CAR-NK cells across a wider spectrum of malignancies, including efficacy against solid tumors, where early studies are already underway. Future efforts will need to address how biomarker-driven strategies may refine patient selection and optimize therapeutic outcomes. Ultimately, it will be compelling to observe how various cell therapy programs address the unmet need in r/r LBCL by targeting distinct indications across different lines of therapy and tailoring treatments to patients with unique disease characteristics.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
AB: Conceptualization, Data curation, Formal Analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. FB: Data curation, Investigation, Resources, Supervision, Validation, Writing – review & editing. JH: Conceptualization, Data curation, Formal Analysis, Investigation, Validation, Writing – review & editing, Visualization, Supervision.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We thank the patients and families who were treated on the selected study protocols. We acknowledge the use of ChatGPT-4o (OpenAI, San Francisco, CA, USA) for assistance with language editing. Original schematics were created in Biorender.
Conflict of interest
JH provided consulting services and received honoraria from Johnson & Johnson, BMS, Kite/Gilead, Abbvie, Servier, Novartis, Roche, Iovance, Imatics, Philogen, Boehringer Ingelheim, Immunocore, Autolus, Kyverna. AB declares the absence of any commercial or financial conflict of interest. FB received honoraria from Johnson & Johnson, BMS, Amgen and GSK.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. We acknowledge the use of ChatGPT-4o (OpenAI, San Francisco, CA, USA) for assistance with language editing.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1585556/full#supplementary-material
Supplementary Figure 1 | Geographical footprint and sponsor type for investigated CAR-IEC products (n=19 unique trials).
References
1. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377(26):2531–44.
2. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380(1):45–56.
3. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet. (2020) 396(10254):839–52.
4. Schuster SJ, Tam CS, Borchmann P, Worel N, McGuirk JP, Holte H, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. (2021) 22(10):1403–15. doi: 10.1016/S1470-2045(21)00375-2
5. Locke FL, Miklos DB, Jacobson CA, Perales M-A, Kersten M-J, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. (2022) 386(7):640–54.
6. Cappell KM and Kochenderfer JN. Long-term outcomes following CAR T cell therapy: What we know so far. Nat Rev Clin Oncol. (2023) 20(6):359–71. doi: 10.1038/s41571-023-00754-1
7. Neelapu SS, Jacobson CA, Ghobadi A, Miklos DB, Lekakis LJ, Oluwole OO, et al. Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. (2023) 141(19):2307–15. doi: 10.1182/blood.2022018893
8. Jacobson CA, Munoz J, Sun F, Kanters S, Limbrick-Oldfield EH, Spooner C, et al. Real-world outcomes with chimeric antigen receptor T cell therapies in large B cell lymphoma: A systematic review and meta-analysis. Transplant Cell Ther. (2024) 30(1):77. doi: 10.1016/j.jtct.2023.10.017
9. Palomba ML, Crombie JL, Nastoupil LJ, Andreadis C, Isufi I, Hunter B, et al. Multicenter, real-world study in patients with R/R large B-cell lymphoma (LBCL) who received lisocabtagene maraleucel (liso-cel) in the United States (US). Transplant Cell Ther. (2024) 30(2):S40–S1. doi: 10.1016/j.jtct.2023.12.071
10. Abramson JS, Palomba ML, Gordon LI, Lunning M, Wang M, Arnason J, et al. Two-year follow-up of lisocabtagene maraleucel in relapsed or refractory large B-cell lymphoma in TRANSCEND NHL 001. Blood. (2024) 143(5):404–16. doi: 10.1182/blood.2023020854
11. Lin Y, Raje NS, Berdeja JG, Siegel DS, Jagannath S, Madduri D, et al. Idecabtagene vicleucel for relapsed and refractory multiple myeloma: Post hoc 18-month follow-up of a phase 1 trial. Nat Med. (2023) 29(9):2286–94. doi: 10.1038/s41591-023-02496-0
12. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384(8):705–16. doi: 10.1056/NEJMoa2024850
13. San-Miguel J, Dhakal B, Yong K, Spencer A, Anguille S, Mateos M-V, et al. Cilta-cel or standard care in lenalidomide-refractory multiple myeloma. N Engl J Med. (2023) 389(4):335–47. doi: 10.1056/NEJMoa2303379
14. Sidana S, Patel KK, Peres LC, Bansal R, Kocoglu MH, Shune L, et al. Safety and efficacy of standard of care ciltacabtagene autoleucel for relapsed/refractory multiple myeloma. Blood. (2024) 145(1):85–97. doi: 10.1182/blood.2024025945
15. Looka A, Qualls DA, Matthews D, Redd RA, Sakellis C, Duffy C, et al. A real-world comparison of commercial-use axicabtagene ciloleucel and lisocabtagene maraleucel in large B-cell lymphoma. Blood Adv. (2025) 9(3):455–62. doi: 10.1182/bloodadvances.2024012992
16. Looka A, Qualls DA, Matthews D, Redd RA, Sakellis C, Duffy C, et al. A real-world comparison of commercial use axicabtagene ciloleucel and lisocabtagene maraleucel in large B-cell lymphoma. Blood Adv. (2024). doi: 10.1182/bloodadvances.2024012992
17. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature. (2022) 602(7897):503–9. doi: 10.1038/s41586-021-04390-6
18. Bobillo MSO, Kambhampati S, Lee D, Hunter BD, Egini O, Patel K, et al. Real-world (RW) outcomes of lisocabtagene maraleucel (liso-cel) as second-line (2L) therapy in patients (pts) with relapsed or refractory (R/R) large B-cell lymphoma (LBCL): First results from the Center for International Blood and Marrow Transplant Research (CIBMTR) registry. Blood. (2024) 144(Suppl 1):470–. doi: 10.1182/blood-2024-199723
19. Portuguese AJ, Albittar A, Huang JJ, Liang EC, Wuliji N, Taheri M, et al. Real-world comparison of lisocabtagene maraleucel (liso-cel) and axicabtagene ciloleucel (axi-cel): Efficacy & toxicity. Transplant Cell Ther. (2024) 30(2 Suppl):S192. doi: 10.1016/j.jtct.2023.12.249
20. Kersten MJ, Saevels K, Beguin Y, Vermaat JSP, Verbruggen N, Spoon M, et al. Seven-day vein-to-vein point-of-care manufactured CD19 CAR T cells (GLPG5101) in relapsed/refractory NHL: Results from the phase 1 ATALANTA-1 trial. Blood. (2023) 142(Suppl 1):2113–. doi: 10.1182/blood-2023-172736
21. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. (2018) 32(2):520–31. doi: 10.1038/leu.2017.226
22. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
23. Morisot N, Wadsworth S, Davis T, Dailey N, Hansen K, Gonzalez D, et al. 127 Preclinical evaluation of NKX019, a CD19-targeting CAR NK cell. J Immunother Cancer. (2020) 8(Suppl 3):A78–A. doi: 10.1136/jitc-2020-SITC2020.0127
24. Laskowski TJ, Biederstädt A, and Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. (2022) 22(10):557–75. doi: 10.1038/s41568-022-00491-0
25. Tohmé M, Davis T, Zhang M, Lemar H, Duong B, Tan J, et al. 902 NKX019, an off-the-shelf CD19 CAR-NK cell, mediates improved anti-tumor activity and persistence in combination with CD20-directed therapeutic mAbs. J Immunother Cancer. (2022) 10(Suppl 2):A940–A. doi: 10.1136/jitc-2022-SITC2022.0902
26. McGuirk JP, Tam CS, Kröger N, Riedell PA, Murthy HS, Ho PJ, et al. CTX110 allogeneic CRISPR-Cas9-engineered CAR T cells in patients (pts) with relapsed or refractory (R/R) large B-cell lymphoma (LBCL): Results from the phase 1 dose escalation CARBON study. Blood. (2022) 140(Suppl 1):10303–6. doi: 10.1182/blood-2022-166432
27. Jacobson CA, Jayawardena T, Yu L, Hallman J, Schmittling R, Cuneo E, et al. Effective cell dose and functional attributes of azercabtagene zapreleucel (azer-cel; PBCAR0191) associate with allogeneic CAR T-cell safety and efficacy in patients with relapsed/refractory B-cell lymphoma. Blood. (2022) 140(Suppl 1):4588–9. doi: 10.1182/blood-2022-165311
28. Dickinson M, Hamad N, Bryant CE, Kothari N, Ojeras P, Vohra A, et al. First-in-human data of NKX019, an allogeneic CAR NK for the treatment of relapsed/refractory (R/R) B-cell malignancies. Hematol Oncol. (2023) 41(S2):526–7. doi: 10.1002/hon.3164_389
29. Locke FL, Lekakis LJ, Eradat H, Munoz J, Tees MT, Vos SD, et al. Phase 1 results with anti-CD19 allogeneic CAR T allo-501/501A in relapsed/refractory large B-cell lymphoma (R/R LBCL). J Clin Oncol. (2023) 41(16 Suppl):2517–. doi: 10.1200/JCO.2023.41.16_suppl.2517
30. Bagri A, Sims A, Williams A, Mbofung R, Morales-Mantilla D, Wang Y, et al. Development of a multiplexed-engineered, off-the-shelf CAR NK cell with unique multi-pathogenic cell targeting capacity for the treatment of autoimmune diseases in the absence of conditioning chemotherapy. Arthritis Rheumatol. (2024).
31. Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+ B cell tumors: A phase 1/2 trial. Nat Med. (2024) 30(3):772–84. doi: 10.1038/s41591-023-02785-8
32. Xiao X, Liu H, Qiu X, Chen P, Li X, Wang D, et al. CD19-CAR-DNT cells (RJMTY19) in patients with relapsed or refractory large B-cell lymphoma: A phase 1, first-in-human study. eClinicalMedicine. (2024) 70:102516. doi: 10.1016/j.eclinm.2024.102516
33. Gergis U, Martin A, Vogel A, Smith J, Klein A, Reshef R, et al. PBCAR19B, an immune-evading allogeneic CAR T stealth cell, demonstrates potent anti-tumor responses and prolonged B cell depletion supporting sustained immune evasion in patients with relapsed/refractory (R/R) B-cell lymphoma (NHL). Blood. (2024) 144(Supplement 1):7175. doi: 10.1182/blood-2024-210499
34. Neelapu SS, Stevens DA, Hamadani M, Frank MJ, Holmes H, Jacobovits A, et al. A phase 1 study of ADI-001: Anti-CD20 CAR-engineered allogeneic gamma delta1 (γδ) T cells in adults with B-cell malignancies. Blood. (2022) 140(Suppl 1):4617–9. doi: 10.1182/blood-2022-157400
35. Hu B, Nastoupil LJ, Holmes H, Hamdan A, Kanate A, Farooq U, et al. A CRISPR-edited allogeneic anti-CD19 CAR-T cell therapy with a PD-1 knockout (CB-010) in patients with relapsed/refractory B cell non-Hodgkin lymphoma (R/R B-NHL): Updated phase 1 results from the ANTLER trial. J Clin Oncol. (2024) 42(16 Suppl):7025–. doi: 10.1200/JCO.2024.42.16_suppl.7025
36. O'Brien S, Nastoupil LJ, Essell J, Dsouza L, Hart D, Matsuda E, et al. A first-in-human phase 1, multicenter, open-label study of CB-010, a next-generation CRISPR-edited allogeneic anti-CD19 CAR-T cell therapy with a PD-1 knockout, in patients with relapsed/refractory B cell non-Hodgkin lymphoma (ANTLER study). Blood. (2022) 140(Suppl 1):9457–8. doi: 10.1182/blood-2022-168128
37. Kim J, Cho J, Lee MH, Yoon SE, Kim WS, and Kim SJ. CAR T cells vs bispecific antibody as third- or later-line large B-cell lymphoma therapy: A meta-analysis. Blood. (2024) 144(6):629–38. doi: 10.1182/blood.2023023419
38. Liang X, Wang Y, Luo B, Lin B, Lu W, Tian S, et al. Comparison of CAR T-cell and bispecific antibody as third-line or later-line treatments for multiple myeloma: A meta-analysis. J Immunother Cancer. (2024) 12(11):e010064. doi: 10.1136/jitc-2024-010064
39. Bachanova V, Cayci Z, Lewis D, Maakaron JE, Janakiram M, Bartz A, et al. Initial clinical activity of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood. (2020) 136(Suppl 1):8–. doi: 10.1182/blood-2020-141606
40. Bachanova V, Ghobadi A, Patel K, Park JH, Flinn IW, Shah P, et al. Safety and efficacy of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood. (2021) 138:823. doi: 10.1182/blood-2021-151185
41. Bachanova V, Deol A, Al-Juhaishi TMS, Lulla PD, Byrne MT, Wong C, et al. Safety and efficacy of FT522, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy with alloimmune defense receptor (ADR) in relapsed/refractory B-cell lymphoma. Blood. (2024) 144(Suppl 1):6543–. doi: 10.1182/blood-2024-198684
42. Klingemann H. The NK-92 cell line—30 years later: Its impact on natural killer cell research and treatment of cancer. Cytotherapy. (2023) 25(5):451–7. doi: 10.1016/j.jcyt.2022.12.003
43. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ. doi: 10.1136/bmj.n71
44. Abramson JS, Ramakrishnan A, Pierola AA, Braunschweig I, Cartron G, Thieblemont C, et al. Preliminary results of NATHALI-01: A first-in-human phase I/IIa study of UCART20x22, a dual allogeneic CAR-T cell product targeting CD20 and CD22, in relapsed or refractory (R/R) non-Hodgkin lymphoma (NHL). Blood. (2023) 142(Suppl 1):2110–. doi: 10.1182/blood-2023-186570
45. Adicet Bio. ADI-001 phase 1 clinical update. Boston, MA: Adicet Bio (2023). Available at: https://www.adicetbio.com.
46. Caribou Biosciences. Transformative genome-edited therapies for patients. Berkeley, CA: Caribou Biosciences (2024). Available at: https://www.cariboubio.com.
47. Sana Biotechnology. Corporate presentation January 2024. Seattle, WA: Sana Biotechnology (2024). p. 39. Available at: https://www.sana.com.
48. Ramos CA, Robinson SN, Dakhova O, Lulla PD, Kamble RT, Carrum G, et al. . Salt Lake City, UT, USA: 2022 Tandem Meetings of ASTCT and CIBMTR.
49. Chen X, Tan B, Xing H, Zhao X, Ping Y, Zhang Z, et al. Allogeneic CAR-T cells with HLA-A/B and TRAC disruption exhibit promising antitumor capacity against B cell malignancies. Cancer Immunol Immunother. (2024) 73(1):13. doi: 10.1007/s00262-023-03586-1
50. Darrah JM, Varadarajan I, Mehta A, Saultz JN, McKinney M, Ghosh M, et al. Efficacy and safety of TAK-007, cord blood-derived CD19 CAR-NK cells, in adult patients with relapsed/refractory (R/R) B-cell non-Hodgkin lymphoma (NHL). Blood. (2024) 144(Suppl 1):95–. doi: 10.1182/blood-2024-194807
51. Dickinson M, Hamad N, Bryant C, Kothari N, Ojeras P, Vohra A, et al. S261: First in human data of NKX019, an allogeneic CAR NK for the treatment of relapsed/refractory (R/R) B-cell malignancies. HemaSphere. (2023) 7(S3):e37234fb.
52. Ghobadi A, Bachanova V, Patel K, Park JH, Flinn I, Riedell PA, et al. Induced pluripotent stem-cell-derived CD19-directed chimeric antigen receptor natural killer cells in B-cell lymphoma: a phase 1, first-in-human trial. Lancet. (2025) 405(10473):127–36. doi: 10.1016/S0140-6736(24)02462-0
53. Ghobadi A, McGuirk JP, Shaughnessy P, Tam CS, Allen M, Pan C, et al. CTX112, a next-generation allogeneic CRISPR-Cas9 engineered CD19 CAR T cell with novel potency edits: Data from phase 1 dose escalation study in patients with relapsed or refractory B-cell malignancies. Blood. (2024) 144(Suppl 1):4829–. doi: 10.1182/blood-2024-203563
54. Imugene Limited. Azer-Cel CD19 CAR T for blood cancer [corporate presentation]. Sydney, NSW: Imugene Ltd (2024). Available at: https://static1.squarespace.com/static/5b63d41b3e2d09b1f56bf483/t/66de4a8310d1656b14f181e2/1725844110899/IMU+Corporate+Deck+-+AUS+NDR+September+2024+ASX.pdf.
55. McGuirk JP, Tam CS, Kröger N, Riedell PA, Murthy HS, Ho PJ, et al. CTX110 allogeneic CRISPR-Cas9–engineered CAR T cells in patients with relapsed or refractory (R/R) large B-cell lymphoma (LBCL): Results from the phase 1 dose escalation CARBON study. EBMT 2023 Annu Meeting. (2023).
56. Liu Y, Shi J, Yang Q, Wang C, Liang F, Wu Z, et al. The interim analysis of a first-in-human phase 1 trial of ET-901, a CRISPR-edited allogeneic immune-cloaked anti-CD19 CAR-T cell therapy in patients with R/R B-NHL. J Clin Oncol. (2024) 42(16_suppl):e19010. doi: 10.1200/JCO.2024.42.16_suppl.e19010
57. Fate Therapeutics. Initial phase 1 clinical data in relapsed/refractory B-cell lymphoma show favorable safety profile, complete responses, and persistence of FT522 live cells. San Diego, CA: Fate Therapeutics (2024). Available at: https://ir.fatetherapeutics.com/news-releases/news-release-details/fate-therapeutics-highlights-ft522-shelf-adr-armed-car-nk-cell.
58. Mehta A, Farooq U, Chen A, McGuirk JP, Ly T, Wong L, et al. Interim phase I clinical data of FT819-101, a study of the first-ever, off-the-shelf, iPSC-derived TCR-less CD19 CAR T-cell therapy for patients with relapsed/refractory B-cell malignancies. Blood. (2022) 140(Suppl 1):4577–8. doi: 10.1182/blood-2022-167194
59. Nishimoto KP, Barca T, Azameera A, Makkouk A, Romero JM, Bai L, et al. Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models. Clin Transl Immunol. (2022) 11(2):e1373. doi: 10.1002/cti2.1373
60. Fate Therapeutics. FT596 phase 1 clinical data in relapsed/refractory B-cell lymphoma. San Diego, CA: Fate Therapeutics (2024). Available at: https://ir.fatetherapeutics.com/news-releases/news-release-details/fate-therapeutics-highlights-ft522-shelf-adr-armed-car-nk-cell.
61. Mehta A, Farooq U, Chen A, McGuirk JP, Jain N, Ly T, et al. Interim phase I clinical data of FT819-101, a study of the first-ever, off-the-shelf, iPSC-derived TCR-less CD19 CAR T-cell therapy for patients with relapsed/refractory B-cell malignancies. 64th ASH Annu Meeting Exposition. New Orleans, LA, United States: Fate Therapeutics (2022).
62. Precision BioSciences. A phase 1/2a, open-label, dose-escalation, dose-expansion, parallel assignment study to evaluate the safety and clinical activity of PBCAR20A in subjects with relapsed/refractory (R/R) non-Hodgkin lymphoma (NHL) or R/R chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) [Internet]. (2019). Available online at: https://clinicaltrials.gov/study/NCT04030195.
63. St. Jude Children's Research Hospital, National Cancer Institute, and Assisi Foundation. Genetically modified haploidentical natural killer cell infusions for B-lineage acute lymphoblastic leukemia [Internet]. (2009). Available at: https://clinicaltrials.gov/show/NCT00995137 (Accessed January 03, 2025).
64. National University Health System. Pilot study of redirected haploidentical natural killer cell infusions for B-lineage acute lymphoblastic leukemia [Internet]. (2013). Available at: https://clinicaltrials.gov/show/NCT01974479 (Accessed 2025 Jan 3).
65. Ciurea SO, Kongtim P, Srour S, Chen J, Soebbing D, Shpall E, et al. Results of a phase I trial with haploidentical MBIL-21 ex vivo expanded NK cells for patients with multiply relapsed and refractory AML. Am J Hematol. (2024) 99(5):890–9. doi: 10.1002/ajh.27281
66. Davies SM, Ruggieri L, DeFor T, Wagner JE, Weisdorf DJ, Miller JS, et al. Evaluation of KIR ligand incompatibility in mismatched unrelated donor hematopoietic transplants. Killer immunoglobulin-like receptor. Blood. (2002) 100(10):3825–7. doi: 10.1182/blood-2002-04-1197
67. Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. (1999) 94(1):333–9.
68. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood. (2007) 110(1):433–40. doi: 10.1182/blood-2006-07-038687
69. Shapiro RM, Birch GC, Hu G, Vergara Cadavid J, Nikiforow S, Baginska J, et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for post-transplant relapse. J Clin Invest. (2022) 132(1):e154334. doi: 10.1172/JCI154334
70. Ciurea SO, Kongtim P, Soebbing D, Trikha P, Behbehani G, Rondon G, et al. Decrease post-transplant relapse using donor-derived expanded NK cells. Leukemia. (2022) 36(1):155–64. doi: 10.1038/s41375-021-01349-4
71. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295(5562):2097–100. doi: 10.1126/science.1068440
72. Bishara A, De Santis D, Witt CC, Brautbar C, Christiansen FT, Or R, et al. The beneficial role of inhibitory KIR genes of HLA class I NK epitopes in haploidentically mismatched stem cell allografts may be masked by residual donor-alloreactive T cells causing GVHD. Tissue Antigens. (2004) 63(3):204–11. doi: 10.1111/j.0001-2815.2004.00182.x
73. Jiang T, Peng Y, Tang X, Chen S, Yang Z, and Liu Y. HIV-related diffuse large B-cell lymphoma influences on T cell exhaustion. Blood. (2023) 142(Suppl 1):3013–. doi: 10.1182/blood-2023-182550
74. Maurer MJ, Ghesquières H, Link BK, Jais J-P, Habermann TM, Thompson CA, et al. Diagnosis-to-treatment interval is an important clinical factor in newly diagnosed diffuse large B-cell lymphoma and has implication for bias in clinical trials. J Clin Oncol. (2018) 36(16):1603–10. doi: 10.1200/JCO.2017.77.6233
75. Pal SK, Tran B, Haanen J, Hurwitz ME, Sacher A, Tannir NM, et al. CD70-targeted allogeneic CAR T-cell therapy for advanced clear cell renal cell carcinoma. Cancer Discovery. (2024) 14(7):1176–89. doi: 10.1158/2159-8290.CD-24-0102
76. Srour SA, Chahoud J, Drakaki A, Curti BD, Pal S, Tang L, et al. 322 Allo-316 in patients with advanced or metastatic clear cell renal cell carcinoma (ccRCC): Updated safety and efficacy from the phase 1 TRAVERSE multicenter study. J Immunother Cancer. (2024) 12(Suppl 2):A373. doi: 10.1136/jitc-2024-SITC2024.0322
77. Iyer SP, Sica RA, Ho PJ, Prica A, Zain J, Foss FM, et al. Safety and activity of CTX130, a CD70-targeted allogeneic CRISPR-Cas9-engineered CAR T-cell therapy, in patients with relapsed or refractory T-cell malignancies (COBALT-LYM): a single-arm, open-label, phase 1, dose-escalation study. Lancet Oncol. (2025) 26(1):110–22. doi: 10.1016/S1470-2045(24)00508-4
78. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. (2019) 25(1):82–8. doi: 10.1038/s41591-018-0290-5
79. Sloas C, Gill S, and Klichinsky M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front Immunol. (2021) 12:783305. doi: 10.3389/fimmu.2021.783305
80. Reiss KA, Yuan Y, Ueno NT, Johnson ML, Gill S, Dees EC, et al. A phase 1, first-in-human (FIH) study of the anti-HER2 CAR macrophage CT-0508 in subjects with HER2 overexpressing solid tumors. J Clin Oncol. (2022) 40(16_suppl):2533–. doi: 10.1200/JCO.2022.40.16_suppl.2533
81. Abdou Y, Dees EC, Mortimer JE, Pohlmann PR, Johnson ML, Maziarz RT, et al. A phase 1, first-in-human (FIH) study of autologous macrophages engineered to express an anti-HER2 chimeric antigen receptor (CAR) in participants (pts) with HER2-overexpressing solid tumors. J Clin Oncol. (2023) 41(16_suppl):TPS2666. doi: 10.1200/JCO.2023.41.16_suppl.TPS2666
82. Abdin SM, Paasch D, and Lachmann N. CAR macrophages on a fast track to solid tumor therapy. Nat Immunol. (2024) 25(1):11–2. doi: 10.1038/s41590-023-01696-7
83. Lei A, Yu H, Lu S, Lu H, Ding X, Tan T, et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat Immunol. (2024) 25(1):102–16. doi: 10.1038/s41590-023-01687-8
84. Shah Z, Tian L, Li Z, Jin L, Zhang J, Li Z, et al. Human anti-PSCA CAR macrophages possess potent antitumor activity against pancreatic cancer. Cell Stem Cell. (2024) 31(6):803–17.e6. doi: 10.1016/j.stem.2024.03.018
85. Srinivasan S, Tan N, Cheng H-Y, Zhang Y, Tacheva-Grigorova S, Van Blarcom T, et al. Investigation of ALLO-316: a fratricide-resistant allogeneic CAR T targeting CD70 as a potential therapy for the treatment of AML. Blood. (2020) 136(Suppl 1):23. doi: 10.1182/blood-2020-142161
86. Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell. (2018) 175(7):1958–71.e15. doi: 10.1016/j.cell.2018.10.024
87. Schmidt R, Ward CC, Dajani R, Armour-Garb Z, Ota M, Allain V, et al. Base-editing mutagenesis maps alleles to tune human T cell functions. Nature. (2024) 625(7996):805–12. doi: 10.1038/s41586-023-06835-6
88. Schmidt R, Steinhart Z, Layeghi M, Freimer JW, Bueno R, Nguyen VQ, et al. CRISPR activation and interference screens decode stimulation responses in primary human T cells. Science. (2022) 375(6580):eabj4008. doi: 10.1126/science.abj4008
89. Freitas KA, Belk JA, Sotillo E, Quinn PJ, Ramello MC, Malipatlolla M, et al. Enhanced T cell effector activity by targeting the mediator kinase module. Science. (2022) 378(6620):eabn5647. doi: 10.1126/science.abn5647
90. Blaeschke F, Chen YY, Apathy R, Daniel B, Chen AY, Chen PA, et al. Modular pooled discovery of synthetic knockin sequences to program durable cell therapies. Cell. (2023) 186(19):4216–34.e33. doi: 10.1016/j.cell.2023.08.013
91. Thieblemont C, Phillips T, Ghesquières H, Cheah CY, Clausen MR, Cunningham D, et al. Epcoritamab, a novel, subcutaneous CD3×CD20 bispecific T-cell–engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol. (2023) 41(12):2238–47. doi: 10.1200/JCO.22.01725
92. Thieblemont C, Karimi YH, Ghesquières H, Cheah CY, Clausen MR, Cunningham D, et al. Epcoritamab in relapsed/refractory large B-cell lymphoma: 2-year follow-up from the pivotal EPCORE NHL-1 trial. Leukemia. (2024) 38(12):2653–62. doi: 10.1038/s41375-024-02410-8
93. Dickinson MJ, Carlo-Stella C, Morschhauser F, Bachy E, Corradini P, Iacoboni G, et al. Glofitamab for relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2022) 387(24):2220–31. doi: 10.1056/NEJMoa2206913
94. Hutchings M, Morschhauser F, Iacoboni G, Carlo-Stella C, Offner FC, Sureda A, et al. Glofitamab, a novel, bivalent CD20-targeting T-cell–engaging bispecific antibody, induces durable complete remissions in relapsed or refractory B-cell lymphoma: a phase I trial. J Clin Oncol. (2021) 39(18):1959–70. doi: 10.1200/JCO.20.03175
95. Abramson JS, Ku M, Hertzberg M, Huang H-Q, Fox CP, Zhang H, et al. Glofitamab plus gemcitabine and oxaliplatin (GemOx) versus rituximab-GemOx for relapsed or refractory diffuse large B-cell lymphoma (STARGLO): a global phase 3, randomised, open-label trial. Lancet. (2024) 404(10466):1940–54. doi: 10.1016/S0140-6736(24)01774-4
96. Salles G, Duell J, González Barca E, Tournilhac O, Jurczak W, Liberati AM, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. (2020) 21(7):978–88. doi: 10.1016/S1470-2045(20)30225-4
97. Caimi PF, Ai W, Alderuccio JP, Ardeshna KM, Hamadani M, Hess B, et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. (2021) 22(6):790–800. doi: 10.1016/S1470-2045(21)00139-X
98. Plaks V, Rossi JM, Chou J, Wang L, Poddar S, Han G, et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood. (2021) 138(12):1081–5.
99. Sakemura RL, Manriquez Roman C, Horvei P, Siegler EL, Girsch JH, Sirpilla OL, et al. CD19 occupancy with tafasitamab increases therapeutic index of CART19 cell therapy and diminishes severity of CRS. Blood. (2024) 143(3):258–71. doi: 10.1182/blood.2022018905
100. Caimi P, Spear MA, Liter J, Largaespada DA, Moriarity BS, and Webber BR. 244 A phase 1 study of novel transposon-based BAFF CAR-T cells (LMY-920) for treatment of relapsed or refractory non-Hodgkin lymphoma (NHL). J Immunother Cancer. (2024) 12(Suppl 2):A282. doi: 10.1136/jitc-2024-SITC2024.0244
101. Biederstädt A, Manzar GS, and Daher M. Multiplexed engineering and precision gene editing in cellular immunotherapy. Front Immunol. (2022) 13:1063303. doi: 10.3389/fimmu.2022.1063303
Keywords: allogeneic CAR-T cells, allogeneic CAR-NK cells, adoptive cell therapy (ACT), cellular engineering, CRISPR gene editing, precision gene editing, large B cell lymphoma, clinical trial
Citation: Biederstädt A, Bassermann F and Hecker JS (2025) Allogeneic CAR-engineered cellular therapy for relapsed and refractory large B cell lymphoma: a systematic review and meta-analysis. Front. Immunol. 16:1585556. doi: 10.3389/fimmu.2025.1585556
Received: 28 February 2025; Accepted: 09 June 2025;
Published: 08 July 2025.
Edited by:
Avichai Shimoni, Sheba Medical Center, IsraelReviewed by:
Arpad Szoor, University of Debrecen, HungaryAbraham Avigdor, Sheba Medical Center, Israel
Copyright © 2025 Biederstädt, Bassermann and Hecker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Biederstädt, YWxleGFuZGVyLmJpZWRlcnN0YWVkdEB0dW0uZGU=
†ORCID: Alexander Biederstädt, orcid.org/0000-0003-2805-8231
Florian Bassermann, orcid.org/0000-0003-4435-2609
Judith S. Hecker, orcid.org/0000-0002-1531-0517