 Rachel L. Creighton1,2
Rachel L. Creighton1,2 Florian Hladik
Florian Hladik Germán G. Gornalusse
Germán G. Gornalusse- 1Department of Obstetrics and Gynecology, School of Medicine, University of Washington, Seattle, WA, United States
- 2Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Center, Seattle, WA, United States
- 3Department of Medicine, Division of Allergy and Infectious Diseases, University of Washington, Seattle, WA, United States
- 4Department of Global Health, Schools of Medicine and Public Health, University of Washington, Seattle, WA, United States
The barrier to HIV cure is the HIV reservoir, which is composed of latently infected CD4+ T cells and myeloid cells that carry stably integrated and replication-competent provirus. The gastrointestinal tract (GIT) contains a substantial part of the HIV reservoir and its immunophysiology could be especially conducive for HIV persistence and reactivation. However, the exact cellular microenvironment and molecular mechanisms that govern the renewal of provirus-harboring cells and proviral reactivation in the GIT remain unclear. In this review, we outline the evidence supporting an overarching hypothesis that interferon activity driven by specialized enterocytes creates a microenvironment that fosters proliferation of latently infected CD4+ T cells and sporadic HIV reactivation from these cells. First, we describe unique immunologic features of the gastrointestinal associated lymphoid tissue (GALT), specifically highlighting IFN activity in specialized enterocytes and potential interactions between these cells and neighboring HIV susceptible cells. Then, we will describe dysregulation of IFN signaling in HIV infection and how IFN dysregulation in the GALT may contribute to the persistence and reactivation of the latent HIV reservoir. Finally, we will speculate on the clinical implications of this hypothesis for HIV cure strategies and outline the next steps.
1 Introduction
Except for a few isolated cases, HIV infection has never been cured (1). This is because HIV integrates into the host genome (becoming a “provirus”), evading the immune response and escaping antiretroviral therapies (ART) (2, 3). When ART is stopped, reactivation of proviruses in some latently infected cells leads to rebound viremia (4, 5). HIV latency is established very early during acute HIV infection, either through direct infection of resting memory CD4+ T cells or through infection of actively replicating CD4+ T cells that are later induced to a resting state (6–10). Latently infected cells are present in numerous microanatomical environments, including the blood, lymph nodes, brain, and gut (11–15). Tissue-specific factors like cell signaling, cell-cell interactions, and local antiretroviral drug concentration are critical to understanding the persistence and reactivation of latent HIV infection (14, 15).
Given its constant exposure to commensal bacteria and pathogens, the GIT is a highly immunologically active site. Previously, we found a population of cells in the intestinal epithelium producing extremely high levels of type I/III interferon (IFN)-stimulated proteins, including IFN-stimulated gene 15 (ISG15) (16). Co-expression of glycoprotein 2 suggests that some of these cells are microfold cells (M cells) (17). In response to viral pathogens, secreted IFNs upregulate the expression of interferon-stimulated genes (ISGs) to inhibit viral replication and prevent further cellular infection (18). However, in chronic HIV, the antiviral effect of the interferon system becomes pathological due to years of overstimulation (19, 20). This dysregulation of the IFN system, termed “interferonopathy,” has been posited to antagonize a potential HIV cure by driving bystander T cell proliferation, including of latently infected cells, thus contributing to HIV reservoir persistence (21–23). CD4+ T cell proliferation is thought to be the most important mechanism sustaining the HIV reservoir (24–38). There is also evidence that IFN efficiently reactivates HIV-1 (39). Therefore, the high IFN signaling activity observed in the intestinal epithelium led us to hypothesize that these immunologically active enterocytes play a role in the persistence and spontaneous reactivation of the HIV reservoir in the GIT (Figure 1).
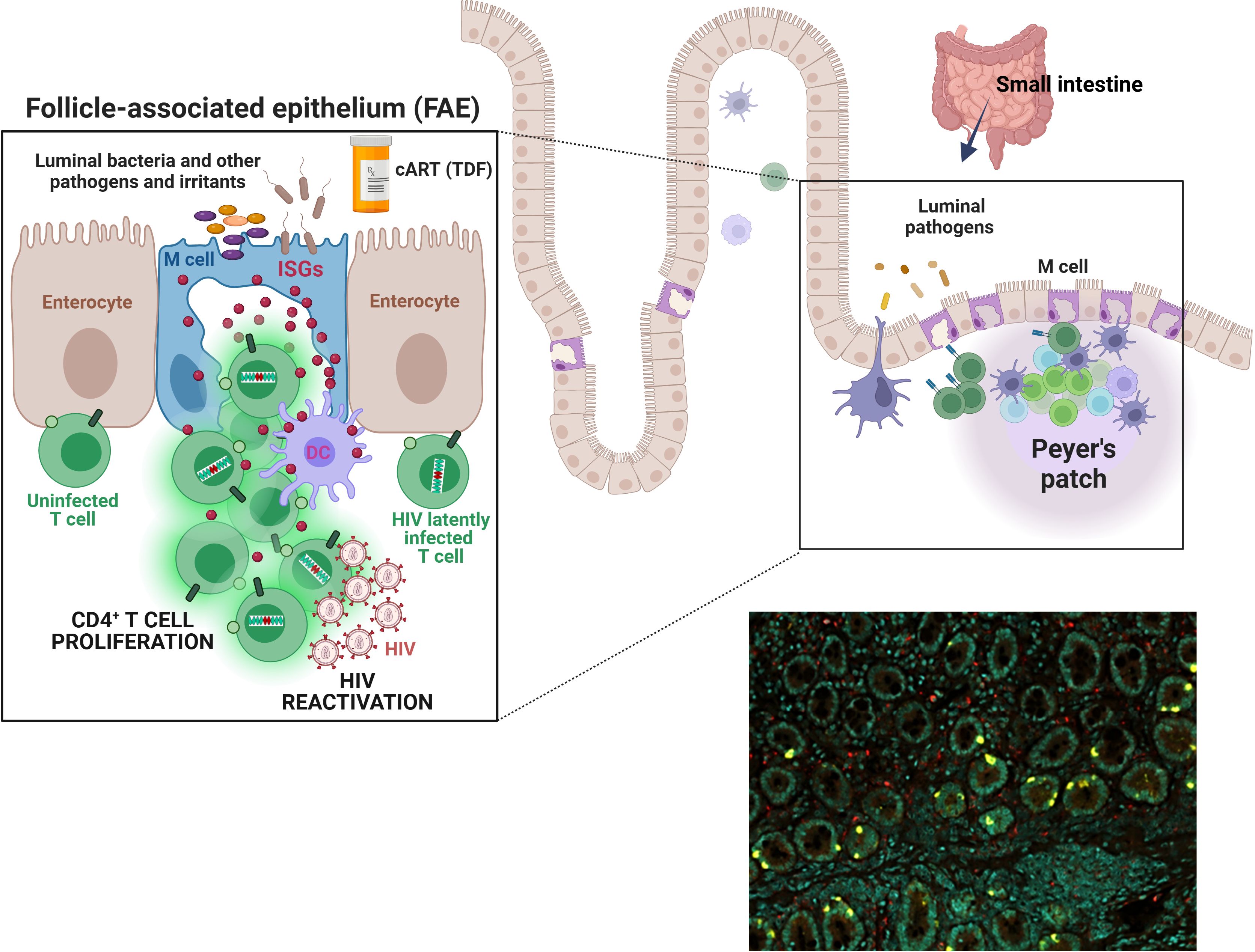
Figure 1. M cells or ISGhigh enterocytes may foster HIV latency and reactivation. Top schema. The epithelium of the intestinal mucosa contains a population of cells expressing extremely high levels of type I/III interferon-stimulated genes (ISGs). Notably, this ISG expression appears independent of direct interferon (IFN) stimulation and also does not coincide with IFN expression by these cells. Co-expression of glycoprotein 2 suggests that many of these enterocytes are microfold cells (M cells). M cells are ~10% of all enterocytes; they are most numerous in the epithelium covering lymphoid follicles (the follicle-associated epithelium, “FAE”) and Peyer’s patches in the small intestine. They transport pathogens and other foreign materials from the lumen to underlying antigen-presenting cells (APCs) to initiate immune responses. They also influence APCs and lymphocytes via cytokines (e.g., IL-1, ISG15) and chemokines (e.g., CCL9 and CXCL16), as well as by expressing HLA-II molecules for direct interaction with T cells. M cells have a large basolateral invagination that amplifies the cell surface and usually contains B and/or T cells. While the significance of ISG15 and other M cell-associated ISGs for the HIV reservoir remains to be investigated, stimulation of type I/III IFN pathways has been shown to play an important role supporting HIV reservoir persistence, likely via T cell proliferation, and HIV-1 reactivation. Our overarching hypothesis is that immunologically hyperactive M cells in the gut create a microenvironment that fosters proliferation of latently infected bystander CD4+ T cells, sporadic HIV reactivation from these cells, or both. Thus, M cells and, more broadly, ISGhigh enterocytes may play a role in HIV persistence and/or viral rebound post-ART cessation. Bottom right image. Immunostaining of a duodenal biopsy for ISG15 (ISGhigh enterocytes, yellow) and CD68 (macrophages, red); DAPI signal denoted in teal. A formalin-fixed paraffin-embedded (FFPE) section was analyzed from a study participant that was exposed to PrEP (a combination of tenofovir disoproxil fumarate and emtricitabine) for two months (16).
2 The gastrointestinal tract, especially the gut-associated lymphoid tissue of the small intestinal tract, contains the largest HIV reservoir
Numerous studies in humans and non-human primates (NHP) have demonstrated that the largest HIV reservoir resides in the GALT. An analysis by Yukl et al. in people living with HIV (PLH) on ART estimated that 83-95% of HIV-infected cells reside in the GIT (40). Likewise, a survey in SIV-infected NHP on ART showed that ~98% of SIV vRNA+ (indicating active transcription and possibility for rebound) cells resided in the GIT (41). This continued low level production of SIV RNA, despite ART, also correlated with the presence of a large pool of SIV DNA+ cells (41). Studies in PLH on ART analyzing only the rectum have consistently found HIV DNA-containing cells (42, 43). The few studies of the upper GIT of PLH consistently identified the small intestinal tract as an important site for harboring HIV DNA+ and RNA+ cells (40, 44–51). HIV DNA (both clade B (46, 48–50) and C (47)) is present at higher levels in the small intestine than the blood. In addition, HIV RNA is more often found in the small intestine than the blood, and the RNA/DNA ratio is higher (40, 49). This site also has higher levels of activated CD4+ T cells than blood, which are suitable targets for infection (40, 49, 50). Thus, a large portion of the HIV reservoir resides in the GIT and HIV reactivation occurs at this site even during treatment.
Additional studies suggest that the HIV reservoir is further compartmentalized within the GIT, although there is some disagreement regarding which section of the GIT harbors the largest reservoir (44–46, 50–52). A recent study by Vellas et al. demonstrated an enrichment of intact proviruses in the ileum and colon compared to the duodenum of virally suppressed PLH (52). Another study identified increased HIV DNA concentrations along the GIT (40), while others found comparable levels in the ileum and rectum (45, 46, 50). With regards to viral transcription, studies agreed that higher levels of HIV RNA are present in the ileum compared to the rectum (40, 45). Further evidence of ongoing productive infection events in the small intestine during ART comes from an ART intensification study, where addition of raltegravir with or without a second antiretroviral drug caused a decrease of unspliced HIV RNA only in the ileum and not in other sites (peripheral blood, duodenum, colon, or rectum) (44). Overall, many studies point to the small intestine as an important and likely functionally unique HIV reservoir site, in particular as a hotspot for viral reactivation.
Several factors could explain the large latent HIV reservoir in the GIT compared to other anatomical sites. The GIT and GALT tissues are seeded rapidly and massively during the initial HIV infection phase, before ART is started (53–55). Compared to blood, a larger proportion of CD4+ T cells in the GIT express the HIV co-receptors CCR5 and CXCR4 and the gut homing receptor α4β7, making them highly susceptible to HIV infection (55–57). Furthermore, GALT contains many B cell follicles, which have been characterized as HIV “sanctuary” sites due to CD8+ T cell depletion, enabling continued productive infection of T follicular helper cells (58). Lastly, once ART is started, some areas of the GIT may experience incomplete tissue penetration of ART drugs (59, 60).
3 GALT immunological function
Here, we review GALT-specific cell types and immunological functions that may contribute to maintaining the HIV reservoir in the GIT. We specifically highlight microfold (M) cells, immunologically active cells that are especially enriched in the small intestine and interact closely with cell types known to harbor latent HIV.
GALT is distributed throughout the GIT and consists of multi-follicular structures (Peyer’s patches, cecal patches, colonic patches) and isolated lymphoid follicles (61–63). The immune structures and cell populations vary substantially along the length of the GIT (reviewed by Mowat et al (61)). Multi-follicular structures are most concentrated in the ileum and consist of germinal centers rich in naïve B cells and follicular dendritic cells surrounded by T cell-rich regions (61, 62, 64). Isolated lymphoid follicles have a similar structure as Peyer’s patches, but are much smaller (a single follicle compared to 10–100 follicles in Peyer’s patches) (63). Unlike multi-follicular structures, isolated lymphoid follicles are distributed along the entire length of the GIT, and their frequency increases 3 fold from the cecum to the rectum (65, 66). T cell populations in follicular structures include naïve CD4+ T cells, central memory CD4+ T cells, FOXP3+ regulatory T cells, and T follicular helper cells (62, 63).
While organized lymphoid structures are specialized for the generation of antigen-specific B cell responses, other immune cells distributed throughout the epithelium and lamina propria are specialized for effector responses and epithelial barrier homeostasis (62, 67–69). Intraepithelial lymphocytes (primarily CD8+) in the intestinal epithelium serve a wide variety of functions, including maintenance of the epithelial barrier, immune regulation, and antigen-specific cytotoxic effector responses (reviewed in (67)). The lamina propria contains CD4+ T cells with effector memory, transitional memory, Th17, and Th22 phenotypes, together with a variety of innate immune cells (62, 68, 69). These CD4+ T cells are of particular relevance due to their susceptibility to HIV infection and ability to harbor latent provirus. The differentiation of these and other immune cell types in the GIT are strongly influenced by dietary components like vitamin A and aryl hydrocarbon receptor ligands and by commensal microbiota and their metabolites (e.g., short chain fatty acids) (70–72).
The epithelium overlying GALT lymphoid follicles contains microfold cells (M cells), which are specialized for uptake and transport of luminal antigens (most eminently studied by Dr. Marian Neutra in the 90 and 00s) (17, 73–78). M cells contain a large basolateral invagination that enables close association with mononuclear phagocytes and intraepithelial B and T cells (73, 75). Antigens are sampled via endocytosis or pinocytosis and transported to the basolateral membrane in vesicles (17, 73, 74). M cells express cytokines (e.g., IL-1 (79), ISG15 (16)) and chemokines (e.g., CCL9 (80) and CXCL16 (81)) to recruit lymphocytes and leukocytes to the basolateral pocket. Some studies also suggest that M cells can express HLA-II molecules (82, 83) for direct interaction with T cells.
Conservatively estimated, under healthy conditions, there are 5×109 M cells or M cell-like enterocytes in the human gut (84). Under pro-inflammatory conditions, such as infection or inflammatory bowel disease, their proportion can increase dramatically, either by trans-differentiation from mature enterocytes or de novo differentiation from crypt stem cells (though the exact mechanisms of M cell formation remain unknown) (73, 85–87). For example, Salmonella typhimurium infection causes increased density of M cells. The mechanism is thought to be via a bacterial effector protein activating RANKL expression in intestinal epithelial cells and inducing epithelial to mesenchymal transition (86).
Furthermore, M cell signaling is influenced by extracellular factors. In a study investigating the effects of two nucleoside/nucleotide reverse transcriptase inhibitors (NRTI)-class drugs in three clinical trials, we found that oral tenofovir disoproxil fumarate (TDF) and emtricitabine (FTC) taken as pre-exposure prophylaxis (PrEP) activated interferon pathways in the intestinal mucosa [Figure 1 and (16)]. The most significantly upregulated genes were IFI6 (IFN α-inducible protein 6), ISG15 (Interferon-Stimulated Protein 15kDa), and MX1 (MX dynamin-like GTPase 1). By co-staining with glycoprotein 2 (GP2) (17) we identified a portion of these cells as mature M cells (16). Similar ISG-expressing enterocytes have been described by others (88–90), with ISG expression modulated by inflammatory conditions like Crohn’s disease, ulcerative colitis, and environmental enteropathy (88, 89).
4 Dichotomous functions of IFN in HIV and the GIT
As indicated in the previous section, our study in people living without HIV demonstrated an immunostimulatory effect of the NRTIs TDF and FTC, which are commonly used as part of ART. Differential gene expression analysis revealed that 13 genes were significantly induced when comparing pretreatment baseline to 60 days of daily oral TDF/FTC PrEP by microarrays (16). Seven of these 13 genes (IFI27, IFI6, IFIT1, ISG15, RSAD2, MX1, and OAS1) are members of the Gene Ontology biological process “type I IFN signaling pathway”; four of the other 6 are known to be induced by type I IFN (DDX60, SAMD9, IFI27L1 and HERC6). Thus, drugs from the NRTI class, which are mainstay ART components, may play a role in the persistent and largely unexplained immune activation seen in PLH whose HIV infection is otherwise well-controlled. In the paragraphs below, we discuss how IFNs, particularly type I (IFN-α and β) and type III (λ), can have contrasting functions, being protective during early events of viral infection and detrimental if their expression is dysregulated in chronic infection.
The antiviral activity of type I IFN is beneficial in acute HIV/SIV infection (91, 92). IFN-α2a administration during early SIV infection in rhesus macaques led to upregulation of ISGs (MX1, MX2, OAS2, IRF7) and a delay in systemic infection (93). Another study in rhesus macaques demonstrated that IL-21 therapy (known to induce NK cell proliferation and maturation (94)) followed by IFNα therapy resulted in a smaller SIV reservoir and delayed viral rebound during ART interruption (95). Meanwhile, in humans, delivery of pegylated IFN-α2b in combination with ART resulted in decreased GALT HIV RNA+ cells and blood HIV DNA+ cells. These changes correlated with increased GALT NK and T cell activation and upregulation of genes related to NK cell mediated immunity and IFN signaling (96, 97). In individuals coinfected with hepatitis C virus and HIV, immunotherapy with pegylated IFN-α2a further reduced proviral HIV DNA levels, which correlated with NK antiviral function (98–100). At the molecular level, type I IFN inhibits HIV-1 virus release through ISG15-mediated inhibition of ubiquitylation of the HIV-1 Gag protein (101), and it induces a number of directly-acting HIV restriction factors, e.g., MX1, TRIM5α, tetherin, and APOBEC3G (102–104).
In the context of chronic HIV infection, stimulation of type I/III IFN pathways can exacerbate the infection rather than clear it (105–107). Two recent studies in a humanized mouse model of chronic HIV infection showed that disrupting IFN-I/III pathways by blocking the IFN-α/β receptor 1 led to less immune activation, a lower HIV reservoir in lymphoid tissues, and delayed HIV rebound following ART interruption (21, 22). These studies were highlighted in a commentary by Deeks et al. titled “The interferon paradox: can inhibiting an antiviral mechanism advance an HIV cure?” (23). Likewise, in an NHP model of chronic ART-treated SIV infection, blockade of IFNα resulted in SIV reservoir reduction and better clinical outcomes during ART interruption (108). In PLH, chronic activation of IFN pathways has been associated with worse disease outcomes (20), partly driven by immune suppression and CD8+ T cell exhaustion and senescence (19, 107, 109). This dichotomous role of IFN in HIV pathogenesis is especially illustrated by studies comparing nonpathogenic to pathogenic SIV. Natural hosts like African Green Monkeys generate robust interferon responses, but the responses rapidly diminish following acute infection and these animals have minimal pathogenic sequelae (110). In contrast, chronic IFN activation occurs in animals like rhesus macaques, which have pathogenic SIV infection (111).
Several molecular mechanisms may underlie the deleterious effect of type I/III pathway activation on HIV persistence and reactivation. Type I IFN drives bystander T cell proliferation (112), which likely includes latently infected cells and thus may contribute to reservoir maintenance. It also facilitates the establishment of viral latency in monocyte-derived macrophages in vitro through the formation of inaccessible chromatin in the HIV provirus (113). In addition, IFN-α can reactivate HIV-1 from latently infected CD4+ T cells, potentially via STAT5 phosphorylation (39).
Most studies of IFN’s antiviral or deleterious effects have been performed with blood immune cells. However, the GIT is where preferential acute HIV-1/SIV replication, massive CD4+ T cell depletion, and microbial translocation occurs (53, 55), and where the largest HIV reservoir in the body resides (40, 41). In a study comparing long term non-progressors to people with high HIV viral loads, HIV-specific IFNγ secretion from GIT CD8+ and CD4+ lymphocytes was higher in the non-progressor group (114). This suggests a protective antiviral function of IFNγ in the GIT. During HIV/SIV infection, plasmacytoid dendritic cells (pDCs) upregulate β7-integrin expression, resulting in accumulation in the GIT (115, 116). pDCs produce large amounts of type I IFN during HIV/SIV infection, but this activity is reduced in natural SIV hosts (117). Blockade of α4β7 reduced the pDC population and immune activation in the colorectum of SIV-infected rhesus macaques (116). Together, these studies suggest that type I IFN produced by pDCs contributes to chronic immune activation in the GIT.
Dendritic cells and intra-epithelial CD45+ leukocytes also participate in type I/III IFN signaling in the GIT. In the absence of infection, commensal bacteria stimulate IFN secretion from these cells, which then leads to an ISG-mediated antiviral state in intestinal epithelial cells (118, 119). In acute HIV infection, CD4+ T cell depletion in the GIT leads to epithelial barrier dysfunction and microbial dysbiosis that persists even after stable ART (120–122). It is unknown how this persistent disruption of the gut epithelium affects IFN signaling, but in PLH on ART, gut ISG levels positively correlated with gut HIV-1 RNA and markers of immune activation, microbial translocation, and inflammation (124). In the next sections, we will speculate on the potential importance of the interaction between ISGhigh enterocytes such as M cells and the HIV reservoir.
5 Potential role of ISG-expressing enterocytes in GALT HIV reservoir maintenance and rebound
The effect of HIV infection on IFN-signaling in GIT enterocytes is unknown, however GIT enterocyte ISG expression has been shown to be increased by small molecule drugs or autoimmune disease. As mentioned in Section 4, we observed an increase in the number of rectal and duodenal enterocytes expressing ISGs after 2 months of TDF/FTC PrEP (16). In vitro experiments have also demonstrated that nucleotide analogues can stimulate dose-dependent secretion of type III IFN (IFN-λ3) in GIT epithelial cells (123). To further validate the potential for ISG upregulation in GIT enterocytes, we explored scRNA-Seq datasets from Kummerlowe et al (89) and Smillie et al (90), in the Broad Institute Single Cell Portal. These datasets were identified based on species (Homo sapiens), organ (gastrointestinal tract), cells (microfold cells), and genes (ISGs from (16)) of interest. In both datasets, we identified a subset of enterocytes in the duodenum that co-express high amounts of ISGs (ISG15, IFI27, IFI6, MX1, IFIT1, etc.) (Figure 2a, denoted with a red arrow) but, notably, not interferons or interferon receptors (IFNL1, IFNL2, IFNL3, IFNG, IFNE, IFN-alpha receptor 1 or 2, or IFN-lambda receptor IFNLR1) (Figure 2b). Results from these studies align well with our tenofovir study (16), in which type I IFNs (IFN-α and -β) and type III IFNs (IFN-λ1–4) were not detectable. In Kummerlowe et al (89) and Smillie et al (90), the ISGhigh subset of cells did not express GP2, suggesting they are not mature M cells but another type of enterocyte (17). Although both studies included samples from participants with gastrointestinal disease, expression of the same ISGs in enterocytes from our TDF/FTC PrEP study suggests that this pattern of ISG expression is not specific to gastrointestinal disease and that further study of enterocyte ISG expression in HIV is warranted.
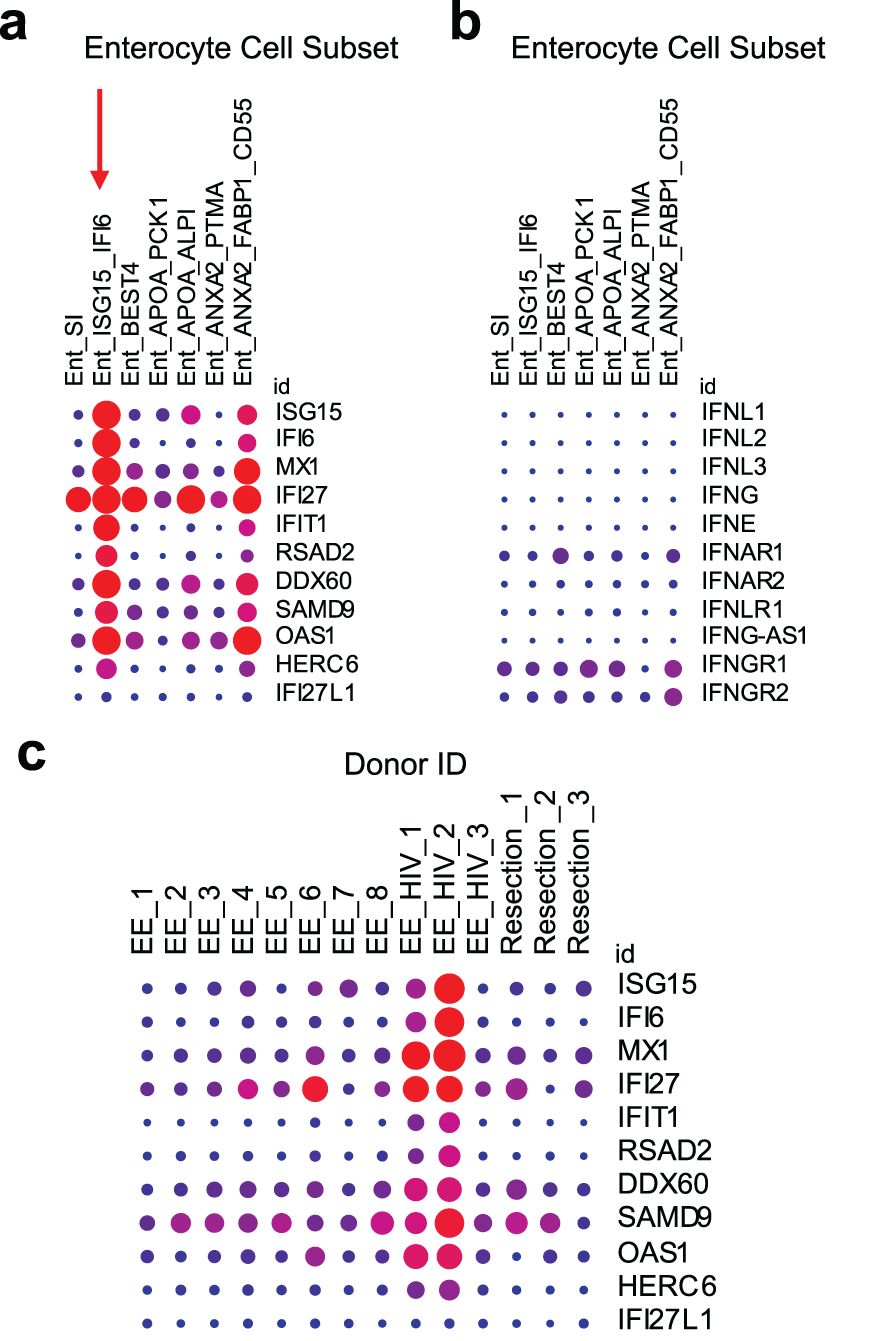
Figure 2. ISG expression is elevated in an enterocyte subset of the small intestine and in HIV infection. Kummerlowe et al (89) performed single cell RNA sequencing of small intestine biopsies from people with environmental enteropathy (EE) (including 3 adults living with HIV, EE_HIV) and people without environmental enteropathy (Resection). (a) Exploration of ISG expression in the data set revealed an enterocyte subset (Ent_ISG15_IFI6) with high ISG expression (denoted with a vertical red arrow), but (b) little or no IFN or IFN receptor expression. (c) A similar pattern of high ISG expression was identified in study participants living with HIV. The data was accessed and the figure was generated using the Single Cell Portal from the Broad Institute (170). In all plots, the dot size represents the percent of the cell population expressing the gene, and the color represents the scaled mean expression from 0 (blue) to 1 (red) across all cell subsets (a, b) or all study participants (c). These plots were generated automatically after searching for the ISGs of interest. Experimental and data analysis details are available in the original paper (89).
The Kummerlowe study also showed an overall increase in ISG expression (Figure 2c) and specifically an increased fractional abundance of the ISGhigh enterocyte subset in ART-treated HIV infection (Supplementary Figure S7 in Kummerlowe et al (89)). The increase in this ISGhigh enterocyte population during chronic HIV infection suggests a role for these cells in HIV persistence and reactivation. It is unknown how HIV-infected cells respond to M cell-derived ISGs and if this interaction can drive HIV reservoir persistence and/or reactivation. Briefly, we will discuss potential effects of ISGs on the HIV reservoir in the GIT using ISG15 as an example.
5.1 ISG15: an M cell-derived ISG with the potential to enhance latent HIV-infected cell proliferation and HIV reactivation
ISG15 is one of the most strongly (125) and rapidly induced (126) ISGs. It has both intracellular innate immune and secreted cytokine-like functions (127, 128). ISG15 is a member of the ubiquitin family and is induced by viral and bacterial infections (129–131), and also directly by IFNs (132, 133). Intracellularly, it binds covalently to target proteins through a process called ISGylation, which serves a key role in innate immunity, specifically inhibiting viral infection and viral release (128). As a cytokine, ISG15 induces lymphocyte proliferation, IFN-γ production, and neutrophil chemotaxis (134, 135). Soluble ISG15 can also stimulate a strong release of pro-inflammatory cytokines such as IL-6, TNF-α, and IL1β (88). These effects are triggered by ISG15 binding the integrin receptor lymphocyte function-associated antigen 1 (LFA1) on NK cells and T cells (136). The specific functions of ISG15 in the GALT and in M cells are unknown.
Regarding HIV-1 pathogenesis, several effects of ISG15 have been reported. ISG15 is upregulated in dendritic cells and macrophages in response to HIV-1 provirus (137), and in PLH, ISG15 mRNA levels in PBMCs correlated with HIV-1 viral load and markers of worse disease outcome (138). ISG15 inhibits HIV-1 virus release by inhibiting ubiquitination of the HIV-1 Gag protein (101). Conversely, intracellular ISG15 was also shown to increase HIV-1 replication in primary CD4+ T cells (139). This could occur via ISG15-mediated stabilization of USP18, a negative regulator of JAK-STAT signaling (140, 141). Overall, despite ISG15 now being intently studied, there is still relatively little known regarding its role in the GIT, HIV infection, and HIV latency.
Taken together, ISG15 expression by enterocytes may affect HIV persistence and reactivation in two ways. First, enterocyte-secreted ISG15 may promote proliferation of neighboring T cells, some of which could be latently infected, thus maintaining or growing the size of the reservoir. It may also recruit and activate CD4+ T cells, which are targets for infection. Second, ISG15 may play a role in viral reactivation of latently infected T cells by stimulating the release of pro-inflammatory cytokines, which then reactivate HIV. Thus, despite its direct intracellular inhibitory effects on HIV, ISG15 expression by enterocytes may drive persistence of the latent HIV reservoir as well as viral reactivation from T cells. Similar effects are likely to result from other ISGs produced by enterocytes.
5.2 IFN-independent induction of ISGs
As mentioned above, ISGhigh enterocytes appear to express few IFN receptors, which suggests that their ISG expression may be independent of IFN stimulation. Transcriptional regulation of ISGs can be activated by IFNs via the classical JAK-STAT pathway or through non-canonical IFN-independent pathways (reviewed in (142)). These non-canonical signaling pathways, including the activation of mitogen-activated protein kinases (MAPKs), mammalian target of rapamycin (mTOR), protein kinase C (PKC), IRF3, or NF-κB, can be activated by cellular stress (e.g., heat shock, DNA damage, oxidative stress) or viral infections (18, 143).
A recent study suggested that ISG induction occurs via NF-κB signaling in an enteroid model of M cells. Ding et al. developed a culture system to generate glycoprotein 2 (GP2) positive M cells in human ileal enteroids using a variety of differentiation factors (RANKL, retinoic acid, and lymphotoxin α2β1) (144). Transcriptomic analysis showed that this lymphotoxin-mediated, IFN protein-independent signaling induced upregulation of several ISGs (IFI6, IFI44, IFITM1, IFIT1, RSAD2) in enteroids with induced M cells (144). This ISG expression prevented rotavirus infection specifically in GP2-positive M cells in the enteroid model (144).
A second IFN-independent mechanism of ISG induction occurs via pattern recognition receptors (PRRs). M cells express PRRs such as Toll-like receptors (TLRs) (145) and nucleotide-binding oligomerization domain-containing proteins (NODs) (146). These receptors recognize microbial molecular motifs, and can trigger the activation of signaling pathways that converge in ISG induction without the need for IFNs. The TLR3 ligand poly(I:C) induced expression of pSTAT1, IRF9, and free ISG15 independently of autocrine or paracrine IFN signaling in an organoid model (88). ISG15 is induced directly by HIV-1 provirus in CD4+ T cells, macrophages, and dendritic cells via MDA5 (147), a RIG-I like receptor that can be expressed by enterocytes (89).
These pathways could be involved in the apparent constitutive ISG expression we observed in enterocytes (16), given their constant exposure to and sampling of the intestinal lumen.
As a caveat, while the ISG-expressing subset of enterocytes did not strongly express IFN or IFN receptors in the studies described at the beginning of Section 5, there were other enterocyte subsets expressing IFN-α receptor 1 and IFN-γ receptors 1 and 2 (89, 90). Additionally, the absence of type I/III IFNs in our tenofovir study (16) could be due to the low sensitivity of the microarray used. Therefore, these previous studies do not exclude the possibility that IFN is involved upstream of the ISG expressing enterocytes.
6 Clinical implications for HIV cure strategies
Understanding how M cells affect the HIV reservoir may enable us to improve experimental HIV cure interventions. Two prominent approaches to curing HIV are “shock and kill” (“sterilizing cure” (148)) and “block and lock” (“functional cure”) (149, 150). “Shock and kill” (also named “kill and kill” or “activation-elimination”) induces HIV reactivation with cytokines and latency reversing agents (LRAs). These LRAs include protein kinase C (PKC) modulators, mitochondrial-derived activators of caspases (SMAC) mimetics, BET-bromodomain inhibitors, histone deacetylase (HDAC) inhibitors, and others (reviewed in (151)). In theory, reactivated cells should be eliminated by viral cytopathic effects or the immune system, but in vitro data and clinical trials have shown that latency reversal alone does not effectively decrease the size of the HIV reservoir (152–155). A combination of LRAs may be more effective (156–158), but such approaches can be toxic. It is possible that studying the immune modulatory effects of ISGhigh enterocytes and M cells could reveal novel and less toxic approaches for latency reversal.
The “block and lock” functional cure strategy (149) is based on inducing deep latency in the HIV reservoir by using latency-promoting agents (LPAs) such as cortistatin A (159, 160), Janus kinase (Jak)-STAT inhibitors (161), and bromodomain-containing protein 4 (BRD4) modulators (162). This approach aims to permanently silence all latent proviruses, preventing the transcription of replication-competent proviruses and blocking actively replicating viruses. Thus, LPAs could maintain functional cure following ART interruption (159, 163). It remains unknown whether immune activation, e.g., by other infections, antagonizes this strategy. Further studies of ISGhigh enterocytes like M cells could be critical to define whether LPAs can overcome endogenous signals that trigger HIV reactivation.
7 Conclusions and next steps
In summary, in this review we argue that HIV reservoir persistence and reactivation in the gut, especially the small intestine, is mediated by ISG expression in M cells or M cell-like cells. Our argument is based on three key facts (1): the GIT contains the majority of the cells in the HIV reservoir (40, 41); (2) microfold (M) cells are uniquely enriched in the mucosa of the small intestine, interact closely with T cells and other mucosal leukocytes (73–75), and express extremely high levels of ISGs (16, 88–90); and (3) IFN signaling can enhance T cell proliferation and HIV reactivation (21, 22, 39, 112). Thus, M cells, or broadly ISGhigh enterocytes, may foster a microenvironment that is especially conducive to maintaining the latent HIV reservoir and/or allowing HIV reactivation in adjacent HIV-infected T cells or macrophages.
M cells are key to the immune environment of the gut. However, their isolation and ex vivo culture is challenging, and there are no available immortalized M-cell lines. Animal models are also of limited utility because M cells are highly variable across species (164, 165). Single-cell transcriptomic data from M cells have been collected from dissociated tissues (89, 90, 166, 167), confirming their high ISG expression, but not yet within their spatial context in situ. Similarly, the effects of M cells on neighboring immune cells in the GALT have not yet been studied because until recently the respective methods had not been available. Today, with the advent of single-cell spatial multiomics in situ (168, 169), this limitation has been overcome. Jointly mapping genomic, epigenomic, transcriptomic, proteomic and metabolic profiles from single cells in their spatial context will shed light on these specialized enterocytes in health and disease. Spatial analyses will be able to address very specific functional questions, namely how T cells and macrophages respond to the influence of neighboring M cells. This may lead to a deeper understanding of HIV latency in the gut, as well as, more broadly, the pathogenesis of enteric infections and autoimmune disorders, and the design of oral vaccines.
Author contributions
RC: Data curation, Investigation, Writing – original draft, Writing – review & editing. SH: Investigation, Writing – review & editing. FH: Conceptualization, Funding acquisition, Project administration, Resources, Writing – review & editing. GG: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the following grants from the National Institutes of Health: R01 AI184122 (NIAID), KL2 TR002317 (NCATS), P30 AI027757 (NIAID), and R01 AI116292 (NIAID). GG also received support from the Royalty Research Fund (RRF) from the University of Washington. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
1. Kalidasan V, Theva Das K. Lessons learned from failures and success stories of HIV breakthroughs: are we getting closer to an HIV cure? Front Microbiol. (2020) 11:46. doi: 10.3389/fmicb.2020.00046
2. Ho DD, Moudgil T, Alam M. Quantitation of human immunodeficiency virus type 1 in the blood of infected persons. N Engl J Med. (1989) 321:1621–5. doi: 10.1056/NEJM198912143212401
3. Pierson T, McArthur J, Siliciano RF. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol. (2000) 18:665–708. doi: 10.1146/annurev.immunol.18.1.665
4. Chun TW, Justement JS, Murray D, Hallahan CW, Maenza J, Collier AC, et al. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: implications for eradication. AIDS. (2010) 24:2803–8. doi: 10.1097/QAD.0b013e328340a239
5. Henrich TJ, Hatano H, Bacon O, Hogan LE, Rutishauser R, Hill A, et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PloS Med. (2017) 14:e1002417. doi: 10.1371/journal.pmed.1002417
6. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. (1997) 94:13193–7. doi: 10.1073/pnas.94.24.13193
7. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. (1997) 278:1295–300. doi: 10.1126/science.278.5341.1295
8. Bitnun A, Samson L, Chun TW, Kakkar F, Brophy J, Murray D, et al. Early initiation of combination antiretroviral therapy in HIV-1-infected newborns can achieve sustained virologic suppression with low frequency of CD4+ T cells carrying HIV in peripheral blood. Clin Infect Dis. (2014) 59:1012–9. doi: 10.1093/cid/ciu432
9. Giacomet V, Trabattoni D, Zanchetta N, Biasin M, Gismondo M, Clerici M, et al. No cure of HIV infection in a child despite early treatment and apparent viral clearance. Lancet. (2014) 384:1320. doi: 10.1016/S0140-6736(14)61405-7
10. Vanhamel J, Bruggemans A, Debyser Z. Establishment of latent HIV-1 reservoirs: what do we really know? J Virus Erad. (2019) 5:3–9. doi: 10.1016/S2055-6640(20)30275-2
11. Chaillon A, Gianella S, Dellicour S, Rawlings SA, Schlub TE, De Oliveira MF, et al. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J Clin Invest. (2020) 130:1699–712. doi: 10.1172/JCI134815
12. De Scheerder MA, Vrancken B, Dellicour S, Schlub T, Lee E, Shao W, et al. HIV rebound is predominantly fueled by genetically identical viral expansions from diverse reservoirs. Cell Host Microbe. (2019) 26:347–58.e7. doi: 10.1016/j.chom.2019.08.003
13. Rothenberger MK, Keele BF, Wietgrefe SW, Fletcher CV, Beilman GJ, Chipman JG, et al. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc Natl Acad Sci U S A. (2015) 112:E1126–34. doi: 10.1073/pnas.1414926112
14. Banga R, Perreau M. The multifaceted nature of HIV tissue reservoirs. Curr Opin HIV AIDS. (2024) 19:116–23. doi: 10.1097/COH.0000000000000851
15. Moron-Lopez S, Xie G, Kim P, Siegel DA, Lee S, Wong JK, et al. Tissue-specific differences in HIV DNA levels and mechanisms that govern HIV transcription in blood, gut, genital tract and liver in ART-treated women. J Int AIDS Soc. (2021) 24:e25738. doi: 10.1002/jia2.25738
16. Hughes SM, Levy CN, Calienes FL, Stekler JD, Pandey U, Vojtech L, et al. Treatment with commonly used antiretroviral drugs induces a type I/III interferon signature in the gut in the absence of HIV infection. Cell Rep Med. (2020) 1:100096. doi: 10.1016/j.xcrm.2020.100096
17. Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature. (2009) 462:226–30. doi: 10.1038/nature08529
18. Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q. Transcriptional regulation of antiviral interferon-stimulated genes. Trends Microbiol. (2017) 25:573–84. doi: 10.1016/j.tim.2017.01.001
19. Sedaghat AR, German J, Teslovich TM, CoFrancesco J, Jie CC, Talbot CC, et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J Virol. (2008) 82:1870–83. doi: 10.1128/JVI.02228-07
20. Fernandez S, Tanaskovic S, Helbig K, Rajasuriar R, Kramski M, Murray JM, et al. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J Infect Dis. (2011) 204:1927–35. doi: 10.1093/infdis/jir659
21. Cheng L, Ma J, Li J, Li D, Li G, Li F, et al. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J Clin Invest. (2017) 127:269–79. doi: 10.1172/JCI90745
22. Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Invest. (2017) 127:260–8. doi: 10.1172/JCI89488
23. Deeks SG, Odorizzi PM, Sekaly RP. The interferon paradox: can inhibiting an antiviral mechanism advance an HIV cure? J Clin Invest. (2017) 127:103–5. doi: 10.1172/JCI91916
24. Reeves DB, Duke ER, Wagner TA, Palmer SE, Spivak AM, Schiffer JT. A majority of HIV persistence during antiretroviral therapy is due to infected cell proliferation. Nat Commun. (2018) 9:4811. doi: 10.1038/s41467-018-06843-5
25. Reeves DB, Duke ER, Hughes SM, Prlic M, Hladik F, Schiffer JT. Anti-proliferative therapy for HIV cure: a compound interest approach. Sci reports. (2017) 7:4011. doi: 10.1038/s41598-017-04160-3
26. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. (2009) 15:893–900. doi: 10.1038/nm.1972
27. Bui JK, Sobolewski MD, Keele BF, Spindler J, Musick A, Wiegand A, et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PloS Pathog. (2017) 13:e1006283. doi: 10.1371/journal.ppat.1006283
28. Wang Z, Gurule EE, Brennan TP, Gerold JM, Kwon KJ, Hosmane NN, et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc Natl Acad Sci U S A. (2018) 115:E2575–e84. doi: 10.1073/pnas.1720665115
29. Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, et al. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. (2014) 345:570–3. doi: 10.1126/science.1256304
30. Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. (2014) 345:179–83. doi: 10.1126/science.1254194
31. Simonetti FR, Sobolewski MD, Fyne E, Shao W, Spindler J, Hattori J, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci U S A. (2016) 113:1883–8. doi: 10.1073/pnas.1522675113
32. Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J Exp Med. (2017) 214(4):959–72. doi: 10.1084/jem.20170193
33. Bull ME, Learn GH, McElhone S, Hitti J, Lockhart D, Holte S, et al. Monotypic human immunodeficiency virus type 1 genotypes across the uterine cervix and in blood suggest proliferation of cells with provirus. J Virol. (2009) 83:6020–8. doi: 10.1128/JVI.02664-08
34. Abana CO, Pilkinton MA, Gaudieri S, Chopra A, McDonnell WJ, Wanjalla C, et al. Cytomegalovirus (CMV) epitope-specific CD4. J Immunol. (2017) 199:3187–201. doi: 10.4049/jimmunol.1700851
35. Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PloS Pathog. (2011) 7:e1002288. doi: 10.1371/journal.ppat.1002288
36. Gantner P, Pagliuzza A, Pardons M, Ramgopal M, Routy JP, Fromentin R, et al. Single-cell TCR sequencing reveals phenotypically diverse clonally expanded cells harboring inducible HIV proviruses during ART. Nat Commun. (2020) 11:4089. doi: 10.1038/s41467-020-17898-8
37. Vandergeeten C, Fromentin R, DaFonseca S, Lawani MB, Sereti I, Lederman MM, et al. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood. (2013) 121:4321–9. doi: 10.1182/blood-2012-11-465625
38. Cohn LB, Chomont N, Deeks SG. The biology of the HIV-1 latent reservoir and implications for cure strategies. Cell Host Microbe. (2020) 27:519–30. doi: 10.1016/j.chom.2020.03.014
39. Van der Sluis RM, Zerbato JM, Rhodes JW, Pascoe RD, Solomon A, Kumar NA, et al. Diverse effects of interferon alpha on the establishment and reversal of HIV latency. PloS Pathog. (2020) 16:e1008151. doi: 10.1371/journal.ppat.1008151
40. Yukl SA, Gianella S, Sinclair E, Epling L, Li Q, Duan L, et al. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J Infect Dis. (2010) 202:1553–61. doi: 10.1086/656722
41. Estes JD, Kityo C, Ssali F, Swainson L, Makamdop KN, Del Prete GQ, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. (2017) 23:1271–6. doi: 10.1038/nm.4411
42. Rueda CM, Velilla PA, Chougnet CA, Montoya CJ, Rugeles MT. HIV-induced T-cell activation/exhaustion in rectal mucosa is controlled only partially by antiretroviral treatment. PloS One. (2012) 7:e30307. doi: 10.1371/journal.pone.0030307
43. Hatano H, Somsouk M, Sinclair E, Harvill K, Gilman L, Cohen M, et al. Comparison of HIV DNA and RNA in gut-associated lymphoid tissue of HIV-infected controllers and noncontrollers. AIDS. (2013) 27:2255–60. doi: 10.1097/QAD.0b013e328362692f
44. Yukl SA, Shergill AK, McQuaid K, Gianella S, Lampiris H, Hare CB, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS. (2010) 24:2451–60. doi: 10.1097/QAD.0b013e32833ef7bb
45. Yukl SA, Shergill AK, Ho T, Killian M, Girling V, Epling L, et al. The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: implications for viral persistence. J Infect Dis. (2013) 208:1212–20. doi: 10.1093/infdis/jit308
46. Horn C, Augustin M, Ercanoglu MS, Heger E, Knops E, Bondet V, et al. HIV DNA reservoir and elevated PD-1 expression of CD4 T-cell subsets particularly persist in the terminal ileum of HIV-positive patients despite cART. HIV Med. (2021) 22:397–408. doi: 10.1111/hiv.13031
47. Liu Z, Julius P, Kang G, West JT, Wood C. Subtype C HIV-1 reservoirs throughout the body in ART-suppressed individuals. JCI Insight. (2022) 7:e162604. doi: 10.1172/jci.insight.162604
48. Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis. (2008) 197:714–20. doi: 10.1086/527324
49. Morón-López S, Navarro J, Jimenez M, Rutsaert S, Urrea V, Puertas MC, et al. Switching from a protease inhibitor-based regimen to a dolutegravir-based regimen: A randomized clinical trial to determine the effect on peripheral blood and ileum biopsies from antiretroviral therapy-suppressed human immunodeficiency virus-infected individuals. Clin Infect Dis. (2019) 69:1320–8. doi: 10.1093/cid/ciy1095
50. Augustin M, Horn C, Ercanoglu MS, Sandaradura de Silva U, Bondet V, Suarez I, et al. CXCR3 expression pattern on CD4+ T cells and IP-10 levels with regard to the HIV-1 reservoir in the gut-associated lymphatic tissue. Pathogens. (2022) 11:483. doi: 10.3390/pathogens11040483
51. Augustin M, Horn C, Ercanoglu MS, Bondet V, de Silva US, Suarez I, et al. From gut to blood: redistribution of zonulin in people living with HIV. Biomedicines. (2024) 12:2316. doi: 10.3390/biomedicines12102316
52. Vellas C, Nayrac M, Collercandy N, Requena M, Jeanne N, Latour J, et al. Intact proviruses are enriched in the colon and associated with PD-1. EBioMedicine. (2023) 100:104954. doi: 10.1016/j.ebiom.2023.104954
53. Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, et al. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. (2004) 200:761–70. doi: 10.1084/jem.20041196
54. Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. (1999) 286:1353–7. doi: 10.1126/science.286.5443.1353
55. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. (2004) 200:749–59. doi: 10.1084/jem.20040874
56. Poles MA, Elliott J, Taing P, Anton PA, Chen IS. A preponderance of CCR5(+) CXCR4(+) mononuclear cells enhances gastrointestinal mucosal susceptibility to human immunodeficiency virus type 1 infection. J Virol. (2001) 75:8390–9. doi: 10.1128/JVI.75.18.8390-8399.2001
57. Cicala C, Martinelli E, McNally JP, Goode DJ, Gopaul R, Hiatt J, et al. The integrin alpha4beta7 forms a complex with cell-surface CD4 and defines a T-cell subset that is highly susceptible to infection by HIV-1. Proc Natl Acad Sci U S A. (2009) 106:20877–82. doi: 10.1073/pnas.0911796106
58. Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. (2015) 21:132–9. doi: 10.1038/nm.3781
59. Asmuth DM, Thompson CG, Chun TW, Ma ZM, Mann S, Sainz T, et al. Tissue pharmacologic and virologic determinants of duodenal and rectal gastrointestinal-associated lymphoid tissue immune reconstitution in HIV-infected patients initiating antiretroviral therapy. J Infect Dis. (2017) 216:813–8. doi: 10.1093/infdis/jix418
60. Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A. (2014) 111:2307–12. doi: 10.1073/pnas.1318249111
61. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. (2014) 14:667–85. doi: 10.1038/nri3738
62. Fenton TM, Jørgensen PB, Niss K, Rubin SJS, Mörbe UM, Riis LB, et al. Immune profiling of human gut-associated lymphoid tissue identifies a role for isolated lymphoid follicles in priming of region-specific immunity. Immunity. (2020) 52:557–70.e6. doi: 10.1016/j.immuni.2020.02.001
63. Mörbe UM, Jørgensen PB, Fenton TM, von Burg N, Riis LB, Spencer J, et al. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. (2021) 14:793–802. doi: 10.1038/s41385-021-00389-4
64. Van Kruiningen HJ, West AB, Freda BJ, Holmes KA. Distribution of Peyer's patches in the distal ileum. Inflammation Bowel Dis. (2002) 8:180–5. doi: 10.1097/00054725-200205000-00004
65. Moghaddami M, Cummins A, Mayrhofer G. Lymphocyte-filled villi: comparison with other lymphoid aggregations in the mucosa of the human small intestine. Gastroenterology. (1998) 115:1414–25. doi: 10.1016/S0016-5085(98)70020-4
66. O'Leary AD, Sweeney EC. Lymphoglandular complexes of the colon: structure and distribution. Histopathology. (1986) 10:267–83. doi: 10.1111/j.1365-2559.1986.tb02481.x
67. Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol. (2011) 11:445–56. doi: 10.1038/nri3007
68. Senda T, Dogra P, Granot T, Furuhashi K, Snyder ME, Carpenter DJ, et al. Microanatomical dissection of human intestinal T-cell immunity reveals site-specific changes in gut-associated lymphoid tissues over life. Mucosal Immunol. (2019) 12:378–89. doi: 10.1038/s41385-018-0110-8
69. Wolff MJ, Leung JM, Davenport M, Poles MA, Cho I, Loke P. TH17, TH22 and Treg cells are enriched in the healthy human cecum. PloS One. (2012) 7:e41373. doi: 10.1371/journal.pone.0041373
70. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. (2007) 317:256–60. doi: 10.1126/science.1145697
71. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. (2011) 13:144–51. doi: 10.1038/ni.2187
72. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
73. Kraehenbuhl JP, Neutra MR. Epithelial M cells: differentiation and function. Annu Rev Cell Dev Biol. (2000) 16:301–32. doi: 10.1146/annurev.cellbio.16.1.301
74. Neutra MR, Mantis NJ, Kraehenbuhl JP. Collaboration of epithelial cells with organized mucosal lymphoid tissues. Nat Immunol. (2001) 2:1004–9. doi: 10.1038/ni1101-1004
75. Amerongen HM, Weltzin R, Farnet CM, Michetti P, Haseltine WA, Neutra MR. Transepithelial transport of HIV-1 by intestinal M cells: a mechanism for transmission of AIDS. J Acquir Immune Defic Syndr (1988). (1991) 4:760–5.
76. Mantis NJ, Cheung MC, Chintalacharuvu KR, Rey J, Corthésy B, Neutra MR. Selective adherence of IgA to murine Peyer's patch M cells: evidence for a novel IgA receptor. J Immunol. (2002) 169:1844–51. doi: 10.4049/jimmunol.169.4.1844
77. Chabot S, Wagner JS, Farrant S, Neutra MR. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol. (2006) 176:4275–83. doi: 10.4049/jimmunol.176.7.4275
78. Chabot SM, Shawi M, Eaves-Pyles T, Neutra MR. Effects of flagellin on the functions of follicle-associated epithelium. J Infect Dis. (2008) 198:907–10. doi: 10.1086/591056
79. Pappo J, Mahlman RT. Follicle epithelial M cells are a source of interleukin-1 in Peyer's patches. Immunology. (1993) 78:505–7.
80. Hase K, Ohshima S, Kawano K, Hashimoto N, Matsumoto K, Saito H, et al. Distinct gene expression profiles characterize cellular phenotypes of follicle-associated epithelium and M cells. DNA Res. (2005) 12:127–37. doi: 10.1093/dnares/12.2.127
81. Hase K, Murakami T, Takatsu H, Shimaoka T, Iimura M, Hamura K, et al. The membrane-bound chemokine CXCL16 expressed on follicle-associated epithelium and M cells mediates lympho-epithelial interaction in GALT. J Immunol. (2006) 176:43–51. doi: 10.4049/jimmunol.176.1.43
82. Nagura H, Ohtani H, Masuda T, Kimura M, Nakamura S. HLA-DR expression on M cells overlying Peyer's patches is a common feature of human small intestine. Acta Pathol Jpn. (1991) 41:818–23. doi: 10.1111/j.1440-1827.1991.tb01624.x
83. Spencer J, Finn T, Isaacson PG. Expression of HLA-DR antigens on epithelium associated with lymphoid tissue in the human gastrointestinal tract. Gut. (1986) 27:153–7. doi: 10.1136/gut.27.2.153
84. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PloS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
85. Bennett KM, Parnell EA, Sanscartier C, Parks S, Chen G, Nair MG, et al. Induction of colonic M cells during intestinal inflammation. Am J Pathol. (2016) 186:1166–79. doi: 10.1016/j.ajpath.2015.12.015
86. Tahoun A, Mahajan S, Paxton E, Malterer G, Donaldson DS, Wang D, et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe. (2012) 12:645–56. doi: 10.1016/j.chom.2012.10.009
87. Borghesi C, Taussig MJ, Nicoletti C. Rapid appearance of M cells after microbial challenge is restricted at the periphery of the follicle-associated epithelium of Peyer's patch. Lab Invest. (1999) 79:1393–401.
88. Østvik AE, Svendsen TD, Granlund AVB, Doseth B, Skovdahl HK, Bakke I, et al. Intestinal epithelial cells express immunomodulatory ISG15 during active ulcerative colitis and crohn's disease. J Crohns Colitis. (2020) 14:920–34. doi: 10.1093/ecco-jcc/jjaa022
89. Kummerlowe C, Mwakamui S, Hughes TK, Mulugeta N, Mudenda V, Besa E, et al. Single-cell profiling of environmental enteropathy reveals signatures of epithelial remodeling and immune activation. Sci Trans Med. (2022) 14:eabi8633. doi: 10.1126/scitranslmed.abi8633
90. Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. (2019) 178:714–30.e22. doi: 10.1016/j.cell.2019.06.029
91. Harper MS, Guo K, Gibbert K, Lee EJ, Dillon SM, Barrett BS, et al. Interferon-α Subtypes in an ex vivo model of acute HIV-1 infection: expression, potency and effector mechanisms. PloS Pathog. (2015) 11:e1005254. doi: 10.1371/journal.ppat.1005254
92. Sutter K, Dickow J, Dittmer U. Interferon α subtypes in HIV infection. Cytokine Growth Factor Rev. (2018) 40:13–8. doi: 10.1016/j.cytogfr.2018.02.002
93. Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. (2014) 511:601–5. doi: 10.1038/nature13554
94. Ojo EO, Sharma AA, Liu R, Moreton S, Checkley-Luttge MA, Gupta K, et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci Rep. (2019) 9:14916. doi: 10.1038/s41598-019-51287-6
95. Harper J, Huot N, Micci L, Tharp G, King C, Rascle P, et al. IL-21 and IFNα therapy rescues terminally differentiated NK cells and limits SIV reservoir in ART-treated macaques. Nat Commun. (2021) 12:2866. doi: 10.1038/s41467-021-23189-7
96. Papasavvas E, Azzoni L, Pagliuzza A, Abdel-Mohsen M, Ross BN, Fair M, et al. Safety, immune, and antiviral effects of pegylated interferon alpha 2b administration in antiretroviral therapy-suppressed individuals: results of pilot clinical trial. AIDS Res Hum Retroviruses. (2021) 37:433–43. doi: 10.1089/aid.2020.0243
97. Papasavvas E, Azzoni L, Kossenkov AV, Dawany N, Morales KH, Fair M, et al. NK response correlates with HIV decrease in pegylated IFN-α2a-treated antiretroviral therapy-suppressed subjects. J Immunol. (2019) 203:705–17. doi: 10.4049/jimmunol.1801511
98. Jiao YM, Weng WJ, Gao QS, Zhu WJ, Cai WP, Li LH, et al. Hepatitis C therapy with interferon-α and ribavirin reduces the CD4 cell count and the total, 2LTR circular and integrated HIV-1 DNA in HIV/HCV co-infected patients. Antiviral Res. (2015) 118:118–22. doi: 10.1016/j.antiviral.2015.03.011
99. Sun H, Buzon MJ, Shaw A, Berg RK, Yu XG, Ferrando-Martinez S, et al. Hepatitis C therapy with interferon-α and ribavirin reduces CD4 T-cell-associated HIV-1 DNA in HIV-1/hepatitis C virus-coinfected patients. J Infect Dis. (2014) 209:1315–20. doi: 10.1093/infdis/jit628
100. Hua S, Vigano S, Tse S, Zhengyu O, Harrington S, Negron J, et al. Pegylated interferon-α-induced natural killer cell activation is associated with human immunodeficiency virus-1 DNA decline in antiretroviral therapy-treated HIV-1/hepatitis C virus-coinfected patients. Clin Infect Dis. (2018) 66:1910–7. doi: 10.1093/cid/cix1111
101. Okumura A, Lu G, Pitha-Rowe I, Pitha PM. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc Natl Acad Sci U S A. (2006) 103:1440–5. doi: 10.1073/pnas.0510518103
102. Goujon C, Malim MH. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J Virol. (2010) 84:9254–66. doi: 10.1128/JVI.00854-10
103. Itell HL, Humes D, Overbaugh J. Several cell-intrinsic effectors drive type I interferon-mediated restriction of HIV-1 in primary CD4. Cell Rep. (2023) 42:112556. doi: 10.1016/j.celrep.2023.112556
104. OhAinle M, Helms L, Vermeire J, Roesch F, Humes D, Basom R, et al. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife. (2018) 7:e39823. doi: 10.7554/eLife.39823
105. Reich NC. Too much of a good thing: Detrimental effects of interferon. Semin Immunol. (2019) 43:101282. doi: 10.1016/j.smim.2019.101282
106. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. (2019) 20:1574–83. doi: 10.1038/s41590-019-0466-2
107. Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, Anderson SA, et al. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc Natl Acad Sci U S A. (2006) 103:7000–5. doi: 10.1073/pnas.0600363103
108. Swainson LA, Sharma AA, Ghneim K, Ribeiro SP, Wilkinson P, Dunham RM, et al. IFN-α blockade during ART-treated SIV infection lowers tissue vDNA, rescues immune function, and improves overall health. JCI Insight. (2022) 7. doi: 10.1172/jci.insight.153046
109. Wang S, Zhang Q, Hui H, Agrawal K, Karris MAY, Rana TM. An atlas of immune cell exhaustion in HIV-infected individuals revealed by single-cell transcriptomics. Emerg Microbes Infect. (2020) 9:2333–47. doi: 10.1080/22221751.2020.1826361
110. Raehtz KD, Barrenäs F, Xu C, Busman-Sahay K, Valentine A, Law L, et al. African green monkeys avoid SIV disease progression by preventing intestinal dysfunction and maintaining mucosal barrier integrity. PloS Pathog. (2020) 16:e1008333. doi: 10.1371/journal.ppat.1008333
111. Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest. (2009) 119:3544–55. doi: 10.1172/JCI40093
112. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. (1996) 272:1947–50. doi: 10.1126/science.272.5270.1947
113. Dickey LL, Martins LJ, Planelles V, Hanley TM. HIV-1-induced type I IFNs promote viral latency in macrophages. J Leukoc Biol. (2022) 112:1343–56. doi: 10.1002/JLB.4MA0422-616R
114. Sankaran S, Guadalupe M, Reay E, George MD, Flamm J, Prindiville T, et al. Gut mucosal T cell responses and gene expression correlate with protection against disease in long-term HIV-1-infected nonprogressors. Proc Natl Acad Sci U S A. (2005) 102:9860–5. doi: 10.1073/pnas.0503463102
115. Lehmann C, Jung N, Förster K, Koch N, Leifeld L, Fischer J, et al. Longitudinal analysis of distribution and function of plasmacytoid dendritic cells in peripheral blood and gut mucosa of HIV infected patients. J Infect Dis. (2014) 209:940–9. doi: 10.1093/infdis/jit612
116. Kwa S, Kannanganat S, Nigam P, Siddiqui M, Shetty RD, Armstrong W, et al. Plasmacytoid dendritic cells are recruited to the colorectum and contribute to immune activation during pathogenic SIV infection in rhesus macaques. Blood. (2011) 118:2763–73. doi: 10.1182/blood-2011-02-339515
117. Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S, et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. (2008) 14:1077–87. doi: 10.1038/nm.1871
118. Stefan KL, Kim MV, Iwasaki A, Kasper DL. Commensal microbiota modulation of natural resistance to virus infection. Cell. (2020) 183:1312–24.e10. doi: 10.1016/j.cell.2020.10.047
119. Van Winkle JA, Constant DA, Li L, Nice TJ. Selective interferon responses of intestinal epithelial cells minimize tumor necrosis factor alpha cytotoxicity. J Virol. (2020) 94:e00603-20. doi: 10.1128/JVI.00603-20
120. Dinh DM, Volpe GE, Duffalo C, Bhalchandra S, Tai AK, Kane AV, et al. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis. (2015) 211:19–27. doi: 10.1093/infdis/jiu409
121. Ancona G, Merlini E, Tincati C, Barassi A, Calcagno A, Augello M, et al. Long-term suppressive cART is not sufficient to restore intestinal permeability and gut microbiota compositional changes. Front Immunol. (2021) 12:639291. doi: 10.3389/fimmu.2021.639291
122. Somsouk M, Estes JD, Deleage C, Dunham RM, Albright R, Inadomi JM, et al. Gut epithelial barrier and systemic inflammation during chronic HIV infection. AIDS. (2015) 29:43–51. doi: 10.1097/QAD.0000000000000511
123. Murata K, Asano M, Matsumoto A, Sugiyama M, Nishida N, Tanaka E, et al. Induction of IFN-lambda3 as an additional effect of nucleotide, not nucleoside, analogues: a new potential target for HBV infection. Gut. (2018) 67:362–71. doi: 10.1136/gutjnl-2016-312653
124. Dillon SM, Guo K, Austin GL, Gianella S, Engen PA, Mutlu EA, et al. A compartmentalized type I interferon response in the gut during chronic HIV-1 infection is associated with immunopathogenesis. AIDS. (2018) 32:1599–611. doi: 10.1097/QAD.0000000000001863
125. Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. (1998) 95:15623–8. doi: 10.1073/pnas.95.26.15623
126. Loeb KR, Haas AL. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J Biol Chem. (1992) 267:7806–13. doi: 10.1016/S0021-9258(18)42585-9
127. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. (2018) 16:423–39. doi: 10.1038/s41579-018-0020-5
128. Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res. (2011) 31:119–30. doi: 10.1089/jir.2010.0110
129. Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. (2001) 20:362–71. doi: 10.1093/emboj/20.3.362
130. Radoshevich L, Impens F, Ribet D, Quereda JJ, Nam Tham T, Nahori MA, et al. ISG15 counteracts Listeria monocytogenes infection. Elife. (2015) 4:e06848. doi: 10.7554/eLife.06848
131. Mackelprang RD, Filali-Mouhim A, Richardson B, Lefebvre F, Katabira E, Ronald A, et al. Upregulation of IFN-stimulated genes persists beyond the transitory broad immunologic changes of acute HIV-1 infection. iScience. (2023) 26:106454. doi: 10.1016/j.isci.2023.106454
132. Korant BD, Blomstrom DC, Jonak GJ, Knight E. Interferon-induced proteins. Purification and characterization of a 15,000-dalton protein from human and bovine cells induced by interferon. J Biol Chem. (1984) 259:14835–9. doi: 10.1016/S0021-9258(17)42679-2
133. Knight E, Cordova B. IFN-induced 15-kDa protein is released from human lymphocytes and monocytes. J Immunol. (1991) 146:2280–4. doi: 10.4049/jimmunol.146.7.2280
134. D'Cunha J, Knight E, Haas AL, Truitt RL, Borden EC. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc Natl Acad Sci U S A. (1996) 93:211–5. doi: 10.1073/pnas.93.1.211
135. Owhashi M, Taoka Y, Ishii K, Nakazawa S, Uemura H, Kambara H. Identification of a ubiquitin family protein as a novel neutrophil chemotactic factor. Biochem Biophys Res Commun. (2003) 309:533–9. doi: 10.1016/j.bbrc.2003.08.038
136. Swaim CD, Scott AF, Canadeo LA, Huibregtse JM. Extracellular ISG15 signals cytokine secretion through the LFA-1 integrin receptor. Mol Cell. (2017) 68:581–90.e5. doi: 10.1016/j.molcel.2017.10.003
137. McCauley SM, Kim K, Nowosielska A, Dauphin A, Yurkovetskiy L, Diehl WE, et al. Intron-containing RNA from the HIV-1 provirus activates type I interferon and inflammatory cytokines. Nat Commun. (2018) 9:5305. doi: 10.1038/s41467-018-07753-2
138. Scagnolari C, Monteleone K, Selvaggi C, Pierangeli A, D'Ettorre G, Mezzaroma I, et al. ISG15 expression correlates with HIV-1 viral load and with factors regulating T cell response. Immunobiology. (2016) 221:282–90. doi: 10.1016/j.imbio.2015.10.007
139. Jurczyszak D, Manganaro L, Buta S, Gruber C, Martin-Fernandez M, Taft J, et al. ISG15 deficiency restricts HIV-1 infection. PloS Pathog. (2022) 18:e1010405. doi: 10.1371/journal.ppat.1010405
140. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. (2015) 517:89–93. doi: 10.1038/nature13801
141. Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun. (2016) 7:11496. doi: 10.1038/ncomms11496
142. Swaraj S, Tripathi S. Interference without interferon: interferon-independent induction of interferon-stimulated genes and its role in cellular innate immunity. mBio. (2024) 15:e0258224. doi: 10.1128/mbio.02582-24
143. Saleiro D, Platanias LC. Interferon signaling in cancer. Non-canonical pathways and control of intracellular immune checkpoints. Semin Immunol. (2019) 43:101299. doi: 10.1016/j.smim.2019.101299
144. Ding S, Song Y, Brulois KF, Pan J, Co JY, Ren L, et al. Retinoic acid and lymphotoxin signaling promote differentiation of human intestinal M cells. Gastroenterology. (2020) 159:214–26.e1. doi: 10.1053/j.gastro.2020.03.053
145. Tyrer P, Foxwell AR, Cripps AW, Apicella MA, Kyd JM. Microbial pattern recognition receptors mediate M-cell uptake of a gram-negative bacterium. Infect Immun. (2006) 74:625–31. doi: 10.1128/IAI.74.1.625-631.2006
146. Lapthorne S, Macsharry J, Scully P, Nally K, Shanahan F. Differential intestinal M-cell gene expression response to gut commensals. Immunology. (2012) 136:312–24. doi: 10.1111/j.1365-2567.2012.03581.x
147. Guney MH, Nagalekshmi K, McCauley SM, Carbone C, Aydemir O, Luban J. IFIH1 (MDA5) is required for innate immune detection of intron-containing RNA expressed from the HIV-1 provirus. Proc Natl Acad Sci U S A. (2024) 121:e2404349121. doi: 10.1073/pnas.2404349121
148. Darcis G, Van Driessche B, Van Lint C. HIV latency: should we shock or lock? Trends Immunol. (2017) 38:217–28. doi: 10.1016/j.it.2016.12.003
149. Vansant G, Bruggemans A, Janssens J, Debyser Z. Block-and-lock strategies to cure HIV infection. Viruses. (2020) 12:84. doi: 10.3390/v12010084
150. Mousseau G, Valente ST. Didehydro-Cortistatin A: a new player in HIV-therapy? Expert Rev Anti Infect Ther. (2016) 14:145–8. doi: 10.1586/14787210.2016.1122525
151. Chou TC, Maggirwar NS, Marsden MD. HIV persistence, latency, and cure approaches: where are we now? Viruses. (2024) 16:1163. doi: 10.3390/v16071163
152. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. (2012) 36:491–501. doi: 10.1016/j.immuni.2012.01.014
153. Delagreverie HM, Delaugerre C, Lewin SR, Deeks SG, Li JZ. Ongoing clinical trials of human immunodeficiency virus latency-reversing and immunomodulatory agents. Open Forum Infect Dis. (2016) 3:ofw189. doi: 10.1093/ofid/ofw189
154. Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. (2013) 155:540–51. doi: 10.1016/j.cell.2013.09.020
155. Kim Y, Anderson JL, Lewin SR. Getting the "Kill" into "Shock and kill": strategies to eliminate latent HIV. Cell Host Microbe. (2018) 23:14–26. doi: 10.1016/j.chom.2017.12.004
156. Reuse S, Calao M, Kabeya K, Guiguen A, Gatot JS, Quivy V, et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PloS One. (2009) 4:e6093. doi: 10.1371/journal.pone.0006093
157. Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, et al. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest. (2015) 125:1901–12. doi: 10.1172/JCI80142
158. Ait-Ammar A, Kula A, Darcis G, Verdikt R, De Wit S, Gautier V, et al. Current status of latency reversing agents facing the heterogeneity of HIV-1 cellular and tissue reservoirs. Front Microbiol. (2019) 10:3060. doi: 10.3389/fmicb.2019.03060
159. Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. The tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio. (2015) 6:e00465. doi: 10.1128/mBio.00465-15
160. Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, Shi J, et al. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell host Microbe. (2012) 12:97–108. doi: 10.1016/j.chom.2012.05.016
161. Gavegnano C, Detorio M, Montero C, Bosque A, Planelles V, SChinazi RF. Ruxolitinib and tofacitinib are potent and selective inhibitors of HIV-1 replication and virus reactivation in vitro. Antimicrob Agents Chemother. (2014) 58:1977–86. doi: 10.1128/AAC.02496-13
162. Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. (2007) 104:13690–5. doi: 10.1073/pnas.0705053104
163. Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, et al. In vivo suppression of HIV rebound by didehydro-cortistatin A, a "Block-and-lock" Strategy for HIV-1 treatment. Cell Rep. (2017) 21:600–11. doi: 10.1016/j.celrep.2017.09.080
164. Jepson MA, Clark MA, Foster N, Mason CM, Bennett MK, Simmons NL, et al. Targeting to intestinal M cells. J Anat. (1996) 189:507–16.
165. Brayden DJ, Baird AW. Microparticle vaccine approaches to stimulate mucosal immunisation. Microbes Infect. (2001) 3:867–76. doi: 10.1016/S1286-4579(01)01445-9
166. Burclaff J, Bliton RJ, Breau KA, Ok MT, Gomez-Martinez I, Ranek JS, et al. A proximal-to-distal survey of healthy adult human small intestine and colon epithelium by single-cell transcriptomics. Cell Mol Gastroenterol Hepatol. (2022) 13:1554–89. doi: 10.1016/j.jcmgh.2022.02.007
167. Elmentaite R, Kumasaka N, Roberts K, Fleming A, Dann E, King HW, et al. Cells of the human intestinal tract mapped across space and time. Nature. (2021) 597:250–5. doi: 10.1038/s41586-021-03852-1
168. Marx V. Method of the Year: spatially resolved transcriptomics. Nat Methods. (2021) 18:9–14. doi: 10.1038/s41592-020-01033-y
169. He S, Bhatt R, Brown C, Brown EA, Buhr DL, Chantranuvatana K, et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol. (2022) 40:1794–806. doi: 10.1038/s41587-022-01483-z
Keywords: HIV latency, interferon, enterocytes, microfold cell (M-cell), interferon stimulated gene (ISG)
Citation: Creighton RL, Hughes SM, Hladik F and Gornalusse GG (2025) The intestinal interferon system and specialized enterocytes as putative drivers of HIV latency. Front. Immunol. 16:1589752. doi: 10.3389/fimmu.2025.1589752
Received: 07 March 2025; Accepted: 23 April 2025;
Published: 14 May 2025.
Edited by:
Gabriella d’Ettorre, Sapienza University of Rome, ItalyReviewed by:
Francesca Cossarini, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2025 Creighton, Hughes, Hladik and Gornalusse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Germán G. Gornalusse, Z2VybWFnQHV3LmVkdQ==