 Solima Sabeel1,2,3
Solima Sabeel1,2,3 Bongani Motaung1,2
Bongani Motaung1,2 Mumin Ozturk1,2
Mumin Ozturk1,2 Trevor S. Mafu1
Trevor S. Mafu1 Robert J. Wilkinson4,5,6
Robert J. Wilkinson4,5,6 Friedrich Thienemann7,8,9*†
Friedrich Thienemann7,8,9*† Reto Guler1,2,4*†
Reto Guler1,2,4*†- 1Department of Pathology, Division of Immunology, Faculty of Health Sciences, Institute of Infectious Diseases and Molecular Medicine (IDM), University of Cape Town, Cape Town, South Africa
- 2International Centre for Genetic Engineering and Biotechnology (ICGEB), Cape Town Component, Cape Town, South Africa
- 3Division of Experimental Medicine, Department of Medicine, Zuckerberg San Francisco General Hospital, University of California, San Francisco, San Francisco, CA, United States
- 4Wellcome Discovery Research Platforms for Infection, Centre for Infectious Diseases Research in Africa, Department of Medicine, Faculty of Health Sciences, Institute of Infectious Disease and Molecular Medicine (IDM), University of Cape Town, Cape Town, South Africa
- 5Francis Crick Institute, London, United Kingdom
- 6Department of Infectious Diseases, Imperial College London, London, United Kingdom
- 7General Medicine & Global Health (GMGH), Department of Medicine and Cape Heart Institute, Faculty of Health Science, University of Cape Town, Cape Town, South Africa
- 8Department of Internal Medicine, University Hospital Zurich, University of Zurich, Zurich, Switzerland
- 9Cape Universities Body Imaging Centre (CUBIC), University of Cape Town, Cape Town, South Africa
Background: Tuberculosis (TB) remains a major global health threat, contributing substantially to high morbidity and mortality rates. This underscores the urgent need for more effective interventions. Recent studies highlight the potential of host-directed therapy approaches to enhance immune defences against TB. Atorvastatin, recognized for both its lipid-lowering properties and its immunomodulatory effects, has emerged as a compelling candidate for host-directed therapy against TB. Here, we investigated the ex vivo efficacy of atorvastatin in inducing immunomodulatory activities (phagosome maturation, autophagy, and apoptosis) and enhancing the mycobacterial killing capacity in Mycobacterium tuberculosis (Mtb)-infected peripheral blood mononuclear cells (PBMCs).
Method: Blood samples from healthy donors were collected for PBMC isolation. PBMCs were then treated overnight with or without atorvastatin, followed by infection with Mtb strains (H37Rv, HN878, and CDC1551) to evaluate intracellular mycobacterial growth by colony-forming units enumeration. Furthermore, co-localization of late endosomal marker (Rab-7), lysosomal markers (Cathepsin-D and LAMP-3), and autophagy marker (LC3B) with GFP-Mtb was investigated in infected PBMCs using laser scanning confocal microscopy. Moreover, multiple apoptotic assays were performed, including the TUNEL assay for DNA fragmentation, quantification of caspase-3 activity, and the expression levels of the pro-apoptotic gene (Bax) and anti-apoptotic gene (Bcl2).
Results: Treatment with atorvastatin significantly reduced intracellular mycobacterial replication compared to untreated controls in Mtb-infected PBMCs. Moreover, atorvastatin enhanced co-localization between Mtb and late endosomal marker (Rab-7), lysosomal markers (Cathepsin-D and LAMP-3), and autophagy marker (LC3B) in Mtb-infected PBMCs. Furthermore, atorvastatin robustly promoted apoptosis in Mtb-infected PBMCs, as demonstrated by TUNEL assay and caspase-3 activation.
Conclusion: Our findings highlight atorvastatin’s potential as a crucial modulator of the immune response in Mtb-infected PBMCs, supporting its role in host-directed therapy.
1 Introduction
Tuberculosis (TB) continues to exert a significant toll as the leading cause of mortality among infectious lung diseases worldwide. According to the World Health Organization’s Global Tuberculosis Report 2024, TB claims an estimated 1.3 million lives annually, with 10.8 million new cases reported in 2023 globally (1). In addition, approximately a quarter of the world’s population is inferred to be infected with Mycobacterium tuberculosis (Mtb), as measured by a positive interferon-gamma release assay or a positive tuberculin skin test. While 5–10% of new infections progress to active disease, this risk increases substantially in immunocompromised individuals, particularly those co-infected with HIV (1, 2).
Treatment of drug-susceptible TB with current drugs requires at least six months to achieve a durable cure, but the prolonged duration significantly increases the risk of non-adherence and treatment failure (3). Low adherence increases the risk of poor outcomes, including treatment failure, relapse, and the development of drug resistance (3). Together with treatment failure and adverse effects, these unintended treatment consequences affect a substantial proportion of patients and have further consequences. For example, adverse treatment effects may lead to severe post-TB lung diseases such as chronic obstructive pulmonary disease, bronchiectasis, and fibrocavitation, among others, which reduce the quality and expectancy of life among treated patients, and increase the likelihood of recurrent TB (4). Advanced therapies, such as targeted therapies, repurposed antimicrobials, and long-acting formulations, are under investigation to improve treatment outcomes (5). Host-directed therapies (HDTs), including immunotherapeutics and non-antimicrobial agents, enhance the host’s immune response against Mtb (6, 7). These approaches leverage specific signaling pathways, existing antibiotics, or reduced dosing frequency (6, 7).
Several studies have demonstrated the potential of statins as HDT for TB (8–11). Statins, known as 3-hydroxy-3-methyl glutaryl coenzyme A reductase (HMG-CoA) inhibitors, are primarily prescribed to reduce serum cholesterol levels by inhibiting HMG-CoA reductase. This enzyme drives the conversion of HMG-CoA into mevalonate, a key step in the cholesterol biosynthesis (12). Inhibiting this pathway decreases cholesterol biosynthesis and reduces the synthesis of isoprenoid intermediates such as farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) (13). In the context of Mtb infection, statins have shown potential to enhance the bactericidal potency of the first-line anti-TB treatments, and thereby shorten the duration of treatment (14). In preclinical studies, mice treated with simvastatin or rosuvastatin exhibited a reduced Mtb burden in the spleens, livers, and lungs when compared to untreated controls (11). Mice treated with simvastatin also showed accelerated clearance of bacilli from the lungs when combined with standard TB therapy (15). Dutta et al. (2020) demonstrated that statins possess potent adjunctive activity against Mycobacterium tuberculosis, significantly reducing lung CFU counts in both the standard mouse model (BALB/c mice) and in C3Heb/FeJ mice, a model that more closely resembles human necrotic TB granulomas (16). More recently, Davuluri et al. (2023) reported that atorvastatin enhances the efficacy of first-line anti-TB regimens and markedly reduces the intracellular bacterial burden in the spleens of guinea pigs (8). Additionally, atorvastatin improves the permeability and distribution of rifampicin and isoniazid within granulomatous lung lesions by decreasing granuloma vascular thickness (8).
In clinical studies, peripheral blood mononuclear cells (PBMCs) isolated from individuals with familial hypercholesterolemia treated with atorvastatin exhibited a significant decrease in the intracellular growth of Mtb compared to PBMCs from healthy controls (11). In a recent clinical trial, Adewole et al. (2023) found that adjunctive atorvastatin therapy accelerates M. tuberculosis clearance in patients with pulmonary TB (17). In contrast, the ROSETTA trial, which evaluated rosuvastatin (10 mg once per day for 8 weeks) as an adjunctive TB therapy, found no significant impact on time to culture conversion and no effect on lung inflammation as measured by PET/CT (18, 19).
Beyond the reported antimicrobial activity, statins also exhibit notable immunomodulatory effects. In a study by Bedi et al. (2017), treatment with 80 mg/day of atorvastatin (considered high-intensive statin therapy) for three months reduced serum levels of interleukin 8 (IL-8), tumor necrosis factor (TNF), intercellular adhesion molecule 1 (ICAM1), C-reactive protein (CRP), and neutrophil counts in patients with bronchiectasis (20). In corroboration, evidence from a pilot study by Mroz et al. (2015) found that atorvastatin 40 mg daily for 12 weeks reduced serum CRP levels, sputum neutrophil count in lung biopsies, and the expression of key genes involved in inflammation and leukocyte activation (21).
Mechanistically, our group previously demonstrated, through the work of Parihar and Guler et al. (2013), that simvastatin promotes phagosome maturation in Mtb-infected PBMCs, as indicated by increased EEA-1 and LAMP-3 expression, as well as enhanced autophagy marked by LC3-II accumulation (11). Similarly, Guerra-De-Blas et al. (2019) showed that simvastatin induces apoptosis in Mtb-infected PBMCs (22). Additional evidence from Dutta et al. (2016) demonstrated that pravastatin enhances phagosome maturation in Mtb-infected mouse bone marrow-derived macrophages (BMDMs) (16). Furthermore, Bruiners et al. (2020) revealed that simvastatin induces autophagy in Mtb-infected human PBMCs via blocked activation of mechanistic target of rapamycin complex 1 (mTORC1), activated AMP-activated protein kinase (AMPK) through increased intracellular AMP: ATP ratios, and favored nuclear translocation of transcription factor EB (TFEB) (10). While most mechanistic studies of Mtb infection have mainly focused on simvastatin, data on the immunomodulatory effects of atorvastatin—an extensively prescribed statin with a distinct pharmacological profile—remain limited.
Herein, we investigated the immunomodulatory activities of atorvastatin, focusing on its ability to enhance phagosome maturation, autophagy, and apoptosis in Mtb-infected PBMCs. Additionally, we assess the mycobacterial killing capacity of atorvastatin-stimulated PBMCs following ex vivo Mtb infection.
2 Material and methods
2.1 Participant samples and ethics
Blood samples were collected from 10 healthy donors (18–65 years, both sexes), according to South African Guidelines for Good Clinical Practice. The study was approved by the University of Cape Town’s Faculty of Health Sciences Human Research Ethics Committee [Reference number: 732/2015].
2.2 Isolation of PBMCs
All blood samples were collected in sodium heparin vacutainer tubes (BD Vacutainer®, BD Diagnostics, Franklin Lakes, NJ, USA) by a qualified and authorized staff member. The detailed protocol for the isolation of PBMCs is provided in the Supplementary Material.
2.3 Ex vivo infection of PBMCs with Mtb
PBMCs were seeded in 96-well plates (Corning®, New York, United States) and treated with varying concentrations of atorvastatin (ATO) at 37°C with 5% CO2. Following overnight incubation, PBMCs were infected with H37Rv, HN878, and CDC1551 Mtb strains with a multiplicity of infection (MOI) of 0.5. Each CFU experiment included three technical replicates per condition. Further details of the experimental protocol can be found in the Supplementary Material.
2.4 Immunofluorescence staining for confocal microscopy
Atorvastatin-treated and untreated PBMCs were seeded in 96-well plates (ibidi ®, Gräfelfing, Germany) with transparent bottoms. Details regarding the confocal microscopy and image-capturing procedures are included in the Supplementary Material. The concentration and dilution of primary and secondary antibodies used for immunofluorescent staining were outlined in Supplementary Table 1.
2.5 DNA fragmentation (TUNEL assay)
In situ apoptosis was detected using Click-It™ Plus TUNEL Assay labelled with Alexa Fluor™ 647 picolyl azide dye (Invitrogen, Massachusetts, United States). All the steps were carried out following the manufacturer’s instructions, as explained in the Supplementary Material. The mean fluorescent intensity (MFI) was calculated in 3–5 fields that contain 25–30 cells.
2.6 Measurement of caspase-3 activity
The proteolytic activity of caspase-3 was determined using the EnzChek® Caspase-3 Assay Kit #2 (Molecular Probes, Leiden, Netherlands), following the manufacturer’s instructions.
2.7 Enzyme-linked immunosorbent assay
Supernatants from ex vivo cell cultures were collected for evaluation of pro-inflammatory cytokines, including IL-1β, IL-6, and IL-8, through ELISA after atorvastatin stimulation and Mtb infection. PBMC were stimulated overnight with different atorvastatin concentrations, including 50 µM, and 100 µM prior to infection with three Mtb strains (H37Rv, HN878, and CDC1551). Culture supernatants were collected at 1, 3, and 6 dpi; ELISA plates were read using a VersaMax™ microplate reader (Molecular Devices, USA).
2.8 RNA extraction and cDNA synthesis
Atorvastatin-treated and Mtb-infected PBMCs were lysed using 2-β-mercaptoethanol (Sigma-Aldrich, St. Louis, Missouri, United States) in lysis buffer (1:100). Total cellular RNA was isolated from cell lysates using the RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s specifications. Total RNA was reverse transcribed using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland) according to the manufacturer’s specifications.
2.9 Real-time quantitative PCR
Quantitative real-time PCR [LightCycler® 480 II system combined with the LightCycler® 480 SYBR Green I Master (Roche, Manheim, Germany)] was performed to measure the relative expression levels of Bcl-2 and Bax using specific primer pairs (Integrated DNA Technologies, Iowa, United States). Hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1) was used as the reference housekeeping gene for normalization. Further details can be found in the Supplementary Material. All primer sequences and cycling conditions for qPCR are shown in Supplementary Tables 2 and 3, respectively.
2.10 Statistical analysis
Statistical analysis was performed using GraphPad Prism version 8 (GraphPad Software, San Diego, California, United States). For each experiment, we used two–three biological replicates (different donors) and performed three technical replicates per condition. Student’s t-test was used to compare differences in quantitative data between two groups, while one-way ANOVA, with Bartlett’s test for homogeneity of variances, was used for comparisons involving three or more groups. Statistically significant values were represented as *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. To account for donor variability, all quantitative data were pooled across donors, and statistical analyses were conducted on biological replicates, using mean values of technical replicates to represent each donor. When applicable, we used one-way ANOVA to determine statistical significance across treatment groups, and all results are presented as mean ± SEM.
3 Results
3.1 Atorvastatin exhibits a dose-dependent suppression of intracellular Mtb growth in ex vivo Mtb-infected PBMCs
We examined the mycobactericidal killing capacity of atorvastatin in Mtb-infected PBMCs. We demonstrated that, compared with untreated controls, atorvastatin significantly reduced the intracellular growth of Mtb in a dose-dependent manner, at 1, 3, and 6 days post-infection (Figures 1A-C).
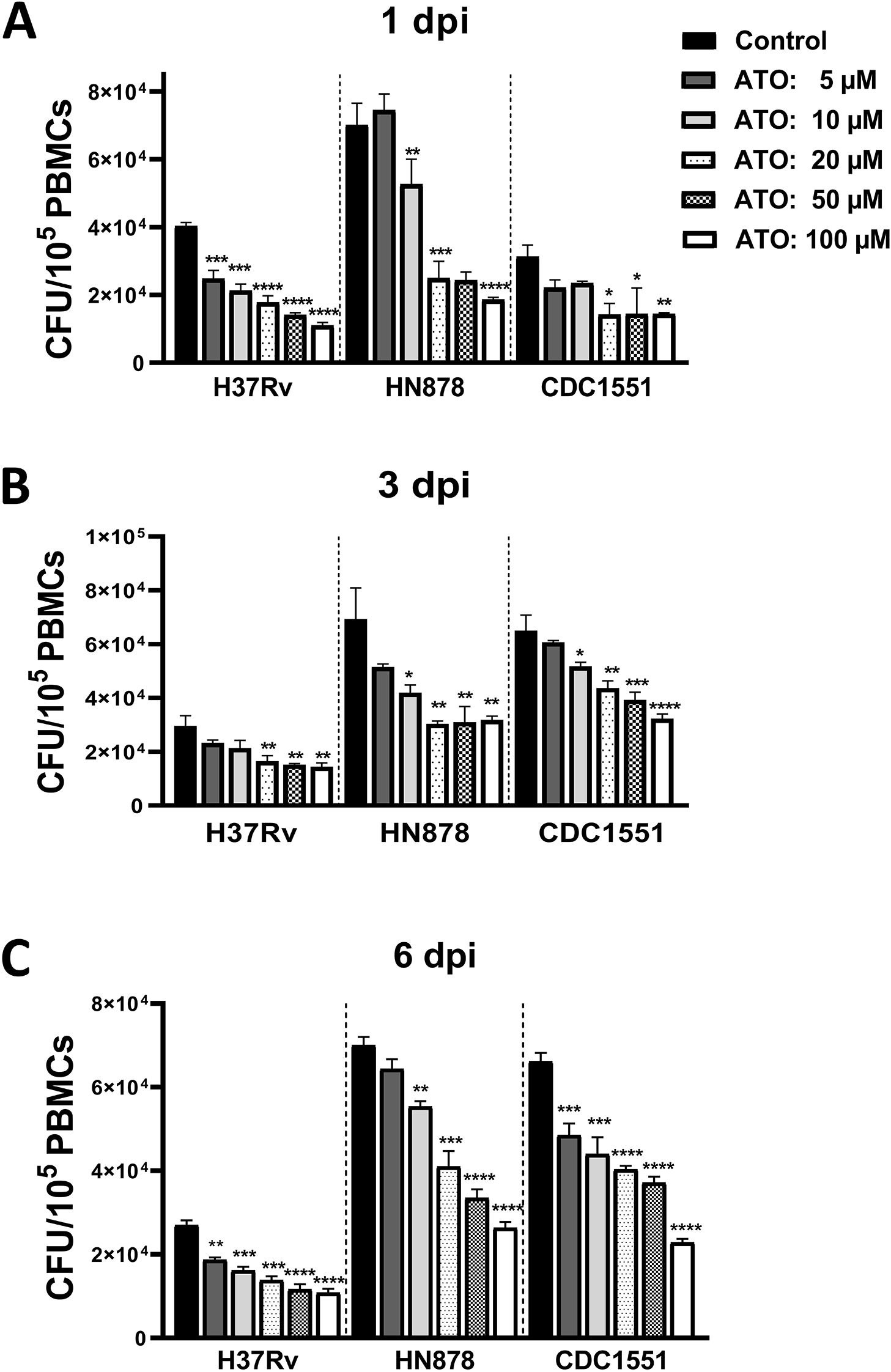
Figure 1. Atorvastatin reduces the intracellular growth of Mycobacterium tuberculosis in PBMC. PBMCs were cultured overnight in the presence of atorvastatin (ATO) or control (DMSO). Subsequently, PBMCs were infected with Mtb strains (H37Rv, HN878, and CDC1551). (A) At 1 day post-infection (dpi), (B) 3 dpi, and (C) 6 dpi, PBMCs were lysed to measure the intracellular growth of Mtb using CFU assay. All data are shown as mean ± SEM using one-way ANOVA with Bartlett’s Test. The results were representative of three independent experiments, *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 compared to the control. ATO, atorvastatin; dpi, days post-infection.
3.2 Atorvastatin enhances phagosome maturation and autophagy in Mtb-infected PBMCs
To evaluate the impact of atorvastatin on these processes, we measured key markers of phagosome (Rab-7), phagolysosome (LAMP-3 and Cathepsin-D), and autophagy (LC3B) maturation in GFP-H37Rv Mtb-infected PBMCs using confocal microscopy and quantitative image analysis. Atorvastatin treatment significantly increased the colocalization of Mtb with the late endosomal marker (Rab-7), phagolysosome markers (LAMP-3 and Cathepsin-D), and the autophagy marker (LC3B) in GFP-H37Rv Mtb-infected PBMCs (Figures 2A-D). Further image quantification using Spearman’s Rank co-localization coefficient confirmed atorvastatin’s ability to increase the colocalization of Mtb with these key phagosome and autophagy markers. Collectively, these findings suggest that atorvastatin is associated with enhanced host protective functions, including increased phagosome maturation and augmented autophagy, which may contribute to the restriction of intracellular Mtb growth.
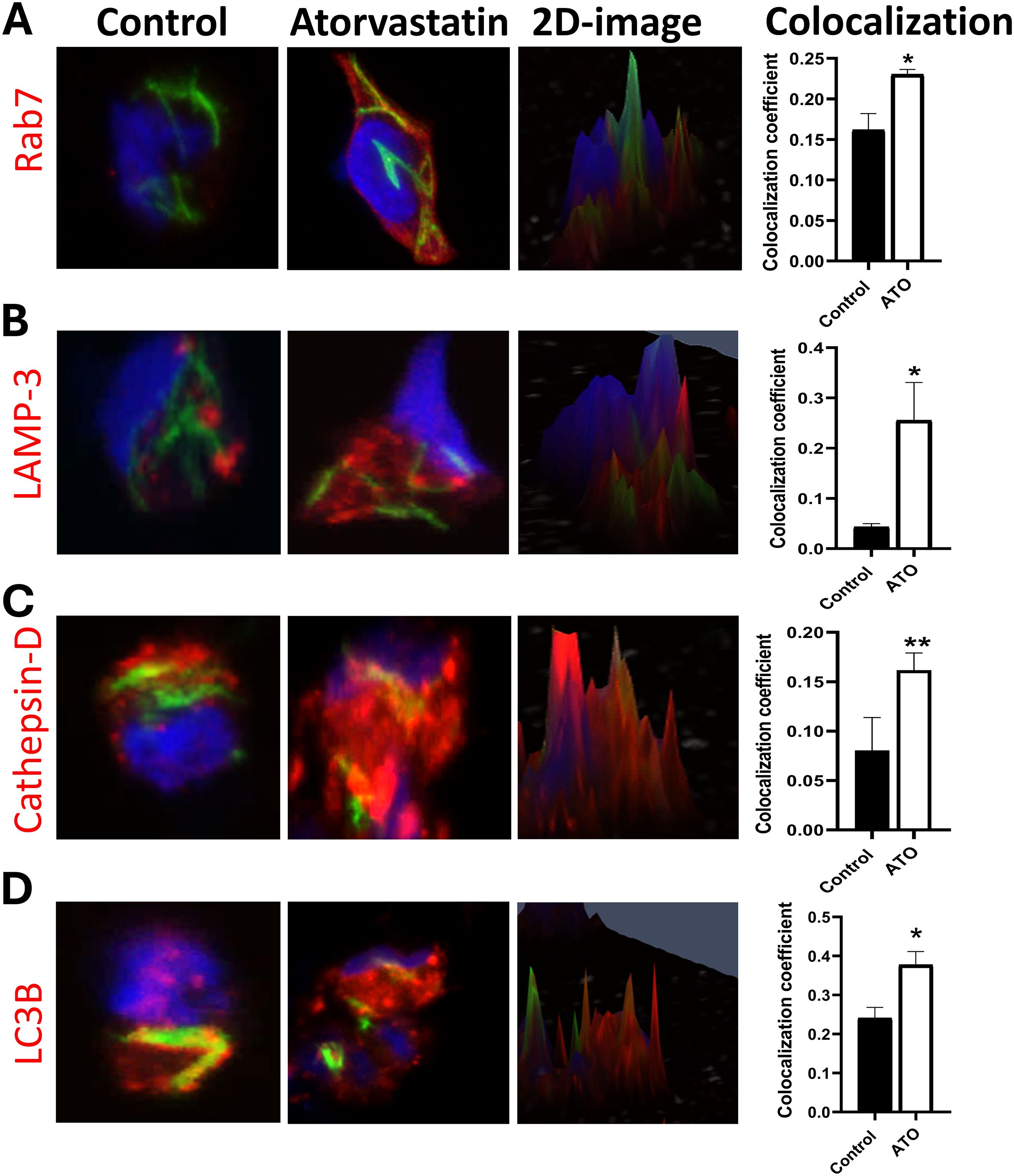
Figure 2. Atorvastatin promotes phagosome maturation and autophagy in PBMCs by increasing colocalization of late endosomal, lysosomal, and autophagy markers with Mtb. Confocal microscopy images and two-dimensional images of atorvastatin-treated PBMC showing enhanced co-localization of GFP-expressing Mtb H37Rv (green) with (A) late endosomes (Rab-7), (B, C) phagolysosome markers (LAMP-3, Cat-D), and (D) autophagy marker LC3B. Quantitative analysis using Spearman’s Rank co-localization coefficient showing increased colocalization in atorvastatin-treated PBMCs following Mtb infection. The yellow color represents the colocalization of green (GFP-Mtb) with red (phagosome and autophagy markers), and blue indicates nuclear stain (Hoechst stain). Data are represented as mean ± SEM (*P<0.05, **P<0.01). ATO, atorvastatin.
3.3 Atorvastatin enhances apoptosis in Mtb-infected PBMCs by inducing in situ DNA fragmentation and activating caspase-3
Macrophages play a crucial role in innate defense against Mtb by inducing apoptosis (23). To investigate atorvastatin’s ability to enhance apoptosis as a host defense mechanism to control Mtb proliferation, we performed three assays on PBMCs: a TUNEL assay, a caspase-3 activity assay, and an expression assay evaluating the levels of pro- and anti-apoptotic markers. Atorvastatin treatment induced pro-apoptotic characteristics, as measured across all three assays. TUNEL-positive cells were significantly increased in atorvastatin-treated PBMCs compared with untreated controls, as evidenced by the overlay of the TUNEL-positive signal (magenta) with the Hoechst-counterstained nuclei (blue) and TUNEL-positive cells (red, Figure 3A). Quantitative image analysis confirmed a threefold increase in apoptotic cells induced by atorvastatin (Figure 3B). Caspase-3 activity was significantly increased in atorvastatin-treated PBMCs compared with untreated controls (Figure 3C). The pro-apoptotic marker (Bax) was significantly upregulated in atorvastatin-treated PBMCs compared with controls (Figure 3D), while the anti-apoptotic marker Bcl-2 was significantly downregulated following atorvastatin treatment (Figure 3E). Taken together, these data suggest that atorvastatin induces apoptosis as a host protective mechanism in Mtb-infected PBMCs.
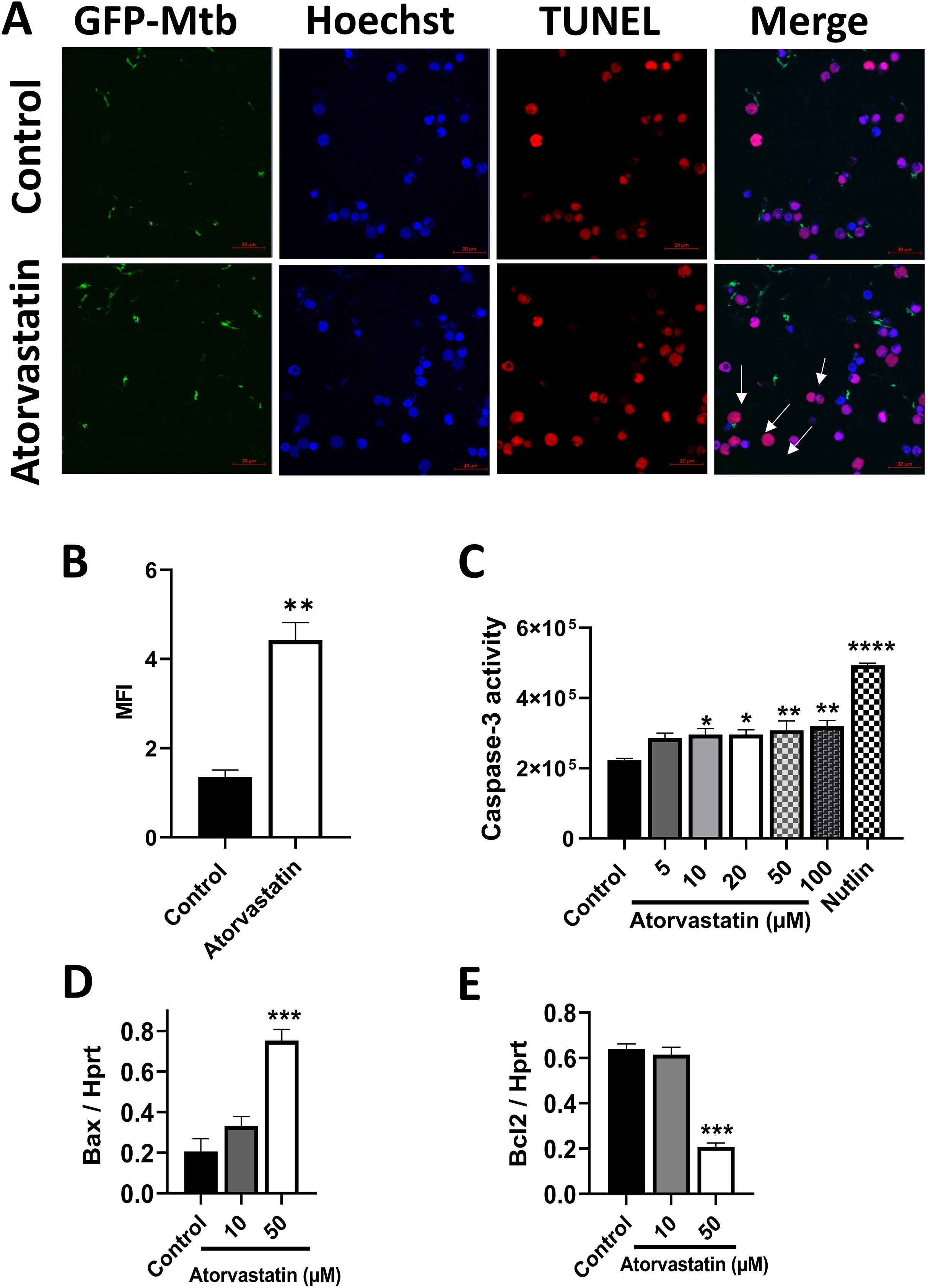
Figure 3. Atorvastatin enhances apoptosis in Mtb-infected PBMCs as measured by TUNEL and caspase-3 activity. PBMCs (5x105/well) were treated with atorvastatin and DMSO (control) overnight, followed by infection with GFP-expressing H37Rv Mtb at an MOI:5 (1 PBMC: 5 Mtb) for 24 hours to assess apoptosis by TUNEL assay. (A) Confocal microscopy images depicting GFP-expressing H37Rv Mtb in green, Hoechst nuclear stain in blue, and TUNEL-positive apoptotic cells in red. White arrows indicate apoptotic cells, which appear magenta in the overlay of nuclear Hoechst stain (blue) and TUNEL-positive cells (red). The scale bar represents 20 μm. Images were acquired using an Airyscan 880 confocal microscopy with a 63x oil immersion lens. (B) Quantitative analysis of the mean fluorescence intensity (MFI) of Tunnel-positive PBMCs. (C) PBMCs (2x106/well) were treated with various concentrations of atorvastatin (5 μM, 10 μM, 20 μM, 50 μM, and 100 μM), DMSO (control), and the apoptosis inducer Nutlin overnight to assess caspase-3 activity using the Ac-DEVD-CHO caspase assay kit, presented as fluorescence unit. PBMC were treated with atorvastatin and infected with Mtb H37Rv for 24 hours to measure (D) the pro-apoptotic marker Bax and (E) the anti-apoptotic marker Bcl-2 using RT-qPCR. Data are presented as mean ± SEM (*P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001).
3.4 Mevalonate supplementation reverses the effect of atorvastatin and increases the intracellular growth of Mtb
To investigate atorvastatin’s ability to directly reduce intracellular growth of Mtb through inhibiting the cholesterol biosynthesis pathway, we supplemented Mtb-infected PBMCs with exogenous mevalonate, a precursor downstream of HMG-CoA reductase in the cholesterol biosynthesis pathway. The addition of mevalonate partially reversed the atorvastatin-induced reduction in CFU, with a notable effect observed in the H37Rv strain at 3 dpi (Figures 4A, B).
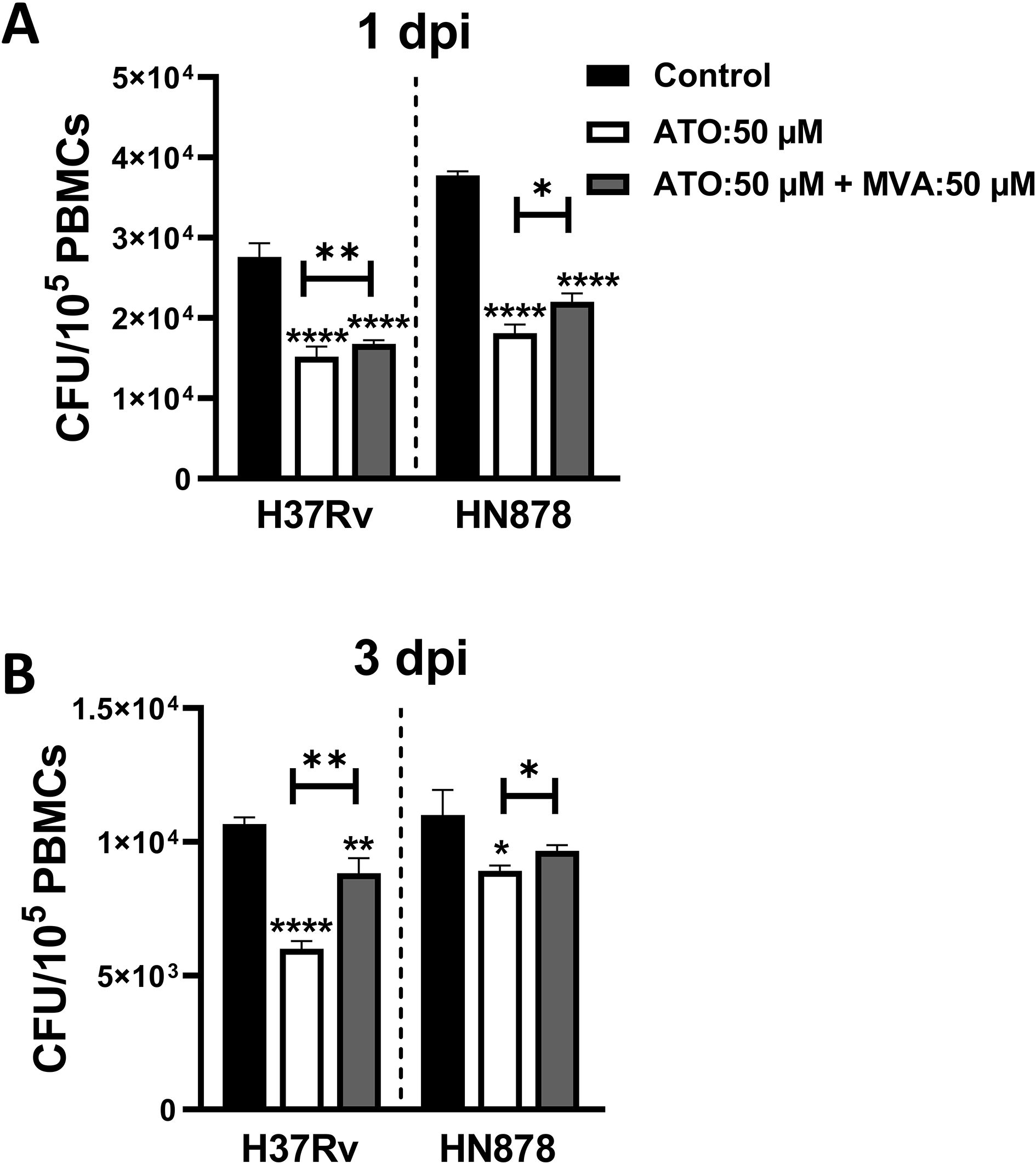
Figure 4. Mevalonate supplementation reverses the effect of atorvastatin and enhances the intracellular growth of Mtb in PBMCs. PBMCs (1x105 cells/well) were treated with either atorvastatin (50 μM), DMSO control, or a combination of atorvastatin (ATO, 50 μM) and mevalonate (MVA, 50 μM). Following overnight treatment, PBMCs were infected with Mtb H37Rv or HN878 strain for (A) 1 dpi and (B) 3 dpi to evaluate the intracellular Mtb growth using CFU assays. Data shown are presented as mean ± SEM (*P<0.05, **P<0.01, ****P<0.0001). ATO, atorvastatin; MVA, mevalonate; dpi, days post-infection.
3.5 Atorvastatin reduces intracellular Mtb growth mainly through inhibition of the geranylgeranyl biosynthesis
The mevalonate pathway is a vital biochemical process responsible for synthesizing cholesterol and metabolites for protein geranylation and farnesylation, critical processes involved in signal transduction and membrane anchoring (24). Atorvastatin inhibits HMG-CoA reductase, the enzyme that catalyzes the conversion of HMG-CoA to mevalonate, thereby reducing cholesterol synthesis. Consequently, atorvastatin also blocks metabolites for protein geranylation and protein farnesylation. To investigate the precise metabolic axis within the mevalonate pathway that contributes to the atorvastatin-mediated reduction of intracellular Mtb growth, we used specific inhibitors of the mevalonate pathway. PBMCs were treated with geranylgeranyl transferase inhibitor (GGTI), farnesyltransferase inhibitor (FTI), or squalene inhibitor (SQI), and then infected with either H37Rv or HN878 Mtb strains to evaluate the intracellular mycobacterial growth (Figure 5A). Early during Mtb infection, at 1-day post-infection, all three inhibitors (GGTI, FTI, and SQI) significantly reduced the intracellular growth of Mtb, even surpassing the efficacy of atorvastatin treatment (Figure 5B). In contrast, at 3 days post-infection, the geranylgeranyl transferase inhibitor exhibited the most substantial reduction in intracellular Mtb growth in PBMCs compared to the other inhibitors (Figure 5C). In summary, atorvastatin’s host-mediated reduction of intracellular Mtb growth is attributed not only to cholesterol inhibition but also to its ability to reduce metabolites in the protein geranylation and protein farnesylation pathway, particularly within the geranylgeranyl biosynthesis pathway.
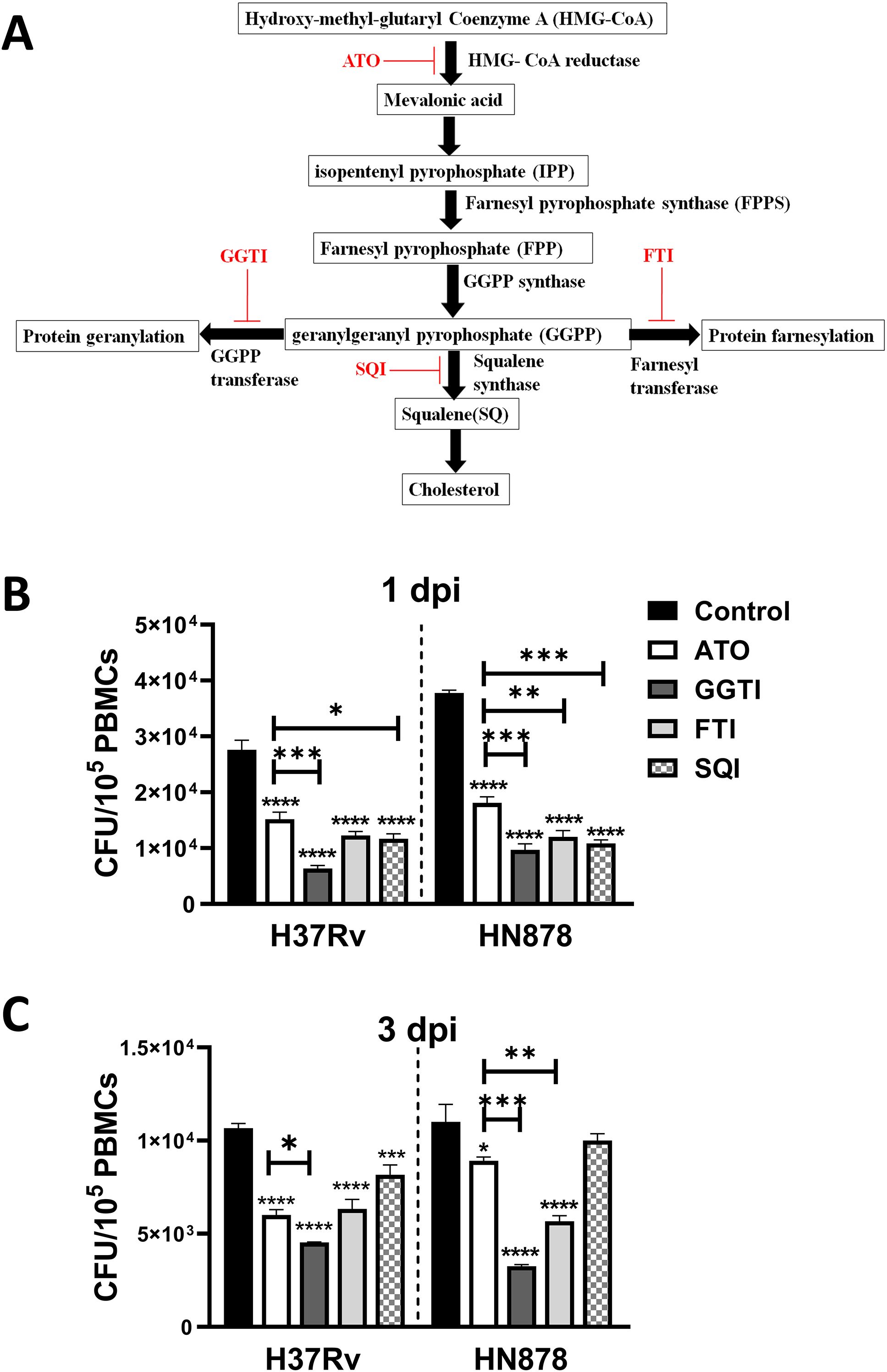
Figure 5. Inhibition of the protein geranylation pathway significantly reduces intracellular growth of Mtb in PBMCs. Schematic representation of the mevalonate pathway highlighting key metabolites, enzymes, and specific inhibitors (ATO, atorvastatin; FTI, farnesyl transferase inhibitors; GGTI, geranylgeranyl transferase inhibitors; SQI, squalene inhibitors). (A) PBMCs were treated overnight with atorvastatin (50 μM) or specific mevalonate pathway inhibitors (GGTI, FTI, and SQI, 50 μM each). Treated PMCs were then infected with Mtb H37Rv or Mtb HN878 strain for (B) 1 dpi and (C) 3 dpi to measure the intracellular Mtb growth using CFU assay. Data are presented as mean ± SEM (*P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001). dpi, days post-infection.
3.6 Atorvastatin reduces IL-1β secretion in a concentration- and time-dependent manner
Atorvastatin treatment reduced IL-1β secretion in a concentration- and time-dependent manner in Mtb-infected human PBMCs, regardless of the infecting strain. In contrast, IL-6 and IL-8 levels remained largely unchanged (Supplementary Figure 5).
4 Discussion
In this study, we investigated the effect of atorvastatin on the ex-vivo intracellular growth of Mtb in PBMCs. Our findings revealed that pre-treatment of Mtb-infected PBMCs with atorvastatin reduces the intracellular growth of Mtb - across three strains: H37Rv, HN878, and CDC1551 - in a dose-dependent manner, compared with untreated controls (Figures 1A-C). This observation is consistent with a previous study conducted by Lobato et al. (2014), which demonstrated that both cholesterol-lowering drugs: atorvastatin and simvastatin, reduce the intracellular growth of Mtb H37Rv in THP-1 derived macrophages (25). Additionally, similar outcomes were reported by Parihar and Guler et al. (2013), who observed a decrease in Mtb H37Rv growth in simvastatin-treated murine macrophages (11). Further supporting of these findings, ex vivo treatment of PBMCs derived from TB patients with persistent lung inflammation (defined as Total Lung Glycolysis [TLG] ≥ 50 SUV*mL) at the end of TB treatment significantly reduced the intracellular growth of Mtb, as measured by CFU assay at 3 dpi. This reinforces the potential of atorvastatin as a host-directed therapy, particularly in TB patients with residual inflammatory activity post-treatment Supplementary Figure 1. At Day 0 (4 hours post infection), all three Mtb strains showed equal bacterial load Supplementary Figure 2.
Macrophages can limit the intracellular growth of Mtb through various mechanisms including phagosome acidification and fusion of phagosomes with lysosomes (26). Phagosome maturation is a complex process involving a series of events within the endocytic pathway, transforming a newly formed phagosome into a phagolysosome capable of degrading engulfed microbes and other particles (27). During phagocytosis, the small GTPase Rab-5 recruits early endosomal antigen-1 (EEA-1) to the membrane of phagosomes (28, 29), then another GTPase, Rab-7, is recruited in the later stages of the endosomal trafficking (30, 31). As phagosomes mature, phagolysosomes occur through fusion with lysosomal vesicles that acquire lysosomal markers, such as lysosome-associated membrane protein (LAMP), and acid hydrolyses, such as cathepsin D (Cat-D) (26, 32). Considering Rab-7’s essential role in endosomal trafficking and lysosome biogenesis (33), we hypothesized that atorvastatin might promote the recruitment of phagosome/phagolysosome maturation markers such as Rab-7, LAMP-3 and Cat-D to Mtb-contained endosomes. We observed a notable enhancement in the co-localization of Rab-7, LAMP-3, and LC3B with Mtb following atorvastatin treatment (Figure 2A). Similarly, we observed increased co-localization of LAMP-3 and Cat-D with GFP-Mtb in atorvastatin-treated PBMCs compared with untreated controls (Figures 2B, C). Our findings are consistent with a previous study by our group, Parihar and Guler et al. (2013), which reported higher co-localization of EEA-1, LAMP-1, and LAMP-3 with GFP-H37Rv Mtb in simvastatin-treated murine macrophages (11). Considerable evidence highlights the critical role of autophagy in the host defense against Mtb infection (34, 35). Autophagy facilitates the delivery of cytosolic macromolecules and organelles containing Mtb to lysosomes for degradation (36). In this context, the role of LC3B, a reliable autophagy marker (37), was assessed. Our data demonstrated an increase in the colocalization coefficient of GFP-Mtb with LC3B in atorvastatin-treated cells when compared to controls (Figure 2D). Together, these results confirm that atorvastatin enhances the host-mediated killing effector mechanisms of PBMCs, thereby reducing the mycobacterial burden. By enhancing phagosome maturation and autophagy, atorvastatin may counteract the inhibition or impairment of these processes by Mtb as an immune evasion strategy (38). To improve transparency and interpretability, the corresponding single-channel images have been included as Supplementary Figure 3.
Apoptosis plays a fundamental role in host defense against Mtb by reducing the viability of intracellular pathogens (23, 39, 40) and preventing the release and spread of Mtb (41). Therefore, we investigated whether atorvastatin induces apoptosis in Mtb-infected PBMCs. One hallmark of apoptosis is chromatin fragmentation and alterations in nuclear organization, which can be visualized using the TUNEL assay (42). Figures 3A, B demonstrate that atorvastatin induces apoptosis in Mtb-infected PBMCs through chromatin fragmentation. This finding is consistent with the observations of Carton et al. (2004), who reported an increase in TUNEL-positive cells following lovastatin treatment in RAW264.7 murine macrophages infected with Salmonella enterica serovar Typhimurium (43). These findings illustrate that atorvastatin induces programmed cell death in Mtb-infected PBMCs, as a crucial host protective immune response against Mtb. Our investigation extended to critical cell signaling events during apoptosis, including caspase-3 activity and regulation of the expression of pro- and anti-apoptosis genes. As shown in Figure 3C, atorvastatin increased caspase-3 activity in Mtb-infected PBMCs. This observation aligns with findings by Guerra et al. (2019), who noted increased caspase-3 expression in PBMCs treated with simvastatin compared to untreated PBMCs (22). Notably, the pro-apoptotic marker, Bax, was significantly overexpressed while the anti-apoptotic marker, Bcl-2, was significantly underexpressed in atorvastatin-treated PBMCs compared with untreated controls (Figures 3D, E). Our findings mirror those of Blanco-Colio et al. (2002), who reported a reduction in Bcl-2 expression in vascular smooth muscle cells cultured with atorvastatin and simvastatin in a time- and dose-dependent manner (44). By enhancing apoptosis via inducing DNA fragmentation and activating caspases, atorvastatin offers a potential strategy to subvert Mtb’s impairment of the apoptotic immune response (45).
To investigate whether the addition of mevalonate reversed the impact of atorvastatin, a metabolic rescue experiment was conducted. As shown in Figures 4A, B, the addition of mevalonate reversed the effects of atorvastatin and enhanced the growth of both H37Rv and HN878 Mtb strains. We next investigated the effects of mevalonate pathway inhibitors on Mtb bacterial load. As shown in Figures 5B, C, the inhibition of geranylgeranyl pyrophosphate by geranylgeranyl transferase inhibitors significantly contributed to reducing the intracellular growth of Mtb in PBMCs. In interpreting our findings on the use of mevalonate pathway inhibitors (Figure 5), it is important to consider their potential off-target effects in PBMCs. Inhibitors such as GGTI and FTI disrupt the post-translational prenylation of small GTPases (e.g., Ras, Rho, Rab), which are critical for immune cell signaling, intracellular trafficking, and apoptosis regulation (46). While such inhibition may contribute to the observed bacterial control effects, it may also unintentionally alter broader PBMC functions, including cytokine production (47) and cell proliferation (48). Moreover, the potential for cross-prenylation—where proteins such as Rho undergo geranylgeranylation as a compensatory response to farnesyltransferase inhibition—may confound the interpretation of FTI-specific effect (49). Additionally, SQI, which targets downstream cholesterol biosynthesis, may impair membrane integrity and disrupt receptor-mediated signaling in PBMCs, potentially influencing immune responses independently of M. tuberculosis infection (50). In summary, our study demonstrates the impact of atorvastatin on enhancing endosomal and phagosome maturation, as well as apoptotic effectors within Mtb-infected PBMCs. These findings contrast with those of Bruiners et al. (2020), who reported that inhibition of protein prenylation did not reduce intracellular Mtb burden (10). Notably, our study extends these earlier observations by utilizing PBMCs, a more heterogeneous immune cell population compared to the macrophage monolayers used previously. Furthermore, we assessed earlier post-infection timepoints (days 1 and 3), in contrast to the 6-day timepoint examined by Bruiners et al. (2020), providing additional insight into the temporal dynamics of atorvastatin’s immunomodulatory effects (10).
Statins are known to modulate inflammatory responses, partly through inhibition of NF-κB activation, with atorvastatin exhibiting one of the strongest effects among the statin class (51). In the current study, we investigated the impact of atorvastatin on cytokine responses in Mtb-infected human PBMCs. Our data show that atorvastatin reduces IL-1β secretion in a concentration- and time-dependent manner. regardless of the infecting Mtb strains. This observation is consistent with findings by Ascer et al. (2004), who reported reduced plasma IL-1 levels in hypercholesterolemic patients following atorvastatin treatment (52). In contrast, the levels of IL-6 and IL-8 remained unaffected under the same conditions, as shown in Supplementary Figure 5. These data suggest that atorvastatin selectively modulates IL-1β responses, indicating a potentially targeted anti-inflammatory mechanism relevant to host–pathogen interactions in tuberculosis.
In summary, our study demonstrates the impact of atorvastatin on enhancing endosome and phagosome maturation, as well as apoptotic effectors within Mtb-infected PBMCs. This combination of mechanisms makes atorvastatin a compelling candidate for host-directed therapy against tuberculosis—the multiple mechanisms exert various effects against Mtb, with the potential to act synergistically and reach dormant reservoirs. When used in combination with standard TB therapy, atorvastatin may reduce the risk of relapse, post-TB lung disease, and associated pulmonary vascular and cardiovascular complications, thereby promoting better lung function and long-term health outcomes.
Nonetheless, our study was not without limitations. While we primarily used PBMCs from healthy donors, further investigation is warranted to explore the immunomodulatory effects of atorvastatin therapy in TB patients, thereby providing more robust evidence for its potential as a host-directed therapy for tuberculosis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Blood samples were collected from 10 healthy donors and six tuberculosis patients (aged 18–65 years, both sexes) in accordance with the South African Guidelines for Good Clinical Practice. The study received ethical approval from the Human Research Ethics Committee of the Faculty of Health Sciences at the University of Cape Town (reference numbers: 732/2015 and 675/2019).
Author contributions
SS: Investigation, Writing – original draft, Visualization, Formal Analysis, Writing – review & editing, Methodology, Conceptualization. BM: Writing – review & editing. MO: Writing – review & editing. TM: Writing – review & editing. RW: Writing – review & editing. FT: Conceptualization, Writing – review & editing. RG: Funding acquisition, Methodology, Conceptualization, Writing – review & editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This publication was produced by StatinTB, which is part of the European and Developing Countries Clinical Trials Partnership 2 (EDCTP2) programme supported by the European Union (grant number RIA2017T-2004-StatinTB). RW receives support from the Francis Crick Institute, which is funded by Wellcome (CC2112), Research Councils Uk (CC2112), and Cancer Research UK (CC2112). He also receives support from Wellcome (226817) and in part from the NIHR Biomedical Medical Research Center of Imperial College Healthcare NHS Trust
Acknowledgments
We would like to express our gratitude to Shandré Pillay for her assistance with RT-qPCR experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Only for minor grammar edits.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1597534/full#supplementary-material
Supplementary Figure 1 | Ex vivo treatment with atorvastatin of PBMC derived from patients with persistent lung inflammation (TLG ≥ 50 SUV*mL) at the end of TB treatment, significantly reduced the intracellular growth of Mtb strains, as measured by CFU assay at 3 days post-infection.
Supplementary Figure 2 | Effect of atorvastatin treatment on intracellular Mtb burden at 4 hours post-infection (hpi). PBMCs were infected with Mtb strains H37Rv, HN878, or CDC1551 and treated with 50 µM and 100 µM of atorvastatin. CFUs were quantified at 4 hours post-infection (4 hpi) to assess bacterial uptake.
Supplementary Figure 3 | Representative single-channel immunofluorescence images. Cells were stained with antibodies against LAMP-3, Rab7, Cathepsin-D (shown in red: Cy3 or Alexa Fluor 594), and nuclei were counterstained with DAPI (blue). GFP-Mtb is shown in green. Overlay images display merged channels to visualize co-localization.
Supplementary Figure 4 | Screenshot from FUJI software illustrating the optimization parameters used for the Spearman’s rank correlation colocalization analysis.
Supplementary Figure 5 | Pro-inflammatory cytokine secretion in Mtb-infected PBMCs following atorvastatin stimulation. Cytokines were measured at 1, 3, and 6 dpi using ELISA; (A) IL-1β, (B) IL-6, and (C) IL-8, where the dotted line indicates detection limits. Data is shown as a representative of three replicates and shown as mean ± SD. Data was analysed using Shapiro-Wilk test for normality, followed by two-way ANOVA with Dunnett’s post hoc test for multiple comparisons against the control; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. ATO, atorvastatin; DPI, Days Post-Infection; LOD, Limit of Detection.
References
2. O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, and Berry MP. The immune response in tuberculosis. Annu Rev Immunol. (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
3. Dobbs TE and Webb RM. Chemotherapy of tuberculosis. Microbiol Spectrum. (2017) 5. doi: 10.1128/microbiolspec.tnmi7-0040-2017
4. Allwood BW, Byrne A, Meghji J, Rachow A, van der Zalm MM, and Schoch OD. Post-tuberculosis lung disease: clinical review of an under-recognised global challenge. Respiration. (2021) 100:751–63. doi: 10.1159/000512531
5. Nair A, Greeny A, Nandan A, Sah RK, Jose A, Dyawanapelly S, et al. Advanced drug delivery and therapeutic strategies for tuberculosis treatment. J Nanobiotechnology. (2023) 21:414. doi: 10.1186/s12951-023-02156-y
6. Mourenza Á, Gil JA, Mateos LM, and Letek M. Novel Treatments against Mycobacterium tuberculosis Based on Drug Repurposing. Antibiotics (Basel). (2020) 9. doi: 10.3390/antibiotics9090550
7. Khameneh B, Iranshahy M, Vahdati-Mashhadian N, Sahebkar A, and Fazly Bazzaz BS. Non-antibiotic adjunctive therapy: A promising approach to fight tuberculosis. Pharmacol Res. (2019) 146:104289. doi: 10.1016/j.phrs.2019.104289
8. Davuluri KS, Singh AK, Singh AV, Chaudhary P, Raman SK, Kushwaha S, et al. Atorvastatin potentially reduces mycobacterial severity through its action on lipoarabinomannan and drug permeability in granulomas. Microbiol Spectr. (2023) 11:e0319722. doi: 10.1128/spectrum.03197-22
9. Alffenaar JC, Akkerman OW, and van Hest R. Statin adjunctive therapy for tuberculosis treatment. Antimicrob Agents Chemother. (2016) 60:7004. doi: 10.1128/AAC.01836-16
10. Bruiners N, Dutta NK, Guerrini V, Salamon H, Yamaguchi KD, Karakousis PC, et al. The anti-tubercular activity of simvastatin is mediated by cholesterol-driven autophagy via the AMPK-mTORC1-TFEB axis. J Lipid Res. (2020) 61:1617–28. doi: 10.1194/jlr.RA120000895
11. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Diseases. (2013) 209:754–63. doi: 10.1093/infdis/jit550
12. Istvan ES and Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. (2001) 292:1160–4. doi: 10.1126/science.1059344
13. Goldstein JL and Brown MS. Regulation of the mevalonate pathway. Nature. (1990) 343:425–30. doi: 10.1038/343425a0
14. Dutta NK, Bruiners N, Pinn ML, Zimmerman MD, Prideaux B, Dartois V, et al. Statin adjunctive therapy shortens the duration of TB treatment in mice. J Antimicrobial Chemotherapy. (2016) 71:1570–7. doi: 10.1093/jac/dkw014
15. Skerry C, Pinn ML, Karakousis PC, Gennaro ML, Bruiners N, and Pine R. Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J Antimicrobial Chemotherapy. (2014) 69:2453–7. doi: 10.1093/jac/dku166
16. Dutta NK, Bruiners N, Zimmerman MD, Tan S, Dartois V, Gennaro ML, et al. Adjunctive host-directed therapy with statins improves tuberculosis-related outcomes in mice. J Infect Dis. (2020) 221:1079–87. doi: 10.1093/infdis/jiz517
17. Adewole OO, Omotoso BA, Ogunsina M, Aminu A, Odeyemi AO, Awopeju OF, et al. Atorvastatin accelerates Mycobacterium tuberculosis clearance in pulmonary TB: a randomised phase IIA trial. Int J Tuberc Lung Dis. (2023) 27:226–8. doi: 10.5588/ijtld.22.0548
18. Cross GB, Sari IP, Kityo C, Lu Q, Pokharkar Y, Moorakonda RB, et al. Rosuvastatin adjunctive therapy for rifampicin-susceptible pulmonary tuberculosis: a phase 2b, randomised, open-label, multicentre trial. Lancet Infect Diseases. (2023) 23:847–55. doi: 10.1016/S1473-3099(23)00067-1
19. Cross GB, Sari IP, Burkill SM, Yap CW, Nguyen H, Quyet D, et al. PET-CT outcomes from a randomised controlled trial of rosuvastatin as an adjunct to standard tuberculosis treatment. Nat Commun. (2024) 15:10475. doi: 10.1038/s41467-024-54419-3
20. Bedi P, Chalmers JD, Graham C, Clarke A, Donaldson S, Doherty C, et al. A randomized controlled trial of atorvastatin in patients with bronchiectasis infected with pseudomonas aeruginosa: A proof of concept study. CHEST. (2017) 152:368–78. doi: 10.1016/j.chest.2017.05.017
21. Mroz RM, Lisowski P, Tycinska A, Bierla J, Trzeciak PZ, Minarowski L, et al. Anti-inflammatory effects of atorvastatin treatment in chronic obstructive pulmonary disease. A controlled pilot study. J Physiol Pharmacol. (2015) 66:111–28.
22. Guerra-De-Blas PDC, Bobadilla-Del-Valle M, Sada-Ovalle I, Estrada-García I, Torres-González P, López-Saavedra A, et al. Simvastatin enhances the immune response against mycobacterium tuberculosis. Front Microbiol. (2019) 10. doi: 10.3389/fmicb.2019.02097
23. Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. (2011) 4:279–87. doi: 10.1038/mi.2011.3
24. Buhaescu I and Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem. (2007) 40:575–84. doi: 10.1016/j.clinbiochem.2007.03.016
25. Lobato LS, Rosa PS, Ferreira Jda S, Neumann Ada S, da Silva MG, do Nascimento DC, et al. Statins increase rifampin mycobactericidal effect. Antimicrob Agents Chemother. (2014) 58:5766–74. doi: 10.1128/AAC.01826-13
26. Vergne I, Chua J, Singh SB, and Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. (2004) 20:367–94. doi: 10.1146/annurev.cellbio.20.010403.114015
27. Kinchen JM and Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. (2008) 9:781–95. doi: 10.1038/nrm2515
28. Gutierrez MG. Functional role(s) of phagosomal Rab GTPases. Small GTPases. (2013) 4:148–58. doi: 10.4161/sgtp.25604
29. Aderem A and Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. (1999) 17:593–623. doi: 10.1146/annurev.immunol.17.1.593
30. Uribe-Querol E and Rosales C. Phagocytosis: our current understanding of a universal biological process. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.01066
31. Fairn GD and Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. (2012) 33:397–405. doi: 10.1016/j.it.2012.03.003
32. Koul A, Herget T, Klebl B, and Ullrich A. Interplay between mycobacteria and host signalling pathways. Nat Rev Microbiol. (2004) 2:189–202. doi: 10.1038/nrmicro840
33. Zhang M, Chen L, Wang S, and Wang T. Rab7: roles in membrane trafficking and disease. Bioscience Rep. (2009) 29:193–209. doi: 10.1042/BSR20090032
34. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
35. Songane M, Kleinnijenhuis J, Netea MG, and van Crevel R. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb). (2012) 92:388–96. doi: 10.1016/j.tube.2012.05.004
36. Ní Cheallaigh C, Keane J, Lavelle EC, Hope JC, and Harris J. Autophagy in the immune response to tuberculosis: clinical perspectives. Clin Exp Immunol. (2011) 164:291–300. doi: 10.1111/j.1365-2249.2011.04381.x
37. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, and Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. (2010) 29:1792–802. doi: 10.1038/emboj.2010.74
38. Chandra P, Grigsby SJ, and Philips JA. Immune evasion and provocation by Mycobacterium tuberculosis. Nat Rev Microbiol. (2022) 20:750–66. doi: 10.1038/s41579-022-00763-4
39. Molloy A, Laochumroonvorapong P, and Kaplan G. Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guérin. J Exp Med. (1994) 180:1499–509. doi: 10.1084/jem.180.4.1499
40. Riendeau CJ and Kornfeld H. THP-1 cell apoptosis in response to Mycobacterial infection. Infect Immun. (2003) 71:254–9. doi: 10.1128/IAI.71.1.254-259.2003
41. Fratazzi C, Arbeit RD, Carini C, and Remold HG. Programmed cell death of Mycobacterium avium serovar 4-infected human macrophages prevents the mycobacteria from spreading and induces mycobacterial growth inhibition by freshly added, uninfected macrophages. J Immunol. (1997) 158:4320–7. doi: 10.4049/jimmunol.158.9.4320
42. Kyrylkova K, Kyryachenko S, Leid M, and Kioussi C. Detection of apoptosis by TUNEL assay. Methods Mol Biol. (2012) 887:41–7. doi: 10.1007/978-1-61779-860-3_5
43. Catron DM, Lange Y, Borensztajn J, Sylvester MD, Jones BD, and Haldar K. Salmonella enterica serovar Typhimurium requires nonsterol precursors of the cholesterol biosynthetic pathway for intracellular proliferation. Infect Immun. (2004) 72:1036–42. doi: 10.1128/IAI.72.2.1036-1042.2004
44. Blanco-Colio LM, Villa A, Ortego M, Hernández-Presa MA, Pascual A, Plaza JJ, et al. 3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of Bcl-2 expression and Rho A prenylation. Atherosclerosis. (2002) 161:17–26. doi: 10.1016/S0021-9150(01)00613-X
45. Miller JL, Velmurugan K, Cowan MJ, and Briken V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PloS Pathog. (2010) 6:e1000864. doi: 10.1371/journal.ppat.1000864
46. Gelb MH, Brunsveld L, Hrycyna CA, Michaelis S, Tamanoi F, Van Voorhis WC, et al. Therapeutic intervention based on protein prenylation and associated modifications. Nat Chem Biol. (2006) 2:518–28. doi: 10.1038/nchembio818
47. Marks RE, Ho AW, Robbel C, Kuna T, Berk S, and Gajewski TF. Farnesyltransferase inhibitors inhibit T-cell cytokine production at the posttranscriptional level. Blood. (2007) 110:1982–8. doi: 10.1182/blood-2006-06-031088
48. Field K, Hayes M, James M, Dieck C, Gaylo A, and Crosby E. Do farnesyltransferase inhibitors modulate immune responses by blocking cytokine secretion from alloreactive T cells? (TRAN3P.899). J Immunol. (2014) 192:202.38–.38. doi: 10.4049/jimmunol.192.Supp.202.38
49. Du W, Lebowitz PF, and Prendergast GC. Cell growth inhibition by farnesyltransferase inhibitors is mediated by gain of geranylgeranylated RhoB. Mol Cell Biol. (1999) 19:1831–40. doi: 10.1128/MCB.19.3.1831
50. Brendolan A and Russo V. Targeting cholesterol homeostasis in hematopoietic Malignancies. Blood. (2022) 139:165–76. doi: 10.1182/blood.2021012788
51. Hilgendorff A, Muth H, Parviz B, Staubitz A, Haberbosch W, Tillmanns H, et al. Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int J Clin Pharmacol Ther. (2003) 41:397–401. doi: 10.5414/CPP41397
Keywords: Mycobacterium tuberculosis, atorvastatin, peripheral blood mononuclear cells, apoptosis, mevalonate
Citation: Sabeel S, Motaung B, Ozturk M, Mafu TS, Wilkinson RJ, Thienemann F and Guler R (2025) Immunomodulatory effects of atorvastatin on peripheral blood mononuclear cells infected with Mycobacterium tuberculosis. Front. Immunol. 16:1597534. doi: 10.3389/fimmu.2025.1597534
Received: 21 March 2025; Accepted: 16 June 2025;
Published: 03 July 2025.
Edited by:
Selvakumar Subbian, The State University of New Jersey, United StatesReviewed by:
Kathirvel Maruthai, Johns Hopkins University, United StatesPetros C. Karakousis, Johns Hopkins University, United States
Copyright © 2025 Sabeel, Motaung, Ozturk, Mafu, Wilkinson, Thienemann and Guler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reto Guler, cmV0by5ndWxlckB1Y3QuYWMuemE=; Friedrich Thienemann, ZnJpZWRyaWNoLnRoaWVuZW1hbm5AdWN0LmFjLnph
†These authors have contributed equally to this work and share senior authorship