 Yuqi Jin1
Yuqi Jin1 Yun Guo1
Yun Guo1 Yohei Kawano1
Yohei Kawano1 Megumi Sasatani2
Megumi Sasatani2 Shun Ohki1
Shun Ohki1 Keita Yamane1
Keita Yamane1 Yusei Ota1
Yusei Ota1 Yumi Tamura1
Yumi Tamura1 Yusuke Sotomaru3
Yusuke Sotomaru3 Yoshihiro Baba4
Yoshihiro Baba4 Tomoharu Yasuda1*
Tomoharu Yasuda1*- 1Department of Immunology, Graduate School of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan
- 2Department of Experimental Oncology, Research Institute for Radiation Biology and Medicine, Hiroshima University, Hiroshima, Japan
- 3Natural Science Center for Basic Research and Development, Hiroshima University, Hiroshima, Japan
- 4Division of Immunology and Genome Biology, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan
Epstein-Barr virus (EBV)-infected B cells effectively induce T cell-mediated immune surveillance that suppresses the proliferation of EBV+ B cells and development of lymphomas. However, it remains unclear whether EBV-specific T cells are involved in the surveillance of EBV-negative general tumors. To address this issue, we induced immune surveillance by expressing key EBV antigens, LMP1 and LMP2A, in germinal center B cells and investigated the formation of non-B cell tumors. LMP1/2A mice showed a significantly reduced incidence of radiation-induced T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) even in the absence of LMP antigens in tumor cells and an extended life-span compared to control mice. LMP1/2A mice showed significantly higher numbers of activated memory T cells in both CD4+ and CD8+ αβT cell fractions compared to controls, suggesting their role in the elimination of tumor cells. Despite nearly absent MHC class I expression, tumor cells were effectively killed by CD8+ T cells activated upon LMP1/2A-expressing B cells. Transcriptome analysis identified upregulation of the NKG2D-NKG2DL pathway, emphasizing the capacity of LMP1/2A-induced T cells in the recognition of common tumor specific antigens. Moreover, not only T-cell tumors, but also intestinal tumors caused by ApcMin mutation were significantly suppressed by the LMP1/2A-induced immune surveillance. These results suggest that LMP1/2A-expression associated with EBV infection contributes to pan-tumor surveillance, implicating a beneficial aspect of EBV infection in humans and providing important insights into cancer prevention.
1 Introduction
Gammaherpesviruses are known to establish lifelong latent infections in their hosts (1) and can systemically enhance immune surveillance not only through antigen-specific responses but also by mechanisms such as bystander T cell activation, cross-presentation, and the stimulation of innate immune pathways (2–4). Among them, EBV a human-specific gammaherpesvirus, is of particular research interest due to its global prevalence and its ability to infect B cells and establish lifelong latency (5).
In healthy individuals, T and NK cell-mediated immune surveillance effectively suppresses EBV-infected B cells which are restricted into a small fraction of circulating B cells with minimal or no viral gene expression (6). However, as immune surveillance weakens, EBV adopts progressively different latency states. In latency I, characteristic of Burkitt lymphoma (BL), viral gene expression is highly restricted; in latency II, seen in Hodgkin’s disease (HD), a few viral genes are expressed; and in latency III, associated with post-transplant lymphoproliferative disorder (PTLD) and AIDS-related B cell lymphoma, all latent viral genes are activated (7, 8). Despite extensive research, the mechanisms by which EBV-infected cells activate immune surveillance and inhibit malignant tumor development remain to be fully elucidated.
Among the EBV-encoded proteins, latent membrane proteins LMP1 and LMP2A expressed in latency II and III are central to B cell activation and transformation. LMP1 mimics an active CD40 receptor, primarily activating the NF-κB and JNK signaling pathways (9, 10). Notably, in vitro studies have identified LMP1 as the only EBV latent protein capable of transforming rodent fibroblasts (11). In transgenic mouse models of EBV pathology, expression of LMP1 in B cells induced B cell proliferation that was subsequently cleared by T cells. Depletion of T cells promoted rapid expansion of LMP1-expressing B cells and lymphomagenesis (12, 13). Conditional LMP1 expression in mouse, where LMP1 is induced in a timed manner and only in a few fractions of B cells is sufficient to model acute EBV infection as seen in human infectious mononucleosis (14, 15). LMP2A, frequently co-expressed with LMP1, complements and amplifies the effect of LMP1 in latent EBV infection mouse models (15, 16). LMP2A substitutes for the B cell receptor (BCR) signaling, rescuing germinal center (GC) B cells from apoptosis caused by the loss of BCR expression due to somatic hypermutation (7, 17, 18). Moreover, LMP2A activates PI3K and MAPK signaling pathways (19), augmenting LMP1’s oncogenic potential while simultaneously eliciting stronger immune surveillance responses (15, 16, 20). These findings highlight the opposing actions of LMP1 and LMP2A, namely their dual roles in oncogenesis and immune surveillance of tumors.
Human studies have demonstrated that EBV-transformed B lymphoblastoid cell lines (LCLs) can induce a population of T cells that recognize EBV-negative B lymphoma cells in vitro, despite lacking viral antigen specificity (21). This suggests that EBV-induced immune surveillance may exert broader antitumor potential beyond targeting viral antigens alone. In mouse models, LMP1 expression during B cell transformation has been shown to induce a variety of non-viral antigens, including tumor-associated antigens (TAAs) and self-antigens, while also enhancing MHC class I antigen presentation and upregulating multiple costimulatory molecules such as CD70, OX40L, and 4-1BBL (12, 22, 23). These effects collectively activate a population of T cells capable of recognizing both viral and tumor-associated antigens. In addition, LMP1 promotes the expression of stress-induced ligands such as NKG2DL and Fas, activating innate immune pathways that enable NK cells and T cells to mediate non-specific cytotoxicity (12). These findings suggest that EBV-induced immune surveillance is not limited to classical MHC-restricted antigen-specific responses but may also involve broader mechanisms such as cross-presentation, bystander activation, and enhanced innate immune signaling. This broadened scope of T cell reactivity potentially enables the recognition and elimination of EBV-negative tumors, representing a cross-protective antitumor mechanism mediated by EBV. Notably, LMP2A acts as a functional cofactor of LMP1, enhancing not only its oncogenic potential but also its immunogenicity, thereby contributing to more robust immune surveillance (14–16, 24). However, direct experimental evidence demonstrating that EBV-induced immune surveillance can eliminate EBV-negative tumors remains lacking. In this study, we investigate whether immune surveillance triggered by the EBV-encoded proteins LMP1 and LMP2A can effectively suppress the development of general tumors independently of EBV antigens.
2 Materials and methods
2.1 Mouse strains
Previously described Cγ1-Cre mice were generated by targeting 129P2-derived embryonic stem (ES) cells and backcrossed to C57BL/6 mice (25). R26-LMP1/2A(iresEYFP)flSTOP mice were generated by targeting C57BL/6-derived ES cells as described detail in the following section (Supplementary Figure 1A). Cγ1-Cre mice were crossed with R26-LMP1/2A(iresEYFP)flSTOP mice to generate Cγ1-Cre/+; R26-LMP1/2A(iresEYFP)flSTOP/+ (GCB-LMP1/2) mice in which LMP1 and LMP2A are expressed with EYFP reporter in GC B cells. Cγ1-Cre/+ mice were used as the control to compare with GCB-LMP1/2 mice. ApcMin/+ mice (26) were crossed with GCB-LMP1/2A mice to generate ApcMin/+; Cγ1-Cre/+; R26-LMP1/2A(iresEYFP)flSTOP/+ (ApcMin/+LMPKI) mice which carry the ApcMin/+ mutation in GCB-LMP1/2A mice. For in vitro cell culture and in vitro killing assays, mice aged 10–20 weeks were used. In radiation treatment and ApcMin/+ induced intestinal tumor formation experiments, the timing of euthanasia was determined based on tumor progression and is specified in each corresponding experiment. All animals were housed under specific pathogen-free conditions and handled according to protocols approved by the Ethics Review Committee for Animal Experimentation of Hiroshima University and the institutional committee at Kyushu University.
2.2 Generation of R26-LMP1/2A(iresEYFP)flSTOP mice
For the targeting vector construction, the 5’ homology arm (1.1 kb) and the 3’ homology arm (1.0 kb) of Gt(ROSA)26Sor were amplified by PCR from C57BL/6 mouse genome. LMP1 and LMP2A sequences from EBV B95–8 strain were tandemly arranged by flanking T2A self-cleaving peptide sequence, followed by EYFP gene with internal ribosome entry site (IRES) and a bovine growth hormone polyadenylation (BGH pA) sequence. The splice acceptor and the loxP flanked neomycin resistant stop cassette was amplified from Rosa26 targeting vector (27) and positioned upstream of LMP1 and LMP2A. PCR fragments were assembled in pBlueScript II plasmid vector using NEBuilder HiFi DNA assembly master mix (NEB) to generate R26-LMP1/2A(iresEYFP)flSTOP targeting vector. PCR amplifications were performed using high fidelity KOD DNA polymerase (TOYOBO). The C57BL/6N ES cells, EGR-101, was kindly provided from Dr. Masahito Ikawa (28). 1x106 C57BL/6N ES cells were electroporated with 100 ng/μl (0.61 μM) recombinant Cas9 Nuclease 3NLS (IDT #1074181), 36 ng/μl (1 μM) gR26–1 crRNA (5’- ACTCCAGTCTTTCTAGAAGA//TGG -3’, IDT): tracrRNA duplex, and 100 ng/μl pR26-LMP1/2A(iresEYFP)flSTOP plasmid DNA in 100 μl of 1xOpti-MEM medium (ThermoFisher) using NEPA 21 super electroporator (NEPA GENE) and 2 mm gap cuvette. The following parameters were used for electroporation; Poring pulse (voltage 125 V; pulse length 2.5 msec; pulse interval 50 msec; number of pulses 2; decay rate 10%; polarity +) and Transfer pulse (voltage 20 V; pulse length 50 msec; pulse interval 50 msec; number of pulses 5; decay rate 40%; polarity ±). The electrical resistivity was typically 0.03 to 0.05 kΩ. Electroporated cells were selected for 6–7 days under ES medium (StemSure D-MEM, 15% fetal calf serum (FCS), L-glutamine, non-essential amino acids, β-mercaptoethanol, and LIF) containing 200 μg/ml G418 (Nacalai tesque). ES cell colonies were subjected to PCR screening to select correct clones. ES cells from selected clones were injected into Balb/c blastocysts. A chimeric male (80-90% chimerism) was crossed to C57BL/6 females to obtain germline transmitted offspring.
2.3 Radiation treatment
Mice (4-week-old) were subjected to total body irradiation (TBI) using a Gamma Cell 40 Exactor Research Irradiator (Best Theratronics), equipped with a 148 TBq 137Cs source. The irradiation was delivered at a dose rate of 770 mGy/min. Each mouse received a total of four weekly fractions of 1.6 Gy, resulting in a cumulative dose of 6.4 Gy. The absorbed dose was calibrated using a GD-302M glass dosimeter (AGC Techno Glass).
2.4 Monitoring and euthanasia criteria
After TBI, mice were monitored every three days for general health status, including activity level, posture, grooming, fur condition, and signs of apparent weight loss. For survival analysis experiments, mice exhibiting signs of distress—such as severe lethargy, hunched posture, ruffled fur, reduced mobility, or visually estimated weight loss—were euthanized according to humane endpoint criteria approved by the institutional animal ethics committee. Organs were collected from these moribund mice and used as end-stage disease samples for further analysis. All end-stage mice were euthanized between 15 and 30 weeks after radiation. For experiments assessing the incidence of T-ALL, all mice were euthanized at a defined time point—14 weeks after the final radiation—prior to the onset of overt clinical symptoms. This endpoint was selected to ensure consistency in evaluation and adherence to ethical standards.
2.5 B cell and T cell purification
Splenic B cells, CD4+ T cells, and CD8+ T cells were isolated by either magnetic bead-based negative selection or FACSAria cell sorter. For B cell purification, splenocytes were incubated with biotinylated anti-CD43 (R2/60) antibodies. For CD4+ T cell and CD8+ T cell purification, splenocytes were incubated with biotinylated antibody cocktail of anti-CD11b (M1/70), CD11c (HL3), CD49b (DX5), I-Ab (KH74), Ter119 (Ter119), Gr-1 (RB6-8C5), and TCRγδ (GL3) antibodies, combined with either anti-CD4 (GK1.5) antibodies for CD8+ T cell enrichment or anti-CD8 (53-6.7) antibodies for CD4+ T cell enrichment. Following 15 minutes incubation at 4°C, cells bound to biotinylated antibodies were captured using BD IMag™ Streptavidin Particles Plus (BD Biosciences) for 10 minutes, and the unbound negative fraction was collected. B cells with at least 95% purity and CD4+ and CD8+ T cells with at least 90% purity were subsequently used for cell culture experiments.
2.6 Preparation of B cells and T-ALL cell line
To establish LMP1 and LMP2A-expressing B cells (BLMP1/2A cells), purified B cells from the spleens of GCB-LMP1/2A mice were cultured in 10% FBS-1640 medium and maintained at 37°C in a 5% CO2 humidified atmosphere for 12 days. Actively proliferating cells were harvested and cryopreserved at a concentration of 2 × 106 cells/ml using a Bambanker (Nippon genetics) at -80°C. For experimental use, cells were thawed and cultured in 10% FBS-1640 medium for three days. For CD40 activated B cells (CD40 act-B cells), purified B cells from wild-type C57BL/6 mice were stimulated with 2 μg/ml anti-CD40 antibody (HM40-3, BioLegend) and 20 ng/ml recombinant mouse IL-4 (Biolegend) in 10% FBS-RPMI-1640 medium for 5 days. To establish a T-ALL cell lines, thymuses were harvested from terminal-stage T-ALL mice (over 20 weeks after the final radiation treatment). The thymic tissues were processed into a single-cell suspension and cultured in 10% FBS-1640 medium. The cells were passaged every three days. While non-malignant cells died within five passages, T cells stably propagating over 10 passages were considered as T-ALL cell line.
2.7 Western blotting
Cells were lysed in 1% NP-40 buffer (150 mM NaCl, 0.5 M NaF, 10 mM Tris pH 7.4, 0.5 mM EDTA, and 2 mM PMSF) containing 2mM phenylmethylsulfonyl fluoride (Nacalai Tesque) supplemented with protease inhibitors (Nacalai Tesque). Protein concentrations were measured using the Pierce™ BCA assay (Thermo Fisher Scientific). Equal amounts of protein were separated on 10% SDS–polyacrylamide gels and transferred to PVDF membranes (Bio-Rad). Membranes were blocked with 6% skim milk in TBS-TweenTM-20 (Thermo Fisher Scientific) and incubated with primary antibodies overnight at 4 °C, followed by HRP-conjugated secondary antibodies. Proteins were visualized using ECL reagents (Cytiva) and detected with ImageQuant LAS 500 (GE Healthcare). The following antibodies were used: mouse anti-EBV LMP1 (S12,Sigma-Aldrich), rat anti-EBV LMP2A (14B7, Santa Cruz Biotechnology), rabbit anti-β-actin (Proteintech), HRP-conjugated goat anti-mouse IgG (Sigma-Aldrich), HRP-conjugated donkey anti-rat IgG (Jackson ImmunoResearch), and HRP-conjugated goat anti-rabbit IgG (Proteintech).
2.8 In vitro cell culture
All cells were cultured in RPMI-1640 (FUJIFILM Wako Chemicals) medium supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 52 μM β-mercaptoethanol, non-essential amino acids, and penicillin-streptomycin (referred to as 10% FBS-1640). For in vitro T cell culture, FACS-purified CD4+ or CD8+ T cells from the spleen were co-cultured with γ-irradiated (6–8 Gy) T-ALL, BLMP1/2A and CD40 act-B cells at a 1:1 T cell to lymphoma cell ratio. The cultures were re-stimulated every three days in 10% FBS-1640 medium supplemented with 10 ng/mL IL-2 (R&D Systems).
2.9 In vitro killing assay
The in vitro killing assay was adapted from previously described methods with modifications (12, 24). Briefly, ex vivo expanded CD8+ T cells were purified by FACS sorting and incubated with target cells at varying effector-to-target (E: T) ratios for 4 hours in U-bottom 96-well plates. Prior to incubation, the effector-target cell mixtures were centrifuged at 200 rpm for 2 minutes to facilitate cell contact. Following incubation, cultures were stained with antibodies against TCRβ, CD8a, and CD24 to distinguish target and effector cells. Cell viability was assessed by propidium iodide (PI, Sigma-Aldrich) staining, where dead cells were identified as PI-positive and viable cells remained PI-negative. Percent-specific killing was calculated using the formula: % specific killing = (% apoptotic target cells in cultures with both effectors and targets) − (% apoptotic target cells in cultures with targets alone). In blocking receptor experiments, ex vivo expanded CD8+ T cells were pre-incubated with NKG2D antibody, 2B4/SLAMF4 antibody, or corresponding isotype controls at a concentration of 20 µg/ml for 60 minutes at 37°C. The killing assay was then performed at an E:T ratio of 20:1 in the continued presence of the blocking antibodies.
2.10 Isolation of lymphocytes from intestinal tumor nodule
To isolate lymphocytes from tumor nodules, mice were euthanized, and the intestines were excised, thoroughly rinsed, and soaked in cold PBS. Tumor nodules were collected from both the small intestine and colon, then minced and digested in a solution containing 1 mg/mL Collagenase D (Roche) and 0.5 mg/mL DNase I (Roche) at 37°C with shaking for 1 hour. The resulting cell suspension was filtered through a 70-μm nylon mesh and treated with Gey’s solution to lyse red blood cells. Cells were then washed with 2% FBS/PBS and resuspended in 33.75% Percoll (GE Healthcare Life Sciences) before centrifugation at 2,300 rpm for 25 minutes at room temperature.
2.11 Flow cytometry
Single-cell suspensions from the spleen, Peyer’s patches (PP), mesenteric lymph nodes (mLN), thymus, and bone marrow were resuspended in Gey’s solutions for red blood cell lysis. Cells were treated with FcBlock (2.4G2, BD Biosciences) followed by staining with fluorochrome-conjugated antibodies or biotinylated antibodies. The cells stained with biotinylated antibodies were detected by fluorochrome-conjugated streptavidin. Anti-B220/CD45R (RA3-6B2), CD19 (6D5), Notch1 (HMN1-12), CD107a (1D4B), NK-1.1 (PK136), Granzyme B (GB11), Perforin (B-D48), PD-1 CD279 (29F.1A12) were purchased from BioLegend; Anti- IFN-γ (XMG1.2), Ly-6A/E (Sca1, D7), CD117 (2B8), CD95 (Jo2), CD4 (RM4-5), CD8a (53-6.7), CD24 (M1/69), TCRβ (H57-597), CD44 (IM7), CD25 (7D4), CD69 (H1.2F3), IgG2a (R35-95) isotype control, TCR γδ (GL3) were purchased from BD Biosciences; Anti- c-Myc (AG1263) were purchased from Proteintech, Anti- CD45 (30-F11), CD314 (CX5), CD244.2 (eBio244F4) were purchased from Thermo Fisher Scientific. Cells were stained with PI (Sigma-Aldrich) to exclude dead cells. For intracellular staining, the cells were fixed and permeabilized using a Foxp3 staining kit (Thermo Fisher Scientific). For cytokine staining, cells were treated with 50 ng/ml of phorbol 12-myristate 13-acetate (PMA) and 1 μg/ml of ionomycin with BD GolgiStop containing monensin for 6 hours followed by staining using cytofix/cytoperm plus fixation/permeabilization solution kit (BD Biosciences). Stained cells were analyzed using FACSCanto II (BD Biosciences) and CytoFlex (BECKMAN COULTER) data were analyzed on FlowJo software (BD Biosciences).
2.12 Histology and immunohistochemistry
Cryosections (6 µm) of spleen, thymus, and intestinal tumor tissues were prepared, air-dried, and either immediately used for hematoxylin and eosin (H&E) staining or dehydrated in acetone, air-dried, and stored at -80°C for subsequent immunohistochemistry. For immunohistochemistry, the following antibodies were used: anti-Keratin 5 (AF138) from Covance Research and anti-Keratin 8 (Ks8.7) from PROGEN; anti-CD8 (53-6.7) and CD90.2 (30-H12) from BioLegend; and anti-CD4 (RM4-5) and CD45R (RA3-6B2) from BD Biosciences.
2.13 Microarray
Gene expression profiling was performed on FACS-sorted T cell samples using Affymetrix GeneChip Mouse Genome 430 2.0 Arrays, according to the manufacturer’s recommendations (Affymetrix, Santa Clara, CA, USA) and data analysis was performed as previously reported (24, 29, 30). Fluorescence ratios were normalized by applying the RMA algorithm using the BRB Array Tools software package (available at https://brb.nci.nih.gov/BRBArrayTools/). These data were then interpreted in comparison to gene expression results obtained in naive T cells. Heatmap were generated by using R-tidyheatmaps. The complete microarray data are available at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/projects/geo; accession number GSE206802).
2.14 Quantitative real time PCR
Total RNA was extracted from normal thymus and spleen, irradiated thymus and spleen, T-ALL cell line, BLMP1/2A cells, and CD8+ T cells activated by T-ALL and BLMP1/2A cells using ISOGEN II (NIPPON GENE) following the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using the ReverTra Ace qPCR Master Mix (TOYOBO) according to the manufacturer’s protocol. Real-time PCR was performed with Thunderbird SYBR qPCR mix (TOYOBO) on a Bio-Rad Real-Time PCR system. The specific primers used were as follows: (Notch1: 5’-GCTGCCTCTTTGATGGCTTCGA-3’ and 5’-CACATTCGGCACTGTTACAGCC-3’; Myc: 5’-TCGCTGCTGTCCTCCGAGTCC-3’ and 5’-GGTTTGCCTCTTCTCCACAGAC-3’; Lmo-2: 5’-TGGGACGGAAATTGTGCAGGAG-3’ and 5’-GGCGCATTTGAAACACTCCAGG-3’; Ptcra: 5’-CTGCAACTGGGTCATGCTTC-3’ and 5’-GTCCAAATTCTGTGGGTGGG-3’; Klrk1: 5’- CCTATCACTGGATGGGACTGGT-3’ and 5’- GCTTGAGCCATAGACAGCACAG-3’; Ulbp1: 5’- GTGCAGGAGACTAACACAACCG-3’ and 5’- TGCCAGTGCTTGTGTCAACACG-3’; Cd244: 5’- CAGTTGCCACAGCAGACTTTCC-3’ and 5’- CTTCCTGGAAGGCTGGACTACT-3’; Cd48: 5’- GCTGCGTGAAACTGAGAACGAG-3’ and 5’- CACACGATAGCCTCAGGTGACA-3’).
2.15 Detection of V–J recombination at the TCRβ locus
Thymocytes harvested 14 weeks after the final radiation treatment were suspended in lysis buffer containing Proteinase K (Roche) and incubated at 55°C for 1 hour, followed by 95°C for 5 minutes to inactivate the enzyme. The lysates were used as templates for PCR amplification. Primers were used as previously reported (31): Dβ1, 5’-TTATCTGGTGGTTTCTTCCAGC-3’; Dβ2, 5’-GCACCTGTGGGGAAGAAACT-3’; Jβ1.6, 5’-GGTAGAAAGGTAGAGGGTTCCAGA-3’; Jβ2.6, 5’-TGAGAGCTGTCTCCTACTATCGATT-3’.
2.16 Statistical analysis
All data analyses were performed with Prism 9 software (GraphPad). The results were presented as mean ± SD. p values were calculated by multiple student`s t-test, multiple t-tests with Holm-Šidák correction, one-way or two-way ANOVA. Statistical Significance was determined with alpha<0.05 and presented as *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
3 Results
3.1 Establishment of immune surveillance in GCB-LMP1/2A mice
To mimic the immune surveillance induced by latent EBV infection in humans as previously reported (15, 16), we generated a genetic mouse model Cγ1-Cre/+; R26-LMP1/2A(iresEYFP)flSTOP/+ referred to hereafter as GCB-LMP1/2A mice in which LMP1 and LMP2A are expressed with EYFP reporter in GC B cells (Figure 1A, Supplementary Figure 1A). To confirm the expression and transforming potential of LMP1/2A, we purified B cells from the spleens of GCB-LMP1/2A mice and cultured them in vitro (Figure 1B). In culture, B cells expressing LMP1 and LMP2A displayed characteristic cluster formation and significant proliferative activity (Figures 1B, C). FACS analysis revealed that these proliferating cells co-expressed Fas, a marker for LMP1 expression (12, 32), and EYFP reporter protein, confirming that expression of LMP1 and LMP2A induces marked proliferation in B cells (Figure 1D). Western blot analysis further confirmed expression of LMP1 and LMP2A in B cells (Supplementary Figure 1B). Hereafter, we refer to ex vivo expanded LMP1/2A-expressing B cells as BLMP1/2A cells. These findings demonstrated that LMP1 and LMP2A are successfully expressed in GC B cells and have potential transforming capacity of B cells. However, no abnormal B cell proliferation or lymphomas were observed in immune-competent GCB-LMP1/2A mice. Instead, the number and percentage of GC B cells in the spleens of GCB-LMP1/2A mice were significantly reduced compared to control mice (Figure 1E), while the proportion of CD8+ effector memory T cells (TEM) defined by CD8+CD44+CD62L- surface phenotype was notably increased (Figure 1F). Similar trends were observed in the mesenteric lymph nodes (mLNs) and Peyer’s Patches (PPs), where GC reactions are consistently observed (Supplementary Figure 1C). To further confirm that the increase in CD8+ TEM cells was directly induced by LMP1/2A-expressing B cells, we conducted an in vitro co-culture assay using CD8+ T cells isolated from control mice. When co-cultured with BLMP1/2A cells or CD40-activated B cells (CD40 act-B), BLMP1/2A cells promoted the proliferation of CD8+ TEM cells, whereas CD40 act-B cells did not (Supplementary Figures 1D, E). These results indicate that immune surveillance against LMP1/2A-expressing B cells successfully established in GCB-LMP1/2A mice, effectively suppressing expansion of those B cells and maintaining CD8+ T cells in a memory state.
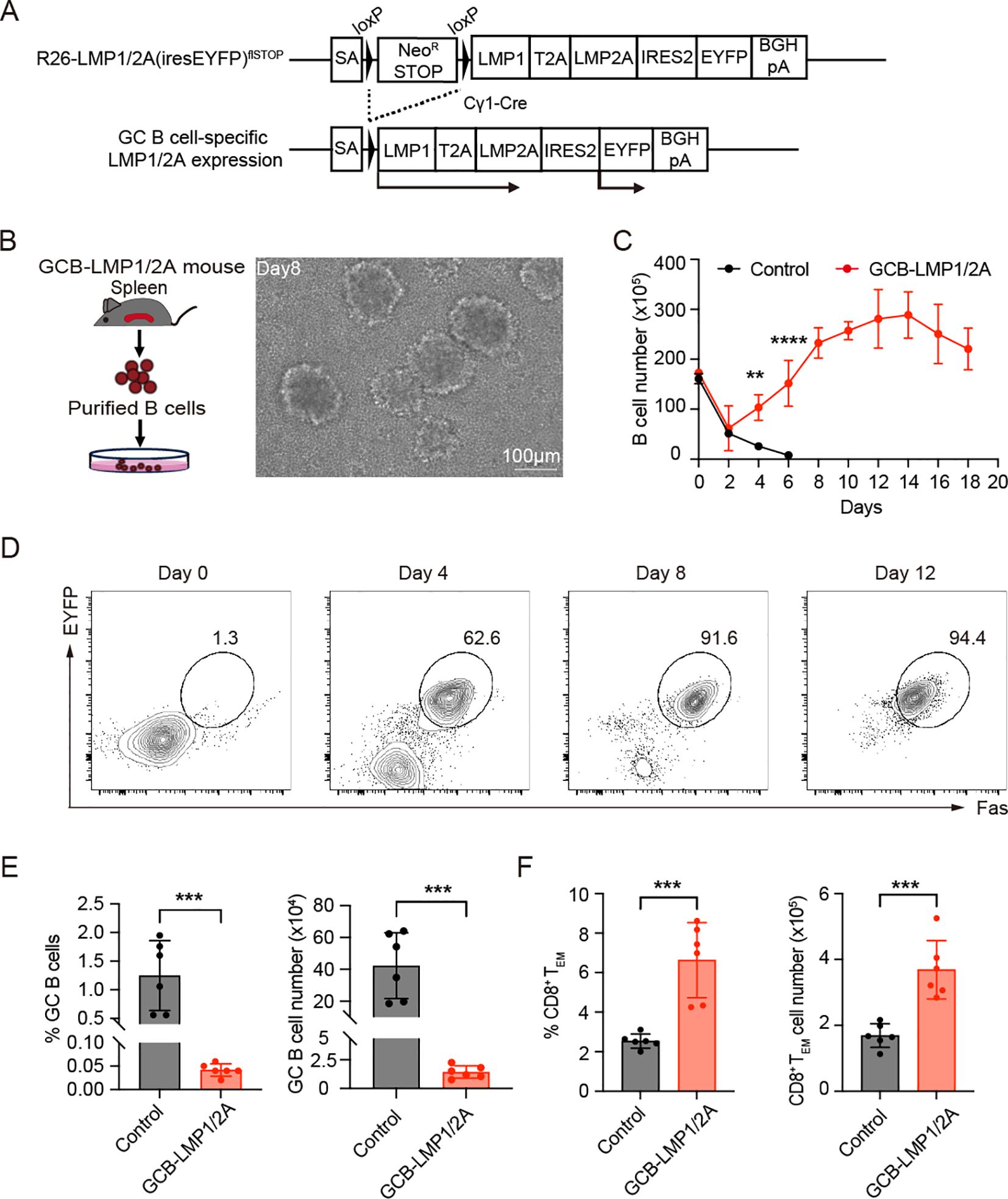
Figure 1. Characterization of germinal center B cell-specific LMP1/2A-expressing mice. (A) Targeting strategy for conditional expression of LMP1 and LMP2A in germinal center (GC) B cells. LMP1, LMP2A, and EYFP reporter genes were targeted into the Rosa26 locus with the neomycin resistance stop cassette enabling constitutive expression of those genes after Cre-mediated excision of stop cassette (R26-LMP1/2A(iresEYFP)flSTOP mouse) and crossed with GC B cell-specific Cre strain, Cγ1-Cre mice. SA, splice acceptor. (B) Workflow for isolating and culturing LMP1/2A+ B cells from GCB-LMP1/2A mice (left). Morphological changes of B cells during culture, showing increased cell size and aggregation into large clumps (right). Scale bar, 100 μm. (C) Growth curves of in vitro cultured B cells from control and GCB-LMP1/2A mice (n=3). Statistical significance tested using two-way ANOVA with Bonferroni’s multiple comparisons test; **p < 0.01; ****p < 0.0001. (D) FACS analysis of in vitro cultured B cells. Proliferating cells expressed Fas, confirming their GC B cell identity. EYFP was used as a reporter for LMP1/2A expression. (E, F) Percentage and cell count of GC B cells (E) and CD8+ TEM cells (F) in the spleens of GCB-LMP1/2A mice compared to control mice (n=6). Statistical significance tested using an unpaired two-tailed Student’s t-test; ***p < 0.001.
3.2 Total body irradiation induces T cell acute lymphoblastic leukemia/lymphoma
EBV-specific cytotoxic T lymphocytes (CTLs) not only exhibit therapeutic effects against EBV-associated lymphomas but have also been found to have potential efficacy against EBV-negative tumors, particularly B cell malignancies (21, 33–36). However, it remains unclear whether it has a similar effect on EBV-negative T-cell-associated tumors. Repeated low-dose gamma-ray radiation exposure has been shown to induces thymic-origin T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) in over 90% of C57BL/6 mice (37–39). In this study, we subjected 4-week-old mice to total body irradiation (TBI), administering 1.6 Gy by weekly four times, 6.4 Gy in total (Figure 2A). Approximately 15 weeks post-radiation, symptoms such as rickets, weight loss, and reduced mobility began to appear, which eventually progressed to T-ALL. Upon dissection, we observed abnormal enlargement of the thymus, mLN, and spleen (Figure 2B), accompanied by an increase in cellularity (Supplementary Figure 2A). Immunofluorescence staining demonstrated the disruption of normal cortical and medullary regions in the thymus, suggesting impaired thymic function and abnormal T-cell development (Figure 2C). The spleen showed T cell hyperplasia, accompanied by a significant reduction in B cell areas (Figure 2D). FACS analysis revealed that these abnormally proliferating and tissue infiltrating T cells were CD24+ immature T cells (Supplementary Figure 2B). Furthermore, no B cell hyper-proliferation was observed in GCB-LMP1/2A mice, suggesting that immune surveillance against LMP1/2A+ B cells remains active even after the onset of T-ALL (Supplementary Figure 2C).
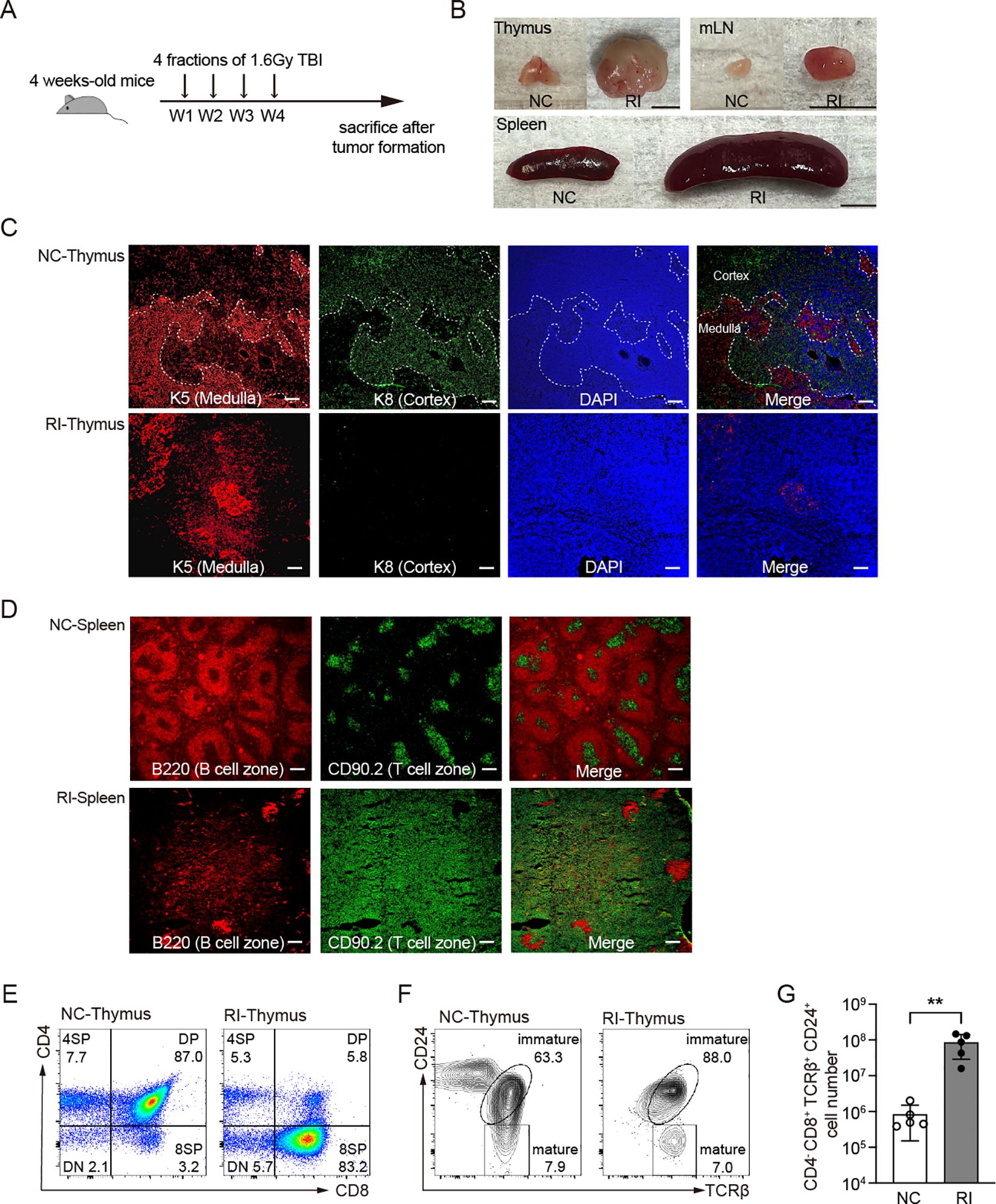
Figure 2. Histological and flow cytometric analysis of mice subjected to total body irradiation. (A) Mice received 1.6 Gy of total body irradiation (TBI) weekly at indicated time points (vertical arrows) for a total four doses of 6.4 Gy. W, week. (B) Representative pictures of thymus and spleen from non-irradiated control mice (NC) and mice after 30 weeks of radiation (RI). mLN, mesenteric lymph nodes. Scale bar=0.5 cm. (C, D) Immunostaining of thymus (C) and spleen (D) from the indicated mice. (C) Thymus were stained with K5 and K8 antibodies to identify thymic epithelial cells. Thymic medulla, K5+; Thymic cortex, K8+. (D) Splenic B cell zone and T cell zone were stained with B220 and CD90.2, respectively. Scale bar=200 μm. (E-F) Representative FACS results of thymus from NC and RI mice with T-ALL. (E) CD4 and CD8 markers distinguish stages of T cell development. (F) CD24 and TCRβ markers distinguish mature and immature stage from 8SP cells. (G) Cell number of T-ALL (CD4-CD8+TCRβ+CD24+) in thymus from NC and RI mice. Each dot indicates one mouse (n=5). Statistical significance tested using an unpaired two-tailed Student’s t-test; **p < 0.01. All RI samples were collected from mice at the end stage of disease progression, between 15 and 30 weeks after radiation, based on humane endpoint criteria detailed in the Materials and methods section.
Compared to the non-irradiated control group, irradiated mice exhibited an abnormally increased percentage of CD8 single-positive (8SP) thymic cells (Figure 2E). These cells displayed an immature phenotype characterized by TCRβ+CD24+ expression (Figure 2F), with a significant increase in their absolute numbers (Figure 2G). The accumulation of CD4-CD8+TCRβ+CD24+ cells in the thymus suggests the onset of T-ALL and migration of thymic tumor cells to the periphery such as spleen (40).
Similar to human pediatric T-ALL, radiation-induced murine T-ALL displayed elevated levels of Notch1, Myc, and Lmo2 mRNAs, a hallmark of human T-ALL (Supplementary Figure 3A) (41, 42). Upregulation of those genes likely contributes to the transformation of normal thymic cells into T-ALL. Furthermore, radiation exposure led to the overexpression of pre-TCRα (Ptcra) mRNAs characteristic in proliferating DN to DP thymocytes, suggesting involvement of pre-TCRα signal in thymic hyperplasia (Supplementary Figure 3B). Sustained pre-TCRα expression may further enhance Notch1 signaling, playing a key role in T-ALL initiation and progression (43). Collectively, these results confirmed that radiation exposure reproducibly induces T-ALL in mice.
3.3 GCB-LMP1/2A mice exhibited a lower incidence of clonal lymphomas and a longer life span after irradiation
Next, we evaluated the incidence of radiation-induced T-ALL. Following radiation exposure, clonal expansion occurs during the rearrangement of the TCRβ receptor locus, serving as one of the key early markers of T-ALL development. By assessing TCRβ rearrangement, we could determine the incidence of early tumor onset. We adopted PCR amplification to detect D-J recombination at TCRβ gene locus (31, 44, 45). Three sets of primers were used: one set for detecting recombination between the Dβ1 and Jβ1 loci, the second set for the Dβ2 and Jβ2 loci, and the third set for the Dβ1 and Jβ2 loci. DNA from the brain exhibited only a single germline band, serving as a control for the absence of recombination. DNA from the normal thymus contains polyclonal thymocytes, which exhibit multiple bands of uniform intensity due to recombination between various Dβ and Jβ loci. In contrast, DNA from the lymphoma exhibited single monoclonal band or a few oligoclonal bands with increased intensity. By assessing TCRβ rearrangements in thymocytes from control and GCB-LMP1/2A mice, we evaluated the incidence of clonal T-ALL expansion in thymus (Figure 3A). At 14-weeks post-radiation exposure, we examined both genotypes and found that the incidence of clonal T-ALL in GCB-LMP1/2A mice was 35.7%, significantly lower than the 64.3% observed in the control mice (Figure 3B). Furthermore, GCB-LMP1/2A mice exhibited a significantly longer lifespan following radiation exposure (Figure 3C). These data indicate that expression of EBV latency genes LMP1 and LMP2A in GC B cells activates host immune surveillance that reduces the incidence of radiation-induced T-ALL despite the absence of EBV antigens in T-ALL cells.
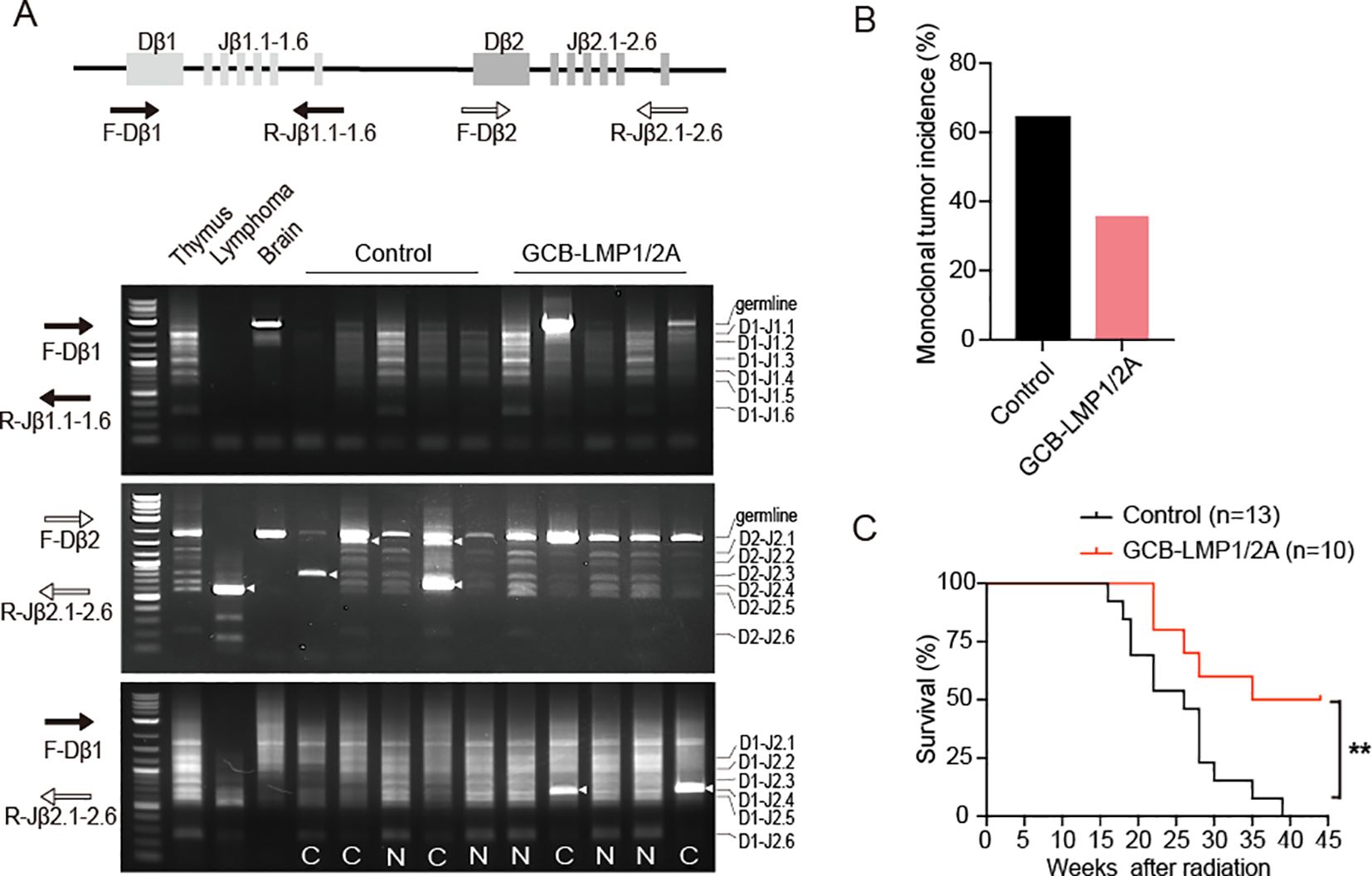
Figure 3. T-ALL incidence and survival curve of total body irradiated mice. (A) Illustration of the TCRβ locus and positions of the PCR primers used for detection of TCRβ rearrangements. The lower panels display gel electrophoresis of PCR products from three primer sets: F-Dβ 1/R-Jβ 1.6 (top), F-Dβ 2/Jβ 2.6 (middle), and F-Dβ 1/R-Jβ 2.6 (bottom). Clonal bands are indicated by arrowheads. N, Normal TCRβ rearrangements; C, Associated with clonal expansion. (B) Monoclonal tumor incidence of T-ALL, n=14 in each group. (C) Kaplan–Meier survival curves for mice after radiation (log-rank test, **p < 0.01). Number of mice are indicated.
3.4 Effector and memory T cells induced by LMP1/2A+ B cells are maintained in GCB-LMP1/2A mice even after irradiation
To uncover the underlying reasons for the lower incidence of T-ALL and a longer lifespan observed in GCB-LMP1/2A mice, we analyzed the acute phase of thymus within 4 weeks following radiation exposure. The thymic cell numbers decreased after radiation, especially the lowest levels occurring in the second week (Figure 4A). The FACS analysis revealed that reduction in DN3 stage was particularly lowest at week 2 and then turned to increase (Figure 4B). Existing studies have shown that when the competitive mechanisms of normal cells in the thymus are disrupted, thymic cells maintain a state of self-renewal, particularly DN3 stage cells, which could lead to the development of leukemia (46–48). Based on this, the reduced incidence of T-ALL in GCB-LMP1/2A mice can be explained by the impaired self-renewing DN3 cells in thymus. However, this seems unlikely because we observed a significantly higher number of DN3 cells in the thymus of GCB-LMP1/2A mice (Figure 4B). Another possible reason is the impaired replenishment of Lineage-Sca1+cKit+ (LSK) cells in the bone marrow (BM), a population of hematopoietic stem/progenitor cells (HSPCs) that can effectively resist radiation damage and improve the survival rates of mice with acute hematopoietic radiation syndrome (49–51). However, we observed no significant differences in the percentage of LSK cells in the BM (Supplementary Figure 4A). Therefore, these data suggest that the lower incidence of T-ALL in GCB-LMP1/2A mice is primarily influenced by environmental factors of T-ALL or its precursor cells within the thymus.
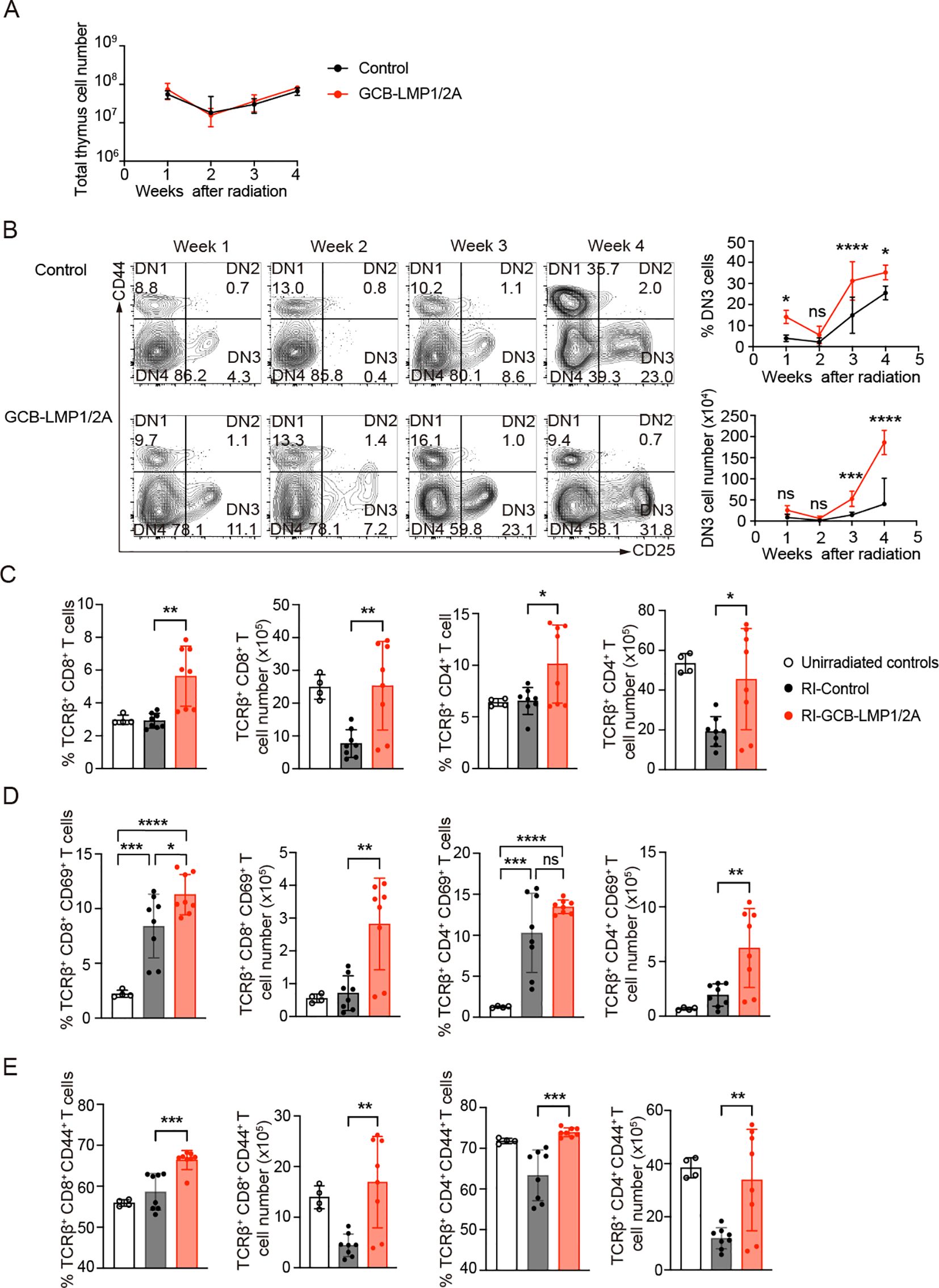
Figure 4. Changes in thymic cell populations following radiation exposure. (A) Total thymic cell counts after the last radiation exposure. Number of mice at week 1 (n=4), week 2 (n=5), week 3 (n=5), and week 4 (n=3). (B) Representative FACS plots of CD4 and CD8 double-negative cells in the thymus at indicate week after the last radiation (left). The percentage in DN fraction (upper right) and absolute number (lower right) of DN3 cells (CD44-CD25+) in thymus at indicated time points. Statistical analysis was performed using two-way ANOVA with Bonferroni’s multiple comparisons test; *p < 0.05; ***p < 0.001; ****p < 0.0001; ns, not significant. (C-E) Percentages and absolute numbers of each T cells in thymus at 3 weeks after the last radiation. (C) The percentage and absolute numbers of TCRβ+CD8+ and TCRβ+CD4+ T cells in thymus. (D) The Percentage and absolute numbers of CD69+ in TCRβ+CD8+ or TCRβ+CD4+ T cell fractions. (E) The Percentage and absolute numbers of CD44+ in TCRβ+CD8+ or TCRβ+CD4+ T cell fractions. Number of mice in unirradiated controls (n=4), RI-Control (n=8), and RI-GCB-LMP1/2A (n=8). Statistical analysis was performed using One-way ANOVA with Bonferroni’s multiple comparisons test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
In the third week after the radiation exposure, where thymic cell numbers began to recover, we analyzed the state of mature T and NK cells in the thymus. GCB-LMP1/2A mice showed significantly higher percentages and numbers of mature αβ T cells in both CD4+ and CD8+ T cells compared to the control mice (Figure 4C). However, no significant increase was observed in NK cells or γδ T cells (Supplementary Figures 4B, C). Therefore, we hypothesized that the mature αβ T cells in the thymus may contribute to reduce the incidence of T-ALL in GCB-LMP1/2A mice. To test this hypothesis, we examined expression of activation markers of T cells, such as CD69 and PD-1. Radiation increased frequency of CD69, an early activation marker, on positive thymic CD4+ and CD8+ T cells compared to non-irradiated mice, where GCB-LMP1/2A mice showed significantly higher number of CD69+ activated CD4+ and CD8+ T cells compared to control mice (Figure 4D). Regarding the late activation marker PD-1, GCB-LMP1/2A mice showed significantly higher number of PD-1+ CD8+ T cells than control mice, whereas no significant increase was observed in cell number of PD-1+ CD4+ T cells between GCB-LMP1/2A mice and control mice (Supplementary Figure 4D).
Next, we evaluated status of memory T cells in GCB-LMP1/2A and control mice. CD44 is one of the most commonly used markers for memory T cells. We previously reported that expression of LMP1 and LMP2A in B cells rapidly upregulates CD44 upon T cell activation and its expression is maintained in memory T cells for more than 37 days (15). Frequency and cell number of CD44+ memory T cells displayed significantly higher levels in both CD4+ and CD8+ T cell fractions from GCB-LMP1/2A mice than control mice (Figure 4E). Additionally, GCB-LMP1/2A mice had more CD8+ TEM cells in the BM compared to control mice (Supplementary Figure 4E). These findings suggest that memory T cells induced by LMP1/2A+ B cells are maintained in thymus and BM even after the radiation that potentially eliminate T-ALL and/or its precursor cells in thymus and other tissues. Such effects of T cell memory may also foster a microenvironment favoring thymic recovery, a process that may promote the proportion of DN3 cells, reduces the incidence of T-ALL, and extends the lifespan of GCB-LMP1/2A mice.
3.5 CD8+ T cells from GCB-LMP1/2A mice exhibit efficient killing of tumor cells
To assess the capacity of T cells to recognize and eliminate tumors, we selected terminal-stage T-ALL mice (over 20 weeks after the last radiation) and cultured thymic cells in vitro, establishing three stable T-ALL cell lines, named TL53, TL54, and TL138. TL53 and TL54 cell lines were established from control mice, whereas TL138 was from GCB-LMP1/2A mice. All three tumor cell lines exhibited downregulation of MHC class I (H2-Kb) and expressing CD24, Notch1, and c-Myc, typical markers for T-ALLs (Figure 5A). To evaluate T cell responses to tumor cells, CD4+ and CD8+ T cells were isolated from the spleens of control and GCB-LMP1/2A mice and co-cultured with each of three tumor cell lines in the presence of IL-2 to support proliferation. T cell number was determined every three days (Figure 5B). CD8+ T cells from GCB-LMP1/2A mice displayed robust proliferative responses to all tumor lines, rapidly expanding from day 3, peaking at day 12, and sustaining proliferation for more than 15 days. In contrast, CD8+ T cells from control mice showed minimal proliferation against the TL53 and TL54 cell lines and only a slight response to the TL138 cell line. CD4+ T cells displayed significantly lower proliferation, with limited response to TL53 and TL54, and only those from GCB-LMP1/2A mice showed modest proliferation in response to the TL138 cell line. The difference in proliferative capacity may be primarily due to CD8+ T cells from GCB-LMP1/2A mice being pre-activated in vivo by BLMP1/2A. As a result, these CD8+ T cells may not only exhibit a higher proportion of TEM but also respond more rapidly to tumor cell stimulation compared to those from control mice.
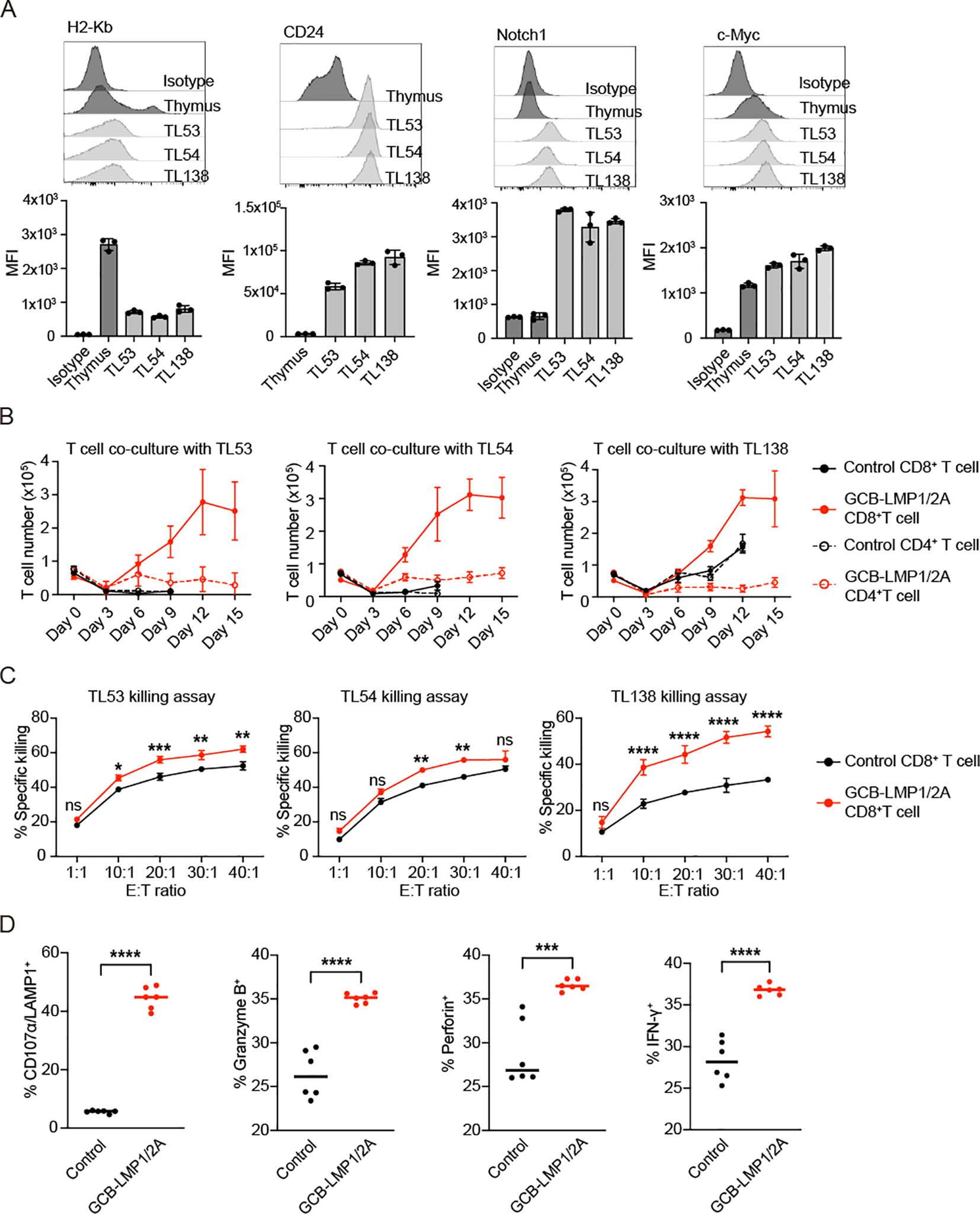
Figure 5. CD8+ T cells exhibit proliferation and cytotoxic activity after co-culture with tumor cells. (A) Flow cytometry of tumor cell lines (TL53, TL54, and TL138) assessed for H2-Kb, CD24, Notch1, and c-Myc expression. Histograms display fluorescence intensity profiles relative to normal thymus and isotype controls. Bar charts below each histogram represent mean fluorescence intensity (MFI) values. All tumor lines exhibit H2-Kb-/low, CD24+, Notch1+, and c-Myc+ phenotypes. (B) Co-culture of control and GCB-LMP1/2A T cells with tumor cell lines TL53, TL54, and TL138 over 15 days. CD8+ and CD4+ T cell number were counted every 3 days, with each point representing the mean ± SD from three replicates. (C) Killing assay of activated CD8+ T cells from control and GCB-LMP1/2A mice against tumor lines TL53, TL54, and TL138. CD8+ T cells were pre-activated in vitro by co-culture with BLMP1/2A for 3 days. Killing efficiency was measured across different effector-to-target (E:T) ratios. Data represent the mean ± SD from three replicates. Statistical analysis was performed using two-way ANOVA with Bonferroni’s multiple comparisons test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. (D) Percentages of CD107α/LAMP1+, Granzyme B+, Perforin+, and IFN-γ+ CD8+ T cells were measured after a 3-day co-culture of CD8+ T cells from the control and GCB-LMP1/2A (n=6 per group) with BLMP1/2A cells. Statistical significance tested using an unpaired two-tailed Student’s t-test; ****p < 0.0001.
To stimulate T cells by BLMP1/2A cells, CD8+ T cells from GCB-LMP1/2A and control mice were co-cultured with BLMP1/2A cells in vitro for three days. The activated CD8+ T cells were then isolated for tumor-killing assays to evaluate their cytotoxic capacity. Results showed that both groups of activated CD8+ T cells demonstrated tumor-killing activity, in particular, CD8+ T cells from GCB-LMP1/2A mice exhibited significantly higher killing efficiency (Figure 5C). Additionally, expression levels of CD107a/LAMP1, Granzyme B, Perforin, and IFN-γ were markedly upregulated in CD8+ T cells from GCB-LMP1/2A mice than in controls (Figure 5D). These results indicate that CD8+ T cells from GCB-LMP1/2A mice exhibit more potent proliferative and cytotoxic responses to T-ALL tumor cell lines, likely due to prior activation by BLMP1/2A cells in vivo, enhancing their antitumor immune function.
We characterized BLMP1/2A cell-specific CD8+ T cells by isolating total RNA and performing microarray analysis to compare their transcriptomes between splenic naive CD8+ T cells and those CD8+ TEM cells activated by LMP1/2A-expressing B-lymphoma cells (22, 24). Representative activation- and cytotoxicity-related genes were ranked by fold changes (Figures 6A, B). The analysis revealed that classical activation markers such as Ifng, Il2, and Il18 were upregulated, indicating an enhanced immune response. The upregulation of chemokine genes Ccl3, Ccl5, Cxcl9, and Cxcl10 suggests that these activated CD8+ T cells possess increased migratory capacity, allowing them to infiltrate the tumor microenvironment more effectively. Transcription factors such as Tbx21, Runx3, Batf3, and Irf8 were also upregulated, indicating that these cells had differentiated into potent effector T cells with robust cytotoxic capabilities. Furthermore, the upregulation of multiple Granzyme genes highlights their remarkable cytotoxic potential. Notably, the expression levels of co-stimulatory receptors Cd244 (2B4/SLAMF4), Tnfrsf9 (4-1BB), Tnfrsf18 (GITR), Tnfrsf4 (OX40), Cd80, and Cd70, along with NK cell-related genes Klrk1 (NKG2D) and Klrc2 (NKG2C), were significantly increased. These molecules provide additional activation signals, enhancing the ability of CD8+ T cells to recognize and eliminate tumor cells with reduced or absent MHC class I expression, thereby reinforcing immune surveillance.
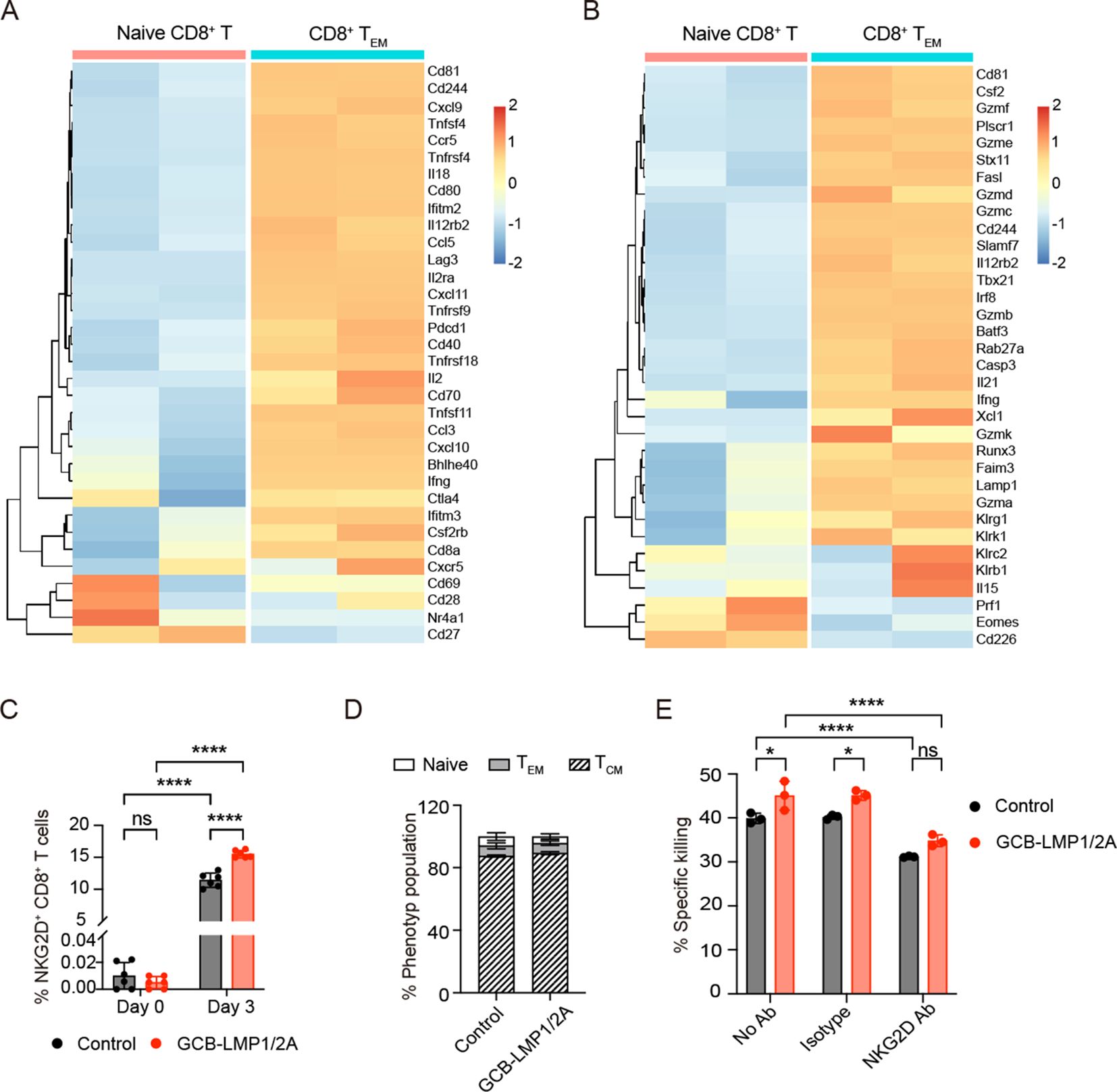
Figure 6. Differentially expressing genes in CD8+ T cells activated by LMP1/2A-expressing B cells. (A, B) CD8+ TEM cells were purified on day 25 of in vitro culture with LMP1/2A-expressing tumor cells and analyzed for mRNA expression by microarray. Data were normalized by RMA normalization and log2 transformed. Heatmaps show the expression of activation-related genes (A) and cytotoxicity-related genes (B) in CD8+ TEM cells compared to naive CD8+ T cells. (C) NKG2D expression levels on CD8+ T cells from GCB-LMP1/2A and control mice before and after co-culture with BLMP1/2A cells on day 3. (D) Population of various phenotypes within the NKG2D+ CD8+ T cells. (E) Killing assay of TL53 cells with no antibody (No Ab), IgG1 isotype antibody (Isotype) and NKG2D antibody (NKG2D Ab). Data represent the mean ± SD from three replicates. Statistical analysis was performed using two-way ANOVA with Bonferroni’s multiple comparisons test; *p < 0.05; ****p < 0.0001; ns, not significant.
Next, we performed qPCR analysis to validate the mRNA expression levels of the cell surface receptors Klrk1 (NKG2D) and Cd244 (2B4/SLAMF4), which are associated with CD8+ T cell activation and cytotoxicity. The results showed that after stimulation with BLMP1/2A cells or T-ALL tumor cells, the expression of Klrk1 and Cd244 was significantly higher in CD8+ T cells from GCB-LMP1/2A mice compared to the control group (Supplementary Figures 5A, B). To further evaluate the roles of these genes in tumor recognition and immune response, we examined the expression levels of their corresponding ligands, Ulbp1 (ligand for NKG2D) and Cd48 (ligand for 2B4/SLAMF4), on BLMP1/2A cells and T-ALL tumor cells. The results indicated that both ligands were highly expressed in these cells (Supplementary Figures 5C, D).
We then measured the protein expression of NKG2D and 2B4 on the surface of CD8+ T cells using specific monoclonal antibodies. After co-culturing CD8+ T cells from both the control and GCB-LMP1/2A groups with BLMP1/2A cells for 3 days, we detected substantial NKG2D+ (Figure 6C) and 2B4+ cells (Supplementary Figure 5E). Notably, CD8+ T cells from GCB-LMP1/2A mice exhibited significantly higher expression of NKG2D and 2B4 compared to the control group, consistent with the qPCR results. Further analysis showed that the majority of NKG2D+ and 2B4+ CD8+ T cells were of the TCM phenotype (Figure 6D, Supplementary Figure 5F).
Next, we investigated whether NKG2D and 2B4 play important role in CD8+ T cell-mediated killing of tumor cells. Blocking of the NKG2D receptor on CD8+ T cells led to a moderate but statistically significant reduction in their cytotoxic activity against TL53 cells, compared with no antibody or isotype control conditions. This suggests that the enhanced killing ability of CD8+ T cells from GCB-LMP1/2A mice is at least partially mediated by NKG2D–NKG2D ligand (NKG2DL) interactions (Figure 6E). However, the incomplete loss of cytotoxicity upon NKG2D blocking indicates that additional mechanisms, such as classical MHC I–mediated recognition or other co-activation pathways, may also contribute to tumor cell elimination. In contrast, blocking of the 2B4 receptor did not lead to a measurable reduction in killing efficiency in this system, suggesting that the 2B4–CD48 interaction may not play a dominant role under these experimental conditions. (Supplementary Figure 5G).
3.6 LMP1/2A-induced immune surveillance suppresses intestinal tumor development
Given the demonstrated suppressing effect of EBV-induced immune surveillance to T-ALL, we further explored its potential effects on other solid tumors. We utilized the ApcMin/+ mouse model, which harbors a mutation in the Apc gene and is prone to developing intestinal tumor (52). By crossing ApcMin/+ mice with GCB-LMP1/2A mice, we generated mice carrying both LMP1/2A antigens and ApcMin/+ mutation (referred to as ApcMin/+LMPKI) to assess whether LMP1/2A-induced immune surveillance could suppress intestinal tumor development. At 22 weeks of age, significant intestinal tumor formation was observed in these mice. Surprisingly, the tumor burden of ApcMin/+LMPKI mice was lower compared to ApcMin/+ mice (Figure 7A). The number of tumor nodules (Figure 7B) and total tumor areas (Figure 7C) were significantly reduced in ApcMin/+LMPKI mice compared to ApcMin/+ controls. As predicted by the lower tumor burden, survival period was extended in ApcMin/+LMPKI mice compared to ApcMin/+ controls (Figure 7D).
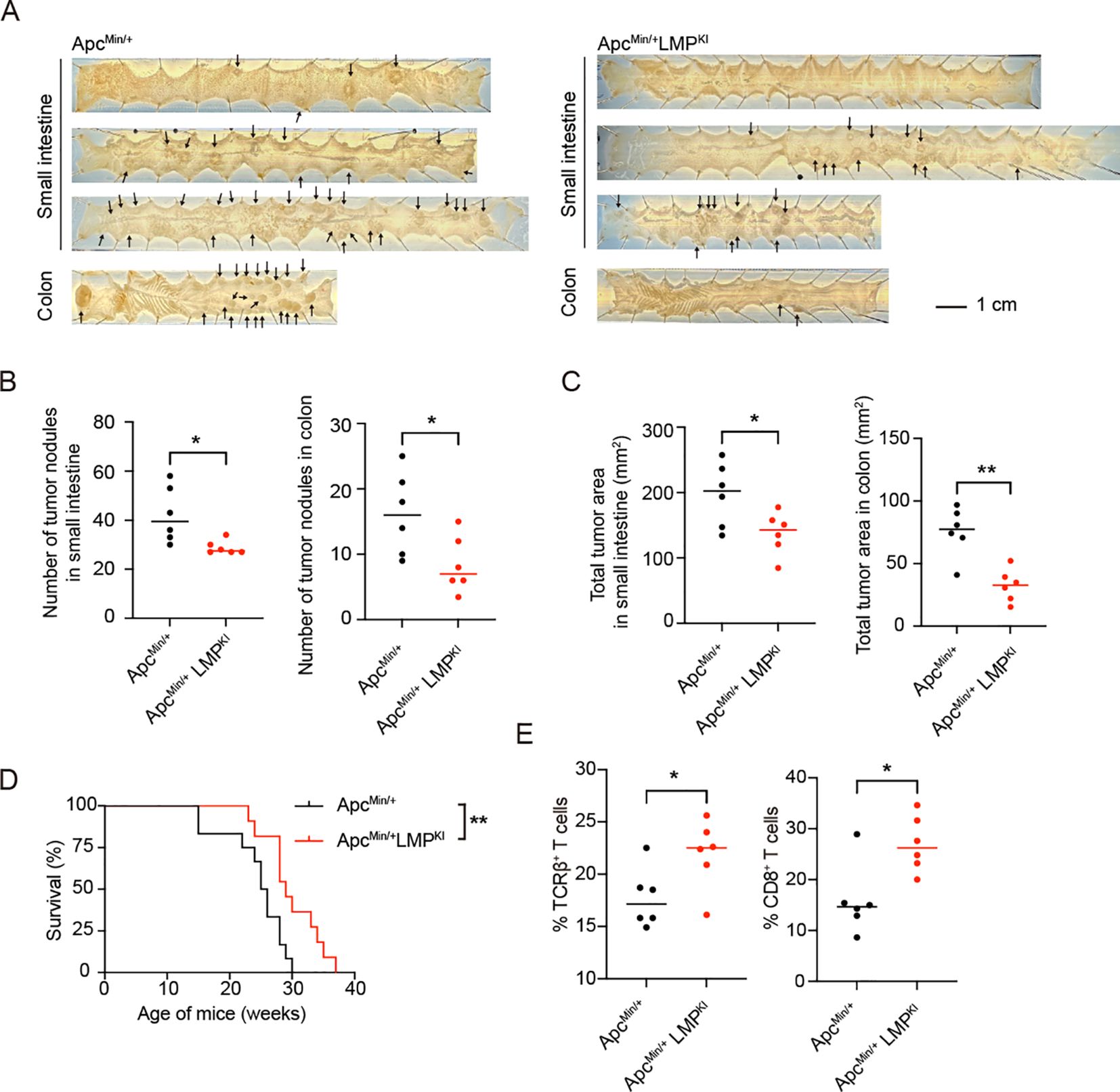
Figure 7. Analysis of LMP1/2A-induced immune surveillance in the ApcMin-induced intestinal tumor formation. (A) Representative macroscopic images of small intestines and colons from ApcMin/+ and ApcMin/+LMPKI mice at 22-week-old with arrows indicating tumor nodules. Scale bar=1 cm. (B, C) Quantification of tumor nodules number in small intestine and colon (B). Quantification of total tumor area in small intestine and colon (C). Each dot represents an individual mouse (n=6 per group). Statistical analysis was performed using an unpaired two-tailed Student’s t test; *p < 0.05; **p < 0.01. (D) Kaplan-Meier survival curves for ApcMin/+ and ApcMin/+LMPKI mice (n=12 per group; log-rank test, **p < 0.01). (E) Flow cytometry analysis of tumor-infiltrating lymphocytes (TILs), showing percentages of TCRβ+ T cells in CD45+ lymphocytes fractions and CD8+ in TCRβ+ T cell fractions in ApcMin/+ and ApcMin/+LMPKI mice (n=6 per group). Statistical analysis was performed using an unpaired two-tailed Student’s t test; *p < 0.05.
To investigate the underlying mechanisms, we isolated intra-tumoral lymphocytes and found a higher proportion of αβ T cells infiltrating the tumors of ApcMin/+LMPKI mice, particularly an increased percentage of CD8+ T cells (Figure 7E). These findings indicate that LMP1/2A-induced immune surveillance enhances both the intra-tumoral infiltration and activation of CD8+ T cells. Thus, beyond its resistance to T-ALL, LMP1/2A-induced immune surveillance also promotes anti-tumor T cell responses in the solid tumor model.
Taken together, our study uncovered a potential mechanism by which LMP1/2A-expression in B cells enhances CD8+ T cell-mediated immune responses to combat both hematological malignancies and solid tumors. These findings highlight beneficial aspects of EBV infection in humans that may contribute to the surveillance of pan-tumor development, providing an important insight into cancer prevention.
4 Discussion
In recent years, growing evidence has shown that viral infections can enhance host immune defense not only through antigen-specific responses but also by activating bystander T cells, promoting cross-presentation, and sustaining innate immune signaling (53). This phenomenon, often referred to as “virus-induced cross-protection (54)” has been demonstrated in various models. For example, latent infection with murine gammaherpesvirus MHV68 has been shown to alleviate allergic asthma (3) and enhance resistance to bacterial infections (2), likely through prolonged innate immune activation. Similarly, EBV, a human-specific gammaherpesvirus, may shape host immunity through comparable mechanisms, as co-infection with EBV and CMV has been reported to expand memory-like NKG2C+ NK cells (55), suggesting its potential role in enhancing immune surveillance. Unlike these studies that primarily focus on innate immunity, our study demonstrates that the EBV latent genes LMP1 and LMP2A, through the transformation of B cells, enhance the expression of stress-induced antigens such as NKG2D ligands and promote the development of a population of CD8+ T cells with broad responsiveness. These T cells are capable of recognizing not only viral antigens but also stress signals, thereby contributing to immune surveillance against non-viral tumors.
In our model, LMP1/2A expression in GC B cells effectively suppressed the development of various EBV-negative cancers through CD8+ T cell-mediated immune surveillance. This complements previous studies on the role of LMP1 in activating CD4+ and CD8+ CTLs, which primarily focused on their responses to LMP1/2A expressing B cell lymphomas (12, 15, 16). Here, we show that LMP1/2A expression in GC B cells leads to robust CD8+ T cell activation, likely in gut-associated lymphoid tissues where GC reactions occur. These activated T cells may differentiate into effector and central memory subsets and exert systemic immune surveillance through peripheral circulation.
Most studies on radiation-induced T cell lymphomas have focused on oncogenic mechanisms, with relatively little focus on tumor clearance. Our findings demonstrated that immune surveillance induced by LMP1/2A expression significantly reduced the incidence of early-stage monoclonal T-ALL development and extended survival after radiation exposure. Although the percentage of hematopoietic stem and progenitor cells, a source of T-ALLs, was not disturbed, GCB-LMP1/2A mice had significantly higher percentages of CD8+ memory T cells in the bone marrow and thymus compared with control mice, suggesting that CD8+ T cell-mediated immune surveillance plays an important role in the suppression of T-ALL.
Further analysis revealed that LMP1/2A-activated CD8+ T cells upregulated various activation and cytotoxicity-related molecules, including IFN-γ, Granzyme B, and Perforin, which likely enhanced their cytotoxic function. Additionally, the upregulation of chemokine genes such as CXCL9 and CXCL10 may facilitate T cell migration to the tumor microenvironment, further strengthening their immune surveillance capabilities. Notably, when T-ALL cells were cultured in vitro and transformed into MHC class I-low/negative tumor cells, LMP1/2A-activated CD8+ T cells still demonstrated effective cytotoxicity against these tumor cells. This suggests that, beyond the MHC class I-mediated pathway, other mechanisms contribute to the activation and effector functions of these T cells.
This study revealed that 2B4/SLAMF4 and NKG2D, key receptors known for regulating NK cell functions, were also significantly upregulated CD8+ T cells activated by LMP1/2A+B cells. Their corresponding ligands, Ulbp1 and CD48, were highly expressed on the tumor cell surface, providing crucial activation signals for CD8+ T cell recognition of MHC class I-low or -negative tumors. In in vitro killing assays, blocking NKG2D moderately reduced cytotoxicity against tumors with high Ulbp1 and extremely low MHC class I expression, suggesting a functional role of this axis under such specific conditions. However, the killing efficiency was not complete and inconsistently reproduced in two additional tumor lines with lower NKG2DL or higher MHC calss I expression. These results indicate that additional mechanisms—such as classical MHC I–dependent recognition or other co-stimulatory pathways—may also contribute to CD8+ T cell-mediated tumor elimination. We thus consider NKG2D–NKG2DL interactions to be one of several pathways involved in this process, but not the only mechanism. Conversely, blocking 2B4/SLAMF4 did not significantly impair killing activity. However, the blocking efficiency of the anti-2B4 antibody used has not been verified in our system, and its potential role may be compensated by alternative cytotoxic pathways. Therefore, we cannot rule out a functional contribution of the 2B4–CD48 axis, although it does not appear to play a dominant role under the current experimental conditions.
While NKG2DLs are stress-induced molecules not typically expressed in normal tissues, many types of tumors express these ligands, enabling NKG2D+ lymphocytes to initiate potent immune responses via cytokine secretion and cytotoxic effector functions (52, 56, 57). The presence of NKG2DLs in tumors, along with evidence from in vitro assays and mouse models, has firmly established the role of the NKG2D–NKG2DL axis in antitumor immunity (58–61). Human studies have further demonstrated that NKG2DLs are upregulated during the lytic phase of EBV infection, enhancing the susceptibility of infected cells to NKG2D+ NK and T cell–mediated clearance (62, 63). Additionally, NKG2D CAR-T cells have shown therapeutic efficacy not only in EBV-associated lymphomas (64) but also in human T-ALL (65), indicating that NKG2D-mediated immune recognition may provide a broadly applicable mechanism for tumor control in both virus-related and unrelated malignancies.
Our study extends this relevance to radiation-induced T-ALL, showing that CD8+ T cells activated by LMP1/2A+B cells exhibit increased NKG2D expression, which enhances their ability to recognize and eliminate tumors lacking MHC class I. In future studies, we plan to use MHC class I-deficient tumor models or MHC class I–neutralizing antibodies to completely block the classical killing pathway, in order to further evaluate the roles of NKG2D, 2B4, and other potential mechanisms. If the MHC I–dependent mechanism remains dominant, we also intend to explore candidate TAAs, aiming for a more comprehensive understanding of how LMP1/2A+B cell–induced CD8+ T cells recognize and eliminate tumors.
In addition to hematological malignancies, LMP1/2A-induced immune surveillance also exhibited anti-tumor effects in intestinal tumor model. By crossing GCB-LMP1/2A mice with ApcMin/+ mice, we found that LMP1/2A-induced immune surveillance significantly reduced the tumor burden of intestinal tumor in ApcMin/+ mice and extended their survival. Further analysis indicated that LMP1/2A likely enhances the infiltration efficiency and activation of CD8+ T cells, promoting anti-tumor immune responses within the tumor microenvironment. These findings suggest that LMP1/2A plays a critical role not only in hematological malignancies but also demonstrates potential anti-tumor capabilities in solid tumors.
This study highlights the critical role of LMP1/2A in suppressing hematological and solid tumors by enhancing CD8+ T cell-mediated immune surveillance, providing a basis for the development of novel therapeutic and preventive strategies targeting MHC class I-negative malignancies across a broad range of cancer types.
Given its strong immunostimulatory properties, LMP1/2A may serve as a molecular tool in future immunotherapy or cancer vaccine strategies. However, its oncogenic potential and ability to modulate the tumor microenvironment underscore the need for careful evaluation of safety and context-specific effects before clinical application. Additionally, although our study highlights the central role of CD8+ T cells in LMP1/2A-induced tumor immune surveillance, we cannot exclude the potential contribution of NK cells or NKT cells. Given the observed activation of the NKG2D pathway, these innate lymphocyte subsets may also exhibit enhanced cytotoxic function through recognition of stress-induced ligands. While we did not observe significant differences in NK cell numbers between tumor-bearing and protected mice, we did not directly assess the functional status of NK or NKT cells, such as activation marker expression, cytokine production, or granule-mediated cytotoxicity. Therefore, their role in the observed immune protection remains unclear. Future studies should investigate the activation and effector profiles of NK and NKT cells to better understand the potential interplay between innate and adaptive immunity in the context of LMP1/2A-mediated tumor control.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by the Ethics Review Committee for Animal Experimentation of Hiroshima University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YJ: Writing – review & editing, Investigation, Data curation, Conceptualization, Writing – original draft, Funding acquisition, Formal Analysis, Visualization. YG: Methodology, Conceptualization, Writing – review & editing, Supervision, Funding acquisition. YK: Resources, Writing – review & editing, Methodology. MS: Writing – review & editing, Methodology, Resources. SO: Visualization, Software, Writing – review & editing. KY: Methodology, Writing – review & editing. YO: Investigation, Visualization, Writing – review & editing. YT: Methodology, Writing – review & editing, Resources. YS: Writing – review & editing, Resources. YB: Resources, Writing – review & editing. TY: Project administration, Writing – review & editing, Funding acquisition, Investigation, Methodology, Supervision, Writing – original draft, Visualization, Conceptualization, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the JSPS KAKENHI under Grant Numbers JP18H02669, JP19K22538, and JP21H02751 to TY; JSPS Program for Forming Japan’s Peak Research Universities (JSPS J-PEAKS) to TY; JST Grant Number JPMJPF2010 to TY; Research Funding for Longevity Sciences from the National Center for Geriatrics and Gerontology (grant 21-2) to TY; the Program of the Network-type Joint Usage/Research Center for Radiation Disaster Medical Science to YG and TY; Program for Developing and Supporting the Next-Generation of Innovative Researchers at Hiroshima University (HU SPRING) to YJ.
Acknowledgments
This work was supported by the Natural Science Center for Basic Research and Development (NBARD-00134) and the Radiation Research Center for Frontier Science at Hiroshima University. This work was partly performed in the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University. We thank M. Ikawa (Osaka University, Japan) for C57BL/6N ES cells (EGR-101); M. Tanaka and K. Kageyama for Technical Support in the Medical Institute of Bioregulation, Kyushu University for animal facility; K. Moriwaki and R. Suzuka for providing antibody; T. Kawaguchi, M. Zhang, and Y. Hayashi for technical assistance; N. Kikkawa, M. Tawa, and Y. Hamano for administrative assistance; All lab members for useful discussion and comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1597731/full#supplementary-material
Supplementary Figure 1 | Generation of knock-in mice conditionally expressing LMP1 and LMP2A in a Cre-dependent manner. (A) Strategy for targeting conditional LMP1 and LMP2A alleles into the Rosa26 locus. (B) Western blot analysis of LMP1 and LMP2A expression in in vitro-expanded B cells from GCB-LMP1/2A mice (BLMP1/2A). CD40-activated B cells (CD40 act-B) from control mice were used as negative controls. β-Actin was used as a loading control. (C) Percentage of GC B cells in the PPs and mLNs of GCB-LMP1/2A mice compared to control mice. Each dot represents an individual mouse (n=4). Statistical significance tested using an unpaired two-tailed Student’s t-test; **p < 0.01; ***p < 0.001. (D) Representative of FACS results of CD8+ T cells gated for effector memory (TEM), central memory (TCM), and naive subsets based on CD44 and CD62L expression at the indicated time points during co-culture with either CD40 act-B cells (T+CD40 act-B) or BLMP1/2A cells (T+BLMP1/2A). (E) CD8+ TEM cell numbers were quantified at the indicated time points.
Supplementary Figure 2 | Post-irradiation thymic and splenic hyperplasia exhibits an immature T cell phenotype. (A) Total cell numbers in the thymus and spleen of non-irradiated control mice (NC) and mice after 15–30 weeks of radiation (RI). Statistical analysis was performed using multiple t-tests, with Holm-Šidák correction for multiple comparisons; **p < 0.01. (B) Representative FACS analysis showing the percentage of CD24+CD8+ immature T cells in the spleen. (C) Representative FACS analysis displaying the percentage of CD19+ B cells and CD3+ T cells in the spleen.
Supplementary Figure 3 | Expression analysis of T-ALL specific genes. (A, B) Quantitative RT-PCR analysis of sorted thymic cell subsets, including DN (CD4-CD8-), DP (CD4+CD8+), 4SP (CD4+CD8-), and 8SP (CD4-CD8+), from mice 14 weeks post-radiation exposure compared to control mice that did not undergo radiation. The expression levels of Notch1, Myc, and Lmo2 are presented in (A). The expression levels of pre-TCRα (Ptcra) presented in (B). Data from three technical replicates is shown. Statistical analysis was performed using multiple t-tests, with Holm-Šidák correction for multiple comparisons; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
Supplementary Figure 4 | Changes in various cell populations in bone marrow and thymus 3 weeks after the last radiation dose. (A) Percentage of LSK cells (Lineage-Sca1+cKit+) in bone marrow (BM). (B) Percentages and absolute numbers of NK cells in thymus. (C) Percentages and absolute numbers of γδ T cells in thymus. (D) The Percentage and absolute numbers of PD-1+ in TCRβ+CD8+ or TCRβ+CD4+ T cell fractions in the thymus. (E) Percentage of CD8+ TEM and CD4+ TEM cells in BM. Statistical significance tested using one-way ANOVA with Bonferroni’s multiple comparisons test; *p < 0.05; **p < 0.01; ****p < 0.0001; ns, not significant.
Supplementary Figure 5 | Expression of cytotoxicity related genes in CD8+ T cells and tumor cells and their impact on cytotoxic function. Relative mRNA expression levels of Klrk1 (A) and Cd244 (B) in CD8+ T cells before stimulation and after 3 days of stimulation with BLMP1/2A cells or tumor cells. Data from three technical replicates is shown. Statistical analysis was performed using multiple t-tests, with Holm-Šidák correction for multiple comparisons; *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. (C, D) Relative mRNA expression levels of Ulbp1 (C) and Cd48 (D) in normal B cells, BLMP1/2A cells, normal DP cells, and tumor cells. (E) 2B4 expression levels on CD8+ T cells from GCB-LMP1/2A and control mice after co-culture with BLMP1/2A cells on day 3. (F) Population of various phenotypes within the 2B4 + CD8+ T cells. (G) Killing assay of TL53 cells with no antibody (No Ab), IgG2a isotype antibody (Isotype) and 2B4 antibody (2B4 Ab). Data represent the mean ± SD from three replicates. Statistical analysis was performed using two-way ANOVA with Bonferroni’s multiple comparisons test and unpaired two-tailed Student’s t-test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
References
1. Lieberman PM. Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat Rev Microbiol. (2013) 11:863–75. doi: 10.1038/nrmicro3135
2. Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. (2007) 447:326–9. doi: 10.1038/nature05762
3. Machiels B, Dourcy M, Xiao X, Javaux J, Mesnil C, Sabatel C, et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat Immunol. (2017) 18:1310–20. doi: 10.1038/ni.3857
4. Dourcy M, Maquet C, Dams L, Gilliaux G, Javaux J, Desmecht D, et al. A gammaherpesvirus licenses cd8 T cells to protect the host from pneumovirus-induced immunopathologies. Mucosal Immunol. (2020) 13:799–813. doi: 10.1038/s41385-020-0293-7
5. Young LS, Yap LF, and Murray PG. Epstein–barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer. (2016) 16:789–802. doi: 10.1038/nrc.2016.92
6. Taylor GS, Long HM, Brooks JM, Rickinson AB, and Hislop AD. The immunology of epstein-barr virus-induced disease. Annu Rev Immunol. (2015) 33:787–821. doi: 10.1146/annurev-immunol-032414-112326
7. Kuppers R. B cells under influence: transformation of B cells by epstein-barr virus. Nat Rev Immunol. (2003) 3:801–12. doi: 10.1038/nri1201
8. Capello D, Cerri M, Muti G, Berra E, Oreste P, Deambrogi C, et al. Molecular histogenesis of posttransplantation lymphoproliferative disorders. Blood. (2003) 102:3775–85. doi: 10.1182/blood-2003-05-1683
9. Rastelli J, Homig-Holzel C, Seagal J, Muller W, Hermann AC, Rajewsky K, et al. Lmp1 signaling can replace cd40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to igg1. Blood. (2008) 111:1448–55. doi: 10.1182/blood-2007-10-117655
10. Soni V, Cahir-McFarland E, and Kieff E. Lmp1 trafficking activates growth and survival pathways. Adv Exp Med Biol. (2007) 597:173–87. doi: 10.1007/978-0-387-70630-6_14
11. Wang D, Liebowitz D, and Kieff E. An ebv membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. (1985) 43:831–40. doi: 10.1016/0092-8674(85)90256-9
12. Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, et al. Immune surveillance and therapy of lymphomas driven by epstein-barr virus protein lmp1 in a mouse model. Cell. (2012) 148:739–51. doi: 10.1016/j.cell.2011.12.031
13. Kulwichit W, Edwards RH, Davenport EM, Baskar JF, Godfrey V, and Raab-Traub N. Expression of the epstein-barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc Natl Acad Sci U.S.A. (1998) 95:11963–8. doi: 10.1073/pnas.95.20.11963
14. Yasuda T, Wirtz T, Zhang B, Wunderlich T, Schmidt-Supprian M, Sommermann T, et al. Studying epstein-barr virus pathologies and immune surveillance by reconstructing ebv infection in mice. Cold Spring Harb Symp Quant Biol. (2013) 78:259–63. doi: 10.1101/sqb.2013.78.020222
15. Wirtz T, Weber T, Kracker S, Sommermann T, Rajewsky K, and Yasuda T. Mouse model for acute epstein-barr virus infection. Proc Natl Acad Sci U.S.A. (2016) 113:13821–6. doi: 10.1073/pnas.1616574113
16. Sommermann T, Yasuda T, Ronen J, Wirtz T, Weber T, Sack U, et al. Functional interplay of epstein-barr virus oncoproteins in a mouse model of B cell lymphomagenesis. Proc Natl Acad Sci U.S.A. (2020) 117:14421–32. doi: 10.1073/pnas.1921139117
17. Mancao C, Altmann M, Jungnickel B, and Hammerschmidt W. Rescue of “Crippled” Germinal center B cells from apoptosis by epstein-barr virus. Blood. (2005) 106:4339–44. doi: 10.1182/blood-2005-06-2341
18. Minamitani T, Yasui T, Ma Y, Zhou H, Okuzaki D, Tsai CY, et al. Evasion of affinity-based selection in germinal centers by epstein-barr virus lmp2a. Proc Natl Acad Sci U.S.A. (2015) 112:11612–7. doi: 10.1073/pnas.1514484112
19. Swart R, Ruf IK, Sample J, and Longnecker R. Latent membrane protein 2a-mediated effects on the phosphatidylinositol 3-kinase/akt pathway. J Virol. (2000) 74:10838–45. doi: 10.1128/jvi.74.22.10838-10845.2000
20. Minamitani T, Ma Y, Zhou H, Kida H, Tsai CY, Obana M, et al. Mouse model of epstein-barr virus lmp1- and lmp2a-driven germinal center B-cell lymphoproliferative disease. Proc Natl Acad Sci U.S.A. (2017) 114:4751–6. doi: 10.1073/pnas.1701836114
21. Long HM, Zuo J, Leese AM, Gudgeon NH, Jia H, Taylor GS, et al. Cd4+ T-cell clones recognizing human lymphoma-associated antigens: generation by in vitro stimulation with autologous epstein-barr virus-transformed B cells. Blood. (2009) 114:807–15. doi: 10.1182/blood-2008-12-194043
22. Choi IK, Wang Z, Ke Q, Hong M, Qian Y, Zhao X, et al. Signaling by the epstein-barr virus lmp1 protein induces potent cytotoxic cd4(+) and cd8(+) T cell responses. Proc Natl Acad Sci U.S.A. (2018) 115:E686–E95. doi: 10.1073/pnas.1713607115
23. Choi IK, Wang Z, Ke Q, Hong M, Paul DW Jr., Fernandes SM, et al. Mechanism of ebv inducing anti-tumour immunity and its therapeutic use. Nature. (2021) 590:157–62. doi: 10.1038/s41586-020-03075-w
24. Tamura Y, Yamane K, Kawano Y, Bullinger L, Wirtz T, Weber T, et al. Concomitant cytotoxic effector differentiation of cd4(+) and cd8(+) T cells in response to ebv-infected B cells. Cancers (Basel). (2022) 14:4118. doi: 10.3390/cancers14174118
25. Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, et al. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci U.S.A. (2006) 103:7396–401. doi: 10.1073/pnas.0602353103
26. Sasatani M, Shimura T, Doi K, Zaharieva EK, Li J, Iizuka D, et al. Morphology dynamics in intestinal crypt during postnatal development affect age-dependent susceptibility to radiation-induced intestinal tumorigenesis in apcmin/+ Mice: possible mechanisms of radiation tumorigenesis. Carcinogenesis. (2023) 44:105–18. doi: 10.1093/carcin/bgac100
27. Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, et al. Canonical nf-kappab activity, dispensable for B cell development, replaces baff-receptor signals and promotes B cell proliferation upon activation. Immunity. (2006) 24:729–39. doi: 10.1016/j.immuni.2006.04.005
28. Fujihara Y, Kaseda K, Inoue N, Ikawa M, and Okabe M. Production of mouse pups from germline transmission-failed knockout chimeras. Transgenic Res. (2013) 22:195–200. doi: 10.1007/s11248-012-9635-x
29. Sander S, Calado DP, Srinivasan L, Kochert K, Zhang B, Rosolowski M, et al. Synergy between pi3k signaling and myc in burkitt lymphomagenesis. Cancer Cell. (2012) 22:167–79. doi: 10.1016/j.ccr.2012.06.012
30. Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, et al. Pi3 kinase and foxo1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity. (2015) 43:1075–86. doi: 10.1016/j.immuni.2015.10.021
31. Kawamoto H, Ohmura K, Fujimoto S, Lu M, Ikawa T, and Katsura Y. Extensive proliferation of T cell lineage-restricted progenitors in the thymus: an essential process for clonal expression of diverse T cell receptor beta chains. Eur J Immunol. (2003) 33:606–15. doi: 10.1002/eji.200323461
32. Le Clorennec C, Youlyouz-Marfak I, Adriaenssens E, Coll J, Bornkamm GW, and Feuillard J. Ebv latency iii immortalization program sensitizes B cells to induction of cd95-mediated apoptosis via lmp1: role of nf-kappab, stat1, and P53. Blood. (2006) 107:2070–8. doi: 10.1182/blood-2005-05-2053
33. Gerdemann U, Katari U, Christin AS, Cruz CR, Tripic T, Rousseau A, et al. Cytotoxic T lymphocytes simultaneously targeting multiple tumor-associated antigens to treat ebv negative lymphoma. Mol Ther. (2011) 19:2258–68. doi: 10.1038/mt.2011.167
34. Linnerbauer S, Behrends U, Adhikary D, Witter K, Bornkamm GW, and Mautner J. Virus and autoantigen-specific cd4+ T cells are key effectors in a scid mouse model of ebv-associated post-transplant lymphoproliferative disorders. PloS Pathog. (2014) 10:e1004068. doi: 10.1371/journal.ppat.1004068
35. Zhang Q and Xu M. Ebv-induced T-cell responses in ebv-specific and nonspecific cancers. Front Immunol. (2023) 14:1250946. doi: 10.3389/fimmu.2023.1250946
36. Zhang B and Choi IK. Facts and hopes in the relationship of ebv with cancer immunity and immunotherapy. Clin Cancer Res. (2022) 28:4363–9. doi: 10.1158/1078-0432.CCR-21-3408
37. Rivina L and Schiestl R. Mouse models for efficacy testing of agents against radiation carcinogenesis-a literature review. Int J Environ Res Public Health. (2012) 10:107–43. doi: 10.3390/ijerph10010107
38. Kaplan HS. The role of radiation on experimental leukemogenesis. Natl Cancer Inst Monogr. (1964) 14:207–20.
39. Boniver J, Humblet C, Rongy AM, Delvenne C, Delvenne P, Greimers R, et al. Cellular aspects of the pathogenesis of radiation–induced thymic lymphomas in C57 bl mice (Review). In Vivo. (1990) 4:41–3.
40. Tan C, Taylor AA, Coburn MZ, Marino JH, Van De Wiele CJ, and Teague TK. Ten-color flow cytometry reveals distinct patterns of expression of cd124 and cd126 by developing thymocytes. BMC Immunol. (2011) 12:36. doi: 10.1186/1471-2172-12-36
41. Weng AP, Ferrando AA, Lee W, Morris J, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of notch1 in human T cell acute lymphoblastic leukemia. Science. (2004) 306:269–71. doi: 10.1126/science.1102160
42. Koch U and Radtke F. Mechanisms of T cell development and transformation. Annu Rev Cell Dev Biol. (2011) 27:539–62. doi: 10.1146/annurev-cellbio-092910-154008
43. Allman D, Karnell FG, Punt JA, Bakkour S, Xu L, Myung P, et al. Separation of notch1 promoted lineage commitment and expansion/transformation in developing T cells. J Exp Med. (2001) 194:99–106. doi: 10.1084/jem.194.1.99
44. Ohi H, Mishima Y, Kamimura K, Maruyama M, Sasai K, and Kominami R. Multi-Step Lymphomagenesis Deduced from DNA Changes in Thymic Lymphomas and Atrophic Thymuses at Various Times after Gamma-Irradiation. Oncogene. (2007) 26:5280–9. doi: 10.1038/sj.onc.1210325
45. Go R, Hirose S, Morita S, Yamamoto T, Katsuragi Y, Mishima Y, et al. Bcl11b heterozygosity promotes clonal expansion and differentiation arrest of thymocytes in gamma-irradiated mice. Cancer Sci. (2010) 101:1347–53. doi: 10.1111/j.1349-7006.2010.01546.x
46. Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V, et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature. (2014) 509:465–70. doi: 10.1038/nature13317
47. Martins VC, Ruggiero E, Schlenner SM, Madan V, Schmidt M, Fink PJ, et al. Thymus-autonomous T cell development in the absence of progenitor import. J Exp Med. (2012) 209:1409–17. doi: 10.1084/jem.20120846
48. Paiva RA, Sousa AGG, Ramos CV, Avila M, Lilue J, Paixao T, et al. Self-renewal of double-negative 3 early thymocytes enables thymus autonomy but compromises the beta-selection checkpoint. Cell Rep. (2021) 35:108967. doi: 10.1016/j.celrep.2021.108967
49. Iwasaki H and Akashi K. Hematopoietic developmental pathways: on cellular basis. Oncogene. (2007) 26:6687–96. doi: 10.1038/sj.onc.1210754
50. Plett PA, Sampson CH, Chua HL, Joshi M, Booth C, Gough A, et al. Establishing a murine model of the hematopoietic syndrome of the acute radiation syndrome. Health Phys. (2012) 103:343–55. doi: 10.1097/HP.0b013e3182667309
51. Hasapis S, Caraballo I, and Lee CL. Transplantation of Unirradiated Bone Marrow Cells after Total-Body Irradiation Prevents the Development of Thymic Lymphoma in Mice through Niche Competition. Radiat Res. (2021) 195:301–6. doi: 10.1667/RADE-20-00221.1
52. McCart AE, Vickaryous NK, and Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract. (2008) 204:479–90. doi: 10.1016/j.prp.2008.03.004
53. Kim TS and Shin EC. The activation of bystander cd8(+) T cells and their roles in viral infection. Exp Mol Med. (2019) 51:1–9. doi: 10.1038/s12276-019-0316-1
54. Unanue ER. Viral infections and nonspecific protection–good or bad? N Engl J Med. (2007) 357:1345–6. doi: 10.1056/NEJMcibr074519
55. Saghafian-Hedengren S, Sohlberg E, Theorell J, Carvalho-Queiroz C, Nagy N, Persson JO, et al. Epstein-barr virus coinfection in children boosts cytomegalovirus-induced differentiation of natural killer cells. J Virol. (2013) 87:13446–55. doi: 10.1128/JVI.02382-13
56. Diefenbach A, Jensen ER, Jamieson AM, and Raulet DH. Rae1 and H60 ligands of the nkg2d receptor stimulate tumour immunity. Nature. (2001) 413:165–71. doi: 10.1038/35093109
57. Prajapati K, Perez C, Rojas LBP, Burke B, and Guevara-Patino JA. Functions of nkg2d in cd8(+) T cells: an opportunity for immunotherapy. Cell Mol Immunol. (2018) 15:470–9. doi: 10.1038/cmi.2017.161
58. Lerner EC, Woroniecka KI, D’Anniballe VM, Wilkinson DS, Mohan AA, Lorrey SJ, et al. Cd8(+) T cells maintain killing of mhc-I-negative tumor cells through the nkg2d-nkg2dl axis. Nat Cancer. (2023) 4:1258–72. doi: 10.1038/s43018-023-00600-4
59. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. Nkg2d-deficient mice are defective in tumor surveillance in models of spontaneous Malignancy. Immunity. (2008) 28:571–80. doi: 10.1016/j.immuni.2008.02.016
60. Cerwenka A, Baron JL, and Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (Rae-1) permits natural killer cell-mediated rejection of a mhc class I-bearing tumor in vivo. Proc Natl Acad Sci U.S.A. (2001) 98:11521–6. doi: 10.1073/pnas.201238598
61. Curio S, Lin W, Bromley C, McGovern J, Triulzi C, Jonsson G, et al. Nkg2d fine-tunes the local inflammatory response in colorectal cancer. Cancers (Basel). (2023) 15:1792. doi: 10.3390/cancers15061792
62. Desimio MG, Covino DA, Cancrini C, and Doria M. Entry into the lytic cycle exposes ebv-infected cells to nk cell killing via upregulation of the micb ligand for nkg2d and activation of the cd56(Bright) and nkg2a(+)Kir(+)Cd56(Dim) subsets. Front Immunol. (2024) 15:1467304. doi: 10.3389/fimmu.2024.1467304
63. Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ Regulates cytotoxic functions of nk and cd8 T cells in chronic ebv infection through nkg2d. Science. (2013) 341:186–91. doi: 10.1126/science.1240094
64. Mai Q, He B, Deng S, Zeng Q, Xu Y, Wang C, et al. Efficacy of nkg2d car-T cells with il-15/il-15ralpha signaling for treating epstein-barr virus-associated lymphoproliferative disorder. Exp Hematol Oncol. (2024) 13:85. doi: 10.1186/s40164-024-00553-z
Keywords: Epstein-Barr virus, LMP1, LMP2A, immune surveillance, radiation-induced tumor, T-ALL, ApcMin, NKG2D
Citation: Jin Y, Guo Y, Kawano Y, Sasatani M, Ohki S, Yamane K, Ota Y, Tamura Y, Sotomaru Y, Baba Y and Yasuda T (2025) T cell-mediated immune surveillance conferred by latent Epstein-Barr virus genes suppresses a broad spectrum of tumor formation through NKG2D-NKG2DL interactions. Front. Immunol. 16:1597731. doi: 10.3389/fimmu.2025.1597731
Received: 21 March 2025; Accepted: 16 May 2025;
Published: 04 June 2025.
Edited by:
Luigi Buonaguro, G. Pascale National Cancer Institute Foundation (IRCCS), ItalyReviewed by:
Ran Wang, Capital Medical University, ChinaPratyusha Mandal, The City University of New York, United States
Copyright © 2025 Jin, Guo, Kawano, Sasatani, Ohki, Yamane, Ota, Tamura, Sotomaru, Baba and Yasuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoharu Yasuda, eWFzdWRhdEBoaXJvc2hpbWEtdS5hYy5qcA==