 Benedetta Elena Di Majo1,2†
Benedetta Elena Di Majo1,2† Chiara Leoni3†
Chiara Leoni3† Eleonora Cartisano1,2Chiara Fossati1
Eleonora Cartisano1,2Chiara Fossati1 Germana Viscogliosi3Valentina Trevisan3
Germana Viscogliosi3Valentina Trevisan3 Lucia Pia Bruno2
Lucia Pia Bruno2 Francesca Conti4
Francesca Conti4 Mattia Moratti4Emilia Monaco3
Mattia Moratti4Emilia Monaco3 Donato Rigante5,6
Donato Rigante5,6 Beatrice Rivalta7,8
Beatrice Rivalta7,8 Caterina Cancrini7,9
Caterina Cancrini7,9 Aleksandra Szczawińska-Popłonyk10
Aleksandra Szczawińska-Popłonyk10 Aleksander Jamsheer11,12
Aleksander Jamsheer11,12 Monika Obara-Moszyńska13Viktoria Zakharova14
Monika Obara-Moszyńska13Viktoria Zakharova14 Anna Shcherbina15Julija Rodina15
Anna Shcherbina15Julija Rodina15 Beyhan Tüysüz16Saumya Shekhar Jamuar17,18Jiin Ying Lim17Jeannette Goh17Anna Cereda19Teresa Agovino19Ilaria Contaldo20Maria Luigia Gambardella20
Beyhan Tüysüz16Saumya Shekhar Jamuar17,18Jiin Ying Lim17Jeannette Goh17Anna Cereda19Teresa Agovino19Ilaria Contaldo20Maria Luigia Gambardella20 Adriana Cristina Balduzzi1,2Alessia Cherubino3Giovanni Antonio Marrocco3
Adriana Cristina Balduzzi1,2Alessia Cherubino3Giovanni Antonio Marrocco3 Silvia Bellesi21Valentina Carusi22
Silvia Bellesi21Valentina Carusi22 Gabriele Rumi23
Gabriele Rumi23 Andrea Biondi24
Andrea Biondi24 Giuseppe Zampino3,6
Giuseppe Zampino3,6 Francesco Saettini1,24*
Francesco Saettini1,24*- 1Pediatria, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy
- 2Dipartimento Di Medicina e Chirurgia, Università Degli Studi Milano-Bicocca, Monza, Italy
- 3Center for Rare Diseases and Birth Defects, Department of Woman and Child Health and Public Health, Fondazione Policlinico A. Gemelli, IRCCS, Rome, Italy
- 4Pediatric Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy
- 5Department of Life Sciences and Public Health, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
- 6Università Cattolica Sacro Cuore, Rome, Italy
- 7Research and Clinical Unit of Primary Immunodeficiencies, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
- 8Clinical Unit of Clinical Immunology and Vaccinology, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
- 9Chair of Pediatrics, Department of Systems Medicine, University of Rome “Tor Vergata”, Roma, Italy
- 10Department of Pediatric Pneumonology, Allergy and Clinical Immunology, Institute of Pediatrics, Poznań University of Medical Sciences, Poznań, Poland
- 11Department of Medical Genetics, Poznań University of Medical Sciences, Poznań, Poland
- 12Diagnostyka GENESIS, Poznań, Poland
- 13Department of Pediatric Endocrinology and Rheumatology, Institute of Pediatrics, Poznan University of Medical Sciences, Poznan, Poland
- 14Clinical Data Analysis Department, National Medical Research Center for Endocrinology, Moscow, Russia
- 15Department of Immunology, Dmitry Rogachev National Medical Research Center of Pediatric Hematology, Oncology and Immunology, Moscow, Russia
- 16Department of Pediatric Genetics, Cerrahpasa Medical School, Istanbul University-Cerrahpasa, Istanbul, Türkiye
- 17Genetics Service, KK Women’s and Children’s Hospital, Singapore, Singapore
- 18International Rare Disease Research Consortium, Paris, France
- 19Department of Pediatric, “Papa Giovanni XXIII” Hospital, Bergamo, Italy
- 20Child Neurology and Psychiatric Unit, Department of Woman and Child Health and Public Health, Fondazione Policlinico Universitario A. Gemelli, IRCCS, Rome, Italy
- 21Dipartimento di Scienze di Laboratorio ed Ematologiche, Fondazione Policlinico Gemelli, IRCCS, Rome, Italy
- 22UOSD Allergologia ed Immunologia Clinica, Dipartimento Scienze Mediche e Chirurgiche, Fondazione Policlinico Universitario A. Gemelli, IRCCS, Rome, Italy
- 23Unità Operativa Semplice Malattie Infiammatorie Croniche Intestinali, Inflammatory Bowel Disease (IBD) Unit, CEMAD, Fondazione Policlinico Universitario A. Gemelli, IRCCS, Rome, Italy
- 24Centro Tettamanti, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy
Introduction: Cardiofaciocutaneous syndrome (CFCS) is a rare syndromic disorder caused by germline mutations affecting the RAS/MAPK pathway. It is characterized by distinctive craniofacial dysmorphism, congenital heart defects, skin abnormalities, gastrointestinal dysfunction, neurocognitive impairment, and epilepsy. Emerging evidence suggests an association with hypogammaglobulinemia, but a comprehensive characterization of immunological abnormalities in CFCS is lacking.
Methods: We conducted a retrospective, multicenter observational study to investigate the immunological phenotype of CFCS. Clinical features, immune-related manifestations, and laboratory parameters were analyzed to delineate the immunological profile of affected individuals.
Results: A total of 56 patients with a confirmed clinical and molecular diagnosis of CFCS were included, with a median age at evaluation of 13 years (range: 1–39 years). Increased susceptibility to infections was reported in 18/56 patients (32%), while autoimmune manifestations were observed in 14/56 patients (25%). Common immunological findings included monocytosis (32%), lymphopenia (21%), and hypogammaglobulinemia, with decreased IgG, IgA, or IgM levels in 21%, 40%, and 35% of patients, respectively. Genotype-phenotype analysis revealed that BRAF mutations were predominantly associated with T-cell lymphopenia, whereas MAP2K1 mutations were linked to monocytosis, reduced naïve and switched-memory B cells, and hypogammaglobulinemia. Immunodeficiency-related treatments, including immunoglobulin replacement therapy, antibiotic prophylaxis, or immunosuppressive therapy, were administered to 6/56 patients (11%).
Conclusions: CFCS is associated with recurrent yet heterogeneous immunological abnormalities, including lymphopenia, hypogammaglobulinemia, and increased infection susceptibility. Given these findings, routine immunological assessment should be considered in CFCS patients to facilitate early detection and appropriate management of immune dysfunction.
1 Introduction
Germline mutations in genes encoding proteins of the RAS/MAPK signaling pathway give rise to a family of clinically related disorders, collectively known as RASopathies. Among these, cardiofaciocutaneous syndrome (CFCS; OMIM #115150, #615278, #615279 and #615280) is a rare autosomal dominant disease that affects 1/800,000 newborns (1) and it is caused by mutations in BRAF, MAP2K1, MAP2K2, KRAS and YWHAZ genes (2). CFCS is characterized by a distinctive combination of dysmorphic craniofacial features, heart and skin abnormalities, gastrointestinal dysfunction, neurocognitive delay and epilepsy (3, 4).
Inborn errors of immunity (IEI) are heterogeneous conditions that share increased susceptibility to infections, autoimmunity, allergy, autoinflammation and predisposition to malignancy (5, 6). Syndromic immunodeficiencies are a subgroup of IEI characterized by involvement of different organ systems simultaneously (7).
Recently, case reports have described the immunological phenotype of CFCS, reporting antibody deficiency in patients affected by MAP2K1 and MAP2K2 mutations (8–10). To date, there are no studies that precisely portray the immunological phenotype of CFCS, its natural history, and treatment for immunological defects.
Here we report the results of a multicenter retrospective observational study on the immunological characterization of patients with clinical and molecular confirmed diagnosis of CFCS. We aim to investigate clinical and immunological features, including rare, severe or novel phenotypes, gather data on employed treatments, and identify correlations with non-immunological traits.
2 Methods
One author (C.E.) carried out a systematic review of all studies published as original articles up to May 31, 2024, in PubMed database. “Cardiofaciocutaneous syndrome”. Languages included were English, Spanish, and French. The corresponding authors of all relevant publications and referring physicians were contacted to obtain updated clinical information on the reported cases. To be included in our study, the criteria were: clinical diagnosis of CFCS, pathogenic or variants of unknown significance in CFCS genes (BRAF, MAP2K1, MAP2K2, KRAS, YWHAZ), completion of the Case Report Form (CRF) and provided written informed consent either from the participant, parents or legal guardians.
Data was obtained from the available medical records and collected using a CRF. Collected data included: demographics, genetic diagnosis, dysmorphisms and organ malformation, other features of CFCS (e.g., intellectual disability, epilepsy), immunological manifestations (increased susceptibility to infections [either severe (11) or recurrent (12)], autoimmunity, lymphoproliferation), immunological tests including complete blood count (CBC) with differential, immunoglobulin levels (IgG, IgA, IgM), lymphocyte subsets, serological response to vaccinations, serum autoantibodies, treatment and need for hospitalization. Lymphocyte subsets distribution and immunoglobulin levels were classified as normal, high or low according to published age-matched reference values (13–15).
The study was approved by the local hospital Ethical Committee (PID-GENMET) and was conducted according to the Helsinki Declaration.
2.1 Statistics
Possible associations between the baseline clinical features and the finding of each one of the laboratory or clinical findings were assessed via Pearson’ chi-square test or Fisher exact test. Correlation among variables included in the study was analyzed through the measurement of Pearson’s correlation coefficient r (ranging from − 1 to 1). The strength of the correlation between the two variables was considered “none” when r value was < 0.3, “weak” when 0.3 ≥ r value < 0.5, “moderate” when 0.5 ≥ r value < 0.7 and “strong” when r value ≥ 0.7. Results were deemed significant for p value < 0.05 with a double-tailed test.
3 Results
3.1 Systematic review
The PubMed search for papers describing the CFCS produced 247 results. Ninety-one papers were excluded as irrelevant (i.e. languages other than the selected ones = 1, papers not describing patients, papers describing different conditions; Supplementary Figure 1). Of the remaining 156 papers, all the 130 corresponding authors were contacted and eight of them provided responses and data. Referring physicians provided updated information (when available) and data on unpublished patients.
3.2 Patients’ characteristics
A total of 56 patients with molecularly confirmed CFCS were included in the final cohort for analysis. Of the 56 patients included, 51 carried pathogenic variants, 4 had likely pathogenic variants, and 1 had a variant of uncertain significance (VUS). In cases with likely pathogenic variants or a VUS, the diagnosis of CFCS was confirmed by an expert clinical geneticist based on the overall phenotype and current variant interpretation guidelines. Demographic data and CFCS-associated anomalies are summarized in Table 1. The median age at the last follow-up was 13 years (range 1–39 years), with a predominance of female patients (females, 62.5%). Molecular diagnosis was primarily achieved through next-generation sequencing (83.6%), while whole exome sequencing (9.1%), Sanger sequencing (5.5%), and whole genome sequencing (1.8%) were used less frequently. In one case, the specific molecular technique could not be retrieved from the patient’s medical records. Among the cohort, 37 patients (66.1%) harbored variants in BRAF, 14 (25.0%) in MAP2K1, 2 (3.6%) in MAP2K2, 2 (3.6%) in KRAS, and 1 (1.8%) in YWHAZ (Figure 1). All the variants were missense mutations detected in heterozygous state. In one patient the variant p.(Phe35Leu) in MAP2K1 was found in a mosaic state (9). In the entire cohort, genetic testing identified BRAF mutations in 37 patients, including 34 with pathogenic variants and 3 with likely pathogenic variants. MAP2K1 mutations were found in 14 patients, all classified as pathogenic. One patient carried a pathogenic variant in MAP2K2, while another patient harbored a variant of uncertain significance (VUS) in the same gene. KRAS mutations were identified in two individuals, both pathogenic. Finally, one patient had a VUS in YWHAZ. Overall, 51/56 variants (91.1%) were classified as pathogenic, 4/56 (7.1%) as likely pathogenic, and 1/56 (1.8%) as a VUS.
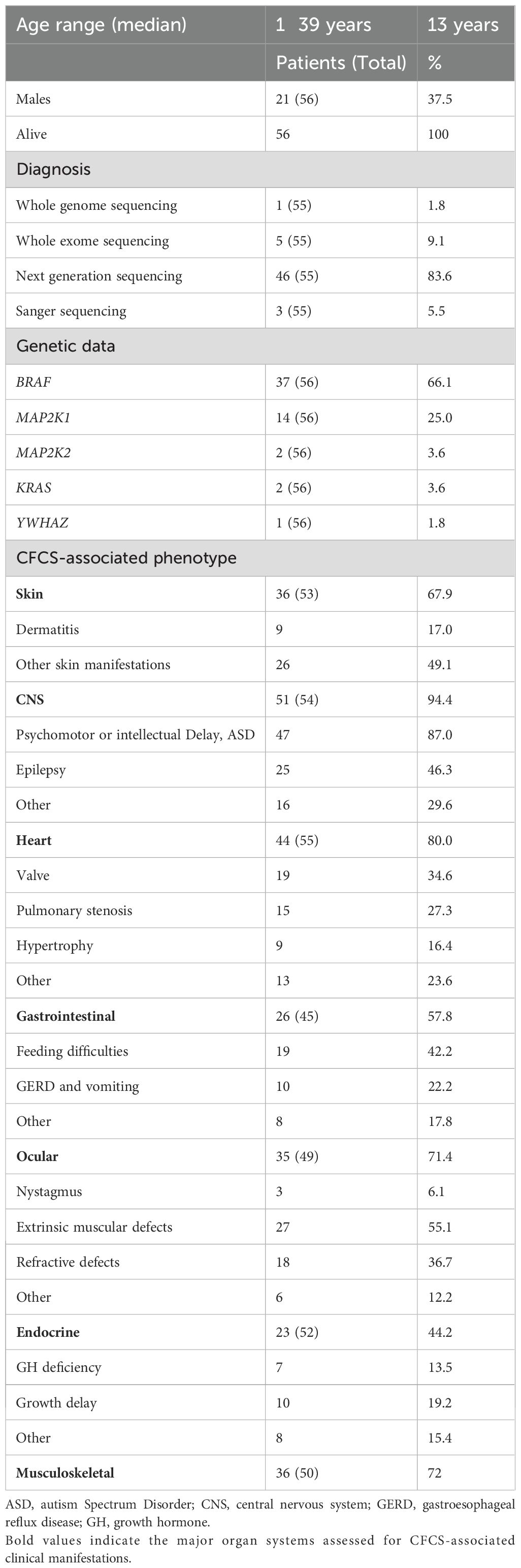
Table 1. Demographic characteristics, genetic data and CFCS associated anomalies in the whole cohort.
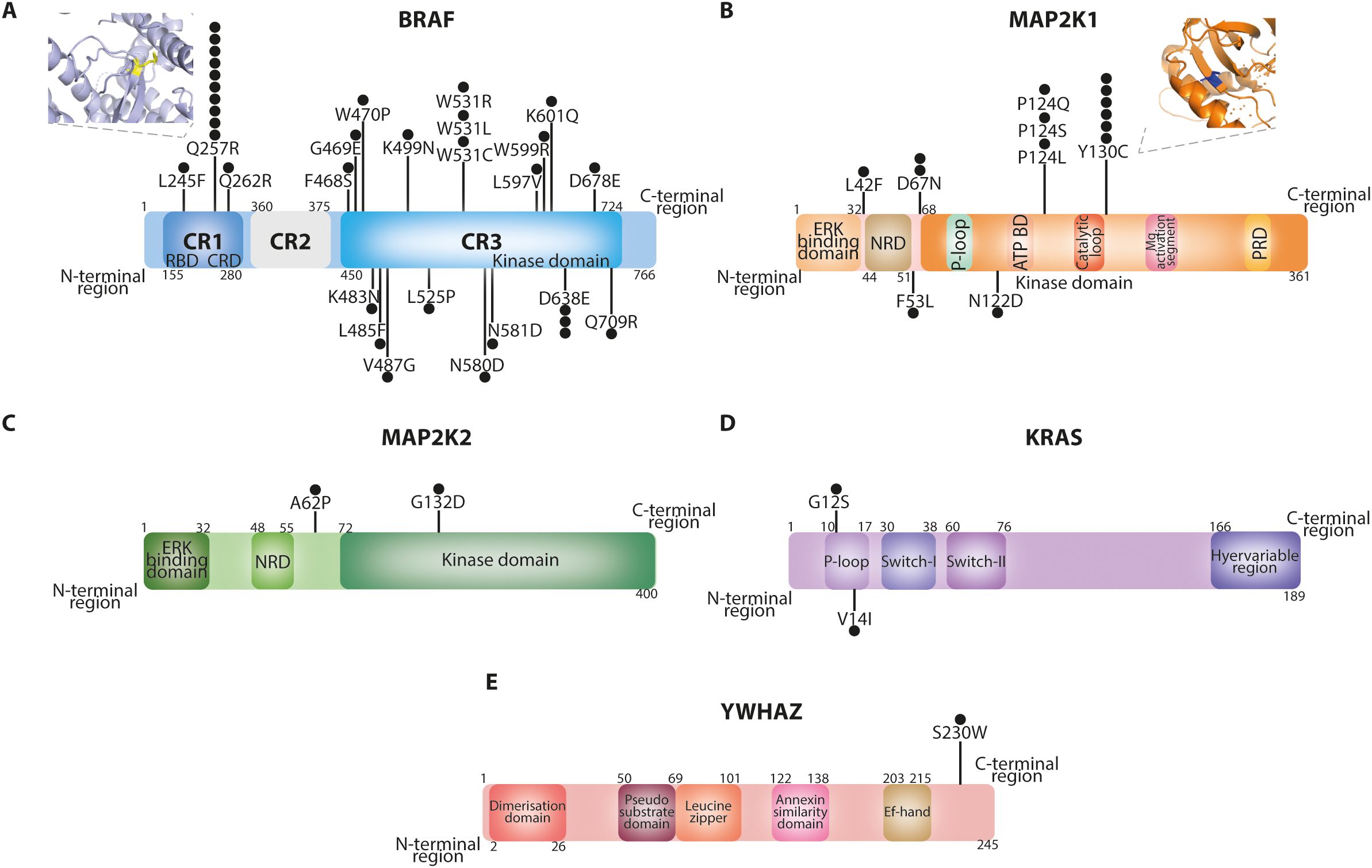
Figure 1. Protein domains of BRAF (A), MAP2K1 (B), MAP2K2 (C), KRAS (D) and YWHAZ (E) proteins highlighting the mutations identified in our cohort. The BRAF Q257R and MAP2K1 Y130C variants were modelled through PyMOL based on the PDB structures 7mfe and 3w8q, respectively. Each black dot indicates an affected patient.
3.3 Infections
Data regarding severe or recurrent infections was available for all patients and is summarized in Table 2; Supplementary Table 1. Eighteen out of 56 patients (32.1%) had a history of severe or recurrent infections, predominantly affecting the upper and lower respiratory tracts (50%) or the gastrointestinal tract (8.9%). Hospitalization for infection management was required in ten individuals (17.9%), primarily due to sepsis, pneumonia or gastroenteritis. Additionally, eleven patients (19.6%) had a history of recurrent infections, including respiratory tract infections (otitis, URTI and LRTI), urinary tract infections and gastroenteritis.
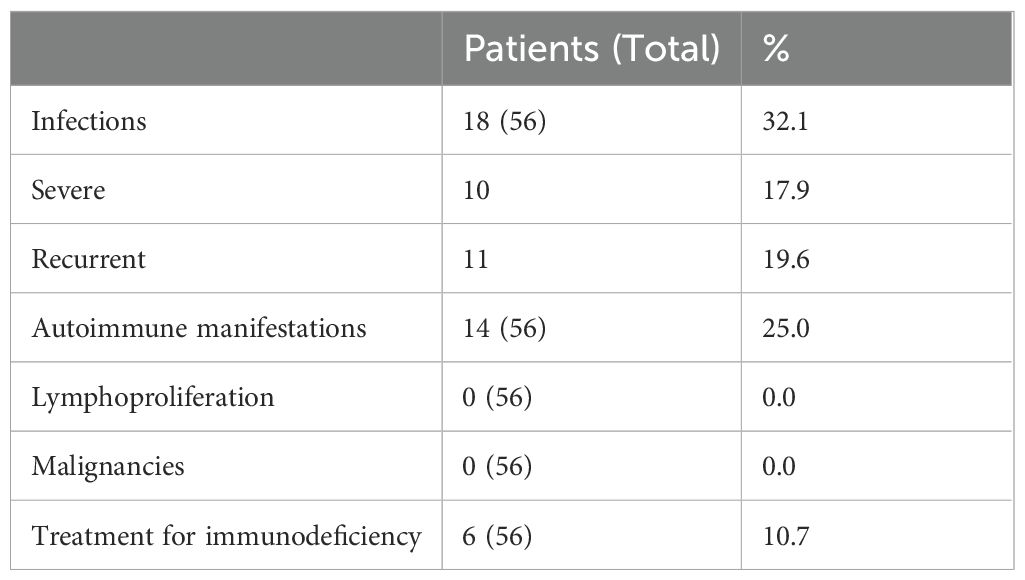
Table 2. Immunological clinical characteristics and treatment strategy in the whole cohort.
3.4 Autoimmune or immune dysregulation manifestations
Features suggestive of autoimmunity or immune dysregulation were identified in 14 patients (25.0%; Supplementary Table 2). These immune-related conditions were observed across all molecular subgroups, recapitulating overall distribution of CFCS-associated mutations. Atopic dermatitis was the most common manifestation, affecting 9 patients (17.0%). Atopic dermatitis was generally mild and managed with topical corticosteroids as needed. However, no additional data, particularly regarding allergic screening, were available. Other reported manifestations included psoriasis and celiac disease, each identified in two individuals (3.6%; one with a BRAF and one with a MAP2K1 mutations, respectively). Less frequent presentations included seronegative autoimmune hepatitis, eosinophilic esophagitis, and thyroid disease, each occurring in one patient each (1.8%). Seronegative Autoimmune hepatitis was confirmed by liver biopsy in one patient with a BRAF mutation. Eosinophilic esophagitis and thyroid disease occurred in two individuals, both carrying BRAF mutations.
Notably, autoimmune cytopenia was not documented in our cohort. Thymectomy (either partial or total) was not reported.
3.5 Lymphoproliferation and malignancies
No cases of malignant neoplasms or polyclonal benign lymphoproliferation were identified in the whole cohort.
3.6 Immunological evaluation
The immune profile of the cohort was investigated by collecting CBC with differential, lymphocyte subsets and immunoglobulin levels (Table 3). Among the CBC results, hemoglobin levels were predominantly normal (86.7%), with a small subset showing low levels (9.4%). White blood cell counts were within normal range in most patients (76.9%), although leukocytosis was observed in 21.2%. Neutrophil counts were normal in nearly all cases (94.4%), while lymphopenia was present in 21.3% of individuals. Monocyte counts showed a trend toward monocytosis (32.4%), and platelet counts were mostly normal (90.4%) with occasional thrombocytosis (9.6%).
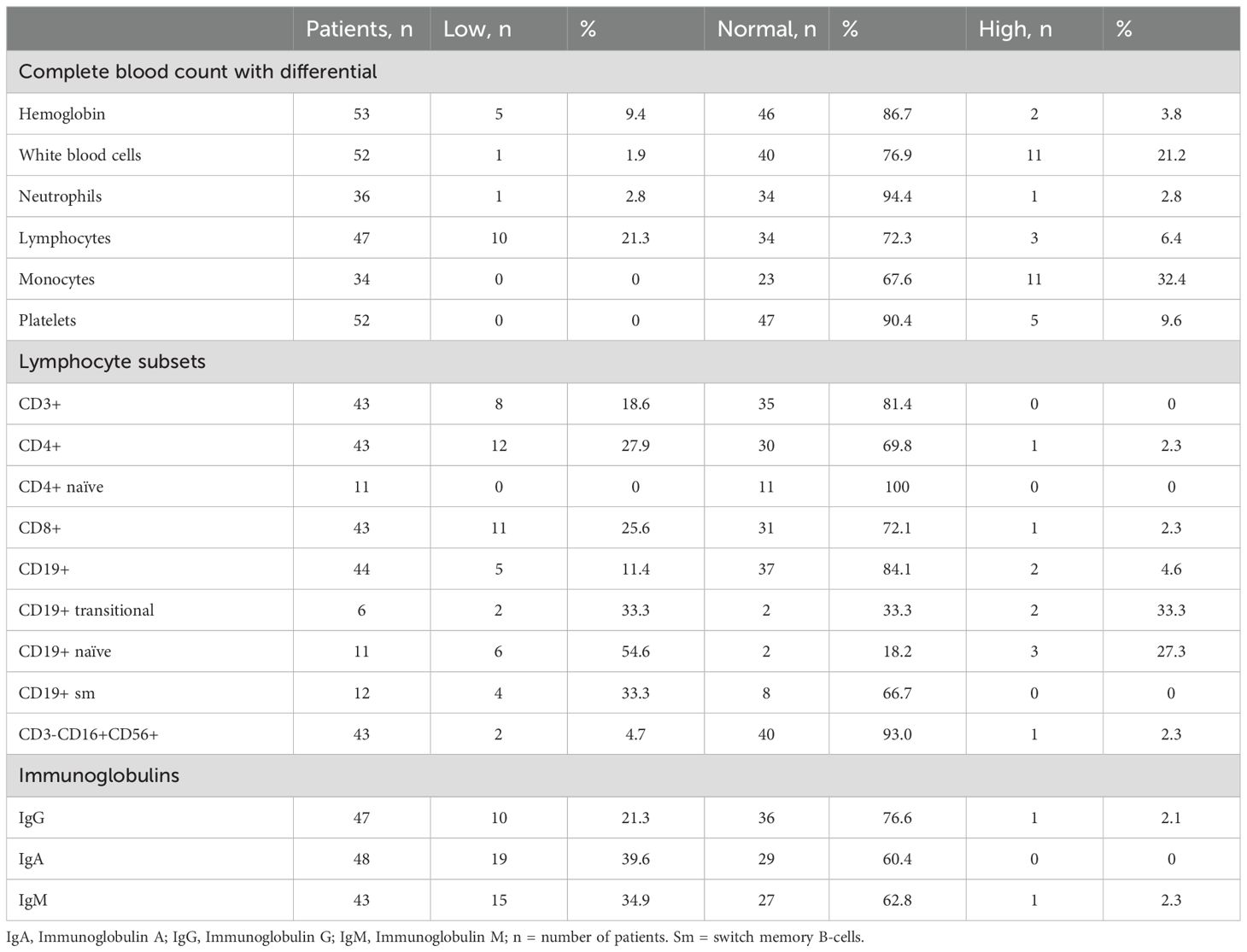
Table 3. Immune-hematological findings in the whole cohort.
Analysis of lymphocyte subsets revealed CD3+ T-cell lymphopenia in 18.6% of patients, with CD4+ lymphopenia in 27.9%, although naïve CD4+ T-cell counts were consistently normal in all tested patients. CD8+ T-cell deficiency was observed in 25.6% of cases. B-cell analysis showed reduced CD19+ cell numbers in 11.4% of patients, with a reduction in naïve B cells (54.6%) and switched memory B cells (33.3%). Natural killer (NK) cell counts were largely normal (93.0%). Twenty-six patients showed at least one decreased immunoglobulin isotype, specifically IgG (21.3%), IgA (39.6%), and IgM (34.9%). Decreased or absent response to vaccines was registered in 6 out of 16 patients (37.5%).
No significant correlation was observed between the absolute number of B cells and the levels of IgG, IgA, or IgM (data not shown). However, a moderate correlation was noted between switched memory B (smB) cells and both IgG (r = 0.56, p = 0.058) and IgA (r = 0.40, p = 0.2), though these findings did not reach statistical significance. Among immunoglobulins, moderate correlations were identified between IgG and IgM (r = 0.48, p = 0.001), IgA and IgM (r = 0.48, p = 0.001), and IgG and IgA (r = 0.62, p < 0.0001), suggesting potential interdependencies among immunoglobulin levels, though the relationship with B-cell subsets warrants further investigation (Figure 2).
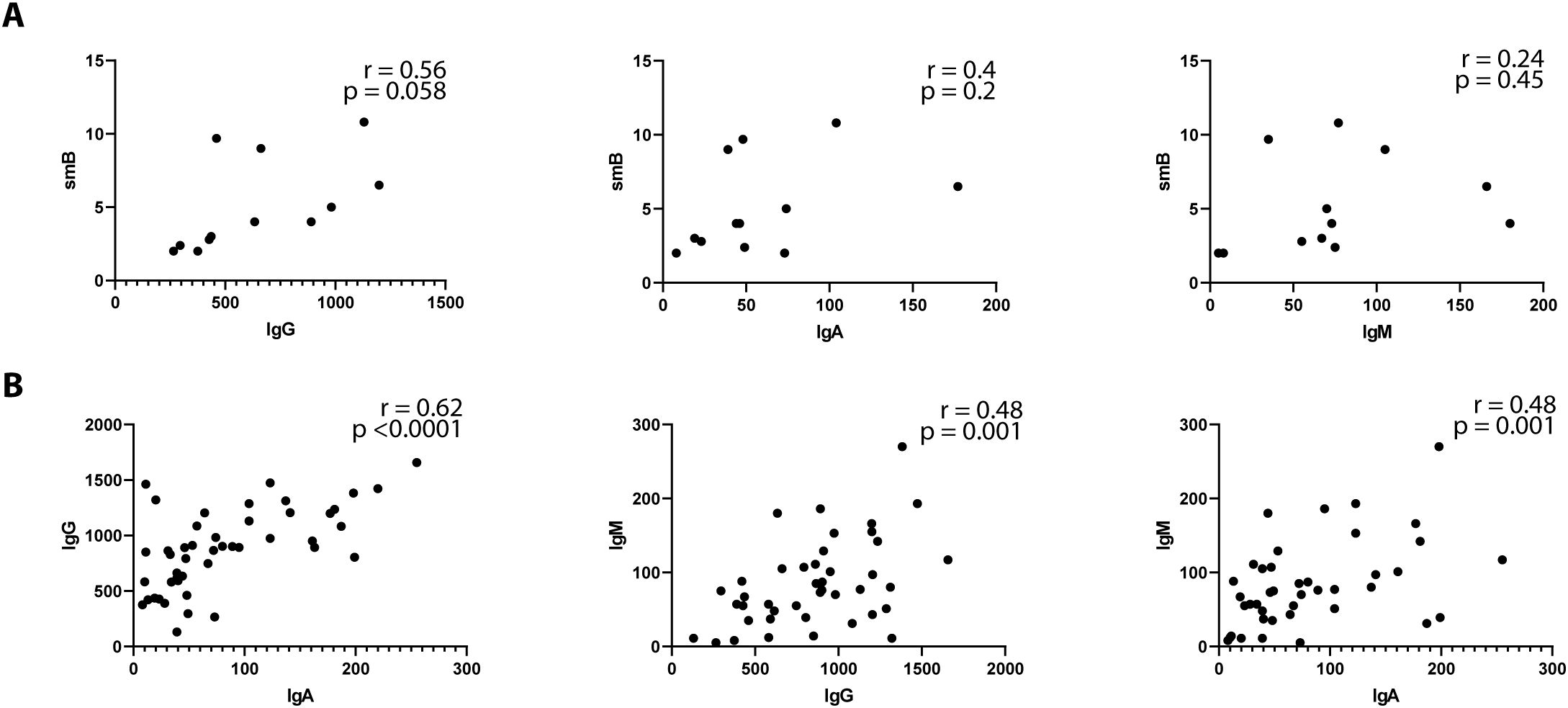
Figure 2. Immunoglobulins correlation profile in Cardiofaciocutaneous syndrome. Spearman’s correlation coefficient was used to compare between serum immunoglobulin isotypes and switched memory B (smB) cells (A) or immunoglobulin isotypes (B). smB are depicted as percentages of total B cells. Immunoglobulin isotypes are depicted as per mg/ml.
No statistically significant associations of hypogammaglobulinemia (either IgG, IgA or IgM), decreased IgG, IgM or IgA, or autoimmunity/immune dysregulation and infections were detected (data not shown). Similarly, no association between absent or partial vaccine responses and hypogammaglobulinemia or low smB was noticed (data not shown).
3.7 Genotype-phenotype correlation
Comparing BRAF- (n = 37) and MAP2K1-mutated (n = 14) patients revealed notable differences in several hematological and immunological parameters (Table 4). In the CBC, leukocytosis was observed more frequently in patients with MAP2K1 (33.3%) compared to BRAF mutations (14.3%). Similarly, lymphocytosis was more common in the MAP2K1 cohort (16.7%) compared to BRAF (3.1%), while lymphopenia was higher in BRAF-mutated patients (25.0%) than in MAP2K1-mutated patients (8.3%). Monocytosis was also more prevalent in MAP2K1 patients (50.0%) compared to BRAF (21.7%).
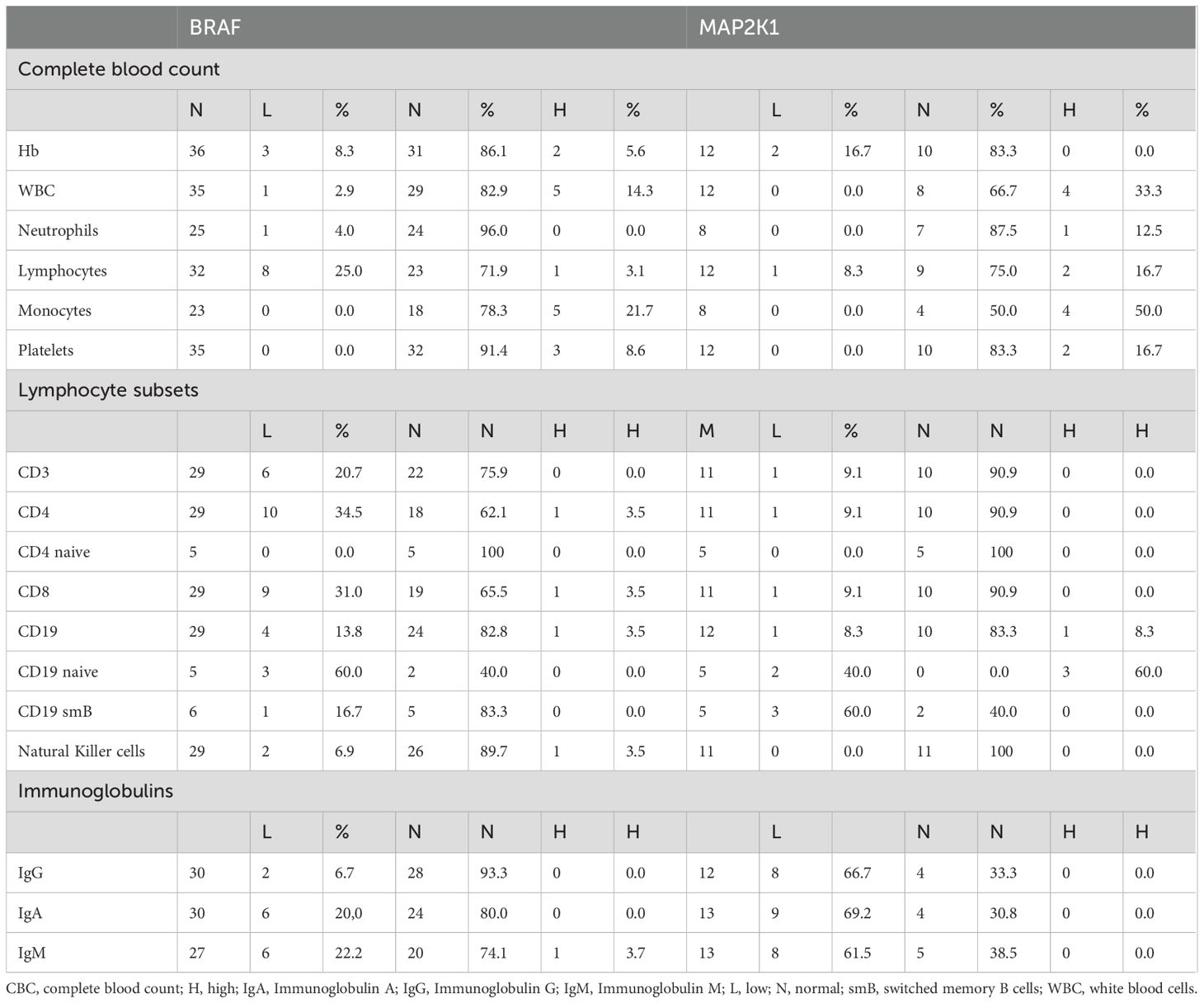
Table 4. Comparison of immunological characteristics of BRAF- and MAP2K1-mutated patients.
Regarding lymphocyte subsets, CD4+ lymphopenia was observed in 34.5% of patients with BRAF mutations, whereas only 9.1% of MAP2K1-mutated individuals exhibited this finding. Similarly, CD8+ lymphopenia was more frequent in BRAF-mutated patients (31.0%) compared to MAP2K1 (9.1%). A higher proportion of abnormalities in smB was found in MAP2K1-mutated individuals, with 60% showing decreased smB cells compared to 16.7% in BRAF-mutated patients.
Regarding immunoglobulin levels, IgG deficiency was observed in 6.7% of BRAF-mutated individuals compared to a much higher 66.7% in MAP2K1 patients. Similarly, deficiencies in IgA (20.0% vs. 69.2%) and IgM (22.2% vs. 61.5%) were more prevalent in MAP2K1-mutated individuals.
Rate of recurrent/severe infections was similar between BRAF- (12/34) while 5 patients were MAP2K1-mutated patients (5/14; p>0.99).
The comparison of immune phenotypes between individuals harboring the Q257R BRAF mutation (n = 10) and those with the Y130C MAP2K1 mutation (n = 6) highlighted notable differences (Supplementary Table 3). Lymphopenia was more common in Q257R patients (30.0%) compared to Y130C patients (20.0%). CD4+ lymphopenia was present in 33.3% of Q257R patients and 20.0% of Y130C patients, with the majority of patients in both groups maintaining normal CD8+ T-cell levels. Decreased B-cell counts were rare in Q257R patients (11.1%) but absent in Y130C patients. Decreased IgG levels were present in 11.1% of Q257R patients compared to 80% of Y130C patients, indicating a more severe defect in antibody production in the latter group. All the Y130C patients (100%) showed IgA deficiency, which was observed in 33.3% of Q257R patients. IgM deficiency was noted in 22.2% of Q257R patients compared to 83.3% of Y130C patients.
3.8 Treatment and overall survival
Given the widespread use of antiepileptic drugs (AEDs) in the CFC cohort, we investigated whether this treatment was associated with hypogammaglobulinemia as a potential side effect. Data regarding immunoglobulin levels was available for 18 out of the 24 patients receiving AEDs at the time of immunological evaluation. Among them, 50% (9/18) exhibited decreased levels of at least one immunoglobulin isotypes (IgG, IgA, or IgM). In the subgroup of patients not receiving AED treatment (32 individuals), immunoglobulin levels were assessed in 28 cases, with hypogammaglobulinemia detected in 17 cases. However, no statistically significant differences in immunoglobulin levels were observed between the AED-treated and non-treated groups (p = 0.55).
Immunodeficiency-related treatments were administered in 6 patients (10.7%). Immunoglobulin replacement therapy (IgRT) was initiated in three individuals, all of whom carried MAP2K1 variants, had decreased IgG levels and a history of recurrent respiratory tract infections. Additionally, three patients received antibiotic prophylaxis, two due to recurrent respiratory infections (one of whom was also receiving IgRT) and due to a reduced absolute count of CD4+ T cells and naïve CD4+ T cells, for which trimethoprim-sulfamethoxazole was administered.
At the last follow-up visit, all the patients were alive.
4 Discussion
CFCS is a complex multisystem disorder with variable phenotypic expression. Building on the recent evidence reporting hypogammaglobulinemia in CFCS (8–10), here we describe a large international multicenter and molecularly defined cohort, providing the most comprehensive analysis of immune abnormalities in CFCS to date. We investigated immune dysregulation, susceptibility to infections, lymphocytes’ and immunoglobulins’ alterations, and treatments in affected individuals.
Our results demonstrate that immunological alterations are common in CFCS. The immunological phenotype is highly heterogeneous, ranging from asymptomatic patients to common variable immunodeficiency-like phenotypes. CFCS should be considered a syndromic immunodeficiency. Syndromic immunodeficiencies are conditions in which immunological defects may be present only in a subset of patients (16). In recent years, there has been growing interest not only in identifying novel syndromic immunodeficiencies (17–19) but also in reassessing known syndromic conditions to determine whether immunological defects have been previously overlooked (7, 20–23).
Lymphopenia, hypogammaglobulinemia, and infections emerged as recurrent features in CFCS. Although increased infection susceptibility, mostly involving the respiratory tract, was observed, opportunistic, viral or fungal severe infections were notably absent. The frequency of infections in our CFCS cohort appears lower than in other syndromic immunodeficiencies, such as in Kabuki syndrome (KS, 69%) (21), Jacobsen syndrome (JS, 50%) (22) or Rubinstein-Taybi Syndrome (RSTS, 72%) (7). While bronchiectasis, a hallmark of primary immunodeficiencies with hypogammaglobulinemia, was not reported in our cohort, it is important to note that pulmonary function testing and chest imaging were not routinely performed. Prospective studies will be needed to define the evolution and lung sequelae of recurrent infections in CFCS patients. Additionally, craniofacial anomalies, which are characteristic of CFCS, may contribute to an increased risk of recurrent upper and lower respiratory tract infections, further complicating the immunological phenotype. Despite the relatively mild impact of immune defects, 11% of our cohort required immunodeficiency-related treatments. Thus, immune evaluations should be considered in the clinical management of CFCS patients.
AEDs were commonly administered in our cohort. Although levetiracetam (24), carbamazepine (25), valproic acid (26) and phenytoin (27) have been implicated in causing hypogammaglobulinemia, we observed no significant differences in the prevalence of hypogammaglobulinemia between AED-treated and non-treated groups.
Despite the high prevalence of skin abnormalities among CFCS patients (21/36, 58.3%), seven of whom presented with nevi, no increased risk of melanoma was observed, consistent with previous reports (2). Unlike other RASopathies, which are classified as cancer-predisposing syndromes, our CFCS cohort did not exhibit malignant lymphoproliferative disorders, further distinguishing CFCS in terms of oncological risk (2).
A noteworthy finding in our study is the high incidence (25%) of immune-mediated manifestations, which have not been previously recognized as common complication of CFCS. Although immune-mediated manifestations could point toward primary immune-regulatory disorders (28), CFCS does not exhibit concomitant autoinflammation, lymphoproliferation, malignancy, and severe atopy. This suggests that, while CFCS features immune regulatory defects, it does not fit the classic definition of a primary immune regulatory disorder.
The diverse molecular landscape of CFCS contributes to the heterogeneity of its immunological phenotype. Oncogenic BRAF and MAP2K1 mutations are functionally classified based on kinase activity, RAS-dependency and dimerization status (BRAF) (29, 30) or their dependence on phosphorylation by RAF for activation (MAP2K1) (31, 32). Germline BRAF mutations affecting the cysteine-rich domain (CRD; residues 234–280), such as the recurrent Q257R in our CFCS cohort, have been classified into CRD Classes A–C based on their ability to relieve autoinhibition and promote membrane binding. These mutations lead to enhanced RAS-dependent and -independent signaling and constitutive ERK activation (33). To date PVs or LPVs in the protein kinase domain of BRAF (F468-K601) (3) and p.Y130C/H/N variants of MAP2K1 (exon 3) have both been associated with severe epileptic phenotype (34). These genotype-phenotype correlations underscore the importance of mutation-specific analyses in CFCS to refine clinical management strategies. Our findings highlight distinct immunological signatures associated with different causative mutations, particularly between BRAF and MAP2K1 variants. BRAF mutations clustering in the CRD, particularly the Q257R variant, were predominantly associated with lymphopenia—especially T-cell lymphopenia—whereas MAP2K1 mutations, particularly Y130C variant, were frequently linked to monocytosis, reduced naïve and switched-memory B cells, and hypogammaglobulinemia, suggesting differential effects on lymphocyte development and immune cell homeostasis driven by dysregulated MAPK signaling.
The mechanism by which a subset of CFCS patients shows immune alteration remains to be elucidated. The RAS/MAPK signaling pathway is fundamental for cellular homeostasis, differentiation, proliferation, and survival. To the best of our knowledge, the role of CFCS-associated proteins in the physiology of the immune system function has not been yet characterized. CFC-causing mutations dysregulate MAPK signaling resulting in increased ERK phosphorylation (35). Similar ERK hyperactivation is observed in other RASopathies, including CBL syndrome, where monocytes exhibit ERK hyperphosphorylation (36). CBL syndrome commonly shows monocytosis, increased transitional B cells and B-cell lymphocytosis (37). Although in our cohort monocytosis was a frequent finding, we were unable to systematically investigate transitional B cells, yet B cell lymphocytosis did not appear as a common feature. In vitro studies of MAP2K1 mutations (K57M, G128V, Y130C) in neuroblastoma cell lines have demonstrated increased cell proliferation and autophagy (38). Given the tightly regulated metabolic demands of highly proliferative cells, including B cells and other hematopoietic cells, metabolic dysfunction is emerging as a key contributor to IEI (7, 19, 23). Notably, mitochondrial dysfunction, affecting oxidative phosphorylation and mitochondrial proteostasis, has been described in fibroblasts from patients with RASopathies, including CFCS (39, 40). These intriguing findings highlight the possibility that RAS/MAPK signaling dysregulation in CFCS may impact immune cell energy metabolism, contributing to the observed immunological abnormalities in CFCS’ hematopoietic cells.
Currently, no targeted therapies exist for CFCS. MEK inhibitors, including trametinib, have shown promising results in treating RASopathy-associated hypertrophic cardiomyopathy (41) and severe lymphatic anomalies (42–44). As MEK inhibitors are likely to be used in additional CFCS patients in the future, it would be valuable to conduct immunological assessments before and after treatment. Studying peripheral blood immune cells from CFCS patients undergoing MEK inhibition could provide critical insights into the role of RAS/MAPK signaling in hematopoietic cell function.
This study has several limitations. Approximately 35% of the patients were identified through literature review, leading to missing or incomplete data. Most published studies focus on pediatric populations, raising the possibility that immune system abnormalities (such as infections, immune dysregulation, and lymphoproliferation) may become more pronounced with age. Consequently, immune defects in CFCS could be underrecognized, and longitudinal studies are needed to fully assess their progression. Microbiological data, including pathogen identification and cultures during infectious episodes, were not systematically collected, limiting our ability to fully characterize the infectious burden and its etiology in CFCS patients.
Additionally, dual molecular diagnoses or multilocus genomic variations have been reported in up to 5% of patients undergoing whole exome sequencing (45) and in 10% of those screened for IEI (46). Since WES was not widely performed in our cohort, we cannot exclude the possibility that some patients carry additional pathogenic variants contributing to their immunological phenotype (47).
5 Conclusions
Until recently, the connection between immunological alterations and CFCS has been reported only in a few cases, and immunodeficiency was not listed within the main symptoms that individuals with CFCS may present. Our study establishes that CFCS is associated with variable but recurring immunological abnormalities, including lymphopenia, hypogammaglobulinemia, and increased infection susceptibility. Although immune defects in CFCS appear mild compared to other syndromic immunodeficiencies, a subset of patients requires immunodeficiency-related treatments. Our findings emphasize the need for routine immunological assessments in CFCS and highlight the potential role of RAS/MAPK signaling in immune system regulation. Future studies should focus on the metabolic and signaling pathways underlying CFCS-associated immune dysfunction, as well as the impact of MEK inhibitors on immune homeostasis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Comitato Etico Brianza (PID-GENMET). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
BEDM: Writing – original draft, Data curation. CL: Writing – review & editing, Writing – original draft. EC: Data curation, Writing – original draft. CF: Writing – original draft. GV: Writing – original draft. VT: Investigation, Writing – original draft, Data curation. LB: Formal Analysis, Writing – original draft. FC: Data curation, Writing – original draft. MM: Writing – original draft, Data curation. EM: Data curation, Writing – original draft. DR: Writing – original draft, Data curation. BR: Writing – original draft. CC: Writing – original draft. AS-P: Writing – original draft, Data curation. AJ: Writing – original draft, Data curation. MO-M: Writing – original draft, Data curation. VZ: Writing – original draft, Data curation. AS: Data curation, Writing – original draft. JR: Data curation, Writing – original draft. BT: Writing – original draft, Data curation. SJ: Writing – original draft, Data curation. JL: Writing – original draft, Data curation. JG: Writing – original draft, Data curation. ACe: Writing – original draft, Data curation. TA: Data curation, Writing – original draft. IC: Writing – original draft, Data curation. MG: Writing – original draft, Data curation. ACB: Writing – review & editing. ACh: Writing – original draft. GM: Writing – original draft. SB: Writing – original draft. VC: Writing – original draft. GR: Writing – review & editing. AB: Writing – review & editing. GZ: Writing – review & editing. FS: Conceptualization, Formal Analysis, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. SJ is supported by National Medical Research Council Clinician Scientist Award (NMRC/CSAINVJun21-0003) and (NMRC/CSAINV24jul-0001). CL is supported by GR-2019-12371203 funded by Italian Ministry of Health.
Conflict of interest
AJ was employed by Diagnostyka GENESIS.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1598896/full#supplementary-material
Supplementary Table 1 | Clinical, molecular and laboratory characteristics of the CFCS patients.
Supplementary Table 2 | Immunological characteristics of BRAF Q257R vs MAP2K1 Y130C mutated patients.
References
1. Pierpont ME, Magoulas PL, Adi S, Kavamura MI, Neri G, Noonan J, et al. Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. (2014) 134:e1149–62. doi: 10.1542/peds.2013-3189
2. Scorrano G, David E, Calì E, Chimenz R, La Bella S, Di Ludovico A, et al. The cardiofaciocutaneous syndrome: from genetics to prognostic-therapeutic implications. Genes. (2023) . 14:2111. doi: 10.3390/genes14122111
3. Battaglia DI, Gambardella ML, Veltri S, Contaldo I, Chillemi G, Veredice C, et al. Epilepsy and BRAF mutations: phenotypes, natural history and genotype-phenotype correlations. Genes. (2021) 12:1316. doi: 10.3390/genes12091316
4. Musto E, Gambardella ML, Perulli M, Quintiliani M, Veredice C, Verdolotti T, et al. Status epilepticus in BRAF-related cardio-facio-cutaneous syndrome: Focus on neuroimaging clues to physiopathology. Epilepsia Open. (2024) . 9:258–67. doi: 10.1002/epi4.12864
5. IJspeert H, Edwards ESJ, O’Hehir RE, Dalm VASH, and van Zelm MC. Update on inborn errors of immunity. J Allergy Clin Immunol. (2024) 155(3):740–51. doi: 10.1016/j.jaci.2024.12.1075
6. Bosch JVWT, Hlaváčková E, Derpoorter C, Fischer U, Saettini F, Ghosh S, et al. How to recognize inborn errors of immunity in a child presenting with a Malignancy: guidelines for the pediatric hemato-oncologist. Pediatr Hematol Oncol. (2023) 40:131–46. doi: 10.1080/08880018.2022.2085830
7. Saettini F, Herriot R, Prada E, Nizon M, Zama D, Marzollo A, et al. Prevalence of immunological defects in a cohort of 97 rubinstein-taybi syndrome patients. J Clin Immunol. (2020) 40:851–60. doi: 10.1007/s10875-020-00808-4
8. Szczawińska-Popłonyk A, Popłonyk N, Niedziela M, Sowińska-Seidler A, Sztromwasser P, Jamsheer A, et al. Case report: The cardio-facio-cutaneous syndrome due to a novel germline mutation in MAP2K1: A multifaceted disease with immunodeficiency and short stature. Front Pediatr. (2022) 10:990111. doi: 10.3389/fped.2022.990111
9. Zakharova V, Raykina E, Mersiyanova I, Deordieva E, Pershin D, Vedmedskia V, et al. Cancer-causing MAP2K1 mutation in a mosaic patient with cardio-facio-cutaneous syndrome and immunodeficiency. Hum Mutat. (2022) 43:1852–5. doi: 10.1002/humu.24463
10. Leoni C, Tedesco M, Onesimo R, Giorgio V, Rigante D, and Zampino G. Immunoglobulin deficiency associated with a MAP2K1-related mutation causing cardio-facio-cutaneous syndrome. Immunol Letters. (2020) . 227:79–80. doi: 10.1016/j.imlet.2020.08.009
11. Farruggia P, Fioredda F, Puccio G, Porretti L, Lanza T, Ramenghi U, et al. Autoimmune neutropenia of infancy: data from the Italian neutropenia registry. Am J Hematol. (2015) 90:E221–2. doi: 10.1002/ajh.24187
12. Schaad UB, Esposito S, and Razi CH. Diagnosis and management of recurrent respiratory tract infections in children: A practical guide. Arch Pediatr Infect Dis. (2016) 4:e31039. doi: 10.5812/pedinfect.31039
13. Schatorjé EJH, Gemen EFA, Driessen GJA, Leuvenink J, van Hout RWNM, van der Burg M, et al. Age-matched reference values for B-lymphocyte subpopulations and CVID classifications in children. Scand J Immunol. (2011) 74:502–10. doi: 10.1111/j.1365-3083.2011.02609.x
14. Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: The Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. (2003) 112:973–80. doi: 10.1016/j.jaci.2003.07.003
15. Schauer U, Stemberg F, Rieger CHL, Borte M, Schubert S, Riedel F, et al. IgG subclass concentrations in certified reference material 470 and reference values for children and adults determined with the binding site reagents. Clin Chem. (2003) 49:1924–9. doi: 10.1373/clinchem.2003.022350
16. Ming JE and Stiehm ER. Syndromic immunodeficiencies. In: Rezaei N, Aghamohammadi A, and Notarangelo L, editors. Primary immunodeficiency diseases. Springer, Berlin, Heidelberg (2017).
17. Verdura E, Rodríguez-Palmero A, Vélez-Santamaria V, Planas-Serra L, de la Calle I, Raspall-Chaure M, et al. Biallelic PI4KA variants cause a novel neurodevelopmental syndrome with hypomyelinating leukodystrophy. Brain. (2021) . 144:2659–69. doi: 10.1093/brain/awab124
18. Saettini F, Guerra F, Mauri M, Salter CG, Adam MP, Adams D, et al. Biallelic PI4KA mutations disrupt B-cell metabolism and cause B-cell lymphopenia and hypogammaglobulinemia. J Clin Immunol. (2024) . 45:15. doi: 10.1007/s10875-024-01793-8
19. Saettini F, Poli C, Vengoechea J, Bonanomi S, Orellana JC, Fazio G, et al. Absent B cells, agammaglobulinemia, and hypertrophic cardiomyopathy in folliculin-interacting protein 1 deficiency. Blood. (2021) 137:493–9. doi: 10.1182/blood.2020006441
20. Lacombe D, Bloch-Zupan A, Bredrup C, Cooper EB, Houge SD, García-Miñaúr S, et al. Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement. J Med Genet. (2024) 61:503–19. doi: 10.1136/jmg-2023-109438
21. Rossini L, Ricci S, Montin D, Azzari C, Gambineri E, Tellini M, et al. Immunological aspects of kabuki syndrome: A retrospective multicenter study of the Italian primary immunodeficiency network (IPINet). J Clin Immunol. (2024) 44:105. doi: 10.1007/s10875-024-01676-y
22. Baronio M, Saettini F, Gazzurelli L, Rossi S, Marzollo A, Ricci S, et al. Immunological evaluation of patients affected with jacobsen syndrome reveals profound not age-related lymphocyte alterations. J Clin Immunol. (2022) 42:365–74. doi: 10.1007/s10875-021-01169-2
23. Saettini F, Guerra F, Fazio G, Bugarin C, McMillan HJ, Ohtake A, et al. Antibody deficiency in patients with biallelic KARS1 mutations. J Clin Immunol. (2023) 43:2115–25. doi: 10.1007/s10875-023-01584-7
24. Ozdemir H, Sumer S, Karabagli H, Akdemir G, Caliskaner AZ, and Artac H. B cell aplasia and hypogammaglobulinemia associated with levetiracetam. Ann Saudi Med. (2018) 38:65–8. doi: 10.5144/0256-4947.2018.09.01.1430
25. Yamamoto T, Uchiyama T, Takahashi H, Himuro K, Kanai K, and Kuwabara S. B cell aplasia and hypogammaglobulinemia after carbamazepine treatment. Internal Med. (2010) 49:707–8. doi: 10.2169/internalmedicine.49.3087
26. Eom TH, Lee HS, Jang PS, and Kim YH. Valproate-induced panhypogammaglobulinemia. Neurological Sci. (2013) 34:1003–4. doi: 10.1007/s10072-012-1153-3
27. Pereira LF and Sanchez JF. Reversible panhypogammaglobulinemia associated with phenytoin treatment. Scand J Infect Dis. (2002) 34:785–7. doi: 10.1080/00365540260348662
28. Chan AY and Torgerson TR. Primary immune regulatory disorders: a growing universe of immune dysregulation. Curr Opin Allergy Clin Immunol. (2020) 20:582–90. doi: 10.1097/ACI.0000000000000689
29. Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. (2015) 28:370–83. doi: 10.1016/j.ccell.2015.08.001
30. Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, et al. Tumors with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. (2017) 548:234–8. doi: 10.1038/nature23291
31. Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger R, et al. Allele-specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov. (2018) 8:648–61. doi: 10.1158/2159-8290.CD-17-1452
32. Mizuno S, Ikegami M, Koyama T, Sunami K, Ogata D, Kage H, et al. High-throughput functional evaluation of MAP2K1 variants in cancer. Mol Cancer Ther. (2023) 22:227–39. doi: 10.1158/1535-7163.MCT-22-0302
33. Spencer-Smith R, Terrell EM, Insinna C, Agamasu C, Wagner ME, Ritt DA, et al. RASopathy mutations provide functional insight into the BRAF cysteine-rich domain and reveal the importance of autoinhibition in BRAF regulation. Mol Cell. (2022) 82:4262–4276.e5. doi: 10.1016/j.molcel.2022.10.016
34. Pierpont EI, Kenney-Jung DL, Shanley R, Zatkalik AL, Whitmarsh AE, Kroening SJ, et al. Neurologic and neurodevelopmental complications in cardiofaciocutaneous syndrome are associated with genotype: A multinational cohort study. Genet Med. (2022) 24:1556–66. doi: 10.1016/j.gim.2022.04.004
35. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. (2006) 311:1287–90. doi: 10.1126/science.1124642
36. Bohlen J, Bagarić I, Vatovec T, Ogishi M, Ahmed SF, Cederholm A, et al. Autoinflammation in patients with leukocytic CBL loss of heterozygosity is caused by constitutive ERK-mediated monocyte activation. J Clin Invest. (2024) 134:e181604. doi: 10.1172/JCI181604
37. Saettini F, Coliva TA, Vendemini F, Galbiati M, Bugarin C, Masetti R, et al. Abnormal B-cell maturation and increased transitional B cells in CBL syndrome. Front Pediatr. (2022) 10:935951. doi: 10.3389/fped.2022.935951
38. Chen J, Che L, Xu C, Zhao S, Yang J, Li M, et al. Cardio-facio-cutaneous syndrome-associated pathogenic MAP2K1 variants activate autophagy. Gene. (2020) 733:144369. doi: 10.1016/j.gene.2020.144369
39. Dard L, Hubert C, Esteves P, Blanchard W, Bou About G, Baldasseroni L, et al. HRAS germline mutations impair LKB1/AMPK signaling and mitochondrial homeostasis in Costello syndrome models. J Clin Invest. (2022) 132:e131053. doi: 10.1172/JCI131053
40. Kleefstra T, Wortmann SB, Rodenburg RJ, Bongers EM, Hadzsiev K, Noordam C, et al. Mitochondrial dysfunction and organic aciduria in five patients carrying mutations in the Ras-MAPK pathway. Eur J Hum Genet. (2011) 19:138–44. doi: 10.1038/ejhg.2010.171
41. Wolf CM, Zenker M, Boleti O, Norrish G, Russell M, Meisner JK, et al. Impact of MEK inhibition on childhood RASopathy-associated hypertrophic cardiomyopathy. J Am Coll Cardiol Basic Trans Sci. (2024) 10(2):152–66. doi: 10.1016/j.jacbts.2024.10.002
42. Pascarella A, Limongelli G, De Falco A, Minale EMP, Di Nardo G, Di Marco GM, et al. Refractory chylothorax and ventricular hypertrophy treated with trametinib in a patient with noonan syndrome: 18-month follow-up. Children. (2024) . 11:1342. doi: 10.3390/children11111342
43. Gazzin A, Fornari F, Cardaropoli S, Carli D, Tartaglia M, Ferrero GB, et al. Exploring new drug repurposing opportunities for MEK inhibitors in RASopathies: A comprehensive review of safety, efficacy, and future perspectives of trametinib and selumetinib. Life. (2024) . 14:731. doi: 10.3390/life14060731
44. Gordon K, Moore M, Van Zanten M, Pearce J, Itkin M, Madden B, et al. Case Report: Progressive central conducting lymphatic abnormalities in the RASopathies. Two case reports, including successful treatment by MEK inhibition. Front Genet. (2022) . 13:1001105. doi: 10.3389/fgene.2022.1001105
45. Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. (2017) 376:21–31. doi: 10.1056/NEJMoa1516767
46. Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. (2017) 139:232–45. doi: 10.1016/j.jaci.2016.05.042
Keywords: primary immunodeficiency, inborn errors of immunity, cardiofaciocutaneous syndrome, rasopathy, hypogammaglobulinemia, BRAF, MAP2K1, Syndromic immunodeficiency
Citation: Di Majo BE, Leoni C, Cartisano E, Fossati C, Viscogliosi G, Trevisan V, Bruno LP, Conti F, Moratti M, Monaco E, Rigante D, Rivalta B, Cancrini C, Szczawińska-Popłonyk A, Jamsheer A, Obara-Moszyńska M, Zakharova V, Shcherbina A, Rodina J, Tüysüz B, Jamuar SS, Lim JY, Goh J, Cereda A, Agovino T, Contaldo I, Gambardella ML, Balduzzi AC, Cherubino A, Marrocco GA, Bellesi S, Carusi V, Rumi G, Biondi A, Zampino G and Saettini F (2025) Cardiofaciocutaneous syndrome and immunodeficiency: data from an international multicenter cohort. Front. Immunol. 16:1598896. doi: 10.3389/fimmu.2025.1598896
Received: 24 March 2025; Accepted: 11 June 2025;
Published: 07 July 2025.
Edited by:
Maurizio Miano, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Federica Pulvirenti, Academic Hospital Policlinico Umberto, ItalyBen Shillitoe, Sheffield Children’s Hospital, United Kingdom
Emma Westermann-Clark, University of South Florida, United States
Copyright © 2025 Di Majo, Leoni, Cartisano, Fossati, Viscogliosi, Trevisan, Bruno, Conti, Moratti, Monaco, Rigante, Rivalta, Cancrini, Szczawińska-Popłonyk, Jamsheer, Obara-Moszyńska, Zakharova, Shcherbina, Rodina, Tüysüz, Jamuar, Lim, Goh, Cereda, Agovino, Contaldo, Gambardella, Balduzzi, Cherubino, Marrocco, Bellesi, Carusi, Rumi, Biondi, Zampino and Saettini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Saettini, Zi5zYWV0dGluaUBnbWFpbC5jb20=
†These authors have contributed equally to this work