 Juan Zhou1,2†Danli Ye3†
Juan Zhou1,2†Danli Ye3† Yuansen Li3†Xuwen Lai3Wenzhi Cui3
Yuansen Li3†Xuwen Lai3Wenzhi Cui3 Wenyuan He3Ling Yu3Jingyi Wu4
Wenyuan He3Ling Yu3Jingyi Wu4 Guangning Yan3*Chengyong Lei5*
Guangning Yan3*Chengyong Lei5* Wei Wang3,6*
Wei Wang3,6*- 1Department of Oncology, General Hospital of Southern Theater Command, People’s Liberation Army of China, Guangzhou, China
- 2The First School of Clinical Medicine, Southern Medical University, Guangzhou, China
- 3Department of Pathology, General Hospital of Southern Theater Command, People’s Liberation Army of China, Guangzhou, China
- 4Graduate School, Guangzhou University of Chinese Medicine, Guangzhou, China
- 5Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, China
- 6School of Basic Medicine, Southern Medical University, Guangzhou, China
Introduction: Circulating tumor DNA (ctDNA) methylation markers show potential for early detection of cancer metastasis. This study aimed to identify ctDNA methylation markers predictive of recurrence and prognosis in colorectal cancer (CRC) patients, and to explore the influence of the tumor immune microenvironment on outcomes.
Methods: We analyzed 603 overlapping methylation markers from both plasma and tissue samples and developed a risk model to predict CRC recurrence and prognosis.
Results: ZNF671 and ZNF132 were identified as key methylation markers. The model predicted relapse risk in stage III CRC patients with an AUC of 0.90 and prognosis in stage IV patients. High-risk patients exhibited a significantly higher early relapse rate (75.4% vs. 20%) and were more likely to have a low Immunoscore (IS), which correlates with poorer prognosis.
Discussion: ZNF671 and ZNF132 methylation levels inversely correlate with Immunoscore and may serve as valuable biomarkers for CRC immunotherapy. These findings provide insights for improved prognostic evaluation and personalized treatment strategies.
Introduction
Colorectal carcinoma (CRC) is the most common malignant tumor with frequent distant metastasis (1). Most of late-stage CRC metastasis sites are liver, while lungs and peritoneum metastasis are common as well. However, patients could still receive resection of the primary and metastatic lesions as long as the metastasis were limited, which may offer the only opportunity for potential cure or long-term survival (2–5). Stage III-IV CRC patients still has high recurrence rate, even if curative surgery and adjuvant chemotherapy was timely applicated (6). To date, despite the enormous efforts that the researchers have already put in, there are still little precise biomarkers to predict the recurrence risk or guide personalized treatment strategies.
Minimal residual disease (MRD) and circulating tumor cell (CTC) clusters are critical factors in cancer relapse and metastasis (7) but cannot be detected by routine imaging techniques (8–10). Research suggests that CTCs may undergo DNA methylation changes that facilitate metastatic seeding (7, 11). The epigenetic modification of DNA methylation has been shown to surpass somatic mutations in clinical applications, particularly in evaluating recurrence risk and developing personalized treatment strategies (12–15). Immunoscore could also predict the recurrence risk for CRC patients and be a predictive tool for cancer treatment (16, 17). Moreover, DNA methylation can interact with various immune cells and stromal components in tumor microenvironment (TME) which may potentially provide new avenues for cancer immunotherapy (18, 19). This study aims to identify novel methylation markers that could facilitate their clinical application and large-scale population screening. Furthermore, we investigate the prognostic significance of immune cell infiltration, as well as the relationship between the methylation model with the tumor immune microenvironment in CRC patients. The ctDNA methylation risk model, integrated with immune infiltrating cells in the TME, may provide valuable insights to predict and stratify CRC patients who are likely to benefit from adjuvant chemotherapy or immunotherapy.
Methods
Ethics approvals and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committees of Southern Hospital (approval number: 2020-010) and General Hospital of Southern Theater Command (approval number: 2024GJJ036). All experimental plans were reviewed and approved by the committees before the clinical study began. Written informed consent was obtained from all participants prior to their enrollment in the study.
Sample collection
Tissue and plasma samples from CRC patients were collected between May 2015 and October 2020 at the Southern Hospital of Southern Medical University and the General Hospital of the Southern Theater Command. The inclusion criteria were as follows: 1) pathologically and radiologically confirmed CRC diagnosis; 2) stage IV CRC patients who underwent liver, lung, or peritoneal metastasectomy with curative intent; 3) availability of tissue and/or blood samples for analysis. The exclusion criteria included: 1) a history of preoperative treatments; 2) incomplete clinical information; 3) a history of other cancers. Paired primary tumor and metastatic lesions from 27 stage IV CRC patients were analyzed to evaluate the correlation between primary tumor and its distant metastases. The healthy control group consisted of individuals with no significant medical history (such as cancer or chronic diseases) and aged over 18 years.
The sample collection followed our previous study protocol (20). In brief, a total of 139 tissue samples were collected, including 40 primary tumor samples, 42 metastatic stage IV tumor samples, and 57 normal mucosa samples from CRC patients. Additionally, 308 preoperative blood samples from CRC patients and 50 blood samples from healthy controls were included. Advanced CRC patients were regularly followed up after completing systemic treatment. During the first 3 years, imaging examinations were conducted every 6 months, followed by annual examinations until 5 years or the first radiological recurrence or death. Outcome data were collected through phone calls and outpatient records. Serum levels of CEA and CA199 had reference ranges of 0-5 μg/mL and 37 U/mL, respectively (21).
Anchor Dx’s proprietary targeted methylation sequencing
All samples were processed using AnchorDx’s proprietary targeted methylation sequencing platform, which focuses on 12,624 cancer-specific CpG regions. Methylation sequencing was then performed using the Illumina HiSeqX Ten Sequencing System, with the detailed methods, including DNA extraction, bisulfite conversion, and construction of the AnchorIRIS™ pre-library, previously described in our published studies (13, 22, 23).
Data processing was performed using Illumina Sequencing Analysis Viewer and FastQC software to assess sequencing quality, and a custom algorithm was applied to trim low-quality bases. The sequencing reads were then aligned to the in silico converted hg19 reference genome using Bismark, and Picard was used to evaluate the sequencing performance, excluding CpG sites with coverage less than 30× or a missing rate greater than 0.20.
Immunohistochemical detection and Immunoscore assay
The Immunoscore assay was conducted as previously reported (24). Briefly, experienced pathologists selected tumor blocks containing both the tumor and invasive margin from CRC patients. Sections of 4 μm were processed for standardized CD3 and CD8 immunohistochemical staining following the manufacturer’s instructions. Digital images of the stained tissue sections were captured at 20× magnification with a resolution of 0.45 µm/pixel. The densities of CD3 and CD8 in both regions of the whole slide were determined using dedicated Immunoscore software and then converted into percentiles by comparison with the Immunoscore database. The mean of the four percentiles was calculated and converted into an Immunoscore. The Immunoscore was typically categorized into three groups: 0%-25% (low), >25%-70% (intermediate), and >70%-100% (high). Immunohistochemical analysis and full scans of staining images were performed at Genecast Laboratory (Wuxi, China). The predictive accuracy of the Immunoscore was evaluated by the integrated area under the ROC curve, and the performance of risk prediction models was compared using the likelihood ratio test.
ZNF671 and ZNF132 expression analysis and association with prognosis and the immune microenvironment
Statistical analysis was conducted using RStudio 4.4.1 with Bioconductor. Group differences were assessed using either Wilcoxon tests or paired t-tests, with a significance threshold set at p < 0.05. ROC curves were used to evaluate predictive accuracy, and Cox regression models were employed to analyze overall survival. The “ESTIMATE” package was used to calculate immune scores, while the “WGCNA” package identified gene co-expression modules. Additionally, the “xCell” package was utilized to examine the relationship between gene expression and immune cell infiltration.
Statistical analysis
All statistical analyses and data visualizations were conducted using R (version 3.6.0) and Prism 8 (GraphPad Software). Pearson’s chi-square test and Fisher’s exact test were used to compare group differences. Survival data were analyzed with the Kaplan-Meier method, and Cox regression identified prognostic factors for recurrence-free survival (RFS). Hazard ratios (HR) and 95% confidence intervals (CI) were calculated using both univariate and multivariate Cox models. ROC curves were generated with the pROC R package (version 1.15.3) to evaluate the methylation model’s performance. LASSO and random forest methods were used for variable selection and risk model construction. Statistical significance was set at a p-value < 0.05.
Results
Patient characteristics and sample collection
A total of 358 plasma samples (including 308 CRC patients and 50 healthy controls) and 139 formalin-fixed paraffin-embedded (FFPE) tissue samples (57 normal mucosa, 40 primary tumors, and 42 metastatic tissues from stage IV CRC patients) were collected in the study. Twelve tissue samples and 45 plasma samples were excluded due to DNA extraction quality control (QC) failure or low library yield. A total of 84 stage IV CRC patients underwent surgery, including 57 with liver metastases, 9 with lung metastases, and 18 with abdominal metastases. Detailed clinical characteristics are provided in Supplementary Table S1. Ultimately, 27 patients had matched preoperative plasma, primary tumor, and metastatic tumor tissue samples. The design and implementation of this study are outlined in Figure 1.
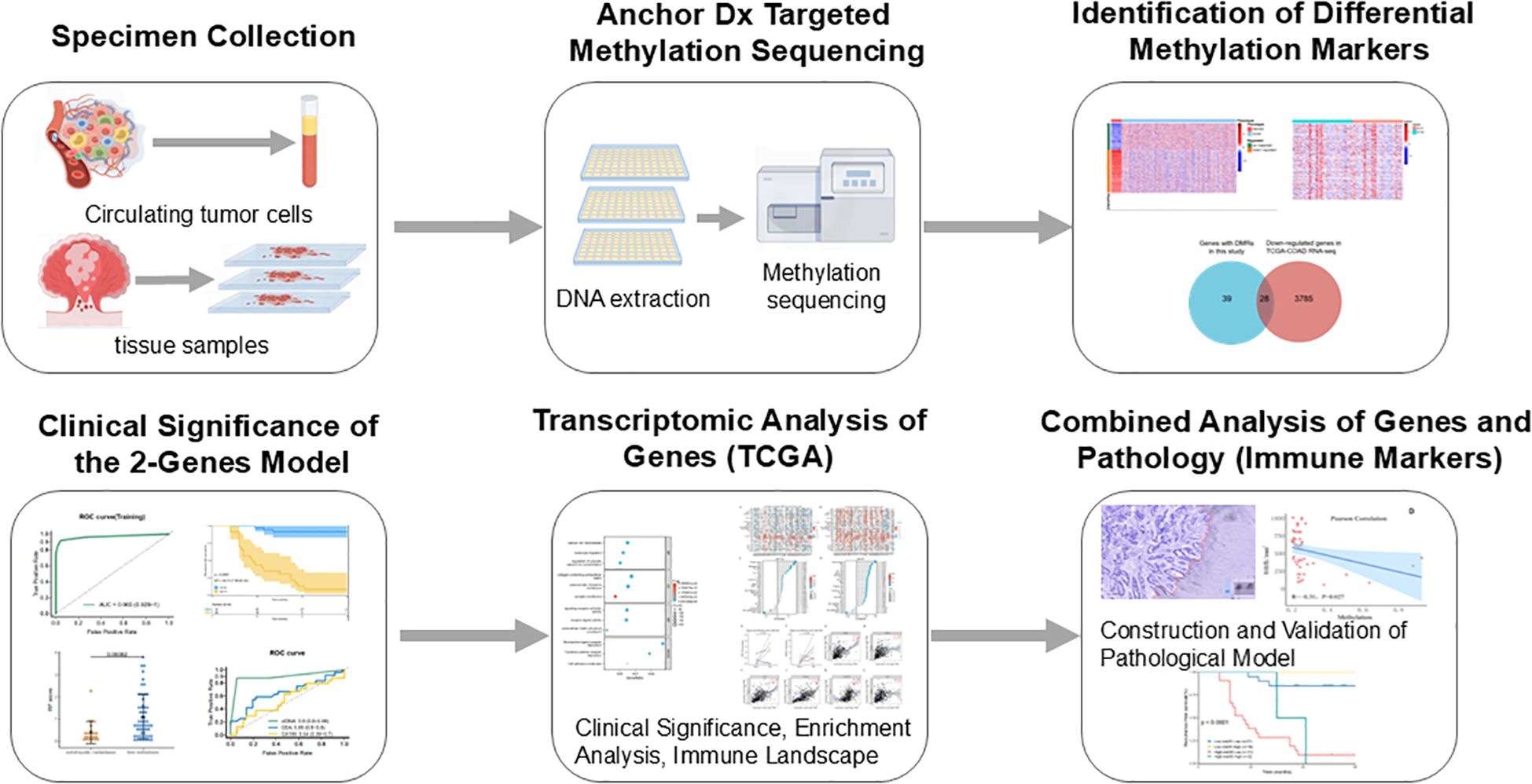
Figure 1. The design and process of our study are shown in the flow chart.
Identification of differentially methylated markers from tissue and plasma samples in stage IV CRC patients
To identify potential methylation markers associated with distant metastasis in CRC, we first analyzed the differentially methylated regions (DMRs) between primary tumors and their metastatic tissues in stage IV CRC. However, few DMRs were detected between primary and metastatic tissues in stage IV disease. The methylation profiles of primary tumors were similar to those of distant metastatic tissues (Supplementary Figures S1A, B). A total of 2,274 DMRs were identified when we further analyzed DMRs between 53 normal mucosa samples and 74 stage IV tumor tissues (including 34 primary and 40 liver metastatic tissues). Differential methylation analysis of plasma samples revealed 981 DMRs that distinguished stage IV CRC patients from stage I/II populations. Finally, 603 DMRs were identified that were shared by CRC tissues and plasma. Nearly all of the CpG sites screened were hypermethylated in CRC tissues compared to normal mucosa, as confirmed in a previous study (22), The final set of 28 overlapping DMRs located within the promoter regions of down-regulated genes may reflect tumor burden in the peripheral blood of advanced CRC patients (Supplementary Figures S1C, D).
Development and verification of a risk probability score for assessing tumor load
Least Absolute Shrinkage and Selection Operator (LASSO) was applied for variable selection from CRC patients plasma samples. Training and validation cohorts at a 3:2 ratio were randomly assigned from 119 stage I/II and 84 stage IV samples. The DMRs with the highest classification Area Under Curve (AUC) were aligned based on the latest UCSC hg19 RefSeq annotation. A methylation risk assay model was constructed, which included cg11977686(ZNF671) and cg19776201(ZNF132), utilizing the random forest algorithm. This model was able to distinguish late-stage patients from early-stage patients in the training cohort, with a sensitivity of 91.84%, specificity of 95.78%, and an AUC score of 0.965 (Figures 2A, C, Supplementary Table S2A). Patients were classified into different groups based on these two methylation markers, and each patient was assigned a risk probability (RP) score derived from the random forest model.
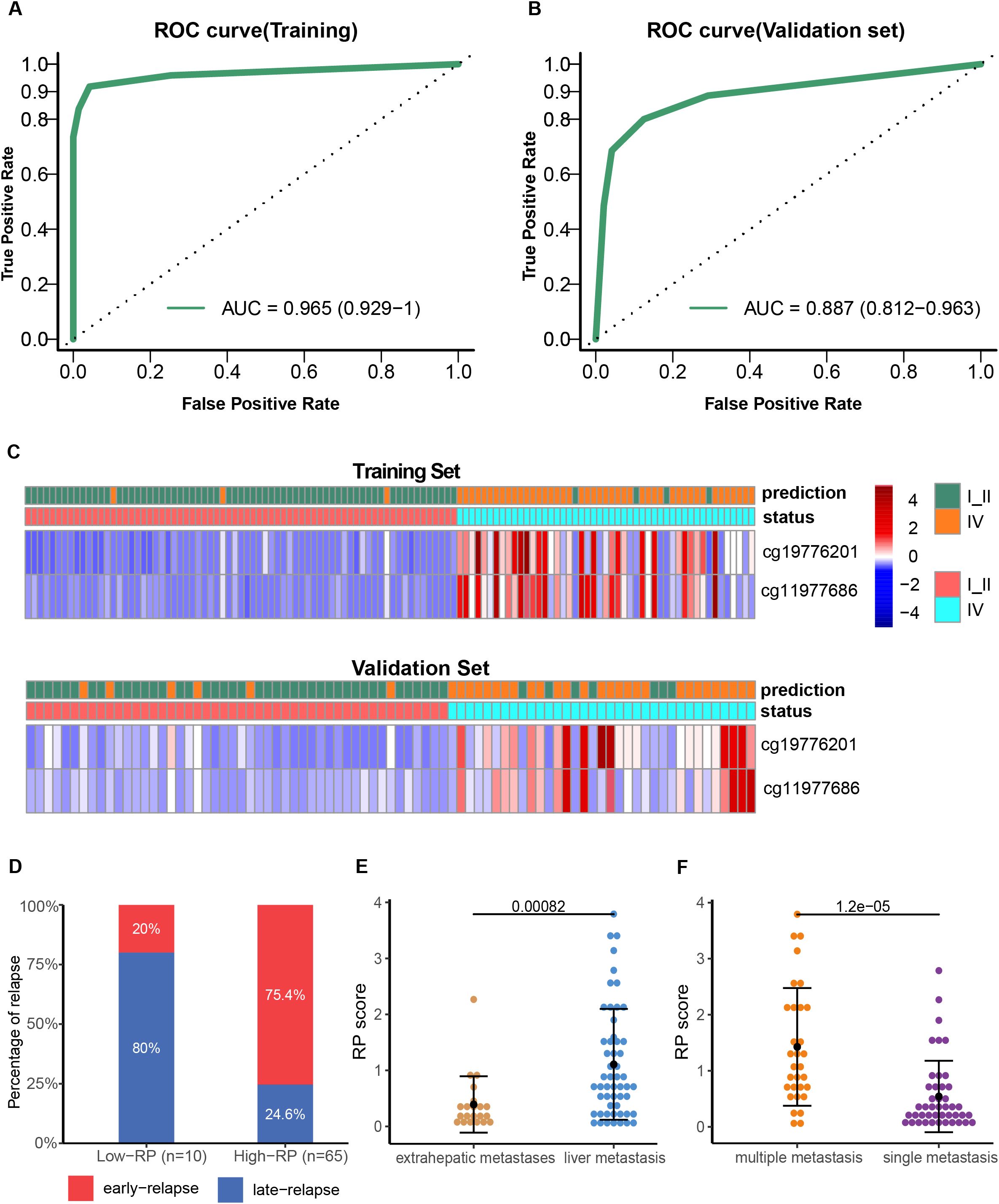
Figure 2. Development and validation of the two-methylation marker model. ROC curves of the risk model for late-stage CRC diagnosis in the training (A) and validation (B) cohorts. Heatmap showing the CpG markers between early-stage and late-stage IV CRC in the training (C) and validation cohorts. Each row represents an individual patient, and each column represents a CpG marker. The blue color represents a low methylation level, whereas the red color indicates a high methylation level. Comparison of early relapse rate for high and low RP score stage IV CRC patients (D); RP scores difference between patients with liver metastasis and those with extrahepatic metastasis (E); RP score difference between patients with multiple metastases and those with a single metastatic focus (F).
The performance of the methylation marker assay in distinguishing stage IV from stage I-II patients was then evaluated in the validation cohort, achieving an AUC score of 0.887 (95% CI: 0.812–0.963), with sensitivity of 80.0% and specificity of 87.5% (Figures 2B, D, Supplementary Table S2B). Further, we divided the stage IV CRC patients with postoperative relapse into an early relapse group (within 12 months) and a late relapse group (after 12 months). Notably, the early relapse rate was significantly higher in patients with high RP scores (75.4%, 49 of 65 patients) compared to those with low RP scores (20%, 8 of 10 patients) (Figure 2D, OR: 12.25; 95% CI: 2.35–63.72; p=0.0012). Additionally, a significant difference was observed between patients with liver metastasis and those with extrahepatic metastases (P<0.001), with higher RP scores detected in patients with multiple metastases compared to those with a single metastatic focus (P<0.001, Figures 2E, F).
The recurrence risk prediction for stage III CRC patients by the methylation risk assay
The performance of the methylation risk model in monitoring recurrence risk was further evaluated in a cohort of 60 stage III CRC patients (Figure 3A). ROC analysis revealed that the risk model outperformed traditional clinical markers, such as preoperative CEA and CA199 levels (Figure 3B). Based on the RP score calculated from the methylation assay, 24 patients were identified as being at high recurrence risk. After a median follow-up of 79.3 months (range: 57-112 months), 21 of these high-risk patients developed liver recurrence and/or distant metastasis (87.5%, 95% CI: 67.6-97.3%; Table 1). In contrast, recurrence occurred in only 2 out of 36 low-risk group patients (5.6%, 95% CI: 0.70-18.7%). The recurrence risk at 5 years was significantly higher in the high-risk group compared to the low-risk group (Figure 3C, HR: 24.15; 95% CI: 7.08-82.42, P<0.0001). Furthermore, patients in the low RP score group had a significantly higher overall survival (OS) rate compared to patients in the high RP score group (Figure 3D, HR: 16.52; 95% CI: 5.67-48.78, P<0.0001).
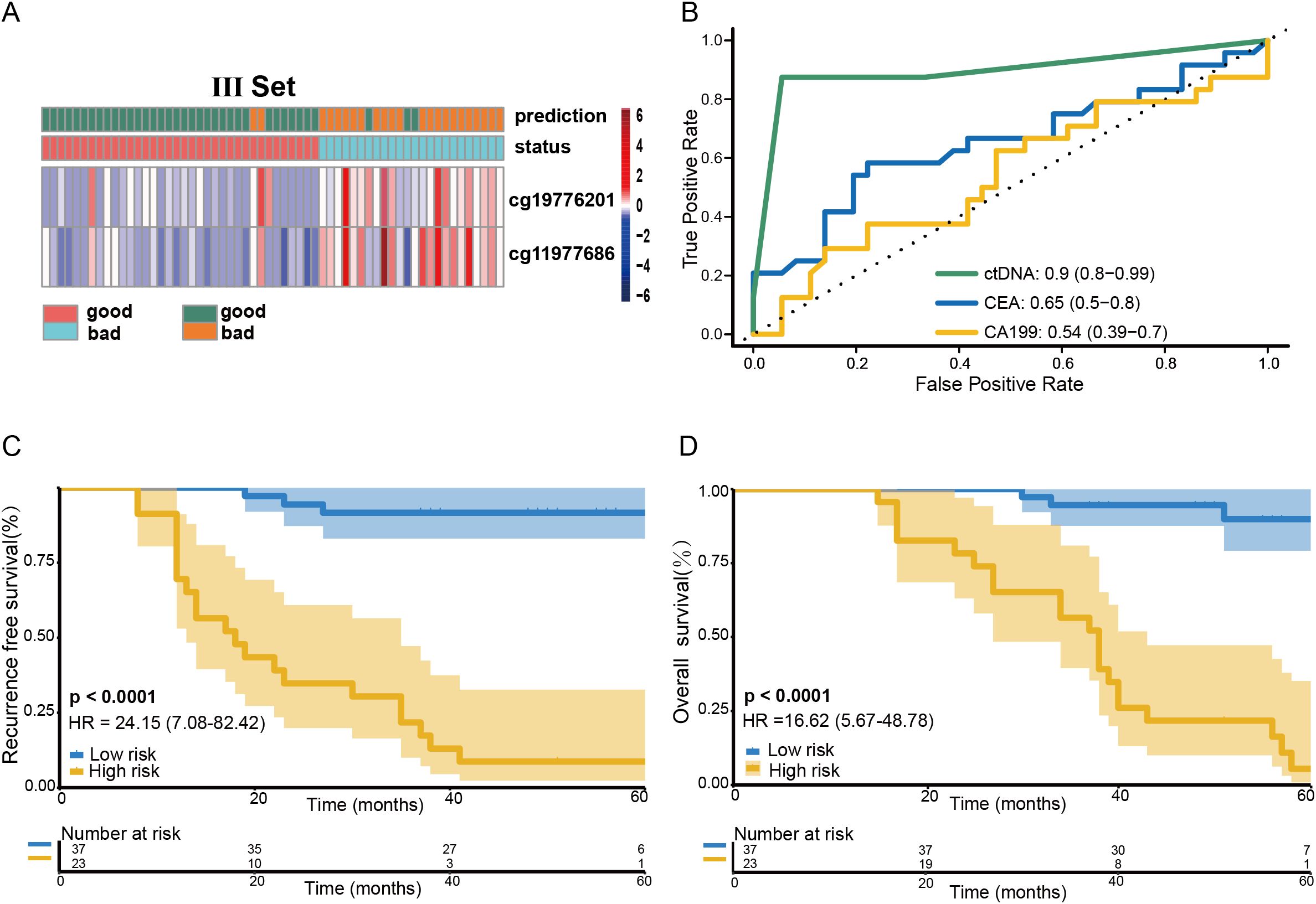
Figure 3. Predictive value of the RP score model on the prognosis of stage III CRC patients. Heatmap showing the methylation markers ZNF132 and ZNF671 in stage III CRC patients (A). The methylation risk assay demonstrated superior performance in predicting recurrence risk compared to preoperative CEA and CA199 levels for stage III CRC patients (B). Patients in the low-risk RP score group exhibited a significantly higher RFS (C) and OS (D) compared to those in the high-risk RP score group.
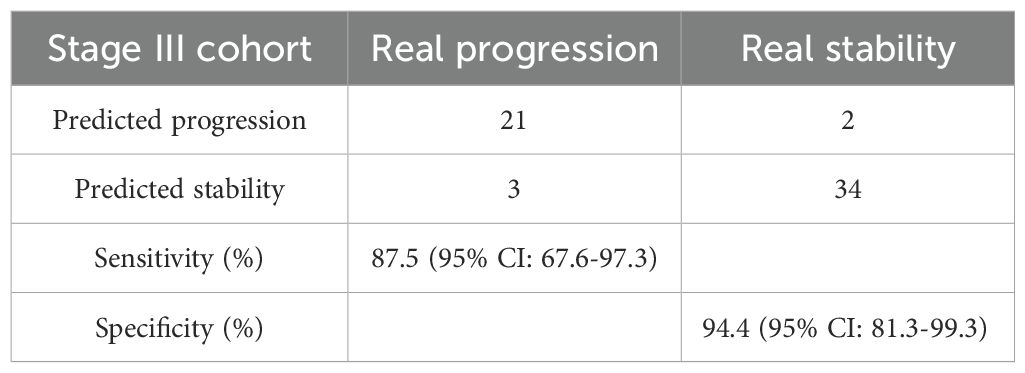
Table 1. Confusion matrices built from the ctDNA-based methylation marker prediction in stage III CRC patients.
Univariate analyses showed that both the methylation risk model and preoperative CEA levels were significantly associated with 5-year RFS) in stage III CRC patients. Notably, preoperative methylation of ZNF132 and ZNF671 was strongly associated with 5-year RFS and emerged as an independent risk factor for recurrence in multivariate analysis (HR: 78.94; 95% CI: 14.53-428.85; P=4.21e-07, Supplementary Table S3).
The association analysis of ZNF671 and ZNF132 gene expression with clinical prognostic factors
We further validated the reliability of the model by analyzing the differential expression of ZNF671 and ZNF132 in common tumors and adjacent normal tissues from the TCGA database. The results showed that the promoter methylation levels of ZNF671 and ZNF132 were significantly higher in CRC tumor tissues compared to normal mucosa (Supplementary Figures S2A, B). ROC analysis revealed that both ZNF671 and ZNF132 exhibited high diagnostic accuracy for CRC, with AUC values of 0.917 and 0.889, respectively (Supplementary Figure S2C).
Survival analysis indicated that low expression of ZNF132 (which corresponds to high methylation) was associated with significantly poorer survival in CRC patients compared to high expression (P < 0.05) (Supplementary Figure S2D). However, no significant survival difference was observed for ZNF671 (P > 0.05).
To further explore the potential prognostic roles and pathways of ZNF671 and ZNF132, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses (Supplementary Figures S2E, F). The enriched pathways included signal transduction, cell adhesion, immune regulation, metabolism, and disease development, underscoring the roles of ZNF671 and ZNF132 in tumor progression and prognosis.
ZNF132 and ZNF671 expression involved in regulating immune and may suggest a therapeutic strategy in CRC patients
Given the significant enrichment of ZNF132 and ZNF671 in immune function and related pathways, we used immune scores from the ESTIMATE algorithm as phenotypic data and applied WGCNA to identify gene modules significantly associated with ZNF132 and ZNF671 expression (Figures 4A–F). The identified modules suggest a potential link between ZNF132 and ZNF671 expression and immune cell infiltration, as well as immune-related pathways, highlighting their involvement in regulating immune responses and interacting with stromal cells.
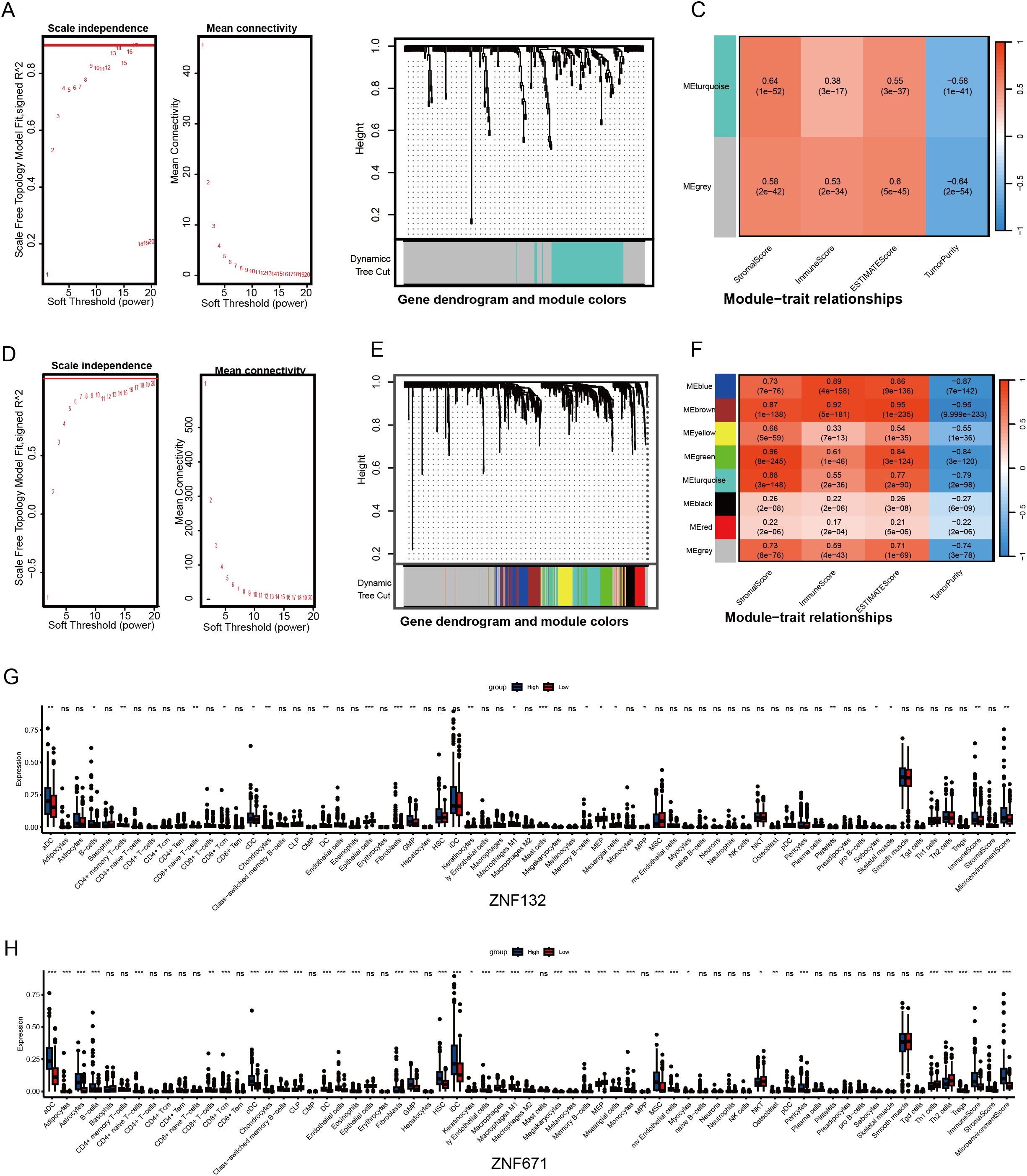
Figure 4. Association analysis of ZNF132 and ZNF671 expression with the immune microenvironment of CRC. Analysis of network topology for various soft-thresholding powers, determining the optimal power for constructing the ZNF132-associated scale-free network (A). Gene cluster tree, showing the module division of ZNF132-associated genes (B). Module-trait associations, illustrating the relationship between ZNF132-associated modules and immune infiltration (C). ZNF671 (D-F). ZNF132 (G) and ZNF671 (H) expression levels in relation to immune cell infiltration in general and the abundance of specific cell types *p < 0.05, **p < 0.01, ****p < 0.001.
Analysis with the xCell package revealed that ZNF132 is primarily associated with the immune infiltration of epithelial cells, fibroblasts, and Granulocyte-Monocyte Progenitor (GMP) cells (Figure 4G). In contrast, ZNF671 exhibited significant correlations with the infiltration of most immune and stromal cell types (Figure 4H). In summary, both ZNF132 and ZNF671 may play crucial roles in regulating immune responses and tumor-stroma interactions.
Further analysis across various cancers showed that ZNF132 and ZNF671 expression was significantly correlated with immune cell infiltration (Figures 5A, B). In CRC, ZNF132 primarily correlated with T helper cells (r=0.31) and T central memory (Tcm) cells (r=0.30) (Figure 5C). Meanwhile, ZNF671 showed the strongest correlation with macrophages (r = 0.5), followed by T effector memory (Tem) cells (r=0.42) (Figure 5D). Additionally, ZNF671 expression was moderately correlated with key immune checkpoint genes, including PD-1, PD-L1, and CTLA-4, in CRC (Figures 5E–J).
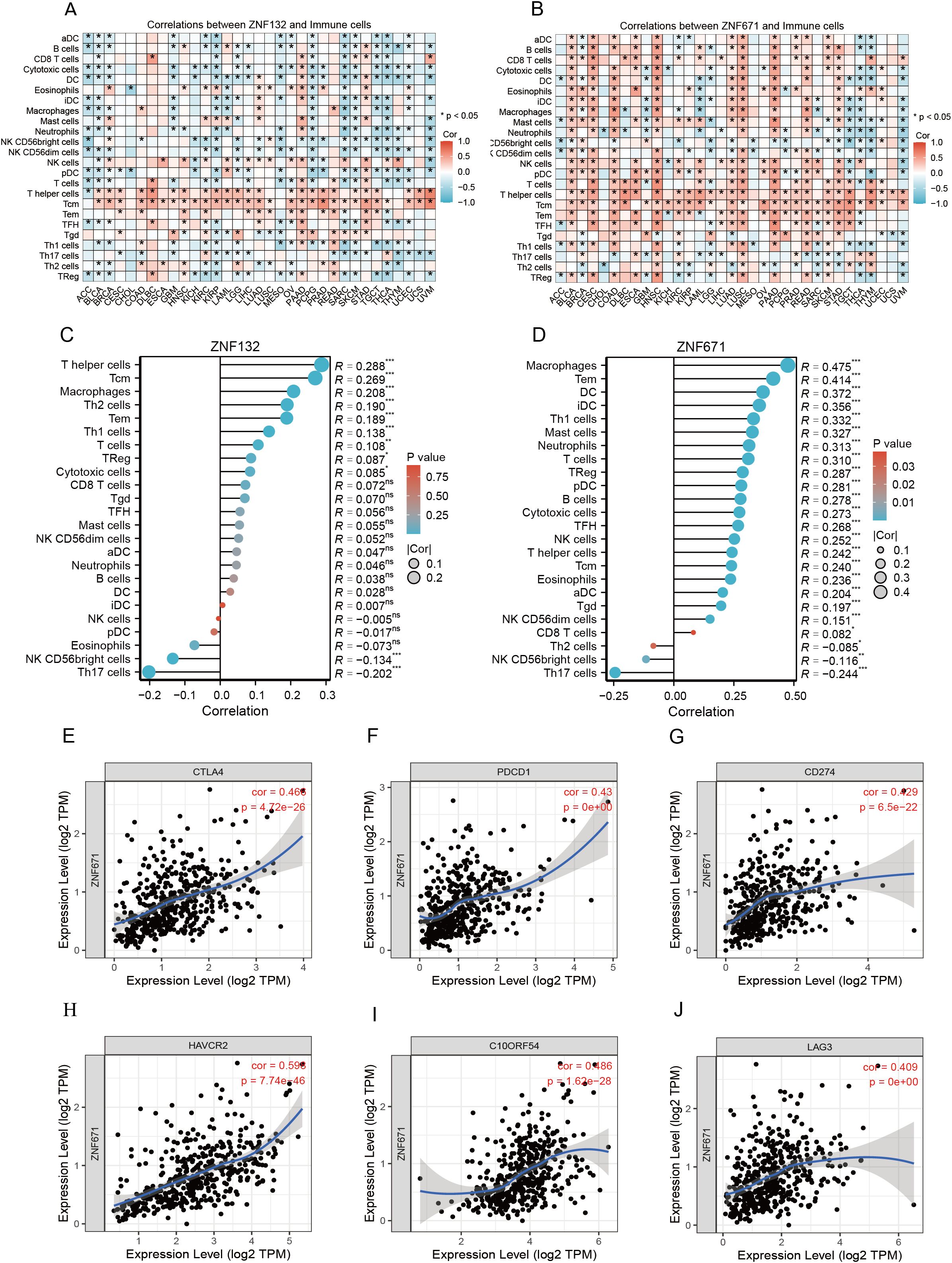
Figure 5. Correlation of ZNF132 and ZNF671 with immune cells and immune checkpoint genes, and changes after chemotherapy. Correlation of ZNF132 (A) and ZNF671 (B) expression with various immune cells in different cancer types. Correlation of ZNF132 (C) and (D) ZNF671 expression with various immune cells in CRC. Correlation of CTLA4 (E), PDCD1 (F), CD274 (G), HAVCR2 (H), C10ORF54 (I), and LAG3 (J) expression with ZNF671 in CRC.
Prognostic value of the Immunoscore in stage III CRC patients and its correlation with the risk model
Building on our previous analysis, which revealed that the expression levels of ZNF132 and ZNF671 were associated with the enrichment of immune-related pathways, we assessed immune scores in a matched cohort of 60 stage III CRC patients. The immune scores were categorized into high, intermediate (Int), and low groups, with 18 (30%), 25 (41.7%), and 17 (28.3%) patients, respectively (Figures 6A, B). The 5-year recurrence rates for IS-high, IS-Int, and IS-low patients were 11.1%, 36%, and 76.5%, respectively (P < 0.001) (Figure 6C). Notably, CRC patients with high immune scores exhibited significantly better 5-year RFS compared to those with intermediate or low immune scores (Figure 6D).
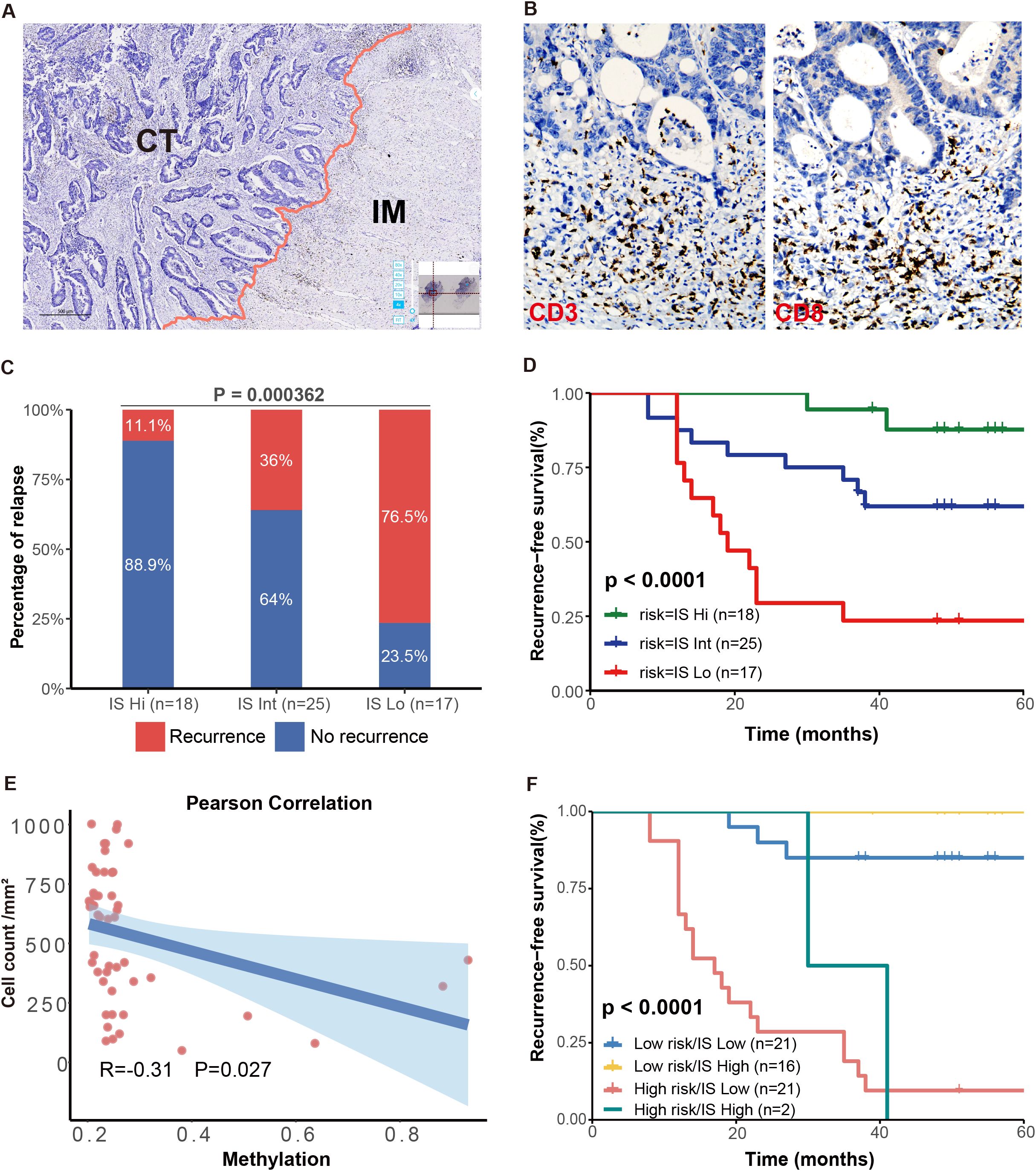
Figure 6. Prognostic significance of IS and association with the methylation risk model. Schematic diagram illustrating the separation of tumor regions into the central tumor (CT) and invasive margin (IM) regions (A). Immunohistochemical staining of CD3 and CD8 in CRC tissues (B). The 5-year RFS were significantly different among patients with high, intermediate, and low IS (C). High IS CRC patients had significantly better 5-year RFS compared to patients with IS-Int or IS-low (D). IS was negatively correlated with the RP score (E). Kaplan-Meier curves for RFS in patients with different risk and IS levels (F).
Additionally, a negative correlation was observed between the immune score and methylation risk score (R = -0.31, P = 0.027, Figure 6E). This finding aligns with previous analyses from the TCGA database, where CRC patients with lower methylation levels of ZNF132 and ZNF671 generally exhibited more lymphocytes infiltration. The combination of high immune scores and low methylation risk scores provided the best prognostic prediction (Figure 6F). These results suggest that the combined assessment of immune score and methylation risk score holds significant clinical prognostic and therapeutic value.
Assistance of the methylation model in the tumor staging of CRC patients
Interestingly, four CRC patients were initially suspected of having liver or lung metastasis based on imaging examination at the time of first diagnosis. Among these, two patients had elevated CEA levels, while the other two had normal CEA levels. However, all four patients were classified into the low recurrence risk group by the methylation assay model. After an average follow-up of 79.3 months, none of these patients experienced tumor progression (Supplementary Figure S3). Further investigation revealed that the suspicious pulmonary or intrahepatic lesions were actually old tuberculosis or hepatic hemangioma.
This finding underscores that, in some advanced-stage CRC patients, imaging examinations alone can be ambiguous and may not accurately determine the tumor stage. The methylation risk model, however, may provide valuable assistance in determining the TNM staging and predicting the outcomes for such equivocal cases.
Discussion
Plasma circulating tumor DNA (ctDNA) has emerged as a promising non-invasive tool for the repeated evaluation of tumor burden and epigenetic profiles. Cancer development is closely associated with epigenetic alterations (25). Aberrant DNA methylation plays an important role in many solid tumors and has been extensively studied as a promising marker for early diagnosis and prognostic evaluation across various tumor types (12–15, 26, 27). In patients with stage III colon cancer, adjuvant chemotherapy prevents recurrence and tumor progression by eradicating minimal residual disease or clusters of tumor cells hidden in the peripheral blood (28, 29). However, limited clinical success has been achieved in identifying patients at high risk of recurrence after surgery or completion of standard adjuvant treatment. For late-stage CRC patients with distant metastatic disease, 70% will experience recurrence within 2 years, despite some being cured following liver resection (6, 30). Identifying late-stage CRC patients with a high risk of rapid progression could help personalize treatment strategies and improve patients’ outcomes. The primary factors underlying cancer relapse and metastasis are minimal residual disease or clusters of tumor cells circulating in the peripheral blood. Postsurgical ctDNA analysis can detect minimal residual disease and recurrence in CRC, while preoperative ctDNA detection is associated with tumor burden and worse disease-specific survival (DSS) (8, 31–33). Preoperative ctDNA methylation levels have been shown to enable prognosis prediction in various cancers, including CRC, hepatocellular carcinoma, and lung cancer, due to the abundant methylated loci in both cancer tissues and cfDNA (12–14, 34).
Due to advances in next-generation sequencing (NGS) technology, aberrant methylation specific to CRC has been identified, facilitating the screening of ctDNA methylation biomarkers for early detection and prognosis prediction of CRC (14, 20, 34). However, most of the established models include too many markers and rely on NGS assays, which involve high economic and time costs, thereby limiting their clinical application and large-scale screening. In this study, based on an increased sample size and improved data analysis, we developed a new methylation model that only includes methylation markers ZNF132 and ZNF671. This model demonstrated diagnostic performance comparable to previous reports, achieving an accuracy of 87.5%. Kaplan-Meier survival analysis revealed that the high-risk group, as determined by the RP score, had significantly lower survival rates compared to the low-risk group. Additionally, for stage IV CRC patients who underwent resection of all visible disease, rapid progression was significantly higher in the high-risk group (91.4%) compared to the low-risk group (13.3%) (p < 0.001), a prediction not addressed in other studies previously.
High methylation levels typically lead to the silencing of tumor suppressor genes. Analyzing the methylation levels of ZNF132 and ZNF671 and their impact on gene expression in CRC patient samples helps clarify the roles of these biomarkers in tumor development and progression. Based on the TCGA database, we found that high methylation of ZNF132 and ZNF671 results in gene silencing or reduced expression, which in turn influences the tumor progression and correlates with the prognosis of CRC patients. Gene enrichment analysis of tumor samples with high ZNF132 and ZNF671 methylation revealed their involvement in immune pathways and cell proliferation, highlighting their biological functions and potential prognostic value.
A previous study proposed that the intra-tumoral immune contexture could be used to evaluate the clinical outcomes of solid tumors (35). Galon et al. summarized and calculated the density of CD3+ and CD8+ T cells within the tumor and its invasive margin to define the Immunoscore (IS), which provides a reliable estimate of the risk of recurrence in CRC patients (24). Our study of 60 stage III CRC patients also confirmed that the IS is a reliable predictor of recurrence risk in CRC. High IS was associated with the lower risk of recurrence and the highest RFS and overall survival (OS). Furthermore, we found a negative correlation between methylation levels and IS. Specifically, patients with high ZNF132 and ZNF671 methylation levels generally exhibited lower immune cells infiltration. Further analysis using WGCNA and xCell to explore the correlation between ZNF132 and ZNF671 expression and immune cell infiltration in the TME indicated that both genes are involved in regulating immune responses and tumor-stroma interactions. ZNF671 expression showed the strongest correlation with macrophages and effector memory T cells (Tem), a moderate correlation with key immune checkpoint genes (PD-1, PD-L1, CTLA-4, etc.) in CRC. ZNF132 exhibited weak correlations with T helper cells and central memory T cells (Tcm). Previous research has shown that ZNF671 inhibits CRC progression by suppressing the Notch signaling pathway (36), which is critically involved in immune cell development and function, including T-cell differentiation (37), macrophage polarization (38), and regulatory T cell maintenance (37). Given the established role of Notch signaling in immune regulation, it is plausible that ZNF671 may modulate the tumor immune microenvironment through this pathway. However, the mechanism of ZNF132 in immune regulation remains largely unexplored. Future studies incorporating mechanistic assays and immune cell functional experiments will be essential to clarify the roles of ZNF132 and ZNF671 in CRC immunity. In addition to the epigenetic and immune-related markers discussed above, other inflammatory or metabolic biomarkers may also offer prognostic value. For example, urinary 11-dehydrothromboxane B2 (11-dehydroTXB2), a stable metabolite of thromboxane A2, has been implicated in tumor-associated inflammation, immunosuppression, and metastasis (39–41). Its measurement may provide non-invasive insight into the systemic immune-inflammatory status of CRC patients.
Finally, an interesting finding in our research is that the methylation risk model can assist in TMN staging of CRC to some extent. For suspicious liver or pulmonary nodules detected at the time of primary cancer diagnosis, a low methylation risk score could help refine CRC tumor staging, potentially avoiding misdiagnosis and excessive treatment.
Overall, we developed a more concise two-methylation-marker model. This model could evaluate the relapse risk of stage III CRC patients and predict the prognosis of late-stage patients. Database analysis further confirms that ZNF132 and ZNF671 play significantly diagnostic and prognostic roles in CRC. Both genes are linked to immune cell infiltration, immune-related pathways, and show a strong correlation with the immune stroma, highlighting their potential value as predictive markers for immunotherapy. Furthermore, our study provides further confirmation that the IS can predict the prognosis of stage III CRC patients and is negatively correlated with the methylation risk model. Previous studies have strongly suggested higher sensitivity to immunotherapy in low-risk Cutaneous Melanoma patients (42). Other studies have also shown a reduced response to anti-PD-L1 therapy in lung adenocarcinoma patients when the risk score is high (43, 44). Whether the IS and methylation risk model could offer a new categorization to guide therapeutic strategy in CRC patients needs to be tested in future studies. Our findings could improve management strategies and facilitate early intervention and personalized adjuvant therapies for CRC patients.
Limitations
There are several potential limitations to our study. First, the plasma samples, which included 119 cases of early-stage CRC and 84 cases of late-stage CRC, were randomly divided into training and validation sets at a 3:2 ratio. This resulted in a relatively small number of samples in the validation set (83 cases). As a result, the robustness of the methylation model should be further validated with a larger sample size.
Data availability statement
The data supporting the findings of this study have been publicly deposited on GitHub at: https://github.com/zxcwuzheng/stopCOAD. This repository contains the complete DNA methylation beta value dataset and grouping information, along with detailed data documentation. All data comply with ethical standards and contain no personally identifiable information. Some datasets presented in this article are not readily available due to the ethical considerations and intellectual property regulations that govern the sharing of clinical data and plasma assay data. Requests to access these datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by General Hospital of Southern Theater Command. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
YL: Writing – review & editing, Formal Analysis, Writing – original draft, Visualization, Validation, Data curation. JZ: Formal Analysis, Validation, Writing – original draft, Supervision, Data curation, Writing – review & editing. DY: Writing – review & editing, Methodology, Writing – original draft, Visualization, Investigation, Validation, Data curation, Supervision. XL: Writing – review & editing, Validation, Visualization, Formal Analysis. WC: Writing – review & editing, Investigation, Validation. WH: Writing – review & editing, Visualization, Validation, Investigation. LY: Writing – review & editing, Investigation, Visualization. JW: Writing – review & editing, Visualization, Formal Analysis. GY: Validation, Investigation, Supervision, Writing – review & editing, Visualization, Formal Analysis, Writing – original draft. CL: Methodology, Validation, Writing – original draft, Supervision, Project administration, Visualization, Formal Analysis, Writing – review & editing. WW: Funding acquisition, Data curation, Resources, Investigation, Validation, Visualization, Conceptualization, Writing – original draft, Writing – review & editing, Supervision, Formal Analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Natural Science Foundation of Guangdong Province of China (NO.2023A1515012384), and Science and Technology Planning Project of Guangzhou (NO.2023A03J0169).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1607548/full#supplementary-material
References
1. Li N, Lu B, Luo C, Cai J, Lu M, Zhang Y, et al. Incidence, mortality, survival, risk factor and screening of colorectal cancer: A comparison among China, Europe, and northern America. Cancer Lett. (2021) 522:255–68. doi: 10.1016/j.canlet.2021.09.034
2. Panni RZ and D’Angelica M. Stage IV rectal cancer and timing of surgical approach. Clinics Colon Rectal Surg. (2024) 37:248–55. doi: 10.1055/s-0043-1770719
3. Kahi CJ, Boland CR, Dominitz JA, Giardiello FM, Johnson DA, Kaltenbach T, et al. United states multi-society task force on colorectal cancer. Colonoscopy surveillance after colorectal cancer resection: recommendations of the US multi-society task force on colorectal cancer. Gastroenterology. (2016) 150:758–768.e11. doi: 10.1053/j.gastro.2016.01.001
4. Bolhuis K, Kos M, Van Oijen MGH, Swijnenburg R-J, and Punt CJA. Conversion strategies with chemotherapy plus targeted agents for colorectal cancer liver-only metastases: A systematic review. Eur J Cancer (Oxford England: 1990). (2020) 141:225–38. doi: 10.1016/j.ejca.2020.09.037
5. Siegel RL, Wagle NS, Cercek A, Smith RA, and Jemal A. Colorectal cancer statistics, 2023. CA: Cancer J Clin. (2023) 73:233–54. doi: 10.3322/caac.21772
6. Abdalla EK, Vauthey J-N, Ellis LM, Ellis V, Pollock R, Broglio KR, et al. Recurrence and outcomes following hepatic resection, radiofrequency ablation, and combined resection/ablation for colorectal liver metastases. Ann Surg. (2004) 239:818–825; discussion 825-827. doi: 10.1097/01.sla.0000128305.90650.71
7. Bolhuis K, Van ‘t Erve I, Mijnals C, Delis-Van Diemen PM, Huiskens J, Komurcu A, et al. Postoperative circulating tumour DNA is associated with pathologic response and recurrence-free survival after resection of colorectal cancer liver metastases. EBioMedicine. (2021) 70:103498. doi: 10.1016/j.ebiom.2021.103498
8. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Trans Med. (2016) 8:346ra92. doi: 10.1126/scitranslmed.aaf6219
9. Wan JCM, Mughal TI, Razavi P, Dawson S-J, Moss EL, Govindan R, et al. Liquid biopsies for residual disease and recurrence. Med (New York NY). (2021) 2:1292–313. doi: 10.1016/j.medj.2021.11.001
10. Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. (2017) 17:223–38. doi: 10.1038/nrc.2017.7
11. Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. (2019) 176:98–112.e14. doi: 10.1016/j.cell.2018.11.046
12. Xu R-H, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. (2017) 16:1155–61. doi: 10.1038/nmat4997
13. Liang W, Chen Z, Li C, Liu J, Tao J, Liu X, et al. Accurate diagnosis of pulmonary nodules using a noninvasive DNA methylation test. J Clin Invest. (2021) 131:e145973, 145973. doi: 10.1172/JCI145973
14. Mo S, Dai W, Wang H, Lan X, Ma C, Su Z, et al. Early detection and prognosis prediction for colorectal cancer by circulating tumour DNA methylation haplotypes: A multicentre cohort study. EClinicalMedicine. (2023) 55:101717. doi: 10.1016/j.eclinm.2022.101717
15. Wang B, Wang M, Lin Y, Zhao J, Gu H, and Li X. Circulating tumor DNA methylation: a promising clinical tool for cancer diagnosis and management. Clin Chem Lab Med. (2024) 62:2111–27. doi: 10.1515/cclm-2023-1327
16. Ascierto PA, Capone M, Urba WJ, Bifulco CB, Botti G, Lugli A, et al. The additional facet of immunoscore: immunoprofiling as a possible predictive tool for cancer treatment. J Trans Med. (2013) 11:54. doi: 10.1186/1479-5876-11-54
17. Bindea G, Mlecnik B, Angell HK, and Galon J. The immune landscape of human tumors: Implications for cancer immunotherapy. Oncoimmunology. (2014) 3:e27456. doi: 10.4161/onci.27456
18. Michaud DS, Ruan M, Koestler DC, Alonso L, Molina-Montes E, Pei D, et al. DNA methylation-derived immune cell profiles, CpG markers of inflammation, and pancreatic cancer risk. Cancer Epidemiol Biomarkers Prev. (2020) 29:1577–85. doi: 10.1158/1055-9965.EPI-20-0378
19. Wang Q, Ma C, Yang B, Zheng W, Liu X, and Jian G. Dysregulation of DNA methylation in colorectal cancer: biomarker, immune regulation, and therapeutic potential. Int Immunopharmacol. (2025) 145:113766. doi: 10.1016/j.intimp.2024.113766
20. Wang W, Zhu X, Zhang X, Lei C, Zeng Z, Lan X, et al. Recurrence risk assessment for stage III colorectal cancer based on five methylation biomarkers in plasma cell-free DNA. J Pathol. (2023) 259:376–87. doi: 10.1002/path.6047
21. Stojkovic Lalosevic M, Stankovic S, Stojkovic M, Markovic V, Dimitrijevic I, Lalosevic J, et al. Can preoperative CEA and CA19-9 serum concentrations suggest metastatic disease in colorectal cancer patients?. Hellenic J Nucl Med. (2017) 20:41–5. doi: 10.1967/s002449910505
22. Wang W, Zhang X, Zhu X, Cui W, Ye D, Tong G, et al. Seven DNA methylation biomarker prediction models for monitoring the Malignant progression from advanced adenoma to colorectal cancer. Front Oncol. (2022) 12:827811. doi: 10.3389/fonc.2022.827811
23. Chen X, Zhang J, Ruan W, Huang M, Wang C, Wang H, et al. Urine DNA methylation assay enables early detection and recurrence monitoring for bladder cancer. J Clin Invest. (2020) 130:6278–89. doi: 10.1172/JCI139597
24. Pagès F, Mlecnik B, Marliot F, Bindea G, Ou F-S, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet (London England). (2018) 391:2128–39. doi: 10.1016/S0140-6736(18)30789-X
25. Herman JG and Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. New Engl J Med. (2003) 349:2042–54. doi: 10.1056/NEJMra023075
26. Kulis M and Esteller M. DNA methylation and cancer. Adv Genet. (2010) 70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2
27. Klutstein M, Nejman D, Greenfield R, and Cedar H. DNA methylation in cancer and aging. Cancer Res. (2016) 76:3446–50. doi: 10.1158/0008-5472.CAN-15-3278
28. Alnajjar S, Shoucair S, Almanzar A, Zheng K, Lisle D, and Gupta V. Predictors of timely initiation and completion of adjuvant chemotherapy in stage II/III colorectal adenocarcinoma. Am Surgeon. (2024) 90:2724–32. doi: 10.1177/00031348241248689
29. Nakamura Y, Watanabe J, Akazawa N, Hirata K, Kataoka K, Yokota M, et al. ctDNA-based molecular residual disease and survival in resectable colorectal cancer. Nat Med. (2024), 30:3272–82. doi: 10.1038/s41591-024-03254-6
30. Angelsen J-H, Viste A, Løes IM, Eide GE, Hoem D, Sorbye H, et al. Predictive factors for time to recurrence, treatment and post-recurrence survival in patients with initially resected colorectal liver metastases. World J Surg Oncol. (2015) 13:328. doi: 10.1186/s12957-015-0738-8
31. Narayan RR, Goldman DA, Gonen M, Reichel J, Huberman KH, Raj S, et al. Peripheral circulating tumor DNA detection predicts poor outcomes after liver resection for metastatic colorectal cancer. Ann Surg Oncol. (2019) 26:1824–32. doi: 10.1245/s10434-019-07201-5
32. Mo S, Ye L, Wang D, Han L, Zhou S, Wang H, et al. Early detection of molecular residual disease and risk stratification for stage I to III colorectal cancer via circulating tumor DNA methylation. JAMA Oncol. (2023) 9:770–8. doi: 10.1001/jamaoncol.2023.0425
33. Zhao Z, Cai S, and Wang Z. Circulating tumor DNA as a prognostic marker in stage III colon cancer. JAMA Oncol. (2020) 6:932. doi: 10.1001/jamaoncol.2020.0283
34. Luo H, Zhao Q, Wei W, Zheng L, Yi S, Li G, et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Trans Med. (2020) 12:eaax7533. doi: 10.1126/scitranslmed.aax7533
35. Fridman WH, Pagès F, Sautès-Fridman C, and Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
36. Wang Y, Chen F-R, Wei C-C, Sun L-L, Liu C-Y, Yang L-B, et al. Zinc finger protein 671 has a cancer-inhibiting function in colorectal carcinoma via the deactivation of Notch signaling. Toxicol Appl Pharmacol. (2023) 458:116326. doi: 10.1016/j.taap.2022.116326
37. Brandstadter JD and Maillard I. Notch signalling in T cell homeostasis and differentiation. Open Biol. (2019) 9:190187. doi: 10.1098/rsob.190187
38. Zhou C, Weng J, Liu C, Liu S, Hu Z, Xie X, et al. Disruption of SLFN11 deficiency-induced CCL2 signaling and macrophage M2 polarization potentiates anti-PD-1 therapy efficacy in hepatocellular carcinoma. Gastroenterology. (2023) 164:1261–78. doi: 10.1053/j.gastro.2023.02.005
39. Yang J, Yamashita-Kanemaru Y, Morris BI, Contursi A, Trajkovski D, Xu J, et al. Aspirin prevents metastasis by limiting platelet TXA2 suppression of T cell immunity. Nature. (2025) 640:1052–61. doi: 10.1038/s41586-025-08626-7
40. Kiely M, Milne GL, Minas TZ, Dorsey TH, Tang W, Smith CJ, et al. Urinary thromboxane B2 and lethal prostate cancer in African American men. J Natl Cancer Institute. (2022) 114:123–9. doi: 10.1093/jnci/djab129
41. Patrignani P, Tacconelli S, Contursi A, Piazuelo E, Bruno A, Nobili S, et al. Optimizing aspirin dose for colorectal cancer patients through deep phenotyping using novel biomarkers of drug action. Front Pharmacol. (2024) 15:1362217. doi: 10.3389/fphar.2024.1362217
42. Xue L, Wu P, Zhao X, Jin X, Wang J, Shi Y, et al. Using immune-related lncRNA signature for prognosis and response to immunotherapy in cutaneous melanoma. Int J Gen Med. (2021) 14:6463–75. doi: 10.2147/IJGM.S335266
43. Gu X, Huang X, Zhang X, and Wang C. Development and Validation of a DNA Methylation-related Classifier of Circulating Tumour Cells to Predict Prognosis and to provide a therapeutic strategy in Lung Adenocarcinoma. Int J Biol Sci. (2022) 18:4984–5000. doi: 10.7150/ijbs.75284
Keywords: ctDNA methylation, CRC, prognosis prediction, tumor immune microenvironment, ZNF671, ZNF132
Citation: Zhou J, Ye D, Li Y, Lai X, Cui W, He W, Yu L, Wu J, Yan G, Lei C and Wang W (2025) Combining the DNA methylation markers of circulating tumor cells with immune infiltrating cells to assess recurrence and prognosis and to suggest a therapeutic strategy in stage III-IV colorectal cancer. Front. Immunol. 16:1607548. doi: 10.3389/fimmu.2025.1607548
Received: 07 April 2025; Accepted: 09 July 2025;
Published: 28 July 2025.
Edited by:
Qun Chen, University of Oklahoma, United StatesReviewed by:
Vaibhavi V. Gujar, National Institutes of Health (NIH), United StatesPaola Patrignani, University of Studies G. d’Annunzio Chieti and Pescara, Italy
Copyright © 2025 Zhou, Ye, Li, Lai, Cui, He, Yu, Wu, Yan, Lei and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Wang, cmljZXdhbmc3OUAxMjYuY29t; Chengyong Lei, bmZ5eWxjeTIwMjRAMTYzLmNvbQ==; Guangning Yan, Y2F3YWZyb2dAaG90bWFpbC5jb20=
†These authors have contributed equally to this work