 Karsten E. Kluge1,2*
Karsten E. Kluge1,2* Sigrun Halvorsen1,2
Sigrun Halvorsen1,2 Geir Ø. Andersen1Charlotte H. Hansen1
Geir Ø. Andersen1Charlotte H. Hansen1 Ingebjørg Seljeflot1
Ingebjørg Seljeflot1 Theis Tønnessen2,3
Theis Tønnessen2,3 Ida G. Lunde1,2,4
Ida G. Lunde1,2,4 Ragnhild Helseth1
Ragnhild Helseth1- 1Oslo Center for Clinical Heart Research, Department of Cardiology, Oslo University Hospital Ullevål, Oslo, Norway
- 2University of Oslo, Oslo, Norway
- 3Department of Cardiothoracic Surgery, Oslo University Hospital, Oslo, Norway
- 4Kristian Gerhard Jebsen Center for Cardiac Biomarkers, University of Oslo, Oslo, Norway
Introduction: Preclinical data indicates reciprocal activation of the complement system and the coagulation cascade. The magnitude of this interaction in patients with acute myocardial infarction is unknown. We aimed to determine associations between circulating markers of complement and coagulation activation in patients with acute ST-elevation myocardial infarction (STEMI), and explore a possible link to myocardial injury and left ventricular dysfunction.
Materials and methods: We included 864 patients with STEMI. Blood was drawn at a median of 18 hours after percutaneous coronary intervention. Complement activation was assessed by the terminal complement complex (TCC), and coagulation activation by prothrombin fragment 1 + 2 (F1 + 2), D-dimer and endogenous thrombin potential (ETP). Myocardial injury was estimated by peak troponin T (TnT), and left ventricular function was quantified on echocardiography by left ventricular ejection fraction (LVEF).
Results: TCC was weakly correlated to F1 + 2 (r=0.086, p=0.012), D-dimer (r=0.176, p<0.001) and ETP (r=0.144, p<0.001). In multivariate binary logistic regression, there was no significant interaction between TCC and the coagulation markers on the risk of having high peak TnT or low LVEF.
Conclusion: In this STEMI cohort, complement activation as measured by TCC was weakly associated with markers of coagulation activation, but the measured markers had no combined relation with the risk of high peak TnT or low LVEF. These findings suggest that while simultaneous activation of complement and coagulation cannot be ruled out, combined high levels of TCC and coagulation markers do not mirror the extent of myocardial injury or dysfunction in STEMI.
1 Introduction
The complement and coagulation systems are cascade systems consisting of inactive precursor proteins, rapidly activated upon stimuli (1, 2). The complement system is activated in response to invading pathogens or cellular damage, and its primary function is to mediate host defense (3). The coagulation system is activated by vascular injury, and mediates hemostasis (4). Beyond their canonical functions, both systems are involved in the pathogenesis of coronary atherosclerosis and ST-elevation myocardial infarction (STEMI) (5, 6). The complement system contributes to atherosclerosis by inducing endothelial dysfunction and triggering inflammatory signaling within the atherosclerotic plaque (7). In STEMI, the complement system contributes to plaque destabilization and ischemia-reperfusion (IR) injury (8, 9). The coagulation system contributes to atherosclerosis by triggering inflammation within the atherosclerotic plaque, as well as contributing to atherothrombosis following plaque rupture or erosion (10, 11). Atherothrombosis might cause STEMI, in which case coagulation contributes to IR injury (12), but atherothrombosis might also remain asymptomatic, where it functions as an important mechanism of plaque growth (13).
Reciprocal activation between the complement system and the coagulation cascade has been reported (2). The anaphylatoxins C3a and C5a, as well as the terminal complement complex (TCC) has been reported to activate platelets and cause them to shed pro-coagulant microparticles (MPs), and serine proteases of the complement system has been reported to activate coagulation factors (14). Conversely, coagulation factors have been reported to activate complement components directly (15), and platelets and MPs might function as platforms for complement activation (16). As a marker of “total” complement activation, TCC is well established (17). We have previously reported associations between both complement and coagulation activation to higher peak troponin T (TnT) and lower left ventricular ejection fraction (LVEF) in the present STEMI cohort (18, 19). In patients with chronic coronary syndrome, we have previously observed a link between TCC and markers of hypercoagulability (20). Associations between TCC and coagulation activation have not been reported in STEMI, where reciprocal activation could cause a self-enforcing cycle of inflammation and coagulation, theoretically resulting in a more rigid coronary thrombus and exacerbated IR injury. On this basis, we aimed to determine the associations between TCC and coagulation activation in patients with STEMI, and explore any combined relation to myocardial injury by peak troponin T (TnT) and myocardial dysfunction by left ventricular ejection fraction (LVEF). We hypothesized that TCC and the coagulation markers would be simultaneously increased, and that combined high levels would be associated with markers of myocardial injury and dysfunction.
2 Materials and methods
2.1 Study population
The study design and population have been described previously (21). In brief, patients admitted to Oslo University Hospital Ullevål with STEMI treated with percutaneous coronary intervention (PCI) were included (n=1028). Patients below 18 years of age and patients unable or unwilling to give written informed consent were excluded. Clinical information was collected from hospital records and questionnaires obtained at inclusion.
For this sub-study, patients using oral anticoagulation were excluded due to its effect on the coagulation cascade (22). A total of 164 patients were excluded, leaving 864 for the final analysis. The study was approved by the Regional Ethics Committee of South East Norway (project ID: 107832).
2.2 Blood sampling
Blood samples were collected in a fasting condition, between 8:00 and 10:00 a.m. the morning after PCI. For patients admitted during the weekend, inclusion was performed the following Monday morning. Blood collection was performed at a median time of 24 h after symptom debut and 18 h after PCI. Serum was prepared by centrifugation for 10 min at 2000 x g, and EDTA plasma and citrated plasma kept on ice, and prepared within one hour by centrifugation for 30 min at 3000 x g at 4 °C. Samples were aliquoted and stored at -80 °C until analyzed. All reported markers were measured in samples collected at the same time.
2.3 Laboratory methods
Peak TnT was defined as the highest measurement during hospitalization. The analysis was performed in serum by electrochemiluminescence technology (3rd generation cTroponinT, Elecsys 2010, Roche, Mannheim, Germany). The inter-assay coefficient of variation (CV) was 7%. Complement activation was measured in EDTA plasma by the TCC, using a commercially available enzyme-linked immunosorbent assay (ELISA) (human TCC, HycultBiotech, Uden, The Netherlands). The inter-assay CV was 8.1%. Coagulation activation was measured in citrated plasma by F1 + 2 and D-dimer, both analyzed by ELISA (Enzygnost F1 + 2; Siemens, Marburg, Germany and Asserachrom D-dimer; Stago Diagnostica, Ansiere, France, respectively), and by the endogenous thrombin potential (ETP), analyzed by the calibrated automated thrombogram assay. Inter-assay CVs were 5.4% for F1 + 2, 6.5% for D-dimer and 6.0% for ETP.
2.4 Echocardiography
LVEF was assessed by echocardiography within three months after the index infarction, by either visual approximation or the Simpson’s biplane method.
2.5 Definitions
STEMI was defined as electrocardiographic ST segment elevation of >2mm in two or more contiguous chest leads, >1mm in two or more limb leads, or new onset of left bundle-branch block, together with chest pain or other typical symptoms and troponin levels above the 99th percentile (23). Previous cardiovascular disease (CVD) was defined as previous myocardial infarction, ischemic stroke, PCI procedure, or coronary artery bypass surgery. Diabetes mellitus and hypertension were defined as treated diabetes and hypertension. Smoking was defined as current smoking or cessation <3 months prior to study inclusion.
2.6 Statistical analysis
Data is presented as mean ± SD, median (25th, 75th percentile), or numbers (%) as appropriate. The unpaired Student t-test and Mann-Whitney U test were used to test differences between groups. Proportional data was compared using the chi-squared test. Correlation analyses were performed using Spearman’s rho. Multivariate binary logistic regression was used to quantify the individual contributions of variables, and their interactions, on risk of an outcome. P-values of ≤0.05 were considered statistically significant, and all statistical analyses were performed using IBM SPSS statistics v.27. Figures were created using Microsoft Excel v.16.x and STATA v.18.
2.6.1 Statistical analysis plan
We aimed to explore associations between TCC and markers of coagulation activation by correlation analysis. To assess the magnitude of the correlation, we compared the distributions of coagulation markers dichotomized at median TCC, using a non-parametric test (Mann-Whitney U test). We prospectively combined TCC and the coagulation markers in binary logistic regression to assess possible interactions. For this analysis, above-median peak TnT and LVEF ≤40% were used as outcomes, and TCC and coagulation markers were divided into quartiles and treated as continuous variables. LVEF ≤40% was chosen as a cut-off because this is the echocardiographic criterion for heart failure with reduced ejection fraction (24). Age and gender were included in the model due to reported associations to complement activation (25).
3 Results
3.1 Study population
The characteristics of the study population are described in Table 1. The mean age was 61 years, 20% were female, 23% had previously diagnosed CVD, and 11% received prehospital thrombolysis.
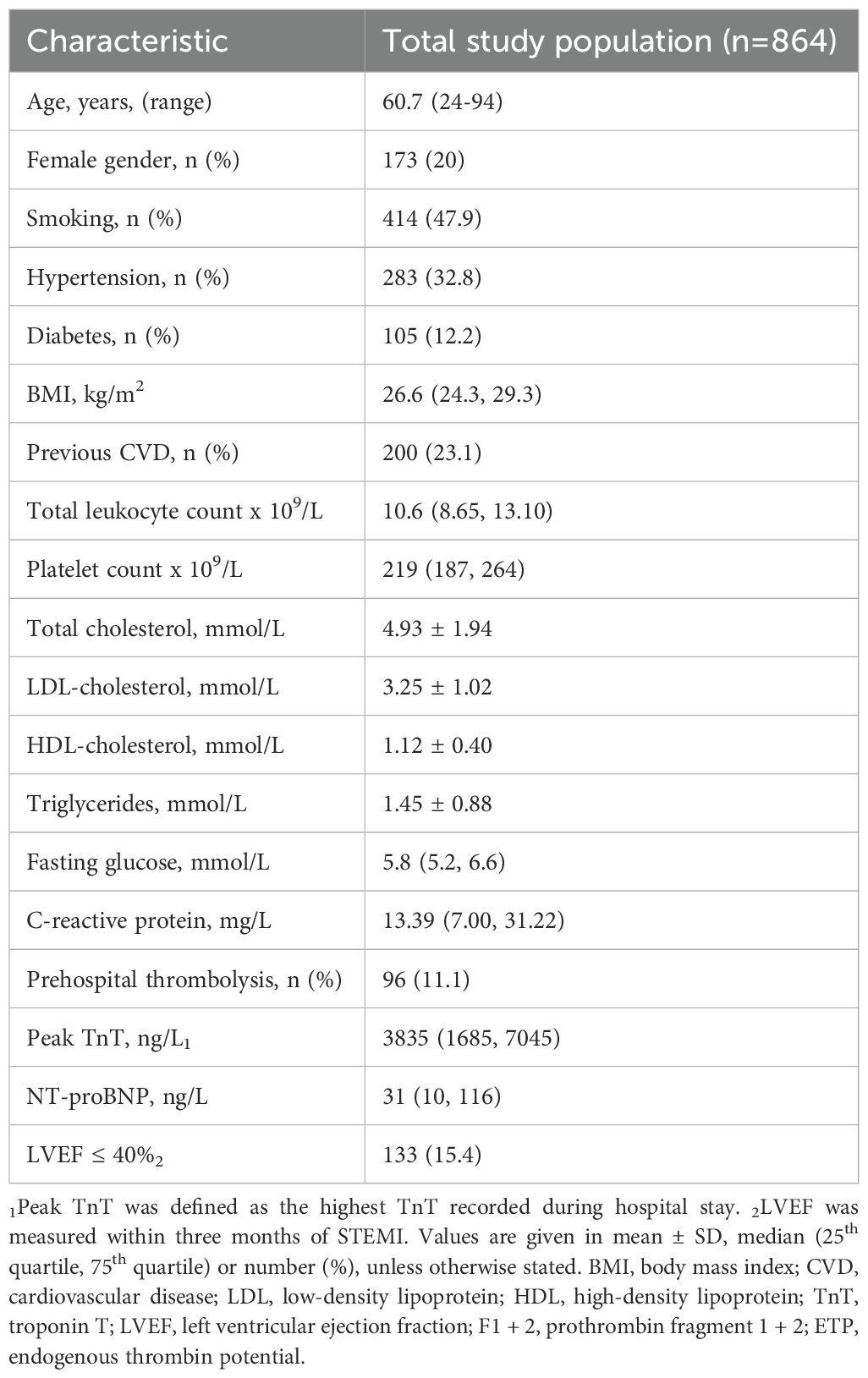
Table 1. Characteristics of the study population.
3.2 Associations between complement and coagulation markers
Statistically significant, but weak associations were observed between TCC and markers of coagulation activation (Table 2). When dichotomizing TCC at the median level (3200 AU), the group with above-median (“high”) TCC had significantly higher D-dimer (p<0.001) and ETP (p=0.002) levels, but no difference in F1 + 2 levels (p=0.106) (Supplementary Figure S1).

Table 2. Correlations (Spearman`s Rho) between TCC and markers of hypercoagulability.
3.3 Associations between combined high complement and coagulation activation and peak TnT and LVEF
The proportion of patients with high peak TnT and LVEF ≤40% according to quartiles of TCC, F1 + 2, D-dimer and ETP are given in Figure 1. Visually, there seemed to be some interaction between complement and coagulation, especially for TCC and F1 + 2 and the risk of high peak TnT (Figure 1a), and for TCC and D-dimer and LVEF ≤40% (Figure 1d). However, in multivariate binary logistic regression adjusting for age and gender, none of these were significant (Table 3).
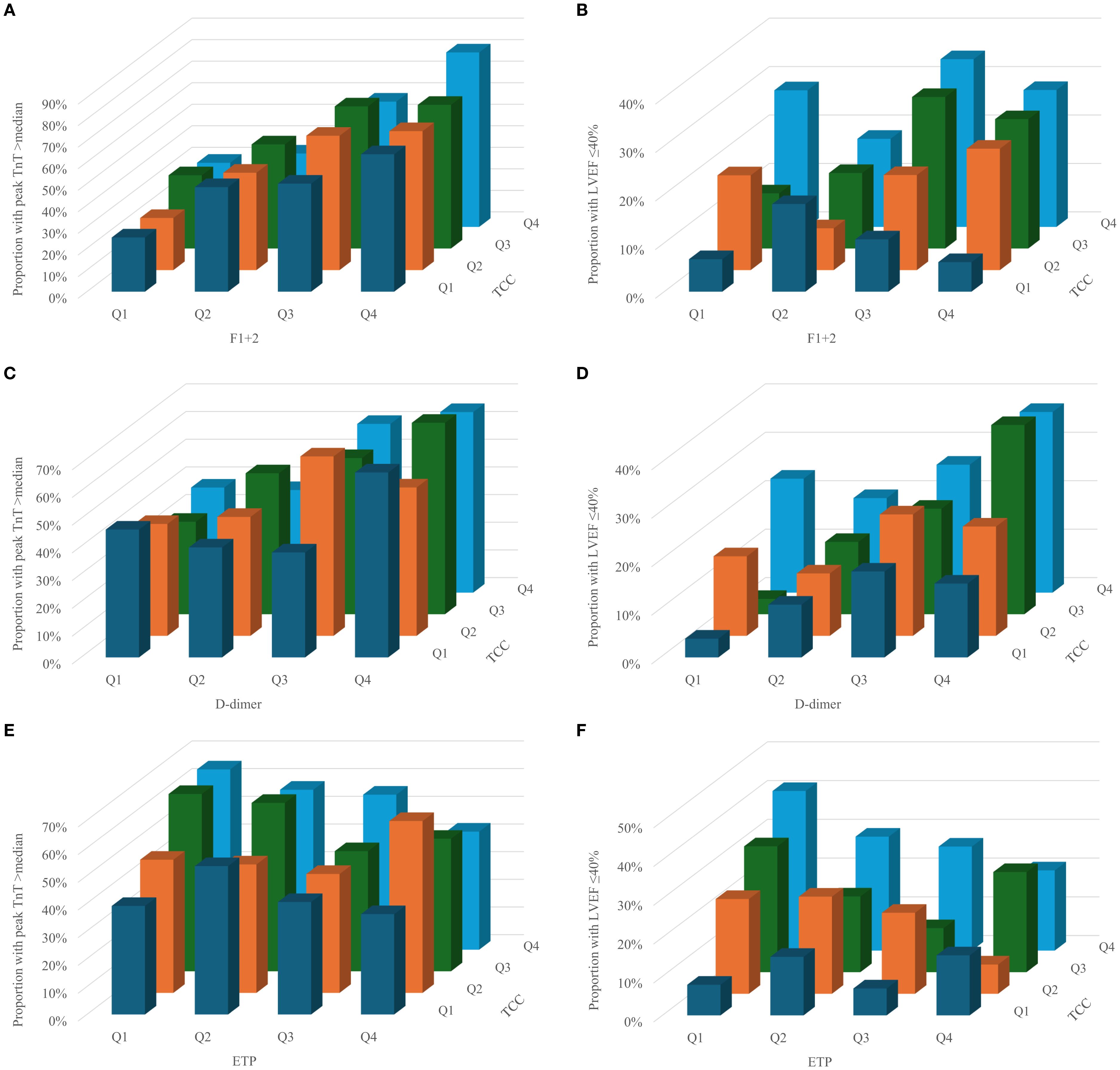
Figure 1. Proportion of patients with high peak TnT and LVEF ≤40% according to quartiles of TCC and F1+2 (a and b), d-dimer (c and d) and ETP (e and f).
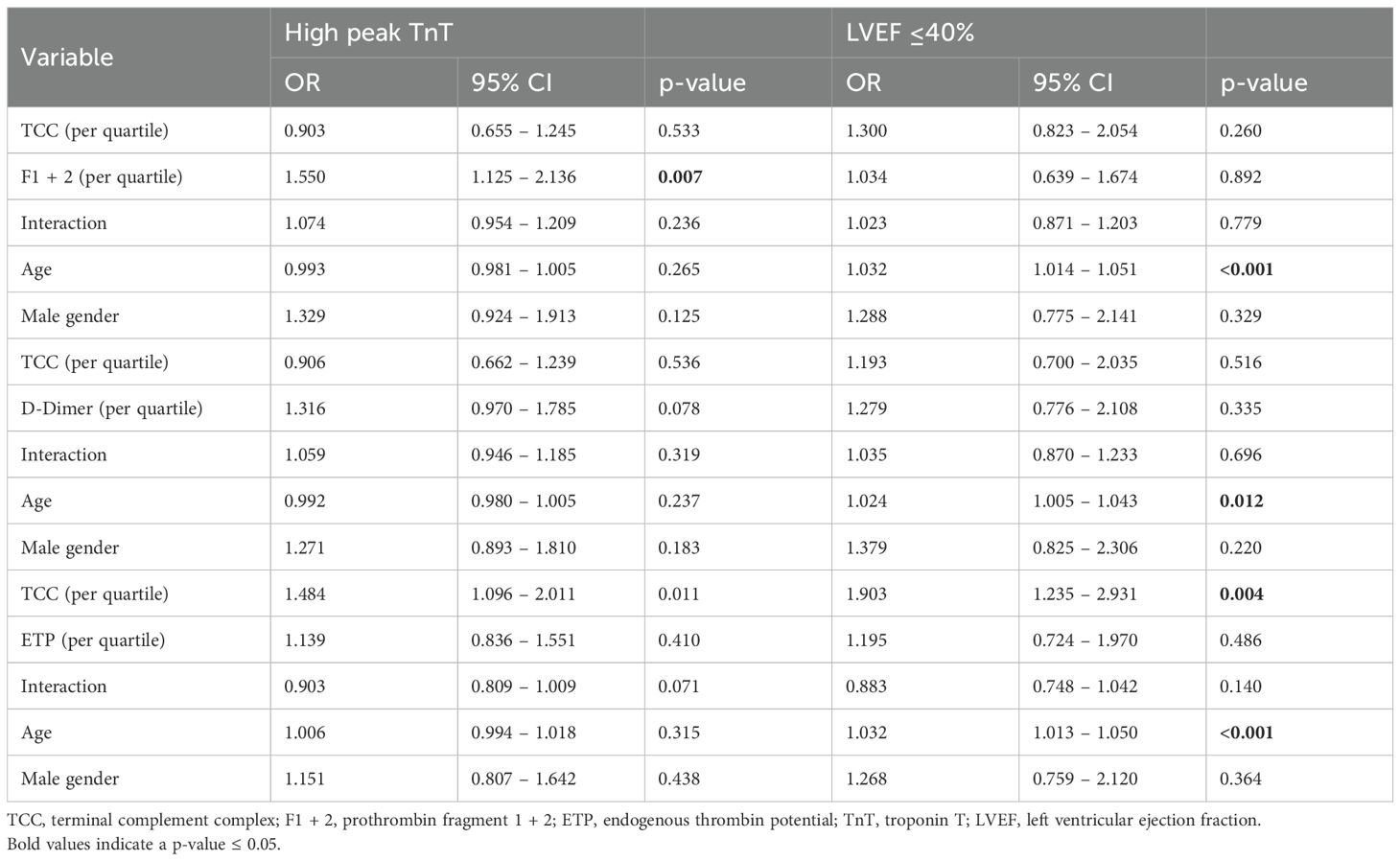
Table 3. Logistic regression of the predictive value of age, gender, TCC and the coagulation markers on peak TnT and LVEF.
4 Discussion
In this large STEMI cohort, we show that complement activation, measured by TCC, was weakly associated with coagulation activation in peripheral blood taken at a median of 18 hours after PCI. There was no significant interaction between the complement and coagulation markers and the risk of high peak TnT or low LVEF.
The participation of complement and coagulation as two distinct processes in patients with STEMI is well-established. Circulating TCC has been reported to be elevated in myocardial infarction, and cell-bound TCC is more heavily deposited in revascularized than non-revascularized myocardium (26, 27). Still, complement inhibition in acute myocardial infarction has so far mediated neutral results (28, 29). As for coagulation, F1 + 2 and D-dimer have been reported to associate with infarct size and mortality in STEMI (30–33). The literature has suggested mutual activation of the two systems through multiple mechanisms, including complement-mediated activation of platelets and direct activation of the complement system by proteins of the coagulation cascade (2).
In this first study on the clinical relevance of combined complement and coagulation activation in STEMI, we found complement activation by TCC to be weakly correlated to the coagulation markers F1 + 2, D-dimer and ETP. Despite two out of three markers of coagulation being significantly higher in patients with high levels of TCC, the associations between complement and coagulation activation were generally weak. This indicates that if there was reciprocal activation between complement and coagulation, it was of modest strength. Both the inflammatory response and coagulation activation have been shown to reflect infarct size (19, 26, 32–34), and we find it likely that other factors, such as the myocardial infarction, PCI procedure, and inflammation following ischemia and reperfusion, affected the respective activation of complement and coagulation in this very acute situation. Unfortunately, the cross-sectional study design did not permit dissection of pathophysiological mechanisms involved in potential interactions between complement and coagulation.
Based on the reported contribution of complement and coagulation on IR injury (6, 35), we hypothesized that activation of both systems would have a negative combined effect on myocardial injury and dysfunction despite the weak association between the systems. We have previously reported that the coagulation markers F1 + 2 and D-dimer were associated with peak TnT and LVEF in this population (19). Combining the complement marker TCC with the coagulation showed no interaction between TCC and F1 + 2 and D-dimer in predicting the risk of high peak TnT or LVEF ≤40%. The interaction between TCC and ETP seemed to be associated with a lower risk of high peak TnT. Although this could indicate that ETP modulates the effect of increasing TCC, it should be interpreted with caution, as the p-value is close to the significance value, which likely makes it a result of multiple comparisons. Taken together, these findings suggest that simultaneous activation of the two systems does not potentiate adverse clinical outcomes in STEMI. This means that despite proposed individual contributions to IR injury, these contributions do not seem to potentiate each other, and breaking this interaction might thus not be a viable pharmacological target.
4.1 Limitations
The cross-sectional design of this study does not permit conclusions regarding causality. A greater infarct size might lead to increased complement and coagulation activation as part of the inflammatory response during myocardial injury and heart failure development – and the proposed mechanism might be the other way around. Another limitation is that markers of complement and coagulation activation were only measured once, and their full interaction is probably not accounted for in the singe blood sample analyzed for this study. Moreover, the variable timing of blood samples after PCI should be highlighted, as time from PCI to blood sampling was correlated to the majority of markers (Supplementary Table S1), and the markers have been reported to peak at differing time points following myocardial infarction (36–40). There might also be differing effects depending on which complement activation pathways are activated, and this is naturally not captured when measuring only TCC. Additionally, as patients received standard pre- and in-hospital antithrombotic medication, this probably affected measured markers, as there are reports of acetylsalicylic acid, thrombolytic agents and heparins affecting complement and coagulation activation (41–48). Also, measuring circulating TCC does not account for cell-bound TCC, and the result might thus not reflect “total complement activation” (49). Finally, as neither time nor method for measuring LVEF was standardized, the LVEF results should be interpreted with caution.
5 Conclusions
In this large STEMI cohort, there were weak associations between circulating markers of complement and coagulation activation. There was no combined effect of complement and coagulation activation on the risk of high peak TnT or low LVEF.
Data availability statement
The data analyzed in this study is subject to the following licenses/restrictions: The dataset used during the current study is not available publicly due to Norwegian legislation about general data protection regulations, but are available from the corresponding author on request. Requests to access these datasets should be directed to Karsten Engseth Kluge, S2Fyc3RlbmVrQGdtYWlsLmNvbQ==.
Ethics statement
The studies involving humans were approved by Regional Committees for Medical Research Ethics South East Norway. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
KK: Writing – original draft, Writing – review & editing, Formal Analysis, Visualization. SH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – review & editing. GA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing – review & editing. CH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing – review & editing. IS: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – review & editing. TT: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – review & editing. IL: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – review & editing. RH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The study was financially supported by The Research Council of Norway through the Medical Student Research Program at the University of Oslo (KEK), Ada og Hagbart Waages humanitære og veldedige stiftelse (KEK), Marie Stenbergs Legat (RH), the Norwegian Health Association (RH), and by unrestricted grants from Stein Erik Hagen’s Foundation for Clinical Heart Research, Oslo Norway.
Acknowledgments
The authors would like to thank the laboratory staff at the Oslo Center for Clinical Heart Research for excellent technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1613603/full#supplementary-material
References
1. Merle NS, Church SE, Fremeaux-Bacchi V, and Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Pryzdial ELG, Leatherdale A, and Conway EM. Coagulation and complement: Key innate defense participants in a seamless web. Front Immunol. (2022) 13:918775. doi: 10.3389/fimmu.2022.918775
3. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, and Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
4. Hoffman M and Monroe DM 3rd. A cell-based model of hemostasis. Thromb Haemost. (2001) 85:958–65. doi: 10.1055/s-0037-1615947
5. Bentzon JF, Otsuka F, Virmani R, and Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114:1852–66. doi: 10.1161/CIRCRESAHA.114.302721
6. Howard MC, Nauser CL, Farrar CA, and Sacks SH. Complement in ischaemia-reperfusion injury and transplantation. Semin Immunopathol. (2021) 43:789–97. doi: 10.1007/s00281-021-00896-3
7. Patzelt J, Verschoor A, and Langer HF. Platelets and the complement cascade in atherosclerosis. Front Physiol. (2015) 6:49. doi: 10.3389/fphys.2015.00049
8. Banz Y and Rieben R. Role of complement and perspectives for intervention in ischemia-reperfusion damage. Ann Med. (2012) 44:205–17. doi: 10.3109/07853890.2010.535556
9. Duehrkop C and Rieben R. Ischemia/reperfusion injury: effect of simultaneous inhibition of plasma cascade systems versus specific complement inhibition. Biochem Pharmacol. (2014) 88:12–22. doi: 10.1016/j.bcp.2013.12.013
10. Ten Cate H, Hackeng TM, and García de Frutos P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost. (2017) 117:1265–71. doi: 10.1160/TH17-02-0079
11. Kremers BMM, Ten Cate H, and Spronk HMH. Pleiotropic effects of the hemostatic system. J Thromb Haemost. (2018) 16(8):1464–1473. doi: 10.1111/jth.14161
12. Eltzschig HK and Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
13. Yin Y, Fang C, Jiang S, Wang J, Wang Y, Guo J, et al. In vivo evidence of atherosclerotic plaque erosion and healing in patients with acute coronary syndrome using serial optical coherence tomography imaging. Am Heart J. (2022) 243:66–76. doi: 10.1016/j.ahj.2021.09.007
14. Dzik S. Complement and coagulation: cross talk through time. Transfus Med Rev. (2019) 33:199–206. doi: 10.1016/j.tmrv.2019.08.004
15. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
16. Martel C, Cointe S, Maurice P, Matar S, Ghitescu M, Théroux P, et al. Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PLoS One. (2011) 6:e18812. doi: 10.1371/journal.pone.0018812
17. Harboe M, Thorgersen EB, and Mollnes TE. Advances in assay of complement function and activation. Adv Drug Delivery Rev. (2011) 63:976–87. doi: 10.1016/j.addr.2011.05.010
18. Kluge KE, Langseth MS, Andersen GØ, Halvorsen S, Opstad TB, Arnesen H, et al. Complement activation in association with clinical outcomes in ST-elevation myocardial infarction. Am Heart J Plus. (2022) 24:100228. doi: 10.1016/j.ahjo.2022.100228
19. Hansen CH, Ritschel V, Halvorsen S, Andersen G, Bjørnerheim R, Eritsland J, et al. Markers of thrombin generation are associated with myocardial necrosis and left ventricular impairment in patients with ST-elevation myocardial infarction. Thromb J. (2015) 13:31. doi: 10.1186/s12959-015-0061-1
20. Kluge KE, Langseth MS, Bratseth V, Pettersen AA, Arnesen H, Tonnessen T, et al. Circulating levels of the terminal complement complex are associated with hypercoagulability in patients with stable coronary artery disease. Thromb Res. (2020) 196:106–8. doi: 10.1016/j.thromres.2020.08.023
21. Ritschel VN, Seljeflot I, Arnesen H, Halvorsen S, Weiss T, Eritsland J, et al. IL-6 signalling in patients with acute ST-elevation myocardial infarction. Results Immunol. (2014) 4:8–13. doi: 10.1016/j.rinim.2013.11.002
22. Larson EA, German DM, Shatzel J, and DeLoughery TG. Anticoagulation in the cardiac patient: A concise review. Eur J Haematol. (2019) 102:3–19. doi: 10.1111/ejh.13171
23. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth universal definition of myocardial infarction (2018). Circulation. (2018) 138:e618–e51. doi: 10.1161/CIR.0000000000000617
24. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. (2021) 42:3599–726. doi: 10.1093/eurheartj/ehab368
25. Gaya da Costa M, Poppelaars F, van Kooten C, Mollnes TE, Tedesco F, Würzner R, et al. Age and sex-associated changes of complement activity and complement levels in a healthy caucasian population. Front Immunol. (2018) 9:2664. doi: 10.3389/fimmu.2018.02664
26. Ørn S, Manhenke C, Ueland T, Damås JK, Mollnes TE, Edvardsen T, et al. C-reactive protein, infarct size, microvascular obstruction, and left-ventricular remodelling following acute myocardial infarction. Eur Heart J. (2009) 30:1180–6. doi: 10.1093/eurheartj/ehp070
27. Nijmeijer R, Krijnen PAJ, Assink J, Klaarenbeek MAR, Lagrand WK, Veerhuis R, et al. C-reactive protein and complement depositions in human infarcted myocardium are more extensive in patients with reinfarction or upon treatment with reperfusion. Eur J Clin Invest. (2004) 34:803–10. doi: 10.1111/j.1365-2362.2004.01425.x
28. Martel C, Granger CB, Ghitescu M, Stebbins A, Fortier A, Armstrong PW, et al. Pexelizumab fails to inhibit assembly of the terminal complement complex in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention. Insight from a substudy of the Assessment of Pexelizumab in Acute Myocardial Infarction (APEX-AMI) trial. Am Heart J. (2012) 164:43–51. doi: 10.1016/j.ahj.2012.04.007
29. Testa L, Van Gaal WJ, Bhindi R, Biondi-Zoccai GGL, Abbate A, Agostoni P, et al. Pexelizumab in ischemic heart disease: A systematic review and meta-analysis on 15,196 patients. J Thorac Cardiovasc Surgery. (2008) 136:884–93. doi: 10.1016/j.jtcvs.2007.12.062
30. Hansen CH, Ritschel V, Andersen GO, Halvorsen S, Eritsland J, Arnesen H, et al. Markers of thrombin generation are associated with long-term clinical outcome in patients with ST-segment elevation myocardial infarction. Clin Appl Thromb Hemost. (2018) 24:1088–94. doi: 10.1177/1076029618764847
31. Granger CB, Becker R, Tracy RP, Califf RM, Topol EJ, Pieper KS, et al. Thrombin generation, inhibition and clinical outcomes in patients with acute myocardial infarction treated with thrombolytic therapy and heparin: results from the GUSTO-I Trial. GUSTO-I Hemostasis Substudy Group. Global Utilization of Streptokinase and TPA for Occluded Coronary Arteries. J Am Coll Cardiol. (1998) 31:497–505. doi: 10.1016/S0735-1097(97)00539-1
32. Choi S, Jang WJ, Song YB, Lima JA, Guallar E, Choe YH, et al. D-dimer levels predict myocardial injury in ST-segment elevation myocardial infarction: A cardiac magnetic resonance imaging study. PLoS One. (2016) 11:e0160955. doi: 10.1371/journal.pone.0160955
33. Biccirè FG, Farcomeni A, Gaudio C, Pignatelli P, Tanzilli G, and Pastori D. D-dimer for risk stratification and antithrombotic treatment management in acute coronary syndrome patients: a systematic review and metanalysis. Thromb J. (2021) 19:102. doi: 10.1186/s12959-021-00354-y
34. Vanhaverbeke M, Veltman D, Pattyn N, De Crem N, Gillijns H, Cornelissen V, et al. C-reactive protein during and after myocardial infarction in relation to cardiac injury and left ventricular function at follow-up. Clin Cardiol. (2018) 41:1201–6. doi: 10.1002/clc.23017
35. Oikonomopoulou K, Ricklin D, Ward PA, and Lambris JD. Interactions between coagulation and complement–their role in inflammation. Semin Immunopathol. (2012) 34:151–65. doi: 10.1007/s00281-011-0280-x
36. Orrem HL, Nilsson PH, Pischke SE, Kleveland O, Yndestad A, Ekholt K, et al. IL-6 receptor inhibition by tocilizumab attenuated expression of C5a receptor 1 and 2 in non-ST-elevation myocardial infarction. Front Immunol. (2018) 9:2035. doi: 10.3389/fimmu.2018.02035
37. Hoffmeister HM, Szabo S, Kastner C, Beyer ME, Helber U, Kazmaier S, et al. Thrombolytic therapy in acute myocardial infarction: comparison of procoagulant effects of streptokinase and alteplase regimens with focus on the kallikrein system and plasmin. Circulation. (1998) 98:2527–33. doi: 10.1161/01.CIR.98.23.2527
38. Solheim S, Seljeflot I, Lunde K, Bratseth V, Aakhus S, Forfang K, et al. Prothrombotic markers in patients with acute myocardial infarction and left ventricular thrombus formation treated with pci and dual antiplatelet therapy. Thromb J. (2013) 11:1. doi: 10.1186/1477-9560-11-1
39. Vila V, Martínez-Sales V, Réganon E, Peris E, Perez F, Ruano M, et al. Effects of unfractionated and low molecular weight heparins on plasma levels of hemostatic factors in patients with acute coronary syndromes. Haematologica. (2001) 86:729–34. doi: 10.3324/%x
40. Bavia L, Lidani KCF, Andrade FA, Sobrinho MIAH, Nisihara RM, and de Messias-Reason IJ. Complement activation in acute myocardial infarction: An early marker of inflammation and tissue injury? Immunol Lett. (2018) 200:18–25. doi: 10.1016/j.imlet.2018.06.006
41. Hänsch GM, Voigtländer V, and Rother U. Effect of aspirin on the complement system in vitro. Int Arch Allergy Appl Immunol. (1980) 61:150–8. doi: 10.1159/000232428
42. Poppelaars F, Damman J, de Vrij EL, Burgerhof JG, Saye J, Daha MR, et al. New insight into the effects of heparinoids on complement inhibition by C1-inhibitor. Clin Exp Immunol. (2016) 184:378–88. doi: 10.1111/cei.12777
43. Agostoni A, Gardinali M, Frangi D, Cafaro C, Conciato L, Sponzilli C, et al. Activation of complement and kinin systems after thrombolytic therapy in patients with acute myocardial infarction. A comparison between streptokinase and recombinant tissue-type plasminogen activator. Circulation. (1994) 90:2666–70. doi: 10.1161/01.CIR.90.6.2666
44. Wallén NH and Ladjevardi M. Influence of low- and high-dose aspirin treatment on thrombin generation in whole blood. Thromb Res. (1998) 92:189–94. doi: 10.1016/S0049-3848(98)00126-1
45. Couturaud F, Kearon C, Bates SM, and Ginsberg JS. Decrease in sensitivity of D-dimer for acute venous thromboembolism after starting anticoagulant therapy. Blood Coagul Fibrinolysis. (2002) 13:241–6. doi: 10.1097/00001721-200204000-00010
46. Hemker HC, Al Dieri R, and Béguin S. Heparins: A shift of paradigm. Front Med (Lausanne). (2019) 6:254. doi: 10.3389/fmed.2019.00254
47. Weitz JI, Fredenburgh JC, and Eikelboom JW. A test in context: D-dimer. J Am Coll Cardiol. (2017) 70:2411–20. doi: 10.1016/j.jacc.2017.09.024
48. Amelsberg A, Zurborn KH, Gärtner U, Kiehne KH, Preusse AK, and Bruhn HD. Influence of heparin treatment on biochemical markers of an activation of the coagulation system. Thromb Res. (1992) 66:121–31. doi: 10.1016/0049-3848(92)90182-A
Keywords: complement system, coagulation system, STEMI, coronary artery disease, revascularization, heart failure
Citation: Kluge KE, Halvorsen S, Andersen GØ, Hansen CH, Seljeflot I, Tønnessen T, Lunde IG and Helseth R (2025) Combined complement and coagulation activation in ST-elevation myocardial infarction: associations with myocardial injury and dysfunction. Front. Immunol. 16:1613603. doi: 10.3389/fimmu.2025.1613603
Received: 17 April 2025; Accepted: 20 October 2025;
Published: 04 November 2025.
Edited by:
Beate E. Kehrel, University Hospital Münster, GermanyReviewed by:
Michael Osthoff, University Hospital of Basel, SwitzerlandAnna Swierzko, Polish Academy of Sciences, Poland
Copyright © 2025 Kluge, Halvorsen, Andersen, Hansen, Seljeflot, Tønnessen, Lunde and Helseth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karsten E. Kluge, S2Fyc3RlbmVrQGdtYWlsLmNvbQ==