Abstract
Although current treatments for autoimmune diseases can effectively control symptoms, they rarely lead to cures and often require lifelong use, accompanied by considerable adverse effects. This emphasizes the urgent need for more targeted therapies that offer long-term efficacy and curative potential. Chimeric antigen receptor (CAR) T-cell therapy presents a promising option by specifically targeting and eliminating autoreactive B cells, with the potential to reset the patient’s immune system and promote long-term immune balance. Originally developed for treating hematologic malignancies, where it has achieved remarkable success, recent studies have demonstrated substantial promise of CAR T-cell therapy, such as systemic lupus erythematosus (SLE) and myasthenia gravis. This article provides an overview of the current progress in CAR T-cell therapy for autoimmune diseases, focusing on five key approaches: CD19-targeted CAR T cells, CAR T cells targeting long-lived plasma cells, CAR T cells targeting specific autoantibodies, organ-specific CAR regulatory T cells (Treg cells), and mRNA-engineered CAR T cells. Additionally, this article discusses strategies for optimizing CAR T-cell therapy, including “off-the-shelf” allogeneic CAR T-cell therapy, combined CAR T-cell therapy, establishing timely consensus guidelines for their application in autoimmune diseases, and risk stratification strategies aimed at enhancing the personalization of treatments and minimizing adverse effects. While current research results are promising, further large-scale clinical trials and long-term follow-up are essential to thoroughly evaluate the safety and efficacy of CAR T-cell therapy in autoimmune diseases.
1 Introduction
Autoimmune diseases are a heterogeneous group of disorders caused by the misdirection of the immune system toward its host, and the presence of autoantibodies is a common feature of autoimmune diseases (1). There are nearly 100 different types of autoimmune diseases in humans, some of these are organ-specific, such as type 1 diabetes (T1D), while others are systemic, such as systemic lupus erythematosus (SLE) (2). Autoimmune diseases, previously considered rare, affect approximately 5%–8% of the world population according to epidemiological studies (3).
Given the limited understanding of the complex molecular mechanisms underlying autoimmune diseases, it is widely accepted that a combination of genetic predisposition and environmental factors disrupts immune tolerance (4–6), This disruption leads to the clonal expansion of autoreactive B and T cells, resulting in the production of autoantibodies that trigger autoimmune inflammation and the pathological attack on the body’s tissues. Current standard approaches offer no cure; rather, they focus on lifetime management. The primary approach for treating autoimmune diseases continues to involve controlling autoreactive T and B cells that attack host tissues, typically through the use of broad immunosuppressive agents such as glucocorticoids, methotrexate, and azathioprine (7–9). Recent advances have introduced a new paradigm of interventions, including biological agents and targeted small molecules, which more specifically inhibit detrimental immune cells and modulate specific inflammatory pathways. However, many patients with autoimmune diseases eventually relapse and may suffer from life-threatening complications (3). In summary, while current treatments can manage symptoms, they generally lack curative potential and are often associated with significant toxic side effects, some of which can be life-threatening. This highlights the urgent need for more optimized and curative therapies.
Chimeric antigen receptor (CAR) is a fusion protein consisting of an antigen recognition domain and a cell activation domain that can redirect T cell specificity, function, and metabolism (10). In oncology, CAR T cells have been used to accurately identify and eliminate cancer, showing remarkable and lasting efficacy in the treatment of certain cancers, particularly leukemia, lymphoma, and multiple myeloma (11–13). A decade-long follow-up study demonstrated that CAR T cells can persist in leukemia patients for up to ten years post-treatment, correlating with sustained disease remission and prolonged B-cell aplasia (14). Originally developed for cancer therapy, this approach mediates durable elimination of autoreactive B cells in autoimmune diseases, potentially eliminating the need for chronic immunosuppression. Importantly, it may also reestablish immune tolerance through secondary modulation of pathogenic T cell responses. Through this mechanism, CAR T-cell therapy, which targets autoreactive immune cells, is increasingly emerging as a potential curative approach for autoimmune diseases (15–18). It should be noted that CAR T-cell therapy is not intended to entirely replace existing treatment strategies. Instead, it is more appropriately positioned as a potential later-line option for patients with autoimmune diseases who exhibit inadequate responses to conventional therapies—including biological agents—or who are unable to tolerate their associated toxicities, particularly in cases of refractory or relapsing diseases. Recent preclinical studies, along with case reports and small-scale clinical trials, have yielded promising results in the treatment of autoimmune diseases using CAR T-cell therapy (19, 20). This review will focus on the clinical needs and applications, advantages and limitations, proposed solutions, and prospects of CAR T-cell therapy in the context of autoimmune diseases.
2 Classification and pathogenesis of autoimmune diseases
For clinical practitioners, autoimmune diseases are generally categorized into two main types: systemic and organ-specific. This classification is straightforward and practical, but its value in guiding treatment is limited, particularly as it does not reflect the underlying causes and immune mechanisms of the diseases. Therefore, a more precise classification based on specific pathogenic immune responses (such as abnormal T cell or B cell-mediated reactions) is beneficial for formulating personalized treatment plans. Characteristics of pathogenic immune responses include selective dysregulation of T cells or B cells, as well as dysregulated immune responses to specific self-antigens. For instance, in organ-specific diseases (such as T1D and thyroiditis), T cells play a crucial role, while in systemic diseases, such as SLE and rheumatoid arthritis (RA), B cells and their antibodies dominate. This classification method is beneficial in clinical treatment, as different immune mechanisms may require distinct therapeutic strategies (Table 1).
Table 1
| Autoimmune disease | Target organ | Autoantigen(s) | Primary mechanism of damage |
|---|---|---|---|
| Organ-specific | |||
| T1D | Pancreatic β cells | Insulin, glutamic acid decarboxylase | T cells |
| Multiple sclerosis | Central nervous system | Myelin basic protein, myelin oligodendrocyte glycoprotein, proteolipid Protein | T cells |
| Pemphigus vulgaris | Skin and mucous membranes | Desmoglein 1, desmoglein 3 | Antibody |
| Primary biliary cholangitis | Liver | Pyruvate dehydrogenase complex E2 component | T cells/Antibody |
| Myasthenia gravis | Neuromuscular junction | Acetylcholine receptor, muscle-specific Kinase | Antibody |
| Thyroiditis (autoimmune) | Thyroid | Thyroid peroxidase, thyroglobulin | T cells/Antibody |
| Autoimmune gastritis | Stomach | H+/K+ ATPase | T cells/Antibody |
| Autoimmune hepatitis | Liver | Cytochrome P450 enzymes | T cells/Antibody |
| Autoimmune oophoritis | Ovaries | Steroid-producing enzymes | T cells/Antibody |
| Autoimmune inner ear disease | Inner ear | Cochlear antigens, proteins in the inner ear structures | T cells/Antibody |
| Autoimmune orchitis | Testes | Sperm-specific proteins, testicular antigens | T cells/Antibody |
| Neuromyelitis optica spectrum disorder | Central nervous system | Aquaporin-4 | Antibody |
| Idiopathic glomerulonephritis | Kidneys | Glomerular basement membrane, podocyte proteins | Antibody |
| Immune thrombocytopenia | Platelets and megakaryocytes | Glycoproteins on the surface of platelets, such as GPIIb/IIIa and GPIb/IX | Antibody |
| Systemic | |||
| SLE | Skin, joints, kidneys, heart, lungs, central nervous system, blood cells, others | Nuclear antigens, others | Antibody |
| RA | Joints, lungs, heart, blood, others | Citrullinated proteins, rheumatoid factor | Antibody |
| Systemic sclerosis | Skin, lungs, gastrointestinal tract, heart, kidneys, others | Topoisomerase I (Scl-70), centromere proteins, RNA polymerase III | Antibody |
| Sjögren’s Syndrome | Exocrine glands, lungs, kidneys, liver,joints,others | Ro/SSA, La/SSB | Antibody |
| Polymyositis and dermatomyositis | Skeletal muscles, skin, lungs, heart, joints, others | Muscle antigens, aminoacyl-tRNA synthetases, other nuclear antigens | T cells/Antibody |
| Antiphospholipid syndrome | Blood vessels, pregnancy-related organs, brain, heart, Kidneys, lungs, others | β2-glycoprotein I, prothrombin | Antibody |
| Systemic vasculitis | Vessels, skin, kidneys, lungs, nervous system, heart, others | Proteinase 3 (PR3), myeloperoxidase (MPO), endothelial cell antigens | Antibody |
Major organ-specific and systemic autoimmune diseases, targets, and mechanisms.
T1D, type 1 diabetes; SLE, systemic lupus erythematosus; RA, rheumatoid arthritis.
The pathogenesis of autoimmune diseases involves a complex interplay between genetic susceptibility and environmental factors, with T cells and B cells collaboratively triggering and maintaining abnormal immune responses, forming an autoimmune cycle that ultimately leads to chronic inflammation and tissue damage (21) (Figure 1). Systemic autoimmune diseases, such as SLE and RA, exhibit widespread activation of the immune system, affecting multiple organs and tissues, and their pathogenic mechanisms involve self-antigen exposure and immune complex deposition (22). In these diseases, dendritic cells (DCs) present self-antigens to T cells (23), leading to the activation of T cells that secrete cytokines such as IL-21 and IFN-γ (24), which further activate B cells. Activated B cells undergo affinity maturation and differentiation into plasma cells, producing autoantibodies targeting a wide range of antigens (25). These autoantibodies bind to free molecules and antigens to form immune complexes, which can deposit in the vascular walls and tissues of multiple organs, triggering inflammation and damage (26). The deposition of immune complexes and the resultant local inflammatory responses establish an autoimmune cycle, continuously stimulating the activation of T cells and B cells, thereby exacerbating tissue damage and inflammation (27). Once this autoimmune cycle is initiated, if not interrupted by pharmacological intervention, the abnormal immune response persists, leading to the long-term maintenance and progression of autoimmune diseases (28).
Figure 1

Illustration of the autoimmune cycle. The interaction between genetic susceptibility and environmental factors leads to the misrecognition of self-antigens. APCs present these self-antigens to T cells, initiating the immune response. Activated CD8+ T cells attack target organs, while CD4+ T cells activate autoimmune B cells to produce autoantibodies. These autoantibodies bind to antigens, forming immune complexes that deposit in organs, causing localized or systemic inflammation and damage. Immune complexes and inflammation continuously activate T and B cells, forming an autoimmune cycle that exacerbates tissue damage. APC, antigen-presenting cell; T1D, type 1 diabetes; PBC, primary biliary cholangitis; SLE, systemic lupus erythematosus; RA, rheumatoid arthritis.
In contrast, organ-specific autoimmune diseases, such as T1D and Graves’ disease, primarily target specific organs and involve immune responses against specific organ self-antigens. In these diseases, specific antigens, such as islet β-cell antigens or thyroid antigens, are presented to T cells by local antigen-presenting cells (APC), including DCs and macrophages. Activated T cells then attack the cells within the target organ by secreting specific cytokines like IL-17 and IFN-γ (29). For instance, in T1D, cytotoxic T cells directly attack islet β-cells, resulting in insufficient insulin secretion; in Graves’ disease, autoantibodies stimulate thyroid receptors, leading to hyperthyroidism (30, 31). These autoantibodies and local inflammatory responses create a specific immune cycle, which, unlike systemic diseases, is primarily confined to the affected organ (32).
In both systemic and organ-specific autoimmune diseases, the mechanism commonly involves the abnormal activation or dysregulation of T cells and B cells. This abnormal activation not only initiates the immune response but also sustains the pathological progression, forming an autoimmune cycle that makes the disease difficult to reverse (33, 34). Understanding this mechanism is crucial for developing effective treatment strategies. Although traditional therapies can alleviate symptoms and control disease activity, completely disrupting the autoimmune cycle remains a challenge (22, 35).
3 Limitations of current treatments for autoimmune diseases: highlighting the urgent need for novel therapeutic approaches
Current treatments for autoimmune diseases mainly include corticosteroids, non-steroidal anti-inflammatory drugs (NSAIDs), disease-modifying anti-inflammatory drugs (DMARDs), biologics, and targeted small molecules. Despite the variety of therapeutic options available, many patients are still predominantly treated with non-specific conventional treatments, with corticosteroids being among the most commonly used drugs. Corticosteroids can rapidly alleviate symptoms due to their powerful and broad anti-inflammatory and immunosuppressive effects. However, corticosteroids are a double-edged sword. Long-term or high-dose use can lead to serious complications, such as secondary infections, gastrointestinal ulcers, and femoral head necrosis, which can significantly impact patients’ quality of life and even be life-threatening (36–39). To reduce the cumulative dose of corticosteroids, DMARDs with steroid-sparing effects are often utilized. These include methotrexate, leflunomide, tacrolimus, azathioprine, mycophenolate mofetil, hydroxychloroquine, and cyclophosphamide (40–45). While DMARDs can help decrease the need for corticosteroids and lower the risk of disease relapse, patients in remission generally require lifelong low-dose maintenance therapy to prevent relapse. Furthermore, DMARDs do not offer targeted immunosuppressive effects; instead, they broadly inhibit various immune pathways, which can affect multiple cells in the body and increase the risk of secondary diseases, such as cancer (46).
Researchers are working to develop new treatments that are more effective, specific, and safer. Recently developed and clinically applied biological agents, such as various monoclonal antibodies and targeted small molecules like Janus kinase (JAK) inhibitors, have shown promise as treatment options for many autoimmune diseases, including SLE, RA, multiple sclerosis, myositis, psoriasis, atopic dermatitis, and pemphigus, without causing life-threatening adverse reactions (47, 48). The emergence of these novel drugs has alleviated some of the current treatment challenges and brought new hope to patients. For example, the anti-CD20 monoclonal antibody rituximab has demonstrated efficacy in several autoimmune diseases and has received marketing authorization for use in patients with SLE, granulomatosis with polyangiitis, microscopic polyangiitis, and pemphigus vulgaris (49). Belimumab, another biological agent used to treat SLE, works by inhibiting B cell survival and differentiation into antibody-producing plasma cells through the blockade of B-cell activating factor (BAFF) (50). Additionally, the targeted small molecule upadacitinib, a JAK inhibitor, blocks the phosphorylation of downstream effector proteins, thereby inhibiting cytokine signaling in key inflammatory pathways in immune cells. It has been approved for the treatment of RA and psoriatic arthritis (51).
Unfortunately, while these novel drugs can effectively attenuate the inflammatory process, they often require continuous treatment over many years or even a lifetime. Despite achieving remission, disease recurrence is common when immunosuppression is discontinued. This may be due to immune escape mechanisms that prevent the complete eradication of autoimmunity in patients with autoimmune diseases. Studies demonstrate that autoreactive B cells persist in specific tissue compartments after rituximab therapy, including SLE tonsils, systemic sclerosis skin lesions, and RA synovium (52–55). These findings suggest that, compared to circulating B cells, memory B cells in tissues are more resistant to depletion. Notably, these remaining tissue-resident B cells are thought to be responsible for disease relapse (53, 56).
4 The emergence of novel immunotherapies and comparison of emerging therapies
Current treatments for autoimmune diseases rely mainly on systemic immunosuppression, which generally reduces disease activity and can slow progression in many patients. However, this approach often requires ongoing medication. Prolonged use of immunosuppressive therapies can lead to side effects and complications, with some patients developing refractory disease states that are difficult to manage effectively.
Researchers have been striving to develop therapies capable of “resetting” the immune system, with the goal of profoundly recalibrating immune balance, terminating disease activity, and allowing patients to achieve long-term remission without the need for continued immunosuppressive therapy. In 1993, Marmont A. M. first proposed hematopoietic stem cell transplantation (HSCT) for severe, refractory lupus patients (57). This idea soon extended to severe autoimmune diseases (58, 59) and has been considered an alternative for SLE patients unresponsive to standard treatments (60). Multiple clinical trials suggest HSCT can induce long-term remission in various autoimmune diseases without maintenance therapy (61, 62). However, despite its advantages, the high rate of adverse effects, such as infections and secondary autoimmune diseases (63, 64), has limited HSCT’s use in autoimmune conditions like SLE (65). Further research is needed to assess the efficacy and safety of HSCT as a promising option for treating autoimmune diseases.
Dendritic cell (DC) therapy, a promising new immunotherapy that has made strides in cancer treatment (66), has recently been applied to autoimmune diseases (67). Through tolerogenic DCs, this approach induces immune tolerance in T cells against self-antigens, thereby suppressing autoreactive T cells in autoimmune disease management (68). Currently, DC therapy is in preclinical and early trials, demonstrating potential in inducing antigen-specific immune tolerance, thereby modulating T-cell responses and alleviating disease progression in RA (69), T1D (70) and multiple sclerosis (71). Despite this potential, challenges remain, including generating stable and functional DCs ex vivo and designing personalized DC vaccines tailored to diverse disease mechanisms. Further large-scale trials are required to validate its long-term efficacy and safety.
The application of natural killer (NK) cell therapy in autoimmune diseases remains in an early exploratory phase. NK cells can regulate the immune system by cytokine secretion and interacting with other immune cells (72, 73), modulating the activity of T cells, DCs, and macrophages, which impacts immune tolerance and inflammatory responses (72, 74). Consequently, NK cells show potential in suppressing autoimmune responses and restoring immune balance. Preclinical studies demonstrate that in vitro expansion and activation of NK cells effectively reduce inflammatory responses and improve clinical symptoms in the experimental autoimmune encephalomyelitis (EAE) model (75, 76). However, challenges persist, including precise control of NK cell activity to avoid damage to normal tissues, ensuring long-term stability, and optimizing applications across different autoimmune diseases. Despite these challenges, NK cell therapy’s immunomodulatory capacity and low toxicity present a promising therapeutic avenue for future treatment strategies.
Significant progress has been made in applying gene editing therapies to autoimmune diseases, primarily by modifying immune cell functions to inhibit pathological immune responses. The CRISPR/Cas9 gene editing technology, with its high efficiency, specificity, and flexibility, has been widely used to regulate T cells, B cells, and regulatory T cells (Treg cells), aiming to reduce their attack on self-tissues and restore immune tolerance. With technological advances, these approaches have shown promise in treating diseases like SLE, RA, and multiple sclerosis (77, 78). By targeting pathogenic genes or immune regulatory pathways, CRISPR/Cas9 holds promise for achieving personalized treatment and offers a novel strategy for precise intervention in autoimmune diseases. However, the long-term follow-up data on CRISPR/Cas9 gene editing technology remain limited, making it impossible to completely rule out potential long-term risks. The challenges of clinical translation and limitations of delivery systems further restrict the application of CRISPR/Cas9 in autoimmune diseases. Additionally, its high cost, ethical concerns, and the complexity of personalized treatment continue to limit widespread clinical application.
These therapies each have their advantages and limitations, and the most suitable option should be selected based on the patient’s disease characteristics, severity, and treatment goals. However, HSCT faces high risks and costs. Both DC and NK cell therapies are still in the early stages, and the long-term sustainability of their efficacy remains to be verified. Gene editing therapies, while precise, face ethical controversies and challenges related to personalized treatment. Recently, the development of engineered receptors has rapidly advanced CAR T-cell therapy (79). Initially applied to hematologic malignancies, CAR T-cell therapy has now been explored for autoimmune diseases. Early clinical applications have shown good safety and efficacy in diseases such as SLE and myasthenia gravis (80–82). Studies suggest that CAR T-cell therapy can target and eliminate pathogenic B cells, effectively control disease activity, and significantly reduce relapse risk, demonstrating potential for a cure (83). These initial successes have laid the foundation for expanding CAR T-cell therapy to a broader range of autoimmune diseases, gradually making it a preferred emerging therapy in the field. This review will focus on the prospects of CAR T-cell therapy in autoimmune diseases.
5 CAR T-cell therapies
5.1 Design and structure of CAR
A typical CAR comprises five functional domains: the antigen recognition domain, hinge region, transmembrane domain, co-stimulatory signaling domain, and T cell activation domain (84, 85). The antigen recognition domain is usually composed of a single-chain variable fragment (scFv) derived from the variable heavy (VH) and light (VL) chains of a monoclonal antibody, conferring high affinity and specificity toward the target antigen. Unlike conventional T cell receptors, the scFv enables direct recognition of cell surface antigens independent of antigen processing or MHC presentation, a feature particularly advantageous in autoimmune diseases with well-defined antigenic targets. The selection of CAR targets in autoimmune diseases must consider antigen specificity, expression patterns, and the risk of off-target effects. Numerous candidate targets have been proposed (Table 2), which provide a basis for the precise application of CAR T-cell therapy.The hinge region connects the scFv to the transmembrane domain, providing structural flexibility and facilitating optimal spatial orientation between the CAR and its cognate antigen. Common hinge elements include sequences from CD8α, IgG1, or IgG4, whose length and conformation can influence CAR surface expression, antigen-binding affinity, and potential cross-reactivity with non-target tissues. The transmembrane domain anchors the CAR to the T cell membrane and contributes to receptor stability and signal transmission. Frequently used transmembrane regions are derived from CD28, CD3ζ, or CD8α. The co-stimulatory signaling domain enhances T cell functionality and persistence. Second-generation CARs incorporate a single co-stimulatory signal, such as CD28 or 4-1BB, whereas third-generation CARs combine multiple co-stimulatory motifs to further amplify immune responses. The choice of co-stimulatory domain critically affects T cell metabolism, differentiation, and the cytokine release profile. Nearly all CAR constructs evaluated in clinical settings utilize the CD3ζ chain as the activation domain, which contains immunoreceptor tyrosine-based activation motifs (ITAMs) that initiate downstream signaling cascades leading to T cell activation, expansion, and cytotoxic function.
Table 2
| Autoimmune disease | Target antigen | CAR strategy |
|---|---|---|
| SLE | CD19, BCMA | CAR-T |
| Multiple Sclerosis | CD19, CD20, XCR1, MOG | CAR-T / CAR-Treg |
| RA | CD19, CD20, BCMA | CAR-T |
| Systemic Sclerosis | CD19, BCMA | CAR-T |
| Dermatomyositis, Polymyositis | CD19 | CAR-T |
| Sjögren’s syndrome | CD19, BCMA | CAR-T |
| Myasthenia gravis | CD19, BCMA, MuSK | CAR-T / CAAR-T |
| ANCA-associated vasculitis | CD19 | CAR-T |
| Neuromyelitis optica spectrum disorders | CD19, BCMA | CAR-T |
| Autoimmune hemolytic anemia | CD19 | CAR-T |
| PV | DSG3 | CAAR-T |
| IBD / Crohn’s disease | IL-23R, FliC | CAR-Treg |
| Anti-NMDAR encephalitis | NMDAR | CAAR-T |
| T1D | Insulin, GAD65, other β-cell antigens | CAR-Treg |
Target antigens of CAR-based therapies in major autoimmune diseases.
AIHA, autoimmune hemolytic anemia; ANCA, anti-neutrophil cytoplasmic antibody; BCMA, B cell maturation antigen; CAAR-T, chimeric autoantibody receptor T cell; CAR-T, chimeric antigen receptor T cell; CAR-Treg, chimeric antigen receptor regulatory T cell; DSG3, Desmoglein 3; FliC, Flagellin C; GAD65, Glutamic acid decarboxylase 65; IBD, inflammatory bowel disease; IL-23R, Interleukin-23 receptor; MOG, myelin oligodendrocyte glycoprotein; MuSK, muscle-specific kinase; NMDAR, N-methyl-D-aspartate receptor; PV, pemphigus vulgaris; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; T1D, type 1 diabetes; XCR1, X-C motif chemokine receptor 1.
With ongoing innovation in CAR design, their safety, efficacy, and durability are continuously optimized across diverse disease settings. In the context of autoimmune diseases, additional considerations include antigen specificity, minimization of off-target effects on healthy tissues, and fine-tuning of activation thresholds to mitigate immune-mediated tissue damage. To address these challenges, novel CAR architectures—such as inducible CARs (e.g., Tet-On systems) (86), bispecific CARs (87), and switchable CARs (88) are under active investigation. These strategies aim to achieve precise temporal and spatial control of CAR T cell activity, thereby enhancing therapeutic precision and safety.
5.2 CAR T cells targeting CD19
Anti-CD19 CAR-T cell therapy can effectively eliminate tumor cells expressing CD19, resulting in significant clinical efficacy. Most patients experience progression-free survival for more than five years after treatment (89, 90). This success has sparked hope for the potential radical cure of autoimmune diseases (91). Given the diverse functions of T cells, such as tissue penetration, immunomodulatory effects, and cytotoxicity, CAR T-cell therapy holds promise for addressing various pathogenic mechanisms in autoimmune diseases. Therefore, identifying a suitable target antigen is crucial. CD19 is predominantly specific to the B cell lineage and is expressed across various stages of B cell differentiation, including in plasmablasts and a subset of plasma cells (92). Research has shown that although CD19 expression decreases in plasma cells, approximately 72% of CD38+CD138+ normal bone marrow plasma cells still express CD19 (93). However, terminally differentiated plasma cells, known as long-lived plasma cells (LLPCs), no longer express CD19 (94). These LLPCs reside in the bone marrow and are responsible for maintaining long-term antibody responses to previous viral infections and vaccinations. As expected, after anti-CD19 CAR T-cell therapy, most plasma cell compartments remain intact, and antibody titers from prior vaccinations remain stable. This preservation of humoral immunity and protective vaccine titers has been observed in both cancer patients and those with autoimmune diseases (95, 96), highlighting a potential immune-protective advantage of anti-CD19 CAR T-cell therapy. Moreover, anti-CD19 CAR T-cell therapy has demonstrated its efficacy in clearing B cells from tissues, even in the presence of large lymphoma masses (97, 98). In addition to CD19, other antigens such as CD20 and CD22 have also been explored as targets for CAR T-cell therapy in SLE and other autoimmune diseases. However, while the expression of these antigens overlaps with that of CD19, it is lower or absent in plasmablasts and plasma cells (Figure 2). Consequently, due to its broad and deep B cell depletion and its potential for preserving immune protection, anti-CD19 CAR T-cell therapy is emerging as a promising option for the treatment of autoimmune diseases.
Figure 2

In the B-cell lineage, B cells at various developmental stages express distinct antigens on their surface. The expression stages of these markers are represented by colored rectangles. BCMA, B-cell maturation antigen.
In preclinical models of SLE, anti-CD19 CAR T-cell therapy has been shown to induce sustained depletion of CD19+ B cells, clear renal inflammation, and significantly extend the lifespan of model mice. These findings highlight the strong therapeutic and preventive potential of CAR T-cell therapy for SLE (99, 100). These milestone achievements in preclinical models provide robust support for the application of CAR T-cell therapy in autoimmune diseases and pave the way for subsequent clinical research and treatment. In 2021, CD19-directed CAR T cells were used for the first time to treat a 20-year-old woman with severe, treatment-refractory SLE (80). The patient exhibited various manifestations of lupus and responded poorly to multiple immunosuppressive agents, including belimumab and rituximab. Following the infusion of autologous anti-CD19 CAR T cells, rapid B cell depletion was observed, accompanied by a swift expansion of CAR T cells in the peripheral blood. Three months after the infusion, the patient achieved complete clinical remission, including the resolution of proteinuria and seroconversion of double-stranded DNA (dsDNA) autoantibodies. Moreover, all immunosuppressive medications, including glucocorticoids, were successfully discontinued, and there were no signs of disease relapse over the 18-month follow-up period. Importantly, the treatment was well tolerated, with no occurrence of cytokine release syndrome (CRS) or neurotoxicity (80).
Building on this promising case report where anti-CD19 CAR T cells successfully treated a patient with severe SLE, Mackensen et al. subsequently reported the use of anti-CD19 CAR T-cell therapy in five young adults diagnosed with SLE (81). Following CAR T cell infusion, the treated patients exhibited consistent expansion dynamics of CAR T cells, peaking around one-week post-infusion, which was accompanied by a marked reduction in disease activity. According to the DORIS criteria, all five patients achieved remission of SLE within three months, with peripheral B cells depleted to undetectable levels shortly after the infusion. Most pathogenic autoantibodies, including those specific to dsDNA, single-stranded DNA, and nuclear antigen Sm, were reduced to normal levels. The CAR T-cell therapy was well-tolerated, with three of the five patients experiencing low-grade CRS (Grade 1: fever), and no instances of neurotoxicity. Although B cell reconstitution was observed in all five patients, the reappearing B cells were predominantly of the IgM subtype, with a very low number of memory B cells. This suggests that the B cell compartment may have undergone a “reset,” potentially leading to the long-term depletion of autoreactive B cell clones. During subsequent long-term follow-up, no relapse of SLE was observed, even after the patients discontinued all SLE-related medications.
These encouraging research findings have led to a promising concept: if deep cellular depletion of autoreactive B cells can induce remission in SLE, it may also be effective for other autoimmune diseases. Following the success of SLE, clinical research has extended the use of anti-CD19 CAR T-cell therapy to other autoimmune conditions, such as systemic sclerosis (101–103), refractory immune-mediated necrotizing myopathy (103), refractory anti-synthetase syndrome (104, 105), and myasthenia gravis (96). These studies have all demonstrated significant efficacy and good tolerability, with no occurrences of CRS or other severe adverse events. While these results are promising, it is important to note that they are based on small cohorts of patients, and the full range of potential safety concerns will only become evident with extended long-term follow-up. Nonetheless, anti-CD19 CAR T-cell therapy is introducing a new treatment paradigm and is increasingly being considered a potentially curative approach for various autoimmune diseases, bringing new hope to patients (Figure 3).
Figure 3

Anti-CD19 CAR T cell therapy for SLE. Anti-CD19 CAR T cells mediate immune modulation in SLE by targeting and eliminating CD19-expressing B cells. The scFv of CAR T cells specifically recognizes the CD19 antigen on the surface of B cells which, through the intracellular CD3ζ signaling domain and the CD28 co-stimulatory molecule, activates the CAR T cells, leading to the release of perforin, granzyme B, and IFN-γ, which induce B cell death. This mechanism inhibits the production of autoantibodies, thereby disrupting the autoimmune loop in SLE and ultimately achieving therapeutic goals. CAR, chimeric antigen receptor; scFv, single-chain variable fragment; SLE, systemic Lupus Erythematosus.
5.3 Optimizing CAR T-cell therapy strategies by targeting long-lived plasma cells
B cell maturation antigen (BCMA), a member of the tumor necrosis factor superfamily, is predominantly expressed on plasmablasts (106), plasma cells (107), and some memory B cells (107) (Figure 2). These cells are responsible for producing autoantibodies that drive the development and progression of autoimmune diseases (108, 109). Anti-CD19 CAR T-cell therapy has shown promising results in treating autoimmune diseases such as SLE. The rapid decline in anti-dsDNA antibody levels observed in SLE patients undergoing anti-CD19 CAR T-cell therapy suggests that CD19+ plasmablasts and CD19+ plasma cells are the primary sources of these autoantibodies. However, unlike CD19+ cell populations, CD19- LLPCs primarily express BCMA and reside in the bone marrow (92, 110). These cells contribute to the production of autoreactive antibodies, such as anti-dsDNA antibodies (81), resist immunosuppressive treatments, and maintain kidney inflammation in mouse models of SLE (111, 112). Given the relatively short clinical follow-up time after anti-CD19 CAR T cell infusion, the possibility of disease relapse due to CD19- LLPCs evading CD19-targeted CAR T-cell therapy cannot be ruled out. Additionally, the severity of SLE and lupus nephritis has been correlated with increased expression of the BCMA surface antigen on LLPCs, making BCMA a promising therapeutic target (113).
A 5-year clinical outcome study on anti-BCMA CAR T-cell therapy in patients with relapsed/refractory multiple myeloma demonstrated significant efficacy and safety, providing important confidence and a reference point for the potential use of BCMA-targeted CAR T cells in the treatment of autoimmune diseases (114). Recently, BCMA-targeted CAR T cells were used to treat patients with myasthenia gravis, and both patients exhibited good safety profiles and sustained clinical improvement over 18 months, suggesting that the therapeutic efficacy may be linked to the reconstruction of the B-cell lineage accompanied by a sustained reduction in pathogenic autoantibodies (82). Additionally, in another clinical study targeting SLE, a bispecific approach simultaneously targeting BCMA and CD19 was employed. The results demonstrated that this strategy was safe and effective, leading to drug-free remission and the clearance of pathogenic autoantibodies (87). Furthermore, Qin et al. conducted an open-label, single-arm phase 1 clinical trial using anti-BCMA CAR T cells to treat neuromyelitis optica spectrum disorder (NMOSD). In this trial, 12 patients received anti-BCMA CAR T cells infusions, and the results indicated controllable safety and therapeutic potential (115). Moreover, Qin et al. performed a single-cell multi-omics analysis of blood and cerebrospinal fluid (CSF) samples from five NMOSD clinical trial participants treated with anti-BCMA CAR T cells. The findings revealed that anti-BCMA CAR T cells, enriched with chemotaxis gene programs, may be capable of crossing the blood-brain-cerebrospinal fluid barrier, eliminating abnormally expanded plasma cells in the CSF, and reducing the pro-inflammatory environment in the central nervous system in NMOSD (116). This suggests that anti-BCMA CAR T-cell therapy may have a natural advantage in treating autoimmune-related encephalopathies, including lupus encephalopathy.
CD38 is a multifunctional cell surface protein that plays a key role in inflammation and autoimmunity. It acts as an enzyme involved in nicotinamide adenine dinucleotide (NAD) consumption and intracellular signal transduction, while also functioning as an adhesion receptor (117). CD38 expression begins at the early stages of B cell development and increases progressively as B cells mature (118–120). It is highly expressed on plasmablasts, plasma cells, and memory B cells, with particularly elevated levels observed on LLPCs in the bone marrow (94) (Figure 2). The distinct expression pattern of CD38, coupled with its dual role in adhesion and ectoenzyme activity, has spurred the development of targeted CD38 antibodies (121). Daratumumab, the first antibody targeting CD38, has been shown to significantly reduce anti-dsDNA antibodies and vaccine-induced antibodies, lower antinuclear antibody titers, and markedly improve disease symptoms, suggesting its effectiveness in depleting LLPCs (122). However, as seen in the treatment of multiple myeloma, the response to daratumumab is often transient (123). SLE patients receiving daratumumab usually require continued immunosuppression or maintenance therapy with belimumab to prevent the regeneration of autoreactive LLPCs (122). Thus, CD38 represents a valuable target antigen for further development and clinical application.
5.4 Selective elimination of autoreactive B cells through targeting pathogenic autoantibody epitopes
Despite the notable successes of CAR T-cell therapy in treating cancers and autoimmune diseases such as SLE, it is important to recognize that these strategies lead to the depletion of both autoreactive and healthy B cells. Following CAR T-cell infusion, patients experience B-cell depletion lasting weeks to months. During this period, they remain in a highly immunocompromised state. To address the issue of non-selective B cell depletion, therapeutic T cells have been engineered to express pathogenic autoantigen as the extracellular domains of chimeric immunoreceptors. This design allows for enhanced selectivity in targeting autoreactive B cells in autoantibody-mediated diseases with defined autoantigens, while preserving normal B cells (124, 125). These “reverse-engineered” T cells, known as chimeric autoantibody receptor (CAAR) T cells, represent a promising strategy for enhancing the specificity and safety of CAR T-cell therapy in autoimmune diseases (Figure 4).
Figure 4

CAAR T cells are engineered by fusing a fragment of the autoantigen with the CD137 and CD3ζ signaling domains, aiming to specifically eliminate pathogenic B cells in autoantibody-mediated autoimmune diseases. Upon binding of the CAAR to the BCR on the B cell surface, CAAR T cells are activated, subsequently proliferate, persist, and selectively eliminate autoantigen-specific B cells in vivo, without affecting other B cell populations. This highly specific immune regulation provides a novel strategy for precision therapy in autoimmune diseases. CAAR, chimeric autoantibody receptor; Abs, autoantibodies; BCR, B cell receptor; MG, myasthenia gravis; AE, autoimmune encephalitis; PV, pemphigus vulgaris.
Pemphigus vulgaris (PV) is a rare blistering skin disease caused by autoantibodies against desmoglein 1 and 3 (DSG1/3). Rituximab treatment significantly reduces anti-DSG antibodies and improves clinical symptoms. These findings underscore the critical role of B cells, particularly DSG-specific B cells, in PV pathogenesis (126–128). Preclinical studies have demonstrated that CAAR T cells engineered to express DSG3, fused to CD137-CD3ζ signaling domains, are capable of expanding, persisting, and selectively depleting DSG3-specific B cells in vivo (124, 129). These encouraging preclinical findings have guided the design of ongoing open-label clinical trials evaluating DSG3-CAAR T cells in patients with mucosal PV (NCT04422912) (Table 3). In the case of myasthenia gravis, anti-muscle-specific tyrosine kinase (MuSK) autoantibodies disrupt neuromuscular junction signaling, leading to muscle weakness (130–132). Preclinical studies in a myasthenia gravis model with circulating anti-MuSK autoantibodies have shown that MuSK-CAAR T cells can reduce anti-MuSK antibody titers without inducing widespread B cell depletion (133). A clinical study investigating MuSK-CAAR T cells for myasthenia gravis is currently underway and actively recruiting patients (NCT05451212) (Table 3).
Table 3
| Clinical trial number | T cell type | CAR target(s) | Autoimmune disease | Phase | Sponsor |
|---|---|---|---|---|---|
| NCT06038474 | CAR T cells | CD19 | SLE | II | Cartesian Therapeutics |
| NCT06294236 | CAR T cells | CD19 | SLE, lupus nephritis, ANCA-associated vasculitis | I | Sana Biotechnology |
| NCT06465147 | CAR T cells | CD19 | SLE | I | Seattle Children's Hospital |
| NCT05798117 | CAR T cells | CD19 | SLE | I/II | Novartis Pharmaceuticals |
| NCT06121297 | CAR T cells | CD19 | SLE | I/II | Cabaletta Bio |
| NCT05869955 | CAR T cells | CD19 | SLE, idiopathic Inflammatory myopathy, systemic sclerosis | I | Juno Therapeutics, Inc., a Bristol-Myers Squibb Company |
| NCT06342960 | CAR T cells | CD19 | SLE, lupus nephritis | I/II | Kyverna Therapeutics |
| NCT06189157 | CAR T cells | CD19 | SLE | I/II | Miltenyi Biomedicine GmbH |
| NCT06347718 | CAR T cells | CD19 | SLE, systemic sclerosis, dermatomyositis, polymyositis | I/II | University of Erlangen-Nürnberg Medical School |
| NCT03030976 | CAR T cells | CD19 | SLE | I | Shanghai GeneChem Co., Ltd. |
| NCT05988216 | CAR T cells | CD19 | SLE | N/A | Bioray Laboratories |
| NCT06106906 | CAR T cells | CD19 | SLE | I/II | Wuhan Union Hospital, China |
| NCT06373991 | CAR T cells | CD19 | SLE | I | EdiGene Inc. |
| NCT06340490 | CAR T cells | CD19 | SLE | I | Guangdong Ruishun Biotech Co., Ltd |
| NCT06106893 | CAR-γδ T cells | CD19 | SLE | I/II | Wuhan Union Hospital, China |
| NCT06420154 | CAR T cells | CD19 | SLE, sjögren's syndrome, systemic sclerosis, inflammatory myopathy, | I | First Affiliated Hospital of Wenzhou Medical University |
| ANCA-associated vasculitis, antiphospholipid syndrome | |||||
| NCT05859997 | CAR T cells | CD19 | SLE, sjögren's syndrome, systemic sclerosis, inflammatory myopathy, | N/A | Bioray Laboratories |
| ANCA-associated vasculitis, antiphospholipid syndrome | |||||
| NCT06222853 | CAR T cells | CD19 | SLE in children | I | The Children's Hospital of Zhejiang University School of Medicine |
| NCT05765006 | CAR T cells | CD19 | SLE | I | Shanghai Ming Ju Biotechnology Co., Ltd. |
| NCT06310811 | CAR T cells | CD19 | SLE | N/A | Wuhan Union Hospital, China |
| NCT06549296 | CAR T cells | CD19 | SLE, systemic sclerosis, dermatomyositis, polymyositis, ANCA-associated vasculitis, idiopathic inflammatory myopathies, sjögren’s syndrome | Early I | Nanjing Bioheng Biotech Co., Ltd. |
| NCT06361745 | CAR T cells | CD19 | SLE, idiopathic inflammatory myopathies, systemic sclerosis, IgG4 related disease, primary sjögren 's syndrome | N/A | PersonGen BioTherapeutics (Suzhou) Co., Ltd. |
| NCT05930314 | CAR T cells | CD19 | SLE, lupus nephritis, immune thrombocytopenia | Peking Union Medical College Hospital | |
| NCT04146051 | CAR T cells | CD19 | Generalized myasthenia gravis | II | Cartesian Therapeutics |
| NCT06359041 | CAR T cells | CD19 | Generalized myasthenia gravis | I/II | Cabaletta Bio |
| NCT05828225 | CAR T cells | CD19 | Myasthenia gravis | I | Zhejiang University |
| NCT06154252 | CAR T cells | CD19 | Idiopathic inflammatory myopathy, dermatomyositis, anti-synthetase syndrome, immune-mediated necrotizing myopathy | I/II | Cabaletta Bio |
| NCT06231368 | CAR T cells | CD19 | Autoimmune hemolytic anemia | I | Institute of Hematology & Blood Diseases Hospital, China |
| NCT06212154 | CAR T cells | CD19 | Autoimmune hemolytic anemia | I | Institute of Hematology & Blood Diseases Hospital, China |
| NCT06138132 | CAR T cells | CD19 | Multiple sclerosis | I | Stanford University |
| NCT06451159 | CAR T cells | CD19 | Multiple sclerosis | I | Bruce Cree |
| NCT06220201 | CAR T cells | CD19 | Multiple sclerosis | I | Juno Therapeutics, Inc., a Bristol-Myers Squibb Company |
| NCT05938725 | CAR T cells | CD19 | Lupus nephritis | I/II | Kyverna Therapeutics |
| NCT06298019 | CAR T cells | CD19 | Dermatomyositis | I | Stanford University |
| NCT06518876 | CAR T cells | CD19 | POEMS syndrome | I | Novatim Immune Therapeutics (Zhejiang) Co., Ltd. |
| NCT6688799 | CAR T cells | CD19 | Autoimmune diseases | I/II | Beijing GoBroad Hospital |
| NCT06056921 | CAR T cells | CD19 | SLE, sjögren’s syndrome, systemic scleroderma, dermatomyositis, ANCA-associated vasculitis | I | Chongqing Precision Biotech Co., Ltd |
| NCT06513429 | CAR T cells | CD19 | SLE | N/A | Peking University Third Hospital |
| NCT06585514 | CAR T cells | CD19 | SLE, lupus nephritis | I/II | Beijing GoBroad Hospital |
| NCT06193889 | CAR T cells | CD19 | Myasthenia gravis | II | Kyverna Therapeutics |
| NCT06544330 | CAR T cells | CD19 | SLE, lupus nephritis | I | Synthekine |
| NCT06588491 | CAR T cells | CD19 | Stiff-person syndrome | II | Kyverna Therapeutics |
| NCT06475495 | CAR T cells | CD19 | RA | I/II | Charite University, Berlin, Germany |
| NCT06333483 | CAR T cells | CD19 | SLE | I | Autolus Limited |
| NCT04561557 | CAR T cells | BCMA | Neuromyelitis optica spectrum disorder, myasthenia gravis | I | Tongji Hospital |
| NCT06277427 | CAR T cells | BCMA | Lupusnephritis, ANCA-associated vasculitis | N/A | Tongji Hospital |
| NCT06497387 | CAR T cells | BCMA | Lupus nephritis, IgG4-related disease | I | Tongji Hospital |
| NCT06633042 | CAR T cells | BCMA | Refractory AQP4 antibody positive neuromyelitis optica spectrum disease | I | Bioray Laboratories |
| NCT06428188 | CAR T cells | BCMA/CD19 | SLE, sjögren’s syndrome | I/II | Essen Biotech |
| NCT05858684 | CAR T cells | BCMA/CD19 | SLE | Early I | RenJi Hospital |
| NCT06350110 | CAR T cells | BCMA/CD19 | SLE, lupus nephritis, ANCA-associated vasculitis | I/II | Essen Biotech |
| NCT05474885 | CAR T cells | BCMA/CD19 | SLE | I | iCell Gene Therapeutics |
| NCT05846347 | CAR T cells | BCMA/CD19 | SLE | I | Zhejiang University |
| NCT06249438 | CAR T cells | BCMA/CD19 | SLE, immune-mediated necrotizing myopathy, neuromyelitis optica spectrum disorders, multiple sclerosis, myasthenia gravis | I | RenJi Hospital |
| NCT06503224 | CAR T cells | BCMA/CD19 | RA, SLE, primary sjögren’s syndrome, systemic sclerosis | N/A | The First Affiliated Hospital of University of Science and Technology of China |
| NCT06371040 | CAR T cells | BCMA/CD19 | Myasthenia gravis | I | Tang-Du Hospital |
| NCT06485232 | CAR T cells | BCMA/CD19 | Neuromyelitis optica spectrum disorders, generalized myasthenia gravis, multiple sclerosis, chronic inflammatory demyelinating polyradiculoneuropathy | I | Xuanwu Hospital, Beijing |
| NCT06419166 | CAR T cells | BCMA/CD19 | Generalized myasthenia gravis | I | Zhejiang University |
| NCT05263817 | CAR T cells | BCMA/CD19 | POEMS syndrome, amyloidosis, autoimmune hemolytic anemia, vasculitis | I | Zhejiang University |
| NCT05085431 | CAR T cells | BCMA/CD19 | Sjögren's syndrome | I | Zhejiang University |
| NCT05085418 | CAR T cells | BCMA/CD19 | Immune nephritis, lupus nephritis | I | Zhejiang University |
| NCT06497361 | CAR T cells | BCMA/CD19 | Lupus nephritis, IgG4-related disease | I | Tongji Hospital |
| NCT05030779 | CAR T cells | BCMA/CD19 | SLE | Early I | Zhejiang University |
| NCT06285279 | CAR T cells | BCMA/CD19 | Lupus nephritis, ANCA-associated vasculitis, membranous nephropathy-PLA2R induced, IgG4-related diseases | I | Nanjing University School of Medicine |
| NCT06733610 | CAR T cells | BCMA/CD19 | Autoimmune hemolytic anemia | I | Institute of Hematology & Blood Diseases Hospital, China |
| NCT06785519 | CAR T cells | BCMA/CD19 | Lupus nephritis | I | Zhejiang University |
| NCT06787989 | CAR T cells | BCMA/CD19 | Refractory immune cytopenia | I | iCell Gene Therapeutics |
| NCT06340750 | CAR T cells | BAFF | SLE | I | Luminary Therapeutics |
| NCT06279923 | CAR T cells | CD19/BAFF | SLE, systemic sclerosis, dermatomyositis, immune nephritis, neuromyelitis optica | I | Zhejiang University |
| NCT06153095 | CAR T cells | CD19/CD20 | SLE, lupus nephritis | I/II | ImmPACT Bio |
| NCT06462144 | CAR T cells | CD19/CD20 | SLE, ANCA-associated sculitis, idiopathic inflammatory myopathy | I | The Affiliated Nanjing Drum Tower Hospital of Nanjing University Medical School |
| NCT06373081 | CAR T cells | CD19/CD3E | SLE, sjögren’s syndrome, systemic sclerosis, inflammatory myopathy | N/A | Shanghai Changzheng Hospital |
| ANCA-associated vasculitis, antiphospholipid syndrome | |||||
| NCT05239702 | CAR T cells | CD7 | Crohn Disease, ulcerative colitis, dermatomyositis, still Disease | I | Zhejiang University |
| NCT04422912 | CAAR T/CAR T cells | DSG3/CD19 | Pemphigus vulgaris | I | Cabaletta Bio |
| NCT05451212 | CAAR T cells | MuSK | Myasthenia gravis | I | Cabaletta Bio |
| NCT05993611 | CAR Treg cells | CD6 | Chronic graft versus host disease, steroid refractory graft versus host disease | I | City of Hope Medical Center |
| NCT05234190 | CAR Treg cells | HLA-A2 | Liver transplant rejection, liver failure | I/II | Quell Therapeutics Limited |
| NCT04817774 | CAR Treg cells | HLA-A2 | Kidney transplant rejection, end stage renal disease | I/II | Sangamo Therapeutics |
Ongoing clinical trials using different CAR T cells for the treatment of autoimmune diseases.
ANCA, anti-neutrophil cytoplasmic antibody; BAFF, B cell activating factor; BCMA, B cell maturation antigen; CAAR, chimeric autoantibody receptor; CAR, chimeric antigen receptor; N/A, not applicable; POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes; SLE, systemic lupus erythematosus; RA, rheumatoid arthritis.
N-methyl-D-aspartate receptor (NMDAR) encephalitis, the most common autoimmune encephalitis, is mediated by pathogenic autoantibodies targeting the NMDAR. To address this, NMDAR-specific chimeric autoantibody receptor (NMDAR-CAAR) T cells have been engineered to selectively target and eliminate B cells producing anti-NMDAR autoantibodies. Preclinical studies have demonstrated that NMDAR-CAAR T cells can effectively deplete anti-NMDAR B cell lines and maintain reduced autoantibody levels, all without inducing significant non-specific toxicity. These findings provide a foundation for future clinical trials (134). Moreover, in the context of immune thrombocytopenia (ITP), researchers have developed a novel chimeric autoantibody receptor targeting glycoprotein (GP) Ibα (GP Ibα-CAAR T cells). Compared to traditional CAR T-cell therapies, this approach enables the precise elimination of autoreactive B cells without causing broad B cell depletion. The efficacy and safety of GP Ibα-CAAR T cells have been validated in both in vitro and in vivo studies, highlighting their potential as a therapeutic option for patients with refractory or relapsed ITP (135).
5.5 Organ-specific CAR Treg therapy: targeted immune regulation approach
Autoimmune diseases arise from a breakdown in immune tolerance to self-antigens, primarily due to inadequate suppression of aberrant immune responses by Treg cells (136). In recent years, significant efforts have been made to explore the therapeutic potential of Treg cells in treating autoimmune diseases (137, 138). However, thus far, these approaches have not yet achieved optimal therapeutic outcomes, primarily due to their inability to maintain long-term immune tolerance and the lack of clinical approval (139, 140). Interestingly, antigen-specific CAR Treg cells have shown the capacity to home to effector T cells (Teffs) and inflammatory sites in disease-relevant tissues, enabling targeted immune suppression in affected tissues or organs (140–142) (Figure 5). This targeted approach has the potential to reduce systemic immunosuppression, thereby minimizing associated side effects. In contrast to conventional CAR T-cell therapies that eliminate autoreactive cells, CAR Treg modulate immune responses by suppressing (but not killing) pathogenic lymphocytes, thereby promoting long-term tolerance (143, 144). Additionally, CAR Treg therapy offers another distinct advantage: stable Treg cells do not produce pro-inflammatory cytokines, which significantly lowers the risk of CRS typically associated with CAR therapies (145, 146).
Figure 5
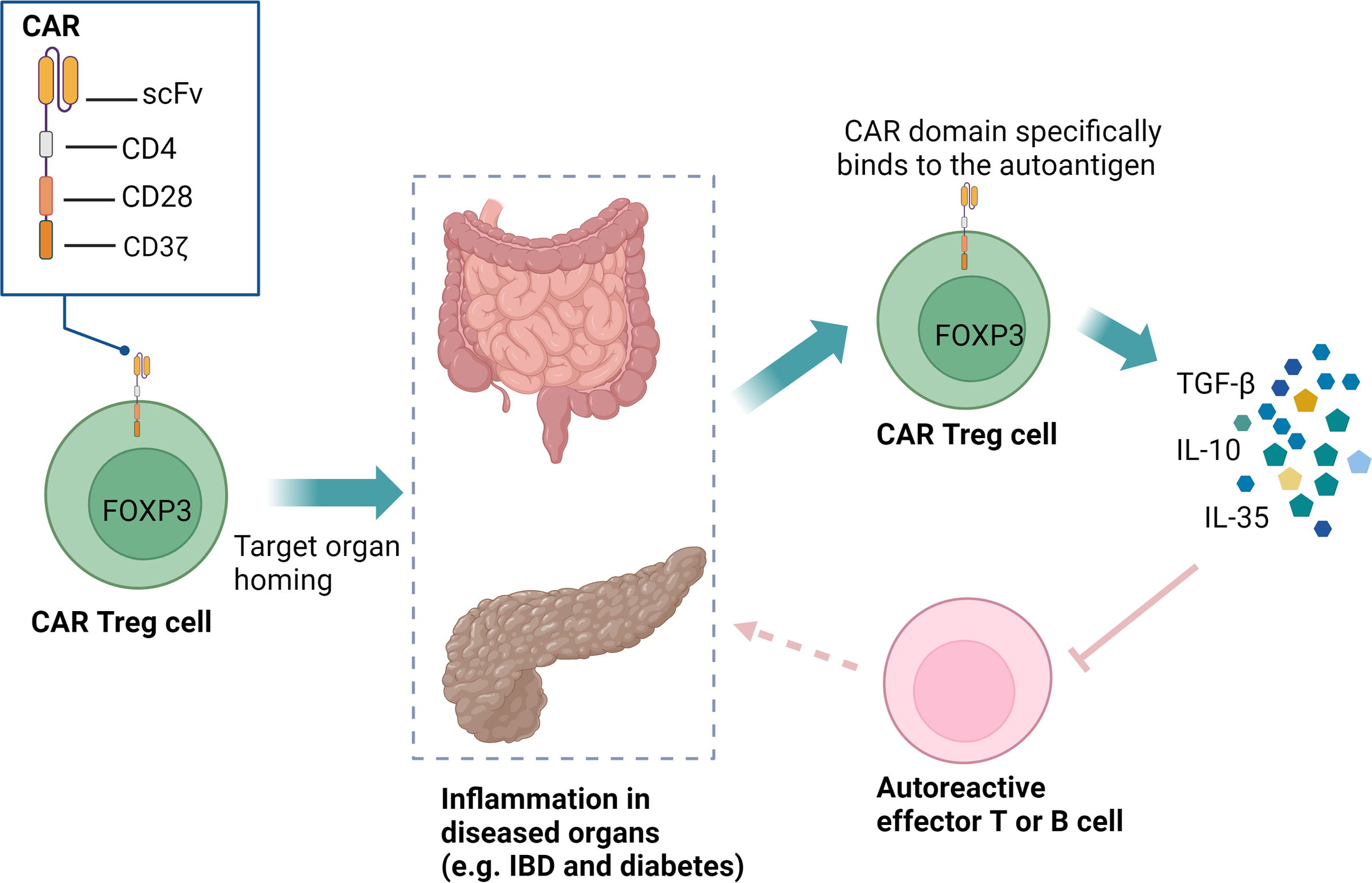
CAR Treg cells can specifically recognize organ-specific autoantigens and home to inflamed microenvironments. These CAR Treg cells, equipped with CD28 and CD3ζ signaling domains, can expand in vivo, persist long-term, and exert immunoregulatory functions through the secretion of TGF-β, IL-10, and IL-35. This precise targeting facilitates localized immunotherapy, effectively reducing the need for systemic immunosuppression and minimizing associated side effects. CAR, chimeric antigen receptor; Treg, regulatory T cells; IBD, inflammatory bowel disease; scFv, single-chain variable fragment; FOXP3, forkhead box P3.
CAR Treg cells have the potential to selectively target disease-associated tissue antigens affected by self-reactive immune responses, thereby enabling localized and organ-specific therapeutic strategies. This makes them highly promising for treating organ-specific autoimmune diseases such as inflammatory bowel disease (IBD) (147), multiple sclerosis (148), and T1D (149). Furthermore, CAR Treg cells are being explored as a strategy to induce immune tolerance and prevent organ rejection in transplantation (150, 151). A novel CAR targeting the flagellin protein derived from Escherichia coli H18 (FliC) has been developed. FliC-CAR Treg cells can recognize and respond to soluble flagellin independently of toll-like receptor 5, promoting intestinal homing in a humanized mouse model and exhibiting potent immunosuppressive effects. These findings support the clinical development of CAR Treg cells for IBD treatment and reveal the potential of CARs targeting microbial antigens (152). A survey by Vent-Schmidt et al. showed that most IBD patients would be willing to try CAR Treg therapy, highlighting its clinical promise (153). In a preclinical study on the treatment of multiple sclerosis, the transfer of X-C motif chemokine receptor 1 (XCR1)-specific CAR Treg cells into EAE, an animal model of multiple sclerosis, successfully inhibited T helper 1 cells (Th1)-driven EAE (154). Tenspolde and colleagues used CAR technology to redirect T cell specificity to insulin and reprogram Teffs into Treg cells through Forkhead box P3 (FOXP3) transduction. The data indicated that these insulin-specific CAR Treg cells were functionally stable, suppressive, and long-lasting in vivo, demonstrating the potential for restoring immune tolerance in T1D (149). In transplant medicine, the primary goal is to establish immune tolerance to prevent organ rejection without lifelong pharmacological immunosuppression. CAR technology has been used to direct human Treg cells toward specific HLA class I molecules. Drawing upon previous studies (155–158), Proics and colleagues further developed a lentiviral vector encoding a humanized single-chain variable fragment (scFv) specific to the HLA-A02 antigen (TX200) for clinical applications and optimized the isolation strategy for autologous initial (CD45RA+) human Treg cells (TR101). The resulting HLA-A02 CAR Treg cells were named TX200-TR101. A first-in-human trial is currently planned, marking the first clinical trial investigating CAR Treg cells (151).
5.6 mRNA-based CAR T-cell therapy: a novel treatment with transient in vivo expression and rapid therapeutic effects
The B cell burden in autoimmune diseases is generally much lower than in cancer patients, allowing CAR T cells to quickly eliminate cells carrying the target antigen in vivo. The loss of the target antigen may trigger early contraction of the CAR T cell population. Deep B cell depletion is sufficient to eliminate disease activity and reset the B cell system (124, 159). The long-term persistence of functional CAR T cells in autoimmune diseases seems to have limited value. Prolonged B cell depletion is not beneficial in autoimmune diseases because B cells contribute to humoral protection and are involved in immune homeostasis (160). Patients with permanent B cell depletion exhibit an increased risk of infections and poorer outcomes, such as in the case of COVID-19 infection (161). Traditional CAR T cells use lentiviral or gamma-retroviral vectors for permanent genetic modification, which poses risks of genotoxicity and regulatory challenges, and may also result in the lifelong persistence of CAR T cells (162, 163).
Currently, mRNA CAR T-cell therapy demonstrates tremendous potential in overcoming the limitations of traditional CAR T treatments. This therapy combines mRNA gene modification technology with CAR T immunotherapy, achieving cell transformation through in vivo targeting and in situ reprogramming of T cells, thus avoiding the complex and time-consuming ex vivo manufacturing process typical of traditional CAR T-cell therapy. By utilizing lipid nanoparticles (LNPs) as efficient delivery vectors, mRNA CAR T cells can precisely deliver mRNA encoding the CAR to T cells, enabling them to rapidly synthesize functional CAR molecules and initiate targeted immune responses (164). However, due to the transient nature of mRNA, CAR expression and immune activity gradually decline within days as the mRNA degrades (165). Thanks to its transient expression mechanism and high flexibility, mRNA CAR T-cell therapy offers the unique advantage of repeatable administration based on treatment needs (166, 167). By precisely adjusting mRNA dosage and administration frequency, the intensity and duration of the treatment can be dynamically controlled to better adapt to changes in the patient’s condition, such as disease relapse or heterogeneity of target antigen expression. This ability for repeated dosing provides significant flexibility for personalized treatment and long-term management. Compared to traditional CAR T-cell therapy, mRNA CAR T-cell therapy effectively reduces long-term toxicity risks due to its transient expression characteristics. The non-persistent expression avoids the issue of CAR genes remaining in T cells for prolonged periods, thereby reducing the occurrence of side effects such as CRS, neurotoxicity, and organ damage. The transient CAR expression may also reduce the risk of antigen escape or tumor resistance. More importantly, because mRNA does not integrate into the T cell genome, its non-integrating nature significantly reduces the potential risk of tumorigenesis. Additionally, the high adjustability of mRNA CAR T-cell therapy makes the treatment regimen more flexible, particularly for high-risk patients or those who need to avoid long-term immunosuppression (168).
In mouse models, injection of CD3-targeted LNPs carrying anti-CD19 CAR mRNA successfully induced CAR expression in T cells. These nanoparticles transduced CAR T cells, which significantly enhanced therapeutic efficacy against B-cell leukemia, as evidenced by a marked reduction in tumor burden and a notable extension of survival in the mice (169). Rurik et al. showed that T cell-targeting LNPs delivered mRNA encoding a CAR against activated fibroblasts in mice, generating functional CAR T cells. In a mouse model of heart failure, a single injection of CD5-targeted LNPs efficiently delivered the modified mRNA to T cells, producing transient yet effective CAR T cells. These CAR T cells accumulated in the spleen, exhibited phagocytic activity, maintained target antigen recognition, and ultimately improved cardiac function while attenuating fibrosis (170). These studies suggest that mRNA CAR T technology, with its transient and controllable immune cell functionality, shows unique potential in modulating immune balance and suppressing aberrant immune responses, indicating its promising applications in the treatment of autoimmune diseases. The first clinical trial of mRNA CAR T-cell therapy in autoimmune diseases targeted myasthenia gravis. The MG-001 clinical trial is a prospective, multi-center, open-label phase 1b/2a study designed to evaluate the efficacy and safety of anti-BCMA mRNA CAR T-cell therapy in treating myasthenia gravis. The trial treated a total of 14 patients using electroporated mRNA-based CAR T cells without lymphocyte-depleting chemotherapy. The follow-up period ranged from 3 to 9 months. The results indicated that the treatment was safe, well-tolerated, and significantly reduced the clinical severity of myasthenia gravis, highlighting the clinical potential of mRNA CAR T-cell therapy for treating myasthenia gravis and other autoimmune diseases (171).
6 CAR T-cell therapy toxicities: CRS, neurotoxicity, and management strategies
While CAR T-cell therapy shows great therapeutic promise, it is also associated with serious toxicities, particularly CRS and immune effector cell-associated neurotoxicity syndrome (ICANS), which have become major safety challenges in clinical applications. CRS is the most common adverse event following CAR T cell infusion. It is triggered by the robust activation of CAR T cells and other immune effector cells, leading to a massive release of inflammatory cytokines such as IL-6, IFN-γ, and TNF-α (172). Clinical manifestations range from fever, hypotension, and hypoxia to severe multi-organ dysfunction. The American Society for Transplantation and Cellular Therapy (ASTCT) has proposed a grading system to standardize the assessment of CRS. The risk and severity of CRS are influenced by tumor burden, CAR construct design, and T cell dose. ICANS is another severe toxicity, which often occurs after or concurrently with CRS but can also develop independently. Its pathogenesis involves cytokine-mediated endothelial activation, increased blood-brain barrier permeability, and neuroinflammation (172). Patients may present with confusion, language impairment, seizures, or even cerebral edema. A hallmark of ICANS is the rapid onset of neurological symptoms, typically within one week after infusion.
Currently, tocilizumab, an IL-6 receptor antagonist, is the first-line treatment for CRS and can rapidly alleviate symptoms without compromising the efficacy of CAR T cells. Glucocorticoids are used for moderate to severe CRS and for ICANS, particularly when tocilizumab proves ineffective. Due to its limited ability to cross the blood-brain barrier, tocilizumab is less effective for ICANS, making glucocorticoids the mainstay of treatment. Current preventive strategies primarily include preemptive IL-6 blockade, individualized adjustment of CAR T cell dosage, and the incorporation of “safety switches” into the CAR construct, such as suicide genes that allow for the elimination of CAR T cells when necessary (173). In addition, to reduce the risk of prolonged immune activation, transient CAR expression systems using mRNA transfection or externally controllable CAR designs represent promising directions for therapeutic optimization (174).
As CAR T-cell therapy is progressively applied to autoimmune diseases, patients in this population often present with long-standing immune dysregulation and chronic inflammatory backgrounds, which may render them more susceptible to CAR T-related toxicities. Therefore, establishing a comprehensive toxicity management system is of critical importance. The integration of risk stratification and early intervention into a refined management framework holds promise for enhancing the overall safety of CAR T-cell therapy while maintaining its therapeutic efficacy.
7 Risk stratification and management for CAR T-cell therapy
For autoimmune diseases treated with CAR T-cell therapy, risk stratification—based on disease type, severity, immune status, and clinical characteristics—enables personalized treatment optimization to maximize efficacy. Since CAR T-cell therapy may trigger CRS and immune effector cell-associated neurotoxicity syndrome, risk stratification would help identify high-risk patients in advance and implement preventive measures to reduce adverse effects (175, 176). Furthermore, stratified management optimizes resource allocation by prioritizing patients with the highest predicted benefit, improving therapeutic success rates while enhancing safety through personalized risk mitigation (177).
For stratified management of CAR T-cell therapy in autoimmune diseases, patients can be categorized into B cell-mediated and T cell-mediated subtypes based on disease pathogenesis. This stratification approach enables precise treatment strategies tailored to distinct autoimmune subtypes, potentially enhancing efficacy while minimizing adverse effects. B-cell-mediated diseases, such as SLE and RA, are primarily driven by autoantibody production from B cells. Anti-CD19 CAR T cells can target and eliminate CD19-positive B cells, thereby reducing autoantibody production and inhibiting disease progression. In SLE patients, anti-CD19 CAR T-cell therapy induces profound B cell depletion, leading to reduced autoantibody titers and subsequent mitigation of tissue damage (81). Furthermore, CAAR T cells are specifically engineered to selectively target pathogenic B cell subsets while preserving the function of normal B cells, thereby reducing the side effects associated with long-term immunosuppressive drugs (124). For T cell-mediated diseases, such as T1D and multiple sclerosis, the pathology is mainly driven by autoreactive T cells. CAR Treg cells promote immune tolerance by suppressing autoreactive T cell activity, thereby attenuating inflammation and halting tissue damage (141).
In CAR T-cell therapy for autoimmune diseases, stratified management based on disease severity might help optimize treatment outcomes and reduce risks. For mild to moderate patients, low-dose CAR T-cell therapy is typically considered only when conventional immunomodulatory strategies prove ineffective or cause significant side effects. This approach could allow for gradual adjustment of the immune response, preventing overt immunosuppression (178). However, for severe or refractory patients who are unresponsive to standard therapies, CAR T-cell therapy might be used as a last resort to control systemic inflammation and organ damage by eliminating pathogenic immune cells. This treatment carries a higher risk of side effects, necessitating strict monitoring to prevent complications such as CRS (179, 180).
In CAR T-cell therapy for autoimmune diseases, individualized stratified management based on patient age and immune status would be crucial. Younger patients are typically able to tolerate higher doses of CAR T-cell therapy, which might result in more significant outcomes. In contrast, elderly patients, facing challenges such as immune senescence and comorbidities, would require lower doses of CAR T cells and strict monitoring for adverse effects to enhance safety (181, 182). Patients with normal immune function would be suited for standard treatment protocols, whereas those with impaired immune function, such as those on long-term immunosuppressive therapy or with immunodeficiency, might need tailored treatment adjustments to optimize efficacy and minimize side effects (183). All patients require continuous monitoring and adjustment of therapeutic strategies to ensure both safety and efficacy.
8 Preparatory conditioning chemotherapy regimens
The conditioning chemotherapy is a critical element in CAR T-cell therapy, designed to maximize the therapeutic potential of CAR T cells by promoting their proliferation, persistence, and efficacy within the patient’s body. It functions by depleting immunosuppressive Treg cells and myeloid-derived suppressor cells, as well as increasing the availability of key serum cytokines, such as IL-15 and IL-7, which further enhance CAR T cells activity and durability (177, 184). Additionally, conditioning chemotherapy reduces the existing lymphocytes in the patient’s body, creating a “space” for the infused CAR T cells to better proliferate and survive (185). In certain cases, it can also contribute to a reduction in the tumor burden, thereby enhancing the antitumor activity of CAR T cells (186).
The conditioning chemotherapy regimens currently used for autoimmune diseases are primarily adapted from those established for cancer treatment. In CAR T-cell therapy for cancer, common regimens include cyclophosphamide monotherapy, a combination of cyclophosphamide with fludarabine, or bendamustine monotherapy. Cyclophosphamide is generally reserved for specific cases or for patients who cannot tolerate more intensive regimens, and it is infrequently used as monotherapy (177). The cyclophosphamide-fludarabine combination chemotherapy regimen has been widely used in multiple clinical trials and has become the standard conditioning regimen for CAR T-cell therapy. The combined use of cyclophosphamide and fludarabine is believed to more effectively reduce immunosuppressive cells, thereby aiding in the expansion and long-term survival of CAR T cells. Compared to cyclophosphamide alone, the cyclophosphamide-fludarabine combination regimen has been shown to improve CAR T cells expansion and clinical outcomes (187, 188). Bendamustine, as an alternative option, is occasionally employed in patients who cannot tolerate the cyclophosphamide-fludarabine combination. Studies suggest that bendamustine can achieve comparable CAR T cells expansion but may reduce adverse effects, such as neutropenia, though its use is less common (189). In the context of autoimmune diseases, conditioning chemotherapy regimens for CAR T-cell therapy are often derived from those used in cancer but are adjusted in terms of drug selection and dosing. The cyclophosphamide-fludarabine combination regimen has shown promise in autoimmune disease patients, such as SLE, where it effectively reduces disease-driving autoreactive B cells and facilitates an immune system “reset” (81, 124). However, bendamustine is generally not recommended for use in autoimmune disease patients due to its potential for high toxicity, severe immunosuppression, and the lack of safety and efficacy data in this population (172, 175). Therefore, the cyclophosphamide-fludarabine combination is considered a safer and more effective conditioning regimen for autoimmune diseases and is more commonly recommended in clinical practice.
The conditioning regimen for CAR Treg therapy in autoimmune diseases differs from that of conventional CAR T-cell therapy, as CAR Treg cells are designed to enhance immune suppression to prevent autoimmune responses, rather than to eliminate B cells. Therefore, the selection of the conditioning regimen must be carefully considered to avoid impairing Treg cells’ function. Kanakry et al. showed that posttransplantation cyclophosphamide selectively preserved and promoted the reconstitution of Treg cells. This effect is primarily attributable to the high expression of aldehyde dehydrogenase (ALDH) in Treg cells, which metabolizes acrolein—a toxic metabolite of cyclophosphamide—thereby conferring resistance to cyclophosphamide in Treg cells (190). This characteristic suggests cyclophosphamide as an ideal preconditioning regimen for CAR-Treg therapy. Nevertheless, in autoimmune disease patients—many of whom are women of reproductive age—cyclophosphamide presents a concern due to its potential impact on fertility, necessitating careful evaluation (191, 192).
Bendamustine, due to its potent immunosuppressive effects, may impede the expansion and persistence of CAR Treg cells, potentially compromising therapeutic efficacy. Consequently, its application in CAR Treg therapy is generally limited (175). However, the necessity of pre-treatment chemotherapy for CAR Treg therapy in autoimmune diseases remains a topic of debate. Some studies suggest that pre-treatment chemotherapy may enhance the expansion and persistence of CAR Treg cells, while others raise concerns about introducing unnecessary toxicity and risk. As a result, pre-treatment chemotherapy is not universally required for all CAR Treg therapies, and decisions regarding its use should be made with caution, tailored to individual patient needs (193, 194).
9 Outlook: potential breakthroughs and future challenges of CAR T-cell therapy
The traditional CAR T-cell therapy process is complex and highly personalized, requiring the collection, genetic modification, and expansion of T cells from the patient, which results in long treatment cycles and may be accompanied by disease progression. In patients with autoimmune diseases undergoing immunosuppressive therapy, T-cell function is often suppressed, limiting the collection and expansion of autologous CAR T cells, thereby affecting the efficiency or yield of the final product, with a manufacturing failure rate of 2-10% (195). Moreover, the time-consuming processes of manufacturing, testing, and release, along with the logistical challenges of transporting cells between the treatment site and production facilities, pose significant risks, especially for patients with rapidly progressing or severe conditions. A pressing concern is that two major challenges currently hinder the clinical translation and large-scale trials of CAR T-cell therapy in autoimmune diseases: limited accessibility and high treatment costs. On the one hand, the therapy heavily relies on individualized cell manufacturing, complex genetic engineering platforms, and advanced cell therapy centers, making it difficult to implement routinely in most healthcare institutions. On the other hand, the high costs associated with production, quality control, and regulatory management result in overall treatment expenses far exceeding those of conventional therapies, imposing a significant burden on patients and healthcare systems. Therefore, reducing costs and improving accessibility have become critical bottlenecks that must be addressed to advance the broader application of CAR T-cell therapy. Therefore, the development of allogeneic CAR T technology offers a potential solution to overcome the limitations of traditional CAR T-cell therapies.
Allogeneic CAR T cells, also known as “off-the-shelf” CAR T cells (196, 197), are generated by genetically modifying T cells from healthy donors, enabling rapid therapeutic availability without the need to collect, process, and expand the patient’s own cells. Compared to the complex and individualized manufacturing process of autologous CAR T cells, allogeneic CAR T cells streamline the introduction of multiple cell modifications and achieve product standardization through donor selection and processing. This production approach significantly reduces costs through industrialization and large-scale manufacturing, allowing the generation of a substantial number of CAR T cells from a single donor. Furthermore, the production method for allogeneic CAR T cells enables the batch cryopreservation of T cells, ensuring treatments are readily available for patients. This approach not only saves resources and time but is particularly critical for patients with severe and acute conditions, as it meets their urgent need for timely therapeutic interventions (196). Additionally, the batch manufacturing of allogeneic CAR T cells facilitates the reuse of products when needed, providing a convenient option for retreatment. Most patients undergoing CAR T-cell therapy have been on long-term immunosuppressive treatments (such as corticosteroids, cyclophosphamide, tacrolimus, and rituximab) to control their condition, which can severely impair T cell quantity and function. As a result, the collection and expansion of autologous CAR T cells may be restricted, leading to the production of insufficient quantities or quality of autologous CAR T cells for treatment (196, 198). In such cases, using T cells from healthy donors to produce allogeneic CAR T cells becomes an ideal alternative. This approach not only provides a sufficient quantity of functionally robust T cells but also avoids the issue of suboptimal treatment outcomes due to the patient’s own T cells being functionally deficient or insufficient in number (199, 200). Compared to the personalized process of producing autologous CAR T cells, the production of allogeneic CAR T cells allows for scaling and standardization, significantly reducing costs, saving resources and time, and enabling coverage of a broader patient population (196). Moreover, allogeneic CAR T cells sourced from healthy donors can reduce the number of autoreactive cells in the patient’s body, thereby lowering the risk of relapse. For patients with autoimmune diseases, using allogeneic CAR T cells may also reduce treatment-related side effects, such as CRS (200, 201).
Although CAR T-cell therapy has demonstrated remarkable efficacy in the treatment of tumors and autoimmune diseases, it still faces challenges such as off-target effects, including non-specific attacks on healthy cells (175, 202). For instance, while eliminating pathogenic immune cells, CAR T cells may inadvertently attack normal immune cells, disrupting immune homeostasis or causing adverse effects such as CRS and organ damage. Off-target effects can also damage normal cells, reducing CAR T cell functionality and viability, thereby compromising therapeutic efficacy and increasing risks. The CRISPR/Cas9 gene-editing technology holds promise for enhancing the safety and efficacy of CAR T-cell therapy by mitigating off-target effects and non-specific actions.
Studies have shown (203–205) that directly delivering pre-assembled Cas9/gRNA ribonucleoprotein (RNP) complexes into cells allows Cas9 to rapidly exhibit activity. The rapid degradation of RNP significantly shortens Cas9’s duration of action, effectively reducing the probability of off-target cleavage while maintaining efficient and precise target editing. The combination of CRISPR/Cas9 gene-editing technology with RNP delivery strategy enables precise genome editing in CAR-T cells, thereby enhancing the specificity of target antigen recognition and reducing the risk of non-target antigen misrecognition that may lead to reduced efficacy or disease relapse. Additionally, this strategy facilitates multiplex gene editing and significantly reduces the risk of CRISPR off-target effects due to the transient activity characteristic of RNP delivery (206). In vitro experiments demonstrated that knocking out the SHP-1 gene in CD133-targeted CAR T cells significantly enhanced their cytolytic efficacy against CD133-positive glioblastoma cells. No Cas9 insertion or notable off-target effects were observed, confirming the potential of CRISPR/Cas9 technology to improve CAR T cell precision and reduce off-target effects (207). In a phase I open-label study targeting B-cell acute lymphoblastic leukemia, CRISPR/Cas9 technology was used to construct universal CD19/CD22 dual-targeted CAR T cells. The study enrolled six patients and employed high-fidelity CRISPR/Cas9 editing to knock out the TRAC and CD52 genes. Results demonstrated that the technology achieved high precision without observable off-target effects, while the treatment exhibited favorable safety and significant efficacy (208). By enhancing the specificity, persistence, and functional potency of CAR T cells, CRISPR/Cas9 gene-editing technology is expected to provide critical scientific evidence and technical support for the development of safe and effective CAR T-cell therapies for autoimmune diseases.
CAR T-cell therapy for autoimmune diseases is still in the early stages of clinical trials. While the results are promising, they are limited to a small number of patients, and long-term follow-up will reveal all potential safety issues. It is important to emphasize that these results are preliminary and require further investigation with longer durations and larger patient populations. A report by Mackensen et al. (81) on the use of anti-CD19 CAR T cells in 5 patients with refractory SLE showed that the patients tolerated the treatment well, with only mild CRS. Immature B cells reappeared on average 110 ± 32 days after CAR T cell infusion, and during a longer follow-up period (median (range) 8 (12) months post-CAR T cell administration), the patients remained in drug-free remission. Although this study report demonstrates that CAR T cells have the potential for long-term remission and good tolerability, it is important to consider that SLE patients may accumulate autoantibodies for several years before showing clinical symptoms. Therefore, even in cases of residual or re-emerging potential immune dysregulation, there could still be persistent clinical remission (209). Current studies have relatively short follow-up periods (typically ranging from a few months to about a year) and involve a small number of cases. To better evaluate the long-term effects and safety of CAR T-cell therapy, extended follow-up (five to ten years) is necessary to observe long-term remission rates, disease relapse rates, and potential delayed side effects. Special attention should be given to the chronic toxicity that CAR T-cell therapy may cause, including damage to internal organs, chronic inflammation, or fibrosis (175, 180). CAR T-cell therapy for autoimmune diseases requires further large-scale clinical studies to validate these preliminary results. Besides, long-term monitoring of patients’ immune system status is essential, with particular emphasis on the quantity, types, and functionality of regenerated B cells, as well as changes in T cell subsets, to assess the duration and quality of immune system reconstitution.
In autoimmune diseases, persistent inflammatory responses are compounded by pathological processes including autoimmune attacks, immune complex deposition, vasculitis, and tissue fibrosis, all of which can cause irreversible organ damage (210, 211). Even successful CAR T-cell therapy may fail to reverse established organ damage in autoimmune diseases, as demonstrated by SLE-related glomerulosclerosis, pulmonary fibrosis in systemic sclerosis, exocrine gland fibrosis in Sjögren’s syndrome, and demyelinating lesions in multiple sclerosis. This limitation highlights the critical importance of early intervention during the reversible disease phase. Initiating CAR T-cell therapy before irreversible structural changes occur can significantly improve treatment response rates and preserve long-term organ function.
Combined CAR T-cell therapy is emerging as an important approach for treating autoimmune diseases. In recent years, clinical research has explored the combined use of CAR T cells targeting different antigens, such as CD19 and BCMA, aiming to enhance therapeutic efficacy and overcome the limitations of single-target therapies. This strategy not only effectively eliminates various types of pathogenic cells but also has the potential to improve durability and breadth of treatment through multiple mechanisms. Preliminary clinical data demonstrate that BCMA-CD19 compound CAR T-cell therapy is safe and effective in patients with SLE (87). It not only induces medication-free remission but also significantly reduces pathogenic autoantibodies, showing potential for reducing relapse risk and achieving immune reconstitution. Additionally, combining CAR T with CAAR T offers new possibilities for future research. Anti-BCMA CAR T-cell therapy targets and eliminates pathogenic plasma cells, while CAAR T precisely target abnormal B cells responsible for autoimmune reactions. Their combination may generate synergistic effects, comprehensively clearing pathogenic cells and restoring B cell homeostasis. Although combination therapies show great promise, current research is still in its early stages. Future studies should focus on evaluating their safety, tolerability, and efficacy, as well as addressing potential immune responses and side effects induced by treatment. Overall, combined CAR T-cell therapy offer a novel therapeutic approach for autoimmune diseases, warranting further exploration and clinical validation.
It is noteworthy that most current CAR T-cell therapies primarily target B cells, particularly CD19+ B cells and their pathogenic plasma cell derivatives. However, the pathogenesis of many autoimmune diseases is not solely B-cell driven but heavily dependent on T cells—especially CD4+ helper T cells reactive to the same autoantigens. The generation of long-lived plasma cells typically requires T-cell help, and the autoantibodies they secrete are antigen-driven. Therefore, even if pathogenic B-cell populations are eliminated, residual autoreactive CD4+ T cells may reinitiate the activation of newly regenerated B cells, leading to the re-emergence of pathogenic autoantibodies and increasing the risk of disease relapse. To address this concern, future CAR T-cell strategies may benefit from incorporating approaches that also target autoreactive T cells—such as the development of CAR T cells specifically directed against pathogenic T-cell subsets, or combinatorial strategies involving CAR-Tregs to promote immune tolerance. Furthermore, longitudinal monitoring of CD4+ T-cell subsets and their antigen specificity following therapy will be essential to assess whether true immune reprogramming has been achieved. Therefore, optimizing CAR T-cell therapies for autoimmune diseases will require advances in target precision, understanding of autoantigen-driven responses, and elimination of immunological memory to improve long-term efficacy and minimize relapse risk.
To ensure the standardization and normalization of the treatment process, and to enhance patient safety and treatment efficacy, it is essential to establish global consensus guidelines for CAR T-cell therapy in autoimmune diseases. Given the complexity of CAR T-cell therapy for autoimmune conditions, which involves extensive multidisciplinary knowledge, detailed guidelines are needed to direct clinical practice. This will help standardize treatment procedures and reduce variability. Moreover, it will provide clear standards for treatment and side-effect management, ensuring patients receive optimal care while minimizing treatment-related risks. By developing consensus guidelines, multidisciplinary collaboration can be promoted, evaluation standards for treatment efficacy and side effects can be unified, and treatment strategies can be optimized, thereby improving overall safety and effectiveness. Furthermore, such guidelines can advance scientific research, help formulate more scientific and forward-looking treatment protocols, and enhance patient and public understanding and acceptance of the treatment options.
In conclusion, CAR T-cell therapy represents a novel approach to the treatment of autoimmune diseases, offering the potential for durable immune reconstitution and even a cure by specifically eliminating autoreactive B cells. Its remarkable efficacy has been demonstrated in refractory autoimmune diseases, such as SLE. Future innovations, including mRNA-engineered CAR T, “off-the-shelf” allogeneic CAR T-cell therapies, and combination CAR T-cell therapy are expected to enhance therapeutic outcomes, reduce toxicity, and lower costs. Additionally, risk stratification strategies will optimize personalized treatment, minimize adverse effects, and improve clinical safety and efficacy. Together, these advances position CAR T-cell therapy as a transformative frontier in autoimmune disease management, with the potential to achieve sustained remission and cures.
Statements
Author contributions
DW: Writing – original draft, Writing – review & editing. ZYXM: Visualization, Writing – review & editing. JZ: Data curation, Writing – original draft. KY: Data curation, Formal Analysis, Writing – review & editing. XW: Formal Analysis, Investigation, Writing – review & editing. YF: Data curation, Formal Analysis, Writing – review & editing. KHY: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Damoiseaux J Andrade LE Fritzler MJ Shoenfeld Y . Autoantibodies 2015: From diagnostic biomarkers toward prediction, prognosis and prevention. Autoimmun Rev. (2015) 14:555–63. doi: 10.1016/j.autrev.2015.01.017
2
Wang L Wang FS Gershwin ME . Human autoimmune diseases: a comprehensive update. J Internal Med. (2015) 278:369–95. doi: 10.1111/joim.12395
3
Fugger L Jensen LT Rossjohn J . Challenges, progress, and prospects of developing therapies to treat autoimmune diseases. Cell. (2020) 181:63–80. doi: 10.1016/j.cell.2020.03.007
4
Kivity S Arango MT Ehrenfeld M Tehori O Shoenfeld Y Anaya JM et al . Infection and autoimmunity in Sjogren’s syndrome: a clinical study and comprehensive review. J Autoimm. (2014) 51:17–22. doi: 10.1016/j.jaut.2014.02.008
5
Peng J Narasimhan S Marchesi JR Benson A Wong FS Wen L . Long term effect of gut microbiota transfer on diabetes development. J Autoimm. (2014) 53:85–94. doi: 10.1016/j.jaut.2014.03.005
6
Sollid LM Pos W Wucherpfennig KW . Molecular mechanisms for contribution of MHC molecules to autoimmune diseases. Curr Opin Immunol. (2014) 31:24–30. doi: 10.1016/j.coi.2014.08.005
7
Invernizzi P Benedetti MD Poli S Monaco S . Azathioprine in multiple sclerosis. Mini Reviews Medicinal Chem. (2008) 8:919–26. doi: 10.2174/138955708785132756
8
Flammer JR Rogatsky I . Minireview: Glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol. (2011) 25:1075–86. doi: 10.1210/me.2011-0068
9
Hashkes PJ Becker ML Cabral DA Laxer RM Paller AS Rabinovich CE et al . Methotrexate: new uses for an old drug. J Pediatr. (2014) 164:231–6. doi: 10.1016/j.jpeds.2013.10.029
10
June CH Sadelain M . Chimeric antigen receptor therapy. New Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
11
Gupta A Gill S . CAR-T cell persistence in the treatment of leukemia and lymphoma. Leuk Lymphoma. (2021) 62:2587–99. doi: 10.1080/10428194.2021.1913146
12
Manier S Ingegnere T Escure G Prodhomme C Nudel M Mitra S et al . Current state and next-generation CAR-T cells in multiple myeloma. Blood Rev. (2022) 54:21. doi: 10.1016/j.blre.2022.100929
13
Young RM Engel NW Uslu U Wellhausen N June CH . Next-generation CAR T-cell therapies. Cancer Discov. (2022) 12:1625–33. doi: 10.1158/2159-8290.CD-21-1683
14
Melenhorst JJ Chen GM Wang M Porter DL Chen C Collins MA et al . Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. (2022) 602:503–9. doi: 10.1038/s41586-021-04390-6
15
Aghajanian H Rurik JG Epstein JA . CAR-based therapies: opportunities for immuno-medicine beyond cancer. Nat Metab. (2022) 4:163–9. doi: 10.1038/s42255-022-00537-5
16
Baker DJ June CH . CAR T therapy extends its reach to autoimmune diseases. Cell. (2022) 185:4471–3. doi: 10.1016/j.cell.2022.10.026
17
Li YR Lyu Z Chen Y Fang Y Yang L . Frontiers in CAR-T cell therapy for autoimmune diseases. Trends Pharmacol Sci. (2024) 45:839–57. doi: 10.1016/j.tips.2024.07.005
18
Schett G Müller F Taubmann J Mackensen A Wang W Furie RA et al . Advancements and challenges in CAR T cell therapy in autoimmune diseases. Nat Rev Rheumatol. (2024) 20:531–44. doi: 10.1038/s41584-024-01139-z
19
Baker DJ Arany Z Baur JA Epstein JA June CH . CAR T therapy beyond cancer: the evolution of a living drug. Nature. (2023) 619:707–15. doi: 10.1038/s41586-023-06243-w
20
Schett G Müller F Taubmann J Mackensen A Wang W Furie RA et al . Advancements and challenges in CAR T cell therapy in autoimmune diseases. Nat Rev Rheumatol. (2024) 6:024–01139. doi: 10.1038/s41584-024-01139-z
21
Matzinger P . The danger model: a renewed sense of self. Science. (2002) 296:301–5. doi: 10.1126/science.1071059
22
Tsokos GC . Systemic lupus erythematosus. New Engl J Med. (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
23
Steinman L . Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. (1996) 85:299–302. doi: 10.1016/S0092-8674(00)81107-1
24
Weaver CT Hatton RD Mangan PR Harrington LE . IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. (2007) 25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557
25
Xiao ZX Miller JS Zheng SG . An updated advance of autoantibodies in autoimmune diseases. Autoimmun Rev. (2021) 20:14. doi: 10.1016/j.autrev.2020.102743
26
Elkon K Casali P . Nature and functions of autoantibodies. Nat Clin Pract Rheumatol. (2008) 4:491–8. doi: 10.1038/ncprheum0895
27
McInnes IB Schett G . The pathogenesis of rheumatoid arthritis. New Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
28
Rosenblum MD Remedios KA Abbas AK . Mechanisms of human autoimmunity. J Clin Invest. (2015) 125:2228–33. doi: 10.1172/JCI78088
29
Atkinson MA Eisenbarth GS . Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. (2001) 358:221–9. doi: 10.1016/S0140-6736(01)05415-0
30
Davidson A Diamond B . Autoimmune diseases. New Engl J Med. (2001) 345:340–50. doi: 10.1056/NEJM200108023450506
31
Bahn RS . Graves’ ophthalmopathy. New Engl J Med. (2010) 362:726–38. doi: 10.1056/NEJMra0905750
32
Bluestone JA Herold K Eisenbarth G . Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. (2010) 464:1293–300. doi: 10.1038/nature08933
33
Goodnow CC Sprent J Fazekas de St Groth B Vinuesa CG . Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. (2005) 435:590–7. doi: 10.1038/nature03724
34
Manson JJ Rahman A . Systemic lupus erythematosus. Orphanet J Rare Dis. (2006) 1:1750–172. doi: 10.1186/1750-1172-1-6
35
Firestein GS McInnes IB . Immunopathogenesis of rheumatoid arthritis. Immunity. (2017) 46:183–96. doi: 10.1016/j.immuni.2017.02.006
36
Lester RS Knowles SR Shear NH . The risks of systemic corticosteroid use. Dermatol Clin. (1998) 16:277–88. doi: 10.1016/S0733-8635(05)70010-3
37
Buchman AL . Side effects of corticosteroid therapy. J Clin Gastroenterol. (2001) 33:289–94. doi: 10.1097/00004836-200110000-00006
38
Weldon D . The effects of corticosteroids on bone: osteonecrosis (avascular necrosis of the bone). “Annals Allergy Asthma Immunol. (2009) 103:91–7. doi: 10.1016/S1081-1206(10)60159-7
39
Money S . The risks of chronic corticosteroid exposure. J Pain Palliative Care Pharmacother. (2017) 31:160–1. doi: 10.1080/15360288.2017.1298689
40
Petri M . Cyclophosphamide: new approaches for systemic lupus erythematosus. Lupus. (2004) 13:366–71. doi: 10.1191/0961203303lu1028oa
41
Sádaba B Azanza JR García Quetglas E Fernández V . Treatment with tacrolimus in autoimmune diseases. Rev Med Univ Navarra. (2004) 48:24–38.
42
Cipriani P Ruscitti P Carubbi F Liakouli V Giacomelli R . Methotrexate: an old new drug in autoimmune disease. Expert Rev Clin Immunol. (2014) 10:1519–30. doi: 10.1586/1744666X.2014.962996
43
Fragoso YD Brooks JB . Leflunomide and teriflunomide: altering the metabolism of pyrimidines for the treatment of autoimmune diseases. Expert Rev Clin Pharmacol. (2015) 8:315–20. doi: 10.1586/17512433.2015.1019343
44
Danza Á Graña D Goñi M Vargas A Ruiz-Irastorza G . Hydroxychloroquine for autoimmune diseases. Rev Med Chile. (2016) 144:232–40. doi: 10.4067/S0034-98872016000200012
45
Şimşek C Wahlin S Efe C . Autoimmune hepatitis: Mycophenolate mofetil vs. azathioprine as first-line therapy. J Hepatol. (2024) 81:e41–2. doi: 10.1016/j.jhep.2024.02.022
46
Solomon DH Kremer JM Fisher M Curtis JR Furer V Harrold LR et al . Comparative cancer risk associated with methotrexate, other non-biologic and biologic disease-modifying anti-rheumatic drugs. Semin Arthritis Rheum. (2014) 43:489–97. doi: 10.1016/j.semarthrit.2013.08.003
47
Andreakos E Taylor PC Feldmann M . Monoclonal antibodies in immune and inflammatory diseases. Curr Opin Biotechnol. (2002) 13:615–20. doi: 10.1016/S0958-1669(02)00355-5
48
Taylor PC Choy E Baraliakos X Szekanecz Z Xavier RM Isaacs JD et al . Differential properties of Janus kinase inhibitors in the treatment of immune-mediated inflammatory diseases. Rheumatology. (2024) 63:298–308. doi: 10.1093/rheumatology/kead448
49
Thomas RM Colon A Motaparthi K . Rituximab in autoimmune pemphigoid diseases: Indications, optimized regimens, and practice gaps. Clinics In Dermatol. (2020) 38:384–96. doi: 10.1016/j.clindermatol.2019.07.023
50
Guerreiro Castro S Isenberg DA . Belimumab in systemic lupus erythematosus (SLE): evidence-to-date and clinical usefulness. Ther Adv Musculoskelet Dis. (2017) 9:75–85. doi: 10.1177/1759720X17690474
51
Mohamed MF Bhatnagar S Parmentier JM Nakasato P Wung P . Upadacitinib: Mechanism of action, clinical, and translational science. Clin Trans Sci. (2024) 17:18. doi: 10.1111/cts.13688
52
Thurlings RM Vos K Wijbrandts CA Zwinderman AH Gerlag DM Tak PP . Synovial tissue response to rituximab: mechanism of action and identification of biomarkers of response. Ann Rheum Dis. (2008) 67:917–25. doi: 10.1136/ard.2007.080960
53
Terrier B Amoura Z Ravaud P Hachulla E Jouenne R Combe B et al . Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum. (2010) 62:2458–66. doi: 10.1002/art.27541
54
Kamburova EG Koenen HJ Borgman KJ ten Berge IJ Joosten I Hilbrands LB . A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am J Transpl. (2013) 13:1503–11. doi: 10.1111/ajt.12220
55
Boonstra M Meijs J Dorjée AL Marsan NA Schouffoer A Ninaber MK et al . Rituximab in early systemic sclerosis. RMD Open. (2017) 3:e000384. doi: 10.1136/rmdopen-2016-000384
56
Merrill JT Neuwelt CM Wallace DJ Shanahan JC Latinis KM Oates JC et al . Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. (2010) 62:222–33. doi: 10.1002/art.27233
57
Marmont AM . Immune ablation with stem-cell rescue: a possible cure for systemic lupus erythematosus? Lupus. (1993) 2:151–6. doi: 10.1177/096120339300200304
58
Burt RK Traynor AE Craig R Marmont AM . The promise of hematopoietic stem cell transplantation for autoimmune diseases. Bone Marrow Transpl. (2003) 31:521–4. doi: 10.1038/sj.bmt.1703868
59
Tyndall A Saccardi R . Haematopoietic stem cell transplantation in the treatment of severe autoimmune disease: results from phase I/II studies, prospective randomized trials and future directions. Clin And Exp Immunol. (2005) 141:1–9. doi: 10.1111/j.1365-2249.2005.02806.x
60
Leone A Radin M Almarzooqi AM Al-Saleh J Roccatello D Sciascia S et al . Autologous hematopoietic stem cell transplantation in Systemic Lupus Erythematosus and antiphospholipid syndrome: A systematic review. Autoimmun Rev. (2017) 16:469–77. doi: 10.1016/j.autrev.2017.03.008
61
Snowden JA Badoglio M Labopin M Giebel S McGrath E Marjanovic Z et al . Evolution, trends, outcomes, and economics of hematopoietic stem cell transplantation in severe autoimmune diseases. Blood Adv. (2017) 1:2742–55. doi: 10.1182/bloodadvances.2017010041
62
Alexander T Greco R Snowden JA . Hematopoietic stem cell transplantation for autoimmune disease. Annu Rev Med. (2021) 72:215–28. doi: 10.1146/annurev-med-070119-115617
63
Loh Y Oyama Y Statkute L Quigley K Yaung K Gonda E et al . Development of a secondary autoimmune disorder after hematopoietic stem cell transplantation for autoimmune diseases: role of conditioning regimen used. Blood. (2007) 109:2643–548. doi: 10.1182/blood-2006-07-035766
64
Daikeler T Labopin M Di Gioia M Abinun M Alexander T Miniati I et al . Secondary autoimmune diseases occurring after HSCT for an autoimmune disease: a retrospective study of the EBMT Autoimmune Disease Working Party. Blood. (2011) 118:1693–8. doi: 10.1182/blood-2011-02-336156
65
de Buys P Khanna D Furst DE . Hemopoietic stem cell transplantation in rheumatic diseases–an update. Autoimmun Rev. (2005) 4:442–9. doi: 10.1016/j.autrev.2005.03.003
66
Kantoff PW Higano CS Shore ND Berger ER Small EJ Penson DF et al . Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
67
Ahmed MS Bae YS . Dendritic cell-based immunotherapy for rheumatoid arthritis: from bench to bedside. Immune Netw. (2016) 16:44–51. doi: 10.4110/in.2016.16.1.44
68
Morelli AE Thomson AW . Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. (2007) 7:610–21. doi: 10.1038/nri2132
69
Benham H Nel HJ Law SC Mehdi AM Street S Ramnoruth N et al . Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Trans Med. (2015) 7:290ra287. doi: 10.1126/scitranslmed.aaa9301
70
Giannoukakis N Phillips B Finegold D Harnaha J Trucco M . Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. (2011) 34:2026–32. doi: 10.2337/dc11-0472
71
Raϊch-Regué D Grau-López L Naranjo-Gómez M Ramo-Tello C Pujol-Borrell R Martínez-Cáceres E et al . Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur J Immunol. (2012) 42:771–82. doi: 10.1002/eji.201141835
72
Schuster IS Coudert JD Andoniou CE Degli-Esposti MA . Natural regulators”: NK cells as modulators of T cell immunity. Front Immunol. (2016) 7:235. doi: 10.3389/fimmu.2016.00235
73
Martinez-Espinosa I Serrato JA Ortiz-Quintero B . Role of IL-10-producing natural killer cells in the regulatory mechanisms of inflammation during systemic infection. Biomolecules. (2021) 12:4. doi: 10.3390/biom12010004
74
Moretta A Marcenaro E Sivori S Della Chiesa M Vitale M Moretta L . Early liaisons between cells of the innate immune system in inflamed peripheral tissues. Trends In Immunol. (2005) 26:668–75. doi: 10.1016/j.it.2005.09.008
75
Hao J Campagnolo D Liu R Piao W Shi S Hu B et al . Interleukin-2/interleukin-2 antibody therapy induces target organ natural killer cells that inhibit central nervous system inflammation. Ann Neurol. (2011) 69:721–34. doi: 10.1002/ana.22339
76
Kamyan D Hassane M Alnaqbi A Souid AK Rasbi ZA Tahrawi AA et al . Ozanimod-mediated remission in experimental autoimmune encephalomyelitis is associated with enhanced activity of CNS CD27(low/-) NK cell subset. Front Immunol. (2024) 15:1230735. doi: 10.3389/fimmu.2024.1230735
77
Mohammadzadeh I Qujeq D Yousefi T Ferns GA Maniati M Vaghari-Tabari M . CRISPR/Cas9 gene editing: A new therapeutic approach in the treatment of infection and autoimmunity. IUBMB Life. (2020) 72:1603–21. doi: 10.1002/iub.2296
78
Israr J Kumar A . Current progress in CRISPR-Cas systems for autoimmune diseases. Prog Mol Biol Trans Sci. (2024) 208:231–59. doi: 10.1016/bs.pmbts.2024.07.011
79
Rosenbaum L . Tragedy, perseverance, and chance - the story of CAR-T therapy. New Engl J Med. (2017) 377:1313–5. doi: 10.1056/NEJMp1711886
80
Mougiakakos D Krönke G Völkl S Kretschmann S Aigner M Kharboutli S et al . CD19-targeted CAR T cells in refractory systemic lupus erythematosus. New Engl J Med. (2021) 385:567–9. doi: 10.1056/NEJMc2107725
81
Mackensen A Müller F Mougiakakos D Böltz S Wilhelm A Aigner M et al . Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
82
Tian DS Qin C Dong MH Heming M Zhou LQ Wang W et al . B cell lineage reconstitution underlies CAR-T cell therapeutic efficacy in patients with refractory myasthenia gravis. EMBO Mol Med. (2024) 16:966–87. doi: 10.1038/s44321-024-00043-z
83
Harrison C . CAR-Ts sweep into autoimmunity. Nat Biotechnol. (2024) 42:995–7. doi: 10.1038/s41587-024-02321-0
84
Rafiq S Hackett CS Brentjens RJ . Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
85
Cappell KM Kochenderfer JN . A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. (2021) 18:715–27. doi: 10.1038/s41571-021-00530-z
86
Gu X He D Li C Wang H Yang G . Development of inducible CD19-CAR T cells with a tet-on system for controlled activity and enhanced clinical safety. Int J Mol Sci. (2018) 19:3455. doi: 10.3390/ijms19113455
87
Wang W He S Zhang W Zhang H DeStefano VM Wada M et al . BCMA-CD19 compound CAR T cells for systemic lupus erythematosus: a phase 1 open-label clinical trial. Ann Rheum Dis. (2024) 83:1304–14. doi: 10.1136/ard-2024-225785
88
Rodgers DT Mazagova M Hampton EN Cao Y Ramadoss NS Hardy IR et al . Switch-mediated activation and retargeting of CAR-T cells for B-cell Malignancies. Proc Natl Acad Sci U S A. (2016) 113:12. doi: 10.1073/pnas.1524155113
89
Chong EA Ruella M Schuster SJ . Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. New Engl J Med. (2021) 384:673–4. doi: 10.1056/NEJMc2030164
90
Neelapu SS Jacobson CA Ghobadi A Miklos DB Lekakis LJ Oluwole OO et al . Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. (2023) 141:2307–15. doi: 10.1182/blood.2022018893
91
Sosnoski HM Posey AD Jr. Therapeutic intersections: Expanding benefits of CD19 CAR T cells from cancer to autoimmunity. Cell Stem Cell. (2024) 31:437–8. doi: 10.1016/j.stem.2024.03.006
92
Mei HE Wirries I Frölich D Brisslert M Giesecke C Grün JR et al . A unique population of IgG-expressing plasma cells lacking CD19 is enriched in human bone marrow. Blood. (2015) 125:1739–48. doi: 10.1182/blood-2014-02-555169
93
Bhoj VG Arhontoulis D Wertheim G Capobianchi J Callahan CA Ellebrecht CT et al . Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. (2016) 128:360–70. doi: 10.1182/blood-2016-01-694356
94
Halliley JL Tipton CM Liesveld J Rosenberg AF Darce J Gregoretti IV et al . Long-lived plasma cells are contained within the CD19(-)CD38(hi)CD138(+) subset in human bone marrow. Immunity. (2015) 43:132–45. doi: 10.1016/j.immuni.2015.06.016
95
Hill JA Krantz EM Hay KA Dasgupta S Stevens-Ayers T Bender Ignacio RA et al . Durable preservation of antiviral antibodies after CD19-directed chimeric antigen receptor T-cell immunotherapy. Blood Adv. (2019) 3:3590–601. doi: 10.1182/bloodadvances.2019000717
96
Haghikia A Hegelmaier T Wolleschak D Böttcher M Desel C Borie D et al . Anti-CD19 CAR T cells for refractory myasthenia gravis. Lancet Neurol. (2023) 22:1104–5. doi: 10.1016/S1474-4422(23)00375-7
97
Kochenderfer JN Dudley ME Feldman SA Wilson WH Spaner DE Maric I et al . B-cell depletion and remissions of Malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
98
Kochenderfer JN Somerville RPT Lu T Shi V Bot A Rossi J et al . Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. (2017) 35:1803–13. doi: 10.1200/JCO.2016.71.3024
99
Kansal R Richardson N Neeli I Khawaja S Chamberlain D Ghani M et al . Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Trans Med. (2019) 11:eaav1648. doi: 10.1126/scitranslmed.aav1648
100
Jin X Xu Q Pu C Zhu K Lu C Jiang Y et al . Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell Mol Immunol. (2021) 18:1896–903. doi: 10.1038/s41423-020-0472-1
101
Bergmann C Müller F Distler JHW Györfi AH Völkl S Aigner M et al . Treatment of a patient with severe systemic sclerosis (SSc) using CD19-targeted CAR T cells. Ann Rheum Dis. (2023) 82:1117–20. doi: 10.1136/ard-2023-223952
102
Merkt W Freitag M Claus M Kolb P Falcone V Röhrich M et al . Third-generation CD19.CAR-T cell-containing combination therapy in Scl70+ systemic sclerosis. Ann Rheum Dis. (2024) 83:543–6. doi: 10.1136/ard-2023-225174
103
Wang X Wu X Tan B Zhu L Zhang Y Lin L et al . Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis. Cell. (2024) 187:4890–904. doi: 10.1016/j.cell.2024.06.027
104
Müller F Boeltz S Knitza J Aigner M Völkl S Kharboutli S et al . CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet. (2023) 401:815–8. doi: 10.1016/S0140-6736(23)00023-5
105
Pecher AC Hensen L Klein R Schairer R Lutz K Atar D et al . CD19-targeting CAR T cells for myositis and interstitial lung disease associated with antisynthetase syndrome. Jama. (2023) 329:2154–62. doi: 10.1001/jama.2023.8753
106
Avery DT Kalled SL Ellyard JI Ambrose C Bixler SA Thien M et al . BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. (2003) 112:286–97. doi: 10.1172/JCI18025
107
Tai YT Mayes PA Acharya C Zhong MY Cea M Cagnetta A et al . Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. (2014) 123:3128–38. doi: 10.1182/blood-2013-10-535088
108
Tarlinton D Good-Jacobson K . Diversity among memory B cells: origin, consequences, and utility. Science. (2013) 341:1205–11. doi: 10.1126/science.1241146
109
Nutt SL Hodgkin PD Tarlinton DM Corcoran LM . The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
110
Cohen AD Garfall AL Stadtmauer EA Melenhorst JJ Lacey SF Lancaster E et al . B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. (2019) 129:2210–21. doi: 10.1172/JCI126397
111
Cassese G Lindenau S de Boer B Arce S Hauser A Riemekasten G et al . Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. (2001) 31:2726–32. doi: 10.1002/1521-4141(200109)31:9<2726::AID-IMMU2726>3.0.CO;2-H
112
Hoyer BF Manz RA Radbruch A Hiepe F . Long-lived plasma cells and their contribution to autoimmunity. Ann New York Acad Sci. (2005) 1050:124–33. doi: 10.1196/annals.1313.014
113
Salazar-Camarena DC Palafox-Sánchez CA Cruz A Marín-Rosales M Muñoz-Valle JF . Analysis of the receptor BCMA as a biomarker in systemic lupus erythematosus patients. Sci Rep. (2020) 10:6236. doi: 10.1038/s41598-020-63390-0
114
Chen D Zhu Y Chen Z Jiang S He H Qiang W et al . A five-year follow-up clinical study of the B cell maturation antigen chimeric antigen receptor-T cell therapy HDS269B in patients with relapsed or refractory multiple myeloma. Clin Cancer Res. (2024) 30:3747–56. doi: 10.1158/1078-0432.CCR-24-0414
115
Qin C Tian DS Zhou LQ Shang K Huang L Dong MH et al . Anti-BCMA CAR T-cell therapy CT103A in relapsed or refractory AQP4-IgG seropositive neuromyelitis optica spectrum disorders: phase 1 trial interim results. Signal Transduct Target Ther. (2023) 8:5. doi: 10.1038/s41392-022-01278-3
116
Qin C Zhang M Mou DP Zhou LQ Dong MH Huang L et al . Single-cell analysis of anti-BCMA CAR T cell therapy in patients with central nervous system autoimmunity. Sci Immunol. (2024) 9:eadj9730. doi: 10.1126/sciimmunol.adj9730
117
Piedra-Quintero ZL Wilson Z Nava P Guerau-de-Arellano M . CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front Immunol. (2020) 11:597959. doi: 10.3389/fimmu.2020.597959
118
Donís-Hernández FR Parkhouse RM Santos-Argumedo L . Ontogeny, distribution and function of CD38-expressing B lymphocytes in mice. Eur J Immunol. (2001) 31:1261–7. doi: 10.1002/1521-4141(200104)31:4<1261::AID-IMMU1261>3.0.CO;2-H
119
Romero-Ramírez H Morales-Guadarrama MT Pelayo R López-Santiago R Santos-Argumedo L . CD38 expression in early B-cell precursors contributes to extracellular signal-regulated kinase-mediated apoptosis. Immunology. (2015) 144:271–81. doi: 10.1111/imm.12370
120
Zeng F Zhang J Jin X Liao Q Chen Z Luo G et al . Effect of CD38 on B-cell function and its role in the diagnosis and treatment of B-cell-related diseases. J Cell Physiol. (2022) 237:2796–807. doi: 10.1002/jcp.30760
121
Benfaremo D Gabrielli A . Is there a future for anti-CD38 antibody therapy in systemic autoimmune diseases? Cells. (2019) 9:77. doi: 10.3390/cells9010077
122
Ostendorf L Burns M Durek P Heinz GA Heinrich F Garantziotis P et al . Targeting CD38 with daratumumab in refractory systemic lupus erythematosus. New Engl J Med. (2020) 383:1149–55. doi: 10.1056/NEJMoa2023325
123
Lokhorst HM Plesner T Laubach JP Nahi H Gimsing P Hansson M et al . Targeting CD38 with daratumumab monotherapy in multiple myeloma. New Engl J Med. (2015) 373:1207–19. doi: 10.1056/NEJMoa1506348
124
Ellebrecht CT Bhoj VG Nace A Choi EJ Mao X Cho MJ et al . Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
125
Chen Y Sun J Liu H Yin G Xie Q . Immunotherapy deriving from CAR-T cell treatment in autoimmune diseases. J Immunol Res. (2019) 2019:5727516. doi: 10.1155/2019/5727516
126
Joly P Maho-Vaillant M Prost-Squarcioni C Hebert V Houivet E Calbo S et al . First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
127
Chen DM Odueyungbo A Csinady E Gearhart L Lehane P Cheu M et al . Rituximab is an effective treatment in patients with pemphigus vulgaris and demonstrates a steroid-sparing effect. Br J Dermatol. (2020) 182:1111–9. doi: 10.1111/bjd.18482
128
Werth VP Joly P Mimouni D Maverakis E Caux F Lehane P et al . Rituximab versus mycophenolate mofetil in patients with pemphigus vulgaris. New Engl J Med. (2021) 384:2295–305. doi: 10.1056/NEJMoa2028564
129
Lee J Lundgren DK Mao X Manfredo-Vieira S Nunez-Cruz S Williams EF et al . Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. (2020) 130:6317–24. doi: 10.1172/JCI138416
130
Yi JS Guptill JT Stathopoulos P Nowak RJ O’Connor KC . B cells in the pathophysiology of myasthenia gravis. Muscle Nerve. (2018) 57:172–84. doi: 10.1002/mus.25973
131
Borges LS Richman DP . Muscle-specific kinase myasthenia gravis. Front Immunol. (2020) 11:707. doi: 10.3389/fimmu.2020.00707
132
Dziadkowiak E Baczyńska D Waliszewska-Prosół M . MuSK myasthenia gravis-potential pathomechanisms and treatment directed against specific targets. Cells. (2024) 13:556. doi: 10.3390/cells13060556
133
Oh S Mao X Manfredo-Vieira S Lee J Patel D Choi EJ et al . Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat Biotechnol. (2023) 41:1229–38. doi: 10.1038/s41587-022-01637-z
134
Reincke SM von Wardenburg N Homeyer MA Kornau HC Spagni G Li LY et al . Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell. (2023) 186:5084–97. doi: 10.1016/j.cell.2023.10.001
135
Zhou J Xu Y Shu J Jiang H Huang L Xu M et al . GPIbα CAAR T cells function like a Trojan horse to eliminate autoreactive B cells to treat immune thrombocytopenia. Haematologica. (2024) 109:2256–70. doi: 10.3324/haematol.2023.283874
136
Scheinecker C Göschl L Bonelli M . Treg cells in health and autoimmune diseases: New insights from single cell analysis. J Autoimm. (2020) 110:102376. doi: 10.1016/j.jaut.2019.102376
137
Marek-Trzonkowska N Mysliwiec M Dobyszuk A Grabowska M Techmanska I Juscinska J et al . Administration of CD4+CD25highCD127- regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care. (2012) 35:1817–20. doi: 10.2337/dc12-0038
138
Dall’Era M Pauli ML Remedios K Taravati K Sandova PM Putnam AL et al . Adoptive treg cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol. (2019) 71:431–40. doi: 10.1002/art.40737
139
Bender E . Creating T cells to guard against autoimmune disease. Nature. (2021) 595:S60–S62. doi: 10.1038/d41586-021-01840-z
140
Beheshti SA Shamsasenjan K Ahmadi M Abbasi B . CAR Treg: A new approach in the treatment of autoimmune diseases. Int Immunopharmacol. (2022) 102:108409. doi: 10.1016/j.intimp.2021.108409
141
Arjomandnejad M Kopec AL Keeler AM . CAR-T regulatory (CAR-treg) cells: engineering and applications. Biomedicines. (2022) 10:287. doi: 10.3390/biomedicines10020287
142
Kahmini FR Shahgaldi S . Harnessing the inherent power of chimeric antigen receptor (CAR)-expressing regulatory T cells (CAR-Tregs) to treat autoimmune-related disorders. Mol Biol Rep. (2022) 49:4069–78. doi: 10.1007/s11033-022-07511-0
143
Eggenhuizen PJ Ng BH Ooi JD . Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci. (2020) 21:7015. doi: 10.3390/ijms21197015
144
Passeri L Marta F Bassi V Gregori S . Tolerogenic dendritic cell-based approaches in autoimmunity. Int J Mol Sci. (2021) 22:8415. doi: 10.3390/ijms22168415
145
Sakaguchi S Yamaguchi T Nomura T Ono M . Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
146
Raffin C Vo LT Bluestone JA . T(reg) cell-based therapies: challenges and perspectives. Nat Rev Immunol. (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
147
Blat D Zigmond E Alteber Z Waks T Eshhar Z . Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. (2014) 22:1018–28. doi: 10.1038/mt.2014.41
148
Fransson M Piras E Burman J Nilsson B Essand M Lu B et al . CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflamm. (2012) 9:1742–2094. doi: 10.1186/1742-2094-9-112
149
Tenspolde M Zimmermann K Weber LC Hapke M Lieber M Dywicki J et al . Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimm. (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
150
Bézie S Charreau B Vimond N Lasselin J Gérard N Nerrière-Daguin V et al . Human CD8+ Tregs expressing a MHC-specific CAR display enhanced suppression of human skin rejection and GVHD in NSG mice. Blood Adv. (2019) 3:3522–38. doi: 10.1182/bloodadvances.2019000411
151
Proics E David M Mojibian M Speck M Lounnas-Mourey N Govehovitch A et al . Preclinical assessment of antigen-specific chimeric antigen receptor regulatory T cells for use in solid organ transplantation. Gene Ther. (2023) 30:309–22. doi: 10.1038/s41434-022-00358-x
152
Boardman DA Wong MQ Rees WD Wu D Himmel ME Orban PC et al . Flagellin-specific human CAR Tregs for immune regulation in IBD. J Autoimm. (2023) 134:102961. doi: 10.1016/j.jaut.2022.102961
153
Vent-Schmidt J Goldsmith LJ Steiner TS . Patients’ Willingness and perspectives toward chimeric antigen receptor T-regulatory cell therapy for inflammatory bowel diseases. Crohns Colitis. (2020) 2:otaa085. doi: 10.1093/crocol/otaa085
154
Moorman CD Yu S Briseno CG Phee H Sahoo A Ramrakhiani A et al . CAR-T cells and CAR-Tregs targeting conventional type-1 dendritic cell suppress experimental autoimmune encephalomyelitis. Front Immunol. (2023) 14:1235222. doi: 10.3389/fimmu.2023.1235222
155
MacDonald KG Hoeppli RE Huang Q Gillies J Luciani DS Orban PC et al . Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
156
Boardman DA Philippeos C Fruhwirth GO Ibrahim MA Hannen RF Cooper D et al . Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transpl. (2017) 17:931–43. doi: 10.1111/ajt.14185
157
Noyan F Zimmermann K Hardtke-Wolenski M Knoefel A Schulde E Geffers R et al . Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transpl. (2017) 17:917–30. doi: 10.1111/ajt.14175
158
Dawson NA Lamarche C Hoeppli RE Bergqvist P Fung VC McIver E et al . Systematic testing and specificity mapping of alloantigen-specific chimeric antigen receptors in regulatory T cells. JCI Insight. (2019) 4:e123672. doi: 10.1172/jci.insight.123672
159
Cohen SB Emery P Greenwald MW Dougados M Furie RA Genovese MC et al . Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. (2006) 54:2793–806. doi: 10.1002/art.22025
160
Mauri C . Regulation of immunity and autoimmunity by B cells. Curr Opin Immunol. (2010) 22:761–7. doi: 10.1016/j.coi.2010.10.009
161
Spanjaart AM Ljungman P de la Camara R Tridello G Ortiz-Maldonado V Urbano-Ispizua A et al . Poor outcome of patients with COVID-19 after CAR T-cell therapy for B-cell Malignancies: results of a multicenter study on behalf of the European Society for Blood and Marrow Transplantation (EBMT) Infectious Diseases Working Party and the European Hematology Association (EHA) Lymphoma Group. Leukemia. (2021) 35:3585–8. doi: 10.1038/s41375-021-01466-0
162
Sadelain M Brentjens R Rivière I . The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
163
Naldini L . Gene therapy returns to centre stage. Nature. (2015) 526:351–60. doi: 10.1038/nature15818
164
Billingsley MM Singh N Ravikumar P Zhang R June CH Mitchell MJ . Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. (2020) 20:1578–89. doi: 10.1021/acs.nanolett.9b04246
165
Billingsley MM Gong N Mukalel AJ Thatte AS El-Mayta R Patel SK et al . In Vivo mRNA CAR T Cell Engineering via Targeted Ionizable Lipid Nanoparticles with Extrahepatic Tropism. Small. (2024) 20:10. doi: 10.1002/smll.202304378
166
Qureischi M Mohr J Arellano-Viera E Knudsen SE Vohidov F Garitano-Trojaola A . mRNA-based therapies: Preclinical and clinical applications. Int Rev Cell Mol Biol. (2022) 372:1–54. doi: 10.1016/bs.ircmb.2022.04.007
167
Wu J Wu W Zhou B Li B . Chimeric antigen receptor therapy meets mRNA technology. Trends Biotechnol. (2024) 42:228–40. doi: 10.1016/j.tibtech.2023.08.005
168
Moretti A Ponzo M Nicolette CA Tcherepanova IY Biondi A Magnani CF . The past, present, and future of non-viral CAR T cells. Front Immunol. (2022) 13:867013. doi: 10.3389/fimmu.2022.867013
169
Parayath NN Stephan SB Koehne AL Nelson PS Stephan MT . In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. (2020) 11:6080. doi: 10.1038/s41467-020-19486-2
170
Rurik JG Tombácz I Yadegari A Méndez Fernández PO Shewale SV Li L et al . CAR T cells produced in vivo to treat cardiac injury. Science. (2022) 375:91–6. doi: 10.1126/science.abm0594
171
Granit V Benatar M Kurtoglu M Miljković MD Chahin N Sahagian G et al . Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. (2023) 22:578–90. doi: 10.1016/S1474-4422(23)00194-1
172
Gauthier J Turtle CJ . Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr Res Transl Med. (2018) 66:50–2. doi: 10.1016/j.retram.2018.03.003
173
Diaconu I Ballard B Zhang M Chen Y West J Dotti G et al . Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. (2017) 25:580–92. doi: 10.1016/j.ymthe.2017.01.011
174
Parayath NN Stephan SB Koehne AL Nelson PS Stephan MT . In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. (2020) 11:020–19486. doi: 10.1038/s41467-020-19486-2
175
Neelapu SS Tummala S Kebriaei P Wierda W Gutierrez C Locke FL et al . Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
176
Santomasso BD Nastoupil LJ Adkins S Lacchetti C Schneider BJ Anadkat M et al . Management of immune-related adverse events in patients treated with chimeric antigen receptor T-cell therapy: ASCO guideline. J Clin Oncol. (2021) 39:3978–92. doi: 10.1200/JCO.21.01992
177
Turtle CJ Hanafi LA Berger C Gooley TA Cherian S Hudecek M et al . CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
178
Porter DL Levine BL Kalos M Bagg A June CH . Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
179
Bonifant CL Jackson HJ Brentjens RJ Curran KJ . Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. (2016) 3:16011. doi: 10.1038/mto.2016.11
180
Brudno JN Kochenderfer JN . Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
181
Shouse G Danilov AV Artz A . CAR T-cell therapy in the older person: indications and risks. Curr Oncol Rep. (2022) 24:1189–99. doi: 10.1007/s11912-022-01272-6
182
Reyes KR Huang CY Lo M Arora S Chung A Wong SW et al . Safety and efficacy of BCMA CAR-T cell therapy in older patients with multiple myeloma. Transpl Cell Ther. (2023) 29:350–5. doi: 10.1016/j.jtct.2023.03.012
183
Seif M Einsele H Löffler J . CAR T cells beyond cancer: hope for immunomodulatory therapy of infectious diseases. Front Immunol. (2019) 10:2711. doi: 10.3389/fimmu.2019.02711
184
Maude SL Teachey DT Porter DL Grupp SA . CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
185
Kochenderfer JN Rosenberg SA . Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. (2013) 10:267–76. doi: 10.1038/nrclinonc.2013.46
186
Davila ML Riviere I Wang X Bartido S Park J Curran K et al . Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Trans Med. (2014) 6:3008226. doi: 10.1126/scitranslmed.3008226
187
Maude SL Frey N Shaw PA Aplenc R Barrett DM Bunin NJ et al . Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
188
Locke FL Neelapu SS Bartlett NL Siddiqi T Chavez JC Hosing CM et al . Phase 1 results of ZUMA-1: A multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
189
Jain MD Davila ML . Concise review: emerging principles from the clinical application of chimeric antigen receptor T cell therapies for B cell Malignancies. Stem Cells. (2018) 36:36–44. doi: 10.1002/stem.2715
190
Kanakry CG Ganguly S Zahurak M Bolaños-Meade J Thoburn C Perkins B et al . Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Trans Med. (2013) 5:211. doi: 10.1126/scitranslmed.3006960
191
Qin X Zhao Y Zhang T Yin C Qiao J Guo W et al . TrkB agonist antibody ameliorates fertility deficits in aged and cyclophosphamide-induced premature ovarian failure model mice. Nat Commun. (2022) 13:914. doi: 10.1038/s41467-022-28611-2
192
Barberino RS Silva RLS Palheta Junior RC Smitz JEJ Matos MHT . Protective effects of antioxidants on cyclophosphamide-induced ovarian toxicity. Biopreserv Biobank. (2023) 21:121–41. doi: 10.1089/bio.2021.0159
193
Elinav E Waks T Eshhar Z . Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology. (2008) 134:2014–24. doi: 10.1053/j.gastro.2008.02.060
194
Schreeb K Culme-Seymour E Ridha E Dumont C Atkinson G Hsu B et al . Study design: human leukocyte antigen class I molecule A(∗)02-chimeric antigen receptor regulatory T cells in renal transplantation. Kidney Int Rep. (2022) 7:1258–67. doi: 10.1016/j.ekir.2022.03.030
195
Martinez-Cibrian N Español-Rego M Pascal M Delgado J Ortiz-Maldonado V . Practical aspects of chimeric antigen receptor T-cell administration: From commercial to point-of-care manufacturing. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.1005457
196
Depil S Duchateau P Grupp SA Mufti G Poirot L . ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
197
Kim DW Cho JY . Recent advances in allogeneic CAR-T cells. Biomolecules. (2020) 10:263. doi: 10.3390/biom10020263
198
Brudno JN Kochenderfer JN . Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
199
Hartmann J Schüßler-Lenz M Bondanza A Buchholz CJ . Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. (2017) 9:1183–97. doi: 10.15252/emmm.201607485
200
Graham C Jozwik A Pepper A Benjamin R . Allogeneic CAR-T cells: more than ease of access? Cells. (2018) 7:155. doi: 10.3390/cells7100155
201
MacKay M Afshinnekoo E Rub J Hassan C Khunte M Baskaran N et al . The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. (2020) 38:233–44. doi: 10.1038/s41587-019-0329-2
202
Lamers CH Sleijfer S van Steenbergen S van Elzakker P van Krimpen B Groot C et al . Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. (2013) 21:904–12. doi: 10.1038/mt.2013.17
203
Kim S Kim D Cho SW Kim J Kim JS . Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. (2014) 24:1012–9. doi: 10.1101/gr.171322.113
204
Lin S Staahl BT Alla RK Doudna JA . Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. (2014) 15:04766. doi: 10.7554/eLife.04766.010
205
Zuris JA Thompson DB Shu Y Guilinger JP Bessen JL Hu JH et al . Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. (2015) 33:73–80. doi: 10.1038/nbt.3081
206
Liu X Zhang Y Cheng C Cheng AW Zhang X Li N et al . CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells[M. Cell Res. (2017) 27:154–7. doi: 10.1038/cr.2016.142
207
Liu M Zhang L Zhong M Long Y Yang W Liu T et al . CRISPR/Cas9-mediated knockout of intracellular molecule SHP-1 enhances tumor-killing ability of CD133-targeted CAR T cells in vitro[M. Exp Hematol Oncol. (2023) 12:88. doi: 10.1186/s40164-023-00450-x
208
Hu Y Zhou Y Zhang M Ge W Li Y Yang L et al . CRISPR/cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. (2021) 27:2764–72. doi: 10.1158/1078-0432.CCR-20-3863
209
Arbuckle MR McClain MT Rubertone MV Scofield RH Dennis GJ James JA et al . Development of autoantibodies before the clinical onset of systemic lupus erythematosus. New Engl J Med. (2003) 349:1526–33. doi: 10.1056/NEJMoa021933
210
Fortuna G Brennan MT . Systemic lupus erythematosus: epidemiology, pathophysiology, manifestations, and management. Dental Clinics North America. (2013) 57:631–55. doi: 10.1016/j.cden.2013.06.003
211
Pisetsky DS . Pathogenesis of autoimmune disease. Nat Rev Nephrol. (2023) 19:509–24. doi: 10.1038/s41581-023-00720-1
Summary
Keywords
CAR T-cell therapy, autoimmune disease, autoreactive B cell targeting, Treg cell, personalized risk stratification
Citation
Wu D, Xu-Monette ZY, Zhou J, Yang K, Wang X, Fan Y and Young KH (2025) CAR T-cell therapy in autoimmune diseases: a promising frontier on the horizon. Front. Immunol. 16:1613878. doi: 10.3389/fimmu.2025.1613878
Received
25 April 2025
Accepted
20 July 2025
Published
12 August 2025
Volume
16 - 2025
Edited by
Melita Irving, Hospitalier Universitaire Vaudois (CHUV), Switzerland
Reviewed by
Gerson D. Keppeke, Universidad Católica del Norte, Chile
Subhash Kumar Tripathi, Seattle Children’s Research Institute, United States
Updates
Copyright
© 2025 Wu, Xu-Monette, Zhou, Yang, Wang, Fan and Young.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ken H. Young, ken.young@duke.edu; Yongsheng Fan, applezjtcm@126.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.