 Haogeng Wang
Haogeng Wang Taixi Huang1
Taixi Huang1- 1School of Acupuncture and Tuina, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2Key Laboratory of Traditional Chinese Medicine Classical Theory, Ministry of Education, Shandong University of Traditional Chinese Medicine, Jinan, China
Ulcerative colitis (UC) is a kind of chronic inflammatory bowel disease, is driven by dysregulated immune responses involving neutrophils (NEUs) and macrophages. NEUs exacerbate mucosal injury through reactive oxygen species (ROS), neutrophil extracellular traps (NETs), proteases, and cytokine interactions, while also exhibiting dual roles in tissue repair. Macrophages contribute to UC progression via M1-mediated pro-inflammatory cytokine release and epithelial barrier disruption, whereas M2 macrophages promote resolution through anti-inflammatory signals (IL-10, TGF-β) and epithelial regeneration. Clinically, NEU-derived biomarkers predict disease activity and therapeutic response, while macrophage-targeted therapies modulate inflammation. This review summairzes current knowledge on the mechanistic roles of these immune cells in UC pathogenesis and their clinical implications, such as NET inhibition, MMP-9 blockade, and M2 polarization, which hold promise for precision medicine in UC.
1 Introduction
Ulcerative colitis (UC), a major subtype of inflammatory bowel disease (IBD), is characterized by chronic, relapsing inflammation of the colorectal mucosa, leading to bloody diarrhea and abdominal pain, which may be life-threatening in severe cases (1). Since the early 21st century, UC has emerged as a global health concern, with rising prevalence imposing a significant socioeconomic burden (2). Therapeutic goals have evolved from clinical to endoscopic and now histological remission, as persistent histologic inflammation despite endoscopic healing is linked to poorer prognosis (3).
The pathogenesis of UC is multifactorial, involving gut microbiota dysbiosis, disruption of the intestinal mucosal barrier, and aberrant immune cell function (4). Among these immune elements, neutrophil (NEU) infiltration is a defining histological feature of UC, with NEU depletion associated with lower relapse risk, and NEU-related biomarkers offering prognostic value (5). Macrophages are essential for phagocytosis and immune modulation. Studies have demonstrated that the number of macrophages in the lamina propria of the colon in patients with active UC is approximately tenfold higher than that in healthy individuals (6), and skew toward a more activated state (7, 8), suggesting their pivotal involvement in UC pathogenesis. This review aims to provide a comprehensive overview of the mechanistic roles of neutrophils and macrophages in the development and progression of UC, as well as their potential clinical applications.
2 NEUs regulate the intestinal inflammation of UC
2.1 NEU-derived ROS and NETs exacerbate intestinal inflammation
NEUs are essential effectors of innate immunity, yet their excessive activation has been implicated in the onset and progression of various autoimmune diseases (9, 10). During maturation, NEUs generate three distinct types of granules: primary granules, which contain enzymes such as myeloperoxidase (MPO) and neutrophil elastase (NE); secondary granules, including collagenases; and tertiary granules, which carry MMP-9 (11). In UC, massive infiltration of NEUs into the intestinal mucosa leads to the release of granule contents and ROS, resulting in epithelial and stromal injury and manifesting as cryptitis, mucosal erosion, and ulceration (12). ROS induces cellular apoptosis and necrosis by oxidatively damaging nucleic acids, proteins, and lipids (13). In UC, excessive ROS production by infiltrating NEUs, coupled with insufficient ROS clearance, leads to ROS accumulation in the mucosa (14, 15). NETs are extracellular mesh-like structures composed of decondensed chromatin, DNA, and antimicrobial peptides, extruded from activated NEUs as part of their antimicrobial defense (16, 17). NETs amplify inflammatory cascades through the release of IL-1β and TNF-α, representing a key trigger of immune dysregulation in UC (18). Angelidou et al. demonstrated that activation of the REDD1/autophagy/NETs/IL-1β axis mediates UC-related inflammation and mucosal injury (19). Moreover, UC is a recognized risk factor for venous thromboembolism, including deep vein thrombosis and pulmonary embolism (20). NEUs also secrete proteinase-3 and cathepsin G, while NE specifically degrades extracellular matrix components such as elastin (21, 22). Serine protease inhibitor B1, an endogenous NE suppressor, inhibits H2O2-induced NE activity and may help preserve epithelial integrity (23). Infliximab, a TNF-α-targeting monoclonal antibody and the first biologic approved for moderate-to-severe UC, effectively induces mucosal healing (24) (Figure 1).
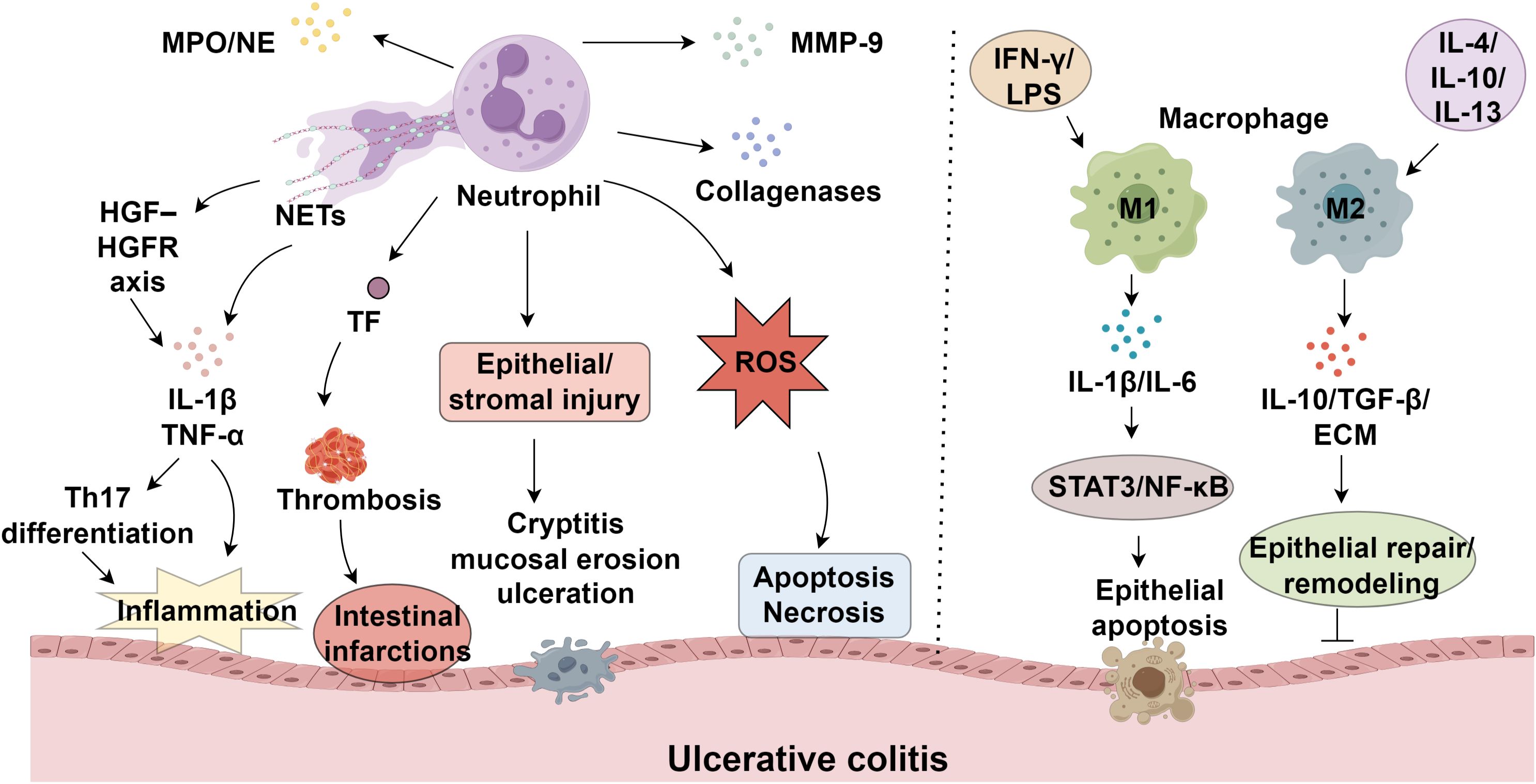
Figure 1. Neutrophils and macrophage polarization in ulcerative colitis progression.
2.2 Cytokine–NEU interactions drive inflammatory activity
Matrix metalloproteinases (MMPs), a family of zinc-requiring endopeptidases, play critical roles in extracellular matrix degradation and tissue remodeling, with their overexpression implicated in immune-mediated tissue damage (25, 26). Within ulcerative colitis, these enzymes drive disease progression through multiple mechanisms, including basement membrane breakdown, enhanced barrier permeability, regulation of epithelial repair, leukocyte migration, and angiogenic modulation (27, 28). Among MMPs, MMP-9 is predominantly secreted by NEUs upon degranulation and serves as a key contributor to UC pathogenesis (29, 30). By compromising epithelial tight junction integrity, MMP-9 exacerbates mucosal permeability and impairs barrier function (31). During active UC, NEUs constitute the predominant immune cell population in the lamina propria, acting as major effectors of mucosal injury (32). The dynamic interaction between NEUs and inflammatory cytokines is fundamental to UC development. Circulating and tissue-infiltrating NEUs produce IL-1β, which amplifies inflammatory responses and tissue destruction via dual mechanisms: NEU-derived serine proteases and inflammasome/caspase-1 activation (33). Hence, targeting NEU serine proteases or caspase-1 may offer novel therapeutic strategies. Stakenborg et al. reported that NEUs promote IL-1β and TNF-α production via the HGF–HGFR tyrosine kinase signaling axis, promoting Th17 differentiation and mucosal inflammation (34). Additionally, antigen-primed NEUs contribute significantly to UC exacerbations; upon re-exposure to antigens, IgG-bound Fcγ receptor I engagement on sensitized NEUs induces TNF-α release, further aggravating inflammation and precipitating disease recurrence (35).
2.3 Microbiome-NEUs crosstalk
NEUs play a paradoxical role in intestinal pathology, contributing to both inflammatory responses and tissue protection (36) While defending against microbial invasion through phagocytosis, NETs, antimicrobial peptides, and ROS. Simultaneously, NEUs also secrete cytokines, chemokines, and growth factors that facilitate mucosal repair and barrier regeneration (37). Notably, specific NEU subpopulations demonstrate enhanced protective functions. For instance, CD177+ neutrophils generate elevated ROS and antimicrobial peptides, strengthening mucosal defense while suppressing pro-inflammatory cytokine expression (38). Furthermore, CD177+ neutrophils produce IL-22, a key mediator in maintaining epithelial homeostasis (36). Research by Leppkes et al. revealed that NEUs accumulating in UC lesions form NETs in a PAD4-dependent manner, transforming blood clots into immune thrombi to reduce hemorrhage and accelerate tissue repair (39). The gut microbiota profoundly regulates NEU behavior in UC through multiple molecular mechanisms (40). Bacterial fermentation of dietary fibers yields short-chain fatty acids (SCFAs), including butyrate, propionate, and acetate, which are crucial in controlling neutrophil function (41). By activating GPR41 and GPR43 receptors on NEUs, SCFAs fine-tune ROS generation and facilitate inflammatory resolution (42). However, microbial dysbiosis in UC diminishes SCFA levels, compromising neutrophil regulation and perpetuating chronic inflammation (43). Additionally, microbial components directly influence NET formation. Pathogens such as Escherichia coli and Clostridium difficile induce NET release by engaging pattern recognition receptors (PRRs), particularly Toll-like receptors (TLRs), which initiate downstream signaling cascades (44, 45). Bacterial products like lipopolysaccharides (LPS) intensify this response by potentiating PRR activation, thereby aggravating UC-associated inflammation (46, 47). Although NETs, comprising DNA, histones, and antimicrobial proteins, worsen tissue injury, they also confine pathogens and restrict dissemination (48). Under homeostatic conditions, NETs aid in infection control without inciting persistent inflammation, whereas dysbiosis disrupts this equilibrium, exacerbating mucosal damage and disease severity (49).
2.4 Dynamic behavior of NEUs
NEUs occupy diverse functional states along a continuum from immune-enhancing/pro-resolving phenotypes to dysfunctional, hyperinflammatory programs often described as “exhausted” (50). Pro-resolving or immune-enhancing states can be experimentally induced—for example, “resolving memory neutrophils” trained with 4-phenylbutyrate show enhanced antimicrobial functions and distinct transcriptional features, while low-dose endotoxin can reprogram neutrophils toward immune-enhancing phenotypes (51). At the opposite end, chronic or excessive stimulation drives neutrophil programs with sustained inflammatory mediator release and impaired resolution capacity, consistent with exhausted-like states noted in single-cell studies and reviews of IBD myeloid heterogeneity (52, 53). These polarized neutrophil states have concrete implications in UC: immune-enhancing/pro-resolving programs may facilitate epithelial repair and hemorrhage control, whereas dysfunctional/exhausted programs amplify tissue injury through persistent NETosis, protease release, and cytokine production (16, 30, 39). Recognizing and therapeutically steering neutrophils toward immune-enhancing trajectories such as pro-resolving training and cautious innate “training” paradigms while restraining exhausted-like, hyperinflammatory activity could help tailor interventions for patients with persistent histologic activity (54–58).
3 Inhibition of NEU in UC
3.1 Inhibition of NEU-derived ROS and pro-inflammatory cytokines
Hesperidin methyl chalcone (HMC), a citrus flavonoid derivative, exerts antioxidative, anti-inflammatory, and analgesic effects by enhancing colonic glutathione levels and antioxidant capacity, thereby limiting NEU infiltration and mucosal damage in UC (15). The sesquiterpenoid compound nerolidol (NRD) demonstrates similar protective effects by suppressing myeloperoxidase (MPO) activity, a key marker of NEU recruitment, while concurrently reducing proinflammatory cytokine secretion and colonic inflammation (59). NRD further enhances cellular defense mechanisms through upregulation of superoxide dismutase and catalase, coupled with decreased ROS generation and lipid peroxidation (59–61). Cyclosporine A (CSA), a calcineurin inhibitor used in refractory UC cases, modulates NEU activity via SIRT6/HIF-1α-dependent metabolic regulation, inhibiting ROS production, MPO release, and antimicrobial peptide expression to prevent excessive neutrophil migration and apoptosis (62). Ursolic acid (UA), a triterpenoid isolated from medicinal plants and fruits, effectively reduces epithelial NEU migration and downregulates IL-6 expression in both systemic circulation and colonic tissues (63, 64). The artemisinin-derived compound SM934 exhibits potent immunosuppressive activity by significantly decreasing MPO levels and attenuating macrophage/NEU accumulation in inflamed colonic regions, leading to reduced IL-1β, IL-6, and TNF-α production (65). Another critical regulatory mechanism involves peptidoglycan recognition protein 1 (PGLYRP-1), which stimulates proinflammatory mediator release (TNF-α, IL-1β, IL-6, MPO) from neutrophils upon interaction with triggering receptor expressed on myeloid cells 1 (TREM-1). Therapeutic targeting of TREM-1 with neutralizing antibodies effectively disrupts this pathway, particularly in UC patients exhibiting heightened PGLYRP-1 expression and neutrophil infiltration (66).
3.2 Inhibition of NET formation and NE activity
Extrachromosomal DNA (ecDNA) is critically involved in the generation of NETs. The enzymatic degradation of ecDNA within the colonic microenvironment by DNases offers a promising therapeutic strategy for UC (67). To achieve site-specific delivery, staphylococcal nuclease (SNase), a highly efficient phosphodiesterase with broad substrate specificity, was formulated into calcium alginate microspheres (ALG-SNase). This targeted intervention facilitated NET disruption, attenuated inflammatory responses in the colon, enhanced epithelial barrier function, and increased expression of key tight junction proteins, including occludin and zonula occludens-1 (68). Peptidylarginine deiminase 4 (PAD4) is essential for histone citrullination during NET formation. Peptidylarginine deiminase 4 (PAD4) plays a crucial role in mediating histone citrullination, a prerequisite for NET formation. Studies demonstrate that NETs activate the cGAS-STING pathway in MC38 cells in a dose- and time-dependent manner, promoting the release of pro-inflammatory cytokines and impairing intestinal barrier integrity. Genetic ablation of STING ameliorates disease severity, as evidenced by improved clinical colitis scores, reduced intestinal inflammation, and restored barrier function. Notably, suppression of NET generation through PAD4 knockout attenuates STING upregulation (69). Pharmacological inhibition of this post-translational modification has shown therapeutic benefits in UC models (70). However, PAD4-deficient UC mice exhibit impaired mucosal healing due to defective remodeling of fibrin clots at wound sites (39).
Furthermore, NE’s proteolytic activity compromises the TNF-neutralizing efficacy of infliximab, lowering clinical response rates. Co-administration of exogenous protease inhibitors may counteract NE-mediated degradation, enhancing the efficacy of biologic therapy (71). Selective blockade of the neonatal Fc receptor (FcRn) alleviates UC pathology by suppressing NET formation in the colon through enhanced clearance of anti-neutrophil cytoplasmic antibodies (ANCAs) (72). Baicalein (BCL) demonstrates efficacy in preventing UC relapse by downregulating FcRn expression via inhibition of NF-κB signaling mediated by the p50/p65 heterodimer. Prolonged BCL treatment in UC mice significantly reduces colonic FcRn levels, serum ANCA titers, neutrophil-activating peptide (NAP) expression, and inflammatory markers (including TNF-α, IL-1β, and CRP), while improving disease activity indices and histological scores, outperforming sulfasalazine (73).
4 The role of macrophages in UC
4.1 The role of M1 macrophages in UC
Macrophages exhibit phenotypic plasticity in response to microenvironmental cues, polarizing into pro-inflammatory (M1) or anti-inflammatory (M2) subsets (74, 75). Polarization toward the M1 phenotype is predominantly induced by IFN-γ, LPS, and TNF-α (76). In UC, compromised intestinal epithelium permits microbial invasion, which is detected by M1 macrophages. These cells subsequently overproduce inflammatory cytokines and chemokines (77), exacerbating inflammation, tissue damage, and impaired healing (78, 79), and driving disease progression through cytokine-dependent mechanisms. In contrast, M2 macrophages, stimulated by IL-4, IL-10, or IL-13, exhibit diminished reactivity to bacterial antigens while maintaining phagocytic and antimicrobial activity (80). Their impaired regulatory function contributes to epithelial barrier dysfunction, a key feature of UC pathology (81). Notably, M1 macrophages impair mucosal integrity via excessive MMP secretion, especially MMP-9, which disrupts the ECM, elevating gut permeability and permitting additional immune cell migration (82). Pro-inflammatory cytokines such as IL-1β and IL-6 predominantly released by M1 macrophages (83, 84). Elevated IL-1β levels in UC patients weaken the intestinal barrier, permitting immune cell influx into the lamina propria and aggravating epithelial injury, thereby accelerating disease initiation (85). Similarly, IL-6 exacerbates mucosal edema, increases epithelial permeability, and triggers NF-κB signaling through STAT3 activation, fostering cytokine imbalance and amplifying tissue damage in UC (86). Collectively, these mechanisms sustain chronic inflammation and perpetuate UC progression by undermining intestinal barrier function (Table 1).
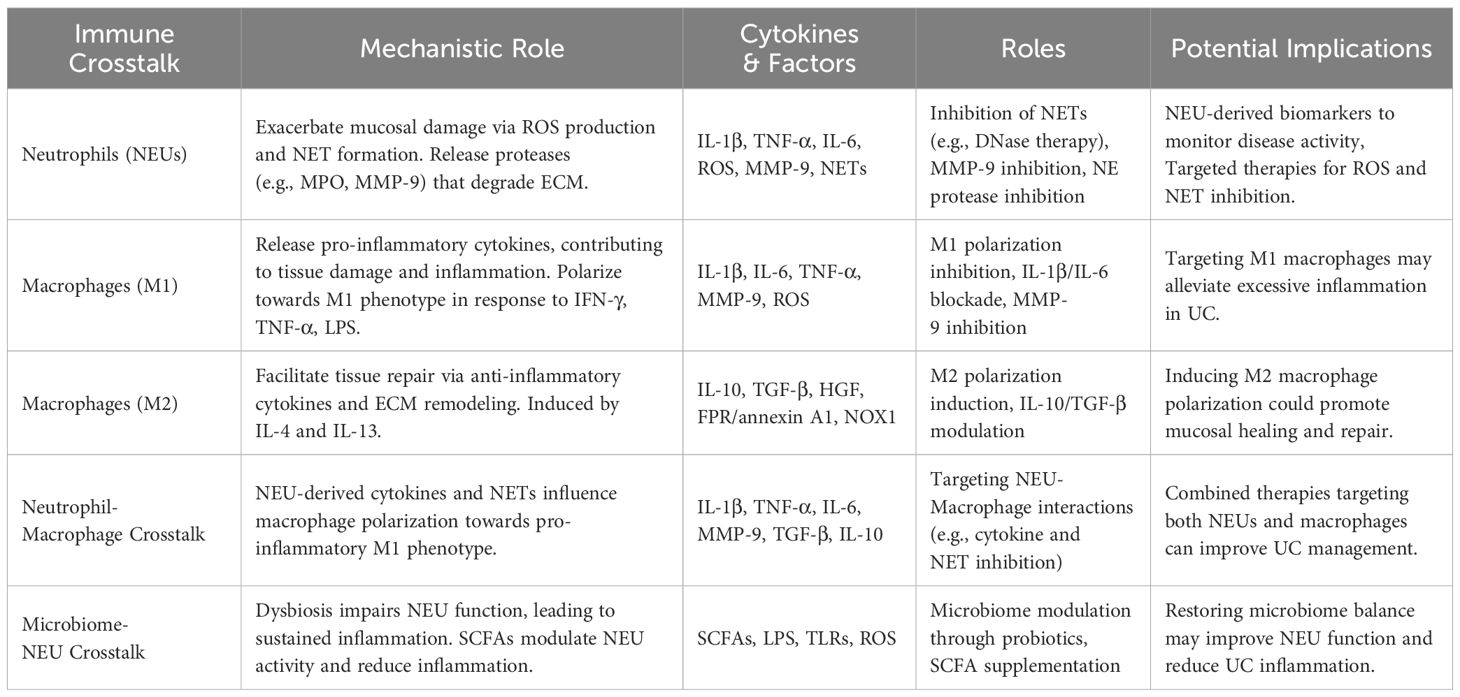
Table 1. Mechanisms and therapeutic strategies targeting neutrophils and macrophages in ulcerative colitis.
4.2 Exacerbation of intestinal inflammation
Under normal physiological conditions, macrophages in the colonic lamina propria express high levels of CX3CR1. However, in UC, microbial invasion or epithelial barrier disruption leads to the recruitment of inflammatory macrophages expressing intermediate CX3CR1 levels (CX3CR1int), derived from circulating CX3CR1low Ly6Chigh CCR2+ monocytes. These macrophages produce substantial pro-inflammatory mediators, drive local inflammation, and enhance effector T cell functions (87). Compared to their counterparts in healthy colonic tissue, macrophages in UC exhibit both phenotypic and functional alterations. Macrophages infiltrating inflamed colonic tissue in UC patients display an activated phenotype, increased TNF-α secretion, and enhanced stimulation of mucosal T cells, which, in turn, produce elevated IFN-γ levels (88). This cytokine interplay promotes epithelial apoptosis, compromises the mucosal barrier, and initiates pathological immune responses, leading to further infiltration of activated macrophages and T cells into the colonic mucosa. The disruption of Th1/Th2 homeostasis ultimately sustains and intensifies mucosal inflammation (89).
4.3 M2 macrophages in UC
Polarization of M2 macrophages is induced by the cytokines IL-4 and IL-13. In patients with IBD, M1-associated markers and pro-inflammatory cytokines are typically elevated, whereas M2-associated markers and IL-1 (90). In a dextran sulfate sodium (DSS)-induced murine model of UC, upregulation of Yes-associated protein (YAP) in macrophages was shown to drive M2 polarization and increase the production of anti-inflammatory cytokines such as IL-10 and IL-13, thereby suppressing intestinal inflammation and promoting mucosal healing (91). Similarly, activation of free fatty acid receptors FFAR1 and FFAR4 reduced lipid accumulation by enhancing fatty acid metabolism and induced M2 macrophage polarization, concomitantly increasing the expression of CD206, carnitine palmitoyltransferase-1α (CPT-1α), and anti-inflammatory cytokines (IL-4, IL-10, IL-13), ultimately ameliorating DSS-induced colitis (92). Concurrently, activation of the IL-4–STAT6 signaling pathway promoted M2 polarization and improved colonic mucosal injury (93), while this process can be suppressed by certain chemokines (94). M2 macrophages release anti-inflammatory mediators such as IL-10 and TGF-β, along with extracellular matrix (ECM) components, which collectively support epithelial repair and tissue remodeling (95). Additionally, they contribute to regenerative processes (96), mediated in part by hepatocyte growth factor (HGF) (97, 98), and initiate reparative mechanisms through pathways involving formyl peptide receptor (FPR)/annexin A1, NADPH oxidase 1 (NOX1), or IL-10/CREB/WISP-1 signaling (99). When exposed to microbial stimuli, M2 macrophages generate TNF-α, which triggers epithelial NF-κB activation, a critical regulator of mucosal homeostasis and inflammatory control (100). Although these cells predominantly display an M2-like phenotype, which appears essential for mucosal healing, their precise role in UC pathogenesis requires further investigation.
Under homeostatic conditions, tolerogenic macrophages are induced by dietary antigens or commensal microbiota, exhibiting a non-inflammatory profile characterized by diminished pro-inflammatory cytokine secretion and nitric oxide production, thereby preserving mucosal equilibrium (101). Following tissue damage, colonic macrophages engage in phagocytic clearance of pathogens and apoptotic cells, supporting microbial defense and epithelial repair. In UC, M2-polarized macrophages demonstrate dual functionality, combining antimicrobial activity and tissue remodeling with anti-inflammatory cytokine release, thereby alleviating intestinal injury (77). Emerging research has identified vessel-associated macrophages (VAMs) localized near colonic blood vessels. Single-cell transcriptomic analyses reveal elevated expression of genes associated with angiogenesis in these cells (102), with features aligning with M2 phenotype. VAMs contribute to a gut-vascular barrier, preventing microbial translocation to liver/systemic circulation (103, 104), effectively serving as vascular sentinels that safeguard microbial containment and vascular stability. Current therapeutic approaches targeting macrophage biology in UC focus predominantly on cytokine signaling modulation and polarization state manipulation (105).
5 The dynamic crosstalk between neutrophils and macrophages in UC
The pathogenesis of UC involves a complex interplay between neutrophils (NEUs) and macrophages, wherein neutrophil-derived mediators, including cytokines and neutrophil extracellular traps (NETs), modulate macrophage behavior (106, 107). IL-1β, TNF-α, and IL-6 released by activated NEUs promote macrophage polarization toward the pro-inflammatory M1 phenotype (108). Consequently, these polarized macrophages enhance the inflammatory response by producing additional cytokines and recruiting more immune cells to damaged tissues, exacerbating mucosal injury and perpetuating disease progression (12). M1 macrophages, in turn, secrete inflammatory cytokines such as IL-6 and IL-12, amplifying mucosal injury (109). NETs contribute to amplify the inflammatory cascade by reinforcing inflammatory signaling and providing a structural framework that facilitates macrophage infiltration (110, 111). However, emerging evidence suggests that NETs and neutrophil-derived signals may also play a role in resolving inflammation. In specific contexts, NETs promote the polarization of macrophages toward an M2 phenotype, characterized by the release of anti-inflammatory cytokines like IL-10 and TGF-β, which facilitate tissue repair (112). This dual functionality of NETs and NEUs underscores the intricate nature of their interactions with macrophages in UC. Given their opposing roles in inflammation and repair, targeting these cellular dynamics may present novel therapeutic opportunities for disease management.
6 Conclusion
The pathogenesis of UC is intricately linked to the dysregulated activities of NEUs and macrophages, which collectively drive inflammation, tissue injury, and impaired healing. NEUs amplify mucosal damage via ROS, NETs, and proteolytic enzymes. However, the protective subsets of NEUs, alongside their reparative cytokines, demonstrate their functional duality. Similarly, macrophages exhibit context-dependent roles: M1 polarization perpetuates inflammation through cytokine storms and barrier disruption, while M2 phenotypes promote microbial defense and epithelial repair.
To achieve histologic remission, which remains the gold standard for UC treatment, targeted therapies directed at NEUs and macrophages must be tailored to individual patient profiles. Specifically, patients with persistent subclinical inflammation despite endoscopic healing may benefit from therapies that more precisely modulate neutrophil activity, such as NET and MMP-9 inhibitors, or macrophage polarization strategies that encourage a shift toward the M2 phenotype. By focusing on these strategies, we may overcome challenges related to therapeutic resistance and the heterogeneity of UC, ultimately improving long-term patient outcomes. Further research into immune-stromal crosstalk and novel therapeutic agents is essential to refine treatment protocols for UC and move toward personalized, immune-centric approaches that can address the underlying mechanisms of persistent disease.
Author contributions
HW: Writing – original draft. TH: Writing – original draft. YM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the High Level Traditional Chinese Medicine Key Disciplines of the State Administration of Traditional Chinese Medicine, External Treatment of Traditional Chinese Medicine (zyyzdxk-2023116), the general program of Shandong Natural Science Foundation (No. ZR2021MH373), the fifth batch of National TCM clinical Excellent Talents Training Program (National TCM Education Word [2022] No.1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pravda J. Ulcerative colitis: Timeline to a cure. World J Gastroenterol. (2025) 31:108375. doi: 10.3748/wjg.v31.i26.108375
2. Buie MJ, Quan J, Windsor JW, Coward S, Hansen TM, King JA, et al. Global hospitalization trends for crohn’s disease and ulcerative colitis in the 21st century: A systematic review with temporal analyses. Clin Gastroenterol Hepatol. (2023) 21:2211–21. doi: 10.1016/j.cgh.2022.06.030
3. D’Amico F, Fasulo E, Jairath V, Paridaens K, Peyrin-Biroulet L, and Danese S. Management and treatment optimization of patients with mild to moderate ulcerative colitis. Expert Rev Clin Immunol. (2024) 20:277–90. doi: 10.1080/1744666X.2023.2292768
4. Li B, Sakaguchi T, Tani H, Ito T, Murakami M, Okumura R, et al. OTUD3 prevents ulcerative colitis by inhibiting microbiota-mediated STING activation. Sci Immunol. (2025) 10:eadm6843. doi: 10.1126/sciimmunol.adm6843
5. Magro F and Estevinho MM. Do neutrophils contribute to development of crohn’s disease and ulcerative colitis? Clin Gastroenterol Hepatol. (2020) 18:2430–1. doi: 10.1016/j.cgh.2020.01.032
6. Cai J, Liu J, Fan P, Dong X, Zhu K, Liu X, et al. Dioscin prevents DSS-induced colitis in mice with enhancing intestinal barrier function and reducing colon inflammation. Int Immunopharmacol. (2021) 99:108015. doi: 10.1016/j.intimp.2021.108015
7. Yuan S, Liu BH, Cheng WW, Meng H, Hou XT, Xue JC, et al. Polyphyllin VI modulates macrophage polarization through autophagy-NLRP3 inflammasome to alleviate inflammatory bowel disease. Phytomedicine. (2025) 143:156640. doi: 10.1016/j.phymed.2025.156640
8. Edwards M and Brockmann L. Microbiota-dependent modulation of intestinal anti-inflammatory CD4(+) T cell responses. Semin Immunopathol. (2025) 47:23. doi: 10.1007/s00281-025-01049-6
9. Li J, Liu Y, Sun Z, Zeng S, and Zheng C. Identification of neutrophil extracellular trap-related biomarkers in ulcerative colitis based on bioinformatics and machine learning. Front Genet. (2025) 16:1589999. doi: 10.3389/fgene.2025.1589999
10. Wéra O, Lancellotti P, and Oury C. The dual role of neutrophils in inflammatory bowel diseases. J Clin Med. (2016) 5:118. doi: 10.3390/jcm5120118
11. Muthas D, Reznichenko A, Balendran CA, Böttcher G, Clausen IG, Kärrman Mårdh C, et al. Neutrophils in ulcerative colitis: a review of selected biomarkers and their potential therapeutic implications. Scand J Gastroenterol. (2017) 52:125–35. doi: 10.1080/00365521.2016.1235224
12. Hu C, Wang Y, Liao S, Zhang L, Li C, Zhou D, et al. Neutrophil-macrophage hybrid membrane-coated pRussian blue nanozyme for ulcerative colitis treatment and mechanistic insights. J Nanobiotechnology. (2025) 23:43. doi: 10.1186/s12951-025-03123-5
13. Wang Z, Wu H, Chang X, Song Y, Chen Y, Yan Z, et al. CKMT1 deficiency contributes to mitochondrial dysfunction and promotes intestinal epithelial cell apoptosis via reverse electron transfer-derived ROS in colitis. Cell Death Dis. (2025) 16:177. doi: 10.1038/s41419-025-07504-4
14. Wei Q, Jiang H, Zeng J, Xu J, Zhang H, Xiao E, et al. Quercetin protected the gut barrier in ulcerative colitis by activating aryl hydrocarbon receptor. Phytomedicine. (2025) 140:156633. doi: 10.1016/j.phymed.2025.156633
15. Guazelli CFS, Fattori V, Ferraz CR, Borghi SM, Casagrande R, Baracat MM, et al. Antioxidant and anti-inflammatory effects of hesperidin methyl chalcone in experimental ulcerative colitis. Chem Biol Interact. (2021) 333:109315. doi: 10.1016/j.cbi.2020.109315
16. Dinallo V, Marafini I, Di Fusco D, Laudisi F, Franzè E, Di Grazia A, et al. Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis. J Crohns Colitis. (2019) 13:772–84. doi: 10.1093/ecco-jcc/jjy215
17. Kałużna A, Olczyk P, and Komosińska-Vassev K. The role of innate and adaptive immune cells in the pathogenesis and development of the inflammatory response in ulcerative colitis. J Clin Med. (2022) 11:400. doi: 10.3390/jcm11020400
18. Wang Y, Wu H, Sun J, Li C, Fang Y, Shi G, et al. Effects of the N-butanol extract of pulsatilla decoction on neutrophils in a mouse model of ulcerative colitis. Pharm (Basel). (2024) 17:1077. doi: 10.3390/ph17081077
19. Angelidou I, Chrysanthopoulou A, Mitsios A, Arelaki S, Arampatzioglou A, Kambas K, et al. REDD1/autophagy pathway is associated with neutrophil-driven IL-1β Inflammatory response in active ulcerative colitis. J Immunol. (2018) 200:3950–61. doi: 10.4049/jimmunol.1701643
20. Sandborn WJ, Panés J, Sands BE, Reinisch W, Su C, Lawendy N, et al. Venous thromboembolic events in the tofacitinib ulcerative colitis clinical development programme. Aliment Pharmacol Ther. (2019) 50:1068–76. doi: 10.1111/apt.15514
21. Gómez-Alonso IS, Martínez-García S, Betanzos-Cabrera G, Juárez E, Sarabia-León MC, Herrera MT, et al. Low concentration of the neutrophil proteases cathepsin G, cathepsin B, proteinase-3 and metalloproteinase-9 induce biofilm formation in non-biofilm-forming staphylococcus epidermidis isolates. Int J Mol Sci. (2022) 23:4992. doi: 10.3390/ijms23094992
22. Zhu HY, Wang HJ, and Liu P. Versatile roles for neutrophil proteinase 3 in hematopoiesis and inflammation. Immunol Res. (2024) 73:1. doi: 10.1007/s12026-024-09578-2
23. Uchiyama K, Naito Y, Takagi T, Mizushima K, Hirai Y, Hayashi N, et al. Serpin B1 protects colonic epithelial cell via blockage of neutrophil elastase activity and its expression is enhanced in patients with ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1163–1170. doi: 10.1152/ajpgi.00292.2011
24. Gilmore R, Tan WL, Fernandes R, An YK, and Begun J. Upadacitinib salvage therapy for infliximab-experienced patients with acute severe ulcerative colitis. J Crohns Colitis. (2023) 17:2033–6. doi: 10.1093/ecco-jcc/jjad115
25. Yan J, Xu X, Zhu Y, Wang Y, and Duan X. Escin Ia ameliorates DSS-induced chronic colitis in mice by inhibiting inflammation and oxidative stress via the LOXL2/MMP-9 pathway. J Ethnopharmacol. (2025) 345:119623. doi: 10.1016/j.jep.2025.119623
26. Opdenakker G, Vermeire S, and Abu El-Asrar A. How to place the duality of specific MMP-9 inhibition for treatment of inflammatory bowel diseases into clinical opportunities? Front Immunol. (2022) 13:983964. doi: 10.3389/fimmu.2022.983964
27. Bui TM, Yalom LK, Ning E, Urbanczyk JM, Ren X, Herrnreiter CJ, et al. Tissue-specific reprogramming leads to angiogenic neutrophil specialization and tumor vascularization in colorectal cancer. J Clin Invest. (2024) 134:e174545. doi: 10.1172/JCI174545
28. Sandborn WJ, Bhandari BR, Fogel R, Onken J, Yen E, Zhao X, et al. Randomised clinical trial: a phase 1, dose-ranging study of the anti-matrix metalloproteinase-9 monoclonal antibody GS-5745 versus placebo for ulcerative colitis. Aliment Pharmacol Ther. (2016) 44:157–69. doi: 10.1111/apt.13653
29. Dong Y, Zhao H, Man J, Fu S, and Yang L. MMP-9-mediated regulation of hypoxia-reperfusion injury-related neutrophil inflammation in an in vitro proximal tubular cell model. Ren Fail. (2021) 43:900–10. doi: 10.1080/0886022X.2021.1930558
30. Long D, Mao C, Xu Y, and Zhu Y. The emerging role of neutrophil extracellular traps in ulcerative colitis. Front Immunol. (2024) 15:1425251. doi: 10.3389/fimmu.2024.1425251
31. Bai X, Bai G, Tang L, Liu L, Li Y, and Jiang W. Changes in MMP-2, MMP-9, inflammation, blood coagulation and intestinal mucosal permeability in patients with active ulcerative colitis. Exp Ther Med. (2020) 20:269–74. doi: 10.3892/etm.2020.8710
32. Ranson N, Veldhuis M, Mitchell B, Fanning S, Cook AL, Kunde D, et al. NLRP3-dependent and -independent processing of interleukin (IL)-1β in active ulcerative colitis. Int J Mol Sci. (2018) 20:57. doi: 10.3390/ijms20010057
33. Zhang D, Duan S, He Z, Zhu Z, Li Z, Yi Q, et al. Sijunzi decoction targets IL1B and TNF to reduce neutrophil extracellular traps (NETs) in ulcerative colitis: evidence from silicon prediction and experiment validation. Drug Des Devel Ther. (2023) 17:3103–28. doi: 10.2147/DDDT.S428814
34. Stakenborg M, Verstockt B, Meroni E, Goverse G, De Simone V, Verstockt S, et al. Neutrophilic HGF-MET signalling exacerbates intestinal inflammation. J Crohns Colitis. (2020) 14:1748–58. doi: 10.1093/ecco-jcc/jjaa121
35. Li Y, Zhang YY, Yang LT, Liu JQ, Zhou C, Liu ZQ, et al. FcγRI plays a critical role in patients with ulcerative colitis relapse. Eur J Immunol. (2021) 51:459–70. doi: 10.1002/eji.202048622
36. El-Zimaity H, Shaffer SR, Riddell RH, Pai RK, and Bernstein CN. Beyond neutrophils for predicting relapse and remission in ulcerative colitis. J Crohns Colitis. (2023) 17:767–76. doi: 10.1093/ecco-jcc/jjac178
37. Danne C, Skerniskyte J, Marteyn B, and Sokol H. Neutrophils: from IBD to the gut microbiota. Nat Rev Gastroenterol Hepatol. (2024) 21:184–97. doi: 10.1038/s41575-023-00871-3
38. Zheng C, Li J, Chen H, Ma X, Si T, and Zhu W. Dual role of CD177 + neutrophils in inflammatory bowel disease: a review. J Transl Med. (2024) 22:813. doi: 10.1186/s12967-024-05539-3
39. Leppkes M, Lindemann A, Gößwein S, Paulus S, Roth D, Hartung A, et al. Neutrophils prevent rectal bleeding in ulcerative colitis by peptidyl-arginine deiminase-4-dependent immunothrombosis. Gut. (2022) 71:2414–29. doi: 10.1136/gutjnl-2021-324725
40. Pai RK, Jairath V, Vande Casteele N, Rieder F, Parker CE, and Lauwers GY. The emerging role of histologic disease activity assessment in ulcerative colitis. Gastrointest Endosc. (2018) 88:887–98. doi: 10.1016/j.gie.2018.08.018
41. Tanabe N, Matsumoto H, Morimoto C, and Hirai T. Sputum short-chain fatty acids, microbiome, inflammation, and mucus plugging in obstructive airway disease. J Allergy Clin Immunol. (2025) 155:1675–80. doi: 10.1016/j.jaci.2025.01.031
42. Wang S, Liu Y, Qin S, and Yang H. Composition of maternal circulating short-chain fatty acids in gestational diabetes mellitus and their associations with placental metabolism. Nutrients. (2022) 14:3727. doi: 10.3390/nu14183727
43. Zhou H, Sun J, Ge L, Liu Z, Chen H, Yu B, et al. Exogenous infusion of short-chain fatty acids can improve intestinal functions independently of the gut microbiota. J Anim Sci. (2020) 98:skaa371. doi: 10.1093/jas/skaa371
44. Vong L, Yeung CW, Pinnell LJ, and Sherman PM. Adherent-invasive escherichia coli exacerbates antibiotic-associated intestinal dysbiosis and neutrophil extracellular trap activation. Inflammation Bowel Dis. (2016) 22:42–54. doi: 10.1097/MIB.0000000000000591
45. Dong D, Su T, Chen W, Wang D, Xue Y, Lu Q, et al. Clostridioides difficile aggravates dextran sulfate solution (DSS)-induced colitis by shaping the gut microbiota and promoting neutrophil recruitment. Gut Microbes. (2023) 15:2192478. doi: 10.1080/19490976.2023.2192478
46. Candelli M, Franza L, Pignataro G, Ojetti V, Covino M, Piccioni A, et al. Interaction between lipopolysaccharide and gut microbiota in inflammatory bowel diseases. Int J Mol Sci. (2021) 22:6242. doi: 10.3390/ijms22126242
47. Abraham C, Abreu MT, and Turner JR. Pattern recognition receptor signaling and cytokine networks in microbial defenses and regulation of intestinal barriers: implications for inflammatory bowel disease. Gastroenterology. (2022) 162:1602–1616.e1606. doi: 10.1053/j.gastro.2021.12.288
48. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
49. Zhang D and Frenette PS. Cross talk between neutrophils and the microbiota. Blood. (2019) 133:2168–77. doi: 10.1182/blood-2018-11-844555
50. Caldwell BA, Wu Y, Wang J, and Li L. Altered DNA methylation underlies monocyte dysregulation and immune exhaustion memory in sepsis. Cell Rep. (2024) 43:113894. doi: 10.1016/j.celrep.2024.113894
51. Lin R, Yi Z, Wang J, Geng S, and Li L. Generation of resolving memory neutrophils through pharmacological training with 4-PBA or genetic deletion of TRAM. Cell Death Dis. (2022) 13:345. doi: 10.1038/s41419-022-04809-6
52. Garrido-Trigo A, Corraliza AM, Veny M, Dotti I, Melón-Ardanaz E, Rill A, et al. Macrophage and neutrophil heterogeneity at single-cell spatial resolution in human inflammatory bowel disease. Nat Commun. (2023) 14:4506. doi: 10.1038/s41467-023-40156-6
53. Gudiño V, Bartolomé-Casado R, and Salas A. Single-cell omics in inflammatory bowel disease: recent insights and future clinical applications. Gut. (2025) 74:1335–45. doi: 10.1136/gutjnl-2024-334165
54. Kalafati L, Hatzioannou A, Hajishengallis G, and Chavakis T. The role of neutrophils in trained immunity. Immunol Rev. (2023) 314:142–57. doi: 10.1111/imr.13142
55. Lajqi T, Köstlin-Gille N, Bauer R, Zarogiannis SG, Lajqi E, Ajeti V, et al. Training vs. Tolerance: the yin/yang of the innate immune system. Biomedicines. (2023) 11:766. doi: 10.3390/biomedicines11030766
56. Filep JG. Targeting neutrophils for promoting the resolution of inflammation. Front Immunol. (2022) 13:866747. doi: 10.3389/fimmu.2022.866747
57. Gupta A, Yu A, Peyrin-Biroulet L, and Ananthakrishnan AN. Treat to target: the role of histologic healing in inflammatory bowel diseases: A systematic review and meta-analysis. Clin Gastroenterol Hepatol. (2021) 19:1800–1813.e1804. doi: 10.1016/j.cgh.2020.09.046
58. Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med. (2021) 27:1970–81. doi: 10.1038/s41591-021-01520-5
59. Bastaki SMA, Amir N, Adeghate E, and Ojha S. Nerolidol, a sesquiterpene, attenuates oxidative stress and inflammation in acetic acid-induced colitis in rats. Mol Cell Biochem. (2021) 476:3497–512. doi: 10.1007/s11010-021-04094-5
60. Dong JR, Chang WW, and Chen SM. Nerolidol inhibits proliferation of leiomyoma cells via reactive oxygen species-induced DNA damage and downregulation of the ATM/Akt pathway. Phytochemistry. (2021) 191:112901. doi: 10.1016/j.phytochem.2021.112901
61. Xie H, Xi X, Lei T, Liu H, and Xia Z. CD8(+) T cell exhaustion in the tumor microenvironment of breast cancer. Front Immunol. (2024) 15:1507283. doi: 10.3389/fimmu.2024.1507283
62. Lu H, Lin J, Xu C, Sun M, Zuo K, Zhang X, et al. Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1α-glycolysis axis to alleviate severe ulcerative colitis. Clin Transl Med. (2021) 11:e334. doi: 10.1002/ctm2.334
63. Sheng Q, Li F, Chen G, Li J, Li J, Wang Y, et al. Ursolic acid regulates intestinal microbiota and inflammatory cell infiltration to prevent ulcerative colitis. J Immunol Res. (2021) 2021:6679316. doi: 10.1155/2021/6679316
64. Wei T, Wu L, Ji X, Gao Y, and Xiao G. Ursolic acid protects sodium dodecyl sulfate-induced drosophila ulcerative colitis model by inhibiting the JNK signaling. Antioxidants (Basel). (2022) 11:426. doi: 10.3390/antiox11020426
65. Yan YX, Shao MJ, Qi Q, Xu YS, Yang XQ, Zhu FH, et al. Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages. Acta Pharmacol Sin. (2018) 39:1633–44. doi: 10.1038/aps.2017.185
66. Brynjolfsson SF, Magnusson MK, Kong PL, Jensen T, Kuijper JL, Håkansson K, et al. An antibody against triggering receptor expressed on myeloid cells 1 (TREM-1) dampens proinflammatory cytokine secretion by lamina propria cells from patients with IBD. Inflammation Bowel Dis. (2016) 22:1803–11. doi: 10.1097/MIB.0000000000000822
67. Maronek M, Gromova B, Liptak R, Konecna B, Pastorek M, Cechova B, et al. Extracellular DNA correlates with intestinal inflammation in chemically induced colitis in mice. Cells. (2021) 10:81. doi: 10.3390/cells10010081
68. Dong W, Liu D, Zhang T, You Q, Huang F, and Wu J. Oral delivery of staphylococcal nuclease ameliorates DSS induced ulcerative colitis in mice via degrading intestinal neutrophil extracellular traps. Ecotoxicol Environ Saf. (2021) 215:112161. doi: 10.1016/j.ecoenv.2021.112161
69. Sun T, Wang P, Zhai X, Wang Z, Miao X, Yang Y, et al. Neutrophil extracellular traps induce barrier dysfunction in DSS-induced ulcerative colitis via the cGAS-STING pathway. Int Immunopharmacol. (2024) 143:113358. doi: 10.1016/j.intimp.2024.113358
70. Salas A. What good can neutrophils do in UC? Gut. (2022) 71:2375–6. doi: 10.1136/gutjnl-2021-326484
71. Curciarello R, Sobande T, Jones S, Giuffrida P, Di Sabatino A, Docena GH, et al. Human neutrophil elastase proteolytic activity in ulcerative colitis favors the loss of function of therapeutic monoclonal antibodies. J Inflammation Res. (2020) 13:233–43. doi: 10.2147/JIR.S234710
72. Wen C, Hu H, Yang W, Zhao Y, Zheng L, Jiang X, et al. Targeted inhibition of FcRn reduces NET formation to ameliorate experimental ulcerative colitis by accelerating ANCA clearance. Int Immunopharmacol. (2022) 113:109474. doi: 10.1016/j.intimp.2022.109474
73. Hu H, Lu F, Guan X, Jiang X, Wen C, and Wang L. Baicalein ameliorates experimental ulcerative colitis recurrency by downregulating neonatal fc receptor via the NF-κB signaling pathway. ACS Omega. (2025) 10:10701–12. doi: 10.1021/acsomega.5c00243
74. Qing J, Zhang Z, Novák P, Zhao G, and Yin K. Mitochondrial metabolism in regulating macrophage polarization: an emerging regulator of metabolic inflammatory diseases. Acta Biochim Biophys Sin (Shanghai). (2020) 52:917–26. doi: 10.1093/abbs/gmaa081
75. Zhai X, Zhang H, Xia Z, Liu M, Du G, Jiang Z, et al. Oxytocin alleviates liver fibrosis via hepatic macrophages. JHEP Rep. (2024) 6:101032. doi: 10.1016/j.jhepr.2024.101032
76. Zareie M, Singh PK, Irvine EJ, Sherman PM, McKay DM, and Perdue MH. Monocyte/macrophage activation by normal bacteria and bacterial products: implications for altered epithelial function in Crohn’s disease. Am J Pathol. (2001) 158:1101–9. doi: 10.1016/S0002-9440(10)64057-6
77. Zhang J, Zhao Y, Hou T, Zeng H, Kalambhe D, Wang B, et al. Macrophage-based nanotherapeutic strategies in ulcerative colitis. J Control Release. (2020) 320:363–80. doi: 10.1016/j.jconrel.2020.01.047
78. Yao Y, Xu T, Li X, Shi X, Wu H, Zhang Z, et al. Selenoprotein S maintains intestinal homeostasis in ulcerative colitis by inhibiting necroptosis of colonic epithelial cells through modulation of macrophage polarization. Theranostics. (2024) 14:5903–25. doi: 10.7150/thno.97005
79. Zhuang H, Lv Q, Zhong C, Cui Y, He L, Zhang C, et al. Tiliroside ameliorates ulcerative colitis by restoring the M1/M2 macrophage balance via the HIF-1α/glycolysis pathway. Front Immunol. (2021) 12:649463. doi: 10.3389/fimmu.2021.649463
80. Lundahl MLE, Mitermite M, Ryan DG, Case S, Williams NC, Yang M, et al. Macrophage innate training induced by IL-4 and IL-13 activation enhances OXPHOS driven anti-mycobacterial responses. Elife. (2022) 11:e74690. doi: 10.7554/eLife.74690.sa2
81. Zhen D, Wang S, Liu Z, Xi Y, Du H, Wang N, et al. Fibroblast growth factor 20 attenuates colitis by restoring impaired intestinal epithelial barrier integrity and modulating macrophage polarization via S100A9 in an NF-κB-dependent manner. Cell Mol Gastroenterol Hepatol. (2025) 19:101486. doi: 10.1016/j.jcmgh.2025.101486
82. Lissner D, Schumann M, Batra A, Kredel LI, Kühl AA, Erben U, et al. Monocyte and M1 macrophage-induced barrier defect contributes to chronic intestinal inflammation in IBD. Inflammation Bowel Dis. (2015) 21:1297–305. doi: 10.1097/MIB.0000000000000384
83. Li J, Luo X, Shiu PH, Cheng Y, Nie X, Rangsinth P, et al. Protective effects of Amauroderma rugosum on dextran sulfate sodium-induced ulcerative colitis through the regulation of macrophage polarization and suppression of oxidative stress. BioMed Pharmacother. (2024) 176:116901. doi: 10.1016/j.biopha.2024.116901
84. Wang S, Cai L, Ma Y, and Zhang H. Shaoyao decoction alleviates DSS-induced colitis by inhibiting IL-17a-mediated polarization of M1 macrophages. J Ethnopharmacol. (2025) 337:118941. doi: 10.1016/j.jep.2024.118941
85. Leonard F, Collnot EM, and Lehr CM. A three-dimensional coculture of enterocytes, monocytes and dendritic cells to model inflamed intestinal mucosa. vitro. Mol Pharm. (2010) 7:2103–19. doi: 10.1021/mp1000795
86. Elhefnawy EA, Zaki HF, El Maraghy NN, Ahmed KA, and Abd El-Haleim EA. Genistein and/or sulfasalazine ameliorate acetic acid-induced ulcerative colitis in rats via modulating INF-γ/JAK1/STAT1/IRF-1, TLR-4/NF-κB/IL-6, and JAK2/STAT3/COX-2 crosstalk. Biochem Pharmacol. (2023) 214:115673. doi: 10.1016/j.bcp.2023.115673
87. Grainger JR, Konkel JE, Zangerle-Murray T, and Shaw TN. Macrophages in gastrointestinal homeostasis and inflammation. Pflugers Arch. (2017) 469:527–39. doi: 10.1007/s00424-017-1958-2
88. Niu Y, Li A, Xu W, Zhang R, Mei R, Zhang L, et al. Platelet activation stimulates macrophages to enhance ulcerative colitis through PF4/CXCR3 signaling. Int J Mol Med. (2025) 55:78. doi: 10.3892/ijmm.2025.5519
89. Zhong Y, Xiao Q, Huang J, Yu S, Chen L, Wan Q, et al. Ginsenoside rg1 alleviates ulcerative colitis in obese mice by regulating the gut microbiota-lipid metabolism-th1/th2/th17 cells axis. J Agric Food Chem. (2023) 71:20073–91. doi: 10.1021/acs.jafc.3c04811
90. Creoli M, Di Paola A, Tarallo A, Aziz S, Miele E, Martinelli M, et al. Effects of CB2 receptor modulation on macrophage polarization in pediatric inflammatory bowel disease. Int J Mol Sci. (2025) 26:3720. doi: 10.3390/ijms26083720
91. Wang S, Zou F, Xu M, Wu Z, Xia P, and Deng F. The YAP/TEAD4 transcriptional complex in intestinal macrophages promotes M2 polarization and alleviates DSS-induced colitis via the regulation of C/EBPβ. Sci Rep. (2025) 15:11796. doi: 10.1038/s41598-025-95933-8
92. Zhang LS, Zhang ZS, Wu YZ, Guo B, Li J, Huang XQ, et al. Activation of free fatty acid receptors, FFAR1 and FFAR4, ameliorates ulcerative colitis by promote fatty acid metabolism and mediate macrophage polarization. Int Immunopharmacol. (2024) 130:111778. doi: 10.1016/j.intimp.2024.111778
93. Xiong K, Deng J, Yue T, Hu W, Zeng X, Yang T, et al. Berberine promotes M2 macrophage polarisation through the IL-4-STAT6 signalling pathway in ulcerative colitis treatment. Heliyon. (2023) 9:e14176. doi: 10.1016/j.heliyon.2023.e14176
94. Lin Y, Yang X, Yue W, Xu X, Li B, Zou L, et al. Chemerin aggravates DSS-induced colitis by suppressing M2 macrophage polarization. Cell Mol Immunol. (2014) 11:355–66. doi: 10.1038/cmi.2014.15
95. Daley JM, Brancato SK, Thomay AA, Reichner JS, and Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol. (2010) 87:59–67. doi: 10.1189/jlb.0409236
96. Morhardt TL, Hayashi A, Ochi T, Quirós M, Kitamoto S, Nagao-Kitamoto H, et al. IL-10 produced by macrophages regulates epithelial integrity in the small intestine. Sci Rep. (2019) 9:1223. doi: 10.1038/s41598-018-38125-x
97. Quiros M, Nishio H, Neumann PA, Siuda D, Brazil JC, Azcutia V, et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J Clin Invest. (2017) 127:3510–20. doi: 10.1172/JCI90229
98. Na YR, Stakenborg M, Seok SH, and Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. (2019) 16:531–43. doi: 10.1038/s41575-019-0172-4
99. Viola MF and Boeckxstaens G. Niche-specific functional heterogeneity of intestinal resident macrophages. Gut. (2021) 70:1383–95. doi: 10.1136/gutjnl-2020-323121
100. Delfini M, Stakenborg N, Viola MF, and Boeckxstaens G. Macrophages in the gut: Masters in multitasking. Immunity. (2022) 55:1530–48. doi: 10.1016/j.immuni.2022.08.005
101. Honda M, Surewaard BGJ, Watanabe M, Hedrick CC, Lee WY, Brown K, et al. Perivascular localization of macrophages in the intestinal mucosa is regulated by Nr4a1 and the microbiome. Nat Commun. (2020) 11:1329. doi: 10.1038/s41467-020-15068-4
102. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science. (2015) 350:830–4. doi: 10.1126/science.aad0135
103. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol. (2019) 71:1216–28. doi: 10.1016/j.jhep.2019.08.005
104. Park MD, Silvin A, Ginhoux F, and Merad M. Macrophages in health and disease. Cell. (2022) 185:4259–79. doi: 10.1016/j.cell.2022.10.007
105. Jia DJ, Wang QW, Hu YY, He JM, Ge QW, Qi YD, et al. Lactobacillus johnsonii alleviates colitis by TLR1/2-STAT3 mediated CD206(+) macrophages(IL-10) activation. Gut Microbes. (2022) 14:2145843. doi: 10.1080/19490976.2022.2145843
106. Tao Y, Xu L, Liu X, Wang P, Wei S, Huang Y, et al. Chitosan-coated artesunate protects against ulcerative colitis via STAT6-mediated macrophage M2 polarization and intestinal barrier protection. Int J Biol Macromol. (2024) 254:127680. doi: 10.1016/j.ijbiomac.2023.127680
107. Wu MM, Wang QM, Huang BY, Mai CT, Wang CL, Wang TT, et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol Res. (2021) 172:105796. doi: 10.1016/j.phrs.2021.105796
108. Korba-Mikołajczyk A. Służalska KD, Kasperkiewicz P: Exploring the involvement of serine proteases in neutrophil extracellular traps: a review of mechanisms and implications. Cell Death Dis. (2025) 16:535. doi: 10.1038/s41419-025-07857-w
109. Yan H and Li G. Inhibition of ca2+ induced macrophage oxidative stress cascade in mice with ulcerative colitis. Altern Ther Health Med. (2024) 30:222–9.
110. Wei X, Zou S, Xie Z, Wang Z, Huang N, Cen Z, et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc Res. (2022) 118:2179–95. doi: 10.1093/cvr/cvab269
111. Jia H, Yue G, Li P, Peng R, Jin R, Chen Y, et al. Neutrophil extracellular traps license macrophage production of chemokines to facilitate CD8+ T cell infiltration in obstruction-induced renal fibrosis. Protein Cell. (2025). doi: 10.1093/procel/pwaf020
Keywords: ulcerative colitis, neutrophil extracellular traps (NETs), macrophage polarization, mucosalimmunity, biomarkers, targeted therapy
Citation: Wang H, Huang T and Ma Y (2025) Mechanisms and therapeutic strategies of macrophages and neutrophils inducing ulcerative colitis progression. Front. Immunol. 16:1615340. doi: 10.3389/fimmu.2025.1615340
Received: 21 April 2025; Accepted: 15 August 2025;
Published: 29 August 2025.
Edited by:
Susetta Finotto, Universitätsklinikum Erlangen, GermanyReviewed by:
Zhijia Xia, Ludwig Maximilian University of Munich, GermanyCopyright © 2025 Wang, Huang and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuxia Ma, bXl4aWExOTc2QDE2My5jb20=