 Fatemeh Fattahi1†
Fatemeh Fattahi1† Jason S. Ellis1†Laura Vallance1Kristin Bahleda1,2Julia Holden1Sarah Socha1Joshua Meier1
Jason S. Ellis1†Laura Vallance1Kristin Bahleda1,2Julia Holden1Sarah Socha1Joshua Meier1 Francisco Gomez-Rivera1
Francisco Gomez-Rivera1 Ulus Atasoy1,3*
Ulus Atasoy1,3*- 1Division of Allergy and Clinical Immunology, Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI, United States
- 2Whitehead Institute for Biomedical Research, Cambridge, MA, United States
- 3Division of Allergy-Immunology, Ann Arbor VA Health System, Ann Arbor, MI, United States
Introduction: The RNA-binding protein HuR (Elavl1), a key post-transcriptional regulator, plays a critical role in T cell activation and function by stabilizing target mRNAs. To investigate the role of HuR in regulatory T cell (Treg) function, we generated the Foxp3YFP/Cre HuRfl/fl mouse model.
Methods: In this model, homozygous females and hemizygous males for Foxp3 developed a scurfy-like phenotype displaying autoimmune features, including failure to thrive, splenomegaly, hair loss, tail stippling, and widespread multi-organ immune cell infiltration. Molecular analysis included direct interaction studies between HuR and Foxp3 mRNA to assess mRNA stability, RNA sequencing of YFP⁺ Tregs, Protein-Protein Interaction (PPI) analysis, qPCR, and Treg functional assays.
Results: To our knowledge, this is the first study demonstrating that HuR directly binds and stabilizes Foxp3 mRNA in Tregs, using a novel Treg-specific HuR-deficient mouse model, with implications for autoimmune regulation. Foxp3 mRNA stability and expression were significantly reduced in Tregs from these HuR KO mice, despite higher frequencies of YFP⁺ Tregs. RNA sequencing revealed significant dysregulation of several pathways, including the T helper differentiation pathway, in which Foxp3 played a central role. PPI analysis showed a direct link between Foxp3 and Rorc (encoding RORγt), connecting Foxp3 to the T cell differentiation pathway via IL-23R. Our qPCR analysis supported these findings. Functional assays demonstrated a reduction in the suppressive capacity of HuR-deficient Tregs.
Conclusion: These findings together suggest that ablation of HuR in Tregs disrupts Foxp3 expression and Treg function, likely through dysregulation of T cell differentiation pathways involving RORγt. This potentially contributes to a disrupted Treg–Th17 axis and autoimmune dysfunction. These data suggest that HuR-mediated post-transcriptional regulation contributes to maintaining Foxp3 expression and immune homeostasis, although compensatory mechanisms such as increased IL-10 expression may also be involved.
1 Introduction
Regulatory T cells (Tregs) are essential for suppressing excessive immune responses and maintaining immune tolerance. In fact, these specialized CD4+ T cells prevent autoimmune diseases by suppressing autoreactive T cells and controlling inflammatory responses (1). The forkhead box P3 (Foxp3), a well-known transcription factor that defines Treg identity and function (2–5), specifically expressed in CD4+CD25+ Tregs and plays a central role in their development, stability, and suppressive function, making it the defining marker of Tregs (2). Foxp3 is critical for preventing excessive immune activation by enabling Tregs to maintain immune homeostasis and self-tolerance (6–8). Mutations or dysregulation of Foxp3 impair Treg function and contribute to the pathogenesis of autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and multiple sclerosis (MS), where the immune system attacks self-antigens (9, 10). Notably, the loss of functional Foxp3 (11–13) or imbalance of its isoforms (14, 15) leads to severe systemic autoimmunity known as immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome in humans and the scurfy phenotype in mice, both characterized by catastrophic immune dysregulation and widespread autoimmunity (16, 17). IPEX, which primarily affects males, with female carriers exhibiting mild symptoms, presents early in life with clinical features such as autoimmune thyroid disease, diabetes, eczema, hemolytic anemia, and failure to thrive, often resulting in a poor prognosis due to severe autoimmune manifestations (11–13, 18, 19).
The stability and expression of many mRNAs are tightly regulated by post-transcriptional mechanisms, including the actions of RNA-binding proteins, microRNAs, and signaling pathways that influence mRNA decay, translation, and degradation (10, 20, 21). Among these RNA-binding proteins, HuR (encoded by Elavl1) plays a key role by stabilizing target mRNAs, such as those in T cells, through binding AU-rich elements (AREs) within the 3′ untranslated region (UTR), thereby enhancing their stability and translation (20, 22). In contrast, other RNA-binding proteins such as the ZFP36 family (ZFP36, ZFP36L1, ZFP36L2) regulate Treg stability indirectly by limiting excessive cytokine signaling, controlling CTLA-4 recycling, and supporting IL-2/IL-7 responsiveness, maintaining immune homeostasis (23), underscoring the importance of post-transcriptional regulation in maintaining Treg stability.
HuR has been extensively studied in T cell activation by our group and others (22, 24–28), including its role in promoting Th17 differentiation by stabilizing critical transcripts such as RORγt, IRF4, and Runx1, which are required for Th17 lineage commitment and autoimmune responses (29–31). Beyond T cells, HuR has been implicated in broader immunoregulatory and disease-associated functions, including modulation of cytokine mRNAs, Th2 differentiation, and inflammatory responses across multiple immune cell types (26, 28). Furthermore, HuR supports oncogenic mRNA networks and profibrotic pathways in cancer, cardiac, and renal fibrosis, and has been targeted therapeutically by small-molecule HuR inhibitors such as KH-3 (32–37). However, to date, no study has directly examined the role of HuR in regulating Foxp3 expression and Treg function.
To address this knowledge gap, we generated a Foxp3YFP/Cre HuRfl/fl mouse in which HuR is selectively deleted in CD4+Foxp3+ cells, a lineage essential for self-tolerance and immune homeostasis. Treg-mediated immune tolerance is critical for suppressing autoimmunity, as shown by studies demonstrating that restoration of Foxp3 in Treg-deficient mice can reverse systemic inflammation (28). However, the molecular mechanisms that maintain Foxp3 expression and Treg stability remain incompletely understood.
Here, we show that Foxp3+ Treg-specific HuR deletion in this model results in abnormal phenotypic features similar to those seen in the scurfy mouse, which lacks functional Tregs due to a Foxp3 missense mutation (16, 38). Our data demonstrate that HuR deficiency leads to reduced Foxp3 expression and stability, partial impairment of Treg function, and disruption of key genes involved in the Treg-Th17 balance, including Rorc and Il23r. This imbalance between Tregs and Th17 cells, which share common precursors, has been suggested to contribute to autoimmune features (38), consistent with our mouse model findings.
2 Materials and methods
2.1 Generation of Foxp3YFP/Cre HuRfl/fl
The generation of the Foxp3YFP/Cre HuRfl/fl, also called HuR-KO Tregs, mice in our laboratory is described in Figure 1A. In brief, HuRfl/fl mice, previously generated in our lab (28), were crossed to Foxp3YFP/Cre mice (Jackson Laboratory, Bar Harbor, ME) to generate Foxp3YFP/Cre HuRfl/fl mice. HuR ablated Foxp3 cells in these mice are labeled with YFP. All mice used were on a C57BL/6 background. All animal procedures were performed under the U.S. National Institutes of Health guidelines and were approved by the University of Michigan Committee on the Use and Care of Animals. Foxp3-GFP mice (Jackson Laboratory) or HuRfl/fl mice were used as control mice where appropriate.
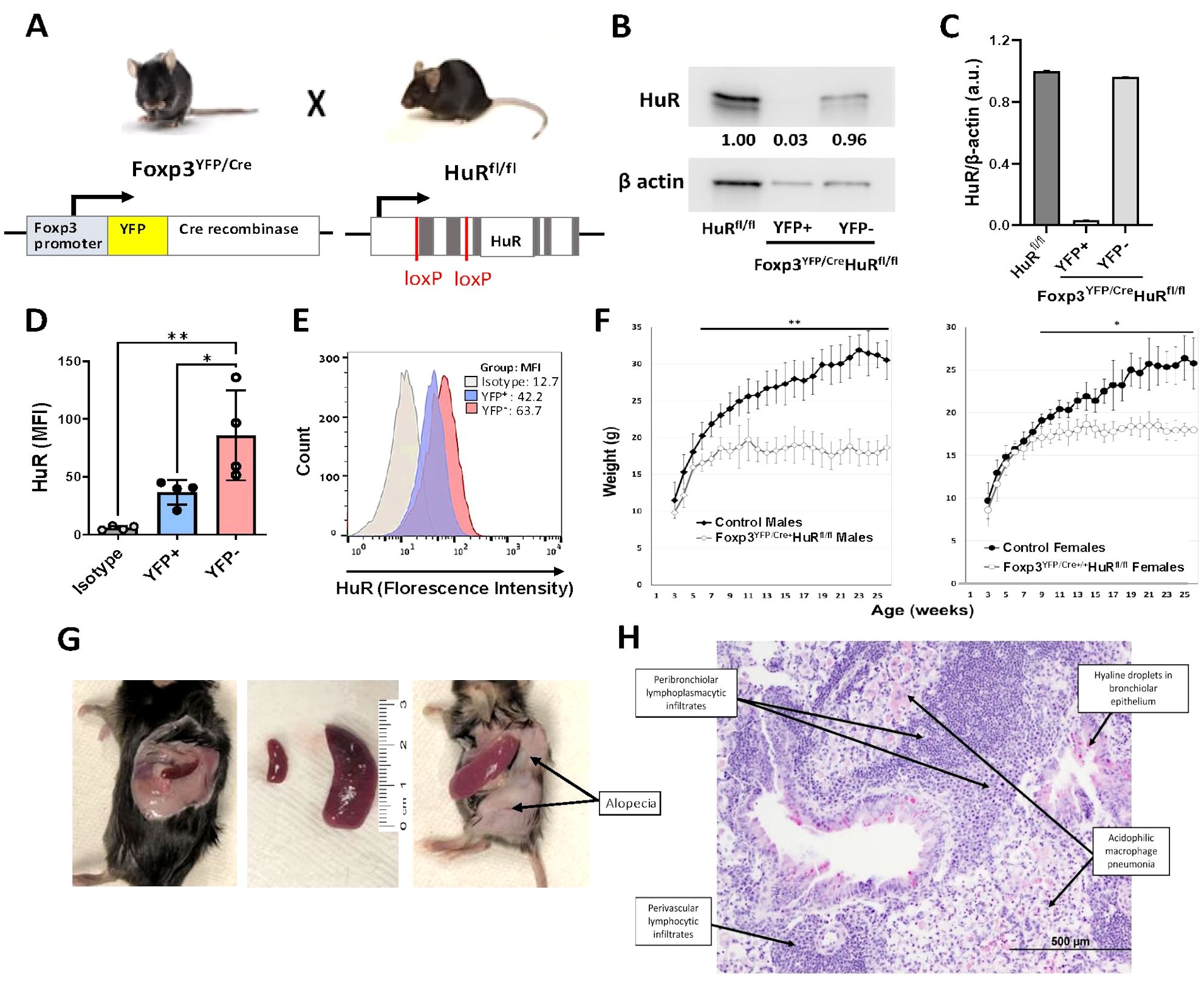
Figure 1. Treg-specific ablation of HuR in Foxp3YFP/Cre HuRfl/fl mice results in severe failure to thrive (FTT) and systemic spontaneous autoimmunity. (A) Schematic illustrating conditional deletion of HuR in Tregs in the Foxp3YFP/Cre, HuRfl/fl (HuR-KO Tregs) mouse model. (B) Western blot of HuR protein in sorted YFP+ (HuR-null) versus YFP− Treg populations showing near-complete HuR deletion in YFP+ cells, with β-actin as loading control. (C) Quantification of HuR/β-actin protein levels from western blots, showing HuR reduction in YFP+ Tregs compared to controls. (D) Bar graph of HuR mean fluorescence intensity (MFI) in YFP+ versus YFP− Tregs and isotype control, showing significant HuR reduction in YFP+ Tregs (mean ± SD, p < 0.05 by unpaired t-test, n = 4). (E) Flow cytometry histogram representative showing HuR expression profiles in YFP+, YFP−, and isotype control populations. (F) Body weight monitored from 3–26 weeks in male and female Foxp3YFP/Cre, HuRfl/fl (open symbols) versus controls (solid symbols). Both male and female HuR-deficient mice exhibited severe growth impairment (males plateaued at 18.5 ± 1.2 g vs. 31.5 ± 1.6 g in controls, p < 0.01; females plateaued at 18.3 ± 1.0 g vs. 26.5 ± 2.4 g in controls, p < 0.05). Two-way repeated measures ANOVA showed significant genotype effect. (G) Clinical phenotype in Foxp3YFP/Cre, HuRfl/fl mice including splenomegaly, alopecia, and tail stippling. (H) Representative H&E lung section from Foxp3YFP/Cre, HuRfl/fl mice demonstrating perivascular and peribronchiolar lymphoplasmacytic infiltrates, acidophilic macrophage pneumonia, and hyaline droplets in bronchiolar epithelium. Representative data are shown from at least 8 mice for panels F-H. The age of the mice at the time of analysis was 6–7 months. *p<0.05, **p<0.01.
2.2 Western blot analysis of HuR expression
CD4+CD25+ cells were first enriched from the pooled spleens of at least five Foxp3YFP/Cre HuRfl/fl mice using magnetic separation with the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec). The enriched population was then subjected to flow cytometric sorting to separate YFP+ (Foxp3-expressing Tregs) and YFP− cells. Sorted cells were lysed in RIPA buffer, and protein extracts were analyzed by SDS-PAGE followed by transfer to PVDF membranes. Membranes were probed with anti-HuR antibodies (Proteintech) to assess HuR expression and confirm the efficiency of HuR deletion specifically in Foxp3YFP/Cre HuRfl/fl Tregs.
2.3 Histological analysis
Tissue samples from various organs, including the lung, spleen, liver, and skin, among others, were collected from HuR-KO Treg mice and their wild-type (WT) controls after perfusion of the blood circulation with PBS. The tissues were fixed in 10% formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin (H&E), and evaluated for immune cell infiltration.
2.4 mRNA stability assay using actinomycin D
RNA stability of Foxp3 mRNA was assessed in splenic Tregs from WT control (non-KO littermate) and Foxp3YFP/Cre HuRfl/fl (HuR-KO Tregs) mice. Tregs were isolated using the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) following the manufacturer’s protocol. The isolated cells were cultured in RPMI 1640 complete medium containing 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate (all from Gibco), 50 µg/ml gentamicin sulfate (IBI Scientific), and 0.05 mM 2-mercaptoethanol (Fisher). Cells were plated on plates pre-coated with anti-CD3 (5 µg/ml) and anti-CD28 (2 µg/ml) antibodies (Invitrogen) and activated for 4 days. On day 4, transcription was inhibited by adding actinomycin D (5 µg/ml; Sigma-Aldrich), and cells were harvested at different time points over a 4-hour period for RNA extraction. Total RNA was isolated, converted to cDNA, and analyzed by quantitative real-time PCR (qRT-PCR) to measure Foxp3 expression. The relative expression of Foxp3 mRNA was quantified using the ΔΔCT method, normalized to Gapdh, with baseline (0 h) values set to 100%. In experiments involving pharmacological inhibition of HuR, WT Tregs were pretreated with either KH-3 (5 µg/ml), a small-molecule HuR inhibitor, or its inactive analog KH-3B (5 µg/ml), for 2 hours prior to activation. After 4 days of activation, cells were treated with actinomycin D as described above, and Foxp3 mRNA stability was evaluated over 3 hours. Decay curves were plotted on a semi-log scale, and mRNA half-lives were calculated using best-fit values.
2.5 RNA-sequencing and pathway analysis
YFP+ Tregs from Foxp3YFP/Cre HuRfl/fl mice were isolated by flow cytometric sorting following magnetic enrichment for CD4+CD25+ cells from at least six individual mice. Post-sort validation by intracellular Foxp3 staining showed around 90% positivity (data not shown). RNA was isolated using standard Trizol reagent procedures and subsequently sequenced on an Illumina HiSeq platform, as previously described by our group (25). The libraries were designed to achieve a depth that allowed comprehensive coverage of all expressed genes. RNA-seq data were analyzed and validated using quality control standards (25), with a focus on capturing comprehensive gene expression profiles. Differentially expressed genes were identified based on stringent criteria: a log fold change (logFC) greater than ±1 and an adjusted p-value threshold of less than 0.05. The details of these genes, including their fold changes and statistical significance, are listed in Supplementary Table 1.
We performed pathway enrichment analysis using Ingenuity Pathway Analysis (IPA). To further explore gene interactions, we used the STRING database via the stringApp plugin in Cytoscape (version 3.x), incorporating both known and predicted PPIs (39, 40). This approach helped us identify key regulatory nodes and build an interaction network among significantly altered genes. Given its essential role in preserving Treg identity and function, FOXP3 was selected as a central focus in our network analysis. To ensure interaction reliability, we applied a confidence score threshold greater than 0.4.
2.6 Quantitative PCR assay
RNA was extracted from isolated Tregs using the RNeasy RNA Extraction Kit (Qiagen) following the manufacturer’s protocol. cDNA synthesis was carried out using SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. Quantitative RT-PCR was then performed using Platinum SYBR Green Universal Master Mix (Invitrogen) on a QuantStudio 3 Real-Time PCR System (Applied Biosystems, Foster City, CA), as previously described (27, 41). Gene expression data were analyzed using the ΔΔCT method to calculate fold changes relative to the Gapdh as the reference gene. The primers used for amplification (Integrated DNA Technologies) are listed in Table 1.
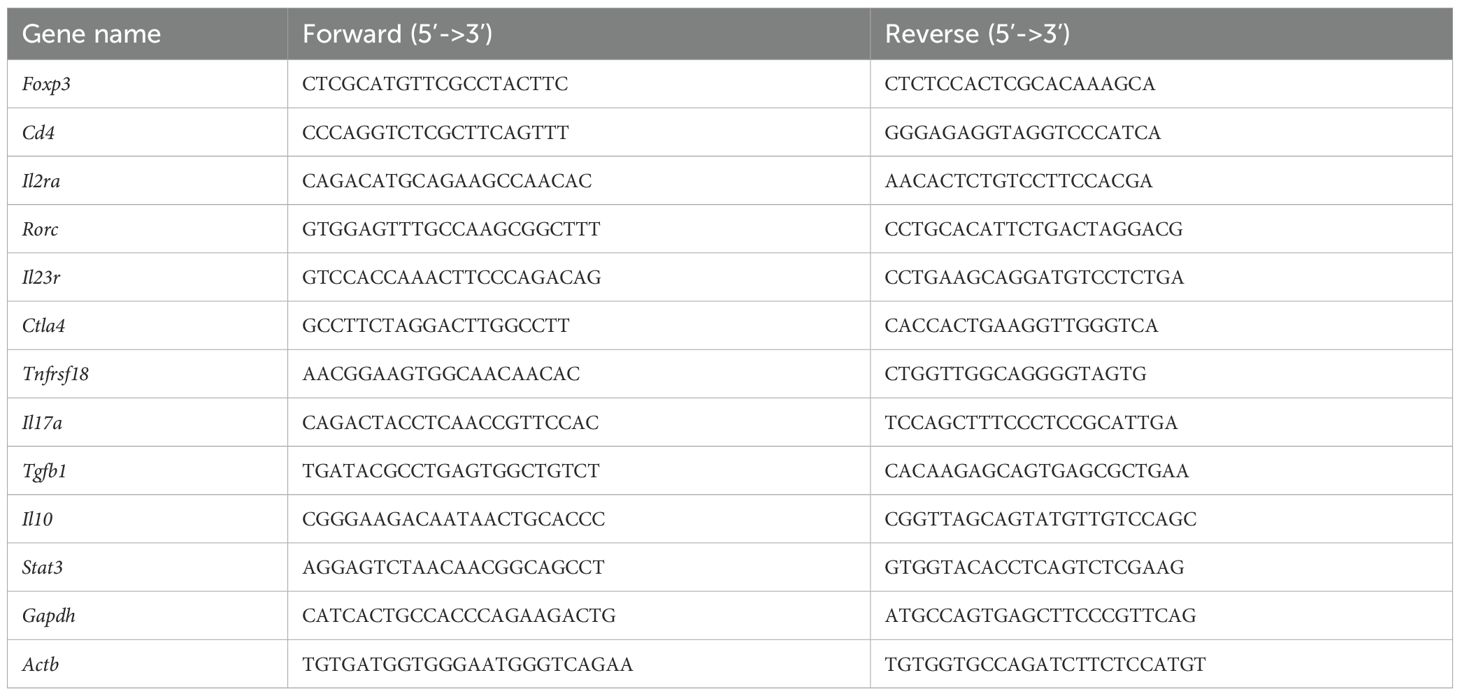
Table 1. Sequences of mouse qPCR primers used in the study.
2.7 RNA immunoprecipitation
RIP was performed on Tregs isolated from WT mice to investigate the interaction of HuR with its mRNA targets. The procedure followed protocols detailed in our previous publications (25, 42, 43). Tregs were isolated and pooled from at least 4 mice per experiment to ensure sufficient cell numbers. The cells were activated in vitro in complete RPMI for 4 days using anti-CD3 (5 µg/ml) and anti-CD28 (2 µg/ml) antibodies from eBioscience™. For the RIP assay, lysates were prepared in polysomal lysis buffer. HuR-associated RNA-protein complexes were immunoprecipitated using an anti-HuR antibody coupled to Protein A/G Sepharose beads. Normal IgG1 was used as an isotype control. RNA was extracted from the immunoprecipitates and analyzed by qRT-PCR. β-actin, a known HuR mRNA target, was used as a positive control, and enrichment of Foxp3 mRNA was quantified. For each RIP experiment, pooled Tregs from four mice were treated as a single biological sample for the immunoprecipitation, and three independent pooled replicates were performed; therefore, statistical testing was not applied to individual mice.
2.8 Treg suppression assay
To assess suppressive function, WT or Foxp3YFP/Cre HuRfl/fl (HuR-KO Tregs) mice were either left untreated or exposed to UVB light (100 mJ/day) for five consecutive days. On Day 5, spleens were harvested, and single-cell suspensions were prepared under sterile conditions by passing the mashed tissue through 70 µm cell strainers (Falcon), followed by centrifugation and resuspension in complete HT-2 medium (DMEM supplemented with 10% FBS, 1% L-glutamine, 1% sodium pyruvate, 0.1% gentamicin, and 50 µM β-mercaptoethanol).
CD4+CD25+ regulatory T cells (Tregs) were isolated from UV-treated mice, and CD4+CD25− effector T cells (Teffs) were isolated from non-UV-treated WT mice using magnetic separation (Miltenyi Biotec). Teffs were labeled with 5 µM Cell Proliferation Dye eFluor™ 670 (Invitrogen) by incubating for 10 minutes at 37 °C in the dark, followed by quenching with cold HT-2 medium, three washes, and resuspension at the desired concentration. In parallel, Tregs were labeled with 5 µM CellTrace™ CFSE (Invitrogen) by incubation for 20 minutes at 37 °C, then quenched with excess HT-2 medium, washed, and resuspended for co-culture.
Labeled Teffs and Tregs were co-cultured at indicated ratios (as described in the figure legend) in 96-well round-bottom plates with Dynabeads™ Mouse T-Activator CD3/CD28 (Gibco) in a final volume of 250 µL per well. Cultures were incubated for 72 hours at 37 °C in 5% CO2. After stimulation, cells were harvested and analyzed by flow cytometry. Teff proliferation was quantified using FlowJo software (Tree Star, Ashland, OR).
2.9 Statistical analyses
For statistical analysis, GraphPad Prism version 10 software (GraphPad Software, La Jolla, CA) was used. All values are expressed as means ± standard deviation. Data were analyzed by two-tailed Student t-test, one-way ANOVA with Tukey’s multiple comparison test, two-way repeated measures ANOVA or two-way ANOVA with Šídák’s multiple comparisons test as appropriate. Differences were considered significant when p < 0.05.
3 Results
3.1 Treg-specific ablation of HuR in Foxp3YFP/Cre HuRfl/fl mice results in a significant failure to thrive and spontaneous systemic autoimmunity
Our previous investigations using a T cell-specific HuR knockout (distal-Lck-Cre HuRfl/fl) revealed a significant reduction in both the frequency and expression levels of Foxp3 in splenic CD4+CD25+ T cells (data not shown). To extend these findings, we generated Foxp3YFP/Cre HuRfl/fl mice to investigate HuR’s role in maintaining Treg stability and function. These mice were produced by crossing our previously established HuRfl/fl strain (28) with the Foxp3YFP/Cre mouse (Figure 1A), resulting in HuR-deficient Tregs marked by YFP expression.
To further validate HuR deletion, HuR protein levels in sorted YFP+ and YFP− Treg populations were analyzed by Western blot. As shown in Figure 1B and quantified in Figure 1C, YFP+ Tregs from HuR-deficient mice exhibited nearly complete loss of HuR protein, whereas YFP− Tregs retained HuR levels comparable to controls. Additionally, flow cytometry studies of sorted YFP+ and YFP− Tregs from four individual mice confirmed significant HuR reduction in the YFP+ population (Figure 1D), with representative plots shown for isotype control, WT, and KO Tregs in Figure 1E. These results confirm efficient and specific deletion of HuR in Tregs.
Body weight was monitored weekly from 3 to 26 weeks in male and female Foxp3YFP/Cre HuRfl/fl (KO) mice (open symbols) and littermate controls (solid symbols). Both homozygous Foxp3YFP/Cre HuRfl/fl female mice and hemizygous male mice developed failure to thrive starting around 6 weeks of age, with significantly impaired weight gain compared to controls. In males, KO mice plateaued at 18.5 ± 1.2 g by week 9, while control males reached 31.5 ± 1.6 g by week 25. In females, KO mice plateaued at 18.3 ± 1.0 g by week 14, compared to 26.5 ± 2.4 g in controls. Two-way repeated measures ANOVA revealed a significant genotype effect across the time course (males p < 0.01; females p < 0.05), with Sidak’s post hoc test confirming differences at multiple time points (Figure 1F). These results indicate that HuR deficiency in Foxp3+ Tregs impairs normal growth in both sexes. Foxp3YFP/Cre HuRfl/fl mice also exhibited splenomegaly, hair loss, and tail stippling (Figure 1G). In contrast, heterozygous Foxp3YFP/Cre+/- HuRfl/fl mice appeared phenotypically normal and were indistinguishable from WT controls (data not shown).
Additionally, Foxp3YFP/Cre HuRfl/fl mice displayed moderate to severe inflammation across multiple organs, including the kidneys, lungs, skin, stomach, liver, and spleen, as summarized in Supplementary Table 2, with scoring performed by a blinded board-certified veterinary pathologist. Notably, the lungs showed dense perivascular and peribronchiolar lymphoplasmacytic infiltrates, acidophilic macrophage pneumonia, and hyaline droplets in the bronchiolar epithelium (Figure 1H). These features are consistent with widespread immune activation and resemble the autoimmune pathology seen in scurfy mice lacking functional Foxp3. In contrast, control lung sections from healthy WT control mice showed no lesions and preserved normal histological architecture (data not shown).
3.2 HuR directly interacts with Foxp3 mRNA and controls its steady-state levels and mRNA stability
To evaluate whether HuR regulates Foxp3 mRNA stability, we conducted actinomycin D chase assays. WT Tregs treated with the small-molecule HuR inhibitor KH-3 exhibited a significant reduction in Foxp3 mRNA stability, with a half-life of 1.9 hours compared to 4.3 hours in sham control-treated cells using its inactive analogue component (KH-3B, p < 0.01) (Figure 2A). A similar analysis in HuR-KO Tregs (Foxp3YFP/Cre HuRfl/fl) revealed a substantial reduction in Foxp3 mRNA stability compared to WT control Tregs, with half-lives of 1.9 and 4.2 hours, respectively (p < 0.001) (Figure 2B). These results indicate that HuR stabilizes Foxp3 mRNA and contributes to maintaining its steady-state levels. Quantification of Foxp3 steady-state mRNA levels in splenic and thymic Tregs further underscores HuR’s essential role in maintaining Foxp3 expression, as Foxp3 mRNA was markedly reduced in KO compared to WT (p < 0.01 in spleen and p < 0.05 in thymus) (Figure 2C). This reduction was specific to the CD4+CD25+ Treg subset, as CD4+CD25− cells exhibited negligible Foxp3 mRNA levels, irrespective of HuR expression. Together, these findings identify HuR as a key post-transcriptional regulator of Foxp3 mRNA in Tregs. RIP studies revealed enrichment of Foxp3 mRNA in the HuR pull-down fraction, showing around 8-fold increase compared to IgG1 controls. As expected, the positive control β-actin mRNA, due to relative abundance of transcript, exhibited high enrichment, validating the RIP assay (Figure 2D). As described in the Methods, RIP experiments were performed on pooled Treg samples, which were treated as single measurements; therefore, formal statistical comparisons were not feasible. These findings demonstrate that HuR directly interacts with Foxp3 mRNA, providing a mechanistic basis for its role in regulating mRNA stability and steady-state levels.
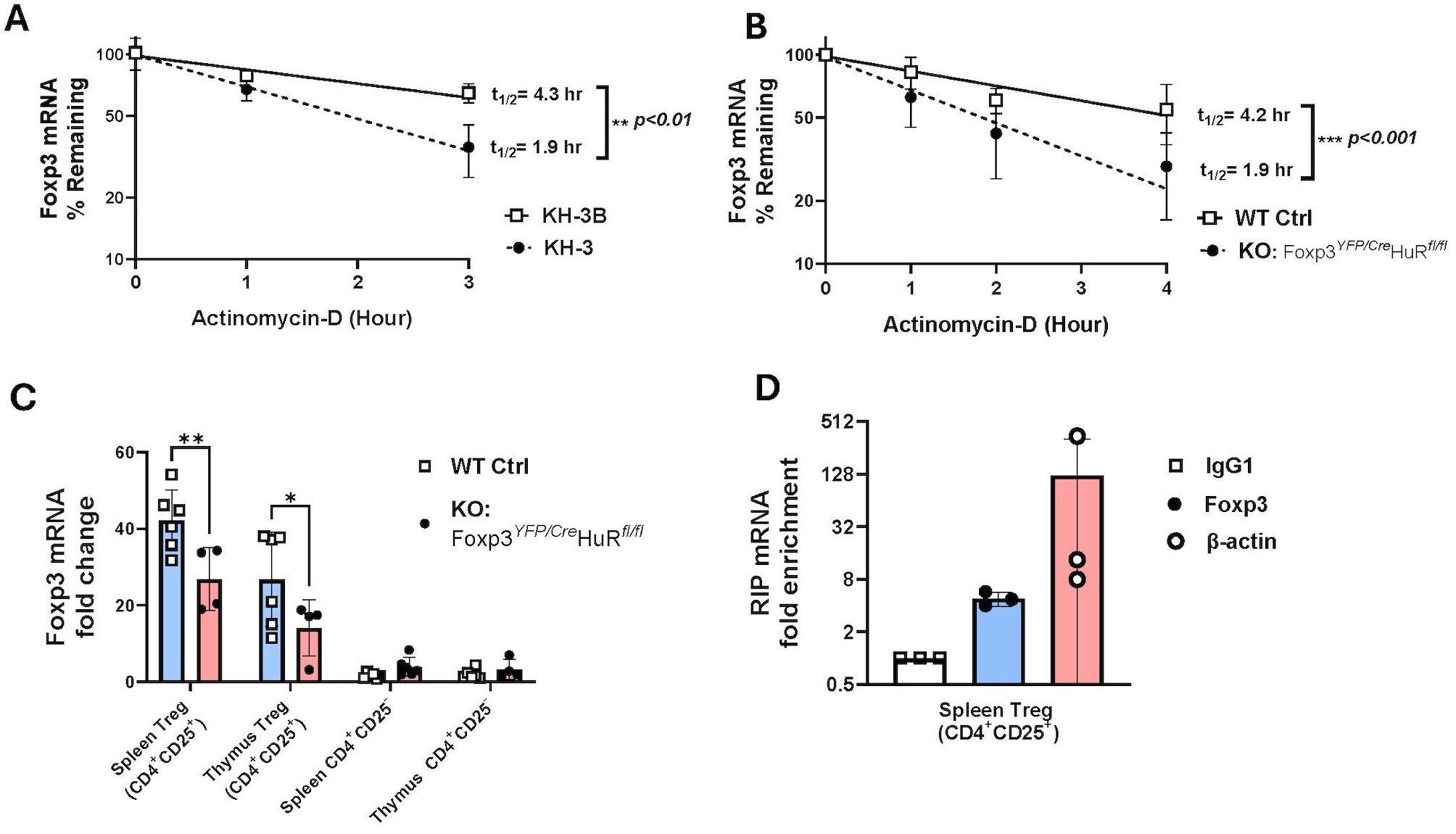
Figure 2. HuR directly interacts with Foxp3 mRNA and controls its steady-state levels and mRNA stability. (A) Foxp3 mRNA stability was evaluated in splenic Tregs (CD4+CD25+) isolated by magnetic column separation from WT mice treated with KH-3 or sham control (KH3-B) for 2 hours, followed by actinomycin (D) HuR inhibition reduced Foxp3 mRNA half-life (1.9 hours with KH-3) compared to control (4.3 hours). Data represents seven WT mice. (B) Foxp3 mRNA stability was assessed in column-isolated splenic Tregs from WT and Foxp3YFP/Cre HuRfl/fl (HuR-KO Tregs) mice. HuR-KO Tregs showed a shorter Foxp3 mRNA half-life (1.9 hours) compared to WT (4.2 hours). Data represents seven WT and four HuR-KO mice. (C) Steady-state Foxp3 mRNA levels were measured in splenic and thymic Tregs. Foxp3 mRNA was significantly lower in HuR-KO Tregs compared to WT control, while CD4+CD25− cells showed minimal expression with no significant differences. Data represent three HuR-KO and three WT mice. (D) RNA immunoprecipitation (RIP) from WT splenic Tregs showed Foxp3 mRNA enrichment in HuR pull-downs compared to IgG1 control, with β-actin as a positive control. Each point represents pooled cells from four WT mice. A total of 12 mice were used across three independent experiments. **p<0.01, ****p<0.0001.
3.3 Gene profiling of RNA-seq and PPI analysis reveals direct interaction between Rorc and Foxp3 in HuR-KO Tregs
To explore the downstream consequences of HuR ablation in Tregs, we performed RNA sequencing (RNA-Seq) on YFP+ Tregs isolated from Foxp3YFP/Cre HuRfl/fl (HuR KO) mice and WT controls. This analysis identified significant transcriptional dysregulation in the HuR-KO Treg population, including key molecules involved in T-helper cell differentiation (Supplementary Table 3).
RNA-Seq analysis identified 2,220 significantly dysregulated genes in YFP+ Tregs from HuR-KO Tregs mice compared to WT controls (Supplementary Table 1). The volcano plot highlights differentially expressed genes, with the top genes visualized (Figure 3A). Among these, Foxp3 itself was significantly downregulated, suggesting the direct regulatory role of HuR in stabilizing Foxp3 mRNA. Similarly, Rorc (encoding RORγt) and Il23R were also significantly downregulated, suggesting a perturbation in the Treg-Th17 axis. This axis is critical for maintaining the balance between regulatory and effector T cell functions, and its dysregulation could contribute to the autoimmune phenotype observed in HuR-KO Tregs mice.
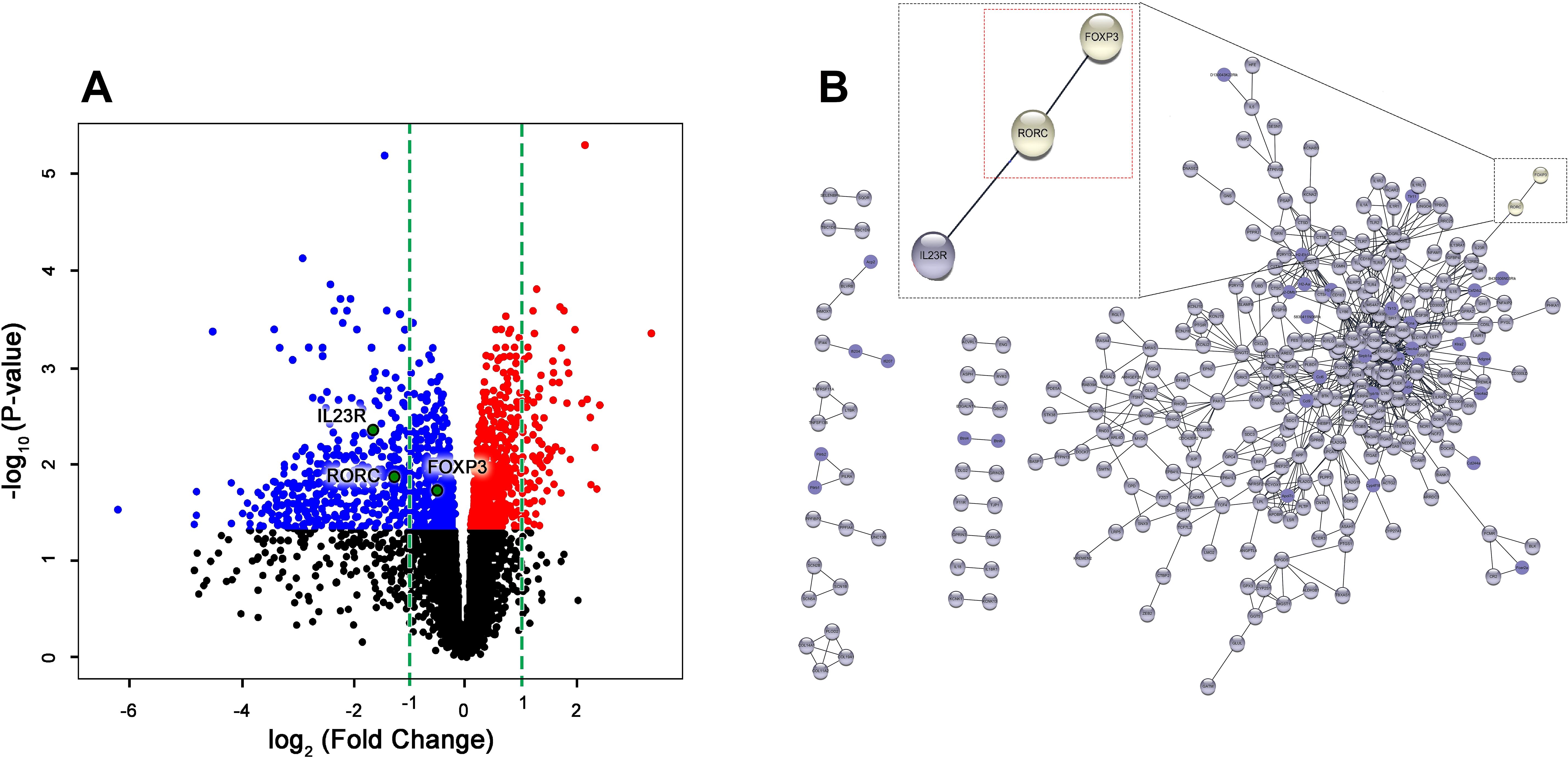
Figure 3. Gene profiling of RNA-seq and PPI analysis reveals direct interaction between Rorc and Foxp3 in HuR-KO Tregs in Foxp3YFP/Cre HuRfl/fl Tregs. (A) Volcano plot visualization of RNA-Seq results from sorted YFP+ (HuR-KO Tregs) isolated from the spleen of Foxp3YFP/Cre HuRfl/fl mice. The plot shows the expression levels of 2220 statistically significant genes in YFP+ (HuR-KO Tregs), with differentially expressed genes highlighted (upregulated in red, downregulated in blue). FOXP3, Rorc, and IL-23R, were identified and labeled. (B) Protein-protein interaction (PPI) network analysis using Cytoscape identified significant gene interactions. Among the differentially expressed genes in YFP+ (HuR-KO Tregs), Rorc (RORγt) was found to directly interact with FOXP3, as shown in the enlarged region (red dashed box). n = 6 mice.
To further explore the molecular pathways disrupted in HuR-deficient Tregs, Ingenuity Pathway Analysis (IPA) was performed on the RNA-Seq data. This analysis identified significant enrichment of genes involved in T helper cell differentiation, with a specific focus on the interplay between Foxp3 and Rorc. Supplementary Table 3 illustrates the involvement of Rorc in the differentiation of Th17 cells, a key effector T cell subset that maintains immune balance alongside Tregs. The role of Foxp3 and Rorc in opposing cellular fates is further emphasized in the pathway. As shown in the enlarged region of Supplementary Table 3, Foxp3 suppresses Rorc to promote Treg identity, while Rorc promotes Th17 differentiation. The simultaneous downregulation of both Foxp3 and Rorc in HuR-deficient Tregs suggests a breakdown in this tightly regulated axis.
PPI analysis further identified a direct interaction between Foxp3 and Rorc (Figure 3B), supporting the hypothesis that HuR plays a critical role in maintaining Treg function by regulating the Treg-Th17 axis. The downregulation of Rorc and Il23r in HuR-KO Tregs suggests a potential mechanism through which HuR regulates immune tolerance and contributes to the autoimmune phenotype observed in the model.
3.4 Altered mRNA expression of selected genes involved in T helper differentiation and Treg function in Foxp3YFP/Cre HuRfl/fl Tregs compared to WT control Tregs
The mRNA quantification of selected genes involved in Treg function and T helper differentiation pathways in Foxp3YFP/Cre HuRfl/fl (HuR-KO Tregs) compared to WT control Tregs is shown in Figure 4. The mRNA levels of Rorc (Figure 4A), which encodes RORγt and is critical for Th17 differentiation, were significantly reduced in HuR-KO Tregs compared to WT Tregs (p < 0.05). Similarly, Il23r (Figure 4B), a receptor involved in Th17 differentiation, also showed a significant decrease in HuR-KO Tregs (p < 0.05), further highlighting the dysregulation of the Treg-Th17 balance. The mRNA expression of Ctla4 (Figure 4C), an important molecule for Treg-mediated immune suppression, was also significantly reduced in HuR-KO Tregs. Il2r (encoding CD25) (Figure 4D), a key receptor for IL-2 signaling in Tregs, showed downregulated levels, though the result approached significance in HuR-KO Tregs (p = 0.054). The mRNA levels of Il10 (Figure 4E), an anti-inflammatory cytokine, were significantly increased in HuR-KO Tregs (p < 0.001). Additionally, Il17a (Figure 4F), a pro-inflammatory cytokine associated with Th17 cells, was significantly downregulated in HuR-KO Tregs (p < 0.05), further supporting the disruption of the Treg-Th17 axis. Genes such as Tnfrsf18 (encoding GITR), Stat3, and Tgfb1, were also assessed but did not show significant differences between the two groups (data not shown). These results suggest that the disruption of HuR leads to impaired genes of Treg function and those involved in the T helper differentiation pathway, particularly affecting the balance between Treg and Th17 cell function.
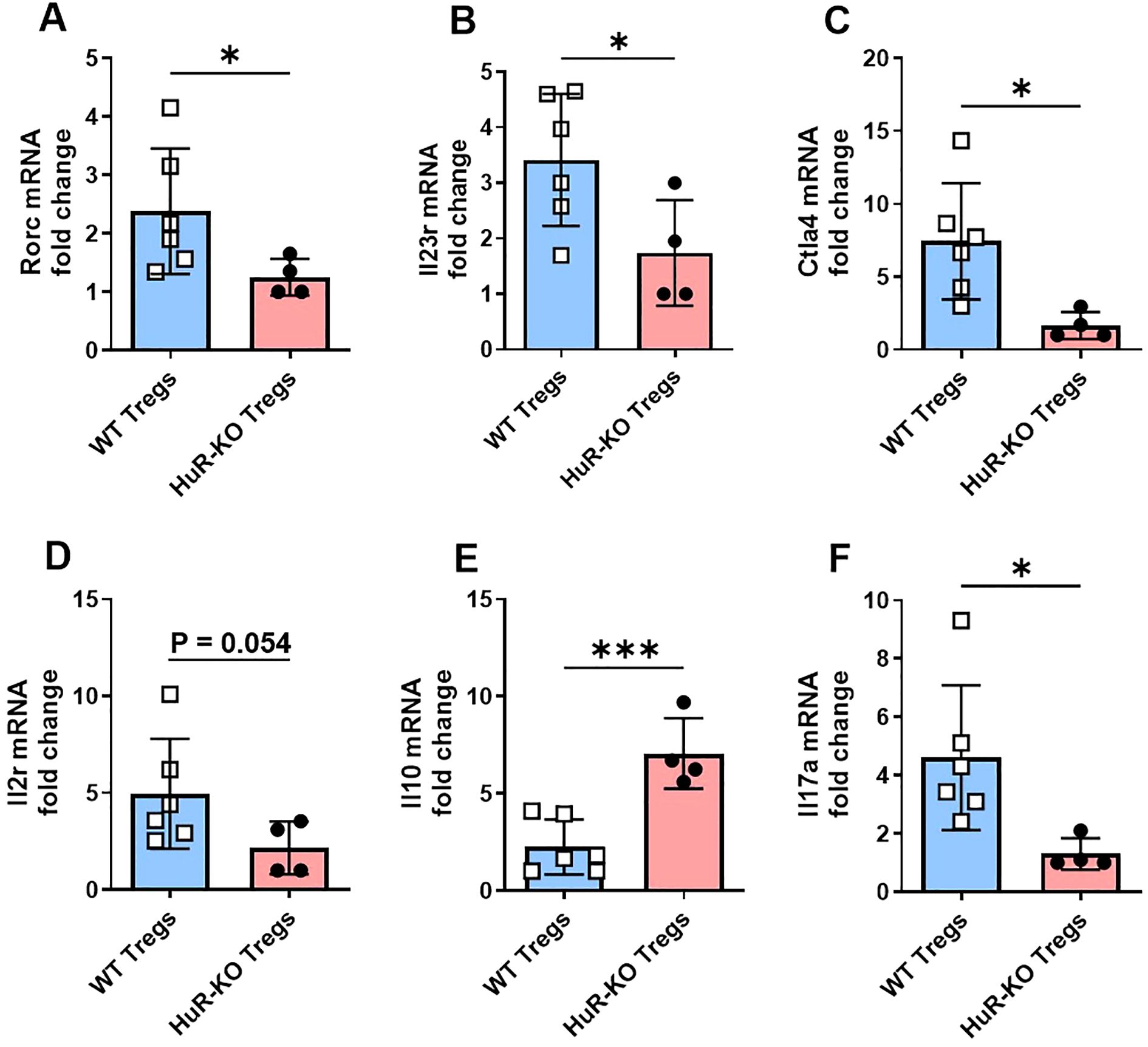
Figure 4. Altered mRNA expression of selected genes involved in T helper differentiation and Treg function in Foxp3YFP/Cre HuRfl/fl Tregs compared to WT control Tregs. (A-F) mRNA expression of key genes involved in T helper differentiation (based on RNA-seq IPA analysis) and Treg function including Rorc, Il23r, Ctla4, Il2r, Il10 and Il17a was measured by qPCR in CD4+CD25+ Tregs isolated from the spleens of HuR-KO and WT control mice, following 4 days of activation with anti-CD3 and anti-CD28. Data were analyzed using unpaired Student’s t-test. Results are representative of four independent HuR-KO mice and a total of six corresponding WT controls, from cohorts distinct from those used in RNA-seq analysis.
3.5 Impaired suppressive function of HuR KO-Tregs (Foxp3YFP/Cre HuRfl/fl) on T effector cell proliferation
We performed an immunosuppression assay to evaluate the suppressive capacity of Tregs isolated from the spleens of Foxp3YFP/Cre HuRfl/fl (KO) and HuRfl/fl (WT) mice. Tregs (CD4+CD25+) were co-cultured with labeled effector T cells (Teff, CD4+CD25−) at varying Treg: Teff ratios (1:2 to 1:512) and activated with anti-CD3/CD28 Dynabeads. The proliferation of Teff cells was measured and expressed as a percentage of proliferating cells.
In Figure 5, panel A summarizes the Teff proliferation percentages across the different Treg: Teff ratios achieved by titrating Treg cell numbers, while panel B shows a representative flow cytometry histogram demonstrating higher Teff proliferation when co-cultured with HuR-KO Tregs compared to WT Tregs, suggesting impaired suppressive function of the HuR-KO Tregs.
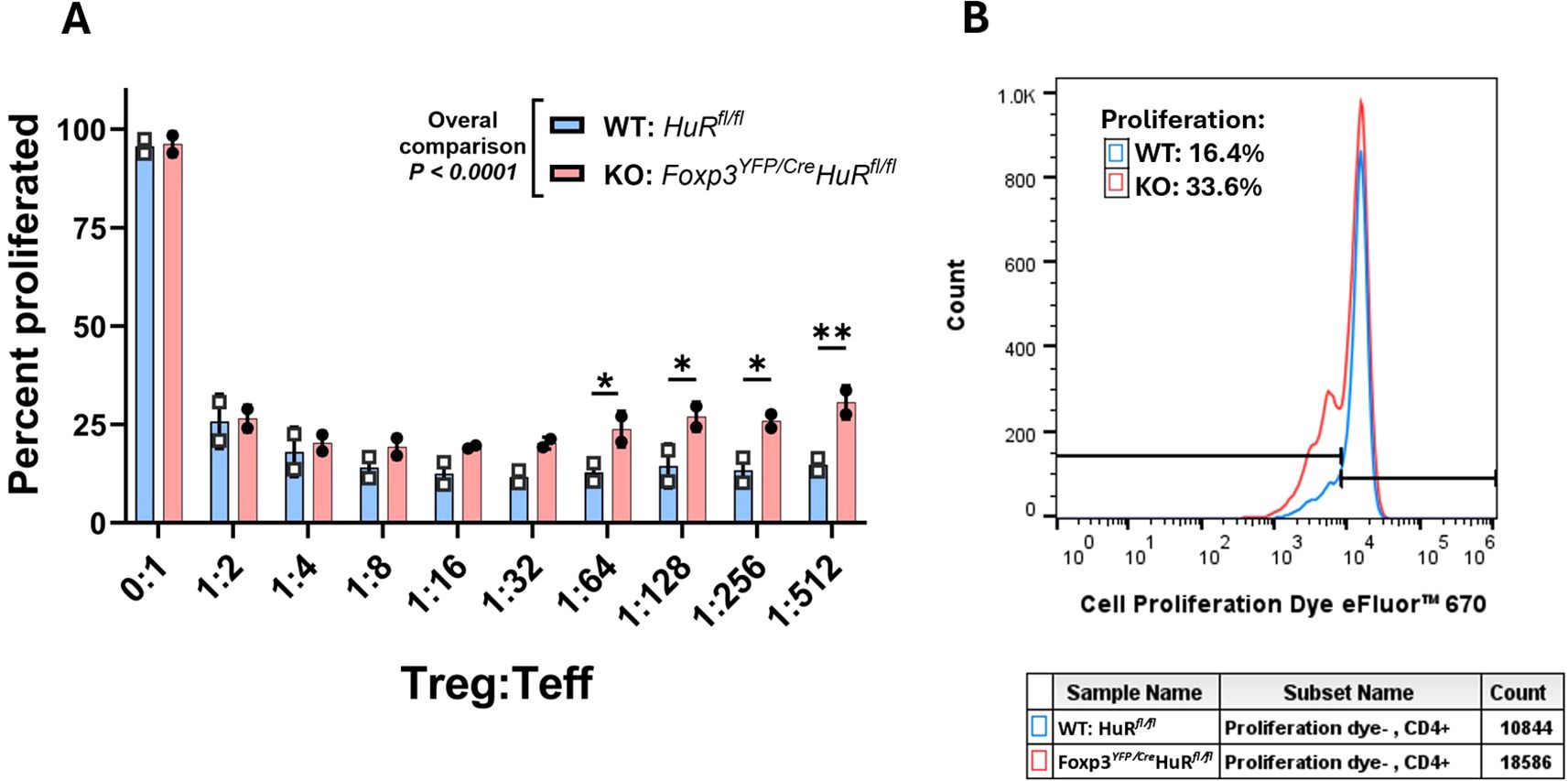
Figure 5. Impaired suppressive function of HuR-KO Tregs (Foxp3YFP/Cre HuRfl/fl) on Teffector cell proliferation. The in vitro suppressive ability of Tregs in regulating Teffector cell (Teff) proliferation was evaluated by flow cytometry. Teff cells were labeled with a proliferation dye prior to co-culture with isolated Tregs (CD4+CD25+) from control HuRfl/fl WT (blue bars) and Foxp3YFP/Cre HuRfl/fl (red bars) mice at varying Treg: Teff ratios, as indicated on the x-axis. (A) Percentage suppression of Teff cell proliferation by CD4+CD25+ Tregs across different Treg: Teff ratios. HuR-KO Tregs showed a significant, progressive decline in suppressive capacity beginning at the 1:64 ratio, whereas WT Tregs maintained stable suppression even at lower Treg frequencies. (B) Representative flow cytometry histograms of Teff proliferation patterns (gated on live cells), demonstrating reduced suppression by HuR-KO Tregs compared to WT Tregs. Overall, HuR-KO Tregs exhibited significantly reduced suppression of Teff cell proliferation (p < 0.0001). Data are from five mice per group, each run-in duplicate. Data were analyzed using two-way ANOVA followed by Šídák’s multiple comparisons test. *p<0.05; **p<0.01.
Overall, HuR-deficient (KO) Tregs exhibited significantly reduced suppressive capacity compared to WT Tregs (p < 0.0001). In WT Tregs, suppression of Teff proliferation reached a plateau from 1:8 to 1:512 Treg: Teff ratios, indicating stable suppressive potential even at lower Treg frequencies. In contrast, KO Tregs showed a progressive decline in suppressive capacity starting at the 1:64 ratio, failing to achieve a comparable plateau. This pattern suggests that HuR may be important for sustaining Treg-mediated suppression under conditions of high Teff challenge and low Treg frequency, where intrinsic regulatory mechanisms, such as post-transcriptional control via HuR, may become limiting. This is conceptually consistent with previous reports highlighting Treg functional thresholds under Teff-dominant conditions (44–46).
4 Discussion
Our data identify a new role for HuR in stabilizing Foxp3 mRNA, which is essential for sustaining Treg function. Using RIP and actinomycin D assays, we showed that HuR binds to Foxp3 mRNA and stabilizes it, preventing degradation. This is in line with earlier studies on other cell types showing HuR promotes mRNA stability by binding to AREs in the 3′ UTRs of target mRNAs expanding our previous work on HuR’s post-transcriptional regulation during T cell activation (22, 25–27). Here, we specifically show that this mechanism is relevant in Tregs. By generating a novel mouse model of Foxp3YFP/Cre HuRfl/fl through conditional ablation of HuR in Tregs, we identified extensive phenotypic and molecular disruptions in Tregs, resulting in an autoimmune phenotype. One of the interesting findings was that Foxp3 expression was reduced in the HuR-KO Tregs, which occurred despite a compensatory increase in Treg populations numbers, as revealed in flow cytometry: ~15% YFP+ CD4+ cells in the spleen, compared to ~10% GFP+ cells in Foxp3-GFP controls (Supplementary Figure 2A).
As stated above, dysregulation of Foxp3 contributes to various autoimmune diseases due to uncontrolled inflammation (2–5). Given that FOXP3 mutations cause IPEX in humans and the scurfy phenotype in mice (16, 17), it is notable that Treg-specific HuR deletion produced a similarly severe inflammatory phenotype, albeit with extended survival. However, unlike scurfy mice, they survived longer, which may make them useful for studying chronic Treg dysfunction and testing therapeutic strategies, even though they remained clinically ill throughout their life. A recent study by Rudensky’s group showed that Foxp3 restoration in Treg-deficient mice reverse systemic autoimmunity even in established inflammation (47). Their data underscores the essential role of Foxp3 in maintaining immune tolerance and preventing autoimmune pathology. While Foxp3 reconstitution can rescue autoimmunity (47), our findings highlight that proper regulation of Foxp3 expression, mediated by HuR, is critical to prevent the onset of autoimmunity (48). Supporting this, deletion of miR-15/16 in Tregs similarly disrupts Foxp3 expression and immune tolerance, underscoring the broader importance of RNA-based post-transcriptional regulation in Treg stability.
Our het females who did not show abnormal phenotype showed down regulation of Foxp3 in their YFP+ population. This is similar to the female carrier for the IPEX syndrome who only express mild form of the disease.
Our RNA-seq analysis revealed significant disruption of immune regulatory pathways, particularly T helper cell differentiation, with Foxp3 emerging as a central regulator of Treg function. HuR-deficient Tregs exhibited downregulation of both Foxp3 and Rorc (encoding RORγt), suggesting a disturbed Treg-Th17 axis, as confirmed by RNA-seq and qPCR. We identified Rorc as a key node connecting Foxp3 to the Th17 pathway via IL-23R in our PPI network analysis, implicating these genes in the maintenance of Treg-Th17 balance. Consistent with this, IL-23R was also significantly downregulated in HuR-KO Tregs, showing a failure in this regulatory axis. An earlier study showed a similar defect in Th17 differentiation in HuR-KO CD4+ T cells in the spleen during experimental autoimmune encephalomyelitis (EAE), where loss of HuR in OX40-Cre HuRfl/fl mice reduced RORγt, IRF4, and RUNX1 expression, key transcription factors essential for Th17 lineage commitment, ultimately impairing Th17 responses and autoimmune neuroinflammation (30). Rorc and Foxp3 are known to antagonize each other to dictate the balance between Tregs and Th17 cells. The simultaneous reduction of Foxp3 and Rorc in HuR-KO Tregs suggests a disrupted regulatory interplay, which likely contributes to increased inflammatory responses. This dysregulation has been implicated in various autoimmune diseases (49–54). Our findings position HuR as an upstream regulator that maintains this axis through post-transcriptional stabilization of Foxp3 and its interacting partners (2–5). By destabilizing Foxp3 and its linked components, such as Rorc and IL-23R, HuR ablation may influence signaling pathways required for Treg differentiation and Th17 regulation. This extends our understanding of the intricate balance between Tregs and Th17 cells and highlight HuR’s role in maintaining immune balance. Although we did not investigate direct evidence of HuR binding to Rorc mRNA in Tregs, and to our knowledge no prior study has confirmed direct HuR-Rorc RIP in T cells, its downregulation may plausibly reflect direct HuR targeting as suggested by our previous work in other Th17-associated pathways (29–31), or indirect regulation through Foxp3-dependent transcriptional networks. Future RIP-seq or CLIP analyses using larger pooled Treg samples could help clarify whether Rorc is a direct HuR target within the Treg compartment.
Interestingly a recent study using bioinformatics analysis of human Crohn’s disease identified ELAVL1 as a top upregulated hub gene in peripheral blood, with enrichment in T cell activation and NOD-like receptor signaling pathways (55), highlighting the importance role of HuR in immune dysregulation and its potential contribution to autoimmune and inflammatory disease contexts.
We found an upregulation of IL-10, a key anti-inflammatory cytokine, which may reflect a compensatory response to the heightened inflammation in HuR-KO Tregs. This is consistent with earlier studies which have shown elevated levels of IL-10 in autoimmune diseases such as SLE, RA, and MS (56), where IL-10 often acts as an ‘alarm cytokine’ connected to the disease severity and progression (57, 58). These findings show the paradoxical role of IL-10 in immune regulation which may contribute to the inflammatory responses observed in HuR-KO Tregs mice.
We found downregulation of Il2ra (encoding CD25) and Ctla4 in HuR-KO Tregs. Since Treg-mediated immunosuppression is critical for controlling effector T cell proliferation and function (59–61), reduced expression of these molecules may contribute to the impaired suppressive capacity of HuR-KO Tregs. CD25, the alpha chain of the IL-2 receptor, is essential for Treg survival and function by allowing efficient uptake of environmental IL-2 during activation (62–65). In typical Tregs, CD25 is abundantly expressed and plays a key role in limiting Teff growth by depleting IL-2 from their surroundings (63). Our findings complement our previously published work which demonstrates that HuR posttranscriptionally regulates IL-2 homeostasis and CD4+ Th2 differentiation (26). This highlights the importance of HuR in maintaining the functional integrity of Tregs.
Lastly, based on immunophenotyping of a limited number of HuR-KO Treg mice (n = 3) (Supplementary Figure 2), we observed dysregulation across various immune cell populations, not restricted to Tregs. These immune changes resemble those observed in models of SLE and RA, where imbalanced inflammation and loss of self-tolerance are well-documented features (66–71). The immune phenotyping data on HuR-KO Tregs mice, although with limited mice, exhibits increase in plasmacytoid dendritic cells (pDCs) but decreased myeloid DCs. This pattern mirrors findings in human SLE, where elevated pDC levels and reduced mDC numbers are reported (72). These cells are implicated in the progression of autoimmune diseases like SLE and psoriasis (73). The altered dendritic cell profile in our model suggests that HuR may play a critical regulatory role in dendritic cell homeostasis and function, potentially offering novel insights into the molecular mechanisms driving autoimmune responses.
We acknowledge several limitations in our study to consider. 1) The number of HuR-KO mice available for experiments was limited due to poor breeding efficiency from using sick male knockouts and carrier female mice, despite optimization efforts such as modified chow and supportive care. As a result, tissue collection was often based on availability rather than tightly age-matched time points, introducing unavoidable variability that may have influenced the immune responses. 2) While we focused on the Treg-Th17 axis, Th1 responses and potential Treg plasticity toward Th1-like phenotypes may also contribute to the observed inflammation and warrant further investigation. 3) In vivo suppression assays, such as adoptive transfer of HuR-deficient Tregs into lymphopenic hosts, would have provided more direct evidence for their functional competence, but these experiments were not feasible in the current study due to limited Treg numbers and poor colony health in the HuR-KO mouse colony. 4) For most of our experimental studies, Tregs were isolated as CD4+CD25+ cells, which may include a small number of activated non-Tregs. 5) Finally, we did not evaluate cytokine production by Teff cells in the co-culture with Tregs due to limited cell numbers; future studies can investigate this to further define the functional consequences of HuR-KO Tregs on Teff responses.
Despite these constraints, our study offers new insights into HuR’s role in immune regulation. Loss of HuR in Tregs led to widespread immune dysregulation, implicating HuR in both Treg function and broader immune homeostasis. Using a novel Treg-specific HuR-deficient mouse model, we demonstrated that HuR binds and stabilizes Foxp3 mRNA in Tregs ex vivo, providing evidence of its regulatory role in maintaining Treg function in vivo. Future studies involving 3′UTR luciferase reporter assays will be important to precisely define the HuR-binding regions within the Foxp3 transcript. Additionally, adoptive transfer experiments using HuR-deficient Tregs in lymphopenic hosts could directly test their in vivo suppressive function and further validate the physiological relevance of our findings. Since HuR (ELAVL1) is also among the top upregulated hub genes in blood samples from Crohn’s disease patients, underscoring its translational relevance (55). With future support, we aim to expand our colony and further investigate HuR’s role across immune compartments and autoimmune disease contexts.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was approved by University of Michigan IACUC Ann Arbor Michigan. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
FF: Methodology, Writing – original draft, Formal analysis, Writing – review & editing. JE: Methodology, Writing – review & editing. LV: Investigation, Writing – review & editing. KB: Investigation, Writing – review & editing. JH: Investigation, Writing – review & editing. JM: Investigation, Writing – review & editing. SS: Investigation, Writing – review & editing. FG-R: Formal analysis, Writing – review & editing. UA: Methodology, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the U.S. Department of Veterans Affairs, Biomedical Laboratory Research and Development Merit Award (VA Merit Award, grant number CX002491), and by the U.S. Department of Health and Human Services, National Institutes of Health (NIH) grant number R21 AI173487.
Acknowledgments
The authors would like to thank Mohi Shakiba at the University of Michigan for his assistance and technical support in preparing the manuscript figures. The authors also extend special thanks to Fahimeh Fattahi at Arak University of Medical Sciences, Arak, Iran, for her valuable guidance and helpful suggestions, particularly with the PPI analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1618677/full#supplementary-material
References
1. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, and Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. (2013) 13:461–7. doi: 10.1038/nri3464
2. Fontenot JD, Gavin MA, and Rudensky AY. Foxp3 programs the development and function of CD4+ss+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
3. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. (2007) 27:786–800. doi: 10.1016/j.immuni.2007.09.010
4. Hori S, Nomura T, and Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
5. Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, and Ramsdell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. (1999) 162:2546–54. doi: 10.4049/jimmunol.162.5.2546
6. Kim CH. FOXP3 and its role in the immune system. Adv Exp Med Biol. (2009) 665:17–29. doi: 10.1007/978-1-4419-1599-3_2
7. Lu L, Barbi J, and Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol. (2017) 17:703–17. doi: 10.1038/nri.2017.75
8. Ochs HD, Ziegler SF, and Torgerson TR. FOXP3 acts as a rheostat of the immune response. Immunol Rev. (2005) 203:156–64. doi: 10.1111/j.0105-2896.2005.00231.x
9. Honing DY, Luiten RM, and Matos TR. Regulatory T cell dysfunction in autoimmune diseases. Int J Mol Sci. (2024) 25:7171. doi: 10.3390/ijms25137171
10. Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, et al. Molecular mechanisms controlling foxp3 expression in health and autoimmunity: from epigenetic to post-translational regulation. Front Immunol. (2019) 10:3136. doi: 10.3389/fimmu.2019.03136
11. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
12. Hou AN, Wang Y, and Pan YQ. A case report of IPEX syndrome with neonatal diabetes mellitus and congenital hypothyroidism as the initial presentation, and a systematic review of neonatal IPEX. J Clin Immunol. (2023) 43:979–88. doi: 10.1007/s10875-023-01456-0
13. Gambineri E, Torgerson TR, and Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. (2003) 15:430–5. doi: 10.1097/00002281-200307000-00010
14. Weinstein KN, Domeier PP, and Ziegler SF. A splice of life: the discovery, function, and clinical implications of FOXP3 isoforms in autoimmune disease. Int Immunol. (2024) 37:83–90. doi: 10.1093/intimm/dxae049
15. Kroger B, Spohn M, Mengel M, Sperhake JP, Ondruschka B, and Mailer RK. Expression of full-length FOXP3 exceeds other isoforms in thymus and stimulated CD4 + T cells. J Clin Immunol. (2024) 44:114. doi: 10.1007/s10875-024-01715-8
16. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27:68–73. doi: 10.1038/83784
17. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. (2001) 27:18–20. doi: 10.1038/83707
18. Bekis Bozkurt H, Bayram Catak F, Sahin A, Yalcin Gungoren E, Gemici Karaarslan B, Yakici N, et al. Diverse clinical and immunological profiles in patients with IPEX syndrome: a multicenter analysis from Turkey. J Clin Immunol. (2024) 45:9. doi: 10.1007/s10875-024-01791-w
19. Lee WQ and Leong KF. Infantile vitiligo and alopecia in immunodysregulation polyendocrinopathy enteropathy X-linked syndrome. Pediatr Dermatol. (2023) 40:886–9. doi: 10.1111/pde.15266
20. Brennan CM and Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. (2001) 58:266–77. doi: 10.1007/PL00000854
21. Lal G and Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. (2009) 114:3727–35. doi: 10.1182/blood-2009-05-219584
22. Atasoy U, Watson J, Patel D, and Keene JD. ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. J Cell Sci. (1998) 111:3145–56. doi: 10.1242/jcs.111.21.3145
23. Saenz-Narciso B, Bell SE, Matheson LS, Venigalla RKC, and Turner M. ZFP36-family RNA-binding proteins in regulatory T cells reinforce immune homeostasis. Nat Commun. (2025) 16:4192. doi: 10.1038/s41467-025-58993-y
24. Stellato C, Gubin MM, Magee JD, Fang X, Fan J, Tartar DM, et al. Coordinate regulation of GATA-3 and Th2 cytokine gene expression by the RNA-binding protein HuR. J Immunol. (2011) 187:441–9. doi: 10.4049/jimmunol.1001881
25. Techasintana P, Davis JW, Gubin MM, Magee JD, and Atasoy U. Transcriptomic-wide discovery of direct and indirect HuR RNA targets in activated CD4+ T cells. PloS One. (2015) 10:e0129321. doi: 10.1371/journal.pone.0129321
26. Techasintana P, Ellis JS, Glascock J, Gubin MM, Ridenhour SE, Magee JD, et al. The RNA-binding protein HuR posttranscriptionally regulates IL-2 homeostasis and CD4+ Th2 differentiation. ImmunoHorizons. (2017) 1:109–23. doi: 10.4049/immunohorizons.1700017
27. Fattahi F, Ellis JS, Sylvester M, Bahleda K, Hietanen S, Correa L, et al. HuR-targeted inhibition impairs Th2 proinflammatory responses in asthmatic CD4(+) T cells. J Immunol. (2022) 208:38–48. doi: 10.4049/jimmunol.2100635
28. Gubin MM, Techasintana P, Magee JD, Dahm GM, Calaluce R, Martindale JL, et al. Conditional knockout of the RNA-binding protein HuR in CD4(+) T cells reveals a gene dosage effect on cytokine production. Mol Med. (2014) 20:93–108. doi: 10.2119/molmed.2013.00127
29. Chen J, Cascio J, Magee JD, Techasintana P, Gubin MM, Dahm GM, et al. Posttranscriptional gene regulation of IL-17 by the RNA-binding protein HuR is required for initiation of experimental autoimmune encephalomyelitis. J Immunol. (2013) 191:5441–50. doi: 10.4049/jimmunol.1301188
30. Chen J, Martindale JL, Abdelmohsen K, Kumar G, Fortina PM, Gorospe M, et al. RNA-binding protein HuR promotes Th17 cell differentiation and can be targeted to reduce autoimmune neuroinflammation. J Immunol. (2020) 204:2076–87. doi: 10.4049/jimmunol.1900769
31. Chen J, Martindale JL, Cramer C, Gorospe M, Atasoy U, Drew PD, et al. The RNA-binding protein HuR contributes to neuroinflammation by promoting C-C chemokine receptor 6 (CCR6) expression on Th17 cells. J Biol Chem. (2017) 292:14532–43. doi: 10.1074/jbc.M117.782771
32. Green LC, Anthony SR, Slone S, Lanzillotta L, Nieman ML, Wu X, et al. Human antigen R as a therapeutic target in pathological cardiac hypertrophy. JCI Insight. (2019) 4:e121541. doi: 10.1172/jci.insight.121541
33. Liu S, Huang Z, Tang A, Wu X, Aube J, Xu L, et al. Inhibition of RNA-binding protein HuR reduces glomerulosclerosis in experimental nephritis. Clin Sci (Lond). (2020) 134:1433–48. doi: 10.1042/CS20200193
34. Wu X, Gardashova G, Lan L, Han S, Zhong C, Marquez RT, et al. Targeting the interaction between RNA-binding protein HuR and FOXQ1 suppresses breast cancer invasion and metastasis. Commun Biol. (2020) 3:193. doi: 10.1038/s42003-020-0933-1
35. Huang Z, Liu S, Tang A, Wu X, Aube J, Xu L, et al. Targeting RNA-binding protein HuR to inhibit the progression of renal tubular fibrosis. J Transl Med. (2023) 21:428. doi: 10.1186/s12967-023-04298-x
36. Ma J, Sun L, Gao W, Li Y, and Dong D. RNA binding protein: coordinated expression between the nuclear and mitochondrial genomes in tumors. J Transl Med. (2023) 21:512. doi: 10.1186/s12967-023-04373-3
37. Majumder M, Chakraborty P, Mohan S, Mehrotra S, and Palanisamy V. HuR as a molecular target for cancer therapeutics and immune-related disorders. Adv Drug Delivery Rev. (2022) 188:114442. doi: 10.1016/j.addr.2022.114442
38. Sharma R, Sung SS, Fu SM, and Ju ST. Regulation of multi-organ inflammation in the regulatory T cell-deficient scurfy mice. J BioMed Sci. (2009) 16:20. doi: 10.1186/1423-0127-16-20
39. Doncheva NT, Morris JH, Gorodkin J, and Jensen LJ. Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res. (2019) 18:623–32. doi: 10.1021/acs.jproteome.8b00702
40. Doncheva NT, Morris JH, Holze H, Kirsch R, Nastou KC, Cuesta-Astroz Y, et al. Cytoscape stringApp 2.0: analysis and visualization of heterogeneous biological networks. J Proteome Res. (2023) 22:637–46. doi: 10.1021/acs.jproteome.2c00651
41. Fan J, Ishmael FT, Fang X, Myers A, Cheadle C, Huang SK, et al. Chemokine transcripts as targets of the RNA-binding protein HuR in human airway epithelium. J Immunol. (2011) 186:2482–94. doi: 10.4049/jimmunol.0903634
42. Dahm GM, Gubin MM, Magee JD, Techasintana P, Calaluce R, and Atasoy U. Method for the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts using RIP-Chip. J Vis Exp: JoVE. (2012) 3851. doi: 10.3791/3851
43. Calaluce R, Gubin MM, Davis JW, Magee JD, Chen J, Kuwano Y, et al. The RNA binding protein HuR differentially regulates unique subsets of mRNAs in estrogen receptor negative and estrogen receptor positive breast cancer. BMC Cancer. (2010) 10:126. doi: 10.1186/1471-2407-10-126
44. Sakaguchi S, Yamaguchi T, Nomura T, and Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
45. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
46. Vignali DA, Collison LW, and Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
47. Hu W, Wang ZM, Feng Y, Schizas M, Hoyos BE, van der Veeken J, et al. Regulatory T cells function in established systemic inflammation and reverse fatal autoimmunity. Nat Immunol. (2021) 22:1163–74. doi: 10.1038/s41590-021-01001-4
48. Johansson K, Gagnon JD, Zhou SK, Fassett MS, Schroeder AW, Kageyama R, et al. An essential role for miR-15/16 in Treg suppression and restriction of proliferation. Cell Rep. (2023) 42:113298. doi: 10.1016/j.celrep.2023.113298
49. Omenetti S and Pizarro TT. The Treg/Th17 axis: A dynamic balance regulated by the gut microbiome. Front Immunol. (2015) 6:639. doi: 10.3389/fimmu.2015.00639
50. Bhaumik S, Mickael ME, Moran M, Spell M, and Basu R. RORgammat promotes Foxp3 expression by antagonizing the effector program in colonic regulatory T cells. J Immunol. (2021) 207:2027–38. doi: 10.4049/jimmunol.2100175
51. Furuyama K, Kondo Y, Shimizu M, Yokosawa M, Segawa S, Iizuka A, et al. RORgammat+Foxp3+ regulatory T cells in the regulation of autoimmune arthritis. Clin Exp Immunol. (2022) 207:176–87. doi: 10.1093/cei/uxab007
52. Gaunt CM, Rainbow DB, Mackenzie RJ, Jarvis LB, Mousa HS, Cunniffe N, et al. The MS remyelinating drug bexarotene (an RXR agonist) promotes induction of human tregs and suppresses Th17 differentiation in vitro. Front Immunol. (2021) 12:712241. doi: 10.3389/fimmu.2021.712241
53. Zhang H, Zhang F, and Li W. Function of intestinal barrier protected by regulating the miR-199a-3p in ulcerative colitis: Modulation of IL-23/IL-17A axis. Fundam Clin Pharmacol. (2021) 35:852–60. doi: 10.1111/fcp.12650
54. Xue H, Yu X, Ma L, Song S, Li Y, Zhang L, et al. The possible role of CD4(+)CD25(high)Foxp3(+)/CD4(+)IL-17A(+) cell imbalance in the autoimmunity of patients with Hashimoto thyroiditis. Endocrine. (2015) 50:665–73. doi: 10.1007/s12020-015-0569-y
55. Li H, Li Q, Sun S, Lei P, Cai X, and Shen G. Integrated bioinformatics analysis identifies ELAVL1 and APP as candidate crucial genes for Crohn’s disease. J Immunol Res. (2020) 2020:3067273. doi: 10.1155/2020/3067273
56. Tian G, Li JL, Wang DG, and Zhou D. Targeting IL-10 in auto-immune diseases. Cell Biochem Biophys. (2014) 70:37–49. doi: 10.1007/s12013-014-9903-x
57. Couper KN, Blount DG, and Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. (2008) 180:5771–7. doi: 10.4049/jimmunol.180.9.5771
58. Sabat R, Grutz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev. (2010) 21:331–44. doi: 10.1016/j.cytogfr.2010.09.002
59. Goldmann O, Nwofor OV, Chen Q, and Medina E. Mechanisms underlying immunosuppression by regulatory cells. Front Immunol. (2024) 15:1328193. doi: 10.3389/fimmu.2024.1328193
60. Schmidt A, Oberle N, and Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
61. Zhang A, Fan T, Liu Y, Yu G, Li C, and Jiang Z. Regulatory T cells in immune checkpoint blockade antitumor therapy. Mol Cancer. (2024) 23:251. doi: 10.1186/s12943-024-02156-y
62. Lokau J, Petasch LM, and Garbers C. The soluble IL-2 receptor alpha/CD25 as a modulator of IL-2 function. Immunology. (2024) 171:377–87. doi: 10.1111/imm.13723
63. Moon BI, Kim TH, and Seoh JY. Functional modulation of regulatory T cells by IL-2. PloS One. (2015) 10:e0141864. doi: 10.1371/journal.pone.0141864
64. Ward NC, Yu A, Moro A, Ban Y, Chen X, Hsiung S, et al. IL-2/CD25: A long-acting fusion protein that promotes immune tolerance by selectively targeting the IL-2 receptor on regulatory T cells. J Immunol. (2018) 201:2579–92. doi: 10.4049/jimmunol.1800907
65. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, and Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64. doi: 10.4049/jimmunol.155.3.1151
66. Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, and Moreau JF. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2005) 52:201–11. doi: 10.1002/art.20745
67. Navegantes KC, de Souza Gomes R, Pereira PAT, Czaikoski PG, Azevedo CHM, and Monteiro MC. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J Transl Med. (2017) 15:36. doi: 10.1186/s12967-017-1141-8
68. Wang X, Qiu L, Li Z, Wang XY, and Yi H. Understanding the multifaceted role of neutrophils in cancer and autoimmune diseases. Front Immunol. (2018) 9:2456. doi: 10.3389/fimmu.2018.02456
69. Abramson SB, Given WP, Edelson HS, and Weissmann G. Neutrophil aggregation induced by sera from patients with active systemic lupus erythematosus. Arthritis Rheumatol. (1983) 26:630–6. doi: 10.1002/art.1780260509
70. Miller SD, McMahon EJ, Schreiner B, and Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. (2007) 1103:179–91. doi: 10.1196/annals.1394.023
71. Liu M, Liang S, and Zhang C. NK cells in autoimmune diseases: protective or pathogenic? Front Immunol. (2021) 12:624687. doi: 10.3389/fimmu.2021.624687
72. Jin O, Kavikondala S, Sun L, Fu R, Mok MY, Chan A, et al. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus. (2008) 17:654–62. doi: 10.1177/0961203308089410
Keywords: regulatory T cells (Tregs), HuR (ELAVL1), Foxp3 stability, systemic autoimmunity, IPEX syndrome, RNA-binding proteins (RBPs), post-transcriptional regulation, RORγt (Rorc)
Citation: Fattahi F, Ellis JS, Vallance L, Bahleda K, Holden J, Socha S, Meier J, Gomez-Rivera F and Atasoy U (2025) HuR ablation destabilizes Foxp3 mRNA and impairs regulatory T cell function, contributing to an autoimmune phenotype. Front. Immunol. 16:1618677. doi: 10.3389/fimmu.2025.1618677
Received: 26 April 2025; Accepted: 12 August 2025;
Published: 26 September 2025.
Edited by:
Xiaoming Zhang, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Mehdi Benamar, Division of Immunology, Boston Children’s Hospital, United StatesSaeed Mohammadi, University of Nizwa, Oman
Copyright © 2025 Fattahi, Ellis, Vallance, Bahleda, Holden, Socha, Meier, Gomez-Rivera and Atasoy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ulus Atasoy, YXRhc295dUBtZWQudW1pY2guZWR1
†These authors have contributed equally to this work