 Lillie Lewis
Lillie Lewis Deepa Valvi
Deepa Valvi Roberto Gedaly
Roberto Gedaly Francesc Marti
Francesc Marti- 1Department of Surgery - Transplant Division, College of Medicine, University of Kentucky, Lexington, KY, United States
- 2Lucillle Parker Markey Cancer Center, College of Medicine, University of Kentucky, Lexington, KY, United States
- 3Alliance Research Initiative (TILT Alliance), College of Medicine, University of Kentucky, Lexington, KY, United States
Age-related conditions, such as neurodegenerative disease, cancer, and autoimmune disorders, are increasingly recognized as closely linked with the gradual deterioration of the immune system. Regulatory T cells (Tregs) are a small, specialized subset of T lymphocytes that play a critical role in maintaining immune homeostasis and self-tolerance. As individuals age, Treg cells demonstrate reduced capacity to suppress some autoreactive immune responses, although they largely retain their capacity to regulate effector antiviral and antitumor immunity. Unlike conventional effector T cells (Teff), which primarily derive energy from glycolysis, Tregs rely more on mitochondrial oxidative phosphorylation to fulfill their energy requirements. This metabolic profile renders them particularly sensitive to mitochondrial dysfunction, underpinning the critical role of mitochondrial protective pathways in preserving the functional integrity of Treg cells. The mitochondrial unfolded protein response (mitoUPR) is gaining special relevance among these protective mechanisms. In this review, we examine the complex interplay between immune aging and mitochondrial dynamics, with particular emphasis on the essential role of mitoUPR in supporting Treg function. We further discuss how targeting mitochondrial stress responses may offer novel therapeutic avenues for age-related diseases characterized by Treg dysfunction.
Introduction
Aging is characterized by a progressive decline in physiological processes across the lifespan of an organism, with the immune system among the most profoundly affected (1). This age-associated immune deterioration, a phenomenon known as immunosenescence, contributes to the higher susceptibility to infections, chronic inflammation, and age-related diseases (2). Notably, different immune cell types exhibit distinct responses to aging, likely reflecting their different metabolic demands and stress response mechanisms.
Regulatory T cells (Tregs) have emerged as a key population in the context of immunosenescence (3–6). This small subset of T cells plays a critical role in maintaining immune homeostasis by promoting tolerance and preventing excessive inflammatory responses in both innate and adaptive immune arms (7, 8). Treg cells possess a distinct metabolic profile. Unlike conventional effector T (Teff) cells, Tregs rely heavily on mitochondrial fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) to sustain their suppressive function (9). As such, Treg cells are particularly vulnerable to agents that compromise mitochondrial function, including reactive oxygen species (ROS), environmental toxins, and genotoxic stressors, which tend to accumulate with age (1, 10, 11). To preserve their functional integrity, Tregs engage different mitochondrial protective mechanisms, including antioxidant pathways, DNA repair mechanisms, and mitochondrial quality control systems (12–16).
One of such mitochondrial quality control systems is the mitochondrial unfolded protein response (mitoUPR). The mitoUPR responds to the accumulation of unfolded or misfolded mitochondrial proteins by upregulating the transcription of chaperones and proteases (17, 18). Disruption of mitoUPR results in the accumulation of dysfunctional mitochondria or misfolded mitochondrial proteins, which may contribute to the onset and progression of a variety of age-related pathologies such as neurodegenerative disorders, metabolic syndromes, and systemic inflammation (19, 20). We have recently demonstrated that Tregs exhibit increased baseline expression of different mitoUPR proteins compared to conventional T cells (Tconv) (21).
This review explores the dynamic interplay between immune metabolism and aging, with a particular focus on Treg cells and the relevance of the mitoUPR in sustaining Treg function and driving their fate.
Immune cells in aging
A properly functioning immune system is essential for health and disease protection. With advancing age, immunosenescence leads to increased vulnerability to chronic inflammation, infectious diseases, and reduced vaccine responsiveness in the elderly population (22).
The innate immune system serves as the body’s first line of defense against infections and injuries, providing a rapid and non-specific response already active from birth (23–25). Key components of the innate immune system include physical barriers such as the skin and mucosal membranes, along with chemical and cellular defenses (23). Most innate immune cells, except for natural killer (NK) cells, derive from myeloid progenitors in the bone marrow (26). Dendritic cells (DCs) and NK cells bridge the innate and adaptive responses by alerting and activating T cells (27, 28). Meanwhile, other innate immune cells neutralize pathogens by releasing cytotoxic chemicals, cytokines, chemokines, and antimicrobial peptides, or by direct pathogen elimination via phagocytosis (29). Traditionally viewed as lacking memory, the innate immune system is now recognized to exhibit trained immunity – a long-term functional reprogramming, leading to an enhanced responsiveness to subsequent, even unrelated, challenge (30). This response is not antigen-specific but rather represents a broad, heightened state of readiness in the cellular innate immune system (30–32).
In aging individuals, innate immune cells exhibit many types of functional impairment, hindering their ability to facilitate tissue repair and properly initiate adaptive responses (33). DCs exhibit impaired antigen uptake and dysfunctional T cell priming, sometimes leading to the activation of T cells in response to self-DNA (34, 35). NK cells in aged individuals secrete less interferon gamma (IFNγ), a key cytokine for T cell differentiation (36, 37). Neutrophils exhibit reduced migration and impaired production of neutrophil extracellular traps (NETs), essential for microbial neutralization and elimination (38–40). Macrophages display impaired chemotaxis and increased ROS production, compromising their ability to clear pathogens (41). Additionally, in the elderly, microglial cells in the central nervous system become sensitized by the chronic, low-grade inflammation that develops with aging, a condition known as inflammaging (42–44).
The adaptive immune system, while slower to respond than the innate immune system, is characterized by its antigen specific response and the generation of immunological memory (25). Activation of adaptive immunity is initiated through the recognition of individual antigens by clonotypic, highly specific receptors expressed on B and T lymphocytes, enabling targeted and long-lasting immune responses (45, 46). In the bone marrow, common lymphoid progenitors develop from hematopoietic stem cells (26). These progenitors give rise to committed T cell, B cell, or NK cell precursors. Once committed to the T cell lineage, T cell precursors migrate to the thymus where they become CD4+ helper T cells, CD8+ cytotoxic T cells, or Treg cells (47). After their thymic release, following antigen exposure, CD4+ and CD8+ T cells differentiate into memory T cells that exhibit enhanced responsiveness upon re-exposure to the same antigen (48). In adulthood, thymic involution leads to a progressive decline in the output of naïve T cells (49); however, the peripheral T cell pool is maintained through homeostatic proliferation and survival within secondary lymphoid tissues (50, 51). This shift from thymic output to peripheral T cell generation dynamics imposes a replicative stress, thereby increasing the risk of replication-induced genomic instability and cellular senescence, and ultimately contributing to functional decline and eventual exhaustion of the T cell population (52, 53).
Unlike other T cell subsets, the Treg cell compartment remains stable or even expands with age (54–56). While aged Tregs retain many of their core suppressive functions, some regulatory mechanisms become selectively impaired (Figure 1). Notably, their ability to control the proliferation of IL-17 producing T helper cells (Th17) is compromised in chronic, but not acute, inflammatory conditions (57, 58). In contrast, aged Tregs continue to effectively suppress antigen-presenting cell function and IFN-γ production (58), which is critical for antiviral and antitumor immunity and promoting the expression of immune checkpoint inhibitors (59). In addition, Tregs in aged mice are less efficient in suppressing IL-2 production of effector T cells (60). This selective dysregulation in Treg function during aging may contribute to the paradoxical combination of weakened immunity against infections and increased autoimmune responses observed in the elderly, underscoring the importance of Treg cells in aging-related immune remodeling and their relevance as targets for immunotherapeutic strategies in aging. The distinct metabolic profiles of glycolytic-driven Teff and mitochondrial-reliant Treg cells (9) has led to extensive investigation into the role of mitochondrial function in maintaining the functional integrity of Tregs.
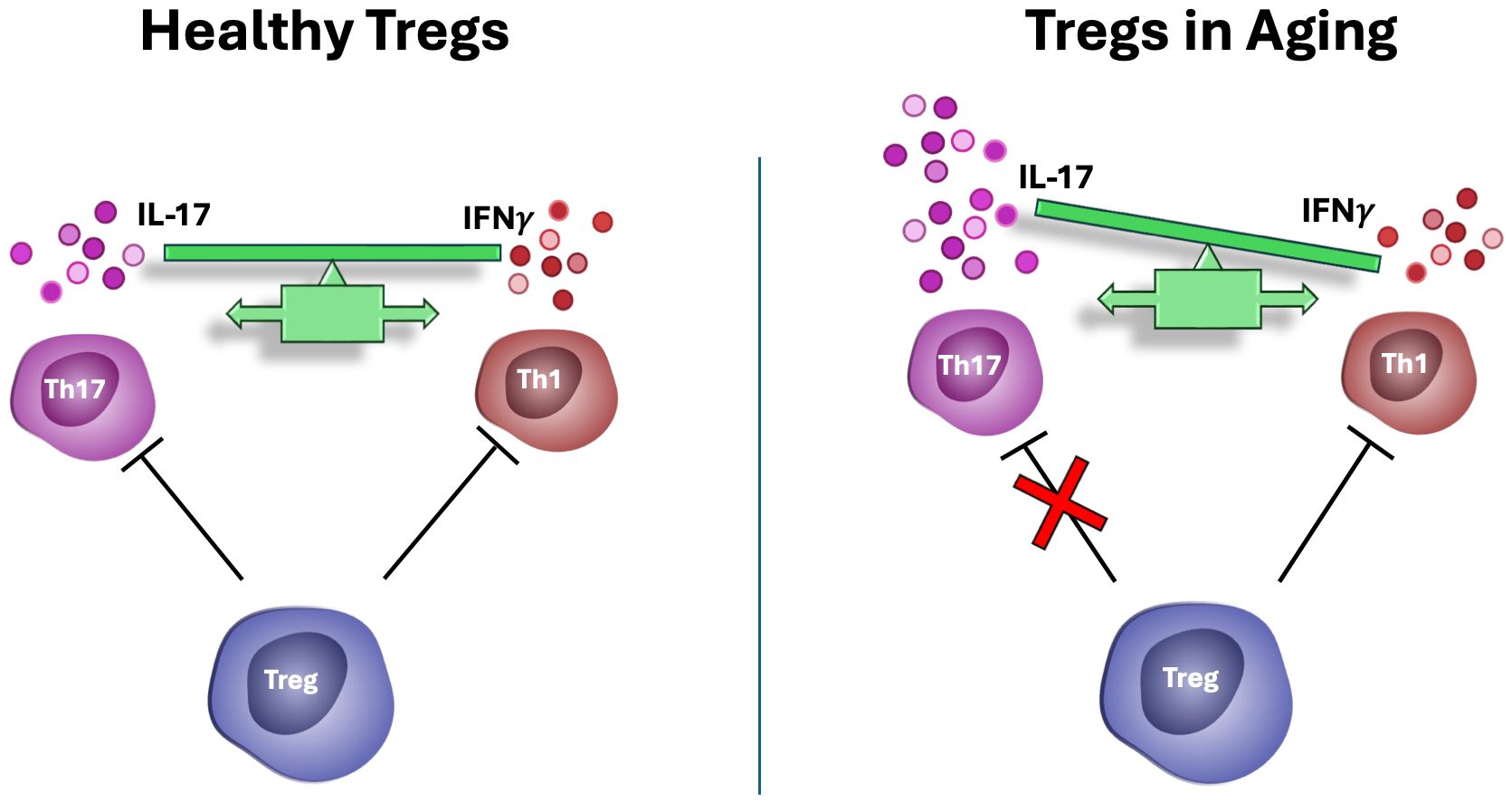
Figure 1. Tregs in aging. In aged individuals, Tregs lose their capacity to suppress IL-17 mediated autoimmunity but retain their ability to suppress antigen presentation, as well as antiviral, and antitumor immune responses. This imbalance may contribute to the simultaneous increased risk for autoimmunity and cancer with aging.
Mitochondrial function in Tregs
Mitochondria are intracellular, membrane-bound organelles with diverse roles in cellular metabolism and homeostasis (61), as outlined in Figure 2. In the immune system, mitochondria are essential for regulating inflammation and determining cell development, fate and function (62). Mitochondria drive inflammatory signaling through the release of mitochondrial damage-associated molecular patterns (mtDAMPs), including mitochondrial ROS (mtROS), mitochondrial N-formyl peptides, and mtDNA (63). These mtDAMPs act as endogenous “alarm signals” that are recognized by pattern recognition receptors (PRRs) such as RIG-1-like receptors (RLRs), NOD-like receptors (NLRs), and Toll-like receptors (TLRs) expressed predominantly in cells of the innate immune system (64). Engagement of PRRs triggers downstream signaling cascades that lead to the induction of pro-inflammatory cytokines and tissue-repair intermediates (65). In addition to playing a major role in initiating immune responses to both microbial pathogens and sterile insults caused by cell death or tissue damage, mitochondrial signaling and metabolism regulate the differentiation and functional programming of immune cells (62, 66–68). Mitochondrial activity also influences both the production of cytokines and the responsiveness of immune cells to cytokine-mediated signals (69, 70). The importance of mitochondrial health in immune regulation is further highlighted by the observation that many immunosenescence-associated alterations are closely linked to progressive mitochondrial dysfunction (71).
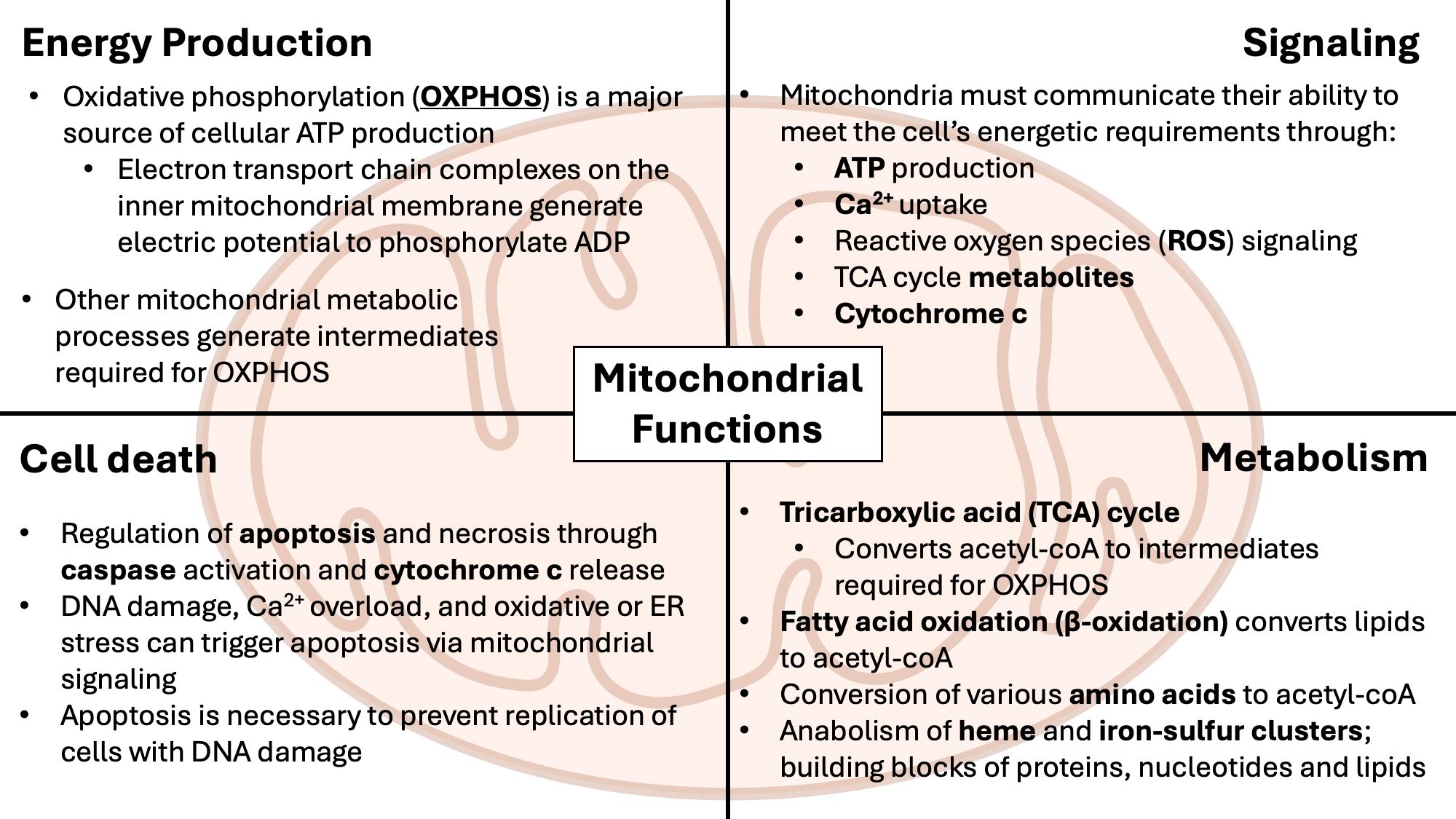
Figure 2. Overview of mitochondrial functions. Mitochondria are membrane-bound metabolic and signaling organelles with their own circular DNA. Within the matrix, separated by the inner mitochondrial membrane, ATP is produced from the tricarboxylic acid (TCA) cycle and the electron transport chain (ETC). β-oxidation contributes to ATP production by breaking down fatty acids into acetyl-CoA required for the TCA cycle. Production of ATP from mitochondria regulates nutrient-sensing via AMPK/mTOR signaling, while mitochondrial reactive oxygen species (mtROS) from the ETC, at low levels, activates the ERK signaling cascade, promoting proliferation and cell growth.
In Treg cells, mitochondria play a unique role in their survival and function (9, 14, 72–75). Mitochondrial metabolism, especially FAO and OXPHOS are essential for the in vitro suppressive abilities of Tregs (9). In contrast, Tconv use glycolytic metabolism for rapid energy production to support their effector functions (9). Genetic studies further underscore the importance of mitochondrial integrity in maintaining the suppressive function of Treg cells, as the deletion of the mitoUPR-related proteins SIRT3 or PGC1-α leads to impaired Treg suppressive activity (72). Different lines of evidence also support a critical role of mitochondrial metabolism in the differentiation of Tregs from naïve T cells (14, 76). Interestingly, in the tumor microenvironment, disruption of OXPHOS and lipid metabolism in tumor-infiltrating Tregs has been shown to paradoxically bolster their suppressive capacity, mediated in part by type I IFN signaling and increased IL-10 production (77), also demonstrating that not all Treg suppressive functions are dependent on lipid metabolism and OXPHOS. Likewise, freshly isolated ex vivo human Tregs exhibit high glycolytic activity (78). However, disruptions in mitochondrial OXPHOS can increase oxidative stress, potentially compromising Treg survival by inducing apoptosis (79). Although excessive oxidative stress is detrimental to Treg cell function, low levels of ROS are essential for intracellular signaling and proper immune regulation (80). mtROS, particularly hydrogen peroxide (H2O2), is an important second messenger that regulates key transcriptional pathways, including nuclear factor kappa B (NFκB) signaling essential for Treg development (80, 81), as well as nuclear factor of activated T cells (NFAT) and activator protein 1 (AP-1), which are involved in T cell signaling and gene expression (82). Additionally, mtROS play pivotal roles in Treg induction by macrophages and in the suppressive function of thymus-derived Tregs (83, 84). These findings illustrate the complex role of ROS in Treg biology, acting as both essential signaling molecules and potential stressors, depending on their levels and cellular context.
Mitochondrial dynamics of fusion and fission are essential for maintaining mitochondrial integrity and function under dynamic cellular conditions (85), and play a pivotal role in Treg cell biology. Mitochondrial fusion is a protective mechanism that mitigates cellular stress by enabling the exchange of mitochondrial DNA, proteins, and metabolites between damaged and healthy mitochondria (86). This process is largely mediated by the GTPases mitofusin 1 (MFN1), mitofusin 2 (MFN2), and optic atrophy protein 1 (OPA1) (85). Mitochondrial fusion is a crucial metabolic checkpoint necessary to promote Treg differentiation, lineage commitment and enhancing their suppressive function (87, 88). Conversely, mitochondrial fission – the division of mitochondria into smaller units – is mediated primarily by dynamin-related protein 1 (DRP1) (89). While mitochondrial fission is not required for Treg differentiation (85), its inhibition increases Treg accumulation in the central nervous system and spleen in animal models of autoimmune disease, such as multiple sclerosis (90). Mitochondrial fission remains essential for maintaining the health of the mitochondrial network (91). When fusion alone is insufficient to ameliorate damage from dysfunctional mitochondrial fragments, fission facilitates the segregation of these fragments, which can then be targeted for degradation through mitophagy, a selective form of autophagy specific to mitochondria (92). Defective mitophagy has been implicated in Treg dysfunction in autoimmune diseases such as myasthenia gravis, underscoring the critical role of mitochondrial quality control in preserving Treg function and immune homeostasis (13, 93).
Mitochondrial dysfunction in aging
Given the unique and critical role of mitochondria in Treg cell proliferation, metabolism and suppressive function, mitochondrial dysfunction poses a significant challenge – particularly in aging populations – by simultaneously driving inflammatory signaling and impairing Treg cell function (19, 93). This dual threat contributes to the age-associated decline in immune homeostasis and the increased prevalence of chronic inflammation. A major source of mitochondrial damage in the elderly is the accumulation of oxidative damage from mtROS generated as byproducts of OXPHOS (94). During OXPHOS, electron leakage from the electron transport chain (ETC) produces highly reactive superoxide anion radicals (O2* −), which are capable of degrading lipids, proteins, and nucleic acids within the cell (95). The enzyme superoxide dismutase (SOD) serves as a key mitochondrial antioxidant defense, converting O2* − into H2O2, a less reactive but still potent signaling molecule (95). H2O2 is essential for the regulation cell cycle signaling by modifying redox-sensitive amino acids such as methionine and cysteine on target proteins (96). However, excessive accumulation of H2O2 can lead to oxidative stress and cellular damage (97). To prevent this, cells rely on a tightly regulated antioxidant system comprising catalase (CAT), glutathione peroxidase (GPx), and peroxiredoxins (Prx) (98, 99), which detoxify H2O2 and maintain redox balance. The relationship between ROS production and mitochondrial function is complex and context dependent. Different studies have reported conflicting correlations between ATP production and ROS levels (100, 101), as well as between ROS production and mitochondrial membrane potential (ΔΨm). These discrepancies likely reflect variations in experimental models, cell types, and physiological conditions, highlighting the nuanced interplay between mitochondrial bioenergetics and redox signaling. Under normal physiological conditions, ATP production, ΔΨm, and ROS levels are positively correlated because the cell can efficiently neutralize ROS. However, under stress conditions, the cell struggles to neutralize ROS, resulting in damaged OXPHOS machinery and a decline in both ΔΨm and ATP production. In conditions like type 2 diabetes and obesity, when ΔΨm is impaired, there is a disruption of the electrochemical gradient necessary for the import of substrates like pyruvate and fatty acids (102–104). As a result, these substrates, along with proteins that fail to be imported, accumulate in the cytosol and activate the mitoUPR) (18, 105). The cytosolic accumulation of ROS and misfolded proteins due to mitochondrial stress leads to various forms of cellular dysfunction, including cellular senescence (Figure 3), which is characterized by the cessation of proliferation and resistance to cell death and apoptosis (106). In addition to mitochondrial dysfunction, other stressors can contribute to cellular senescence, including oncogenic stress, telomere attrition, replicative stress, and irradiation – all of which accumulate with age and create a cycle of increasing toxicity within the intracellular environment of the senescent cell (107). A key hallmark of mitochondrial dysfunction and cellular senescence is genomic instability, which contributes to the accumulation of DNA mutations and is associated with an increased risk of malignancy (10). Telomere shortening, a defining feature of both aging and cellular senescence, is also closely linked to mitochondrial dysfunction (1, 10, 108). Although the precise causal relationship between telomere attrition and mitochondrial impairment remains unclear, growing evidence suggests the bidirectional crosstalk between these two processes, wherein telomere damage can influence mitochondrial function and vice versa (10, 108–110). The deleterious effects of senescence are not confined to the senescent cells themselves. Senescent cells actively secrete a range of pro-inflammatory cytokines, chemokines, growth factors, and proteases that can induce secondary senescence in surrounding healthy cells and tissues – a phenomenon known as the senescence-associated secretory phenotype (SASP) (111, 112), which can contribute to amplify the chronic inflammation that drives aging and its associated pathologies.
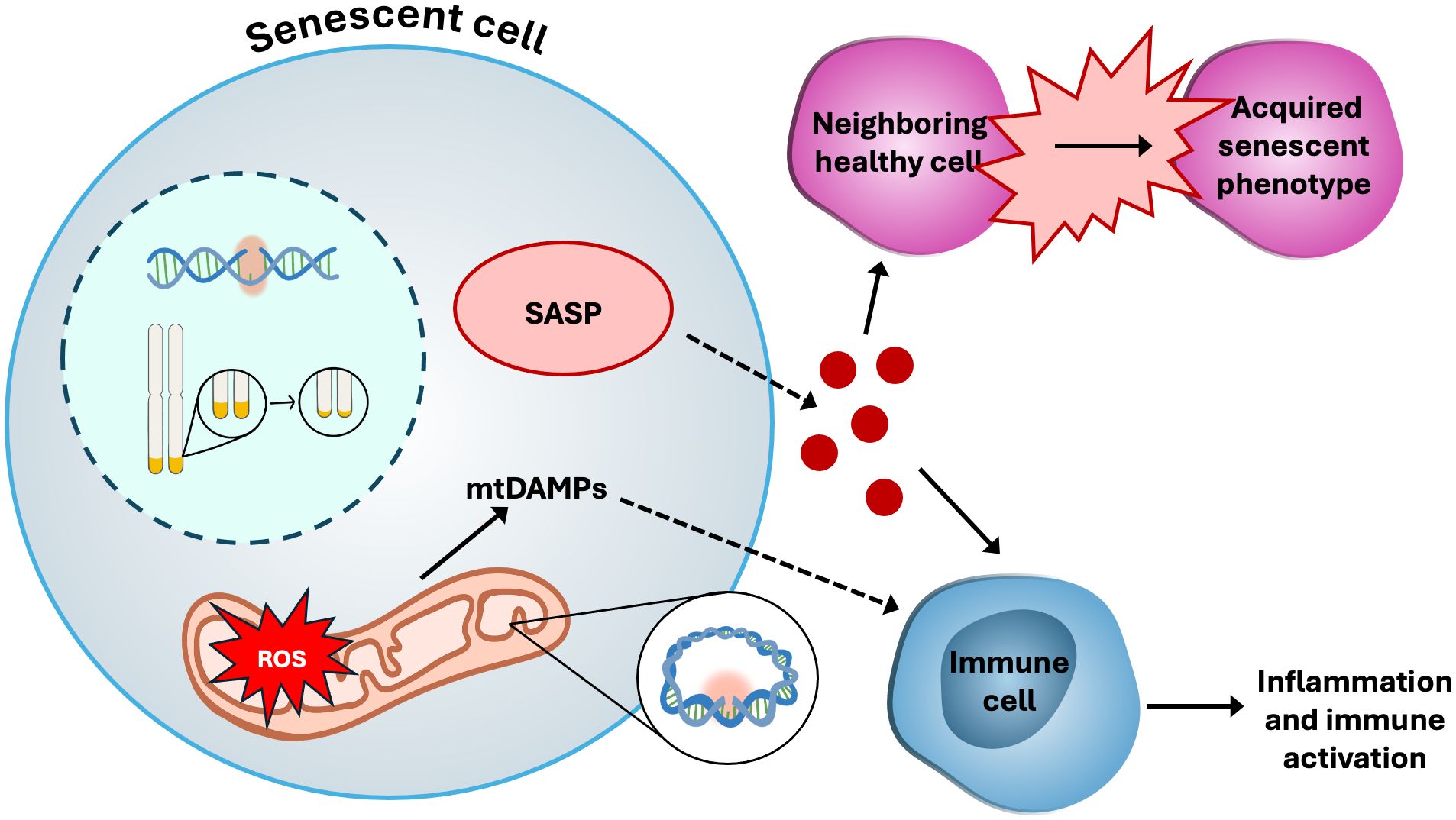
Figure 3. Cellular senescence. Senescent cells display many dysfunctional characteristics, such as: hyper-fused and elongated mitochondria; increased production of ROS, driving genomic instability in mitochondrial DNA (mtDNA) as well as nuclear DNA; release of mitochondrial damage-associated molecular patterns (mtDAMPs) such as mtROS and mtDNA, which stimulate inflammatory responses from immune cells; telomere shortening; and the senescence-associated secretory phenotype (SASP), which includes growth factors and pro-inflammatory signaling molecules.
Given the complex, self-reinforcing interplay among mitochondrial dysfunction, cellular senescence and inflammatory responses, the regulatory role of Treg cells in controlling these inflammatory loops is critical for maintaining immune and tissue homeostasis. In centenarians, Tregs maintain a strongly anti-inflammatory secretory profile, accompanied by reduced levels of pro-inflammatory cytokines compared to those observed in the general aging population (4). This observation suggests a potential role for Tregs in mitigating inflammaging. However, recent studies have shown evidence that senescent Tregs can accumulate with age and may, paradoxically, contribute to the progression of inflammaging and age-related pathologies (3, 5, 113). Because Tregs are particularly vulnerable to mitochondrial dysfunction, they may be more susceptible to mitochondria-driven senescence than Tconv, as demonstrated in mice (5).
Chronic inflammation in elderly populations, often driven by mitochondrial dysfunction, is a key contributor to the process of inflammaging. In this context, Treg activity is essential for suppressing the excessive inflammatory responses and managing the low-grade, subclinical inflammation associated with aging. This underscores the great therapeutic potential of targeting mitochondrial pathways to enhance Treg function and mitigate inflammatory signaling. Among these pathways, the mitoUPR stands out as a particularly promising target, given its central role in preserving mitochondrial integrity and cellular homeostasis under stress conditions.
mitoUPR in Treg cells
Activation of the mitoUPR
The UPR was initially identified in the endoplasmic reticulum (ER) as a protective mechanism activated by ER stress (ERS) (114). ERS occurs upon accumulation of unfolded or misfolded proteins in the ER due to homeostatic disruptions such as oxidative stress or Ca2+ imbalance. More recently, a similar pathway -the mitochondrial UPR (mitoUPR)- has been identified in mitochondria, where stress or damage initiates the transcriptional activation of genes encoding molecular chaperones and proteases to restore mitochondrial function (18, 115).
Any process that compromises mitochondrial protein translation, synthesis, proteostasis, import or degradation can trigger the mitoUPR (116–120). Among these triggers are the mtROS accumulation, the development of the mitochondrial permeability transition pore (mPTP), mito-nuclear protein imbalance, reduced ΔΨm, the overactivation or depletion of mitochondrial chaperones and proteases, and the depletion of mitochondrial prohibitins (121–124). One key activating signal for the mitoUPR is the activating transcription factor associated with stress 1 (ATFS-1) in C. elegans and its mammalian homolog, activating transcription factor 5 (ATF5) (115, 125). When the mitochondrial protein import machinery is functional, ATF5 is efficiently translocated into the mitochondria and subsequently degraded. In contrast, when protein import is compromised, ATF5 is redirected to the nucleus where it activates the transcription of mitoUPR genes (Figure 4), including chaperones and proteases such as heat shock proteins (HSPs) like mitochondrial Hsp70 (mtHsp70 or GRP75) and Hsp60, Lon protease (LonP1), and caseinolytic mitochondrial matrix peptidase proteolytic subunit (ClpP) (125, 126). Chromatin remodeling also regulates the nuclear transcription of mitoUPR genes (18). Under mitochondrial stress, chromatin is compacted by histone methyltransferases, but specific DNA regions encoding mitoUPR genes are protected from compaction by demethylases (115). Notably, disruptions in Ca2+ homeostasis and mitophagy by gene knockdown do not trigger the mitoUPR, suggesting that both processes are required for mitoUPR activation (116, 127, 128).
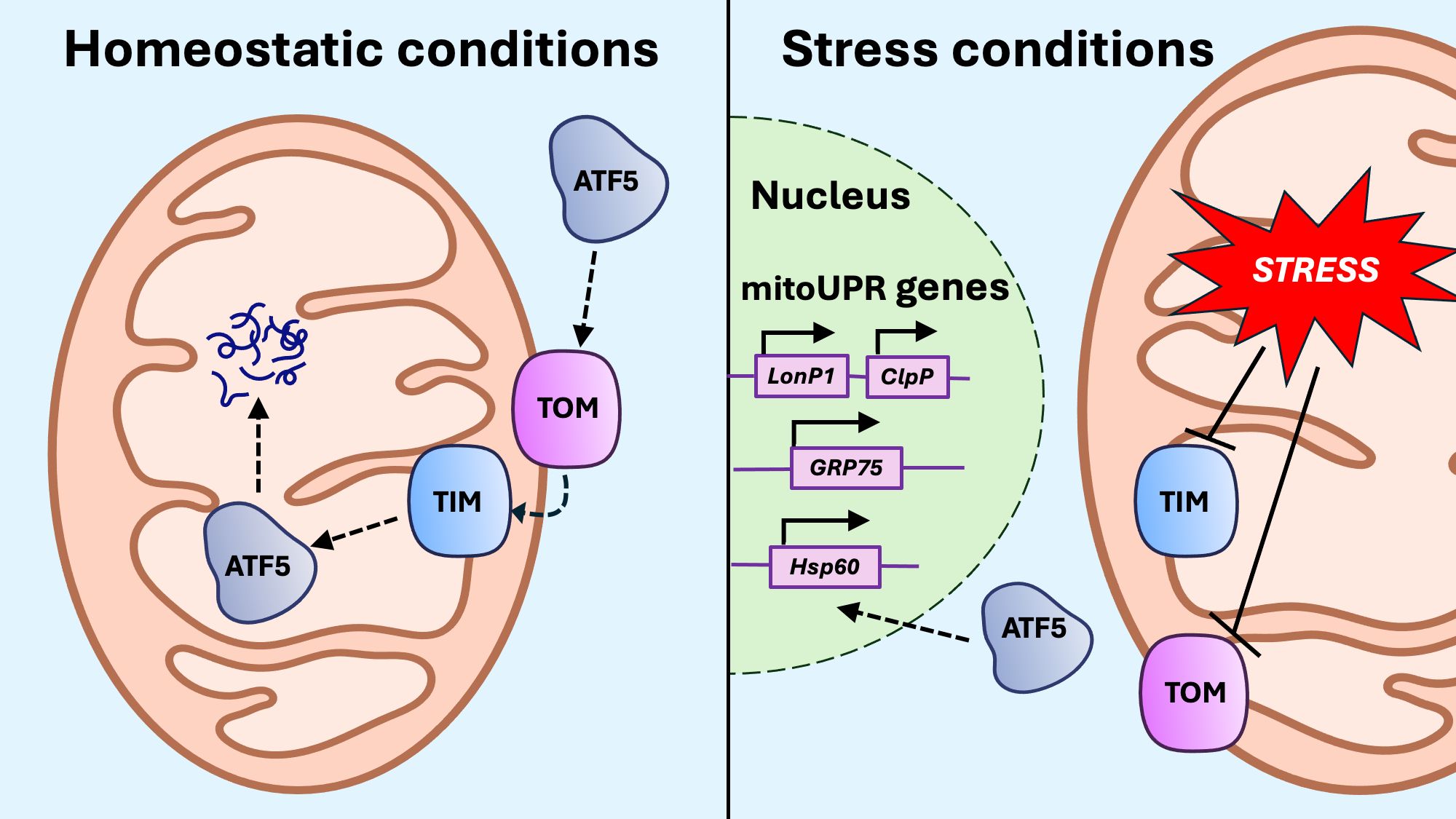
Figure 4. Activation of the mitoUPR. Under conditions of mitochondrial stress, protein import to the mitochondria through the translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM) is impaired, preventing the import of activating transcription factor 5 (ATF5). When ATF5 cannot be imported into the mitochondria, it translocates to the nucleus, where it upregulates transcription of mitoUPR genes, including proteases LonP1 and ClpP, as well as chaperones GRP75 and Hsp60.
Activation of the mitoUPR in mammals is closely linked to the integrated stress response (ISR), a conserved pathway activated by a variety of cellular stressors (129). ISR is initiated by a set of eukaryotic initiation factor 2 alpha (eIF2α) kinases, including protein kinase R-like endoplasmic reticulum kinase (PERK), protein kinase R (PKR), heme-regulated inhibitor (HRI), and general control nonderepressible 2 (GCN2) (130). These kinases phosphorylate eIF2α, promoting the translation of stress-responsive transcription factors such as ATF4, CHOP, and ATF5, which in turn upregulate genes encoding mitochondrial chaperones and proteases involved in the mitoUPR (115, 131). During ISR activation, stress granules (SG) facilitate the nuclear relocation of ATF5 (132). However, while ISR-related transcription factors are essential for mitoUPR activation, the ISR alone is not sufficient to fully activate the mitoUPR (122) and additional mitochondrial-specific signals are required to initiate a complete stress response. One of these additional signals is mtROS. mtROS contribute to the mitoUPR coordination by oxidizing heat shock protein 40 (HSP40). In this oxidized form, HSP40 interacts with mitochondrial preproteins in the cytosol before being imported into the mitochondria, where it associates with HSP70. The accumulation of HSP40-HSP70 complexes with mitochondrial protein precursors promotes the nuclear translocation of the transcription factor heat shock factor 1 (HSF1), where it upregulates the transcription of mitoUPR genes. Although HSF1 has long been recognized as a central regulator of cellular stress responses, its specific role in mitoUPR activation has only recently been identified (133–135).
Interestingly, and similar to the non-cell autonomous nature of the senescence-related SASP phenomenon, mitoUPR activation can extend beyond the stressed cell in neighboring cells, suggesting a form of paracrine mitochondrial communication. In C. elegans, mitochondrial stress in neurons not only activates mitoUPR within the neurons, but also in distal intestinal cells (136–139) by the secretion of serotonin and metabolic signaling molecules from stressed neuronal mitochondria (137, 140, 141). Whether this paracrine mitoUPR signaling occurs in other cellular systems is not yet known.
mitoUPR signaling
Research over the last decade has revealed that mitoUPR operates as a multi-axis system in coordination with other cellular processes and stress responses. This multi-axis regulation includes the canonical axis, the sirtuin axis, the intermembrane space/estrogen receptor alpha (ERα) axis, and the translation axis (129). These different axes work together to preserve mitochondrial function in response to both cellular and mitochondrial stress.
The canonical axis of the mitoUPR is defined by the upregulation of mitochondrial molecular chaperones and proteases that function to disassemble, refold, or degrade misfolded or aggregated proteins (Figure 5). The two main protein-folding systems that operate in the mitochondrial matrix are the chaperone glucose-regulated protein 75 (GRP75, also known as mtHsp70) and the chaperonin Hsp60 (142–144). GRP75 plays a central role during the import and folding of proteins translocated into the matrix, whereas Hsp60 functions primarily to refold misfolded or partially unfolded proteins within the matrix. GRP75 associates with multiple protein complexes along the inner mitochondrial membrane (IMM), contributing not only to protein assembly, folding and import, but also to the regulation of mitoUPR signaling (145). GRP75 cooperates Hsp40 (DNAJA3) to create an efficient ATP-dependent chaperone system that ensures the fidelity of mitochondrial protein folding and assembly (144). Proteolytic degradation of irreversibly damaged proteins during mitoUPR is mediated by protease complexes such as ClpXP. The ClpX subunit recognizes and binds to specific degradation motifs on substrate proteins, partially unfolding them for translocation into the ClpP protease chamber, where proteolysis occurs (146). Overexpression of ClpX, but not ClpP, during myogenesis, increases de expression of mitoUPR components and enhances OXPHOS activity via induction of the transcription factor C/EBP homologous protein (CHOP) (147). This finding suggests that ClpX may exert regulatory control over mitoUPR signaling and mitochondrial metabolism independent of its proteolytic partner. Emerging evidence also indicates that the mitoUPR intersects with broader cellular developmental programs. Components of the canonical mitoUPR have been implicated in the regulation of follicular cell development, supporting a model in which mitochondrial proteostasis pathways are integrated into signaling networks that govern cellular differentiation and function (148, 149). These insights expand the functional scope of the mitoUPR beyond stress adaptation, positioning it as a key regulator of both mitochondrial and cellular homeostasis.
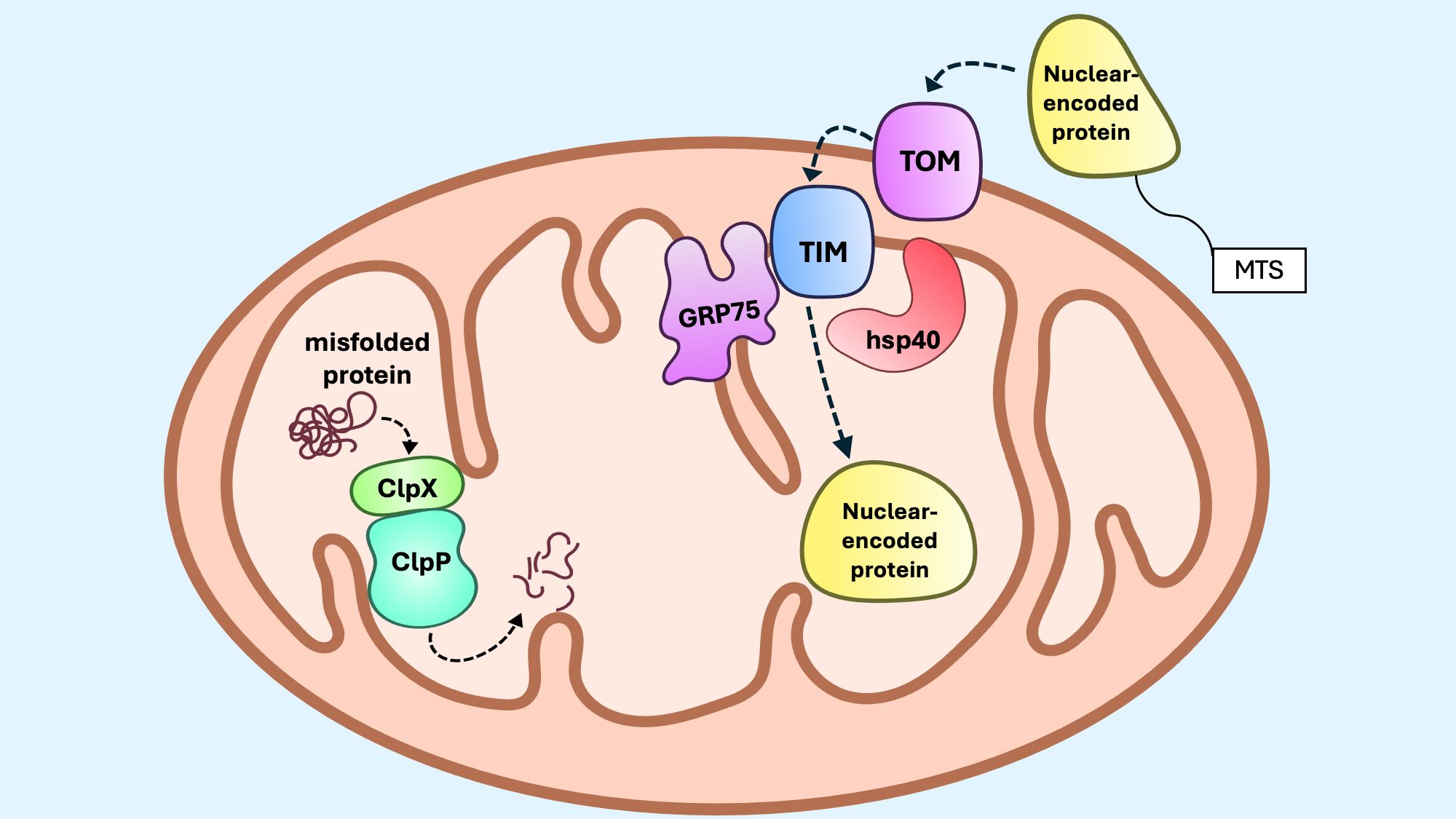
Figure 5. The mitoUPR canonical axis. The canonical mitoUPR consists of chaperones and proteases that control protein folding and degradation within mitochondria. GRP75, along with Hsp40, assist with protein folding during import of nuclear-encoded proteins with mitochondrial targeting sequences (MTS) to the matrix. When misfolded proteins or protein aggregates accumulate, ClpP and ClpX form a protease complex that degrades misfolded proteins. During proteotoxic stress, the expression of these chaperones and proteases are upregulated within mitochondria.
The sirtuin axis (Figure 6) increases resistance to mtROS by upregulating antioxidants and coordinating the mitophagy response (150, 151). The NAD+-dependent deacetylases of the sirtuin family SIRT3 and SIRT1, have been identified as mitoUPR regulators (129). SIRT1 locates in the nucleus and cytosol, while SIRT3 is primarily found in the mitochondria and also, more recently, in the nucleus, though its nuclear function remains unclear (152, 153). mtROS stimulates AMP-activated protein kinase (AMPK), which activates peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), leading to upregulation of SIRT3 via estrogen-related receptor alpha (ERRα) (154–156). Interestingly, the interplay between SIRT3 and PGC-1α is reciprocal, since SIRT3 may also promote PGC-1α activity through multiple mechanisms, including the activation of upstream regulators such as CREB (155, 157), or the deacetylation of forkhead box class O 3a (FOXO3a) (155, 158). PGC-1α and FOXO3a interact to upregulate antioxidants SOD2 and CAT, which neutralize mtROS in a rapid two-step reaction (159) whereby SOD2, along with the cofactor manganese, efficiently converts O2* − into H2O2 and oxygen (160), and CAT rapidly converts H2O2 into water and oxygen (161, 162). The bidirectional SIRT3/PGC-1α regulation forms a positive feedback loop that amplifies the mitochondrial adaptive response under conditions of metabolic stress. Although not directly indicative of mitoUPR engagement, the activation of the SIRT3/PGC-1α/FOXO axis complements the canonical mitoUPR during proteotoxic stress, contributing to the overall mitochondrial stress response. Disruption of this pathway has been implicated in metabolic disorders, including those associated with aging (163–166).
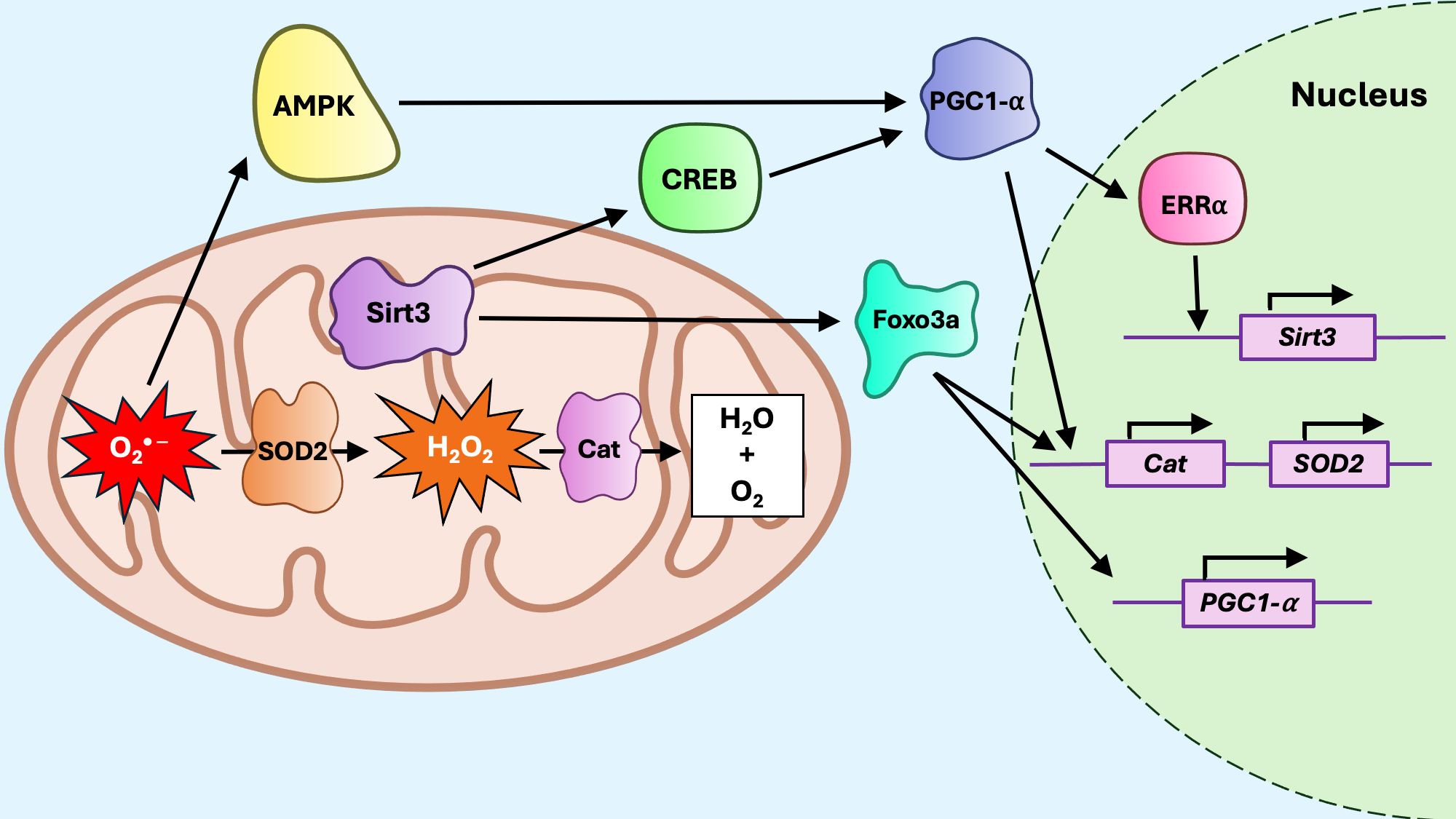
Figure 6. The mitoUPR sirtuin axis. Reactive oxygen species, including superoxide anion (O2* −) and hydrogen peroxide (H2O2) promote activation of Sirt3. Sirt3 deacetylates CREB, which activates PGC1a, and Foxo3a. Foxo3a promotes transcription of PGC1a, but also interacts with PGC1a to promote transcription of antioxidant genes such as Catalase (Cat) and superoxide dismutase (SOD2). PGC1a can also upregulate Sirt3 expression by activating estrogen-related receptor alpha (ERRa), which promotes Sirt3 transcription. Reactive oxygen species are neutralized by upregulation of SOD2, which converts O2* − to H2O2, and Cat, which converts H2O2 to water and oxygen.
The ERα axis of the mitoUPR (Figure 7) in the intermembrane space (IMS) activates estrogen receptor alpha (ERα) via AKT signaling and ROS production, leading to increased levels of nuclear respiratory factor 1 (Nrf1) and the protease OMI1 (167). Overall, ERα activation increases proteasome and protease expression levels, thereby regulating protein quality control within the IMS. Notably, small heat shock proteins (sHSPs) act as chaperones in the IMS, both under homeostatic conditions and in response to protein aggregation (168). In cells that do not express ERα, CHOP and Hsp60 are induced in response to IMS stress, suggesting that, although the ERα axis operates independently of the canonical mitoUPR, the canonical axis may compensate when the ERα axis is insufficient to neutralize proteotoxic stress (169).
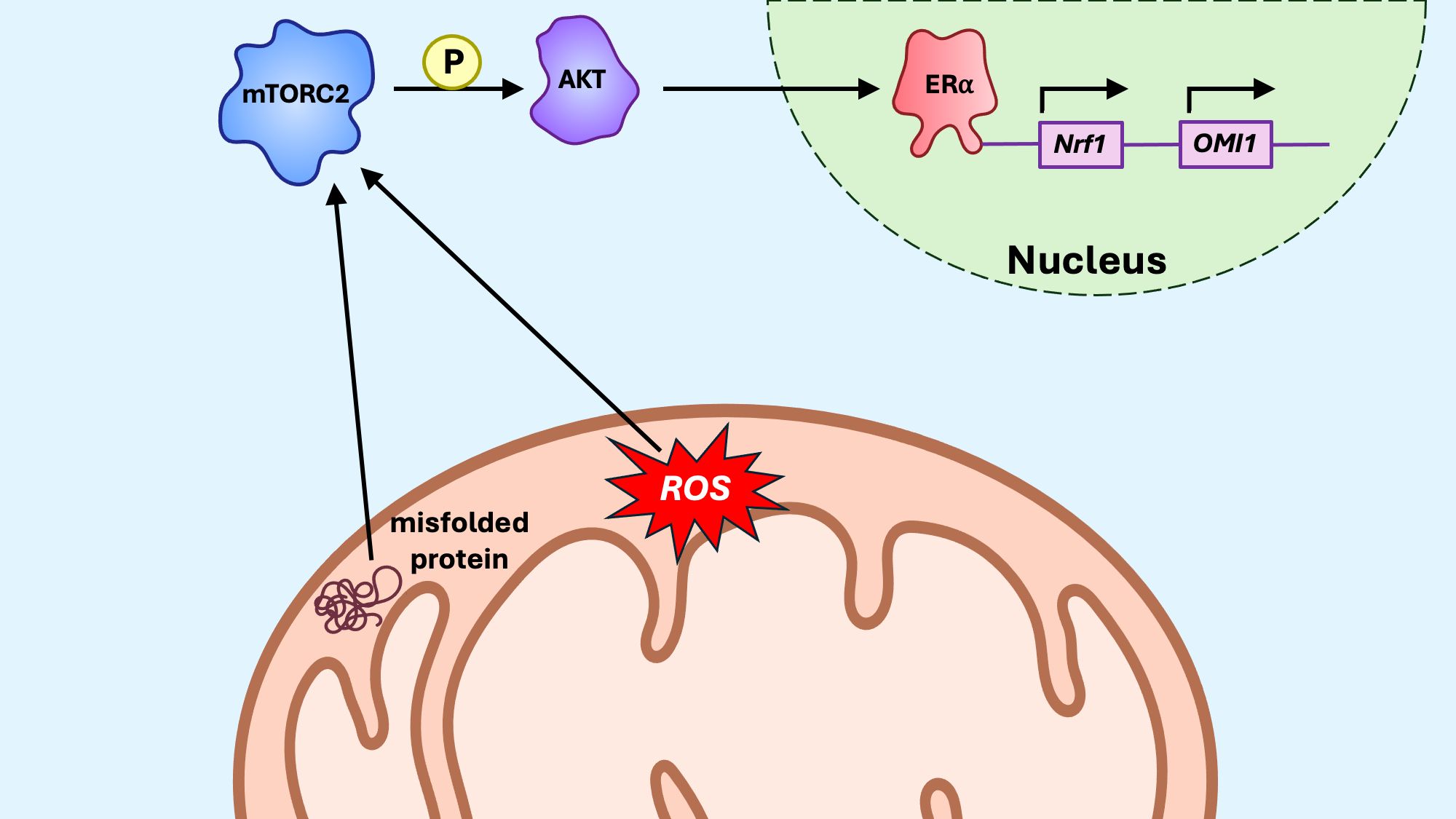
Figure 7. The mitoUPR ERα axis. In response to oxidative stress in the intermembrane space (IMS), estrogen receptor alpha (ERα), via mTORC2/AKT signaling, promotes transcription of the protease OMI1, which degrades misfolded proteins within the IMS. ERα also promotes transcription of Nrf1, a transcription factor promoting transcription of nuclear-encoded components of the electron transport chain (ETC).
The translational axis of the mitoUPR helps reduce the protein synthesis in the matrix, thereby mitigating the burden on the matrix protein-folding machinery (Figure 8). The mitochondrial matrix protease LonP1 is critical for maintaining protein homeostasis in the mitochondrial matrix (170). Upon mitoUPR induction, LonP1 specifically degrades the mitochondrial ribonuclease P catalytic subunit 3 (MRPP3), a key nuclease involved in mitochondrial RNA processing (171). This degradation of MRPP3 leads to a reduced translation of mtDNA-encoded proteins, providing a mechanism to limit the accumulation of unfolded proteins during mitochondrial stress. Conversely, LonP activity can also promote healthy levels of translation by preventing accumulation of misfolded proteins through its protease activity implicated in the canonical axis (172). Another complementary mechanism for mitoUPR activation is the mitochondrial-nuclear protein imbalance (173), which occurs when there is a stoichiometric disparity between nuclear-encoded and mitochondrial-encoded OXPHOS subunits. While disruptions in mitochondrial protein import typically lead to reduced levels of nuclear-encoded proteins within the organelle, a mito-nuclear protein imbalance may also occur from decreased expression or translation of mitochondrial-encoded proteins, often observed when mitochondrial ribosomal proteins (MRPs) are silenced or otherwise impaired (173). In this context, microRNAs (miRNAs) have emerged as potential regulators of mitochondrial proteostasis. Evidence suggests that miRNAs may influence mitoUPR activation by modulating the expression of MRPs or other components involved in mitochondrial translation and protein folding, thus adding an additional layer of post-transcriptional regulation to the stress response machinery (174).
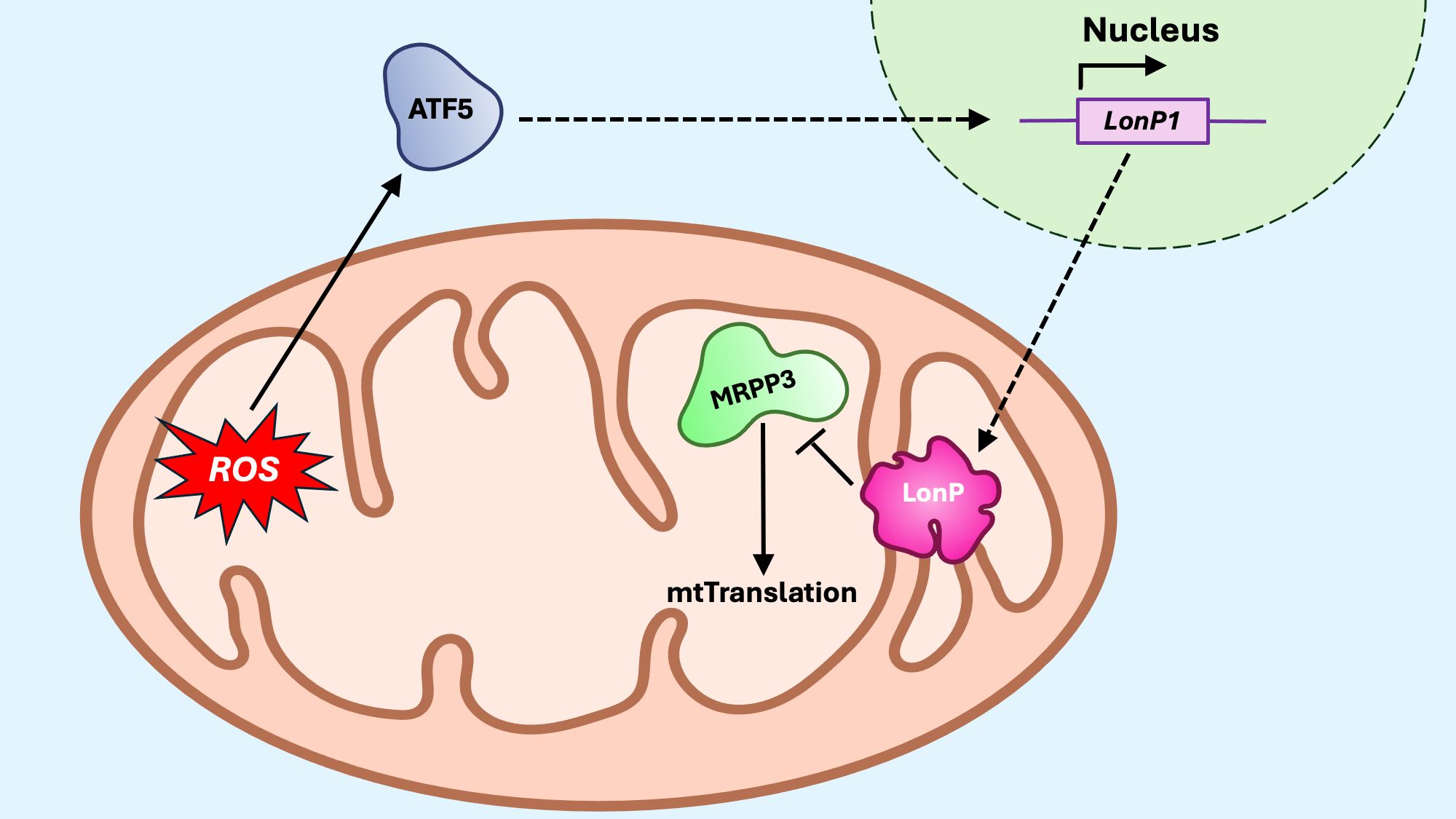
Figure 8. The mitoUPR translational axis. The translational axis responds to mito-nuclear protein imbalances by decreasing the synthesis of mitochondrial proteins, thereby reducing the protein-folding burden within mitochondria. Proteotoxic or oxidative stress activates transcription factor 5 (ATF) to promote transcription of Lon protease (LonP). Once in the mitochondria, LonP degrades the mitochondrial ribosomal protein MRPP3, thus reducing the protein translation within the matrix.
mTOR-mediated regulation of mitoUPR in Tregs
The mammalian target of rapamycin (mTOR) is a master regulator of cellular metabolism and homeostasis, functioning through two distinct complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Both complexes integrate nutrient availability, growth factor signals, and cellular energy status to orchestrate cell growth, differentiation, and survival (175, 176). A recent study in C. elegans demonstrated that mTORC1 is essential for activating the mitoUPR by sensing mitochondrial stress and increasing ATF5 transcription (177). In Treg cells, mTOR signaling plays a pivotal role in shaping metabolic programming and immune function, including mitochondrial homeostasis via the mitoUPR (15, 178, 179). Recent evidence has revealed a complex and dynamic crosstalk between mTOR signaling and the mitoUPR in Tregs, with mTORC1 and mTORC2 exerting distinct and sometimes opposing effects on this stress response pathway (15). Activation of mTORC1 enhances the expression of mitoUPR components by engaging downstream effectors such as heat shock factor 1 (HSF1) and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) (180–182). These transcriptional and translational regulators promote the expression of mitochondrial chaperones and proteases, including CHOP, ATF4, SOD2, and ClpP, facilitating the clearance and refolding of damaged proteins. This effect is sensitive to rapamycin, a specific mTORC1 inhibitor, which reverses mTORC1-driven mitoUPR activation (181, 183). In contrast, mTORC2 contributes to mitoUPR regulation primarily by modulating the activity of FOXO transcription factors. mTORC2 inhibits FOXO proteins via AKT-mediated phosphorylation, thereby restricting their nuclear localization and transcriptional activity (176, 180). This has significant implications for Treg biology, as FOXO factors – particularly FOXO1 and FOXO3 – are essential for the expression of FoxP3, the master transcription factor of Treg cells, and for the induction of key antioxidant enzymes such as superoxide dismutase 2 (SOD2) and CAT (180, 184–186). The FOXO3 axis is further modulated by SIRT1 and SIRT3, suggesting a feedback loop in which mitochondrial stress reinforces FOXO-mediated antioxidant and mitoUPR gene expression (187). Our recent studies have shown that, while short-term inhibition of mTORC1 by rapamycin can reverse acute mitochondrial damage in Tregs, prolonged metabolic stress leads to a progressive decline in both the proliferative capacity and suppressive function of Treg cells (188). These findings illustrate the metabolic plasticity of Treg cells and highlight the potential therapeutic benefit of targeting mTOR-mitoUPR signaling to enhance Treg-mediated immunoregulation in the settings of chronic inflammation, aging, or mitochondrial dysfunction.
Potential therapeutic targeting of the mitoUPR in Tregs
Activation of mitoUPR has been shown to extend lifespan in C. elegans and ameliorate damage from acute injuries such as toxin exposure, ischemia, seizure-related injuries, cardiac injury, and brain trauma (128, 189–198). In contrast, targeting the mitoUPR in chronic disease and aging is likely more complex, since activation of mitoUPR alone may not be sufficient to counteract prolonged stress conditions (192, 194, 199–205). Translation of the mitoUPR targeting strategies into clinical applications will require overcoming key challenges, including (1): the limited understanding of long-term in vivo effects of such approaches, whether transient or toxic, which will require further pre-clinical animal models and long-term clinical studies, and (2) current lack of efficient methods for tissue-specific drug delivery. Several studies have been exploring the potential of developing mitochondria-specific drugs for therapeutic applications. Advances in mitochondrial structural biology are continuously identifying new targets for mitochondriotropic drug delivery carriers, including delocalized lipophilic cations (DLCs), Szeto–Schiller (SS) peptides, vesicle-like aggregates such as dequalinium (DQA) or triphenylphosphonium (TPP) and the use of the mitochondrial protein import machinery for therapeutic interventions (206, 207).
To date, the direct targeting of the mitoUPR to modulate Treg cell function has not been explored in clinical settings. However, substantial pre-clinical evidence support this possibility, including the improvement of Treg function and reduction of autoimmune responses after scavenging mtROS in Tregs (93), the reduced suppressor activity in Treg cells devoid of SIRT3 (72), the higher expression of mitoUPR proteins in Treg cells compared to effector Tconv (15), and the upregulation of these proteins in response to mitochondrial redox induced stress (188).
Because of the prevalent mitochondrial reliance of Treg cells compared to effector Tconv, the modulation of the mitoUPR has emerged as a potential strategy to shift the Treg/effector Tconv balance in disease-specific contexts. Therapeutic strategies targeting the mitoUPR can exert bidirectional effects depending on the clinical objective: either prevent mitochondrial repair, leading to the accumulation of damage and selective depletion of Tregs, or support mitochondrial function and improve Treg cell survival and function (190, 198, 208). This dual potential has significant therapeutic implications. In the context of cancer, where Tregs often suppress anti-tumor immunity and contribute to immune evasion, disrupting mitochondrial integrity and inhibiting mitoUPR in Tregs may amplify anti-tumor effector responses. Conversely, in transplantation, and aging-related conditions like autoimmune or neurodegenerative disorders, where excessive effector responses drive pathology, enhancing mitoUPR to support Treg stability and function may restore immune tolerance and mitigate tissue damage. Symptoms of inflammaging may be alleviated by systemic targeting of the mitoUPR to counteract mitochondrial dysfunction and restore immune homeostasis, or by selective targeting of the mitoUPR in Treg cells to reduce inflammation.
In the context of Treg cell-based adoptive immunotherapy and the optimization of ex vivo expansion protocols for Treg manufacturing, monitoring mitoUPR markers may offer a valuable strategy for assessing and ensuring mitochondrial health and redox balance throughout the production process. Tracking the expression of mitoUPR components during Treg expansion may offer critical insights into the oxidative stress status of the cells and the protective mechanisms engaged to maintain mitochondrial function under in vitro culture conditions. Comparative analyses of mitoUPR markers across different expansion conditions – including nutrient composition, oxygen tension, and pharmacologic modulators – can support the development of standardized, clinically scalable protocols that preserve Treg identity, stability, and suppressive function. Such approaches can serve as functional quality control metrics to predict in vivo performance and therapeutic efficacy.
While the short-term integration of mitoUPR monitoring may improve manufacturing outcomes for clinical applications, a comprehensive understanding of Treg mitochondrial bioenergetics and stress adaptation mechanisms remains essential. Deeper characterization of mitochondrial metabolism, including OXPHOS dynamics, redox signaling, and mitoUPR regulation, will be necessary to fully exploit the therapeutic potential of Tregs, particularly in chronic inflammatory diseases, autoimmunity, and transplant tolerance. Ultimately, integrating mitochondrial profiling into Treg manufacturing workflows may not only enhance cell product consistency and potency but also pave the way for next-generation Treg-based therapies with improved durability and clinical impact.
Author contributions
LL: Writing – original draft, Writing – review & editing. DV: Writing – review & editing. RG: Writing – review & editing. FM: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, and Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
2. Liu Z, Liang Q, Ren Y, Guo C, Ge X, Wang L, et al. Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther. (2023) 8:200. doi: 10.1038/s41392-023-01451-2
3. Fessler J, Raicht A, Husic R, Ficjan A, Schwarz C, Duftner C, et al. Novel senescent regulatory T-cell subset with impaired suppressive function in rheumatoid arthritis. Front Immunol. (2017) 8:300. doi: 10.3389/fimmu.2017.00300
4. Zhou L, Ge M, Zhang Y, Wu X, Leng M, Gan C, et al. Centenarians alleviate inflammaging by changing the ratio and secretory phenotypes of T helper 17 and regulatory T cells. Front Pharmacol. (2022) 13:877709. doi: 10.3389/fphar.2022.877709
5. Guo Z, Wang G, Wu B, Chou WC, Cheng L, Zhou C, et al. Dcaf1 regulates treg senescence via the ros axis during immunological aging. J Clin Invest. (2020) 130:5893–908. doi: 10.1172/JCI136466
6. Deng B, Zhang W, Zhu Y, Li Y, Li D, and Li B. Foxp3+ Regulatory T cells and age-related diseases. FEBS J. (2021) 289:319–35. doi: 10.1111/febs.15743
7. O’Garra A and Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. (2004) 10:801–5. doi: 10.1038/nm0804-801
8. Plitas G and Rudensky AY. Regulatory T cells: differentiation and function. Cancer Immunol Res. (2016) 4:721–5. doi: 10.1158/2326-6066.CIR-16-0193
9. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory cd4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
10. Liu L, Trimarchi JR, Smith PJ, and Keefe DL. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell. (2002) 1:40–6. doi: 10.1046/j.1474-9728.2002.00004.x
11. Sanderson SL and Simon AK. In aged primary T cells, mitochondrial stress contributes to telomere attrition measured by a novel imaging flow cytometry assay. Aging Cell. (2017) 16:1234–43. doi: 10.1111/acel.12640
12. Zhang J, Chen L, Xiong F, Zhang S, Huang K, Zhang Z, et al. Autophagy in regulatory T cells: A double-edged sword in disease settings. Mol Immunol. (2019) 109:43–50. doi: 10.1016/j.molimm.2019.02.004
13. Wang N, Yuan J, Karim MR, Zhong P, Sun YP, Zhang HY, et al. Effects of mitophagy on regulatory T cell function in patients with myasthenia gravis. Front Neurol. (2020) 11:238. doi: 10.3389/fneur.2020.00238
14. Zhao X, Zhang J, Li C, Kuang W, Deng J, Tan X, et al. Mitochondrial mechanisms in treg cell regulation: implications for immunotherapy and disease treatment. Mitochondrion. (2025) 80:101975. doi: 10.1016/j.mito.2024.101975
15. Gedaly R, Orozco G, Ancheta AP, Donoho M, Desai SN, Chapelin F, et al. Metabolic disruption induced by mtor signaling pathway inhibition in regulatory T-cell expansion for clinical application. Cells. (2023) 12. doi: 10.3390/cells12162066
16. Gedaly R, Orozco G, Lewis LJ, Valvi D, Chapelin F, Khurana A, et al. Effect of mitochondrial oxidative stress on regulatory T cell manufacturing for clinical application in transplantation: results from a pilot study. Am J Transplant. (2025) 25:720–33. doi: 10.1016/j.ajt.2024.10.024
17. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, and Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. (2002) 21:4411–9. doi: 10.1093/emboj/cdf445
18. Melber A and Haynes CM. Upr(Mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. (2018) 28:281–95. doi: 10.1038/cr.2018.16
19. Geto Z, Molla MD, Challa F, Belay Y, and Getahun T. Mitochondrial dynamic dysfunction as a main triggering factor for inflammation associated chronic non-communicable diseases. J Inflammation Res. (2020) 13:97–107. doi: 10.2147/JIR.S232009
20. Hernansanz-Agustin P and Enriquez JA. Generation of reactive oxygen species by mitochondria. Antioxid (Basel). (2021) 10. doi: 10.3390/antiox10030415
21. Gedaly R, Marti F, Vilchez V, and Coquillard C. Mtor signaling in regulatory T cell differentiation and expansion. SOJ Immunol. (2015) 3:1–10. doi: 10.15226/soji/3/1/00122
22. Fuentes E, Fuentes M, Alarcon M, and Palomo I. Immune system dysfunction in the elderly. Acad Bras Cienc. (2017) 89:285–99. doi: 10.1590/0001-3765201720160487
23. Turvey SE and Broide DH. Innate immunity. J Allergy Clin Immunol. (2010) 125:S24–32. doi: 10.1016/j.jaci.2009.07.016
24. Tsafaras GP, Ntontsi P, and Xanthou G. Advantages and limitations of the neonatal immune system. Front Pediatr. (2020) 8:5. doi: 10.3389/fped.2020.00005
25. Yatim KM and Lakkis FG. A brief journey through the immune system. Clin J Am Soc Nephrol. (2015) 10:1274–81. doi: 10.2215/CJN.10031014
26. Fang P, Li X, Dai J, Cole L, Camacho JA, Zhang Y, et al. Immune cell subset differentiation and tissue inflammation. J Hematol Oncol. (2018) 11:97. doi: 10.1186/s13045-018-0637-x
27. Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, and Schultze JL. The myeloid cell compartment-cell by cell. Annu Rev Immunol. (2019) 37:269–93. doi: 10.1146/annurev-immunol-042718-041728
28. Crouse J, Xu HC, Lang PA, and Oxenius A. Nk cells regulating T cell responses: mechanisms and outcome. Trends Immunol. (2015) 36:49–58. doi: 10.1016/j.it.2014.11.001
29. Tosi MF. Innate immune responses to infection. J Allergy Clin Immunol. (2005) 116:241–9. doi: 10.1016/j.jaci.2005.05.036
30. Vetvicka V, Sima P, and Vannucci L. Trained immunity as an adaptive branch of innate immunity. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms221910684
31. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
32. Hu Z, Lu SH, Lowrie DB, and Fan XY. Trained immunity: A yin-yang balance. MedComm (2020). (2022) 3:e121. doi: 10.1002/mco2.121
33. Shaw AC, Joshi S, Greenwood H, Panda A, and Lord JM. Aging of the innate immune system. Curr Opin Immunol. (2010) 22:507–13. doi: 10.1016/j.coi.2010.05.003
34. Gupta S. Role of dendritic cells in innate and adaptive immune response in human aging. Exp Gerontol. (2014) 54:47–52. doi: 10.1016/j.exger.2013.12.009
35. Agrawal A, Tay J, Ton S, Agrawal S, and Gupta S. Increased reactivity of dendritic cells from aged subjects to self-antigen, the human DNA. J Immunol. (2009) 182:1138–45. doi: 10.4049/jimmunol.182.2.1138
36. Teixeira LK, Fonseca BP, Vieira-de-Abreu A, Barboza BA, Robbs BK, Bozza PT, et al. Ifn-gamma production by cd8+ T cells depends on nfat1 transcription factor and regulates th differentiation. J Immunol. (2005) 175:5931–9. doi: 10.4049/jimmunol.175.9.5931
37. Wakil AE, Wang ZE, Ryan JC, Fowell DJ, and Locksley RM. Interferon gamma derived from cd4(+) T cells is sufficient to mediate T helper cell type 1 development. J Exp Med. (1998) 188:1651–6. doi: 10.1084/jem.188.9.1651
38. Chen MM, Palmer JL, Plackett TP, Deburghgraeve CR, and Kovacs EJ. Age-related differences in the neutrophil response to pulmonary pseudomonas infection. Exp Gerontol. (2014) 54:42–6. doi: 10.1016/j.exger.2013.12.010
39. Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. (2014) 123:239–48. doi: 10.1182/blood-2013-08-519520
40. Drew W, Wilson DV, and Sapey E. Inflammation and neutrophil immunosenescence in health and disease: targeted treatments to improve clinical outcomes in the elderly. Exp Gerontol. (2018) 105:70–7. doi: 10.1016/j.exger.2017.12.020
41. Moss CE, Phipps H, Wilson HL, and Kiss-Toth E. Markers of the ageing macrophage: A systematic review and meta-analysis. Front Immunol. (2023) 14:1222308. doi: 10.3389/fimmu.2023.1222308
42. Perry VH and Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. (2014) 10:217–24. doi: 10.1038/nrneurol.2014.38
43. Perry VH and Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. (2013) 35:601–12. doi: 10.1007/s00281-013-0382-8
44. Cevenini E, Monti D, and Franceschi C. Inflamm-ageing. Curr Opin Clin Nutr Metab Care. (2013) 16:14–20. doi: 10.1097/MCO.0b013e32835ada13
45. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. (2010) 125:S3–23. doi: 10.1016/j.jaci.2009.12.980
46. den Haan JM, Arens R, and van Zelm MC. The activation of the adaptive immune system: cross-talk between antigen-presenting cells, T cells and B cells. Immunol Lett. (2014) 162:103–12. doi: 10.1016/j.imlet.2014.10.011
47. Bonilla FA and Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. (2010) 125:S33–40. doi: 10.1016/j.jaci.2009.09.017
48. Lam N, Lee Y, and Farber DL. A guide to adaptive immune memory. Nat Rev Immunol. (2024) 24:810–29. doi: 10.1038/s41577-024-01040-6
49. Palmer DB. The effect of age on thymic function. Front Immunol. (2013) 4:316. doi: 10.3389/fimmu.2013.00316
50. Zhang H, Weyand CM, and Goronzy JJ. Hallmarks of the aging T-cell system. FEBS J. (2021) 288:7123–42. doi: 10.1111/febs.15770
51. Westera L, van Hoeven V, Drylewicz J, Spierenburg G, van Velzen JF, de Boer RJ, et al. Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover. Aging Cell. (2015) 14:219–27. doi: 10.1111/acel.12311
52. Mittelbrunn M and Kroemer G. Hallmarks of T cell aging. Nat Immunol. (2021) 22:687–98. doi: 10.1038/s41590-021-00927-z
53. Pangrazzi L and Weinberger B. T cells, aging and senescence. Exp Gerontol. (2020) 134:110887. doi: 10.1016/j.exger.2020.110887
54. Gregg R, Smith CM, Clark FJ, Dunnion D, Khan N, Chakraverty R, et al. The number of human peripheral blood cd4+ Cd25high regulatory T cells increases with age. Clin Exp Immunol. (2005) 140:540–6. doi: 10.1111/j.1365-2249.2005.02798.x
55. Sharma S, Dominguez AL, and Lustgarten J. High accumulation of T regulatory cells prevents the activation of immune responses in aged animals. J Immunol. (2006) 177:8348–55. doi: 10.4049/jimmunol.177.12.8348
56. Chiu BC, Stolberg VR, Zhang H, and Chensue SW. Increased foxp3(+) treg cell activity reduces dendritic cell co-stimulatory molecule expression in aged mice. Mech Ageing Dev. (2007) 128:618–27. doi: 10.1016/j.mad.2007.09.002
57. Jagger A, Shimojima Y, Goronzy JJ, and Weyand CM. Regulatory T cells and the immune aging process: A mini-review. Gerontology. (2014) 60:130–7. doi: 10.1159/000355303
58. Sun L, Hurez VJ, Thibodeaux SR, Kious MJ, Liu A, Lin P, et al. Aged regulatory T cells protect from autoimmune inflammation despite reduced stat3 activation and decreased constraint of il-17 producing T cells. Aging Cell. (2012) 11:509–19. doi: 10.1111/j.1474-9726.2012.00812.x
59. Jorgovanovic D, Song M, Wang L, and Zhang Y. Roles of ifn-gamma in tumor progression and regression: A review. biomark Res. (2020) 8:49. doi: 10.1186/s40364-020-00228-x
60. Zhao L, Sun L, Wang H, Ma H, Liu G, and Zhao Y. Changes of cd4+Cd25+Foxp3+ Regulatory T cells in aged balb/C mice. J Leukoc Biol. (2007) 81:1386–94. doi: 10.1189/jlb.0506364
61. Osellame LD, Blacker TS, and Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. (2012) 26:711–23. doi: 10.1016/j.beem.2012.05.003
62. Weinberg SE, Sena LA, and Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. (2015) 42:406–17. doi: 10.1016/j.immuni.2015.02.002
63. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. (2011) 32:157–64. doi: 10.1016/j.it.2011.01.005
64. Grazioli S and Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol. (2018) 9:832. doi: 10.3389/fimmu.2018.00832
65. Shen H, Kreisel D, and Goldstein DR. Processes of sterile inflammation. J Immunol. (2013) 191:2857–63. doi: 10.4049/jimmunol.1301539
66. Shin B, Benavides GA, Geng J, Koralov SB, Hu H, Darley-Usmar VM, et al. Mitochondrial oxidative phosphorylation regulates the fate decision between pathogenic th17 and regulatory T cells. Cell Rep. (2020) 30:1898–909 e4. doi: 10.1016/j.celrep.2020.01.022
67. Chen Y, Colello J, Jarjour W, and Zheng SG. Cellular metabolic regulation in the differentiation and function of regulatory T cells. Cells. (2019) 8. doi: 10.3390/cells8020188
68. Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, et al. Constitutive glycolytic metabolism supports cd8(+) T cell effector memory differentiation during viral infection. Immunity. (2016) 45:1024–37. doi: 10.1016/j.immuni.2016.10.017
69. Kiritsy MC, McCann K, Mott D, Holland SM, Behar SM, Sassetti CM, et al. Mitochondrial respiration contributes to the interferon gamma response in antigen-presenting cells. Elife. (2021) 10. doi: 10.7554/eLife.65109
70. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
71. McGuire PJ. Mitochondrial dysfunction and the aging immune system. Biol (Basel). (2019) 8. doi: 10.3390/biology8020026
72. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in foxp3(+) T-regulatory cell function and allograft survival. FASEB J. (2015) 29:2315–26. doi: 10.1096/fj.14-268409
73. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex iii is essential for suppressive function of regulatory T cells. Nature. (2019) 565:495–9. doi: 10.1038/s41586-018-0846-z
74. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance T(Reg) cell anabolic metabolism for suppression. Nat Immunol. (2016) 17:1459–66. doi: 10.1038/ni.3577
75. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter glut1 is selectively essential for cd4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
76. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. (2014) 20:1327–33. doi: 10.1038/nm.3704
77. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for treg suppressive function. Cell Metab. (2020) 31:422–37 e5. doi: 10.1016/j.cmet.2019.11.021
78. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity. (2016) 44:406–21. doi: 10.1016/j.immuni.2016.01.028
79. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and pd-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
80. Averill-Bates D. Reactive oxygen species and cell signaling. Review Biochim Biophys Acta Mol Cell Res. (2024) 1871:119573. doi: 10.1016/j.bbamcr.2023.119573
81. Hovelmeyer N, Schmidt-Supprian M, and Ohnmacht C. Nf-kappab in control of regulatory T cell development, identity, and function. J Mol Med (Berl). (2022) 100:985–95. doi: 10.1007/s00109-022-02215-1
82. Peng HY, Lucavs J, Ballard D, Das JK, Kumar A, Wang L, et al. Metabolic reprogramming and reactive oxygen species in T cell immunity. Front Immunol. (2021) 12:652687. doi: 10.3389/fimmu.2021.652687
83. Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci U.S.A. (2010) 107:17686–91. doi: 10.1073/pnas.1012016107
84. Kelley TW, Efimova O, Schumacher J, and Szankasi P. Contrasting requirement for reactive oxygen species for the suppressive function of naturally occurring regulatory T cells but not induced regulatory T cells. Blood. (2012) 120:2146–. doi: 10.1182/blood.V120.21.2146.2146
85. Adebayo M, Singh S, Singh AP, and Dasgupta S. Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. FASEB J. (2021) 35:e21620. doi: 10.1096/fj.202100067R
86. Ono T, Isobe K, Nakada K, and Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. (2001) 28:272–5. doi: 10.1038/90116
87. Fang Y, Zhang Q, Lv C, Guo Y, He Y, Guo P, et al. Mitochondrial fusion induced by transforming growth factor-beta1 serves as a switch that governs the metabolic reprogramming during differentiation of regulatory T cells. Redox Biol. (2023) 62:102709. doi: 10.1016/j.redox.2023.102709
88. McDonald-Hyman C, Aguilar EG, Rhee SY, Mohamed FA, Compeer E, Sparwassar T, et al. Acc1 inhibition enhances treg gvhd treatment efficacy through regulation of mitochondrial fusion and elongation. Blood. (2023) 142:4819. doi: 10.1182/blood-2023-187306
89. Taguchi N, Ishihara N, Jofuku A, Oka T, and Mihara K. Mitotic phosphorylation of dynamin-related gtpase drp1 participates in mitochondrial fission. J Biol Chem. (2007) 282:11521–9. doi: 10.1074/jbc.M607279200
90. Li YH, Xu F, Thome R, Guo MF, Sun ML, Song GB, et al. Mdivi-1, a mitochondrial fission inhibitor, modulates T helper cells and suppresses the development of experimental autoimmune encephalomyelitis. J Neuroinflamm. (2019) 16:149. doi: 10.1186/s12974-019-1542-0
91. Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PloS One. (2008) 3:e3257. doi: 10.1371/journal.pone.0003257
92. Ding W-X and Yin X-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. bchm. (2012) 393:547–64. doi: 10.1515/hsz-2012-0119
93. Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab. (2020) 32:591–604 e7. doi: 10.1016/j.cmet.2020.07.001
94. Shields HJ, Traa A, and Van Raamsdonk JM. Beneficial and detrimental effects of reactive oxygen species on lifespan: A comprehensive review of comparative and experimental studies. Front Cell Dev Biol. (2021) 9:628157. doi: 10.3389/fcell.2021.628157
95. Labunskyy VM and Gladyshev VN. Role of reactive oxygen species-mediated signaling in aging. Antioxid Redox Signal. (2013) 19:1362–72. doi: 10.1089/ars.2012.4891
96. Heo S, Kim S, and Kang D. The role of hydrogen peroxide and peroxiredoxins throughout the cell cycle. Antioxid (Basel). (2020) 9. doi: 10.3390/antiox9040280
97. Duan J, Duan J, Zhang Z, and Tong T. Irreversible cellular senescence induced by prolonged exposure to H2o2 involves DNA-damage-and-repair genes and telomere shortening. Int J Biochem Cell Biol. (2005) 37:1407–20. doi: 10.1016/j.biocel.2005.01.010
98. Karplus PA and Poole LB. Peroxiredoxins as molecular triage agents, sacrificing themselves to enhance cell survival during a peroxide attack. Mol Cell. (2012) 45:275–8. doi: 10.1016/j.molcel.2012.01.012
99. Andrés CMC, Pérez de la Lastra JM, Juan CA, Plou FJ, and Pérez-Lebeña E. Chemistry of hydrogen peroxide formation and elimination in mammalian cells, and its role in various pathologies. Stresses. (2022) 2:256–74. doi: 10.3390/stresses2030019
100. Korshunov SS, Skulachev VP, and Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. (1997) 416:15–8. doi: 10.1016/s0014-5793(97)01159-9
101. Lebiedzinska M, Karkucinska-Wieckowska A, Giorgi C, Karczmarewicz E, Pronicka E, Pinton P, et al. Oxidative stress-dependent P66shc phosphorylation in skin fibroblasts of children with mitochondrial disorders. Biochim Biophys Acta. (2010) 1797:952–60. doi: 10.1016/j.bbabio.2010.03.005
102. Patti ME and Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. (2010) 31:364–95. doi: 10.1210/er.2009-0027
103. Samuel VT and Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. (2012) 148:852–71. doi: 10.1016/j.cell.2012.02.017
104. Petersen KF and Shulman GI. Etiology of insulin resistance. Am J Med. (2006) 119:S10–6. doi: 10.1016/j.amjmed.2006.01.009
105. Schafer JA, Bozkurt S, Michaelis JB, Klann K, and Munch C. Global mitochondrial protein import proteomics reveal distinct regulation by translation and translocation machinery. Mol Cell. (2022) 82:435–46 e7. doi: 10.1016/j.molcel.2021.11.004
106. Martini H and Passos JF. Cellular senescence: all roads lead to mitochondria. FEBS J. (2023) 290:1186–202. doi: 10.1111/febs.16361
107. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
108. Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, et al. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc Natl Acad Sci U.S.A. (2019) 116:18435–44. doi: 10.1073/pnas.1910574116
109. Schank M, Zhao J, Wang L, Li Z, Cao D, Nguyen LN, et al. Telomeric injury by kml001 in human T cells induces mitochondrial dysfunction through the P53-pgc-1alpha pathway. Cell Death Dis. (2020) 11:1030. doi: 10.1038/s41419-020-03238-7
110. Wang L, Lu Z, Zhao J, Schank M, Cao D, Dang X, et al. Selective oxidative stress induces dual damage to telomeres and mitochondria in human T cells. Aging Cell. (2021) 20:e13513. doi: 10.1111/acel.13513
111. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the P53 tumor suppressor. PloS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
112. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. (2013) 15:978–90. doi: 10.1038/ncb2784
113. Soto-Heredero G, Gabande-Rodriguez E, Carrasco E, Escrig-Larena JI, Gomez de Las Heras MM, Delgado-Pulido S, et al. Klrg1 identifies regulatory T cells with mitochondrial alterations that accumulate with aging. Nat Aging. (2025) 5:799–815. doi: 10.1038/s43587-025-00855-9
114. Senft D and Ronai ZA. Upr, autophagy, and mitochondria crosstalk underlies the er stress response. Trends Biochem Sci. (2015) 40:141–8. doi: 10.1016/j.tibs.2015.01.002
115. Shpilka T and Haynes CM. The mitochondrial upr: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. (2018) 19:109–20. doi: 10.1038/nrm.2017.110
116. Rolland SG, Schneid S, Schwarz M, Rackles E, Fischer C, Haeussler S, et al. Compromised mitochondrial protein import acts as a signal for upr(Mt). Cell Rep. (2019) 28:1659–69 e5. doi: 10.1016/j.celrep.2019.07.049
117. Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature. (2015) 524:485–8. doi: 10.1038/nature14951
118. Oliveira AN and Hood DA. Effect of tim23 knockdown in vivo on mitochondrial protein import and retrograde signaling to the upr(Mt) in muscle. Am J Physiol Cell Physiol. (2018) 315:C516–C26. doi: 10.1152/ajpcell.00275.2017
119. Xin N, Durieux J, Yang C, Wolff S, Kim HE, and Dillin A. The uprmt preserves mitochondrial import to extend lifespan. J Cell Biol. (2022) 221. doi: 10.1083/jcb.202201071
120. Soo SK, Traa A, Rudich PD, Mistry M, and Van Raamsdonk JM. Activation of mitochondrial unfolded protein response protects against multiple exogenous stressors. Life Sci Alliance. (2021) 4. doi: 10.26508/lsa.202101182
121. Berry BJ, Nieves TO, and Wojtovich AP. Decreased mitochondrial membrane potential activates the mitochondrial unfolded protein response. MicroPubl Biol. (2021) 2021. doi: 10.17912/micropub.biology.000445
122. Sutandy FXR, Gossner I, Tascher G, and Munch C. A cytosolic surveillance mechanism activates the mitochondrial upr. Nature. (2023) 618:849–54. doi: 10.1038/s41586-023-06142-0
123. Angeli S, Foulger A, Chamoli M, Peiris TH, Gerencser A, Shahmirzadi AA, et al. The mitochondrial permeability transition pore activates the mitochondrial unfolded protein response and promotes aging. Elife. (2021) 10. doi: 10.7554/eLife.63453
124. Hernando-Rodriguez B, Erinjeri AP, Rodriguez-Palero MJ, Millar V, Gonzalez-Hernandez S, Olmedo M, et al. Combined flow cytometry and high-throughput image analysis for the study of essential genes in caenorhabditis elegans. BMC Biol. (2018) 16:36. doi: 10.1186/s12915-018-0496-5
125. Deng P and Haynes CM. Mitochondrial dysfunction in cancer: potential roles of atf5 and the mitochondrial upr. Semin Cancer Biol. (2017) 47:43–9. doi: 10.1016/j.semcancer.2017.05.002
126. Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, and Haynes CM. The transcription factor atf5 mediates a mammalian mitochondrial upr. Curr Biol. (2016) 26:2037–43. doi: 10.1016/j.cub.2016.06.002
127. Ji H, Wang J, Muid D, Song W, Jiang Y, and Zhou H. Fundc1 activates the mitochondrial unfolded protein response to preserve mitochondrial quality control in cardiac ischemia/reperfusion injury. Cell Signal. (2022) 92:110249. doi: 10.1016/j.cellsig.2022.110249
128. Wang Y, Jasper H, Toan S, Muid D, Chang X, and Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. (2021) 45:102049. doi: 10.1016/j.redox.2021.102049
129. Munch C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. (2018) 16:81. doi: 10.1186/s12915-018-0548-x
130. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM. The integrated stress response. EMBO Rep. (2016) 17:1374–95. doi: 10.15252/embr.201642195
131. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. (2000) 6:1099–108. doi: 10.1016/s1097-2765(00)00108-8
132. Lin N, Sun L, Chai J, Qi H, Zhao Y, Ma J, et al. Stress granules affect the dual pi3k/mtor inhibitor response by regulating the mitochondrial unfolded protein response. Cancer Cell Int. (2024) 24:38. doi: 10.1186/s12935-024-03210-x
133. Katiyar A, Fujimoto M, Tan K, Kurashima A, Srivastava P, Okada M, et al. Hsf1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio. (2020) 10:1135–48. doi: 10.1002/2211-5463.12863
134. Masser AE, Kang W, Roy J, Mohanakrishnan Kaimal J, Quintana-Cordero J, Friedlander MR, et al. Cytoplasmic protein misfolding titrates hsp70 to activate nuclear hsf1. Elife. (2019) 8. doi: 10.7554/eLife.47791
135. Sorger PK and Pelham HR. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell. (1988) 54:855–64. doi: 10.1016/s0092-8674(88)91219-6
136. Durieux J, Wolff S, and Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. (2011) 144:79–91. doi: 10.1016/j.cell.2010.12.016
137. Berendzen KM, Durieux J, Shao LW, Tian Y, Kim HE, Wolff S, et al. Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell. (2016) 166:1553–63 e10. doi: 10.1016/j.cell.2016.08.042
138. Shao LW, Niu R, and Liu Y. Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. (2016) 26:1182–96. doi: 10.1038/cr.2016.118
139. Prahlad V and Morimoto RI. Neuronal circuitry regulates the response of caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci U.S.A. (2011) 108:14204–9. doi: 10.1073/pnas.1106557108
140. Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y, et al. The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent wnt signaling. Cell. (2018) 174:870–83 e17. doi: 10.1016/j.cell.2018.06.029
141. Li X, Li J, Zhu D, Zhang N, Hao X, Zhang W, et al. Protein disulfide isomerase pdi-6 regulates wnt secretion to coordinate inter-tissue upr(Mt) activation and lifespan extension in C. Elegans Cell Rep. (2022) 39:110931. doi: 10.1016/j.celrep.2022.110931
142. Horwich AL and Fenton WA. Chaperonin-assisted protein folding: A chronologue. Q Rev Biophys. (2020) 53:e4. doi: 10.1017/S0033583519000143
143. Bahr T, Katuri J, Liang T, and Bai Y. Mitochondrial chaperones in human health and disease. Free Radic Biol Med. (2022) 179:363–74. doi: 10.1016/j.freeradbiomed.2021.11.015
144. Hartl FU, Bracher A, and Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. (2011) 475:324–32. doi: 10.1038/nature10317
145. Larburu N, Adams CJ, Chen CS, Nowak PR, and Ali MMU. Mechanism of hsp70 specialized interactions in protein translocation and the unfolded protein response. Open Biol. (2020) 10:200089. doi: 10.1098/rsob.200089
146. Baker TA and Sauer RT. Clpxp, an atp-powered unfolding and protein-degradation machine. Biochim Biophys Acta. (2012) 1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007
147. Al-Furoukh N, Ianni A, Nolte H, Holper S, Kruger M, Wanrooij S, et al. Clpx stimulates the mitochondrial unfolded protein response (Uprmt) in mammalian cells. Biochim Biophys Acta. (2015) 1853:2580–91. doi: 10.1016/j.bbamcr.2015.06.016
148. Ergun Y, Imamoglu AG, Cozzolino M, Demirkiran C, Basar M, Garg A, et al. Mitochondrial unfolded protein response gene clpp is required for oocyte function and female fertility. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25031866
149. Guo C, Xiao Y, Gu J, Zhao P, Hu Z, Zheng J, et al. Clpp/clpx deficiency impairs mitochondrial functions and mtorc1 signaling during spermatogenesis. Commun Biol. (2023) 6:1012. doi: 10.1038/s42003-023-05372-2
150. Papa L and Germain D. Sirt3 regulates the mitochondrial unfolded protein response. Mol Cell Biol. (2014) 34:699–710. doi: 10.1128/MCB.01337-13
151. Bause AS and Haigis MC. Sirt3 regulation of mitochondrial oxidative stress. Exp Gerontol. (2013) 48:634–9. doi: 10.1016/j.exger.2012.08.007
152. Zhao L, Cao J, Hu K, He X, Yun D, Tong T, et al. Sirtuins and their biological relevance in aging and age-related diseases. Aging Dis. (2020) 11:927–45. doi: 10.14336/AD.2019.0820
153. Wang T, Wang Y, Liu L, Jiang Z, Li X, Tong R, et al. Research progress on sirtuins family members and cell senescence. Eur J Med Chem. (2020) 193:112207. doi: 10.1016/j.ejmech.2020.112207
154. Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, et al. Ampk maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. (2017) 21:1–9. doi: 10.1016/j.celrep.2017.09.026
155. Shen Y, Wu Q, Shi J, and Zhou S. Regulation of sirt3 on mitochondrial functions and oxidative stress in parkinson’s disease. BioMed Pharmacother. (2020) 132:110928. doi: 10.1016/j.biopha.2020.110928
156. Weir HJ, Lane JD, and Balthasar N. Sirt3: A central regulator of mitochondrial adaptation in health and disease. Genes Cancer. (2013) 4:118–24. doi: 10.1177/1947601913476949
157. Shi T, Wang F, Stieren E, and Tong Q. Sirt3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. (2005) 280:13560–7. doi: 10.1074/jbc.M414670200
158. Rius-Perez S, Torres-Cuevas I, Millan I, Ortega AL, and Perez S. Pgc-1alpha, inflammation, and oxidative stress: an integrative view in metabolism. Oxid Med Cell Longev. (2020) 2020:1452696. doi: 10.1155/2020/1452696
159. Olmos Y, Valle I, Borniquel S, Tierrez A, Soria E, Lamas S, et al. Mutual dependence of foxo3a and pgc-1alpha in the induction of oxidative stress genes. J Biol Chem. (2009) 284:14476–84. doi: 10.1074/jbc.M807397200
160. Azadmanesh J and Borgstahl GEO. A review of the catalytic mechanism of human manganese superoxide dismutase. Antioxid (Basel). (2018) 7. doi: 10.3390/antiox7020025
161. Gaetani GF, Ferraris AM, Rolfo M, Mangerini R, Arena S, and Kirkman HN. Predominant role of catalase in the disposal of hydrogen peroxide within human erythrocytes. Blood. (1996) 87:1595–9. doi: 10.1182/blood.V87.4.1595.bloodjournal8741595
162. Alfonso-Prieto M, Biarnes X, Vidossich P, and Rovira C. The molecular mechanism of the catalase reaction. J Am Chem Soc. (2009) 131:11751–61. doi: 10.1021/ja9018572
163. Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, et al. Sirtuin 3, a new target of pgc-1alpha, plays an important role in the suppression of ros and mitochondrial biogenesis. PloS One. (2010) 5:e11707. doi: 10.1371/journal.pone.0011707
164. Li Y, Wang Q, Li J, Shi B, Liu Y, and Wang P. Sirt3 affects mitochondrial metabolic reprogramming via the ampk-pgc-1alpha axis in the development of benign prostatic hyperplasia. Prostate. (2021) 81:1135–48. doi: 10.1002/pros.24208
165. Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, et al. Sirt3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. (2011) 44:177–90. doi: 10.1016/j.molcel.2011.07.019
166. Kincaid B and Bossy-Wetzel E. Forever young: sirt3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci. (2013) 5:48. doi: 10.3389/fnagi.2013.00048
167. Papa L and Germain D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci. (2011) 124:1396–402. doi: 10.1242/jcs.078220
168. Adriaenssens E, Asselbergh B, Rivera-Mejias P, Bervoets S, Vendredy L, De Winter V, et al. Small heat shock proteins operate as molecular chaperones in the mitochondrial intermembrane space. Nat Cell Biol. (2023) 25:467–80. doi: 10.1038/s41556-022-01074-9
169. Kenny TC and Germain D. From discovery of the chop axis and targeting clpp to the identification of additional axes of the uprmt driven by the estrogen receptor and sirt3. J Bioenerg Biomembr. (2017) 49:297–305. doi: 10.1007/s10863-017-9722-z
170. Peter B, Waddington CL, Olahova M, Sommerville EW, Hopton S, Pyle A, et al. Defective mitochondrial protease lonp1 can cause classical mitochondrial disease. Hum Mol Genet. (2018) 27:1743–53. doi: 10.1093/hmg/ddy080
171. Munch C and Harper JW. Mitochondrial unfolded protein response controls matrix pre-rna processing and translation. Nature. (2016) 534:710–3. doi: 10.1038/nature18302
172. Pareek G and Pallanck LJ. Inactivation of lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition. Cell Death Dis. (2018) 9:1168. doi: 10.1038/s41419-018-1213-6
173. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. (2013) 497:451–7. doi: 10.1038/nature12188
174. Dahlmans D, Houzelle A, Andreux P, Wang X, Jorgensen JA, Moullan N, et al. Microrna-382 silencing induces a mitonuclear protein imbalance and activates the mitochondrial unfolded protein response in muscle cells. J Cell Physiol. (2019) 234:6601–10. doi: 10.1002/jcp.27401
175. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mtor kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
176. Saxton RA and Sabatini DM. Mtor signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
177. Li TY, Gao AW, Li X, Li H, Liu YJ, Lalou A, et al. V-atpase/torc1-mediated atfs-1 translation directs mitochondrial upr activation in C. Elegans. J Cell Biol. (2023) 222. doi: 10.1083/jcb.202205045
178. Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mtor kinase activity sets regulatory T cell responsiveness. Immunity. (2010) 33:929–41. doi: 10.1016/j.immuni.2010.11.024
179. Chapman NM and Chi H. Mtor signaling, tregs and immune modulation. Immunotherapy. (2014) 6:1295–311. doi: 10.2217/imt.14.84
180. Feehan RP and Shantz LM. Negative regulation of the foxo3a transcription factor by mtorc2 induces a pro-survival response following exposure to ultraviolet-B irradiation. Cell Signal. (2016) 28:798–809. doi: 10.1016/j.cellsig.2016.03.013
181. Dastidar SG, Pham MT, Mitchell MB, Yeom SG, Jordan S, Chang A, et al. 4e-bp1 protects neurons from misfolded protein stress and parkinson’s disease toxicity by inducing the mitochondrial unfolded protein response. J Neurosci. (2020) 40:8734–45. doi: 10.1523/JNEUROSCI.0940-20.2020
182. Chou SD, Prince T, Gong J, and Calderwood SK. Mtor is essential for the proteotoxic stress response, hsf1 activation and heat shock protein synthesis. PloS One. (2012) 7:e39679. doi: 10.1371/journal.pone.0039679
183. Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, et al. Mtorc1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. (2017) 26:419–28 e5. doi: 10.1016/j.cmet.2017.07.007
184. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, et al. The nad(+)/sirtuin pathway modulates longevity through activation of mitochondrial upr and foxo signaling. Cell. (2013) 154:430–41. doi: 10.1016/j.cell.2013.06.016
185. Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, and Li MO. Foxo proteins cooperatively control the differentiation of foxp3+ Regulatory T cells. Nat Immunol. (2010) 11:618–27. doi: 10.1038/ni.1884
186. Zhao Y, Zhao C, Guo H, Zhang Z, Xu H, Shi M, et al. Mtorc2 orchestrates monocytic and granulocytic lineage commitment by an atf5-mediated pathway. iScience. (2023) 26:107540. doi: 10.1016/j.isci.2023.107540
187. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of foxo transcription factors by the sirt1 deacetylase. Science. (2004) 303:2011–5. doi: 10.1126/science.1094637
188. Gedaly R, Cornea V, Turcios L, Edmisson JS, Harris DD, Watt DS, et al. Anti-neoplastic sulfonamides alter the metabolic homeostasis and disrupt the suppressor activity of regulatory T cells. Sci Rep. (2022) 12:19112. doi: 10.1038/s41598-022-23601-2
189. Xiaowei X, Qian X, and Dingzhou Z. Sirtuin-3 activates the mitochondrial unfolded protein response and reduces cerebral ischemia/reperfusion injury. Int J Biol Sci. (2023) 19:4327–39. doi: 10.7150/ijbs.86614
190. Wang X, Yu X, Li Y, Liu F, Du L, Xie N, et al. Atf5 attenuates apoptosis in hippocampal neurons with seizures evoked by mg(2+)-free medium via regulating mitochondrial unfolded protein response. Neurochem Res. (2023) 48:62–71. doi: 10.1007/s11064-022-03702-0
191. Smyrnias I, Gray SP, Okonko DO, Sawyer G, Zoccarato A, Catibog N, et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol. (2019) 73:1795–806. doi: 10.1016/j.jacc.2018.12.087
192. Shi L, Tan Y, Zheng W, Cao G, Zhou H, Li P, et al. Ctrp3 alleviates mitochondrial dysfunction and oxidative stress injury in pathological cardiac hypertrophy by activating uprmt via the sirt1/atf5 axis. Cell Death Discov. (2024) 10:53. doi: 10.1038/s41420-024-01813-x
193. Xin T and Lu C. Sirt3 activates ampk-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging (Albany NY). (2020) 12:16224–37. doi: 10.18632/aging.103644
194. Xu M, Xue RQ, Lu Y, Yong SY, Wu Q, Cui YL, et al. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and uprmt through sirt3-ampk pathway. Cardiovasc Res. (2019) 115:530–45. doi: 10.1093/cvr/cvy217
195. Zhang S, Zhang J, Wu L, Chen L, Niu P, and Li J. Glutamine supplementation reverses manganese neurotoxicity by eliciting the mitochondrial unfolded protein response. iScience. (2023) 26:107136. doi: 10.1016/j.isci.2023.107136
196. Yu X, Wang X, Xie Y, Peng T, Lian Y, Wang C, et al. Activating transcription factor 4-mediated mitochondrial unfolded protein response alleviates hippocampal neuronal damage in an in vitro model of epileptiform discharges. Neurochem Res. (2023) 48:2253–64. doi: 10.1007/s11064-023-03910-2
197. Sun GW, Ding TY, Wang M, Hu CL, Gu JJ, Li J, et al. Honokiol reduces mitochondrial dysfunction and inhibits apoptosis of nerve cells in rats with traumatic brain injury by activating the mitochondrial unfolded protein response. J Mol Neurosci. (2022) 72:2464–72. doi: 10.1007/s12031-022-02089-5
198. Wang J, Wang X, Du W, Xue Z, Huang W, Guan Z, et al. Bi-1 ameliorates myocardial injury by activating the mitochondrial unfolded protein response and fundc1-related mitophagy in cardiorenal syndrome type 3. Cell Signal. (2022) 91:110218. doi: 10.1016/j.cellsig.2021.110218
199. Zhao R, Zarabi S, Nii T, Halgas O, Jitkova Y, Ruvolo V, et al. Dual targeting of mitochondrial unfolded protein response and bcl2 in acute myeloid leukemia. Blood. (2019) 134:2562. doi: 10.1182/blood-2019-127101
200. Zhou Q, Zhu L, Qiu W, Liu Y, Yang F, Chen W, et al. Nicotinamide riboside enhances mitochondrial proteostasis and adult neurogenesis through activation of mitochondrial unfolded protein response signaling in the brain of als sod1(G93a) mice. Int J Biol Sci. (2020) 16:284–97. doi: 10.7150/ijbs.38487
201. Kumar R, Chaudhary AK, Woytash J, Inigo JR, Gokhale AA, Bshara W, et al. A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via hsp60. J Clin Invest. (2022) 132. doi: 10.1172/JCI149906
202. Cai G, Lin F, Wu D, Lin C, Chen H, Wei Y, et al. Rosmarinic acid inhibits mitochondrial damage by alleviating unfolded protein response. Front Pharmacol. (2022) 13:859978. doi: 10.3389/fphar.2022.859978
203. Liu M, Yu S, Wang J, Qiao J, Liu Y, Wang S, et al. Ginseng protein protects against mitochondrial dysfunction and neurodegeneration by inducing mitochondrial unfolded protein response in drosophila melanogaster pink1 model of parkinson’s disease. J Ethnopharmacol. (2020) 247:112213. doi: 10.1016/j.jep.2019.112213
204. Suarez-Rivero JM, Pastor-Maldonado CJ, Romero-Gonzalez A, Gomez-Fernandez D, Povea-Cabello S, Alvarez-Cordoba M, et al. Pterostilbene in combination with mitochondrial cofactors improve mitochondrial function in cellular models of mitochondrial diseases. Front Pharmacol. (2022) 13:862085. doi: 10.3389/fphar.2022.862085
205. Suarez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Alvarez-Cordoba M, Villalon-Garcia I, Talaveron-Rey M, et al. Upr(Mt) activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J Rare Dis. (2022) 17:204. doi: 10.1186/s13023-022-02331-8
206. Armstrong JS. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br J Pharmacol. (2007) 151:1154–65. doi: 10.1038/sj.bjp.0707288
207. Khan T, Waseem R, Zehra Z, Aiman A, Bhardwaj P, Ansari J, et al. Mitochondrial dysfunction: pathophysiology and mitochondria-targeted drug delivery approaches. Pharmaceutics. (2022) 14. doi: 10.3390/pharmaceutics14122657
Keywords: regulatory T-cells, cell metabolism, cellular stress, oxidative stress, unfolded protein response, immunosenescence, aging
Citation: Lewis L, Valvi D, Gedaly R and Marti F (2025) Mitochondrial unfolded protein response in regulatory T cell function: a protective mechanism in immune aging. Front. Immunol. 16:1621759. doi: 10.3389/fimmu.2025.1621759
Received: 01 May 2025; Accepted: 16 June 2025;
Published: 30 June 2025.
Edited by:
Juliana Escher Toller-Kawahisa, Trinity College Dublin, IrelandReviewed by:
Fabrizia Bonacina, University of Milan, ItalyJonas Aakre Wik, Oslo University Hospital, Norway
Copyright © 2025 Lewis, Valvi, Gedaly and Marti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesc Marti, Zm1hcnQzQHVreS5lZHU=
†ORCID: Francesc Marti, orcid.org/0000-0002-2066-6884