 Chelsey Skeete1
Chelsey Skeete1 Gabriel Sgambettera2Aldana D. Gojanovich2Xianbao He3
Gabriel Sgambettera2Aldana D. Gojanovich2Xianbao He3 Daniel Bryant3Mengwei Yang2Shreya Banerjee1
Daniel Bryant3Mengwei Yang2Shreya Banerjee1 Andrés A. Quiñones-Molina1
Andrés A. Quiñones-Molina1 Hisashi Akiyama1
Hisashi Akiyama1 Gustavo Mostoslavsky2
Gustavo Mostoslavsky2 Andrew J. Henderson3
Andrew J. Henderson3 Suryaram Gummuluru1*
Suryaram Gummuluru1*- 1Department of Virology, Immunology, and Microbiology, Boston University Chobanian and Avedisian School of Medicine, Boston, MA, United States
- 2Boston University Chobanian and Avedisian School of Medicine, Center for Regenerative Medicine, Boston Medical Center, Boston, MA, United States
- 3Section of Infectious Diseases, Boston Medical Center, Boston, MA, United States
People living with HIV-1 (PWH) and chronically using opioids have elevated risks of developing HIV-associated neurological disorders (HAND) that are often correlated with persistent inflammation. Microglia, innate immune cells in the brain, are the principal HIV-1 reservoir in the central nervous system and regulate neuroinflammation. Our group previously showed that HIV-1 infection of induced pluripotent stem cell (iPSC)-derived microglia and viral intron-containing RNA (icRNA) expression triggers inflammatory responses. Microglia express μ opioid receptor, MOR, yet the immunomodulatory effects of opioids on HIV-1 infection in microglia are unclear. Here, we report that MOR activation impacts HIV-1 infection establishment and HIV-1-induced innate responses in microglia. Morphine pretreatment enhanced reverse transcription (RT), integration, viral transcription, and p24Gag secretion in HIV-1-infected iPSC-derived microglia, which was blocked by treatment with naloxone, a MOR antagonist. In contrast, morphine treatment did not impact HIV-1 infection in MOR-deficient monocyte-derived macrophages, although, induced exogenous expression of MOR in macrophages conferred morphine-mediated enhancement of HIV-1 infection. Interestingly, viral transcriptome analysis by digital-drop PCR revealed selective enhancement of HIV-1 icRNA expression in morphine-exposed iPSC-derived microglia, which correlated with enhanced HIV-1 icRNA-induced secretion of IP-10 in MOR+ cells. Further, PI3K inhibitor, wortmannin, blocked morphine-mediated enhancement of HIV-1 replication and HIV-1 icRNA-induced IP-10 secretion, suggesting that MOR signaling and HIV-1 icRNA expression synergistically activate the PI3K-Akt signaling pathway in microglia to exacerbate virus-induced inflammatory responses.
Introduction
HIV-1 remains a public health threat across the globe, with over 39.9 million people living with HIV-1 in 2023 (1). While new infections have declined by 39% as of 2023, primarily due to the expansion of antiretroviral therapy (ART) use, 1.3 million people were still newly diagnosed with HIV in 2023. Injection drug use is highly associated with HIV-1 infection, with intravenous drug use being a major route for HIV-1 infection (2). Globally, there are 11 million people who inject drugs (PWID), and 1 in 8 of these people are living with HIV-1 (3). The most common drug to be injected during non-medical usage is heroin, which is derived from morphine (4, 5). Heroin usage significantly influences opioid-dependency behaviors and raises the risk of developing opioid use disorders (OUD) (6), thus contributing to the establishment of the OUD and HIV-1 syndemic (7). In addition to intravenous routes, opioids such as morphine or fentanyl are also ingested via the oral route in pill form at higher rates in PWH than people without HIV (8). HIV-associated neurocognitive disorders (HAND) are a spectrum of neurocognitive impairments that are estimated to impact 30-50% of people with HIV-1 (PWH) globally (9). PWH who use injection drugs, such as opioids, are at higher risk for various co-morbidities, including neurocognitive disorders (10–12). Further, chronic opioid use has been shown to worsen neurocognition in PWH (13, 14). Therefore, understanding the underlying mechanisms by which opioids can exacerbate HAND is essential to the development of HAND treatments (15).
Microglia are the principal long-term cellular reservoir of HIV-1 in the CNS (16, 17). Despite ART, persistently infected microglia have been observed in PWH (18), and their longevity as brain resident macrophages might contribute to viral persistence (19). Microglia are regulators of neuroinflammation and secrete interferons and inflammatory cytokines such as interferon gamma-induced protein 10 (IP-10) and IL-1β in response to HIV-1 infection (15, 20–22). Secretion of inflammatory cytokines and interferons by microglia is suspected to damage surrounding neurons, contributing to the risk of HAND development (23–25). Specifically, type I interferons have been suggested to contribute to cognitive impairment (26), and IP-10 is associated with poor prognosis in PWH with HAND (27). Persistent neuroinflammation is highly associated with HAND (28–30). Further, opioids have been shown to worsen neuroinflammation by promoting microglial activation, lymphocyte infiltration by chemokine signaling, and blood-brain barrier (BBB) disruption in PWID (31). Concurrently, PWH and using injection opioids have higher levels of systemic inflammation than those who do not use injection drugs (32–34). However, the effects of opioid signaling on HIV infection and virus-induced inflammatory responses in the CNS are not well understood.
Human microglia express the three most common classes of opioid receptors, Mu (MOR), Delta (DOR), and Kappa (KOR) (35), as opposed to non-CNS resident myeloid cells such as macrophages, which have minimal opioid receptor expression (36–38). Opioid receptors are class A G-protein coupled receptors (GPCRs) activated by alkaloid opiates such as morphine (39, 40) that play a role in both reward and analgesic effects. Agonist binding results in receptor phosphorylation and activation that aids the recruitment of other signal transduction cascades, including mitogen-activated protein kinases (MAPKs), protein kinase C, and PI3-kinase (PI3K) dependent pathways (41–45). MOR expression is widespread in the CNS on both neurons and glial cells in the nucleus accumbens, hippocampus, amygdala, and spinal cord, though MOR activation in these cell types can contribute to divergent outcomes (46). For instance, opioid receptor activation in GABAergic neurons inhibits GABA release, which promotes dopamine release from dopaminergic neurons in CNS regions associated with reward, such as the ventral tegmental area (47–49). Opioid receptor activation in astrocytes is suggested to have anti-proliferative effects (50, 51), as well as inducing glutamate release (52). In contrast, opioid receptor activation in microglia, specifically by MOR agonists morphine and D-Ala (2)-mephe(4)-gly-ol(5))enkephalin (DAMGO), has been shown to enhance inflammatory effects (53, 54). MOR displays the highest binding affinity for morphine compared to DOR or KOR (55), and interestingly, ligation of MOR by morphine fails to promote receptor endocytosis and tolerance (56), which might contribute to the induction of prolonged signaling cascades. To date, the effects of morphine-induced MOR signaling on HIV-1 infection kinetics have not been defined in primary human microglia. Previous attempts to unravel mechanisms of HIV-1 interactions with microglia have either utilized murine microglia (57) or human cell lines with an unclear non-human origin that require transformation for maintenance in culture, which resulted in substantial loss in HIV-1 infectivity (58).
Here we investigated the role of morphine on HIV-1 infection in human microglia by using human induced pluripotent stem cell (iPSC) derived microglia. Recent studies by us and others have described that iPSC-derived microglia are permissive to HIV-1 infection, and the successful establishment of HIV-1 infection and expression of HIV-1 intron-containing RNA (HIV-1 icRNA) in iPSC-derived microglia induces inflammatory responses (59–63). In this report, we demonstrate that morphine enhances HIV-1 infection and viral icRNA expression in MOR+ iPSC-derived microglia, which is blocked by the opioid receptor antagonist, naloxone. In contrast, morphine does not significantly impact HIV-1 infection in monocyte-derived macrophages (MDMs), which have undetectable MOR expression. Significantly, overexpression of MOR in a human macrophage cell model, THP-1/PMA macrophages, rescues morphine-dependent enhancement of HIV-1 infection and subsequent HIV-1-infection-induced IP-10 secretion, suggesting that MOR expression is required for morphine-dependent modulation of HIV-1 infection in macrophages and microglia. Further PI3K inhibition suppresses morphine-mediated enhancement of HIV-1 infection and icRNA-induced inflammatory responses, suggesting that morphine-induced activation of MOR signaling synergizes with HIV-1 at the PI3K-Akt pathway to enhance infection kinetics and viral infection-induced inflammatory responses.
Methods
Cells
HEK293T cells (ATCC) and TZM-bl cells (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HRP-8129, contributed by Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc.) (Derdeyn, Decker et al., 2000) were maintained in culture with DMEM/10% FBS and 1% pen/strep (complete D10 media). THP-1 monocytic cells (ATCC, catalog #TIB-202) were cultured in RPMI1640/10% FBS/1% pen/strep (complete R10 media). To generate THP-1 macrophages, THP-1 monocytes were differentiated with PMA (100 nM, SIGMA, catalog # P8139) for 24h, washed, and returned to culture. PBMCs were isolated from fresh leukopaks obtained from NYBiologics from the fluffy top layer of Ficoll Paque Plus (Fisher, catalog# 45-001-750). CD14+ monocytes were isolated from PBMCs using CD14 bead isolation (Miltenyi, catalog# 130-045-101).
Generation of MDMs
Human monocyte-derived macrophages (MDMs) were differentiated from CD14+ peripheral blood monocytes by culturing in RPMI 1640 containing 10% heat-inactivated human AB serum (Sigma, catalog # H4522) and recombinant human M-CSF (Peprotech, catalog # 300-25) (20 ng/mL) for 5 days before culturing in complete R10 media.
Generation of iPSC-derived microglia
To generate iPSC-derived microglia, iPSCs are first differentiated toward a mesodermal, hematopoietic lineage using a protocol that consists of plating iPSCs at low confluency on Matrigel (Corning, catalog #354234)-coated plates in mTeSR (StemCell Technologies, catalog# 100-0276) for 2 days. Hematopoietic differentiation was initiated by replacing mTeSR with StemPro34 (Thermo Fisher Scientific, catalog# 10639011) media supplemented with Glutamax (Thermo Fisher Scientific, catalog#35050061), 450 nM alpha monothioglycerol (α-MTG) (Millipore Sigma, catalog# M6145-25ML), 88 ug/ml ascorbic acid (Sigma Aldrich, catalog #A4403-100MG), 200 g/ml transferrin (Millipore Sigma, catalog #10652202001), 0.1 mg/ml primocin (Fisher Scientific, catalog# ANT-PM-2), 5 ng/ml bone morphogenetic protein-4 (BMP4) (Peprotech, catalog #120-05ET-100UG), 50 ng/ml vascular endothelial growth factor (VEGF) (Peprotech, catalog # 100-20A-100UG) and 2µM CHIR99021 (Fisher Scientific, catalog #44-231-0), and transferring cells to hypoxic culture conditions (5% O2). On days 2 and 4, the media was changed, with 2µM CHIR99021 being switched out for 20 ng/ml basic fibroblast growth factor (bFGF) (StemCell Technologies, catalog #78003). On days 6 through 12, media was replaced every other day with StemPro34, containing Glutamax, 450 nM α-MTG, 50 μg/ml ascorbic acid, 150 g/ml transferrin, 0.1 mg/ml primocin, 10 ng/ml BMP4, 5 ng/ml VEGF, 5 ng/ml bFGF, 100 ng/ml SCF, 10 ng/ml FMS like tyrosine kinase 3 ligand (FLT3L) (R&D Systems, catalog #308-FKHB-050), 30 ng/ml thyroid peroxidase (TPO) (Peprotech, catalog #300-18-100UG), 30 ng/ml IL-3 (R&D Systems, catalog #203-IL-050/CF), 10ng/ml IL-6 (Peprotech, catalog #200-06), 5 ng/ml IL-11 (Peprotech, catalog #200-11), 25 ng/ml insulin like growth factor-1 (IFG1) (R&D Systems, catalog #291-G1-200) and 20 ng/ml sonic hedgehog recombinant protein (SHH) (Peprotech, catalog #100-45). Cells were switched back to normoxia conditions at day 8. At the end of the 12-day protocol, hematopoietic cells contain 25 - 65% (average 43%) CD34+CD45+ progenitor cells (64). Subsequently, the hematopoietic cells were harvested and differentiated toward microglia lineage using STEMdiff™ Microglia Differentiation Kit (StemCell Technologies, catalog #100-0019) and STEMdiff™ Microglia Maturation Kit (StemCell Technologies, catalog #100-0020). This protocol consists of 24 days of microglia differentiation followed by 4–10 days of microglia maturation, resulting in a highly pure population of microglia (> 90% CD45/CD11b-positive, 90% TREM2-positive microglia).
Viruses
HIV-1 proviral plasmids, Lai/YU2-env (replication-competent, CCR5-tropic), and LaiΔenv/GFP (single-round GFP-expressing HIV-1 reporter virus) have been described previously (65, 66). Viruses were derived via calcium phosphate-mediated transient transfection of HEK293T cells with proviral plasmids, packaging plasmid (psPAX2), and VSV-G-expression plasmid (H-CMVG). As previously described, SIVmac Vpx containing VLPs were generated from HEK293T cells by co-transfection with SIV3+ packaging plasmid and H-CMVG (Goujon, Jarrosson-Wuillème et al., 2006). The plasmid pcDNA3.1/OPRM1, which expresses a flag-epitope-tagged MOR, was a gracious gift of the Ferré lab (67). To generate a flag-MOR-expressing retroviral expression plasmid, flag-OPRM1 orf was cloned into pLNCX using Hind III and Apa I restriction enzymes. LNC-MOR or empty LNCX retroviral particles were generated by calcium phosphate-mediated co-transfection of HEK293T cells with LNCX or LNC-MOR retroviral plasmids with packaging plasmid (pCL10A1) and envelope-expression plasmid (H-CMVG). Virus particles were harvested from HEK293T supernatants at two days post-transfection, filtered through a 0.45 µm filter, and concentrated via ultracentrifugation at 24,000 rpm for 1.5 hours at 4 °C over 20% sucrose using a SW32Ti rotor (Beckman Coulter). Virus pellets were resuspended in PBS and stored at -80 °C. To generate CD4+ T cell-derived Lai/YU-2env, CD4+ T cells were infected with Lai/YU2-env at MOI 0.1 by spinoculation at 2300 rpm for 1 hour, washed with PBS, and cultured in R10 media supplemented with 50U/mL IL-2. CD4+ T cell supernatants were harvested at days 3, 6, and 9 post-infection using the same ultracentrifugation steps described above. Infectious virus titers were measured on TZM-bI cells by measuring β-galactosidase activity as described previously in (68).
Generation of MOR-expressing THP-1s
THP-1 monocytes were transduced with flag-MOR-expressing retrovirus particles and cultured in geneticin (Gibco, 10-131-027) (1 mg/mL)-containing complete R10 media. Surface expression of MOR was confirmed by flow cytometry and immunofluorescence analysis using anti-MOR polyclonal antibody (Novus Biologicals, catalog # 31180).
Opioid treatment
Cells were treated with varying concentrations of morphine (NDC 00216-1307-08) or DMSO (vehicle control) for 24h before infection initiation. In some experimental conditions, cells were treated with 1µM naloxone (Sigma-Aldrich, catalog #BP548) for 1 hour before morphine was added.
Infection
iPSC-derived microglia and MDMs were infected with CD4+ T cell-passaged Lai/YU-2env, and THP-1/PMA macrophages were infected with LaiΔenv/GFP/G. To increase the permissiveness of THP-1/PMA macrophages and MDMs to HIV-1 infection, infection conditions included either SIVmac Vpx-VLPs (5 ng of p27) or deoxyribonucleosides (dNs, 2.5mM, Sigma-Aldrich, catalog# D8668, D0776, T1895, D0901) for THP1/macrophages and MDMs, respectively. In some experiments, cells were pretreated with efavirenz (EFV, 1 µM, NIH AIDS Reagent Program) or raltegravir (Ral, 30 µM, Selleck Chemicals, catalog #50-615-1). DMSO (Sigma Aldrich, catalog #BP231-1) was used as a vehicle control. Cells were spinoculated with virus-containing supernatants for 1 hr at 2300 rpm at room temperature with various multiplicities of infection (MOI) ranging from 0.2 to 1. Cells were washed 2 hours after spinoculation to remove unbound virus. Cells were harvested 3 days post-infection, and cells and cell-free supernatants were processed for measurements of infection establishment (flow cytometry for GFP expression or p24gag ELISA).
RNA analysis
Total mRNA (100 ng), isolated from 5x105 cells using a RNeasy Plus kit (Qiagen, catalog #74136), was reverse transcribed (Superscript IV, Invitrogen, catalog #18-090-010) using oligo(dT)20, and random hexamer primers. Target mRNA was quantified using Maxima SYBR Green (Thermo Scientific, catalog #FERK0242) for the following targets: OPRM1, HIV-1 msRNA, HIV-1 gRNA. The threshold cycle (Cq) was normalized to that of GAPDH using the ΔΔCt method (Rao, Huang et al., 2013). Primers for RT-PCR are listed in Table 1. For RT-ddPCR, FAM-conjugated probes were designed to distinguish HIV transcripts. RT-ddPCR probe design was based on those previously reported (69) and multiplexed to measure the frequency of probe sites simultaneously in ddPCR assays. Cycling conditions for ddPCR were described previously (70). Signals were identified using a Bio-Rad QX200 Droplet Digital PCR System and Quantasoft Data Analysis Software. The lists of primers and probes for RT-ddPCR are listed in Tables 2 and 3, respectively.
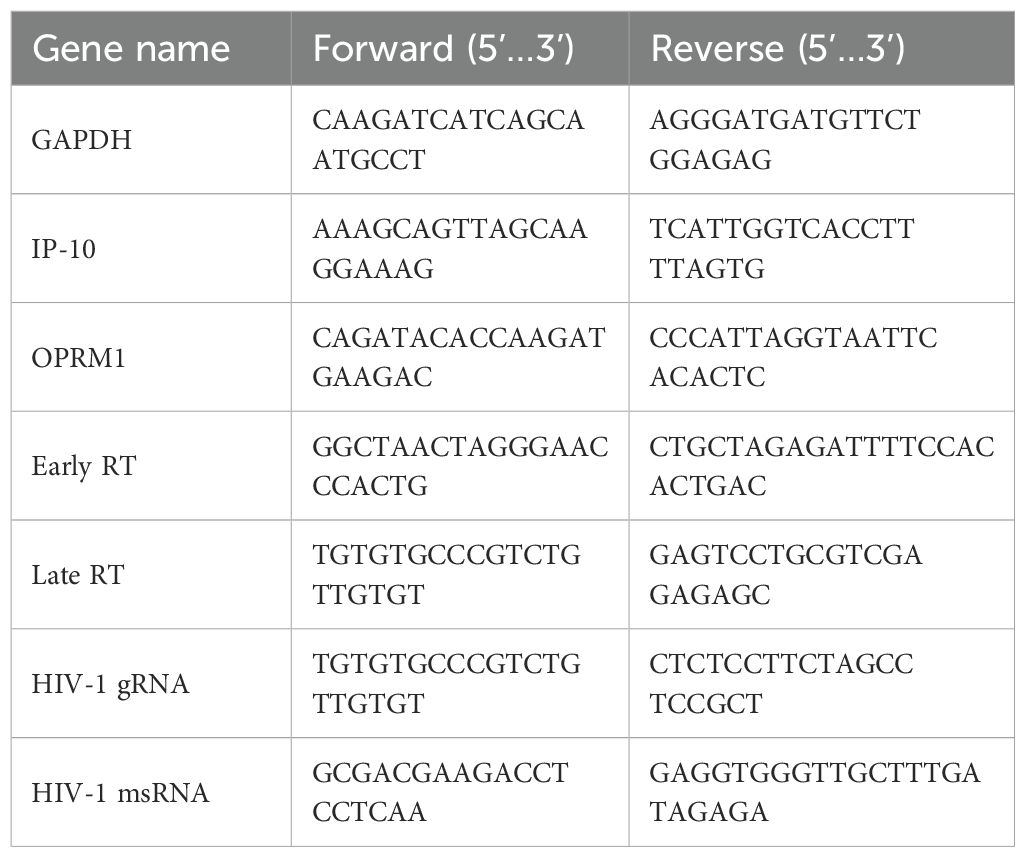
Table 1. List of primers for RT-PCR.
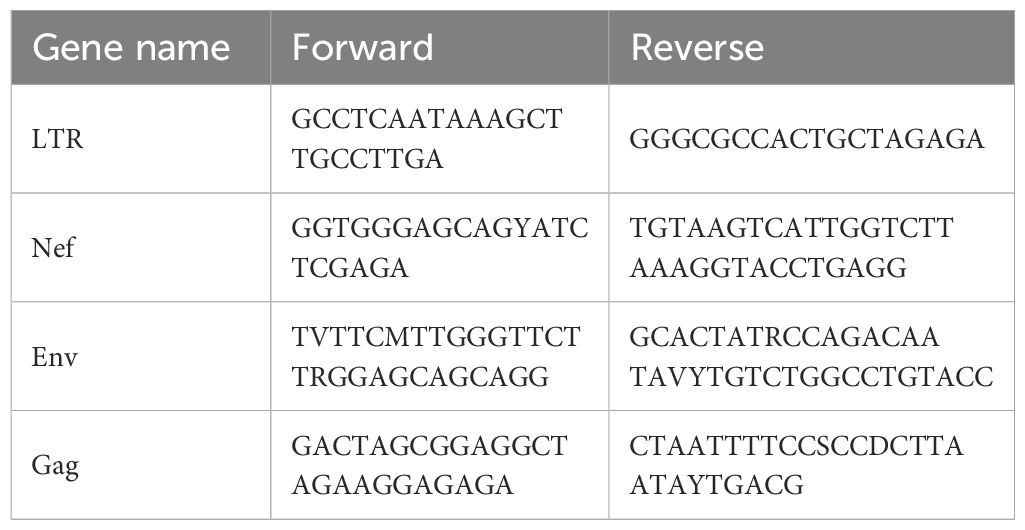
Table 2. List of primers for RT-ddPCR.

Table 3. List of probes for RT-ddPCR.
DNA analysis
Cells were lysed with DNA lysis buffer containing 10 mM EDTA (Bioworld, catalog #40120337, 100 mM NaCl (Fisher Scientific, catalog #S271-3), 0.5% (W/V) SDS (Boston Bioproducts, catalog #BM-230), and 10 mM Tris-Cl (pH 8.0) (Boston Bioproducts, catalog #BBT-75). Lysates were treated with proteinase K (New England Biolabs, catalog #P8107) for 15min at 56 °C. DNA was isolated from proteinase K-treated cell lysates using phenol: chloroform: isoamyl alcohol (Fisher Scientific, 15593031) extraction, precipitated with sodium acetate and ethanol, and resuspended in water. Total cell DNA was used for quantification of early and late RT products by qPCR (primers are listed in Table 1).
Intact proviral DNA assay
Primers and probes targeting the 5’ (psi sequence) and 3’ regions (RRE/env) of HIV-1 provirus, conjugated to FAM and VIC fluorophores, respectively, were used as previously described (70, 71). The lists for IPDA primers and probes are listed in Tables 4 and 5, respectively. The dark probe recognized hypermutated Env sequences to define hypermutated provirus sequences. Hypermutated signals were detected as single positive droplets with FAM signals. RPP30 primers and probes were used to determine the cell number and DNA shearing index. Infections in the presence of EFV or RAL were used as controls to confirm that IPDA signals were dependent on reverse transcription and derived from integrated genomes. Cell numbers are estimated from the RPP30 and DNA shearing quantification.
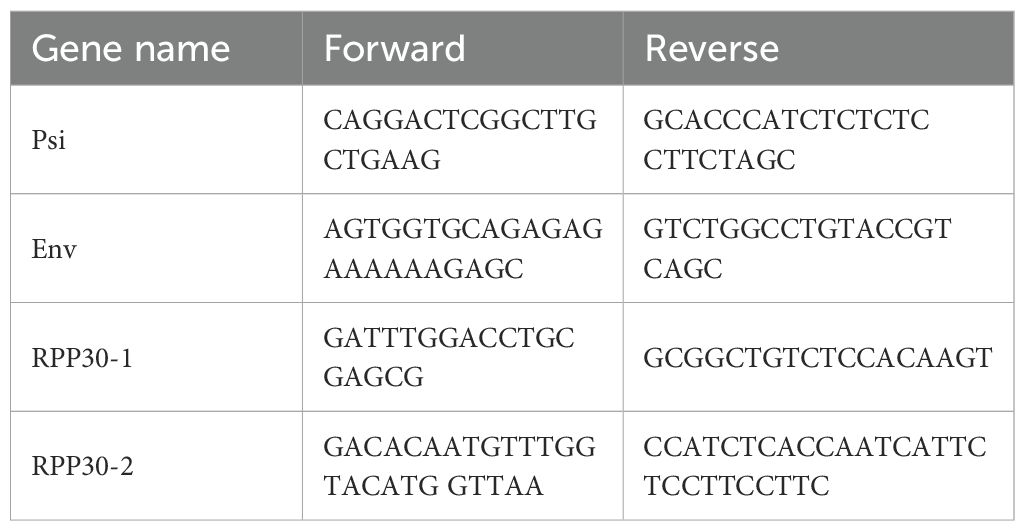
Table 4. List of primers for IPDA.

Table 5. List of probes for IPDA.
Western blot analysis
THP-1/PMA macrophages were pretreated with 1 µM naloxone for 20min before 1 µM morphine or DMSO treatment for 7min. Cells were washed with ice-cold PBS and lysed with RIPA buffer (Boston Bioproducts, catalog #116TX) supplemented with protease inhibitors (Sigma-Aldrich, catalog # 4693159001). Lysates were clarified by centrifugation for 20min at 13,000 rpm. Protein concentrations were determined using Bradford reagent (Fisher Scientific, catalog #PI23246). Cell lysates containing 30 µg of total protein were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with Li-Cor Blocking Buffer (Fisher Scientific, catalog #NC1660556) before being probed with rabbit anti-phospho-Akt (Cell Signaling, catalog # 4060, 1:1000) or rabbit anti-phospho-MOR (Cell Signaling, catalog #3451, 1:1000) and mouse anti-β-actin antibody (Thermo-Fisher, catalog #AM4302, 1:5000). Staining was visualized with secondary antibodies: goat anti-mouse-IgG-DyLight 680 (Pierce, catalog #35568) and goat anti-rabbit-IgG-DyLight 800 (Pierce, catalog #35518). Membranes were stripped with stripping buffer (Invitrogen, catalog #46430) for 30min at room temperature and probed again for rabbit anti-pan-Akt (Cell Signaling, catalog #4691, 1:1000) and mouse anti-actin. Membranes were scanned with Odyssey CLx scanner (Li-Cor).
Imaging
For THP-1/PMA macrophages, MDMs, and iPSC-derived microglia, cells cultured on coverslips were stained with Alexa594-conjugated wheat germ agglutinin (Thermo-Fisher, catalog #32464, 1:250) for 20 minutes at 4 °C to label the plasma membrane. Cells were washed with PBS and stained with a rabbit anti-MOR antibody (Novus Biologics, catalog #NBP1-31180 1:500) followed by an Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, catalog #A11070, 1:200). Cells were fixed with 4% paraformaldehyde (Boston Bioproducts, catalog#BM-155) followed by DAPI (49,6-diamidino-2-phenylindole; Sigma-Aldrich, catalog #D9542) staining to visualize nuclei. Stained cells were imaged with a Nikon SP5 confocal microscope. Images were analyzed with ImageJ (NIH).
ELISAs
IP-10 secretion in culture supernatants was measured with a BD human IP-10 ELISA set (BD, catalog #B550926). IL-8 secretion in culture supernatants was measured with a BD human IL-8 ELISA set (BD, catalog# B555244). MCP-1 secretion in culture supernatants was measured with a BD human MCP-1 ELISA set (BD, catalog #B555179). The p24Gag content in cell supernatant secretion was measured by an ELISA, as previously described (Akiyama, Miller et al., 2018). SIVmac Vpx VLPs were tittered using a commercial p27 ELISA (XpressBio, catalog #SK845).
Cell viability assays
Cell viability was quantified by lactate dehydrogenase (LDH) measurements in cell culture supernatants using a commercial cytotoxicity assay (Fisher Scientific, catalog #PR-G1780).
Results
Morphine enhances HIV-1 infection establishment in microglia in a MOR-dependent manner
Microglia express opioid receptors, though the consequences of opioid exposure on HIV-1 infection establishment in microglia have not been mechanistically characterized, primarily due to limited access to primary human microglia. To overcome problems with cell availability, we chose to determine opioid effects on iPSC-derived microglia, which are phenotypically similar to CNS-resident microglia. (Pandya, Shen et al., 2017) Importantly, we have previously shown that iPSC-derived microglia are susceptible to HIV-1 infection in vitro (60). We used an iPSC-derived microglia differentiation protocol that utilized hematopoietic progenitors on two donor-derived cell lines, BU1 and BU3. Following cell differentiation protocol, > 99% of the CD45+ cells expressed microglial markers CD11b, TREM2, P2RY12, and TMEM119 (Figure 1A). Similar to CNS-resident microglia (Maduna, Audouard et al., 2019), iPSC-derived microglia expressed MOR mRNA (Figure 1B), and cell surface MOR expression was confirmed by confocal immunofluorescence microscopy (Figure 1C). MOR expression overlapped with plasma membrane staining and was also located at subcellular compartments proximal to the plasma membrane (Figure 1C). iPSC-derived microglia were treated with morphine prior to infection with CCR5-tropic replication-competent HIV-1 (Lai-YU2/env). Interestingly, morphine pretreatment enhanced HIV-1 replication in MOR+ iPSC-derived microglia. We observed ~3-fold enhancement in p24Gag secretion at 3 days post-infection (Figure 1D). Morphine-mediated enhancement of p24Gag secretion was suppressed upon naloxone pre-treatment, a MOR antagonist, suggesting that pro-viral effects of morphine required MOR activation in iPSC-derived microglia. The expression of opioid receptors on human macrophages has remained controversial. While some studies have documented MOR mRNA expression in peripheral blood mononuclear cells (PBMCs) and monocytes (Chuang, Killam et al., 1995), expression of MOR protein has not been observed in human macrophages. In agreement with some of the previously published studies (36, 37), monocyte-derived macrophages (MDMs) did not express MOR mRNA or protein (Figures 1E, F). In correlation with the lack of MOR expression, morphine pre-treatment did not significantly impact p24Gag production from MOR-deficient MDMs (Figure 1G). Therefore, morphine increases HIV-1 virion release in microglia but not from macrophages, suggesting that morphine enhancement of HIV-1 infection is dependent on cell-surface MOR expression.
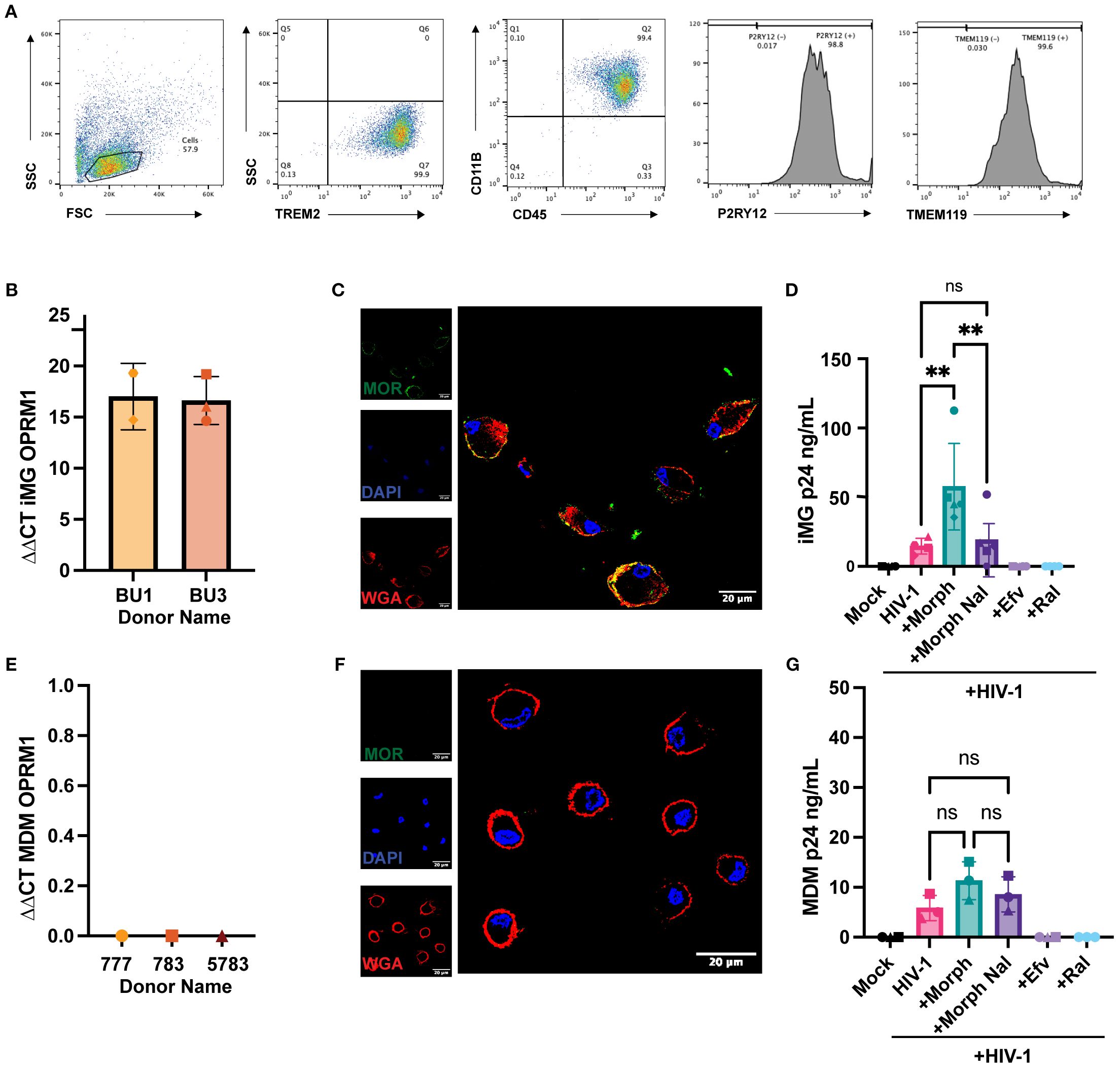
Figure 1. Morphine enhances HIV-1 infection in microglia. (A) Representative flow cytometry plots of TREM2, CD45, CD11B, P2RY12, and TMEM119 expression in iPSC-derived microglia. (B) OPRM1 mRNA expression was measured by RT-qPCR and normalized to GAPDH in iPSC-derived microglia (C) Representative immunofluorescence images of MOR surface expression: stained nucleus (DAPI, blue), plasma membrane (wheat germ agglutinin, red), and MOR (green) in iPSC-derived microglia. (D) iPSC-derived microglia were pretreated with DMSO (vehicle control) or naloxone (1µM) for 1 hr before treatment with morphine (1 µM) for 24hr. Cells were washed and infected with HIV-1(Lai/YU2-env, MOI 0.2). Supernatants were harvested 3 days post-infection for p24Gag measurements by ELISA. (E) OPRM1 mRNA expression was measured by RT-PCR and normalized to GAPDH in MDMs. (F) Representative immunofluorescence images of MOR surface expression: stained nucleus (DAPI, blue), plasma membrane (wheat germ agglutinin, red), and MOR (green) in MDMs. (G) MDMs were pretreated with DMSO (vehicle control) or naloxone (1 µM) for 1 hr before 1 µM morphine was treated for 24 hr. Cells were washed and infected with HIV-1 (Lai/YU2-env, MOI 1, with SIVmac Vpx VLPs (5ng p27)). Supernatants were harvested 3 days post-infection for p24Gag measurements by ELISA in MDMs. The means +/- SEM are shown, and each symbol represents an independent experiment for iPSC-derived microglia. P values: one-way ANOVA followed by the Tukey-Kramer posttest; **, p<0.01 (A-G).
Exogenous expression of MOR confers morphine-mediated enhancement of HIV-1 infection in macrophages
To confirm the requirement of MOR expression for morphine-dependent enhancement of HIV-1 infection, we retrovirally transduced THP-1 monocytes to constitutively express MOR and differentiated cells to macrophages by phorbol 12-myristate 13-acetate (PMA) treatment. While parental THP-1/PMA macrophages do not express MOR mRNA (Supplementary Figure 1A) or protein (Figure 2A), exogenous MOR expression by retroviral transduction in THP-1/PMA macrophages resulted in robust cell surface MOR expression (Figure 2B). We confirmed functional MOR expression in THP-1/PMA macrophages as morphine treatment induced phosphorylation of MOR, which was blocked by naloxone (Figures 2C, D). To determine the effects of morphine on HIV-1 infection, cells were pre-treated with increasing concentrations of morphine prior to virus exposure. While morphine did not affect p24Gag production in parental THP-1/PMA macrophages (Figure 2E), morphine pre-treatment of MOR-THP-1/PMA macrophages significantly enhanced p24Gag secretion in a dose-dependent manner, with the induction peaking between 0.1 and 1 µM morphine (Figure 2F). Hence, we used a dose of 1 µM morphine for all subsequent infection experiments. Morphine at 1 µM did not impact p24Gag secretion in THP-1/PMA macrophages (Figure 2G). Importantly, morphine-mediated induction of p24Gag secretion in MOR-THP-1/PMA macrophages was suppressed by pre-treatment with the MOR antagonist, naloxone (Figure 2H). These findings suggest that morphine enhances HIV-1 infection and virion release in a MOR-dependent manner.
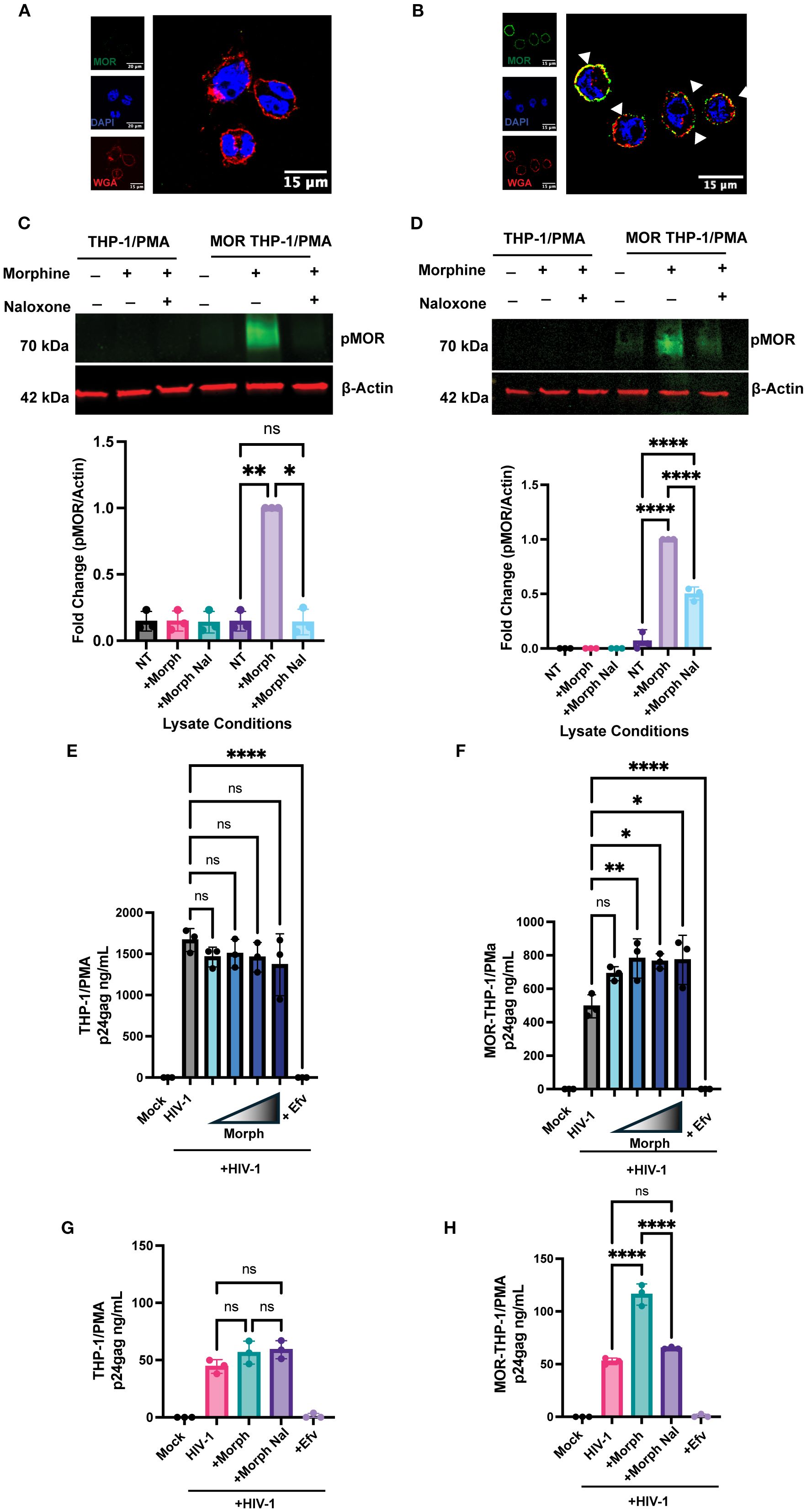
Figure 2. Exogenous MOR expression confers MOR-dependent enhancement of HIV-1 infection in macrophages. (A, B) Representative immunofluorescence images of MOR surface expression: stained nucleus (DAPI, blue), plasma membrane (wheat germ agglutinin, red), and flag-MOR (green) in (A) parental THP-1/PMA and (B) MOR-THP-1/PMA macrophages. Phospho-MOR (pMOR) expression in parental THP-1/PMA macrophages and MOR-THP-1/PMA macrophages with 7 min (C) and 24h (D) morphine treatment. pMOR band intensities were quantified and normalized to actin and to the no treatment (NT) group. THP1/PMA (E) or MOR-THP1/PMA (F) macrophages were pretreated with DMSO (vehicle control) or increasing concentrations (0.01, 0.1, 1, or 10 µM) of morphine for 24 hours. (G, H) Alternatively, cells were pretreated with naloxone (1 µM) prior to morphine (1 µM) treatment. Cells were washed and co-infected with HIV-1 (LaiΔEnvGFP/G, MOI 1, in (E, F) and MOI 0.3 in (G, H)) and SIVmac Vpx VLPs (5 ng p27). Supernatants were harvested 3 days post-infection for p24Gag measurements by ELISA. The means +/- standard error of the SEM are shown, and each symbol represents an independent experiment. p values: one-way ANOVA followed by the Tukey-Kramer post-test; *, p< 0.05; **, p< 0.01; ****, p< 0.0001 (A-H).
Morphine enhances HIV-1 reverse transcription in MOR-expressing cells
Since MOR activation enhanced HIV-1 replication in MOR-expressing cells, we sought to systematically determine the specific step of the HIV-1 life cycle that was modulated by morphine. First, we assessed the effects of MOR activation on early steps of reverse transcription in iPSC-derived microglia and MOR-expressing or MOR-deficient (parental) THP1/PMA macrophages. Morphine pretreatment enhanced early and late RT in iPSC-derived microglia, though only increase in late RT was statistically significant (Figures 3A, B). Further, morphine treatment of MOR/THP-1/PMA macrophages significantly enhanced both early and late RT which was blocked by naloxone pretreatment (Figures 3C, D). In contrast, morphine pretreatment did not significantly impact early (Figure 3E) or late RT steps (Figure 3F) in parental THP-1/PMA macrophages. These findings suggest that MOR activation by morphine enhances HIV-1 reverse transcription, which can be blocked by a MOR antagonist.
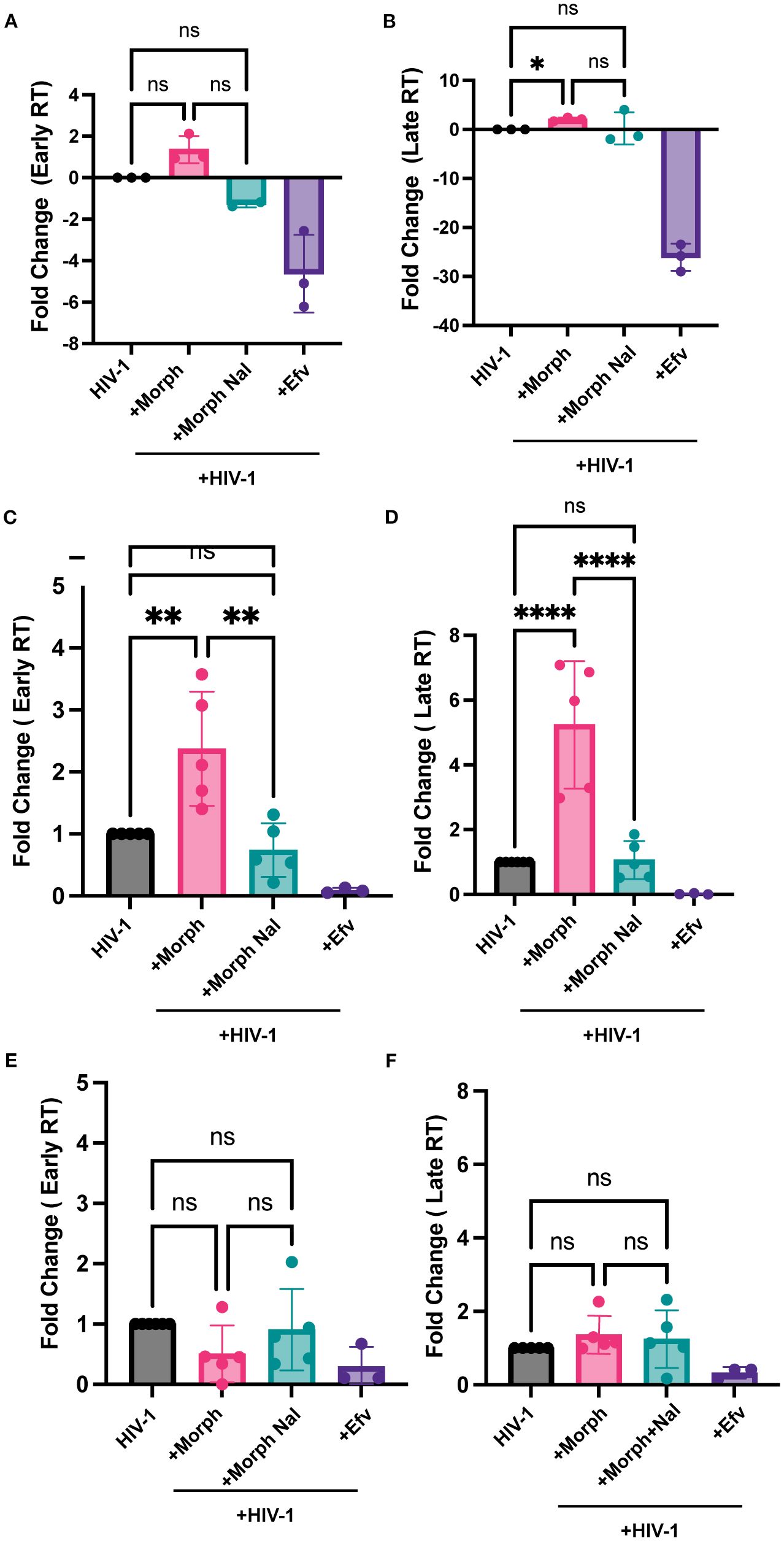
Figure 3. Morphine enhances HIV-1 reverse transcription in a MOR-dependent manner. iPSC-derived microglia (A, B), MOR-THP-1/PMA (C, D), or THP-1/PMA (E, F) macrophages were pretreated with DMSO (vehicle control) or naloxone (1µM) for 1 hr prior to morphine (1µM) treatment for 24 hr. Cells were washed and co-infected with HIV-1 (LaiΔEnvGFP/G, MOI0.3) and SIVmac Vpx VLPs (5 ng p27). THP/PMA macrophages were lysed at 8h and 24h post-infection for measurement of early (C, E) and late RT (D, F) products by qPCR. iPSC-derived microglia were lysed 3 days post-infection for measurement of early (A) and late RT (B). The means +/- SEM are shown, and each symbol represents an independent experiment. p values: one-way ANOVA followed by the Tukey-Kramer posttest; *, p< 0.05; **, p< 0.01; ****, p< 0.0001 (A-D).
Morphine enhances intact proviral establishment in MOR-expressing cells
Since MOR activation enhanced reverse transcription efficiency in MOR-expressing cells, we assessed the effects of MOR activation on the next step, namely, provirus establishment. We utilized Intact Proviral DNA Assay (IPDA) using digital droplet PCR (ddPCR) analysis (70, 72) to determine the number of proviruses in both MOR-expressing (iPSC-derived microglia and MOR- THP-1/PMA macrophages) and cells lacking MOR expression (parental THP-1/PMA macrophages) upon HIV-1 infection in the presence or absence of morphine and naloxone. Genomic DNA, harvested 3 days post-infection, served as a template for IPDA to quantify both intact and defective (lacking or mutated detection of env or Psi sequences) proviruses. Morphine pre-treatment significantly enhanced the numbers of intact proviruses (2-fold) in HIV-1-infected iPSC-derived microglia (Figure 4A) and MOR-THP1/PMA macrophages (Figure 4B), which was suppressed by treatment with naloxone. However, morphine treatment did not impact intact proviral establishment in parental THP-1/PMA macrophages (Figure 4C), suggesting that MOR expression and activation were required for morphine-induced HIV-1 reverse transcription and integration enhancements.
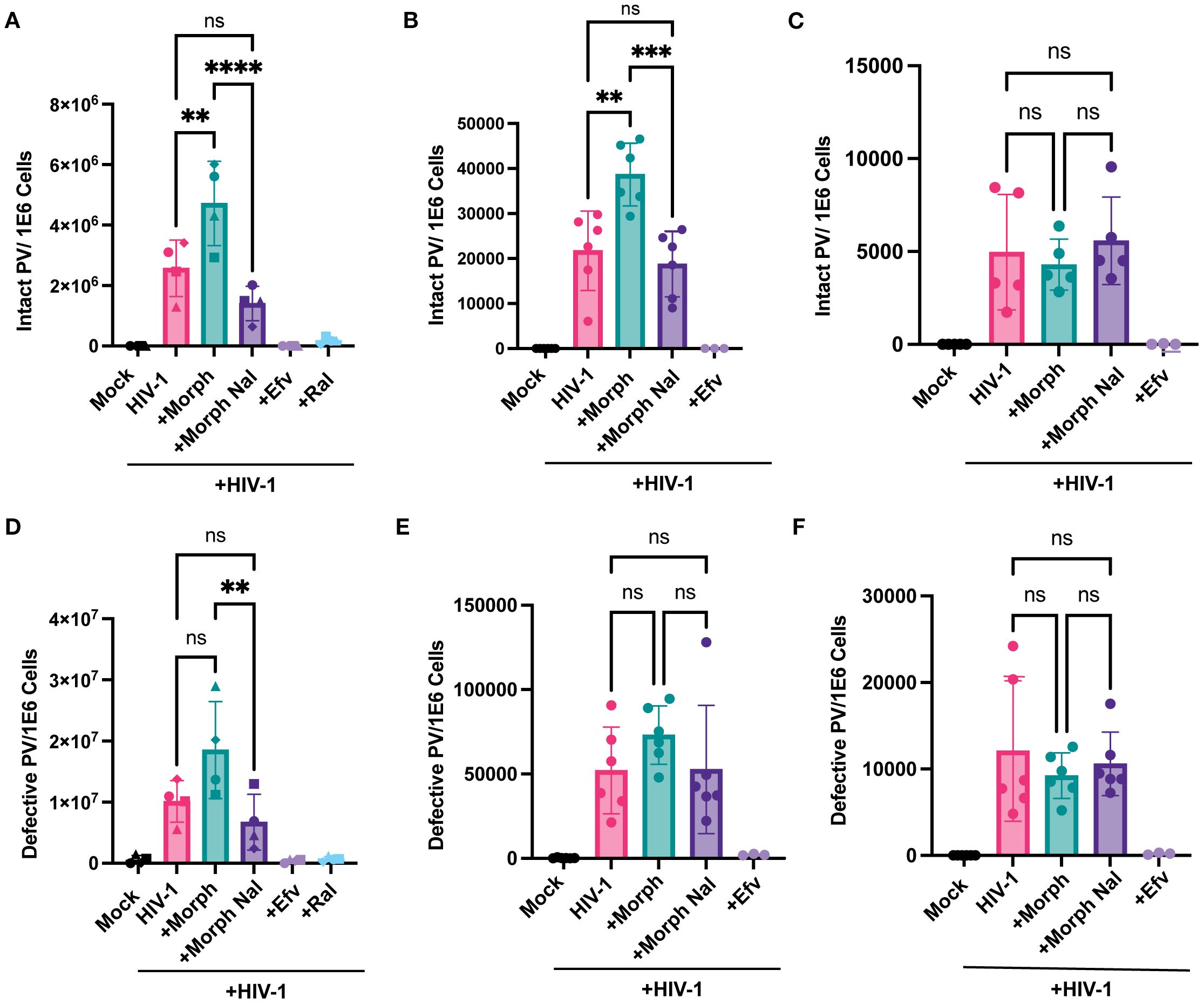
Figure 4. Morphine enhances intact but not defective provirus establishment in a MOR-dependent manner. iPSC-derived microglia and THP-1/PMA macrophages, pretreated with DMSO (vehicle control) or naloxone (1 μM) for 1 hr prior to morphine (1 µM) treatment for 24hr, were infected with HIV-1 (Lai-YU2-env, MOI0.2) for iPSC-derived microglia or LaiΔEnvGFP/G, MOI0.3, with SIVmac Vpx VLP (5 ng p27) for THP1/PMA macrophages. Cells were lysed for DNA extraction 3 days and 1 day post-infection for iPSC-derived microglia and THP-1/PMA macrophages, respectively, and used for measurement of intact and defective proviruses by IPDA. A parallel reaction to detect the host cell gene RPP30 was used as a correction for DNA shearing. Quantification of intact HIV-1 proviruses in (A) iPSC-derived microglia, (B) MOR-THP-1/PMA macrophages, and (C) THP-1/PMA macrophages. Quantification of defective HIV-1 proviruses in (D) iPSC-derived microglia, (E) MOR-THP-1/PMA, (F) THP-1/PMA macrophages. The means +/- SEM are shown, and each symbol represents an independent experiment for iPSC-derived microglia and THP-1/PMA. p values: one-way ANOVA followed by the Tukey-Kramer posttest; **, p< 0.01; ***, p< 0.001; ****, p< 0.0001 (A–F).
HIV-1 infection generates both intact and defective proviruses, with the defective proviruses encompassing by far the predominant fraction of the provirus load in vitro and in vivo (70, 72, 73). Interestingly, although statistically not significant, there was a trend towards enhanced defective proviral establishment in iPSC-derived microglia and MOR-THP-1/PMA upon morphine treatment, which was inhibited by naloxone (Figures 4D, E), while morphine had no impact on defective proviral establishment in THP-1/PMA macrophages (Figure 4F). In fact, the ratio of intact to defective proviruses was not affected by morphine treatment in iPSC-derived microglia and MOR THP-1/PMA macrophages (Supplementary Figures S2 B, F, J). These results indicate that morphine treatment did not significantly impact error rates of HIV-1 integration steps. Rather, these findings suggest that morphine exposure enhances the establishment of intact proviruses in MOR-expressing cells.
Morphine enhances abundance of HIV-1 intron-containing transcripts in MOR-expressing cells
Several studies have demonstrated the extensive diversity of the viral transcriptome in HIV-1 infected cells (74, 75), though only a subset of these viral transcripts encodes for viral proteins essential for the completion of the viral life cycle. Drugs-of-abuse, including opioids, can regulate HIV-1 gene expression via diverse mechanisms, including activation of transcription factors, NF-kB (76) and AP-1 (77), and alleviating RNA polymerase II pausing to promote polymerase activity (78), though the direct requirement for MOR activation and signaling has not been addressed. Hence, we next sought to determine the effects of acute morphine treatment on proviral transcription in MOR-expressing cells. We quantified multiply spliced, partially spliced, and full-length unspliced transcripts by using a previously described RT-ddPCR assay (79). RT-ddPCR simultaneously quantifies both spliced and unspliced LTR-containing transcripts by multiplexing probes for transcripts spanning the 5’ LTR, nef, env, or gag regions. While morphine treatment modestly enhanced levels of multiply spliced transcripts in iPSC-derived microglia (Figure 5A), there was a significant enhancement in unspliced RNA (which we term HIV-1 icRNA) expression by ~5.7 fold, which was blocked by naloxone treatment (Figure 5B). Furthermore, morphine-induced HIV-1 icRNA expression remained significantly elevated even after accounting for an increased number of proviruses in morphine-treated iPSC-derived microglia (Figure 5C), suggesting that acute morphine exposure in microglia selectively enhances HIV-1 icRNA expression. In addition to assessing the abundance of LTR-containing transcripts, analysis of viral transcript diversity in HIV-infected iPSC-derived microglia revealed that morphine selectively enhanced the ratio of full-length unspliced HIV-1 icRNA to that of the total viral transcriptome by ~3-fold (Figure 5D). In contrast to increases in longer transcripts such as full-length HIV-1 icRNA and env deficient 5’LTR+Nef+Gag transcript, morphine pretreatment decreased the abundance of shorter LTR-containing transcripts such as 5’LTR and 5’LTR+Gag in iPSC-derived microglia (Figure 5E). However, ratios of all LTR-containing transcripts remained similar across the no-treatment and morphine+naloxone conditions (Figure 5E). These results suggest that morphine enhances HIV-1 full-length unspliced icRNA abundance by not only modulating splicing efficiency but also alleviating pausing at promoter-proximal regions of viral LTR to favor transcriptional elongation and expression of full-length transcripts in microglia.
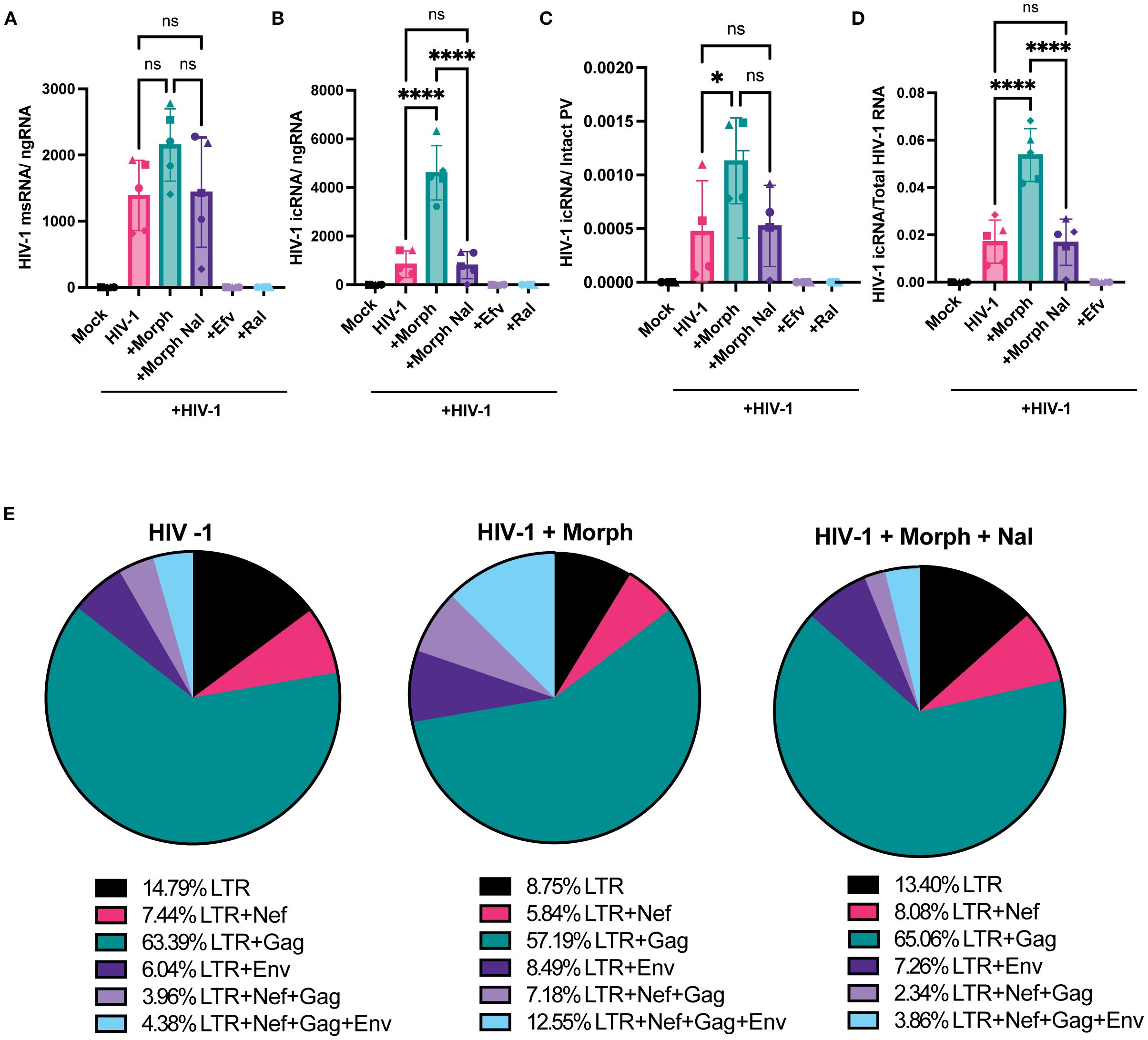
Figure 5. Morphine enhances HIV-1 full-length icRNA transcript abundance and alters transcriptomic diversity in microglia. iPSC-derived microglia were pretreated with DMSO (vehicle control) or naloxone (1 µM) for 1 hr prior to morphine (1 µM) treatment for 24 h, and then infected with HIV-1 (Lai-YU2-env, MOI 0.2). Cells were lysed for RNA extraction 3 days post-infection. (A) Quantification of msRNA (LTR+Nef), and (B) icRNA (LTR+Env+Nef+Gag, full-length transcripts) in iPSC-derived microglia were determined by RT-ddPCR and reported as viral RNA copies/ng of RNA input. (C) HIV-1 icRNA copy numbers were normalized to those of intact proviruses in iPSC-derived microglia. (D) Quantification of the ratio of full-length icRNA transcripts out of all transcripts detected in iPSC-derived microglia. (E) Pie charts displaying the mean percentages of LTR containing transcripts in iPSC-derived microglia infected in the absence (HIV-1) or presence of morphine (HIV-1+ Morph) or both morphine and naloxone (HIV-1+ Morph + Nal). The means +/- SEM are shown, and each symbol represents an independent experiment (iPSC-derived microglia and THP/PMA macrophages) or cells from an independent donor (MDMs). p values: one-way ANOVA followed by the Tukey-Kramer posttest; *, p< 0.05; ****, p< 0.0001 (A–D).
We next assessed if morphine modulated HIV-1 transcription in macrophages in the presence or absence of MOR expression. While morphine-mediated enhancement of multiply-spliced (Figure 6A) and unspliced viral transcripts (Figure 6B) was not observed in parental THP-1/PMA (MOR-deficient) macrophages, morphine significantly enhanced full-length icRNA expression in MOR-THP-1/PMA macrophages (2.5 fold, Figure 6D). Surprisingly, msRNA expression was not impacted by morphine treatment in MOR-THP-1/PMA macrophages (Figure 6C). Importantly, morphine-induced enhancement of icRNA expression in MOR-THP-1/PMA macrophages was suppressed by naloxone (Figure 6D), suggesting that MOR expression and activation is responsible for the enhanced HIV icRNA expression.
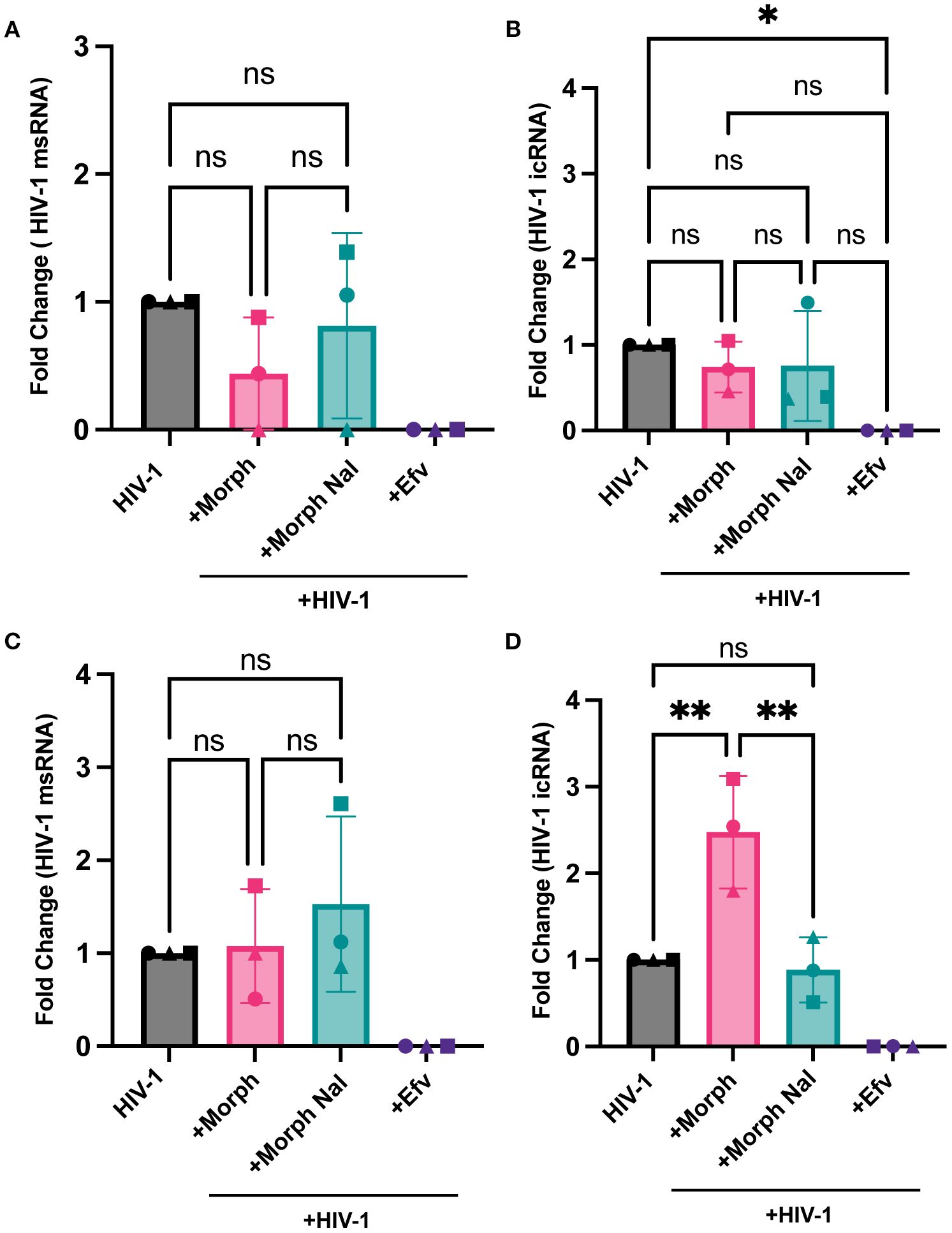
Figure 6. MOR overexpression promotes morphine-dependent enhancement of HIV-1 icRNA expression in macrophages. THP-1/PMA (A, B) or MOR-THP-1/PMA (C, D) macrophages were pretreated with DMSO (vehicle control) or naloxone (1µM) for 1 hr prior to morphine (1µM) treatment for 24hr. Cells were washed and infected with HIV-1 ((LaiΔEnvGFP/G, MOI0.3, with SIVmac Vpx VLP (5 ng p27)). Cells were lysed for RNA extraction 3 days post-infection. Levels of msRNA (A, C) and icRNA (B, D) in THP-1/PMA (A, B) or MOR-THP-1/PMA (C, D) macrophages were quantified by RT-qPCR. The means +/- SEM are shown, and each symbol represents an independent experiment. p values: one-way ANOVA followed by the Tukey-Kramer posttest; *, p< 0.05; **, p< 0.01 (A-D).
Morphine treatment enhances innate immune responses
Our previous studies have shown that de novo transcribed HIV-1 icRNA in both macrophages and microglia induce inflammatory responses, such as the interferon γ-inducible protein 10 (IP-10) (80). Hence, we sought to determine if morphine enhances IP-10 secretion in a MOR-dependent manner. THP-1/PMA macrophages were pretreated with increasing concentrations of morphine (0.01, 0.1, 1, or 10 µM) before infection with HIV-1, and IP-10 secretion was measured by ELISA on supernatants 3 days post-infection. While morphine modestly reduced IP-10 secretion in parental THP-1/PMA macrophages (Figure 7A), significant enhancement was observed in MOR-THP1/PMA macrophages in a dose-dependent manner, peaking at 1µM morphine (Figure 7B). Morphine treatment at 1µM modestly lowered IP-10 secretion in THP-1/PMA macrophages (Figure 7C). Moreover, the enhancement in IP-10 secretion (~3.4 fold) observed in morphine (1 µM) treated HIV-1 infected MOR-THP-1/PMA macrophages was blocked by naloxone (Figure 7D). These results suggest that MOR expression promotes morphine-dependent enhancement of IP-10 secretion during HIV-1 infection.
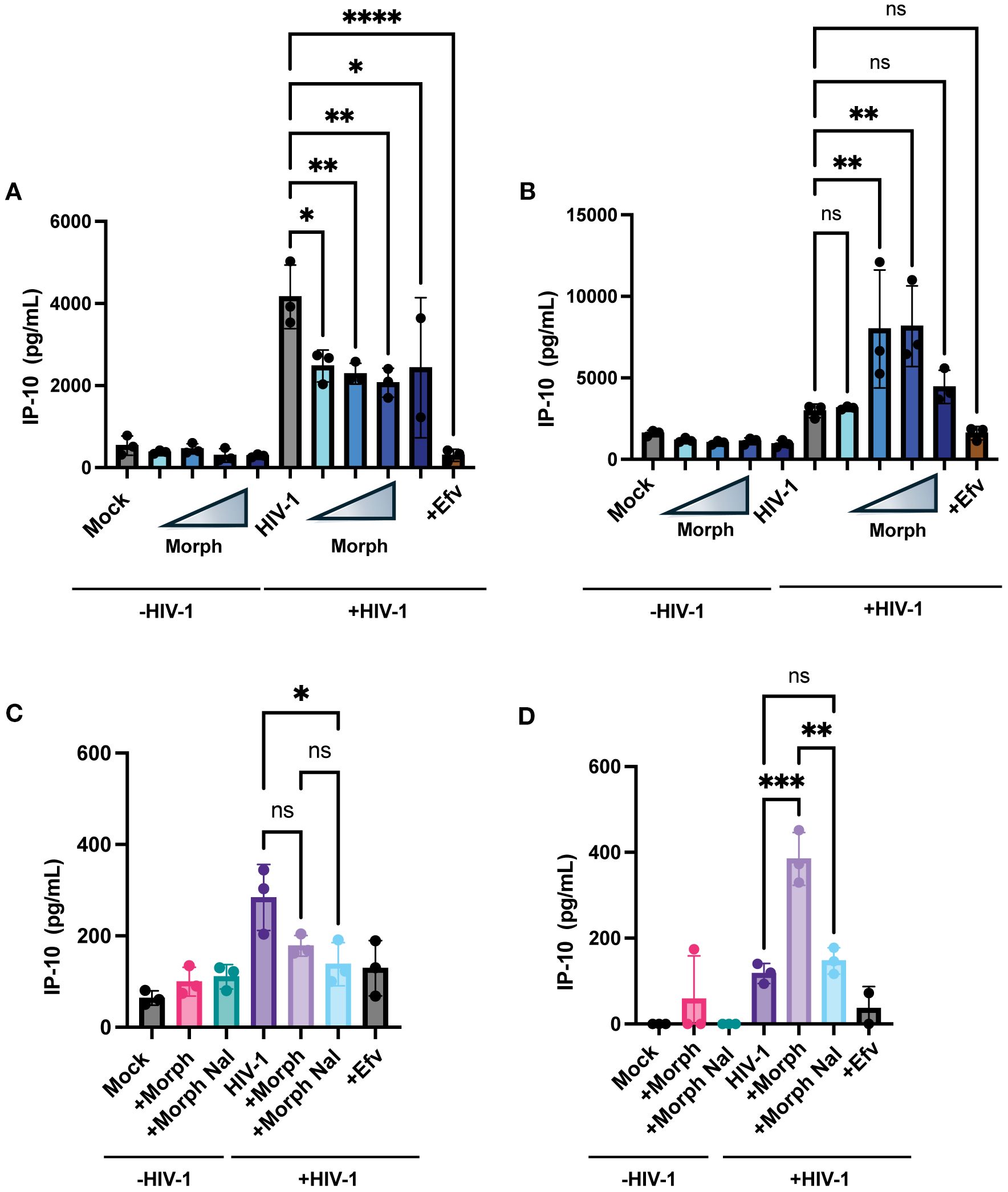
Figure 7. Exogenous MOR expression promotes morphine-dependent enhancement of HIV-1-induced IP-10 secretion in macrophages. THP-1/PMA (A) and MOR-THP-1/PMA (B) macrophages were pretreated with DMSO (vehicle control) or with increasing concentrations of morphine (0.01, 0.1, 1, and 10 µM) for 24hr. Cells were washed and co-infected with HIV-1 (LaiΔEnvGFP/G, MOI 1) and SIVmac Vpx VLPs (5 ng p27). IP-10 content in supernatants harvested 3 days post-infection was determined by an ELISA. Alternatively, cells were pretreated with naloxone (1 µM) prior to morphine (1 µM) treatment in THP-1/PMA (C) and MOR-THP-1/PMA macrophages (D). The means +/- SEM are shown, and each symbol represents an independent experiment. p values: one-way ANOVA followed by the Tukey-Kramer posttest; *, p< 0.05; **, p< 0.01; ***, p< 0.001; ****, p< 0.0001 (A–D).
HIV-1 infection and opioid signaling synergize at the PI3K/Akt pathway
Next, we sought to identify the pathway by which opioid signaling and HIV-1 infection synergized to enhance infection and inflammatory responses. MOR signaling has previously been shown to activate the PI3K/Akt pathway (81, 82). Additionally, HIV-1 infection has been shown to engage the PI3K/Akt pathway in macrophages (83) and T cells (84). Therefore, we sought to determine whether ligation of MOR by morphine induces PI3K/Akt activation during HIV-1 infection. Morphine had no significant impact on pAkt expression in HIV-infected THP-1/PMA macrophages (Figure 8A). In contrast, morphine significantly enhanced pAkt expression in HIV-infected MOR-THP-1/PMA macrophages, which was suppressed by both naloxone and the PI3K inhibitor, wortmannin (Figure 8B). Morphine modestly enhanced pAkt expression in iPSC-derived microglia which was reversed by naloxone and wortmannin (Figure 8C). We next investigated if inhibition of the PI3K/Akt pathway would abrogate morphine-induced HIV-1 infection enhancement. THP-1/PMA macrophages and iPSC-derived microglia were pretreated with wortmannin in the presence or absence of morphine for 24 hours before initiating HIV-1 infection. Importantly, wortmannin treatment did not significantly impact cell viability in iPSC-derived microglia (Supplementary Figure S3A), THP-1/PMA (Supplementary Figure S3B), and MOR-THP-1/PMA macrophages (Supplementary Figure S3C). Though impact of wortmannin on p24Gag secretion in the absence of morphine was minimal and not significant in all cell types (Figures 8D-F), in the presence of morphine, wortmannin treatment significantly reduced p24Gag production in MOR-THP-1/PMA macrophages (Figure 8E) and iPSC-derived microglia (Figure 8F), suggesting that enhanced p24Gag production by morphine in MOR expressing cells is mediated by the PI3K/Akt pathway.
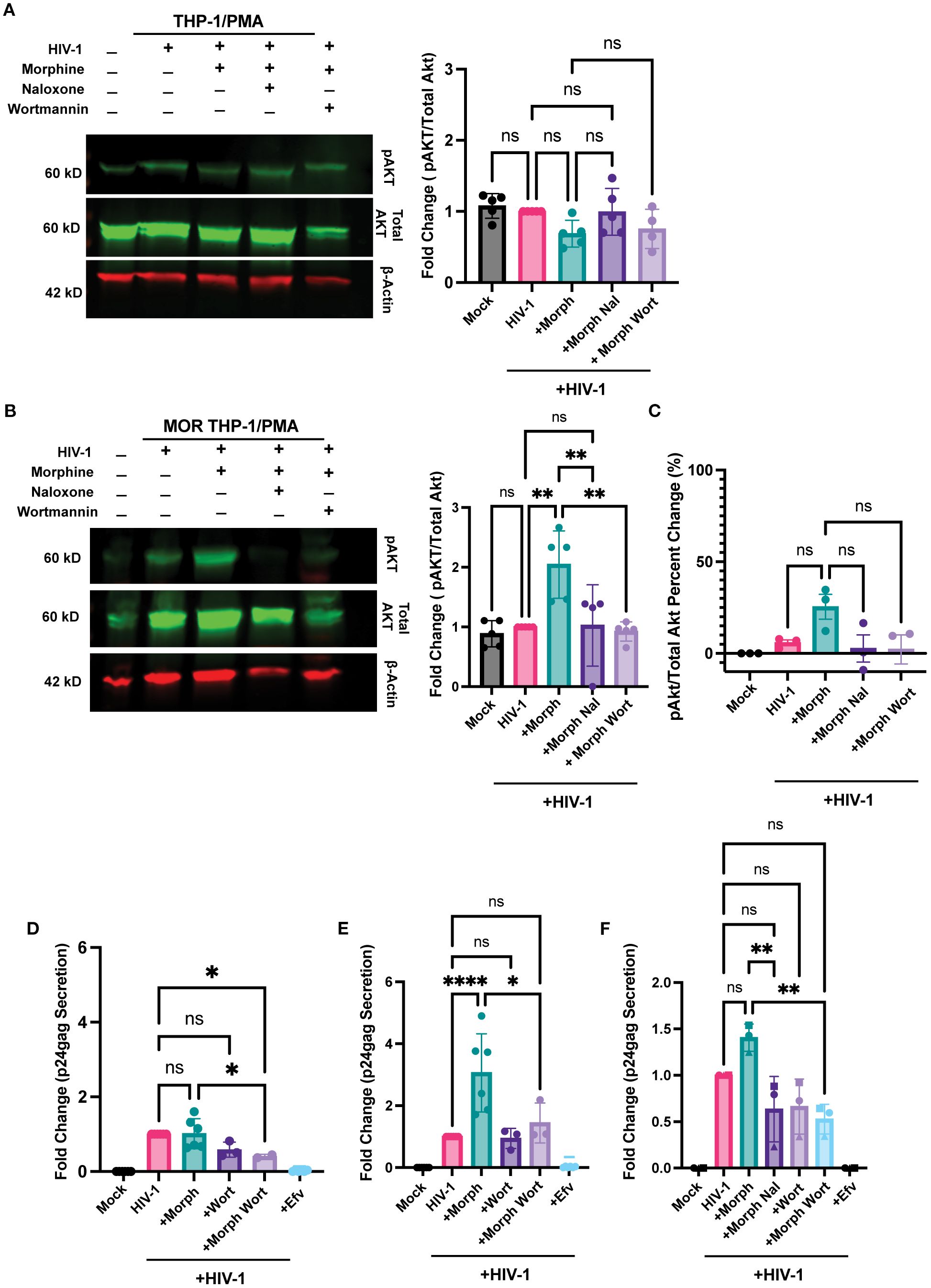
Figure 8. PI3K inhibition abrogates morphine-mediated enhancement of HIV-1 infection. (A, B) Western blot analysis of phosphorylated Akt (pAkt), total Akt, and β-actin on lysates from THP-1/PMA (A) or MOR-THP-1/PMA (B) macrophages. pAkt quantification on lysates from iPSC derived microglia by ELISA (C). THP-1/PMA macrophages (D), MOR-THP-1/PMA macrophages (E) and iPSC-derived microglia (F) were pretreated with combinations of morphine (1 µM) naloxone (1 µM) or wortmannin (0.1 µM) before infection with HIV-1. THP-1/PMA macrophages were co-infected with LaiΔEnvGFP/G (MOI 1) and SIVmac Vpx VLPs (5 ng p27), while iPSC-derived microglia were infected with Lai/YU2-env (MOI 0.2). The p24Gag content in cell supernatants, harvested 3 days post-infection, was determined by an ELISA. The means +/- SEM are shown, and each symbol represents an independent experiment. p values: one-way ANOVA followed by the Tukey-Kramer posttest; *, p< 0.05; **, p< 0.01; ****, p< 0.0001 (A–E).
Finally, we investigated whether PI3K/Akt inhibition affects morphine and virus-induced inflammatory responses. Wortmannin pre-treatment significantly inhibited morphine-mediated enhancement of IP-10 secretion by ~7.7-fold in HIV-infected MOR-THP-1/PMA macrophages (Figure 9B) and ~2.8-fold in iPSC-derived microglia (Figure 9C) but had a negligible impact on IP-10 secretion in HIV-1-infected THP-1/PMA macrophages (Figure 9A). Additionally, wortmannin pre-treatment attenuated IL-8 and MCP-1 secretion (Figures 9D, E) in morphine-treated HIV-1-infected iPSC-derived microglia. Collectively, these findings suggest that opioids promote the induction of inflammatory responses in HIV-infected microglia via MOR activation and that PI3K inhibitors can suppress the synergistic enhancement of inflammatory responses by morphine-induced activation of MOR signaling pathways and HIV-1 infection.
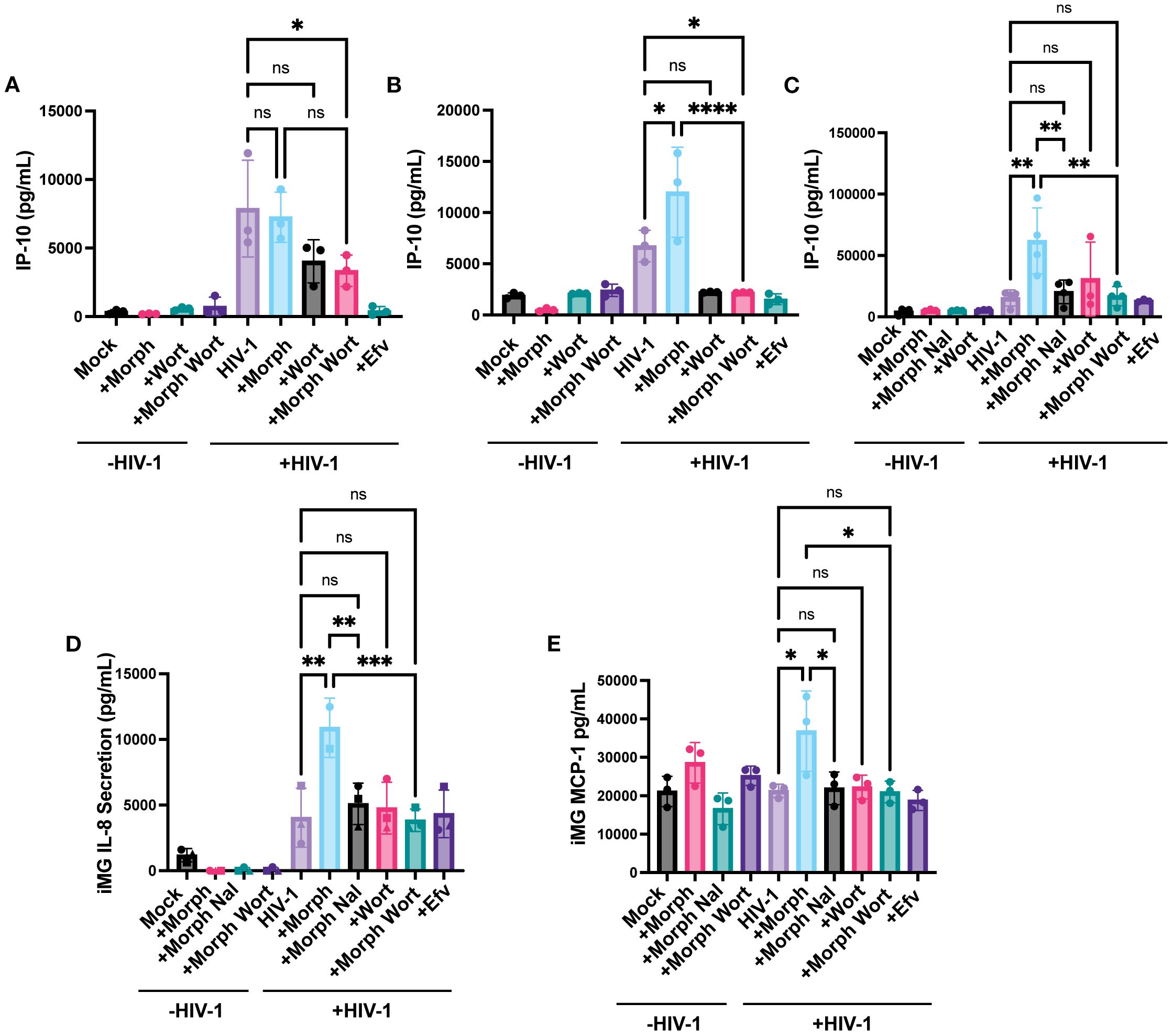
Figure 9. PI3K Inhibition suppresses HIV and morphine-induced inflammatory responses. THP-1/PMA (A), MOR-THP-1/PMA (B) macrophages and iPSC-derived microglia (C-E), were pretreated with DMSO (vehicle control) or naloxone (1 µM) or wortmannin (0.1 µM) for 1 hr prior to morphine (1 µM) treatment for 24hr, and infected with either LaiΔEnvGFP/G (MOI 1), in the presence of SIVmac Vpx VLPs (5 ng p27) or Lai-YU2-env (MOI 1), respectively. Supernatants were harvested 3 days post-infection for quantification of IP-10 secretion in (A) THP-1/PMA, (B) MOR-THP-1/PMA macrophages, and (C) iPSC-derived microglia, or (D) IL-8 secretion and (E) MCP-1 secretion in iPSC-derived microglia by ELISA. The means +/- SEM are shown, and each symbol represents an independent experiment. p values: one-way ANOVA followed by the Tukey-Kramer posttest; *, p< 0.05; **, p< 0.01; ***, p< 0.001; ****, p< 0.0001 (A–E).
Discussion
In this study, we utilized multiple cell models to demonstrate that morphine enhances HIV-1 infection and promotes HIV-1 infection-induced inflammatory responses in MOR-expressing cells. We report cell surface expression of MOR in iPSC-derived microglia, similar to that observed in CNS-resident microglia (35, 85), which correlated with the ability of morphine to enhance HIV-1 infection in MOR+ iPSC-derived microglia but not in MOR-deficient macrophages. Exogenous expression of MOR in THP-1/PMA macrophages promoted morphine-mediated enhancement of HIV-1 infection. Importantly, morphine-dependent enhancement of HIV-1 infection was attenuated upon pre-treatment with a MOR antagonist, naloxone. Taken together, these data suggest that morphine may trigger MOR-dependent signaling in modulating HIV-1 infection in microglia. Previous studies have shown that morphine can modulate CXCR4 and CCR5 expression in myeloid cells (86, 87) and lymphocytes (88). Similarly, endogenous ligands such as endomorphins and endorphins that activate MOR may have similar effects of enhancing HIV-1 infection in microglia in vivo (89, 90). While morphine may enhance CCR5 expression in iPSC-derived microglia and promote HIV-1 entry, proviral effects of morphine were also observed at post-entry steps of revere transcription and integration since morphine also induced VSV G-pseudotyped HIV-1 infection in MOR THP-1/PMA macrophages (Figures 2, 3). As opposed to MOR-THP1/PMA macrophages, there was a trend but not statistically significant enhancement of early RT in iPSC-microglia by morphine (Figure 3A), suggesting that morphine might also modulate post RT steps to enhance HIV-1 replication in iPSC-microglia. In contrast, morphine significantly enhanced reverse transcription in MOR THP-1/PMA macrophages (5-fold increase in late RT products, Figure 3), thus contributing to the observed increases in the numbers of intact proviruses (Figure 4) in these cells. Previous studies have reported conflicting findings on the immuno-modulatory effects of opioids and their ability to impact HIV-1 infection in monocytes and macrophages (87, 91, 92). Though MOR mRNA has been detected at low levels in human MDMs (38, 93), none of the studies have definitively shown functional cell surface MOR expression in macrophages (37, 38, 94–96). Further, our findings show that MDMs do not express MOR at detectable levels (Figure 1). It remains possible that the previously reported immune-modulatory effects of morphine in macrophages might be attributed to TLR4 or non-classical opioid receptor signaling upon morphine exposure (97).
Clinically, people who have frequent ART interruptions have higher HIV-1 proviral DNA reservoirs than people with fewer ART interruptions (98). People with HIV-1 who inject drugs have higher incidences of ART interruption (99) and might have a higher risk of elevated HIV proviral DNA levels. Studies in PWH who inject heroin, which is metabolized into morphine, reported minor increases in intact HIV proviral DNA in PBMCs (98). Additionally, another study has shown that morphine increases the size of SIV reservoirs in brain resident CD11b+ macrophages in rhesus macaques (100), though the effects of opioids on viral reservoirs in the CNS have not been well-characterized in PWH. Previous studies have suggested that opioids can suppress type I IFN responses and expression of antiviral restriction factors (38), such as SAMHD1 and APOBEC3. Suppression of these anti-viral responses might enhance the efficiency of HIV-1 reverse transcription and provirus establishment in MOR-expressing microglia. It should be noted that the ratio of defective to intact HIV-1 proviruses was considerably lower in iPSC-derived microglia, compared to that observed in human viremic brain tissues (>90% of HIV-1 genomes are defective in vivo). In vivo findings likely reflect chronic HIV-1 infection for several months to years in the presence of ART, whereas our data reflect only acute infection conditions in the absence of ART.
Morphine alters HIV-1 transcriptomic landscape in a MOR dependent manner
Morphine exposure led to the increased expression of LTR-containing HIV-1 transcripts, though interestingly, enhancement of unspliced icRNA expression was greater than that observed with multiply-spliced transcripts in microglia and MOR-THP1/PMA macrophages. HIV-1 LTR contains numerous binding sites for transcription factors such as NF-kB, NFAT, AP-1, and CREB (101). Previous studies have suggested that opioids enhance gene expression by activating transcription factors such as NF-κB (102–105) and CREB (103), which may enhance HIV-1 transcription rates in infected microglia, though such a mechanism would not account for the selective enhancement of HIV-1 icRNA expression in morphine-treated microglia. Additionally, since morphine enhanced the ratio of unspliced full-length icRNA and 5’LTR+Nef+Gag transcripts compared to all other LTR-containing transcripts in microglia, morphine might have an additional impact on post-transcriptional mechanisms that control HIV-1 transcript diversity.
Complex alternative splicing of HIV-1 RNA in the nucleus results in the over 40 differentially spliced viral transcripts, some of which encode for the viral accessory proteins and env (106). Spliced RNAs are exported through the NXF1-dependent nuclear export pathway (107). To facilitate nuclear export of unspliced full-length viral RNA, which is utilized for translation of Gag and Gag-Pol polyproteins or used as viral genomic RNA packaged into assembling virions, HIV-1 employs the Rev-CRM1 pathway to suppress RNA splicing and promote nuclear export (106). Morphine may potentially impact either one or multiple of these post-transcriptional RNA processing steps to skew the viral transcript population towards expression of immunostimulatory unspliced icRNA transcripts. For instance, morphine has been shown to modulate the abundance of m6A epitranscriptomic modifications (108, 109) by downregulating the expression of RNA demethylases or m6A erasers, FTO and Alkbh5 (109). Some studies have shown that m6A methylation at 3’ splice sites of various cellular genes inhibits RNA splicing (110). RNA methylation at the major splice donor site in HIV-1 5’UTR has been suggested to contribute to reduced splicing efficiency and increased nuclear abundance of HIV-1 unspliced RNA (111–114). Thus, the ability of morphine to modulate both transcriptional and post-transcriptional regulatory mechanisms might contribute to the selective enhancement of full-length unspliced HIV-1 icRNA expression in microglia.
Morphine treatment enhances innate immune responses during HIV-1 infection
Previous studies by us and others have demonstrated that sensing of HIV-1 icRNA by MDA5 triggers MAVS-dependent innate immune responses and inflammatory cytokine secretion in macrophages and microglia (80, 115–117). Despite ART, HIV-1 RNA has been detected in CNS and CSF of PWH (118–122). Innate immune sensing of persistent HIV-1 icRNA expression and morphine-induced MOR signaling in microglia might contribute to the synergistic increases in inflammatory responses. Our results (Figure 7) and previously published studies (123, 124) suggest that opioid signaling through MOR activates the PI3K/Akt pathway. Interestingly, PI3K inhibitor wortmannin suppressed morphine-induced enhancement of p24Gag secretion (Figure 8) and IP-10 expression (Figure 9) in HIV-infected microglia and MOR-THP-1/PMA macrophages. We hypothesize that the putative synergy between morphine – MOR signaling and HIV icRNA-induced proinflammatory responses might define the molecular basis for HIV and opiate co-exposure-induced neuroinflammation and neurotoxicity. Importantly, opioid antagonists or PI3K/Akt pathway inhibitors might be novel therapeutic modalities to suppress chronic neuroinflammation in PWH using injection drugs (22, 125–128).
Limitations of the study
The experimental model under study in this report addresses consequences of morphine pre-treatment on HIV-1 infection establishment and effects in HIV-1 infection-induced innate immune responses, and most likely represents the scenario of individuals with opioid use disorder who later acquire HIV. This study does not address the consequences of morphine exposure post-infection establishment that might be reflective of PWH who use opioids. Such experimental conditions can be investigated in the future by adding morphine during and after infection establishment in microglia. Finally, there are some discrepancies between the cell models where morphine significantly enhances reverse transcription in THP-1/PMA macrophages, but only trends to enhance reverse transcription in iPSC-derived microglia in a MOR-dependent manner. This discrepancy reflects that morphine may have other reverse transcription-independent effects on HIV-1 infection enhancement in iPSC-derived microglia that differ from the MOR THP-1 cell line.
Conclusions
This study demonstrated for the first time that morphine treatment in human microglia enhances HIV-1 infection. Morphine enhancement of HIV-1 infection is specific to CNS-derived microglia and MOR-expressing cells. Morphine had an additional selective impact on enhanced HIV-1 icRNA expression, sensing of which leads to the induction of inflammatory responses. These findings highlight how opioid use contributes to elevated neuroinflammation and risk for neurodegenerative disorders in people with HIV-1. Therapeutics targeting the PI3K/Akt pathway may reduce neurocognitive disorders in PWH.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Boston University Medical Center IRB. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin because leukopaks from commercial blood center was obtained for isolation of peripheral blood mononuclear cells. These were from anonymous donors, and no information was given to the investigators.
Author contributions
CS: Writing – original draft, Formal analysis, Methodology, Investigation, Writing – review & editing. GS: Methodology, Investigation, Writing – review & editing. AG: Investigation, Writing – review & editing, Methodology. XH: Writing – review & editing, Data curation, Investigation, Methodology. DB: Data curation, Methodology, Writing – review & editing. MY: Methodology, Writing – review & editing. SB: Methodology, Writing – review & editing, Data curation. AQ: Writing – review & editing, Methodology. HA: Methodology, Writing – review & editing. GM: Funding acquisition, Writing – review & editing, Supervision, Resources. AH: Supervision, Writing – review & editing, Resources, Funding acquisition. SG: Supervision, Funding acquisition, Resources, Conceptualization, Writing – review & editing, Project administration, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by NIH grants R01DA059952 (SG and AH), R01AI187175 (SG and AH), R01DA055488 (SG and AH), R01DA051889 (SG and GM), and 5P30AI042853 (SG, AH, and HA).
Acknowledgments
We thank the BUMC Flow Cytometry Core and the Cellular Imaging Core for technical assistance. We thank the NIH/NIAID HIV Reagent Program and associated investigators for reagents used in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1628872/full#supplementary-material
References
1. Van Schalkwyk C, Mahy M, Johnson LF, and Imai-Eaton JW. Updated data and methods for the 2023 UNAIDS HIV estimates. JAIDS J Acquired Immune Deficiency Syndromes. (2024) 95:e1–4. doi: 10.1097/QAI.0000000000003344
2. Wang SC and Maher B. Substance use disorder, intravenous injection, and HIV infection: A review. Cell Transpl. (2019) 28:1465–71. doi: 10.1177/0963689719878380
3. Sarker MS. People who inject drugs: testing and treatment of HIV and HCV. Lancet Glob Health. (2023) 11:e1832–e3. doi: 10.1016/S2214-109X(23)00487-4
4. Bluthenthal RN, Wenger L, Chu D, Bourgois P, and Kral AH. Drug use generations and patterns of injection drug use: Birth cohort differences among people who inject drugs in Los Angeles and San Francisco, California. Drug Alcohol Dependence. (2017) 175:210–8. doi: 10.1016/j.drugalcdep.2017.04.001
5. Degenhardt L, Webb P, Colledge-Frisby S, Ireland J, Wheeler A, Ottaviano S, et al. Epidemiology of injecting drug use, prevalence of injecting-related harm, and exposure to behavioural and environmental risks among people who inject drugs: a systematic review. Lancet Global Health. (2023) 11:e659–e72. doi: 10.1016/S2214-109X(23)00057-8
6. Cicero TJ, Ellis MS, and Kasper ZA. Increased use of heroin as an initiating opioid of abuse. Addictive Behaviors. (2017) 74:63–6. doi: 10.1016/j.addbeh.2017.05.030
7. Des Jarlais DC, Kerr T, Carrieri P, Feelemyer J, and Arasteh K. HIV infection among persons who inject drugs. AIDS. (2016) 30:815–26. doi: 10.1097/QAD.0000000000001039
8. West BS, Diaz JE, Philbin MM, and Mauro PM. Past-year medical and non-medical opioid use by HIV status in a nationally representative US sample: Implications for HIV and substance use service integration. J Subst Use Addict Treat. (2023) 147:208976. doi: 10.1016/j.josat.2023.208976
9. Ellis RJ, Marquine MJ, Kaul M, Fields JA, and Schlachetzki JCM. Mechanisms underlying HIV-associated cognitive impairment and emerging therapies for its management. Nat Rev Neurol. (2023) 19:668–87. doi: 10.1038/s41582-023-00879-y
10. Altice FL, Kamarulzaman A, Soriano VV, Schechter M, and Friedland GH. Treatment of medical, psychiatric, and substance-use comorbidities in people infected with HIV who use drugs. Lancet. (2010) 376:367–87. doi: 10.1016/S0140-6736(10)60829-X
11. Storholm ED, Silverberg MJ, and Satre DD. Racial and ethnic differences in substance use diagnoses, comorbid psychiatric disorders, and treatment initiation among HIV-positive and HIV-negative women in an integrated health plan. J Psychoactive Drugs. (2016) 48:377–83. doi: 10.1080/02791072.2016.1242180
12. Cernasev A, Veve MP, Cory TJ, Summers NA, Miller M, Kodidela S, et al. Opioid use disorders in people living with HIV/AIDS: A review of implications for patient outcomes, drug interactions, and neurocognitive disorders. Pharmacy. (2020) 8:168. doi: 10.3390/pharmacy8030168
13. Byrd DA, Fellows RP, Morgello S, Franklin D, Heaton RK, Deutsch R, et al. Neurocognitive impact of substance use in HIV infection. J Acquir Immune Defic Syndr. (2011) 58:154–62. doi: 10.1097/QAI.0b013e318229ba41
14. Meyer VJ, Rubin LH, Martin E, Weber KM, Cohen MH, Golub ET, et al. HIV and recent illicit drug use interact to affect verbal memory in women. J Acquir Immune Defic Syndr. (2013) 63:67–76. doi: 10.1097/QAI.0b013e318289565c
15. Murphy A, Barbaro J, Martinez-Aguado P, Chilunda V, Jaureguiberry-Bravo M, and Berman JW. The effects of opioids on HIV neuropathogenesis. Front Immunol. (2019) 10:2445. doi: 10.3389/fimmu.2019.02445
16. Wallet C, De Rovere M, Van Assche J, Daouad F, De Wit S, Gautier V, et al. Microglial cells: the main HIV-1 reservoir in the brain. Front Cell Infect Microbiol. (2019) 9:362. doi: 10.3389/fcimb.2019.00362
17. Wahl A and Al-Harthi L. HIV infection of non-classical cells in the brain. Retrovirology. (2023) 20:1. doi: 10.1186/s12977-023-00616-9
18. Tang Y, Chaillon A, Gianella S, Wong LM, Li D, Simermeyer TL, et al. Brain microglia serve as a persistent HIV reservoir despite durable antiretroviral therapy. J Clin Invest. (2023) 133. doi: 10.1172/JCI167417
19. Alliot F, Godin I, and Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Dev Brain Res. (1999) 117:145–52. doi: 10.1016/S0165-3806(99)00113-3
20. Portilla I, Reus S, Leon R, van-der Hofstadt C, Sanchez J, Lopez N, et al. Neurocognitive impairment in well-controlled HIV-infected patients: A cross-sectional study. AIDS Res Hum Retroviruses. (2019) 35:634–41. doi: 10.1089/aid.2018.0279
21. Burlacu R, Umlauf A, Marcotte TD, Soontornniyomkij B, Diaconu CC, Bulacu-Talnariu A, et al. Plasma CXCL10 correlates with HAND in HIV-infected women. J Neurovirol. (2020) 26:23–31. doi: 10.1007/s13365-019-00785-4
22. Jalloh S, Hughes IK, Akiyama H, Gojanovich A, Quiñones A, Yang M, et al. Expression of intron-containing HIV-1 RNA induces NLRP1 inflammasome activation in myeloid cells. (2024). doi: 10.1101/2024.12.23.630103
23. Levine AJ, Miller JA, Shapshak P, Gelman B, Singer EJ, Hinkin CH, et al. Systems analysis of human brain gene expression: mechanisms for HIV-associated neurocognitive impairment and common pathways with Alzheimer’s disease. BMC Med Genomics. (2013) 6:4. doi: 10.1186/1755-8794-6-4
24. Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, et al. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology. (2014) 11:35. doi: 10.1186/1742-4690-11-35
25. Rani V, Verma R, Kumar K, and Chawla R. Role of pro-inflammatory cytokines in Alzheimer’s disease and neuroprotective effects of pegylated self-assembled nanoscaffolds. Curr Res Pharmacol Drug Discov. (2023) 4:100149. doi: 10.1016/j.crphar.2022.100149
26. Rho MB, Wesselingh S, Glass JD, McArthur JC, Choi S, Griffin J, et al. A potential role for interferon-α in the pathogenesis of HIV-associated dementia. Brain Behavior Immunity. (1995) 9:366–77. doi: 10.1006/brbi.1995.1034
27. Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister H-W, and Fontana A. Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-γ inducible protein 10. J Neuroimmunol. (1999) 93:172–81. doi: 10.1016/S0165-5728(98)00223-9
28. Sathekge M, Maes A, Van De Wiele C, and Dadachova E. Effect of AIDS on women who have sex-determined health issues. Semin Nucl Med. (2014) 44:489–98. doi: 10.1053/j.semnuclmed.2014.06.001
29. Bandera A, Taramasso L, Bozzi G, Muscatello A, Robinson JA, Burdo TH, et al. HIV-associated neurocognitive impairment in the modern ART era: are we close to discovering reliable biomarkers in the setting of virological suppression? Front Aging Neurosci. (2019) 11:187. doi: 10.3389/fnagi.2019.00187
30. Chen X, Wei J, Zhang Y, Zhang Y, and Zhang T. Crosstalk between gut microbiome and neuroinflammation in pathogenesis of HIV-associated neurocognitive disorder. J Neurological Sci. (2024) 457:122889. doi: 10.1016/j.jns.2024.122889
31. Hauser KF, Ohene-Nyako M, and Knapp PE. Accelerated brain aging with opioid misuse and HIV: New insights on the role of glially derived pro-inflammation mediators and neuronal chloride homeostasis. Curr Opin Neurobiol. (2023) 78:102653. doi: 10.1016/j.conb.2022.102653
32. Salter ML, Lau B, Mehta SH, Go VF, Leng S, and Kirk GD. Correlates of elevated interleukin-6 and C-reactive protein in persons with or at high risk for HCV and HIV infections. J Acquir Immune Defic Syndr. (2013) 64:488–95. doi: 10.1097/QAI.0b013e3182a7ee2e
33. Dang CM, Nelson CM, Feaster DJ, Kizhner A, Forrest DW, Nakamura N, et al. Opioids exacerbate inflammation in people with well-controlled HIV. Front Immunol. (2023) 14:1277491. doi: 10.3389/fimmu.2023.1277491
34. Hileman CO, Durieux JC, Janus SE, Bowman E, Kettelhut A, Nguyen TT, et al. Heroin use is associated with vascular inflammation in human immunodeficiency virus. Clin Infect Dis. (2023) 76:375–81. doi: 10.1093/cid/ciac812
35. Maduna T, Audouard E, Dembele D, Mouzaoui N, Reiss D, Massotte D, et al. Microglia express mu opioid receptor: insights from transcriptomics and fluorescent reporter mice. Front Psychiatry. (2018) 9:726. doi: 10.3389/fpsyt.2018.00726
36. Gavériaux C, Peluso J, Simonin F, Laforet J, and Kieffer B. Identification of κ- and δ-opioid receptor transcripts in immune cells. FEBS Letters. (1995) 369:272–6.
37. Williams JP, Thompson JP, McDonald J, Barnes TA, Cote T, Rowbotham DJ, et al. Human peripheral blood mononuclear cells express nociceptin/orphanin FQ, but not μ, δ, or κ Opioid receptors. Anesth Analgesia. (2007) 105:998–1005. doi: 10.1213/01.ane.0000278865.11991.9d
38. Karagiannis TT, Cleary JP Jr., Gok B, Henderson AJ, Martin NG, Yajima M, et al. Single cell transcriptomics reveals opioid usage evokes widespread suppression of antiviral gene program. Nat Commun. (2020) 11:2611. doi: 10.1038/s41467-020-16159-y
39. Jordan B. Opioids and their complicated receptor complexes. Neuropsychopharmacology. (2000) 23:S5–S18. doi: 10.1016/S0893-133X(00)00143-3
40. Lamberts J and Traynor J. Opioid receptor interacting proteins and the control of opioid signaling. Curr Pharm Design. (2014) 19:7333–47. doi: 10.2174/138161281942140105160625
41. Bruchas MR, Macey TA, Lowe JD, and Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. (2006) 281:18081–9. doi: 10.1074/jbc.M513640200
42. Macey TA, Lowe JD, and Chavkin C. Mu opioid receptor activation of ERK1/2 is GRK3 and arrestin dependent in striatal neurons. J Biol Chem. (2006) 281:34515–24. doi: 10.1074/jbc.M604278200
43. Melief EJ, Miyatake M, Bruchas MR, and Chavkin C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci. (2010) 107:11608–13. doi: 10.1073/pnas.1000751107
44. Olianas MC, Dedoni S, and Onali P. Regulation of PI3K/Akt signaling by N-desmethylclozapine through activation of δ-opioid receptor. Eur J Pharmacol. (2011) 660:341–50. doi: 10.1016/j.ejphar.2011.04.012
45. Gopalakrishnan L, Chatterjee O, Ravishankar N, Suresh S, Raju R, Mahadevan A, et al. Opioid receptors signaling network. J Cell Communication Signaling. (2022) 16:475–83. doi: 10.1007/s12079-021-00653-z
46. Valentino RJ and Volkow ND. Untangling the complexity of opioid receptor function. Neuropsychopharmacology. (2018) 43:2514–20. doi: 10.1038/s41386-018-0225-3
47. Johnson S and North R. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. (1992) 12:483–8. doi: 10.1523/JNEUROSCI.12-02-00483.1992
48. Bull FA, Baptista-Hon DT, Lambert JJ, Walwyn W, and Hales TG. Morphine activation of mu opioid receptors causes disinhibition of neurons in the ventral tegmental area mediated by β-arrestin2 and c-Src. Sci Rep. (2017) 7. doi: 10.1038/s41598-017-10360-8
49. Ben Hamida S, Boulos LJ, McNicholas M, Charbogne P, and Kieffer BL. Mu opioid receptors in GABAergic neurons of the forebrain promote alcohol reward and drinking. Addict Biol. (2019) 24:28–39. doi: 10.1111/adb.12576
50. Gurwell JA, Duncan MJ, Maderspach K, Stiene-Martin A, Elde RP, and Hauser KF. κ-Opioid receptor expression defines a phenotypically distinct subpopulation of astroglia: relationship to Ca2+ mobilization, development, and the antiproliferative effect of opioids. Brain Res. (1996) 737:175–87. doi: 10.1016/0006-8993(96)00728-7
51. Sargeant TJ, Miller JH, and Day DJ. Opioidergic regulation of astroglial/neuronal proliferation: where are we now? J Neurochemistry. (2008) 107:883–97.
52. Woo DH, Han K-S, Shim JW, Yoon B-E, Kim E, Bae JY, et al. TREK-1 and best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell. (2012) 151:25–40. doi: 10.1016/j.cell.2012.09.005
53. Yang Y, Sun Y, Hu R, Yan J, Wang Z, Li W, et al. Morphine promotes microglial activation by upregulating the EGFR/ERK signaling pathway. PLoS One. (2021) 16:e0256870. doi: 10.1371/journal.pone.0256870
54. Cuitavi J, Andrés-Herrera P, Meseguer D, Campos-Jurado Y, Lorente JD, Caruana H, et al. Focal mu-opioid receptor activation promotes neuroinflammation and microglial activation in the mesocorticolimbic system: Alterations induced by inflammatory pain. Glia. (2023) 71:1906–20. doi: 10.1002/glia.24374
55. Kristensen K, Christensen CB, and Christrup LL. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. (1994) 56:45–50. doi: 10.1016/0024-3205(94)00937-6
56. He L, Fong J, Von Zastrow M, and Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. (2002) 108:271–82. doi: 10.1016/S0092-8674(02)00613-X
57. Bokhari SM, Yao H, Bethel-Brown C, Fuwang P, Williams R, Dhillon NK, et al. Morphine enhances Tat-induced activation in murine microglia. J Neurovirol. (2009) 15:219–28. doi: 10.1080/13550280902913628
58. Garcia-Mesa Y, Jay TR, Checkley MA, Luttge B, Dobrowolski C, Valadkhan S, et al. Immortalization of primary microglia: a new platform to study HIV regulation in the central nervous system. J NeuroVirol. (2017) 23:47–66. doi: 10.1007/s13365-016-0499-3
59. Rai MA, Hammonds J, Pujato M, Mayhew C, Roskin K, and Spearman P. Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology. (2020) 17. doi: 10.1186/s12977-020-00544-y
60. Akiyama H, Jalloh S, Park S, Lei M, Mostoslavsky G, and Gummuluru S. Expression of HIV-1 intron-containing RNA in microglia induces inflammatory responses. J Virol. (2021) 95. doi: 10.1128/JVI.01386-20
61. Boreland AJ, Stillitano AC, Lin H-C, Abbo Y, Hart RP, Jiang P, et al. Sustained type I interferon signaling after human immunodeficiency virus type 1 infection of human iPSC derived microglia and cerebral organoids. iScience. (2024) 27:109628. doi: 10.1016/j.isci.2024.109628
62. Kong W, Frouard J, Xie G, Corley MJ, Helmy E, Zhang G, et al. Neuroinflammation generated by HIV-infected microglia promotes dysfunction and death of neurons in human brain organoids. PNAS Nexus. (2024) 3:pgae179. doi: 10.1093/pnasnexus/pgae179
63. Narasipura SD, Zayas JP, Ash MK, Reyes AF, Shull T, Gambut S, et al. Inflammatory responses revealed through HIV infection of microglia-containing cerebral organoids. J Neuroinflammation. (2025) 22. doi: 10.1186/s12974-025-03353-2
64. Heinze D, Park S, McCracken A, Haratianfar M, Lindstrom J, Villacorta-Martin C, et al. Notch activation during early mesoderm induction modulates emergence of the T/NK cell lineage from human iPSCs. Stem Cell Rep. (2022) 17:2610–28. doi: 10.1016/j.stemcr.2022.10.007
65. Wiley RD and Gummuluru S. Immature dendritic cell-derived exosomes can mediate HIV-1 trans infection. Proc Natl Acad Sci. (2006) 103:738–43. doi: 10.1073/pnas.0507995103
66. Hatch SC, Archer J, and Gummuluru S. Glycosphingolipid composition of human immunodeficiency virus type 1 (HIV-1) particles is a crucial determinant for dendritic cell-mediated HIV-1 trans-infection. J Virol. (2009) 83:3496–506. doi: 10.1128/JVI.02249-08
67. Cai NS, Quiroz C, Bonaventura J, Bonifazi A, Cole TO, Purks J, et al. Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. J Clin Invest. (2019) 129:2730–44. doi: 10.1172/JCI126912
68. Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, et al. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. (2000) 74:8358–67. doi: 10.1128/JVI.74.18.8358-8367.2000
69. Cassidy NAJ, Fish CS, Levy CN, Roychoudhury P, Reeves DB, Hughes SM, et al. HIV reservoir quantification using cross-subtype multiplex ddPCR. iScience. (2022) 25:103615. doi: 10.1016/j.isci.2021.103615
70. Kuniholm J, Armstrong E, Bernabe B, Coote C, Berenson A, Patalano SD, et al. Intragenic proviral elements support transcription of defective HIV-1 proviruses. PloS Pathog. (2021) 17:e1009982. doi: 10.1371/journal.ppat.1009982
71. Bruner KM, Wang Z, Simonetti FR, Bender AM, Kwon KJ, Sengupta S, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature. (2019) 566:120–5. doi: 10.1038/s41586-019-0898-8
72. Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med. (2016) 22:1043–9. doi: 10.1038/nm.4156
73. Imamichi H, Smith M, Adelsberger JW, Izumi T, Scrimieri F, Sherman BT, et al. Defective HIV-1 proviruses produce viral proteins. Proc Natl Acad Sci. (2020) 117:3704–10. doi: 10.1073/pnas.1917876117
74. Karn J and Stoltzfus CM. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harbor Perspect Med. (2012) 2:a006916–a. doi: 10.1101/cshperspect.a006916
75. Knoener R, Evans E, Becker JT, Scalf M, Benner B, Sherer NM, et al. Identification of host proteins differentially associated with HIV-1 RNA splice variants. eLife. (2021) 10. doi: 10.7554/eLife.62470
76. Dave RS, Ali H, Sil S, Knight LA, Pandey K, Madduri LSV, et al. NF-κB duplications in the promoter-variant HIV-1C LTR impact inflammation without altering viral replication in the context of simian human immunodeficiency viruses and opioid-exposure. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00095
77. Sharma AL, Shaffer D, Netting D, and Tyagi M. Cocaine sensitizes the CD4+ T cells for HIV infection by co-stimulating NFAT and AP-1. iScience. (2022) 25:105651. doi: 10.1016/j.isci.2022.105651
78. Sharma AL, Tyagi P, Khumallambam M, and Tyagi M. Cocaine-induced DNA-dependent protein kinase relieves RNAP II pausing by promoting TRIM28 phosphorylation and RNAP II hyperphosphorylation to enhance HIV transcription. Cells. (2024) 13:1950. doi: 10.3390/cells13231950
79. Yukl SA, Kaiser P, Kim P, Telwatte S, Joshi SK, Vu M, et al. HIV latency in isolated patient CD4 + T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci Trans Med. (2018) 10:eaap9927. doi: 10.1126/scitranslmed.aap9927
80. Akiyama H, Miller CM, Ettinger CR, Belkina AC, Snyder-Cappione JE, and Gummuluru S. HIV-1 intron-containing RNA expression induces innate immune activation and T cell dysfunction. Nat Commun. (2018) 9:3450. doi: 10.1038/s41467-018-05899-7
81. Polakiewicz RD, Schieferl SM, Gingras A-C, Sonenberg N, and Comb MJ. μ-opioid receptor activates signaling pathways implicated in cell survival and translational control. J Biol Chem. (1998) 273:23534–41. doi: 10.1074/jbc.273.36.23534
82. Ji S and Wang L. μ-Opioid receptor signalling via PI3K/Akt pathway ameliorates lipopolysaccharide-induced acute respiratory distress syndrome. Exp Physiol. (2019) 104:1555–61. doi: 10.1113/EP087783
83. Chugh P, Bradel-Tretheway B, Monteiro-Filho CM, Planelles V, Maggirwar SB, Dewhurst S, et al. Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology. (2008) 5:11. doi: 10.1186/1742-4690-5-11
84. Hamada K, Maeda Y, Mizutani A, and Okada S. The phosphatidylinositol 3-kinase p110α/PTEN signaling pathway is crucial for HIV-1 entry. Biol Pharm Bulletin. (2019) 42:130–8. doi: 10.1248/bpb.b18-00801
85. Reiss D, Maduna T, Maurin H, Audouard E, and Gaveriaux-Ruff C. Mu opioid receptor in microglia contributes to morphine analgesic tolerance, hyperalgesia, and withdrawal in mice. J Neurosci Res. (2022) 100:203–19. doi: 10.1002/jnr.24626
86. Li Y, Merrill JD, Mooney K, Song L, Wang X, Guo C-J, et al. Morphine enhances HIV infection of neonatal macrophages. Pediatr Res. (2003) 54:282–8. doi: 10.1203/01.PDR.0000074973.83826.4C
87. Guo C-J, Li Y, Tian S, Wang X, Douglas SD, and Ho W-Z. Morphine enhances HIV infection of human blood mononuclear phagocytes through modulation of β-chemokines and CCR5 receptor. J Invest Med. (2002) 50:435–42. doi: 10.1136/jim-50-06-03
88. Miyagi T, Chuang LF, Doi RH, Carlos MP, Torres JV, and Chuang RY. Morphine induces gene expression of CCR5 in human CEM x174 lymphocytes. J Biol Chem. (2000) 275:31305–10. doi: 10.1074/jbc.M001269200
89. McConalogue K, Grady EF, Minnis J, Balestra B, Tonini M, Brecha NC, et al. Activation and internalization of the μ-opioid receptor by the newly discovered endogenous agonists, endomorphin-1 and endomorphin-2. Neuroscience. (1999) 90:1051–9. doi: 10.1016/S0306-4522(98)00514-4
90. Bond C, Laforge KS, Tian M, Melia D, Zhang S, Borg L, et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters β-endorphin binding and activity: Possible implications for opiate addiction. Proc Natl Acad Sci. (1998) 95:9608–13. doi: 10.1073/pnas.95.16.9608
91. Wang Y, Wang X, Ye L, Li J, Song L, Fulambarkar N, et al. Morphine suppresses IFN signaling pathway and enhances AIDS virus infection. PLoS One. (2012) 7:e31167. doi: 10.1371/journal.pone.0031167
92. Wang MR, Wu DD, Luo F, Zhong CJ, Wang X, Zhu N, et al. Methadone inhibits viral restriction factors and facilitates HIV infection in macrophages. Front Immunol. (2020) 11:1253. doi: 10.3389/fimmu.2020.01253
93. Chuang TK, Killam KF, Chuang LF, Kung HF, Sheng WS, Chao CC, et al. Mu opioid receptor gene expression in immune cells. Biochem Biophys Res Commun. (1995) 216:922–30. doi: 10.1006/bbrc.1995.2709
94. Philippe D. Mu opioid receptor expression is increased in inflammatory bowel diseases: implications for homeostatic intestinal inflammation. Gut. (2006) 55:815–23. doi: 10.1136/gut.2005.080887
95. Borner C, Warnick B, Smida M, Hartig R, Lindquist JA, Schraven B, et al. Mechanisms of opioid-mediated inhibition of human T cell receptor signaling. J Immunol. (2009) 183:882–9. doi: 10.4049/jimmunol.0802763
96. Campana G, Sarti D, Spampinato S, and Raffaeli W. Long-term intrathecal morphine and bupivacaine upregulate MOR gene expression in lymphocytes. Int Immunopharmacol. (2010) 10:1149–52. doi: 10.1016/j.intimp.2010.06.016
97. Xie F, Kitagawa Y, Ogata H, Yasuhara S, You Z, and Jeevendra Martyn JA. Morphine induces inflammatory responses via both TLR4 and cGAS-STING signaling pathways. Cytokine. (2024) 183:156737. doi: 10.1016/j.cyto.2024.156737
98. Kirk GD, Astemborski J, Mehta SH, Ritter KD, Laird GM, Bordi R, et al. Nonstructured treatment interruptions are associated with higher human immunodeficiency virus reservoir size measured by intact proviral DNA assay in people who inject drugs. J Infect Dis. (2021) 223:1905–13. doi: 10.1093/infdis/jiaa634
99. McNeil R, Kerr T, Coleman B, Maher L, Milloy MJ, and Small W. Antiretroviral therapy interruption among HIV postive people who use drugs in a setting with a community-wide HIV treatment-as-prevention initiative. AIDS Behavior. (2017) 21:402–9. doi: 10.1007/s10461-016-1470-2
100. Acharya A, Olwenyi OA, Thurman M, Pandey K, Morsey BM, Lamberty B, et al. Chronic morphine administration differentially modulates viral reservoirs in a simian immunodeficiency virus SIVmac251-infected rhesus macaque model. J Virol. (2021) 95:JVI.01657–20. doi: 10.1128/JVI.01657-20
101. D’Orso I. The HIV-1 transcriptional program: from initiation to elongation control. J Mol Biol. (2025) 437:168690. doi: 10.1016/j.jmb.2024.168690
102. Hou Y-N, Vlaskovska M, Cebers G, Kasakov L, Liljequist S, and Terenius L. A μ-receptor opioid agonist induces AP-1 and NF-κB transcription factor activity in primary cultures of rat cortical neurons. Neurosci Letters. (1996) 212:159–62. doi: 10.1016/0304-3940(96)12799-3
103. Zhang J, Guo X, Cai Z, Pan Y, Yang H, Fu Y, et al. Two kinds of transcription factors mediate chronic morphine-induced decrease in miR-105 in medial prefrontal cortex of rats. Transl Psychiatry. (2022) 12:458. doi: 10.1038/s41398-022-02222-3
104. West MJ, Lowe AD, and Karn J. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J Virol. (2001) 75:8524–37. doi: 10.1128/JVI.75.18.8524-8537.2001
105. Hokello J, Lakhikumar Sharma A, and Tyagi M. AP-1 and NF-kappaB synergize to transcriptionally activate latent HIV upon T-cell receptor activation. FEBS Lett. (2021) 595:577–94. doi: 10.1002/1873-3468.14033
106. Emery A and Swanstrom R. HIV-1: to splice or not to splice, that is the question. Viruses. (2021) 13:181. doi: 10.3390/v13020181
107. Emery A, Zhou S, Pollom E, and Swanstrom R. Characterizing HIV-1 splicing by using next-generation sequencing. J Virol. (2017) 91:JVI.02515–16. doi: 10.1128/JVI.02515-16
108. Korostynski M, Piechota M, Kaminska D, Solecki W, and Przewlocki R. Morphine effects on striatal transcriptome in mice. Genome Biol. (2007) 8:R128. doi: 10.1186/gb-2007-8-6-r128
109. Dabrowski KR and Daws SE. Morphine-driven m6A epitranscriptomic neuroadaptations in primary cortical cultures. Mol Neurobiol. (2024) 61:10684–704. doi: 10.1007/s12035-024-04219-z
110. Mendel M, Delaney K, Pandey RR, Chen K-M, Wenda JM, Vågbø CB, et al. Splice site m6A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell. (2021) 184:3125–42.e25. doi: 10.1016/j.cell.2021.03.062
111. Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol. (2016) 1:16011. doi: 10.1038/nmicrobiol.2016.11
112. Tirumuru N, Zhao BS, Lu W, Lu Z, He C, and Wu L. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife. (2016) 5. doi: 10.7554/eLife.15528
113. Pereira-Montecinos C, Toro-Ascuy D, Ananias-Saez C, Gaete-Argel A, Rojas-Fuentes C, Riquelme-Barrios S, et al. Epitranscriptomic regulation of HIV-1 full-length RNA packaging. Nucleic Acids Res. (2022) 50:2302–18. doi: 10.1093/nar/gkac062
114. Baek A, Lee GE, Golconda S, Rayhan A, Manganaris AA, Chen S, et al. Single-molecule epitranscriptomic analysis of full-length HIV-1 RNAs reveals functional roles of site-specific m(6)As. Nat Microbiol. (2024) 9:1340–55. doi: 10.1038/s41564-024-01638-5
115. McCauley SM, Kim K, Nowosielska A, Dauphin A, Yurkovetskiy L, Diehl WE, et al. Intron-containing RNA from the HIV-1 provirus activates type I interferon and inflammatory cytokines. Nat Commun. (2018) 9:5305. doi: 10.1038/s41467-018-07753-2
116. Guney MH, Nagalekshmi K, McCauley SM, Carbone C, Aydemir O, and Luban J. IFIH1 (MDA5) is required for innate immune detection of intron-containing RNA expressed from the HIV-1 provirus. Proc Natl Acad Sci. (2024) 121. doi: 10.1073/pnas.2404349121
117. Ramaswamy S, Akiyama H, Berrigan J, Quiñones A, Olson A, Chen Y, et al. Macrophage-intrinsic MDA5-IRF5 axis drives HIV-1 icRNA-induced inflammatory responses. (2024). doi: 10.1101/2024.09.06.611547
118. Garvey LJ, Everitt A, Winston A, Mackie NE, and Benzie A. Detectable cerebrospinal fluid HIV RNA with associated neurological deficits, despite suppression of HIV replication in the plasma compartment. AIDS. (2009) 23:1443–4. doi: 10.1097/QAD.0b013e32832d077c
119. Canestri A, Lescure FX, Jaureguiberry S, Moulignier A, Amiel C, et al. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin Infect Diseases. (2010) 50:773–8. doi: 10.1086/650538
120. Peluso MJ, Ferretti F, Peterson J, Lee E, Fuchs D, Boschini A, et al. Cerebrospinal fluid HIV escape associated with progressive neurologic dysfunction in patients on antiretroviral therapy with well controlled plasma viral load. AIDS. (2012) 26:1765–74. doi: 10.1097/QAD.0b013e328355e6b2
121. Dahl V, Peterson J, Fuchs D, Gisslen M, Palmer S, and Price RW. Low levels of HIV-1 RNA detected in the cerebrospinal fluid after up to 10 years of suppressive therapy are associated with local immune activation. AIDS. (2014) 28:2251–8. doi: 10.1097/QAD.0000000000000400
122. Demarino C, Denniss J, Cowen M, Norato G, Dietrich DK, Henderson L, et al. HIV-1 RNA in extracellular vesicles is associated with neurocognitive outcomes. Nat Commun. (2024) 15. doi: 10.1038/s41467-024-48644-z
123. Madishetti S, Schneble N, König C, Hirsch E, Schulz S, Müller JP, et al. PI3Kγ integrates c AMP and Akt signalling of the μ-opioid receptor. Br J Pharmacol. (2014) 171:3328–37. doi: 10.1111/bph.12698
124. Gu F, Zhou Y, Tian L, Chen J, Zhang C, Huang Z, et al. Morphine promotes non-small cell lung cancer progression by downregulating E-cadherin via the PI3K/AKT/mTOR pathway. Sci Rep. (2024) 14. doi: 10.1038/s41598-024-72198-1
125. Bell JE, Arango JC, and Anthony IC. Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol. (2006) 1:182–91. doi: 10.1007/s11481-006-9018-2
126. McArthur JC and Johnson TP. Chronic inflammation mediates brain injury in HIV infection: relevance for cure strategies. Curr Opin Neurol. (2020) 33:397–404. doi: 10.1097/WCO.0000000000000807
127. Sonti S, Tyagi K, Pande A, Daniel R, Sharma AL, and Tyagi M. Crossroads of drug abuse and HIV infection: neurotoxicity and CNS reservoir. Vaccines (Basel). (2022) 10. doi: 10.3390/vaccines10020202
Keywords: HIV-1, opioids, inflammation, microglia, µ-opioid receptor
Citation: Skeete C, Sgambettera G, Gojanovich AD, He X, Bryant D, Yang M, Banerjee S, Quiñones-Molina AA, Akiyama H, Mostoslavsky G, Henderson AJ and Gummuluru S (2025) Mu opioid receptor activation in microglia enhances HIV-1 infection and HIV-infection-induced inflammatory responses. Front. Immunol. 16:1628872. doi: 10.3389/fimmu.2025.1628872
Received: 14 May 2025; Accepted: 22 September 2025;
Published: 06 October 2025.
Edited by:
Michael H. Lehmann, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Guochun Jiang, University of North Carolina at Chapel Hill, United StatesSamuel Martinez-Meza, Feinstein Institute for Medical Research, United States
Copyright © 2025 Skeete, Sgambettera, Gojanovich, He, Bryant, Yang, Banerjee, Quiñones-Molina, Akiyama, Mostoslavsky, Henderson and Gummuluru. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suryaram Gummuluru, cmd1bW11bHVAYnUuZWR1