 Huiting Zhang
Huiting Zhang Ning Kong
Ning Kong Zhimin Liao
Zhimin Liao Jiawu Fu
Jiawu Fu Jiangang Pan
Jiangang Pan Wangtao Zhong
Wangtao Zhong- 1Department of Neurology, Affiliated Hospital of Guangdong Medical University, Zhanjiang, China
- 2First School of Clinical Medicine, Guangdong Medical University, Zhanjiang, China
Autoimmune encephalitis (AE) associated with anti-neurexin-3α antibodies, an uncommon variant of AE, exhibits particularly low frequency when coexisting with systemic immune dysregulation. The heterogeneous clinical manifestations of this neuroinflammatory disorder hinder timely diagnosis. We describe a 55-year-old male diagnosed with anti-neurexin-3α-associated AE, complicated by systemic lupus erythematosus (SLE) and antiphospholipid antibodies. The case presented with pyrexia persisting for 3 days, followed by acute-onset vertigo and encephalopathy evolving over 7 h. The clinical course was marked by visual obscuration, cephalalgia with associated emetic episodes, and concurrent neuropsychiatric manifestations, including psychomotor agitation and urinary incontinence. Anti-neurexin-3α antibody was detected in the cerebrospinal fluid. The patient tested positive for anti-neutrophil cytoplasmic antibody, anti-SS-A/RO60KD, anti-SS-A/RO52, anti-SnRNP, and lupus anticoagulant. Plasma complement levels (C3 and C4) were decreased, while elevated titers were observed for antinuclear antibodies, anti-double-stranded DNA, and anti-β2 glycoprotein I IgG antibodies. Immunomodulatory therapy with pulse methylprednisolone and intravenous immunoglobulin infusion elicited marked neurological recovery. This case underscores the imperative to investigate AE in the differential diagnosis of acute neuropsychiatric decompensation, and it is important to consider changes related to the aforementioned pathologies during physical examination and imaging evaluation.
Introduction
Autoimmune encephalitis (AE) refers to rare neurological conditions involving brain inflammation due to immune system dysfunction. It has been demonstrated that the incidence of AE is about 1–2 per 100,000 people per year (1). AE is pathogenically linked to autoantibodies directed against neuronal surface antigens, synaptic adhesion molecules, voltage-gated ion channels, or neurotransmitter receptors. Antibody specificity dictates distinct neuroanatomical targets, with distinct epitope targeting driving pleomorphic clinical phenotypes through region-specific neurophysiological disruptions (2). In 2016, Gresa-Arribas et al. first included anti-neurexin-3α in the list of autoantibody targets for encephalitis (3). To our knowledge, only 12 cases of this encephalitis have been reported (3–10). Therefore, raising awareness of anti-neurexin-3α-associated AE remains highly valuable. The main clinical manifestations of anti-neurexin-3α-associated AE are epileptic seizures, memory and cognitive deficits, movement disorders, autonomic nerve dysfunction, and consciousness disturbance (3). Approximately half of the reported cases have presented systemic autoimmune abnormalities (11). In this study, we reported a novel case of anti-neurexin-3α AE with concurrent systemic lupus erythematosus (SLE) and abnormal antiphospholipid antibodies, thus broadening the clinical spectrum of diseases associated with neurexin-3α antibodies.
Case presentation
On 1 January 2025, a 55-year-old male was urgently transferred to our tertiary care center following a 72 h history of pyrexia and acute-onset disequilibrium with progressive encephalopathic deterioration over 7 h. Three days prior to admission, the patient developed low-grade pyrexia (maximum 37.5 °C) secondary to a preceding upper respiratory tract infection. During initial management at a county hospital, transient focal motor seizures involving bilateral extremities were observed, characterized by absence of autonomic involvement and spontaneous resolution within 5 min. Recurrent stereotyped episodes (n = 3) were documented prior to escalation of care. The patient developed dizziness, blurred vision, headache, disorganized speech, irritability, unfamiliarity with family members, and nausea and vomiting, along with urinary incontinence, without limb weakness, dysphagia, or dysarthria. There were no complaints of ocular dryness, reduced lacrimation, or xerostomia. There was no history of arteriovenous thrombosis events. No rash was present on the face. The cranial nerve examination was unremarkable, with normal muscle strength and tone in all four limbs, absent bilateral pathological reflexes, and no signs of meningeal irritation. Non-contrast cranial computed tomography (CT) demonstrated multifocal calcific deposits within the left basal ganglia nuclei. Subsequent transfer to our tertiary neurosciences center was initiated for further diagnosis and therapy. The patient’s medical history was unremarkable, with negative documentation of allergic sensitization, prior surgical interventions, transfusion requirements, heritable neurological disorders, or chronic ethanol exposure.
Upon initial neurological evaluation at our center, the patient demonstrated preserved arousal (Glasgow Coma Scale = 15) with profound cognitive–linguistic dissociation: fluent but dysarthric speech production, paraphasic errors, and impaired semantic processing manifesting as anosognosia for familial relationships. Neuropsychiatric assessment revealed a complex behavioral phenotype featuring (1): affective dysregulation with episodic vocal outbursts (2); multimodal hallucinations (auditory/visual) (3); sleep–wake cycle fragmentation; and (4) systematized paranoid ideation. Formal neurocognitive testing, including the Mini-Mental State Examination (MMSE) and Montreal Cognitive Assessment (MoCA), was precluded due to profound executive dysfunction. Laboratory tests were as follows: rheumatoid factor: 97 IU/ml (0–20 IU/ml), C-reactive protein (CRP): 26.01 mg/L (0–5 mg/L); erythrocyte sedimentation rate (ESR): 35 mm/h (0–26 mm/h); and procalcitonin: 0.051 ng/mL (<0.05 ng/mL). Complete blood count, serum ammonia, coagulation, and urinary tests showed no abnormalities. Serum cytomegalovirus, and herpes simplex virus type I antibody IgG antibodies were positive. Antinuclear antibodies (ANAs) were elevated at a titer of 1:640 (reference range: <1:80). Anti-neutrophil cytoplasmic antibody (ANCA), anti-SS-A/RO60KD, anti-SS-A/RO52, anti-SnRNP, and lupus anticoagulant were positive. Complement 3 (C3) was 0.524 g/L (reference range: 0.79–1.52 g/L), and complement 4 (C4) was 0.0832 g/L (reference range: 0.16–0.38 g/L). Anti-double-stranded DNA (anti-dsDNA) and anti-β2 glycoprotein I antibody IgG antibodies were elevated to 44.249 IU/mL and 25.7 CU, respectively. Serum tumor markers (alpha-fetoprotein, carcinoembryonic antigen, carbohydrate antigen-199, ferritin), anti-cardiolipin antibody, glycated hemoglobin, and thyroid function were normal. The patient met the 2012 Systemic Lupus International Collaborating Clinics (SLICC) criteria (12) and the 2019 European League Against Rheumatism/American College of Rheumatology classification criteria (13) for SLE, and the neuropsychiatric SLE (NPSLE) could not be ruled out. Brain CT showed a patchy high-density shadow is observed in the left basal region, with a CT value of approximately 146 Hounsfield units, suggesting a high likelihood of a calcification (Figure 1A). Computed tomography angiography (CTA) of the head showed no significant abnormalities (Figure 1B). Brain magnetic resonance imaging (MRI) revealed a few punctate abnormal signals adjacent to the lateral ventricles, with no enhancement. The sulci, fissures, and cisterns of the brain were widened (Figure 1C–F). Chest CT showed no abnormalities. Cerebrospinal fluid (CSF) appearance was clear and transparent. Lumbar puncture pressure was 200 mmH2O. Total cell count was 50×106/L (reference range 0-8×106/L), and white blood cell count was 30×106/L (reference range 0-8×106/L). Glucose and chloride levels were within the standard range. CSF bacterial culture, acid-fast bacilli staining, India ink staining, bacterial and viral cultures, immunoglobulins, and syphilis serology were unremarkable. The results of CSF metagenomic next-generation sequencing (mNGS) and serum paraneoplastic antibodies (Hu-IgG, Yo-IgG, Ri-IgG, CV2-IgG, Amphiphysin-IgG, Ma1-IgG, Ma2-IgG, SOX1-IgG, Tr/DNER-IgG, Zic4-IgG, PKCγ-IgG, Recoverin-IgG, Titin-IgG, GAD65-IgG) tested by dot enzyme-linked immunosorbent assays (Euroimmun, cat# D241024AP) were negative. CSF autoimmune encephalitis antibodies assessed using the tissue-based assay (TBA) and cell-based assay (CBA) methods (Hangzhou Sainting Medical Technology Co., Ltd., cat# 00116-240517) revealed positive anti-neurexin-3α IgG antibody (1:32; Figure 2A, B; Supplementary Table S1), while serum autoimmune encephalitis antibodies were negative.

Figure 1. Cranial computed tomography (CT) and brain magnetic resonance imaging (MRI) scans of the patient. (A) Calcification in the left basal region. (B) Head computed tomography angiography (CTA) was unremarkable. (C-F) Brain magnetic resonance imaging (MRI) revealed a few punctate abnormal signals adjacent to the lateral ventricles and the sulci, fissures, and cisterns of the brain were widened.
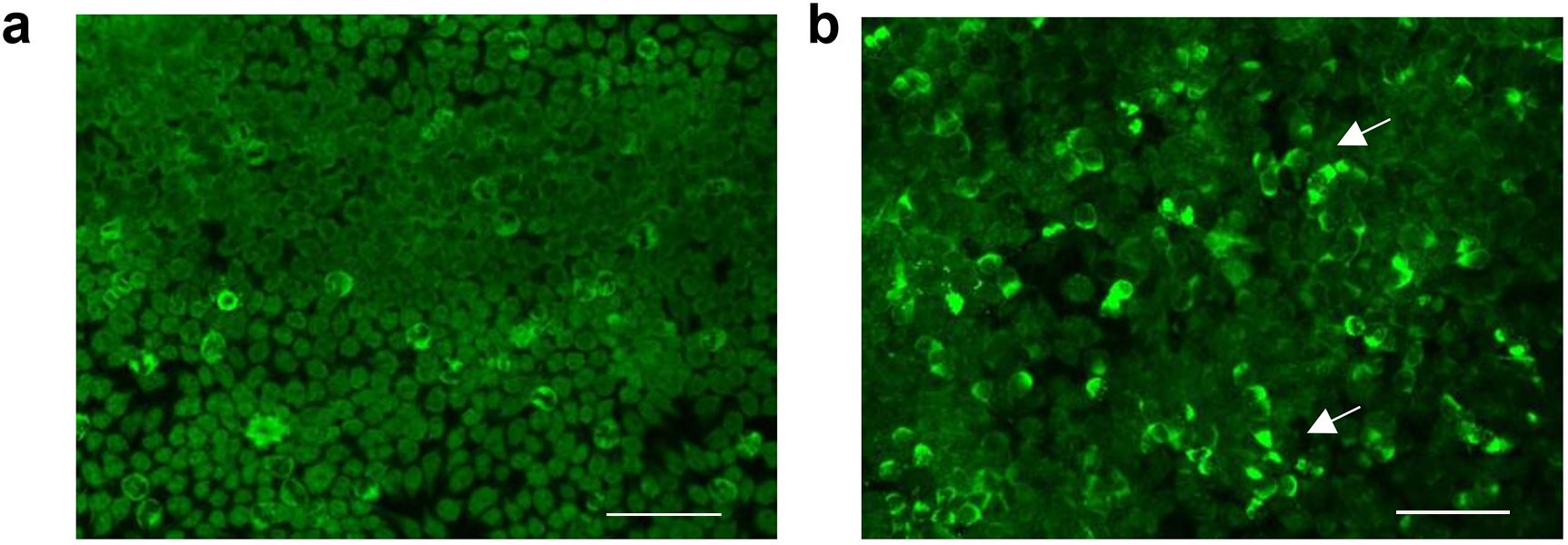
Figure 2. Anti-neurexin-3α antibody analyzed by cell-based assay (CBA) methods. (A) Immunofluorescence analysis revealed positivity in the experimental group (patient CSF co-cultured with HEK293T cells), as evidenced by green fluorescence, in contrast to control specimens (B). Scale bar: 200μm.Scale bar: 200μm.
The patient could not cooperate with the electroencephalographic monitoring. Definitive diagnoses of anti-neurexin-3α AE and SLE were established after rigorous exclusion of infectious, paraneoplastic, and metabolic etiologies (14, 15).
Initial empirical antiviral therapy with acyclovir was discontinued upon serological confirmation of anti-neurexin-3α antibody-mediated encephalitis. The immunomodulatory protocol comprised pulse methylprednisolone therapy (1,000 mg/d iv×3d → 500 mg/d×2d) with concurrent intravenous immunoglobulin (0.4 g/kg/d×5d), followed by a corticosteroid tapering protocol: methylprednisolone 240 mg/d×3d transitioned to prednisone acetate (60 mg/d×14d) with subsequent biweekly 5 mg decrements. Psychomotor agitation was controlled using midazolam infusion initiated at 2 mg, with titration based on clinical response. Hemodynamic parameters and metabolic homeostasis were closely monitored and maintained within physiological ranges. The patient gradually improved, with complete cessation of perceptual disturbances, including auditory/visual hallucinations, and restoration of logical thought processes. The midazolam sedation protocol was successfully discontinued on day 7 post-immunotherapy initiation. On the 40th day of follow-up, the patient’s speech was clear, he was able to recognize his family members, and he showed no mental symptoms or insomnia. Maintenance immunosuppression with prednisone 40 mg/d continues per AE Consortium guidelines (15). The timeline of the patient’s condition is illustrated in Figure 3.

Figure 3. Diagnostic and therapeutic pathway of the patient.
As shown in Table 1, we have summarized the characteristics of thirteen cases of anti-neurexin-3α-associated autoimmune encephalitis identified to date (including this case). The mean age of the patients was 44 years (range, 23–74), and 53.8% were female. More than half of the patients (9/13) presented with prodromal symptoms (headache, fever, nausea, or diarrhea) and experienced rapid progression (1–7 days, median 3) to memory decline, disability, seizure, and decreased level of consciousness. The majority of the patients (12/13) had underlying hematologic diseases or were found to have immune abnormalities, with the exception of one patient who had no other abnormal laboratory findings. Brain MRI revealed approximately 31% of cases with unremarkable structural neuroimaging, and 46% of the cases exhibited abnormalities in the hippocampus or temporal lobe. Among the thirteen patients, twelve patients exhibited neurexin-3α antibody positivity (CSF/serum) and concurrent CSF abnormalities, and one patient had no CSF data available (Table 1). All patients underwent first-line immunotherapy. Regarding to the therapeutic effect, there were three patients died (two from encephalitis/brain herniation secondary to encephalitis, and one died from sepsis), four patients had partial recovery, and four patients achieved full remission, and two patients experienced no marked improvement.

Table 1. Main clinical features of thirteen patients with anti-Neurexin-3α-associated autoimmune encephalitis.
Discussion
Since the neurexin-3α antigenic epitope was identified in 2016, a total of 12 cases of anti-neurexin-3α AE have been documented worldwide (3–10). Among them, approximately half of the cases indicated the presence of systemic autoimmune abnormalities. The patient exhibited acute-onset encephalopathy with a prodromal febrile episode. Clinical manifestations included rapidly progressive cognitive dysfunction and neuropsychiatric disturbances, culminating in impaired consciousness and limb rigidity, suggestive of encephalitis. CSF analysis revealed IgG antibodies against neurexin-3α, fulfilling diagnostic criteria for AE (16, 17). The patient also met the diagnostic criteria for SLE, with concomitant Sjögren’s syndrome and antiphospholipid antibodies; a definitive diagnosis of anti-neurexin-3α AE associated with SLE was therefore established.
Neurexin-3α is a novel antibody target in AE, characterized as the predominant isoform within the synaptic vesicle glycoprotein superfamily, constituting 74% of total alternative splicing variants (18). Neurexins serve as critical presynaptic-postsynaptic interface mediators, modulating voltage-gated calcium channel activity and synaptic vesicle exocytosis via their evolutionarily conserved C-terminal domains (11, 19). Thereby, neurexin-3α demonstrates bidirectional neuromodulatory capacity through presynaptic vesicular release regulation and differential postsynaptic signaling integration, with region-specific functional heterogeneity observed across distinct brain areas (20). In the hippocampus, the extracellular sequence of presynaptic synaptic protein-3α affects memory and cognitive functions by mediating the trans-synaptic regulation of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor (AMPAR). AMPAR functions as a pivotal molecular determinant in orchestrating enduring synaptic plasticity and mnemonic consolidation processes (21–23). Research has demonstrated that antibody-induced depletion of neuromodulin-3a disrupts synaptogenic maturation while preserving synaptic density, ultimately influencing the inhibitory and excitatory functions of synapses (24, 25). In this case, the patient exhibited memory decline and the difficulty in recognizing family members, which might be related to the interaction between neurexin-3a and AMPAR and synaptic function.
Anti-neurexin-3α-associated AE generally manifests with mental and behavioral disturbances, seizures, and cognitive decline. This distinct neuroimmune entity warrants critical diagnostic prioritization due to its frequent clinical overlap with alternative encephalitic pathologies, particularly anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis and viral encephalitis. The definitive diagnosis of anti-neurexin-3α-associated AE necessitates rigorous integration of neurodiagnostic modalities, including clinical symptomatology, electroencephalographic anomalies, and brain MRI, with confirmatory serological and CSF autoantibody positivity. Notably, beyond established predictors such as advanced age and CSF protein elevation, dynamic prognostic determinants including refractory status epilepticus and neuroinflammatory-mediated cerebral edema may represent modifiable contributors to adverse clinical trajectories in neuroimmune encephalopathies (26–28). Previous studies have demonstrated that patients with positive systemic autoantibodies, particularly lupus antibodies, rheumatoid arthritis antibodies, and other related antibody markers, were prone to worsening conditions (3, 29). Notably, this patient manifested progressive worsening of clinical symptoms concurrent with systemic autoimmune abnormalities. Low complements (C3, C4), anti-SS-A/RO60KD, anti-SS-A/RO52, anti-SnRNP, and anti-dsDNA are highly indicative of SLE. Anti-SS-A/Ro52 and anti-SS-A/Ro60 are classic characteristics of Sjögren’s syndrome and are also are frequently present in SLE. Positive lupus anticoagulant and elevated anti-β2 glycoprotein I IgG are cardinal antibodies for antiphospholipid syndrome (APS). In the absence of clinical symptoms of Sjögren’s syndrome or thrombosis, we suppose that the positive anti-SS-A/Ro52, anti-SS-A/Ro60, and anti-β2 glycoprotein I IgG are considered concomitant serologic findings of SLE. These serological findings indicate that, rather than an isolated idiopathic AE, this case underscores the complex interplay between central nervous system (CNS)-specific and systemic autoimmunity. This permissive autoimmune environment may allow for the accidental generation of antibodies against neuronal surface antigens like neurexin-3α, a protein critical for synaptic adhesion and signaling. Secondly, systemic inflammation, a hallmark of active SLE, can disrupt the integrity of the blood–brain barrier (BBB). This breach allows the passage of pre-existing systemic autoantibodies and autoreactive immune cells into the CNS parenchyma. Anti-neurexin-3α antibodies can directly bind to their target on the neuronal surface, leading to internalization of the receptor and reversible synaptic dysfunction, which manifests as the clinical syndrome of encephalitis (seizures, psychosis, cognitive deficits). Thirdly, the initial inflammatory response driven by cell death and the release of nuclear antigens (e.g., DNA) can lead to ‘epitope spreading. Lastly, there is a fundamental loss of immune tolerance, leading to hyperreactivity of B and T cells against a wide array of self-antigens, both nuclear and cytoplasmic. As B cells, T cells, and the complement system are greatly involved in the pathological mechanisms of autoimmune diseases (30, 31). B-cell responses could be initiated against neuronal targets like neurexin-3α in a susceptible individual. We suppose that B cells, T cells, and the complement system might play a crucial role in the immune regulation targeting synapsin-3α. In summary, the finding of anti-neurexin-3α antibodies in a patient with this serological profile is best interpreted as a severe neurological manifestation of an underlying systemic autoimmune diathesis, most likely SLE with overlapping features of Sjögren’s syndrome and APS. Therefore, management must include acutely treating the encephalitis with immunotherapies (e.g., steroids, immunoglobulin) and concurrently managing the systemic disease with appropriate immunosuppressive regimens to achieve long-term remission and prevent further complications. Future research is awaited to explore the immunological mechanism of encephalitis, providing a theoretical basis for investigating additional monoclonal antibody therapies. First-line treatment for autoimmune encephalitis commonly includes intravenous immunoglobulin (IVIG), plasma exchange (PLEX), and corticosteroids. Monoclonal antibodies are part of the second line of treatment, so far. It is reported that approximately 60% patients with AE achieved a favorable prognosis following the aforementioned treatments (7). In this study, the patient achieved complete clinical remission following treatment with corticosteroids and human immunoglobulin.
In conclusion, anti-neurexin-3α AE can be associated with systemic autoimmune abnormalities, with concurrent autoimmune seropositivity portending adverse prognostic trajectories. The clinical manifestations of anti-neurexin-3α AE may be diverse, including mental and behavioral abnormalities, epileptic seizures, consciousness attenuation, and extrapyramidal dysfunction. Furthermore, the cranial MRI of anti-neurexin-3α AE may be negative, contributing to the difficulty in diagnosing this disease. Consequently, in addition to medical history, clinical manifestations, and MRI, the diagnosis of AE should focus on comprehensive screening of the immune antibody spectrum (e.g., neuronal surface antibodies, paraneoplastic antibodies) and and detection of systemic immune indicators. Relevant immunotherapy should be provided promptly, and second-line immunosuppressive treatment should be initiated when necessary.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Affiliated Hospital of Guangdong Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
HZ: Writing – original draft, Investigation, Writing – review & editing, Funding acquisition, Methodology, Data curation, Validation, Conceptualization. NK: Conceptualization, Methodology, Data curation, Investigation, Writing – original draft. ZL: Conceptualization, Investigation, Writing – original draft, Validation, Methodology, Data curation. JF: Investigation, Methodology, Writing – original draft, Validation, Conceptualization. JP: Investigation, Methodology, Writing – original draft, Conceptualization. WZ: Investigation, Supervision, Funding acquisition, Conceptualization, Writing – review & editing, Data curation, Validation.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by Guangdong Medical Research Fund (B2018048), Science and technology research project of Zhanjiang (2021A05074) and Doctor Project of Affiliated Hospital of Guangdong Medical University (10801B20200004), Affiliated Hospital of Guangdong Medical University Clinical Research Program (LCYJ2022B003, LCYJ2023B007) and Affiliated Hospital of Guangdong Medical University High-level Personnel Research Project (2022018).
Acknowledgments
The author thanks the staff and the participants of this study for their valuable contributions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1630292/full#supplementary-material
References
1. Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol. (2018) 83:166–77. doi: 10.1002/ana.25131
2. Uy CE, Binks S, and Irani SR. Autoimmune encephalitis: clinical spectrum and management. Pract Neurol. (2021)21:412–23. doi: 10.1136/practneurol-2020-002567
3. Gresa-Arribas N, Planagumà J, Petit-Pedrol M, Kawachi I, Katada S, Glaser CA, et al. Human neurexin-3α antibodies associate with encephalitis and alter synapse development. Neurology. (2016) 86:2235–42. doi: 10.1212/wnl.0000000000002775
4. Costa A, Silva-Pinto A, Alves J, Neves N, Martínez-Hernández E, Abreu P, et al. Postmalaria neurologic syndrome associated with neurexin-3α antibodies. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e392. doi: 10.1212/nxi.0000000000000392
5. Loehrer PA, Bien CI, Dusoi AE, Timmermann L, and Simon OJ. Neurexin-3α-associated autoimmune encephalitis: a case report of full recovery after rituximab therapy. Eur J Neurol. (2020) 27:e91–3. doi: 10.1111/ene.14481
6. Hansen N, Lange C, Maass F, Hassoun L, Bouter C, Stöcker W, et al. Mild amnestic cognitive impairment and depressive symptoms in autoimmune encephalitis associated with serum anti-neurexin-3α Autoantibodies. Brain Sci. (2021) 11(6):673. doi: 10.3390/brainsci11060673
7. Zhihua. Z, Riming. H, Hong. L, Jianjun. L, Langhua. F, Xiaolong. Z, et al. Neurexin−3α IgG mediated autoimmune encephalitis: a case report. Chin J Neurol. (2021) 54:258–62.
8. Heqing. Z, Lanxiang. W, Sheng. T, Pan. L, Mingxu. L, and Wei. W. Anti−neurexin−3α antibody−associated encephalitis with parkinsonism as the main manifestation: a case report. Chin J Neurol. (2022) 55:497–500.
9. Zhang C, Hao Y, Shao H, Xin M, Bai S, Guan Y, et al. Neurexin-3α-associated autoimmune encephalitis with intracranial diffuse large B lymphoma diagnosed on FDG and TSPO PET/MRI. Eur J Nucl Med Mol Imaging. (2023) 50:1270–2. doi: 10.1007/s00259-022-06054-7
10. Zhu L, Shang Q, Zhao CW, Dai S, and Wu Q. Case report: Anti-neurexin-3α-associated autoimmune encephalitis secondary to contrast-induced encephalopathy. Front Neurol. (2023) 14:1060110. doi: 10.3389/fneur.2023.1060110
11. Dalmau J, Geis C, and Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev. (2017) 97:839–87. doi: 10.1152/physrev.00010.2016
12. Lerkvaleekul B, Chobchai P, Rattanasiri S, and Vilaiyuk S. Evaluating performance of the 2019 EULAR/ACR, 2012 SLICC, and 1997 ACR criteria for classifying adult-onset and childhood-onset systemic lupus erythematosus: A systematic review and meta-analysis. Front Med (Lausanne). (2022) 9:1093213. doi: 10.3389/fmed.2022.1093213
13. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European league against rheumatism/american college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. (2019) 71:1400–12. doi: 10.1002/art.40930
14. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. (2016) 15:391–404. doi: 10.1016/s1474-4422(15)00401-9
15. Hahn C, Budhram A, Alikhani K, AlOhaly N, Beecher G, Blevins G, et al. Canadian consensus guidelines for the diagnosis and treatment of autoimmune encephalitis in adults. Can J Neurol Sci. (2024), 1–21. doi: 10.1017/cjn.2024.16
16. Dutra LA, Abrantes F, Toso FF, Pedroso JL, Barsottini OGP, and Hoftberger R. Autoimmune encephalitis: a review of diagnosis and treatment. Arq Neuropsiquiatr. (2018) 76:41–9. doi: 10.1590/0004-282x20170176
17. Flanagan EP, Geschwind MD, Lopez-Chiriboga AS, Blackburn KM, Turaga S, Binks S, et al. Autoimmune encephalitis misdiagnosis in adults. JAMA Neurol. (2023) 80:30–9. doi: 10.1001/jamaneurol.2022.4251
18. Zhang R, Jiang H, Liu Y, and He G. Structure, function, and pathology of Neurexin-3. Genes Dis. (2023) 10:1908–19. doi: 10.1016/j.gendis.2022.04.008
19. Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. (2008) 455:903–11. doi: 10.1038/nature07456
20. Aoto J, Földy C, Ilcus SM, Tabuchi K, and Südhof TC. Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat Neurosci. (2015) 18:997–1007. doi: 10.1038/nn.4037
21. Diering GH and Huganir RL. The AMPA receptor code of synaptic plasticity. Neuron. (2018) 100:314–29. doi: 10.1016/j.neuron.2018.10.018
22. Lee J and Pak DTS. Amyloid precursor protein combinatorial phosphorylation code regulates AMPA receptor removal during distinct forms of synaptic plasticity. Biochem Biophys Res Commun. (2024) 709:149803. doi: 10.1016/j.bbrc.2024.149803
23. Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, et al. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest. (2015) 125:234–47. doi: 10.1172/jci77888
24. Scheiffele P, Fan J, Choih J, Fetter R, and Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. (2000) 101:657–69. doi: 10.1016/s0092-8674(00)80877-6
25. Sheng M and Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. (2011) 3(12):a005678. doi: 10.1101/cshperspect.a005678
26. Thakur KT, Motta M, Asemota AO, Kirsch HL, Benavides DR, Schneider EB, et al. Predictors of outcome in acute encephalitis. Neurology. (2013) 81:793–800. doi: 10.1212/WNL.0b013e3182a2cc6d
27. Nosadini M, Eyre M, Molteni E, Thomas T, Irani SR, Dalmau J, et al. Use and safety of immunotherapeutic management of N-methyl-d-aspartate receptor antibody encephalitis: A meta-analysis. JAMA Neurol. (2021) 78:1333–44. doi: 10.1001/jamaneurol.2021.3188
28. Venkatesan A. Epidemiology and outcomes of acute encephalitis. Curr Opin Neurol. (2015) 28:277–82. doi: 10.1097/wco.0000000000000199
29. McKeon A, Lesnick C, Vorasoot N, Buckley MW, Dasari S, Flanagan EP, et al. Utility of protein microarrays for detection of classified and novel antibodies in autoimmune neurologic disease. Neurol Neuroimmunol Neuroinflamm. (2023) 10(5):e200145. doi: 10.1212/nxi.0000000000200145
30. Flemming A. NMDAR-directed CAAR T cells show promise for autoimmune encephalitis. Nat Rev Immunol. (2023) 23:786. doi: 10.1038/s41577-023-00969-4
Keywords: anti-neurexin-3α-associated autoimmune encephalitis, systemic autoimmune, abnormal behavior, consciousness disturbance, case report
Citation: Zhang H, Kong N, Liao Z, Fu J, Pan J and Zhong W (2025) Coexistence of anti-neurexin-3α-associated autoimmune encephalitis and systemic lupus erythematosus in an adult patient: a case report. Front. Immunol. 16:1630292. doi: 10.3389/fimmu.2025.1630292
Received: 28 May 2025; Accepted: 10 November 2025; Revised: 05 November 2025;
Published: 04 December 2025.
Edited by:
Honghao Wang, Guangzhou First People’s Hospital, ChinaReviewed by:
Barnali Das, Kokilaben Dhirubhai Ambani Hospital & Medical Research Institute, IndiaZoilo Morel, National University of Asunción, Paraguay
Copyright © 2025 Zhang, Kong, Liao, Fu, Pan and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wangtao Zhong, MTU4OTgwMTE2QHFxLmNvbQ==; Jiangang Pan, ZGlwcGVyMjgwQGFsaXl1bi5jb20=
†These authors have contributed equally to this work