 Guadalupe Fernanda Godinez-Zamora1,2
Guadalupe Fernanda Godinez-Zamora1,2 Patricia Baeza-Capetillo1
Patricia Baeza-Capetillo1 Omar Josué Saucedo-Ramírez3
Omar Josué Saucedo-Ramírez3 Blanca Estela Del-Río-Navarro3
Blanca Estela Del-Río-Navarro3 Sara Elva Espinosa-Padilla4Verónica Fabiola Morán-Barroso5
Sara Elva Espinosa-Padilla4Verónica Fabiola Morán-Barroso5 Jesus Aguirre-Hernandez1*
Jesus Aguirre-Hernandez1*- 1Laboratorio de Genómica, Genética y Bioinformática, Hospital Infantil de México Federico Gómez, Mexico City, Mexico
- 2Programa de Maestría y Doctorado en Ciencias Médicas Odontológicas y de la Salud, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
- 3Servicio de Alergia e Inmunología Clínica, Hospital Infantil de México Federico Gómez, Mexico City, Mexico
- 4Unidad de Investigación en Inmunodeficiencias, Instituto Nacional de Pediatría, Mexico City, Mexico
- 5Departamento de Genética, Hospital General de México Dr. Eduardo Liceaga, Mexico City, Mexico
Inborn errors of immunity (IEI) number more than 500 diseases, with most affected patients being children. Their precise diagnosis is hampered by overlapping phenotypes, and by their ample and varied phenotypic spectrum. We analyzed the contribution of next generation sequencing to the diagnosis of IEI in a cohort of 157 children in a referral hospital in Mexico City. Following the classification of the International Union of Immunological Societies (IUIS), patients were assigned to an IEI group before sequencing, or to an “undefined” group, if it was not possible to assign them to any of them. Patients were again classified in the IUIS groups after sequencing. The diagnostic yield was 32.48%. Before sequencing, the largest group was comprised by patients that could not be assigned to a specific IUIS group (38.35% of the cohort), while after sequencing the largest group was made by the patients where no likely molecular diagnosis was found (67.52% of the cohort). Patients that were assigned to an IUIS group were confirmed to have a disease of that same group in 31.25% of the cases, while in 10.42% the molecular diagnosis corresponded to an immunodeficiency of a different group to the one initially suggested. In 18.03% of the children that could not be assigned to an immunodeficiency group before sequencing, a molecular diagnosis was reached after sequencing. In the patients that remained without a molecular diagnosis, the possibility of new IEI genes was explored by analyzing the variants, first in a curated set of immune related genes, and then across the whole exome. However, after filtering the variants, by frequency, predicted consequence, and known biology, no new IEI candidate genes were identified. This results underscore the large impact of next generation sequencing for the correct diagnosis of IEI, and also points to the need to better understand their genetic architecture in order to increase the diagnostic yield.
Introduction
Inborn errors of immunity (IEI), formerly known as primary immunodeficiencies, are a group of monogenic diseases characterized by dysregulation of the immune system that may affect the innate and the adaptive systems, as well as multiple cellular functions (1). Symptoms may present themselves from a very early age, or during childhood (2). However, recent studies suggest that in the adult population these diseases may be more common than previously thought, and their incidence, across all age groups, may be as high as 1:5,000 (3, 4).
IEI have a broad phenotypic spectrum and variable expressivity (5). These diseases may stem from alterations in a large number of genes (6). Different alterations in the same gene may lead to different phenotypes, and sometimes to different diseases. On the other hand, alterations in different genes may result in different diseases that nevertheless show overlapping phenotypes, making it difficult to distinguish between them (7, 8). Due to these characteristics, next generation sequencing (NGS) has been increasingly used for diagnostic purposes as it enables the simultaneous interrogation of a large number of genes, with the concomitant savings in time and money this entails (5, 9, 10).
Since 2014, an increasing number of research groups have been studying cohorts of IEI patients using NGS, both for diagnostic purposes, and to identify new genes associated with these diseases. In some studies, a panel of candidate genes was sequenced, while in others the whole coding exome (WES), and even the whole genome (WGS), were studied. More than 35 cohorts, studied by NGS, have been reported in the literature (3, 11–19). Most of these studies have been done with a diagnostic goal in mind, while only a small number have included a search for new IEI genes. Up to 2018, most of these studies involved sequencing of gene panels (20–22), while later on WES became increasingly important (11, 12, 14). More recently, particularly in well-resourced institutions, WGS has also been deployed (3, 9, 23, 24). Most NGS studies of large cohorts have been undertaken in European countries and the USA, although other parts of the world are also represented (12, 16–18, 21, 22, 25–27). In these cohort studies, the diagnostic yield varied widely, from 15 to 79% (9, 13, 19). These differences in diagnostic yield are the result of both, the NGS approach used (e.g., gene panels, WES, or WGS), and the criteria employed for selecting the patients to be sequenced. In this regard, some cohorts comprised patients of a single IEI group, while in other studies a suspicion of an IEI was enough to include the patient in the cohort. In the former type of studies, the diagnostic yield is higher than in studies with more heterogeneous diseases. On average, the diagnostic yield is 30% (11). There are various reasons for this relatively low diagnostic yield. Among others, the fact that genes underlying these highly genetically heterogenous diseases still remain to be identified, as underscored by the regular addition of new genes to the literature (6, 9, 14, 28–35). The most recent update of the IUIS contains 555 conditions and 504 genes (6).
Some groups have studied large cohorts of patients with IEI diseases, both to arrive at the molecular diagnosis and to search for new candidate genes. Following this approach, Stray-Pedersen et al. (14) identified six new candidate genes after using WES to study a large number of sporadic cases. With WGS and a Bayesian approach in a large cohort of sporadic cases, Thaventhiran et al. (3) also identified several new candidate genes and pointed to the interplay between high-penetrance rare monogenic variants and common variants. Itan and Casanova (36) used a different approach to search for new genes associated with IEI; they studied the human connectome of IEI genes to propose new candidate genes. In subsequent years, some of these have indeed been found to underly new IEI.
In this paper, we present the first large cohort of IEI patients in Mexico, studied by NGS for diagnostic purposes, followed by the search for new candidate genes: first in a curated list of possible IEI genes, and then by studying variants across the whole exome.
Materials and methods
We studied a cohort of 157 pediatric patients, from Hospital Infantil de México Federico Gómez, between June 2015 and May 2024. These patients were suspected of having an IEI whose identity could not be precisely determined by laboratory tests; in other cases, a molecular diagnosis was required before deciding on the appropriate treatment. The information available for each patient was variable. In some, it was mostly limited to the clinical history (e.g., repeated infections at a very early age); for some others, results of a few laboratory tests were available.
DNA was extracted from peripheral blood samples, or from buccal swabs. In nine patients the TruSightOne Targeted Regions panel v1.1 (Illumina) was used to obtain the sequencing library. From 2016 to 2023, WES was done in 125 patients with the Nextera Rapid Capture Exome Targeted Regions v1.2 kit (Illumina). The Twist exome probes, which were developed more recently, seem to provide better coverage of the targeted regions through better uniformity (37), so starting in 2023 we used Twist ILMN Exome 2.0 or 2.5 Plus Panel kits (Illumina) to sequence the exome of 23 patients. Paired end sequencing (150 bases per end) was done in a NextSeq 500 instrument (Illumina; RRID: SCR_014983). Read alignment and variant calling were undertaken with BaseSpace Enrichment Workflow (Illumina). Quality control was performed by filtering variants with “PASS” in the FILTER field, with a minimum depth of 10X, and a variant allele fraction of 0.20 or higher; this was done with bcftools (RRID: SCR_005227) (38) and the command: bcftools view -O z -f “PASS” -e ‘INFO/DP<10’ -e ‘(AD[0:1]/(AD[0:0]+AD[0:1]))<2/10’. Exomiser (RRID: SCR_002192) was used to annotate, filter, and prioritize genes and variants (39), with human phenotype ontology (HPO, RRID: SCR_006016) terms obtained from the information provided by the clinicians (40). To carry out the analysis, a list of IEI genes was provided, comprising all the genes in the most current update of the IUIS at the time of the analysis (4, 10, 41, 42). The list was supplemented with the genes in the Primary Immunodeficiency panel in PanelApp (43, 44), plus any new IEI genes added to OMIM (RRID: SCR_006437) (45). From this list, three virtual subpanels were created, one for each mode of inheritance (biallelic, monoallelic, and X-linked). For each patient, Exomiser was run separately with each one of these subpanels, using the default parameters set for each mode of inheritance. If variants and genotypes of interest were found, they were reannotated with Variant Effect Predictor (VeP, RRID: SCR_007931) (46), reads were visually examined with Integrative Genomics Viewer (IGV, RRID: SCR_011793) (47, 48), and variants were searched in an in-house database. The possibility of digenic causation was also considered, and Exomiser was run with a set of genes obtained from the DIDA database and the literature (49, 50).
If no molecular diagnosis was reached, the possibility of the patient not having an IEI was considered, and Exomiser was run with a larger set of genes made by merging the Pediatric Disorders, Severe Pediatric Disorders, and Pediatric Disorders Additional Genes panels of PanelApp. If still no likely diagnosis was identified, a further analysis was done with all variants in genes known to be associated with a disease.
In patients where a molecular diagnosis was unsuccessful, a search for new IEI candidate genes was attempted. For this purpose, a list of 3,938 genes was made, containing all the genes with ontology related to the immune system in the AmiGO2 database (51, 52), and all protein-coding genes interacting with known IEI genes, with a maximum distance of 2, according to the Human Reference Protein Interactome (RRID: SCR_01567) (53), the KEGG database (RRID: SCR_012773) (54), and the Human Genome Connectome (RRID: SCR_003490) (Supplementary Table 1). Variants in these genes were annotated and prioritized as described above, and then filtered and ranked based on criteria such as type and effect of the variant, gene function, signaling pathways and immune functions in which they take part, variant frequency in public databases, variant frequency in an in-house database, possible correlation with the phenotype of the patient, information on the role of the gene derived from laboratory models, and any published information related to the genes and their variants. For X-linked and autosomal recessive analyses, all variants and genotypes returned by the analyses were examined in detail; for the autosomal dominant analysis, the top 15 results were reviewed.
For the final stage, all variants in the exome were annotated and prioritized (Supplementary Table 2). All variants and genotypes were analyzed in detail for the X-linked and autosomal recessive modes of inheritance, and the top 25 variants and genes were analyzed when a monoallelic change was assumed.
All statistical tests were done with R (RRID: SCR_001905), version 4.3.1 (55).
Results
From June 2015 to May 2024, 157 pediatric patients, with a suspicion of an IEI, were studied by NGS (Supplementary Table 2). The cohort had an average age of 6 years 10 months, and a median age of 5 years. As a requirement for sequencing, the paperwork accompanying the biological samples included a list of symptoms, a short clinical summary, laboratory test results, and a list of likely clinical diagnoses or genes of interest. The type, amount, and detail of this information varied widely from patient to patient. Examination of this information led to determine that 78.34% of the patients (123 of the 157) met the criteria for suspecting an IEI, according to the Jeffrey Modell Foundation (JMF) (Supplementary Table 2). The most common feature was infectious diseases. In 33 of the remaining patients the information was very limited and, lastly, another patient did not have at least two of the JMF criteria.
Initially, the TruSightOne (TSO) panel was used to obtain the sequencing library of nine patients. However, given the pace at which new disease genes were being reported, this panel was rapidly becoming outdated, and did not allow the re-analysis of the data for newly added IEI genes. For this reason, for the rest of the patients WES was done. In 45, of the 157 patients, a molecular diagnosis for an IEI was reached, representing a diagnostic yield of 28.66%. For the rest of the patients, all genes associated with pediatric disorders were analyzed, and a non-IEI disease was determined in five of them. In another patient, with an atypical mycobacterial infection, examination of the sequencing data, laboratory tests, clinical information, and follow-up, led to suggest that the patient might not have an IEI. Including these six patients in the calculation rises the diagnostic yield to 32.48%. Among the patients with a molecular diagnosis, a female had two diseases, DCLRE1C (Artemis) deficiency, and ichthyosis vulgaris. Also, in a patient with a non-IEI diagnosis, a digenic Alport syndrome was found.
While females comprised 38.22% of sequenced patients, they amounted to only 23.53% of the patients with a molecular diagnosis. In male patients the figures were 61.78% and 76.47%, respectively (Figure 1). This difference in diagnostic yield between sexes is statistically significant (P-value = 0.01422, chi-square test). Ten of the molecular diagnoses involved hemizygous genotypes in genes on the X chromosome in male patients; even when removing these patients, the proportion of molecular diagnoses in females was 29.27%, which is below the 38.22% proportion of females in the cohort. In males, the percentages, excluding X-linked diseases, were 70.73% and 61.78%, respectively.
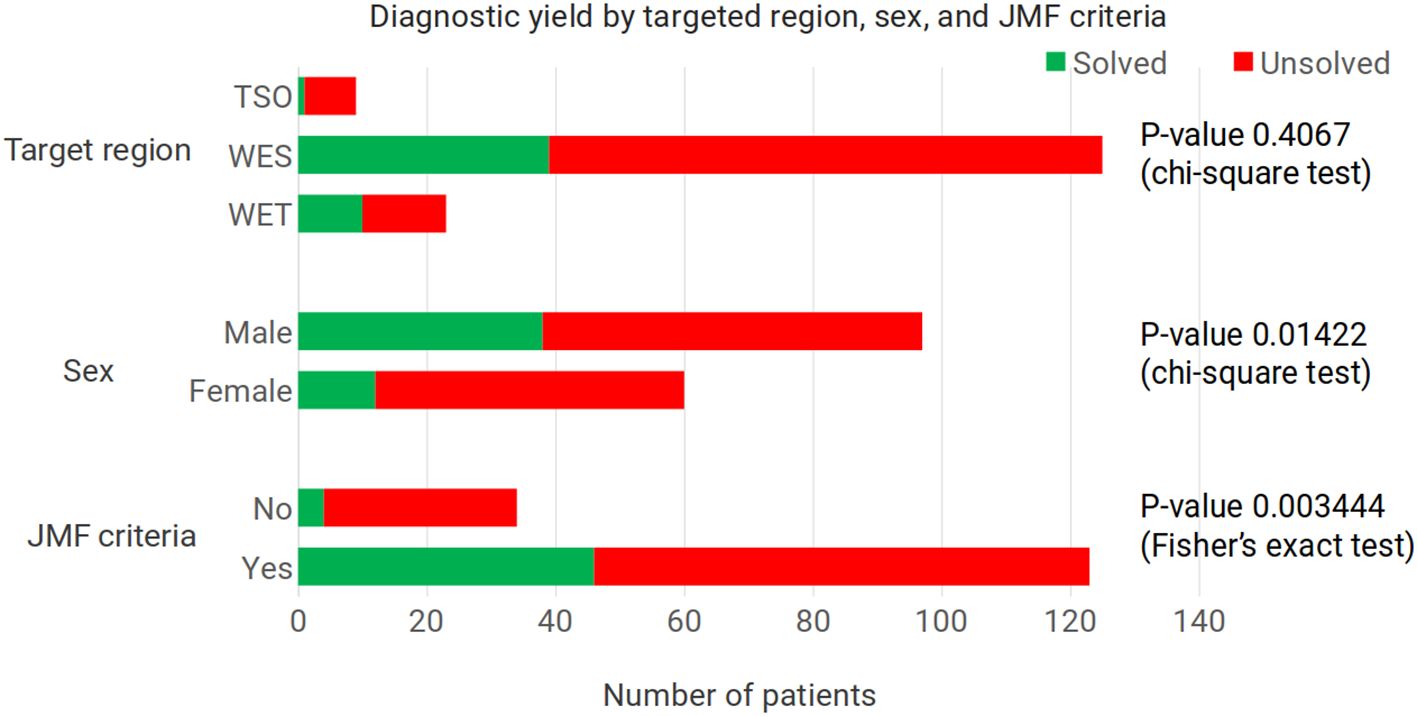
Figure 1. Diagnostic yield according to the enrichment kit used, the patients’ sex, and whether the patients met the criteria for an inborn error of immunity according to the Jeffrey Modell Foundation. For the target region, the P-value corresponds to the comparison of the two whole exome enrichment kits used. With all three enrichment kits, P-value = 0.2347, Fisher’s exact test. TSO – TruSightOne Targeted Regions panel v1.1, WES – Nextera Rapid Capture Exome Targeted Regions v1.2, WET – Twist ILMN Exome 2.0 or 2.5 Plus Panel, JMF - Jeffrey Modell Foundation.
With WES, the diagnostic yield was three times as high as with the TSO panel (33.78% versus 11.11%), although the number of patients sequenced with the latter panel was small (nine patients, versus 148 WES) (Figure 1). Separating the results for the two WES panels, with the Nextera Rapid Capture Exome Targeted Regions v1.2 the diagnostic yield was 32.00%, and with the Twist ILMN Exome 2.0 or 2.5 Plus Panel kits it was 43.48%. This difference was not statistically significant (P-value = 0.4067, chi-square test). We also checked whether the variants found using the Twist enrichment kit could have been found with the Illumina Nextera kit. Two of the variants found after using the Twist kit correspond to the homozygous loss of exons in genes where this defect is known to occur, NCF1 and RAB27A (56–61); the complete absence of reads would have been apparent also with the Illumina Nextera enrichment kit. The remaining eight variants found in patients sequenced with the Twist kit were single nucleotide variants. We examined the coverage of these eight positions in 500 exomes sequenced with the Illumina Nextera kit, and we found good coverage for all of them. The position with the lowest depth in these 500 exomes corresponded to KMT2D chr12:49049834, with a mean depth of 51.19X (st dev 31.61); while the position with the highest depth in these 500 Exomes was found in BTK chrX:100611120, with a mean depth of 165.76 (st dev 86.56).
Results of the patients meeting the criteria of the JMF were compared against the set of patients where the information was insufficient to determine whether they met the criteria. In the group of patients meeting the criteria, 34.96% had a molecular diagnosis of an IEI. This contrasts with only 5.88% of IEI molecular diagnoses among the patients not meeting the JMF criteria (P-value = 0.0034444, Fisher’s exact test) (Figure 1).
Following the IEI classification (6), 27 diseases were found in our cohort. Also, 27 different genes were associated with those diseases (Figure 2), although there is no biunivocal correspondence between the diseases and the genes (e.g., in STAT3, gain of function variants lead to STAT3 GOF, while loss of function variants are associated with AD-HIES STAT3 deficiency (Job syndrome); also, osteopetrosis appears as a single disease in the classification, but it may be caused by defects in any one of seven different genes).
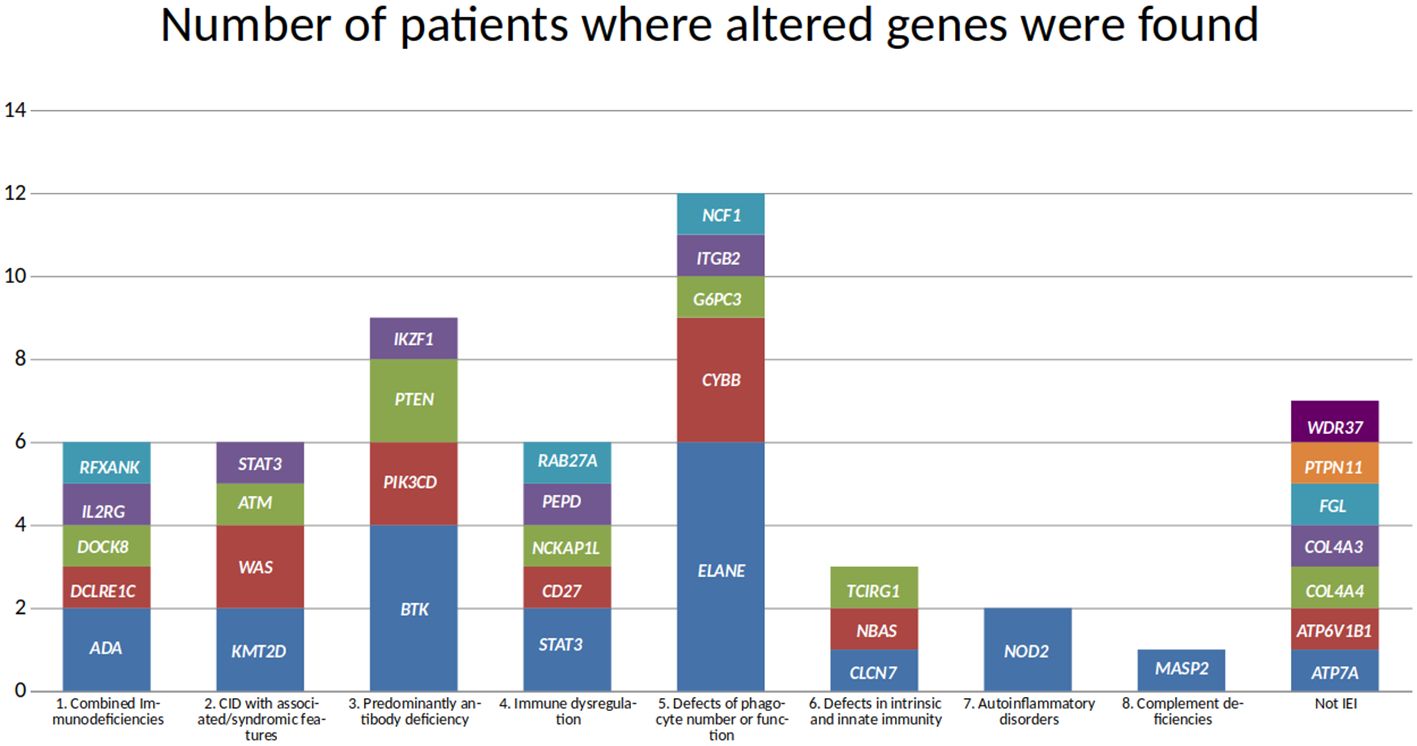
Figure 2. Genes found in the molecular diagnoses in the cohort. Genes are grouped according to the immunodeficiency disease group they are associated with, and the height of each block reflects the number of patients where the altered gene was found.
The most common mode of inheritance of the molecularly diagnosed IEI was autosomal dominant (40.00%), closely followed by autosomal recessive (37.78%). X-linked diseases accounted for 22.22% of the molecular diagnoses.
The disease and gene most frequently seen in this cohort is neutropenia, caused by alterations in ELANE, in six patients. This was followed by variants in the BTK gene, associated with X-linked agammaglobulinemia, in four patients. CYBB and STAT3 defects were each seen in three patients. However, only two of the patients with CYBB variants had X-linked chronic granulomatous disease associated with a missense variant. The third patient had a large deletion spanning approximately 1.8 Mbp, leading to the complete loss of many genes, including CYBB and XK, and a partial loss of CFAP47 and SYTL5. Variants in XK have been associated with McLeod syndrome (OMIM 300842), while variants in CFAP47 may be associated with Spermatogenic failure, X-linked 3 (OMIM 301059). Regarding the three patients with variants in STAT3, in two of them the phenotype corresponded to a gain of function, while in the third it was a loss of function. Even though several patients had variants in ELANE, BTK, CYBB, and STAT3, no two patients shared the same variant. Among the 27 IEI genes, only one variant was observed in more than one patient; a missense variant in PIK3CD.
Missense variants were the most frequent type of change (27 variants, including the PIK3CD variant observed twice), followed by eight stop gain variants. Copy number variants (CNV) were identified in three patients; one of them corresponding to the large hemizygous deletion affecting CYBB and several other genes, mentioned above. Another CNV involved a homozygous deletion in NCF1, and the third CNV comprised the homozygous loss of four exons of RAB27A.
Even though no two patients shared the same variant, apart from the PIK3CD missense variant, homozygous genotypes accounted for the great majority (70.59%) of the genotypes in autosomal recessive diseases (12 out of 17 patients).
Using the clinical and laboratory information attached to the samples, plus the clinically suspected disease, or the genes suspected as relevant, samples were assigned to one of the ten IEI groups recognized by the IUIS (6) (Supplementary Table 2). This is referred to as the “pre-NGS classification”. An extra group (labelled “undefined”) was added to accommodate samples with insufficient information to ascribe them to one of the IUIS groups. In some cases, the information associated with a sample pointed to, or was consistent with, more than one of the IUIS groups; these samples were also placed in the “undefined” group. This group was the largest, with 38.85% of the samples, followed by groups 2 (combined immunodeficiencies with associated or syndromic features; 14.01% of the samples), group 5 (congenital defects affecting phagocytes; 12.10% of the patients), and group 4 (immune dysregulation; 10.19%).
With the results of the WES study, patients were again classified in the IUIS groups; this is the “post-NGS classification” (Figure 3; Supplementary Tables 2, 3). Two additional groups were considered: not-IEI, and “unresolved” cases. The largest group was formed by these unresolved cases (67.52%), followed by group 5 (congenital defects affecting phagocytes; 7.64% of the patients), and group 3 (antibody deficiencies, 5.73%). Some patients that were initially in the “undefined” group could be assigned to IUIS groups after NGS. However, most of the patients in the pre-NGS “undefined” group, ended in the post-NGS “unresolved” group. Patients in every group failed to have a molecular diagnosis after sequencing, and were assigned to the “unresolved” group. In some cases, such as in groups 1 to 4, most patients originally assigned to those groups failed to receive a molecular diagnosis.
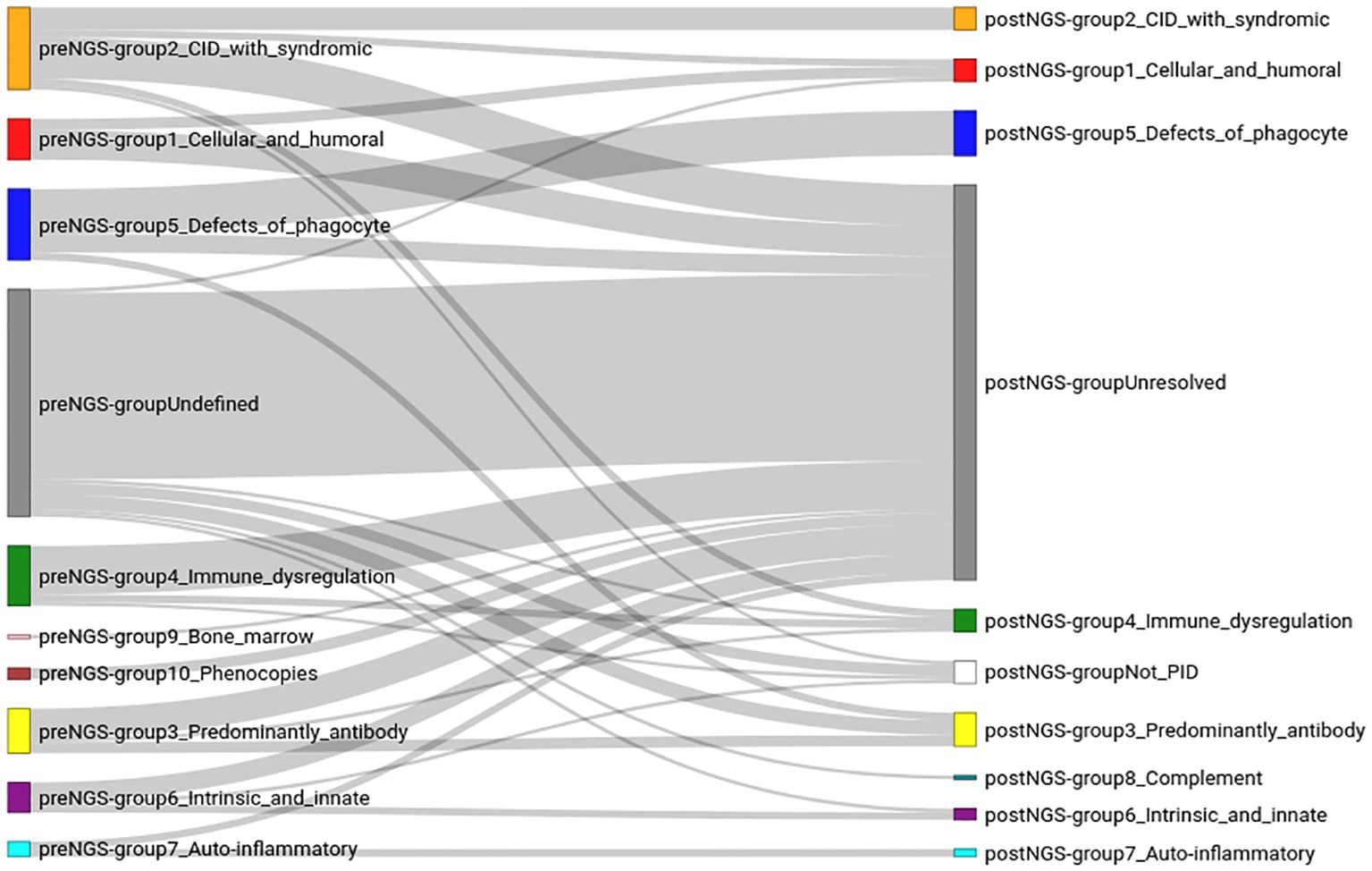
Figure 3. Classification of the patients in the cohort, before (preNGS) and after (postNGS) sequencing. The assignment of the immunodeficiency group before sequencing (left of the figure) was done based on the information provided by the clinicians (the clinical summary, results of some laboratory tests, and the clinician’s assessment of the likely disease of the patient). The right side of the figure corresponds to the immunodeficiency group after sequencing. The lines connecting the groups before and after sequencing reflect how the sequencing data changed the diagnosis of each patient.
The largest IUIS group, both pre-NGS and post-NGS, which also showed the highest concordance (63.16%) in group assignment, before and after sequencing, was group 5 (congenital defects affecting phagocytes), followed by group 7 (autoinflammatory disorders), although only four patients were initially thought to have a disease belonging to this group. Groups 2, 3 and 1 had a high pre-NGS number of patients (22, 12, and 11, respectively), and the concordance after sequencing was similar for all three of them (27.27%, 25.00%, and 27.27%, respectively). Group 4 also had an initial high number of patients, but the concordance after sequencing was lower (12.5%). No patients were initially thought to have a complement deficiency disease (group 8) but, after sequencing, one such patient was identified, who had initially been assigned to the “undefined” group.
Table 1 summarizes the consequences of NGS in the diagnosis of IEI diseases. More than half of the patients who were initially assigned to an IEI group did not receive a molecular diagnosis. However, in slightly under a third of the patients the initial classification was confirmed by sequencing, and in ten percent of the patients the initial classification proved to be wrong, and a molecular diagnosis for a different IUIS group was determined. The table also shows that most (81.97%) of the patients who could not be assigned to an IEI group based on the information accompanying the sample, remained without diagnosis after being sequenced. Nevertheless, in 8% of the “undefined” patients sequencing proved useful in finding the correct disease.
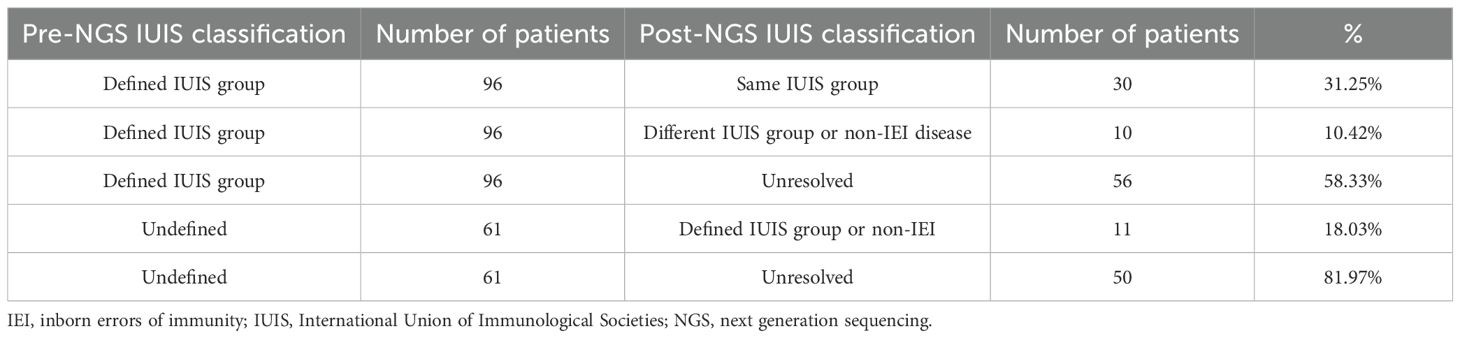
Table 1. Contribution of next generation sequencing to the diagnosis of pediatric patients. IUIS classification of patients before and after sequencing.
As shown in Table 1, 40 patients had an assigned IEI group both pre-NGS and post-NGS; 50 patients were undefined/unresolved pre-NGS and post-NGS; 56 patients were assigned a defined IEI group pre-NGS but were “unresolved” post-NGS; and 11 patients had an undefined IEI pre-NGS, but could be assigned to an IUIS group post-NGS. These changes in patient classification after NGS are statistically significant (P-value = 7.639x10-8, McNemar test).
To search for new IEI candidate genes among the patients where no molecular diagnosis had been found, sixty patients were selected, based on the quality of their clinical and laboratory information. Variants in a curated list of 3,938 candidate genes were annotated, and then prioritized based on the HPO terms of the patients, and the information available for each gene. No strong new IEI candidate genes emerged. The reasons for this were varied. For example, some variants had a higher-than-expected frequency in public databases, or in our in-house database; for other variants, algorithms predicting the effect of the variant on the protein considered them as benign or likely benign, or the variants were in genes with functional information inconsistent with the phenotype of the patient. As a final step, variants were annotated, prioritized, and examined across the whole exome, and not only in the list of curated candidate genes. No strong new candidate genes were found after considering their frequency, the predicted consequence on the protein, and the known role of the gene.
Discussion
We hereby present the results of the first large cohort of pediatric patients, in Mexico, with a suspected IEI, to be studied by next generation sequencing. In all 157 patients, except nine, the whole exome was sequenced. The diagnostic yield, just under 30%, was similar to those reported for other cohorts with heterogeneous IEI (9, 13, 19). The cohort comprised 1.6 times as many male as female patients. However, the proportion among those where a molecular diagnosis was determined was 3.25 diagnosed males for each diagnosed female. The source of this wide gap is unclear, since it cannot be accounted for by X-linked diagnoses. Even excluding all patients with diseases having this mode of inheritance, which were all hemizygous males, there were 2.42 diagnosed males per diagnosed female.
Even though the diagnostic yield in IEI is relatively low, WES is clearly valuable. Almost one in five of the patients that lacked a defined immune disease when they were submitted for sequencing, received a molecular diagnosis, while one in ten of the patients with an IEI of a specific group before sequencing, received a molecular diagnosis of a disease belonging to a different group after sequencing. In addition to this, slightly under a third of the patients that were submitted with a suggested immune disease, had a disease belonging to the same group, according to the IUIS classification, once the molecular diagnosis was determined.
From the clinical information accompanying the samples that were received for sequencing, slightly more than two thirds of the patients qualified as having an IEI according to the criteria of the JMF. The molecular diagnosis was found in approximately one in three of these patients, compared to only one in twenty in the group of patients where the JMF criteria were not met. The reason for this difference is unclear. It does not necessarily mean that the latter group of patients lacked an IEI, given that for almost all of them, failure to meet the criteria for an IEI, according to the JMF, was due to a lack of available information, rather than the phenotype being inconsistent with an IEI. Had this last possibility been the case, it would have been expected to find more non-IEI diagnoses when analyzing variants across the whole exome. This was not the case and, in fact, more non-immune diseases were found among the patients meeting the JMF criteria, than among those that did not.
Twenty-seven different IEI were found, and the same number of different genes. The most common disease was neutropenia due to monoallelic variants in ELANE, in six patients, followed by X-linked agammaglobulinemia in four patients, and X-linked chronic granulomatous disease in three. Even though some patients had the same disease, only two patients in the cohort shared the same variant, a missense change in PIK3CD. This underscores the rarity of the variants associated with these diseases in our population. However, in contrast to this, homozygous genotypes were observed in most of the patients with an autosomal recessive disease; most of these children came from small, and relatively isolated, rural communities.
Missense variants accounted for the vast majority of the genetic defects, while only three CNVs were found. It is possible that these latter variants are underrepresented, given the difficulty of reliably identifying them with exome sequencing (62). It was possible to find the three CNVs because they were homozygous deletions in two of the patients (involving NCF1 and RAB27A), while the third, affecting CYBB, was hemizygous. Deletions affecting CYBB and NCF1 are well known, even if the breakpoints differ between affected patients (56, 58, 63–65).
In our patients with neutropenia associated with variants in ELANE, all variants, except one, occurred in exons four and five. This is consistent with the findings reported in the literature (66). It is known that there is some genotype-phenotype correlation, with some variants found in severe congenital neutropenia, and others in cyclic neutropenia, although a few variants have been found in both diseases (67). Among the six variants we identified, Gly214Arg has been observed repeatedly in patients with neutropenia. This variant is in a mutation hotspot and is associated with poor prognosis (66–68). Patients with this variant are at increased risk of presenting myelodysplastic syndrome and acute lymphoblastic leukemia. The Gly214 residue is evolutionary conserved, and the change affects the conformation and stability of the polypeptide chain (69). All possible changes in this position (c.640G>A, c.640G>C, c.640G>T) have been reported in patients with neutropenia (70). The Gly214Arg change has been the subject of functional studies, and it is known that the misfolded protein triggers the unfolded protein response, and results in apoptosis (71–73). The possibility of using inhibitors of this misfolded protein has been studied in vitro (74). In our cohort, the patient with this variant received the molecular diagnosis when she was 4 months old. She has seen again a few months later, but she did not come back to the hospital so it is unknown how her disease developed. She would be almost seven years old today. The rest of the ELANE variants in our cohort have not been the object of detailed studies. Two of them, Leu227SerfsTer13 and Tyr228Ter (70), are towards the C-terminus of the protein, and it is predicted that they would escape the nonsense-mediated decay process (75, 76). We found two ELANE inframe deletions, one in exon two leading to the predicted loss of six aminoacid residues, and the other in exon four, predicted to result in the loss of two residues. Another variant was a missense change in exon 5 (Gly210Trp) (70); this variant, as well as the former four mentioned above, has not been studied in detail. In our group of neutropenia patients associated with ELANE alterations, the most frequent clinical manifestation was infections, and all patients had at least one elevated Ig class. In all of them, the first symptoms appeared during the first months of life and they comprised recurrent pneumonia, otitis media, and neonatal sepsis. The most common infections were associated with Pseudomonas aeruginosa and Escherichia coli. No differences of note were observed between the six patients. From the very beginning, based on the clinical examination and laboratory tests, a clinical diagnosis of neutropenia was suggested, and this diagnosis was confirmed by sequencing.
Four patients in our cohort had BTK variants. Three of them were stop codon gains, and the fourth was a missense variant. One of the stop codons was in the TH domain of the protein and the affected patient showed a more acute clinical presentation of the disease than the other three cases. This patient had very severe infections and depletion of all lymphocyte subpopulations. Initially, he also had hemophagocytic lymphohistiocytosis. Due to the severity of the clinical signs, chronic granulomatous disease and severe combined immunodeficiency were suspected. The rest of patients with BTK alterations had variants in the kinase domain of the protein. All three of them had typical symptoms of the disease, including agammaglobulinemia, and severe infections as the initial symptoms.
The three patients with CYBB variants, and a diagnosis of chronic granulomatous disease, shared the same features, with a severe presentation, BCGitis, recurrent pneumonia, sinopulmonary infections, and adenitis. Two of the patients had stop gain variants, in codons 157 and 226. The third patient had a large deletion, leading to the complete absence of CYBB sequences, in addition to the loss of at least 30 other genes, including 11 protein coding genes. One breakpoint was located between exons 35 and 36 of CFAP47, and the other between exons 15 and 16 of SYTL5. Only a few of the genes in this region have been associated with a disease. Deletions in this region, leading to the complete loss of CYBB and other genes, are known (77). The extent of the deletion may differ from one patient to another and the phenotype of the patient depends on which genes are lost. This may cause patients to have more than one disease (63, 64, 78). However, in our patient, even though many genes besides CYBB were missing, his disease showed no differences when compared to our other two patients, who had single nucleotide variants causing a stop codon gain. No other phenotypes or diseases, stemming from the loss of genes besides CYBB, were observed. Nevertheless, it is still possible that additional phenotypes may appear at a later age. For example, in this patient the first exons of CFAP47 are missing, and alterations in this gene have been associated with azoospermia (79). Also, alterations, most frequently deletions, in the XK gene have been associated with McLeod syndrome, including late onset myopathy and neuropathy (80–83). In some patients with McLeod syndrome due to large deletions, VPS13B and DMD may be lost. However, these two genes were present in our patient.
Looking at the frequency of the different types of IEI in this cohort, both before and after sequencing, points to a predominance of patients with diseases belonging to group 5 (congenital defects of phagocyte number or function), group 3 (predominantly antibody deficiencies), and group 2 (CIDs with associated or syndromic features). However, this distribution does not necessarily reflect the frequency of the different types of IEI in our country, or even in our hospital. In our institution, there is some bias in the patients submitted for sequencing. Clinicians tend to send patients where they struggle to identify the immune disease; they also send patients requiring a precise molecular diagnosis to proceed with the correct treatment, or very ill inpatients where a precise diagnosis is required as quickly as possible to decide on the best course of action. In the cohort we report, one in ten children had an IEI of a different group than initially thought, while close to 40% had an “undefined” immune disease before sequencing. These figures point to the difficulty of correctly identifying these diseases where symptoms are non-specific and overlap between diseases. In addition to this, in this hospital, there is a very limited number of laboratory tests and studies available to perform on these patients. These factors are an impediment to a precise and correct diagnosis. In this regard, NGS has become an indispensable tool. On the other hand, the proposition that patients, in our hospital, for which no sequencing is sought, have a correct diagnosis, is something that needs to be tested, by sequencing them, and by assessing the concordance between the diagnoses suggested by the clinicians relying on the limited set of laboratory tests available at the institution, and the molecular diagnoses after sequencing (pre-NGS versus post-NGS comparison).
Among the patients where no molecular diagnoses were found, new IEI candidate genes were searched. However, no candidates were identified, first by analyzing variants and genes in a curated set of immune related genes, and then by studying the variants across the whole exome. This negative result is not entirely unexpected. In WES, many factors may be responsible for negative results, without necessarily implying that new genes are involved. CNVs, segmental duplications, structural variants, intronic alterations and intergenic variants, are missed when sequencing exonic coding regions in patients with immune diseases (62, 65, 84–86). In addition to this, GC-rich regions, often found in gene regulatory regions, are difficult to sequence, and sequence drop out (regions with zero coverage) may occur. CNVs have been reported in a number of IEI (62). In our cohort, seven patients suspected of having neutropenia remain unresolved. This disease may sometimes be the result of copy number variants, due to deletions, in ELANE (66, 70, 87), so this possibility should be explored, as well as the possibility of variants in non-coding regions, leading to altered transcripts and proteins (88). It has also been found than some patients with neutropenia have somatic variants in ELANE, as opposed to germline alterations (87). Our cohort also includes a patient with Wiskott-Aldrich where no variants were found after carefully scanning the reads covering all coding exons. In this patient, sequencing of the gene’s regulatory region, analysis of the gene’s transcript, by qRT-PCR, or protein studies by Western blot, may allow to determine if there is a variant affecting not the protein sequence, but the expression of the gene.
Even though the literature mentions that the PIK3CD pathogenic variant E102K may be missed due to a duplicated sequence (84), we had no difficulty finding this variant in our cohort. It was, in fact, the only variant that we found in more than one patient.
Our cohort also includes eight patients where hypereosinophilic syndrome was suspected. One of them was found to have a STAT3 gain of function variant. In the remaining seven, no alterations could be identified. It is known that hypereosinophilic syndrome is associated with structural variants, including chromosome translocations, leading to gene fusions, with FIP1L1 and PDGFRA being the genes most frequently implicated (89, 90). In addition to this, somatic variants in JAK1 and STAT5B have been reported. In these patients, the molecular alterations could have been missed because exome sequencing is unable to find structural variants, unless the breakpoints occur within the coding exons, and they could also have been missed if the disease was caused by somatic alterations.
Among the patients with no molecular diagnosis in our cohort, there are nine where CVID is suspected. The diagnostic yield is low in patients with these diseases, and it has been suggested that it might be necessary to consider non-monogenic genetic architectures, such as digenic, oligogenic, the participation of several common variants affecting specific pathways, or even genetic models corresponding to complex diseases where an external triggering factor plays a role (3, 85, 86). Additionally, epigenetic modifications may also be implicated in IEI, although this area appears to have received scant attention (91). These studies, and the fact that with NGS the diagnostic yield in IEI is lower than for other monogenic diseases, suggest that there is indeed a difference in the assumed genetic architecture of immune diseases (92, 93).
In our hospital, WES is the technique of choice to determine the correct disease in patients with IEI, and to identify the precise variant underlying the disease. We have shown here that this approach leads to the molecular diagnosis of patients where the clinical history, the clinical examination, and laboratory tests are insufficient to suggest a specific IEI disease. We have also shown that patients where a clinical diagnosis has been suggested may, in fact, have a different disease to the one initially considered. Even though WES has proven its usefulness, it has limitations and pathogenic variants may be missed. Relying on WGS, instead of exome sequencing, would likely lead to a higher diagnostic yield, mainly through the detection of CNVs and structural variants. While variants in non-coding regions can also be found by WGS, the difficulty in interpreting these variants means they do not in fact contribute very much to increasing the diagnostic yield. This is true even though bioinformatic tools, such as REMM (RRID: SCR_023095) (94) and CADD (RRID: SCR_018393) (95), have been developed to aid in the interpretation of non-coding variants. For this type of variants, transcriptome sequencing may be preferrable (96). Transcriptome sequencing has the added advantage of providing functional information, on top of the sequencing data itself. Up to now, we have limited ourselves to WES mainly due to cost constraints. However, in order to increase the diagnostic yield in patients with IEI, we are expecting to implement a stratified approach, where WES would be the first technique to use, followed by WGS in patients where no molecular diagnosis is found. This would make it possible to increase the diagnostic yield by 5-10% by enabling the detection of CNVs and structural variants (62). WGS, if performed with long read sequencing, would also enable phasing, and solve segmental duplications that might be difficult to resolve correctly with short read sequencing. In patients where no molecular cause is found after WGS, we would use transcriptome sequencing to look for variants in non-coding regions affecting splicing. Combining transcriptome and WGS data should also aid in the interpretation of non-coding variants affecting gene expression (97).
We conclude that, even though NGS has proven its usefulness in the diagnosis of IEI, more needs to be done if we are to increase the currently low diagnostic yield, and this means broadening the technological and bioinformatics tools in use, and making more progress in understanding the genetic architecture of these diseases.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found in ClinVar submission SUB15651872 (https://www.ncbi.nlm.nih.gov/clinvar/?term=SUB15651872), accession numbers SCV006554572 to SCV006554623.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Hospital Infantil de Mexico. The studies were conducted in accordance with the national legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
GG-Z: Investigation, Writing – original draft, Methodology, Writing – review & editing, Conceptualization, Formal analysis. PB-C: Project administration, Methodology, Writing – review & editing, Supervision. OS-R: Resources, Validation, Writing – review & editing. BD-R-N: Writing – review & editing, Resources. SE-P: Supervision, Validation, Writing – review & editing. VM-B: Supervision, Validation, Writing – review & editing. JA-H: Supervision, Conceptualization, Writing – review & editing, Software, Investigation, Funding acquisition, Project administration, Visualization, Data curation.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by grants HIM/2015/072 and HIM/2017/103 awarded by Hospital Infantil de México Federico Gómez to JA-H. GG-Z was supported by scholarship CVU 850637 awarded by Consejo Nacional de Humanidades, Ciencia y Tecnología, (CONAHCyT).
Acknowledgments
We are grateful to María de los Ángeles Rangel-Téllez and Pablo Miguel Valencia-Segura for their administrative, logistical, and technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1638544/full#supplementary-material
References
1. Rezaei N, Aghamohammadi A, and Notarangelo L. Primary immunodeficiency diseases: Definition, Diagnosis and Management. 2nd edition. New York, NY: Springer Berlin Heidelberg (2017).
2. Walkovich K and Connelly JA. Primary immunodeficiency in the neonate: Early diagnosis and management. Semin Fetal Neonatal Med. (2016) 21:35–43. doi: 10.1016/j.siny.2015.12.005
3. Thaventhiran JED, Lango-Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. (2020) 583:90–5. doi: 10.1038/s41586-020-2265-1
4. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
5. Raje N and Dinakar C. Overview of immunodeficiency disorders. Immunol Allergy Clin North Am. (2015) 35:599–623. doi: 10.1016/j.iac.2015.07.001
6. Poli MC, Aksentijevich I, Bousfiha AA, Cunningham-Rundles C, Hambleton S, Klein C, et al. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Hum Immun. (2025) 1(1):e20250003. doi: 10.70962/jhi.20250003
7. Notarangelo LD, Bacchetta R, Casanova JL, and Su HC. Human inborn errors of immunity: An expanding universe. Sci Immunol. (2020) 5(49):eabb1662. doi: 10.1126/sciimmunol.abb1662
8. Casanova JL, Conley ME, Seligman SJ, Abel L, and Notarangelo LD. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. (2014) 211:2137–49. doi: 10.1084/jem.20140520
9. Yska HAF, Elsink K, Kuijpers TW, Frederix GWJ, van Gijn ME, and van Montfrans JM. Diagnostic yield of next generation sequencing in genetically undiagnosed patients with primary immunodeficiencies: a systematic review. J Clin Immunol. (2019) 39:577–91. doi: 10.1007/s10875-019-00656-x
10. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2022) 42:1473–507. doi: 10.1007/s10875-022-01289-3
11. Arts P, Simons A, AlZahrani MS, Yilmaz E, AlIdrissi E, van Aerde KJ, et al. Exome sequencing in routine diagnostics: a generic test for 254 patients with primary immunodeficiencies. Genome Med. (2019) 11:38. doi: 10.1186/s13073-019-0649-3
12. Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. (2018) 141:1450–8. doi: 10.1016/j.jaci.2017.06.049
13. Vorsteveld EE, Hoischen A, and van der Made CI. Next-generation sequencing in the field of primary immunodeficiencies: current yield, challenges, and future perspectives. Clin Rev Allergy Immunol. (2021) 61:212–25. doi: 10.1007/s12016-021-08838-5
14. Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. (2017) 139:232–45. doi: 10.1016/j.jaci.2016.05.042
15. Suspitsin EN, Guseva MN, Kostik MM, Sokolenko AP, Skripchenko NV, Levina AS, et al. Next generation sequencing analysis of consecutive Russian patients with clinical suspicion of inborn errors of immunity. Clin Genet. (2020) 98:231–9. doi: 10.1111/cge.13789
16. El Hawary RE, Meshaal SS, Abd Elaziz DS, Alkady R, Lotfy S, Eldash A, et al. Genetic testing in Egyptian patients with inborn errors of immunity: a single-center experience. J Clin Immunol. (2022) 42:1051–70. doi: 10.1007/s10875-022-01272-y
17. Belaid B, Lamara Mahammed L, Drali O, Oussaid AM, Touri NS, Melzi S, et al. Inborn errors of immunity in Algerian children and adults: A single-center experience over a period of 13 years (2008-2021). Front Immunol. (2022) 13:900091. doi: 10.3389/fimmu.2022.900091
18. Moundir A, Ouair H, Benhsaien I, Jeddane L, Rada N, Amenzoui N, et al. Genetic diagnosis of inborn errors of immunity in an emerging country: a retrospective study of 216 moroccan patients. J Clin Immunol. (2023) 43:485–94. doi: 10.1007/s10875-022-01398-z
19. Vorsteveld EE, van der Made CI, Smeekens SP, Schuurs-Hoeijmakers JH, Astuti G, Diepstra H, et al. Clinical exome sequencing data from patients with inborn errors of immunity: Cohort level diagnostic yield and the benefit of systematic reanalysis. Clin Immunol. (2024) 268:110375. doi: 10.1016/j.clim.2024.110375
20. Stoddard JL, Niemela JE, Fleisher TA, and Rosenzweig SD. Targeted NGS: A cost-effective approach to molecular diagnosis of PIDs. Front Immunol. (2014) 5:531. doi: 10.3389/fimmu.2014.00531
21. Al-Mousa H, Abouelhoda M, Monies DM, Al-Tassan N, Al-Ghonaium A, Al-Saud B, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. (2016) 137:1780–7. doi: 10.1016/j.jaci.2015.12.1310
22. Kojima D, Wang X, Muramatsu H, Okuno Y, Nishio N, Hama A, et al. Application of extensively targeted next-generation sequencing for the diagnosis of primary immunodeficiencies. J Allergy Clin Immunol. (2016) 138:303–5 e3. doi: 10.1016/j.jaci.2016.01.012
23. Ye X, Maglione PJ, Wehr C, Li X, Wang Y, Abolhassani H, et al. Genomic characterization of lymphomas in patients with inborn errors of immunity. Blood Adv. (2022) 6:5403–14. doi: 10.1182/bloodadvances.2021006654
24. Morup SB, Nazaryan-Petersen L, Gabrielaite M, Reekie J, Marquart HV, Hartling HJ, et al. Added value of reanalysis of whole exome- and whole genome sequencing data from patients suspected of primary immune deficiency using an extended gene panel and structural variation calling. Front Immunol. (2022) 13:906328. doi: 10.3389/fimmu.2022.906328
25. Okano T, Imai K, Naruto T, Okada S, Yamashita M, Yeh TW, et al. Whole-exome sequencing-based approach for germline mutations in patients with inborn errors of immunity. J Clin Immunol. (2020) 40:729–40. doi: 10.1007/s10875-020-00798-3
26. Venkatachari IV, Chougule A, Gowri V, Taur P, Bodhanwala M, Prabhu S, et al. Monogenic inborn errors of immunity in autoimmune disorders. Immunol Res. (2023) 71:771–80. doi: 10.1007/s12026-023-09391-3
27. Al-Mousa H and Barbouche MR. Genetics of Inborn Errors of Immunity in highly consanguineous Middle Eastern and North African populations. Semin Immunol. (2023) 67:101763. doi: 10.1016/j.smim.2023.101763
28. Buchbinder D, Seppanen M, Rao VK, Uzel G, and Nugent D. Clinical challenges: identification of patients with novel primary immunodeficiency syndromes. J Pediatr Hematol/Oncol. (2018) 40:e319–e22. doi: 10.1097/MPH.0000000000001003
29. IJspeert H, Edwards ESJ, O’Hehir RE, Dalm V, and van Zelm MC. Update on inborn errors of immunity. J Allergy Clin Immunol. (2025) 155:740–51. doi: 10.1016/j.jaci.2024.12.1075
30. Takeda Y, Ueki M, Matsuhiro J, Walinda E, Tanaka T, Yamada M, et al. A de novo dominant-negative variant is associated with OTULIN-related autoinflammatory syndrome. J Exp Med. (2024) 221(6):e20231941. doi: 10.1084/jem.20231941
31. Davidson S, Shibata Y, Collard S, Zheng H, Kong K, Sun JM, et al. Dominant negative OTULIN-related autoinflammatory syndrome. J Exp Med. (2024) 221(6):e20222171. doi: 10.1084/jem.20222171
32. Ogishi M, Yang R, Aytekin C, Langlais D, Bourgey M, Khan T, et al. Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat Med. (2021) 27:1646–54. doi: 10.1038/s41591-021-01388-5
33. Nascimento A, Bruels CC, Donkervoort S, Foley AR, Codina A, Milisenda JC, et al. Variants in DTNA cause a mild, dominantly inherited muscular dystrophy. Acta Neuropathol. (2023) 145:479–96. doi: 10.1007/s00401-023-02551-7
34. Materna M, Delmonte OM, Bosticardo M, Momenilandi M, Conrey PE, Charmeteau-De Muylder B, et al. The immunopathological landscape of human pre-TCRalpha deficiency: From rare to common variants. Science. (2024) 383:eadh4059. doi: 10.1126/science.adh4059
35. Momenilandi M, Levy R, Sobrino S, Li J, Lagresle-Peyrou C, Esmaeilzadeh H, et al. FLT3L governs the development of partially overlapping hematopoietic lineages in humans and mice. Cell. (2024) 187:2817–37 e31. doi: 10.1016/j.cell.2024.04.009
36. Itan Y and Casanova JL. Novel primary immunodeficiency candidate genes predicted by the human gene connectome. Front Immunol. (2015) 6:142. doi: 10.3389/fimmu.2015.00142
37. Yaldiz B, Kucuk E, Hampstead J, Hofste T, Pfundt R, Corominas Galbany J, et al. Twist exome capture allows for lower average sequence coverage in clinical exome sequencing. Hum Genomics. (2023) 17:39. doi: 10.1186/s40246-023-00485-5
38. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. (2021) 10:giab008. doi: 10.1093/gigascience/giab008
39. Robinson PN, Kohler S, Oellrich A, Wang K, Mungall CJ, Lewis SE, et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. (2014) 24:340–8. doi: 10.1101/gr.160325.113
40. Kohler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, Gourdine JP, et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. (2019) 47:D1018–D27. doi: 10.1093/nar/gky1105
41. Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J Clin Immunol. (2015) 35:696–726. doi: 10.1007/s10875-015-0201-1
42. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
43. Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. (2019) 51:1560–5. doi: 10.1038/s41588-019-0528-2
44. Stark Z, Foulger RE, Williams E, Thompson BA, Patel C, Lunke S, et al. Scaling national and international improvement in virtual gene panel curation via a collaborative approach to discordance resolution. Am J Hum Genet. (2021) 108:1551–7. doi: 10.1016/j.ajhg.2021.06.020
45. Amberger JS, Bocchini CA, Scott AF, and Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. (2019) 47:D1038–D43. doi: 10.1093/nar/gky1151
46. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
47. Thorvaldsdottir H, Robinson JT, and Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings Bioinf. (2013) 14:178–92. doi: 10.1093/bib/bbs017
48. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. (2011) 29:24–6. doi: 10.1038/nbt.1754
49. Gazzo AM, Daneels D, Cilia E, Bonduelle M, Abramowicz M, Van Dooren S, et al. DIDA: A curated and annotated digenic diseases database. Nucleic Acids Res. (2016) 44:D900–7. doi: 10.1093/nar/gkv1068
50. Sharfe N, Karanxha A, Dadi H, Merico D, Chitayat D, Herbrick JA, et al. Dual loss of p110delta PI3-kinase and SKAP (KNSTRN) expression leads to combined immunodeficiency and multisystem syndromic features. J Allergy Clin Immunol. (2018) 142:618–29. doi: 10.1016/j.jaci.2017.10.033
51. Gene Ontology C, Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, et al. The gene ontology knowledgebase in 2023. Genetics. (2023) 224(1):iyad031. doi: 10.1093/genetics/iyad031
52. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontol Consort Nat Genet. (2000) 25:25–9. doi: 10.1038/75556
53. Luck K, Kim DK, Lambourne L, Spirohn K, Begg BE, Bian W, et al. A reference map of the human binary protein interactome. Nature. (2020) 580:402–8. doi: 10.1038/s41586-020-2188-x
54. Kanehisa M, Furumichi M, Sato Y, Matsuura Y, and Ishiguro-Watanabe M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. (2025) 53:D672–D7. doi: 10.1093/nar/gkae909
55. R Core Team. R: A language and environment for statistical computing. Computing R, editor. Vienna, Austria: R Foundation for Statistical Computing. Vienna, Austria (2023). Available online at: http://www.R-project.org, ISBN: ISBN 3-900051-07-0 (Accessed July 21, 2025).
56. Hayrapetyan A, Dencher PC, van Leeuwen K, de Boer M, and Roos D. Different unequal cross-over events between NCF1 and its pseudogenes in autosomal p47(phox)-deficient chronic granulomatous disease. Biochim Biophys Acta. (2013) 1832:1662–72. doi: 10.1016/j.bbadis.2013.05.001
57. Chanock SJ, Roesler J, Zhan S, Hopkins P, Lee P, Barrett DT, et al. Genomic structure of the human p47-phox (NCF1) gene. Blood Cells Mol Dis. (2000) 26:37–46. doi: 10.1006/bcmd.2000.0274
58. Kuhns DB, Hsu AP, Sun D, Lau K, Fink D, Griffith P, et al. NCF1 (p47(phox))-deficient chronic granulomatous disease: comprehensive genetic and flow cytometric analysis. Blood Adv. (2019) 3:136–47. doi: 10.1182/bloodadvances.2018023184
59. Cay E, Sezer A, Karakulak V, Serbes M, Ozcan D, Bisgin A, et al. Hemophagocytic lymphohistiocytosis in children with Griscelli syndrome type 2: genetics, laboratory findings and treatment. Am J Clin Exp Immunol. (2023) 12:140–52.
60. Mamishi S, Modarressi MH, Pourakbari B, Tamizifar B, Mahjoub F, Fahimzad A, et al. Analysis of RAB27A gene in griscelli syndrome type 2: novel mutations including a deletion hotspot. J Clin Immunol. (2008) 28:384–9. doi: 10.1007/s10875-008-9192-5
61. Vincent LM, Gilbert F, DiPace JI, Ciccone C, Markello TC, Jeong A, et al. Novel 47.5-kb deletion in RAB27A results in severe Griscelli Syndrome Type 2. Mol Genet Metab. (2010) 101:62–5. doi: 10.1016/j.ymgme.2010.05.015
62. Wan R, Schieck M, Caballero-Oteyza A, Hofmann W, Cochino AV, Shcherbina A, et al. Copy number analysis in a large cohort suggestive of inborn errors of immunity indicates a wide spectrum of relevant chromosomal losses and gains. J Clin Immunol. (2022) 42:1083–92. doi: 10.1007/s10875-022-01276-8
63. Francke U, Ochs HD, de Martinville B, Giacalone J, Lindgren V, Disteche C, et al. Minor Xp21 chromosome deletion in a male associated with expression of Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosa, and McLeod syndrome. Am J Hum Genet. (1985) 37:250–67.
64. Watkins CE, Litchfield J, Song E, Jaishankar GB, Misra N, Holla N, et al. Chronic granulomatous disease, the McLeod phenotype and the contiguous gene deletion syndrome-a review. Clin Mol Allergy. (2011) 9:13. doi: 10.1186/1476-7961-9-13
65. Batlle-Maso L, Riviere JG, Franco-Jarava C, Martin-Nalda A, Garcia-Prat M, Parra-Martinez A, et al. Molecular challenges in the diagnosis of X-linked chronic granulomatous disease: CNVs, intronic variants, skewed X-chromosome inactivation, and gonosomal mosaicism. J Clin Immunol. (2023) 43:1953–63. doi: 10.1007/s10875-023-01556-x
66. Makaryan V, Zeidler C, Bolyard AA, Skokowa J, Rodger E, Kelley ML, et al. The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol. (2015) 22:3–11. doi: 10.1097/MOH.0000000000000105
67. Xiao Y, Wang N, Jin X, Liu A, and Zhang Z. Clinical relevance of SCN and CyN induced by ELANE mutations: a systematic review. Front Immunol. (2024) 15:1349919. doi: 10.3389/fimmu.2024.1349919
68. Rosenberg PS, Zeidler C, Bolyard AA, Alter BP, Bonilla MA, Boxer LA, et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol. (2010) 150:196–9. doi: 10.1111/j.1365-2141.2010.08216.x
69. Thusberg J and Vihinen M. Bioinformatic analysis of protein structure-function relationships: case study of leukocyte elastase (ELA2) missense mutations. Hum Mutat. (2006) 27:1230–43. doi: 10.1002/humu.20407
70. Germeshausen M, Deerberg S, Peter Y, Reimer C, Kratz CP, and Ballmaier M. The spectrum of ELANE mutations and their implications in severe congenital and cyclic neutropenia. Hum Mutat. (2013) 34:905–14. doi: 10.1002/humu.22308
71. Nustede R, Klimiankou M, Klimenkova O, Kuznetsova I, Zeidler C, Welte K, et al. ELANE mutant-specific activation of different UPR pathways in congenital neutropenia. Br J Haematol. (2016) 172:219–27. doi: 10.1111/bjh.13823
72. Nanua S, Murakami M, Xia J, Grenda DS, Woloszynek J, Strand M, et al. Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane. Blood. (2011) 117:3539–47. doi: 10.1182/blood-2010-10-311704
73. Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. (2007) 110:4179–87. doi: 10.1182/blood-2006-11-057299
74. Makaryan V, Kelley M, Fletcher B, Archibald I, Poulsen T, and Dale D. Comparison of gene editing versus a neutrophil elastase inhibitor as potential therapies for ELANE neutropenia. J Cell Immunol. (2022) 4:19–28. doi: 10.1097/MOH.0000000000000105
75. Bhuvanagiri M, Schlitter AM, Hentze MW, and Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J. (2010) 430:365–77. doi: 10.1042/BJ20100699
76. Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. (2018) 103:171–87. doi: 10.1016/j.ajhg.2018.06.009
77. Arai T, Oh-ishi T, Yamamoto H, Nunoi H, Kamizono J, Uehara M, et al. Copy number variations due to large genomic deletion in X-linked chronic granulomatous disease. PloS One. (2012) 7:e27782. doi: 10.1371/journal.pone.0027782
78. Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. (2010) 45:246–65. doi: 10.1016/j.bcmd.2010.07.012
79. Liu C, Tu C, Wang L, Wu H, Houston BJ, Mastrorosa FK, et al. Deleterious variants in X-linked CFAP47 induce asthenoteratozoospermia and primary male infertility. Am J Hum Genet. (2021) 108:309–23. doi: 10.1016/j.ajhg.2021.01.002
80. Malandrini A, Fabrizi GM, Truschi F, Di Pietro G, Moschini F, Bartalucci P, et al. Atypical McLeod syndrome manifested as X-linked chorea-acanthocytosis, neuromyopathy and dilated cardiomyopathy: report of a family. J Neurol Sci. (1994) 124:89–94. doi: 10.1016/0022-510X(94)90016-7
81. Roulis E, Hyland C, Flower R, Gassner C, Jung HH, and Frey BM. Molecular basis and clinical overview of mcLeod syndrome compared with other neuroacanthocytosis syndromes: A review. JAMA Neurol. (2018) 75:1554–62. doi: 10.1001/jamaneurol.2018.2166
82. Ying Y, Yu S, Zhang J, He J, Xu X, Hong X, et al. A case of McLeod syndrome caused by a nonsense variation c.942G>A in the XK gene: A case report. Front Genet. (2023) 14:1073139. doi: 10.3389/fgene.2023.1073139
83. Braun AA and Jung HH. Systematic review of phenotypes in McLeod syndrome and case report of a progressive supranuclear palsy in a female carrier. Orphanet J Rare Dis. (2024) 19:312. doi: 10.1186/s13023-024-03309-4
84. Bucciol G, Van Nieuwenhove E, Moens L, Itan Y, and Meyts I. Whole exome sequencing in inborn errors of immunity: use the power but mind the limits. Curr Opin Allergy Clin Immunol. (2017) 17:421–30. doi: 10.1097/ACI.0000000000000398
85. de Valles-Ibanez G, Esteve-Sole A, Piquer M, Gonzalez-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the genetics of common variable immunodeficiency: monogenetic model and beyond. Front Immunol. (2018) 9:636. doi: 10.3389/fimmu.2018.00636
86. Edwards ESJ, Bosco JJ, Ojaimi S, O’Hehir RE, and van Zelm MC. Beyond monogenetic rare variants: tackling the low rate of genetic diagnoses in predominantly antibody deficiency. Cell Mol Immunol. (2021) 18:588–603. doi: 10.1038/s41423-020-00520-8
87. Horwitz MS, Corey SJ, Grimes HL, and Tidwell T. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am. (2013) 27:19–41, vii. doi: 10.1016/j.hoc.2012.10.004
88. Shu Z, Deng M, Han T, and Mao H. De novo deep intron ELANE mutation resulting in severe congenital neutropenia. J Clin Immunol. (2024) 44:183. doi: 10.1007/s10875-024-01783-w
89. Shomali W and Gotlib J. World Health Organization and International Consensus Classification of eosinophilic disorders: 2024 update on diagnosis, risk stratification, and management. Am J Hematol. (2024) 99:946–68. doi: 10.1002/ajh.27287
90. Valent P, Klion AD, Roufosse F, Simon D, Metzgeroth G, Leiferman KM, et al. Proposed refined diagnostic criteria and classification of eosinophil disorders and related syndromes. Allergy. (2023) 78:47–59. doi: 10.1111/all.15544
91. Rodriguez-Cortez VC, Del Pino-Molina L, Rodriguez-Ubreva J, Lopez-Granados E, and Ballestar E. Dissecting epigenetic dysregulation of primary antibody deficiencies. J Clin Immunol. (2016) 36 Suppl 1:48–56. doi: 10.1007/s10875-016-0267-4
92. Akalu YT and Bogunovic D. Inborn errors of immunity: an expanding universe of disease and genetic architecture. Nat Rev Genet. (2024) 25:184–95. doi: 10.1038/s41576-023-00656-z
93. Wright CF, FitzPatrick DR, and Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. (2018) 19:253–68. doi: 10.1038/nrg.2017.116
94. Smedley D, Schubach M, Jacobsen JOB, Kohler S, Zemojtel T, Spielmann M, et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in mendelian disease. Am J Hum Genet. (2016) 99:595–606. doi: 10.1016/j.ajhg.2016.07.005
95. Schubach M, Maass T, Nazaretyan L, Roner S, and Kircher M. CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. (2024) 52:D1143–D54. doi: 10.1093/nar/gkad989
96. Lee H, Huang AY, Wang LK, Yoon AJ, Renteria G, Eskin A, et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet Med. (2020) 22:490–9. doi: 10.1038/s41436-019-0672-1
Keywords: inborn errors of immunity (IEI), whole exome sequencing (WES), molecular diagnosis, novel genes, international union of immunological societies (IUIS), diagnostic yield, children
Citation: Godinez-Zamora GF, Baeza-Capetillo P, Saucedo-Ramírez OJ, Del-Río-Navarro BE, Espinosa-Padilla SE, Morán-Barroso VF and Aguirre-Hernandez J (2025) Contribution of next generation sequencing to the diagnosis of inborn errors of immunity in a pediatric cohort. Front. Immunol. 16:1638544. doi: 10.3389/fimmu.2025.1638544
Received: 30 May 2025; Accepted: 04 August 2025;
Published: 20 October 2025.
Edited by:
Giorgia Santilli, University College London, United KingdomReviewed by:
Alessia Cavazza, University College London, United KingdomNathan White, University College London, United Kingdom
Copyright © 2025 Godinez-Zamora, Baeza-Capetillo, Saucedo-Ramírez, Del-Río-Navarro, Espinosa-Padilla, Morán-Barroso and Aguirre-Hernandez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesus Aguirre-Hernandez, amFndWlycmVAaGltZmcuZWR1Lm14