 Amy P. Hsu1*
Amy P. Hsu1* Eric Karlins2
Eric Karlins2 Justin Lack3,4T. Joseph Pepper5Karen Lau6Kimberly R. Marshall-Batty6Debra Long Priel6Joie Davis1Danielle L. Fink6
Justin Lack3,4T. Joseph Pepper5Karen Lau6Kimberly R. Marshall-Batty6Debra Long Priel6Joie Davis1Danielle L. Fink6 Christa S. Zerbe1John I. Gallin7Harry L. Malech8
Christa S. Zerbe1John I. Gallin7Harry L. Malech8 Steven M. Holland1
Steven M. Holland1 Douglas B. Kuhns6
Douglas B. Kuhns6- 1Immunopathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), Bethesda, MD, United States
- 2Bioinformatics and Computational Biosciences, Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
- 3National Institute of Allergy and Infectious Diseases (NIAID) Collaborative Bioinformatics Resource, National Institute of Allergy and Infectious Diseases (NIAID) National Institutes of Health (NIH), Bethesda, MD, United States
- 4Advanced Biomedical Computational Science, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research, Inc., Frederick, MD, United States
- 5Department of Mathematics, University of Maryland College Park, College, Park, MD, United States
- 6Neutrophil Monitoring Laboratory, Applied/Developmental Research Directorate, Leidos Biomedical Research, Inc, Frederick National Laboratory for Cancer Research, Frederick, MD, United States
- 7Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), Bethesda, MD, United States
- 8Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), Bethesda, MD, United States
Introduction: Chronic granulomatous disease is caused by mutations in any of the 6 components of the phagocytic NADPH oxidase complex including gp91phox, p47phox, p22phox, p40phox, p67phox, or EROS. Functional assays include reactive oxygen species (ROS) production, flow cytometry, and immunoblotting for NADPH proteins. The advent of high-throughput sequencing allows genetic diagnosis for all components except NCF1 (p47phox) due to two, nearly identical, pseudogenes (NCF1B, NCF1C). The majority of NCF1-CGD patients carry a 2-base deletion caused by crossover between NCF1 and NCF1B or NCF1C. Currently, NCF1 deficiency is diagnosed functionally: a characteristic DHR with low levels of residual ROS, loss of p47phox on immunoblot, or digital droplet PCR or Gene-scan to enumerate intact (GTGT) or deleted (ΔGT). While this provides patients a clinical CGD diagnosis, for the 20% of NCF1-CGD patients with a non-ΔGT mutation a definitive genetic diagnosis is still lacking.
Methods: We developed a bioinformatic method using existing short or long-read sequencing data from 48 NCF1-CGD patients or carriers.
Results: We identified both ΔGT and non-ΔGT NCF1 gene mutations. Additionally, we confirm that the presence of ΔGT in NCF1 is due to pseudogene copy into the NCF1 locus. We compare NCF1 sequence from NCF1-CGD patients to cohorts of non-NCF1-CGD and healthy controls (1000Genomes), demonstrating pseudogene replacement of NCF1 in NCF1-CGD as well as the reciprocal replacement of NCF1B or NCF1C by NCF1 in some healthy controls.
Discussion: With this method, reanalysis of existing sequence data may provide genetic diagnosis to NCF1-CGD patients. This technique may be modified for other diagnostically relevant pseudogenes.
Introduction
Chronic granulomatous disease (CGD) is caused by mutations in any of the 6 subunits of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (phox). Patients frequently, but not always, present as young children with recurrent bacterial and/or fungal infections. The majority (68%) of patients in Western countries carry mutations in the X-linked CYBB, encoding gp91phox (1, 2), while bi-allelic NCF1 mutations, encoding p47phox, occur in 25% (2), although this number is significantly larger in regions with high levels of consanguinity (3, 4). Access to large scale diagnostic sequencing, including targeted panel, whole exome, and whole genome, has enabled identification of mutations for the majority of CGD patients. However, due to the presence of two highly homologous pseudogenes (NCF1B, NCF1C, together, ΨNCF1), identification of specific NCF1 mutations remains challenging. Given the 99.5% homology, sequence reads fail to uniquely align to the reference genome, causing lack of coverage and inability to make variant calls. Additionally, the presence of the two pseudogenes essentially creates 6 copies of NCF1, limiting reliability of Sanger sequencing.
Patients suspected to have CGD undergo functional testing including flow cytometry or immunoblotting for NADPH oxidase proteins, ROS production by dihydrorhodamine (DHR), cytochrome C reduction, and/or nitroblue tetrazolium (NBT) reduction assay (Figure 1). While some larger hospital settings perform DHR or NBT assays, frequently patient samples are sent to commercial diagnostic laboratories. Comprehensive neutrophil studies encompassing all of these assays are only available in a handful of dedicated research laboratories. Reduced or absent DHR indicates a defective NADPH complex; loss of one component protein, demonstrated by flow cytometry or immunoblot, may indicate the mutated subunit, however none of these assays reveals underlying genetics. Whole exome, whole genome, or targeted capture panel sequencing provides genetic diagnosis for the other 5 NADPH oxidase components but not NCF1. NCF1 genetics are currently limited to enumeration of pseudogene and NCF1 copy number by Gene-scan (5) or droplet digital PCR (ddPCR) (2), or Sanger sequencing with anchored primers (6).
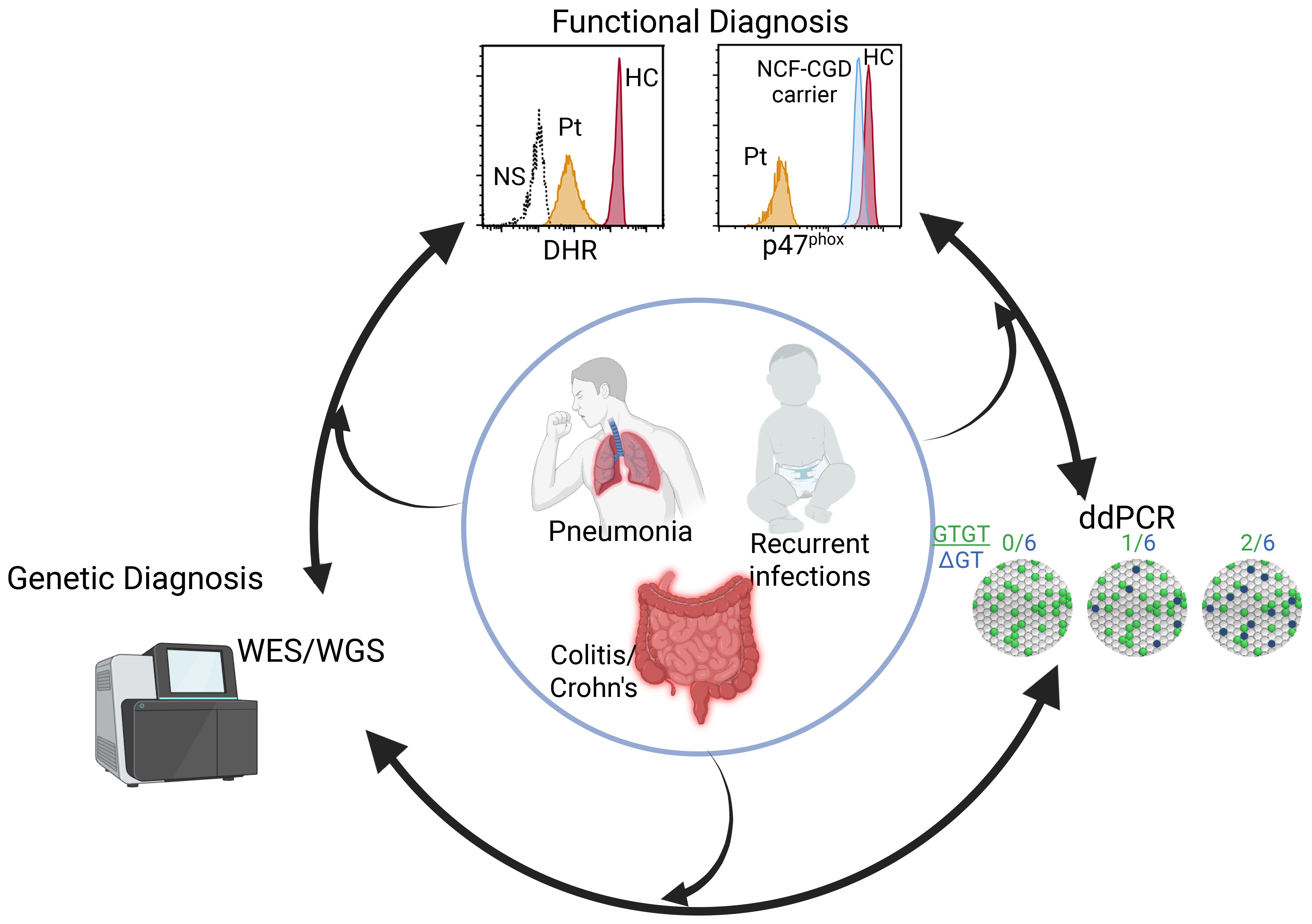
Figure 1. Diagnostics of NCF1-CGD. Patients (inner circle) presenting with disparate phenotypes may enter the diagnostic pipeline at different points including functional studies, sequencing, or for those with a family history, ddPCR or Gene-scan. Identification of genomic variant requires functional assessment while abnormal neutrophil functional studies leads to sequencing and a genetic diagnosis. Figure created in BioRender.
There are several nucleotides differentiating NCF1, NCF1B, and NCF1C, with the crucial difference being a two-base deletion at the start of exon 2 (ΔGT) in ΨNCF1, resulting in frameshift and premature termination (7). Recombination between NCF1 and NCF1B or NCF1C has been posited as the cause of the most common genetic variation, incorporation of ΔGT in NCF1 (8). Currently, the Gene-scan assay (5) is the most common method for ΔGT identification; recently, assays to determine the ratio of intact GTGT vs NCF1/NCF1B/NCF1C copy number by ddPCR (2) or restriction fragment length polymorphism (9) were reported, which provide a ΔGT genetic diagnosis. All three methods are research assays not commercially or commonly available. CGD patients lacking NCF1 protein, yet carrying 1 or 2 intact GTGT alleles, remain without a genetic diagnosis. Herein we describe a bioinformatic pipeline to enable re-analysis of short- and long-read sequence data to identify non-ΔGT NCF1 mutations in NCF1-CGD patients and NCF1-CGD carriers previously lacking genetic diagnoses. This analysis supports the crossover theory between pseudogene and NCF1 and demonstrates multiple discrete recombinations indicating recurring events. Using 1000 Genomes (1000G) data as controls, we also demonstrate the presence of reciprocal replacement of pseudogenes by NCF1.
Methods
Patient cohort
Patients with previously diagnosed chronic granulomatous disease (CGD) (n = 45) and first-degree relatives (n = 3) followed at, or referred to, the National Institutes of Health were included in the study after being consented to IRB-approved protocols NCT00001355, NCT00404560, NCT00001467, or NCT00128973. Routine functional analysis included DHR assay for ROS production, immunoblot and/or flow cytometry to determine the presence of specific NADPH oxidase component proteins, and ddPCR for quantification of ΔGT as previously published (2). Illumina short-read whole exome (n = 21) or whole genome (n = 21) sequencing was performed at commercial laboratories (Johns Hopkins Genomics, Baltimore, MD; Baylor College of Medicine Human Genome Sequencing Center, Houston, TX). Oxford Nanopore long-read whole genome sequencing (n = 6) was performed at Johns Hopkins Genomics, Genetics Resource Core Facility, Baltimore, MD.
Non NCF1-CGD patients included 48 patients with X-linked CYBB mutations (including 4 skewed female patients), 4 bi-allelic CYBA, and 1 bi-allelic NCF2, recruited under the same protocols.
Reference sequences
NCBI reference sequences for NCF1 (NM_00236.7; NP_000256.4), NCF1B (NR_003186), and NCF1C (NR_003187) were aligned using Sequencher (GeneCodes, Ann Arbor, MI).
Bioinformatic pipeline
To effectively map sequencing reads to this region we created an hg38 reference fasta file by masking the pseudogene sequence (chr7:73220639–73235945 and chr7:75156639-75172044) with “N”s using “bedtools maskfasta” (10, 11). Reads were mapped to our masked-reference using bwa-mem (https://bio-bwa.sourceforge.net/bwa.shtml). Variant calling was performed using GATK Best Practices (12), altering the ploidy to 6 to adjust for the tripling of reads in our region of interest. A custom script (https://github.com/niaid/NCF1_variant_calling), adapted from Almeida de Jesus, et al (13), was used to emit putative variant sites when 2 or more reads with alternate alleles were present, regardless of read balance. These sites were then fed to GATK for genotyping. The result of these methods is a list of predicted variants, most of which we were unable to discover using standard methods. We also calculate the alternate allele balance (AltAB) defined as the frequency of variant allele reads versus total read depth from NCF1 plus NCF1B/NCF1C at any given nucleotide. While AltAB is equivalent to variant allele frequency (VAF) used for high throughput sequencing data, given the 6 alleles present in the NCF1/NCF1B/NCF1C locus, we use AltAB to distinguish from normal, heterozygous loci.
1000 Genome reference table
Variants identified using publicly accessible 1000G short read sequence data (https://ftp.sra.ebi.ac.uk/vol1/run/) were parsed by ancestry to provide a reference dataset of masked NCF1 variant frequency in a healthy control population (Supplementary Table 1, 1000_Genomes_NCF1_variants.xlsx).
PCR for P5/P6 large deletion
Primers were selected from outside the deleted region and used to amplify DNA from P5, P6, and the father of P6 (P6-F). Primers used were NCF1del 169F 5’- AAGATAAACCCAAACTAAGGGACATTCTACAAGG– 3’ and NCF1del 5960R 5’ – ATTTTATTTTGAGATGGAGTTTTGTCCTTGTTGC – 3’. Amplification was performed in 15 μl reactions using Platinum Taq HiFi (ThermoFisher) and cycling conditions 95°C 3 min, (95°C 20 sec, 62.3°C 10 sec, 68°C 30 sec) x35, 68°C 3 min; product was visualized on 1.2% agarose gel.
Results
Cohort description
Patients were referred to the NIH for clinical evaluation or functional testing for suspected chronic granulomatous disease. All patients (n=48; 45 NCF1-CGD plus 3 NCF1-carriers) (Table 1) were characterized by functional assays including flow cytometric DHR, immunoblots and/or flow cytometry for gp91phox (CYBB), p22phox (CYBA), p47phox (NCF1), p67phox (NCF2), and p40phox (NCF4). All NCF1-CGD patients had diminished PMA-induced neutrophil superoxide production and DHR (Supplementary Table 2) and undetectable p47phox by immunoblot or flow cytometry. Frequency of ΔGT for each individual was performed using ddPCR (Table 1). Genetic sequencing included short-read whole exome (WES) (n=21) or whole genome (WGS) (n=21) or long-read WGS (n=3 NCF1-CGD patients and 3 NCF1 carriers).
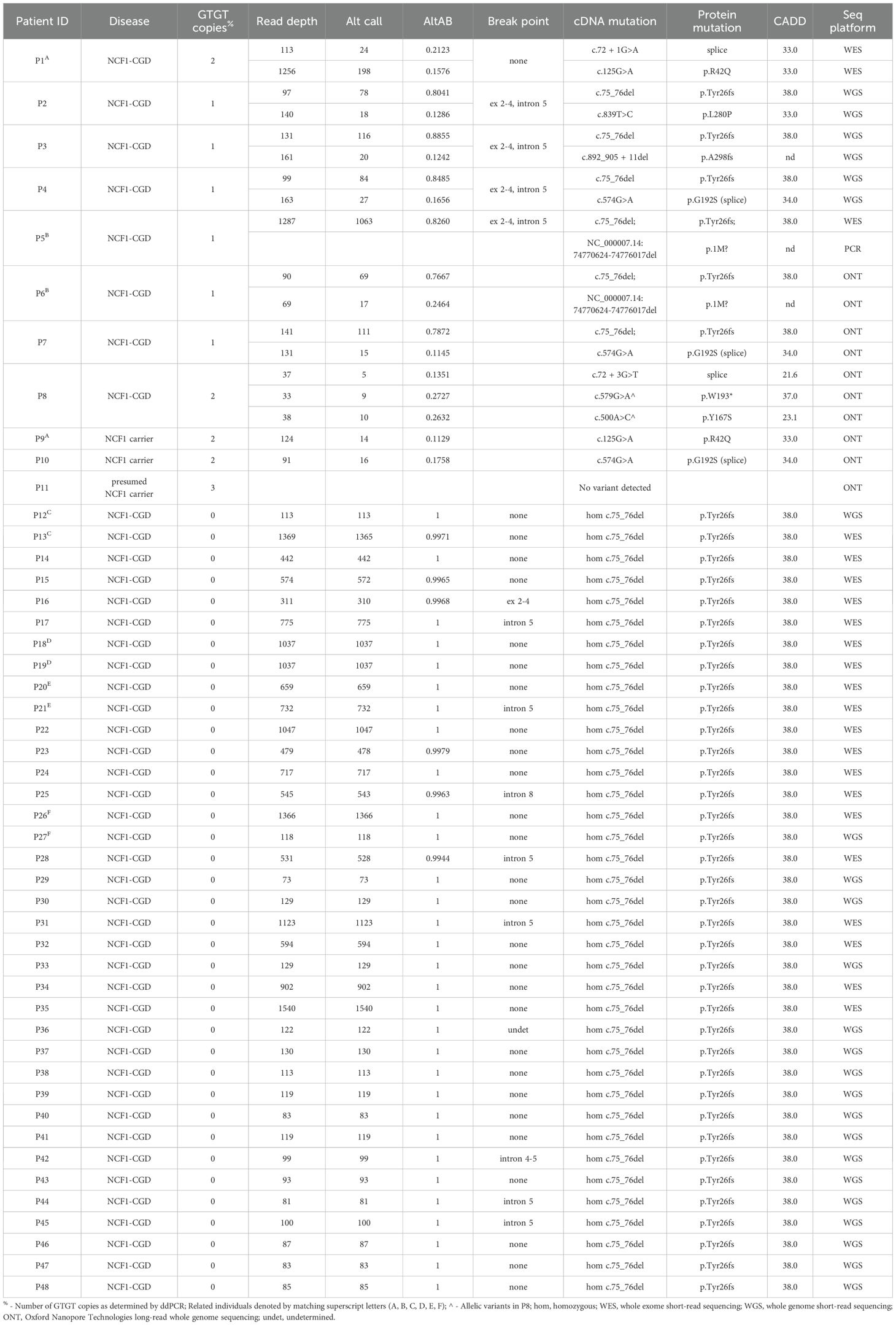
Table 1. NCF1-CGD patients.
Laboratory testing among the NCF1-CGD cohort resembled previously reported patients (2, 14) with all patients displaying residual ROS after PMA stimulation and absent p47phox protein (2). Among NCF1-CGD patients, ddPCR results demonstrated 37/45 (82.2%) had no intact GTGT alleles, 6/45 (13.3%) had 1 GTGT allele, while 2/45 (4.4%) had 2 intact GTGT alleles.
Assignment of genotype from pseudogene masking
The presence of NCF1B/NCF1C (ΨNCF1) prevents unique alignment of sequencing reads. Therefore, we developed a bioinformatic pipeline to mask ΨNCF1, thereby aligning all sequence reads to NCF1 (Figure 2A). Utilizing this pipeline we then performed variant calling using standard methods. Adjusting the ploidy parameter from 2 to 6 allowed identification of variants from NCF1 reference sequence. For each variant, the ratio of variant calls to read depth was used to establish the alternate allele balance (AltAB) from NCF1B and NCF1C. Given that 3 unique autosomal genomic regions were included, AltAB increments approximate 1/6, with variants occurring in both ΨNCF1 loci having AltAB ~0.66 and those occurring in only NCF1B or NCF1C having AltAB ~0.33. We then used AltAB of ΔGT to compare 1000G, NCF1, and non-NCF1 CGD patients. Both 1000G and non-NCF1 patients had median AltAB of 0.66 indicating 4 copies of ΔGT from ΨNCF1 and two intact NCF1 GTGT alleles; by contrast, NCF1 patients had a median AltAB of 1.0 (indicating no remaining GTGT alleles) with only 11 individuals deviating from that (Figure 2B). Since we had previously performed ddPCR to determine ΔGT copies in these patients, we compared AltAB to ddPCR results (Figure 2C). Each of the 11 samples with ΔGT AltAB<1 (P1 through P11) was from patients with 1 (n=6) or 2 (n=2) copies of GTGT; also included were 3 NCF1 mutation carriers, with 2 (n=2) or 3 (n=1) copies of GTGT. Plotting ddPCR-determined GTGT copy number versus AltAB demonstrated concordance between the two methods (R2 = 0.980), validating the use of AltAB to detect the most common NCF1 mutation.
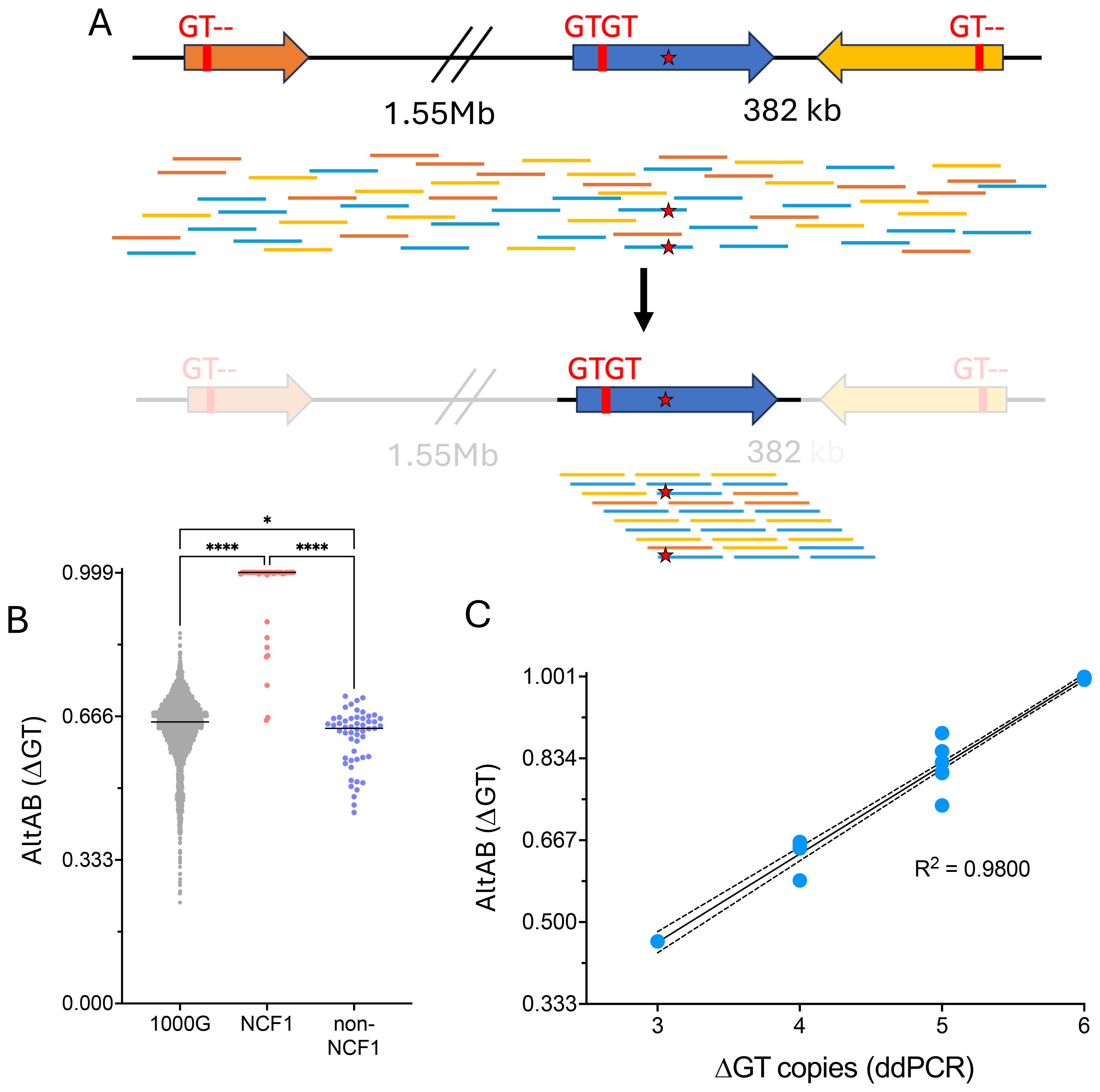
Figure 2. Utilization of masked genome to identify the common NCF1 ΔGT mutation. (A) NCF1 (blue) is flanked by 2 pseudogenes, NCF1B upstream (orange) and NCF1C downstream in reverse orientation (yellow). Both NCF1B and NCF1C have a deletion at the start of exon 2, noted “GT–” while NCF1 has intact, GTGT sequence. Short read, unmapped sequences for NCF1/NCF1B/NCF1C are shown scattered across the loci (top). Mutation within NCF1 is denoted by red star. Using a masked genome to prevent alignment to NCF1B or NCF1C (bottom), all reads align to NCF1 allowing calling of non-ΔGT mutation. (B) Variant allele frequency (AltAB) for ΔGT from 1000G, NCF1-CGD, and non-NCF1-CGD cohorts. (****P<0.0001, *P=0.0211 ANOVA with Kruskal-Wallis test for multiple comparisons) (C) Simple linear regression curve ddPCR determined ΔGT copies compared to AltAB of ΔGT variant per individual.
Variant occurrences in 1000G
Using NCBI reference sequences for the 3 loci we established a variant table, allowing assignment of variants to NCF1B, NCF1C or both (ΨNCF1) (Supplementary Table 3). We then examined the AltAB distribution of these variants among 1000G. ΨNCF1 variants (Supplementary Figure 1) mostly displayed a median AltAB (mAltAB) of 0.66, consistent with 4/6 copies of ΔGT, although the distribution was broad with 3/6 or 5/6 copies not uncommon and several other variants displaying 2/6 copies. For 9 loci, mAltAB was 1.0 indicating incorporation of the variant in NCF1 or incorrect NCF1 reference sequence. It is noteworthy that, despite a normal distribution and mAltAB ~0.66, 12 individuals have AltAB=1.0 for c.269G>A encoding p.R90H (Supplementary Figure 1, blue).
Examining NCF1C variants, in which mAltAB should be ~0.33, 3/14 variants have mAltAB>0.33 with multimodal distribution; an additional 4 variants have mAltAB<0.33, 2 of which have bimodal distributions of 1/6 or 2/6 while the remaining 2 variants have mAltAB=0.26 (Supplementary Figure 2). A similar NCF1B variant analysis reveals 2/14 variants with mAltAB~0.66, 4/14 variants with multimodal distributions and mAltAB>0.33 (Supplementary Figure 3). Collectively, this analysis identifies a subset of variants having frequencies consistent with their presence in one or both pseudogenes (noted in Supplementary Table 3) available for further analyses of the locus.
Using these data, we next examined distribution of AltAB across NCF1 among NCF1 patients, 1000G, and non-NCF1 CGD patients. It was previously suggested that inclusion of ΔGT arises from meiotic crossover between NCF1 and one of the pseudogenes (7). Supporting this, NCF1-CGD patients have higher AltAB values throughout the NCF1 locus compared to 1000G controls and non-NCF1 patients (Figure 3A) indicating a higher proportion of ΨNCF1 variants among patients. NCF1 patients displayed a clear skewing of AltAB with mAltAB=0.66 while both 1000G and non-NCF1 patients had mAltAB of 0.52 and 0.49 respectively. This suggests larger incorporation of ΨNCF1 than solely the ΔGT-containing exon 2 (Figure 2B). To explore this, we plotted AltAB across 4 variants specific to NCF1C or NCF1B. NCF1 patients display increased AltAB across NCF1C variants compared to 1000G or non-NCF1 patients (Figure 3B); by contrast, there is no significant increase in AltAB across NCF1B SNPs (Figure 3C), although some individual patients have increased NCF1B SNP AltAB. Together, these data suggest NCF1 replacement with NCF1C occurs more frequently than with NCF1B.
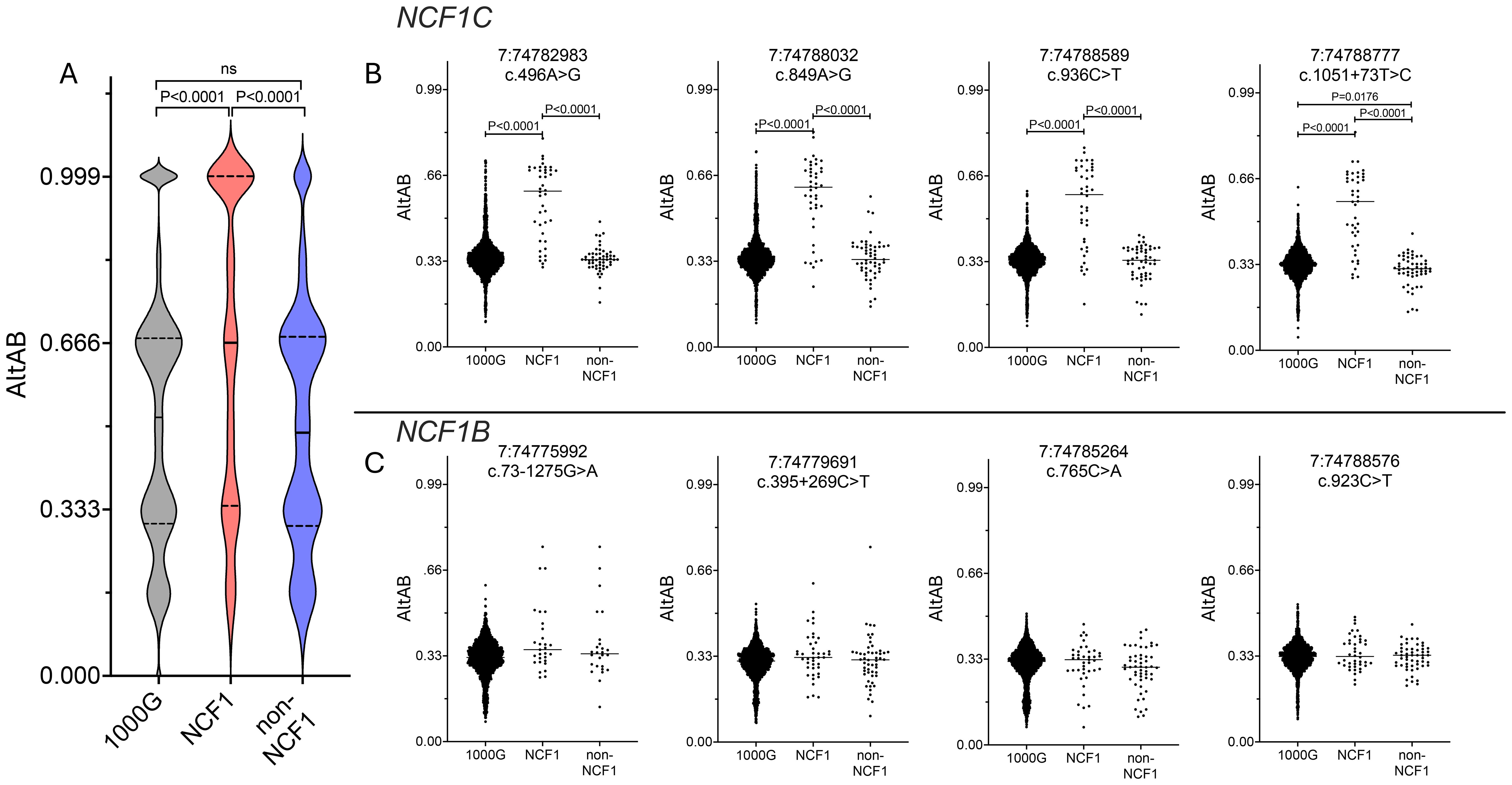
Figure 3. NCF1-CGD patients have more pseudogene copies and these are more frequently NCF1C. (A) AltAB across NCF1 locus for 1000G (grey), NCF1 (red) and non-NCF1 (blue) CGD patients. Violin plots with median (solid line) and quartiles (dotted lines) indicated. Groups compared using Ordinary one-way ANOVA with Tukey’s multiple comparisons test. B and (C) AltAB of NCF1C (B) or NCF1B (C) specific variants across three cohorts. Groups compared using Ordinary one-way ANOVA with Tukey’s multiple comparisons test. Only comparisons with adjusted P<0.05 are shown.
Mapping NCF1 loci recombination
These data suggested full or partial replacement of NCF1 with NCF1C in the majority of ΔGT NCF1 alleles, which we sought to confirm. Using variants demonstrated to have normal distribution among 1000G, we normalized AltAB at each site to 1000G mAltAB and plotted the normalized value across the locus. For patients with no intact GTGT, there would be 6 copies of ΔGT rather than 4, giving a normalized AltAB of 1.5. In the majority of patients (27/37 homozygous ΔGT, 73.0%), we observe normalized AltAB of 1.5 at ΨNCF1 SNPs across the locus indicating full replacement of NCF1 with a pseudogene (Figures 4A, B); additionally, 2/4 heterozygous ΔGT patients replaced one copy of NCF1 with a pseudogene resulting in normalized AltAB of ~1.25. Using the NCF1B or NCF1C specific variants, we observed cases of full NCF1 replacement by NCF1C as demonstrated by a normalized NCF1C AltAB of 2 across the locus indicating 4 copies of NCF1C and 2 copies of NCF1B (P43, Figure 4A). Alternative arrangements are present as well, including replacement of NCF1 in the setting of 5 copies of NCF1C and only 1 NCF1B (P46, Figure 4B).
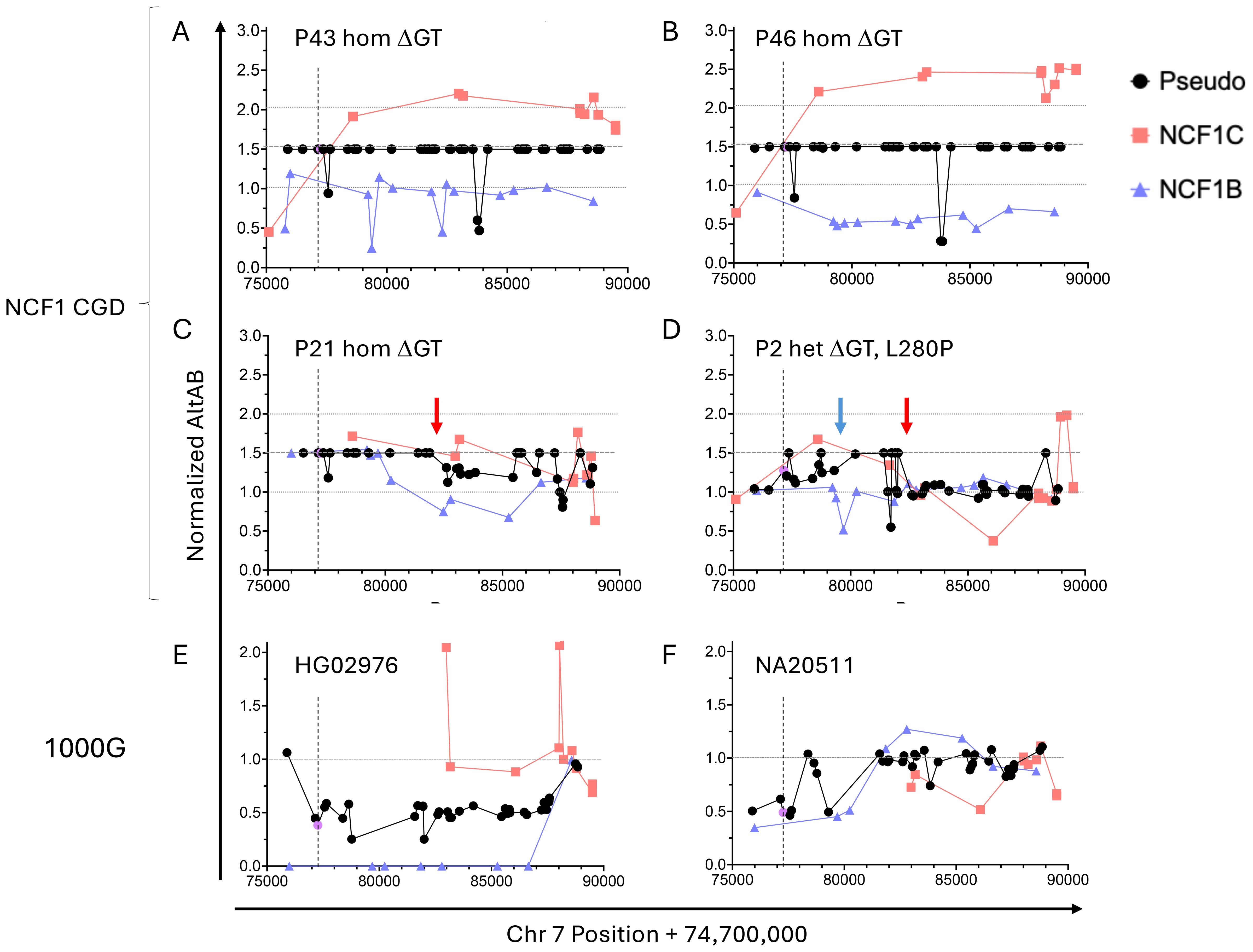
Figure 4. Normalized AltAB shows NCF1/NCF1B/NCF1C rearrangement among NCF1-CGD patients and healthy controls. Variants present in both pseudogenes (black) should have AltAB~0.66, variants present in NCF1C (red) or NCF1B (blue) should have AltAB~0.33; ΔGT is noted in purple. Each patient’s AltAB was normalized (nAltAB) for expected values based on 1000G data. Vertical line denotes location of ΔGT variant. (A) P43, homozygous ΔGT, with nAltAB of 1.5 for pseudogene variants while NCF1C-specific variants have nAltAB = ~2 indicating NCF1 replacement by NCF1C; NCF1B maintains nAltAB~1.0. (B) Similar analysis of P17, homozygous ΔGT, nAltAB =1.5 for pseudogene variants, nAltAB for NCF1C variants ~2. 5 while nAltAB NCF1B variants ~ 0.5 indicating 5 copies of NCF1C and only 1 copy of NCF1B. (C) P25, homozygous ΔGT, with only 5’ end of NCF1 replaced by pseudogene but normalized AltAB after intron 5 breakpoint (red arrow). (D) P2, compound heterozygous for ΔGT and L280P, demonstrating two breakpoints between exons 2-4 (blue arrow) and intron 5 (red arrow). E and (F) 1000G controls with nAltAB < 1.0. (E) HG02976 has pseudogene nAltAB ~ 0.5 indicating only 2 copies of pseudogene present; nAltAB for NCF1B variants = 0 indicating both pseudogene copies present are NCF1C. (F) NA20511 has pseudogene nAltAB ~ 0.75 indicating loss of 1 pseudogene copy, nAltAB for NCF1C ~ 0.5 suggests only 1 copy of NCF1C.
Not all patients replaced the full gene, there was a frequent crossover point in intron 5 (Figures 4C, D red arrows) corresponding to consecutive AluJr and AluSx1 repeats present in all three loci. Among NCF1-CGD patients with short-read sequencing (n=42), we could identify recurrent breakpoints between exons 2-4 (5/42, 11.9%) (Figure 4D, blue arrow) and within intron 5 (10/42, 23.8%); 4 patients within these two groups exhibited both breakpoints (Figure 4D), one patient each had breaks between introns 4–5 and within intron 8; one patient was indeterminate due to lack of informative SNPs (Table 1). In P21, NCF1 was replaced by NCF1C on one allele and only the 5’ portion of NCF1B on the other (Figure 4C). Lastly, P2 carried a missense mutation, (c.839T>C, p.L280P) on one allele, and ΔGT on the other. In this patient the mAltAB was ~1.25 with increased NCF1C in the 5’ region, and normalization of AltAB to 1 after intron 5 (Figure 4D). We validated this approach in two NCF1-CGD siblings, in whom the pattern of normalized AltAB for NCF1B, NCF1C, and ΨNCF1 was similar, as would be expected given the same parental alleles (Supplementary Figure 4).
Having confirmed NCF1 replacement by NCF1C or NCF1B in ΔGT patients we looked for the inverse allele in a control population – that is, replacement of NCF1C or NCF1B by NCF1. Examination of ΔGT frequency in 1000G revealed 24 individuals (24/2504; 1%) with ΔGT AltAB<0.4, indicating fewer than 4 copies of ΔGT. In contrast to the rearrangements seen in CGD patients, these individuals had had NCF1 replacement of a pseudogene. One individual had AltAB = 0.38 for ΔGT, indicating only 2 remaining copies of a pseudogene. SNP analysis across NCF1B and NCF1C revealed a total loss of NCF1B but maintenance of NCF1C SNPs, suggesting replacement of NCF1B by NCF1 (Figure 4E). Another individual displayed apparent loss of at least one copy of NCF1C (Figure 4F), although exact determination was difficult due to the lack of NCF1C specific variants with normal distribution in the 5’ region.
Mutation identification
Having demonstrated AltAB as a valid metric for ΔGT, we used AltAB to screen for non-ΔGT mutations among patients with available short-read sequencing data. Similar to examination of other genes, we first established a variation reference. Analyzing 1000G data with our pipeline identified all variants with an AltAB frequency > 0.08. We determined the number of individuals with each variant as well as minimum, maximum, mean, median and standard deviation of AltAB for each variant; those data were further split by ancestry (Supplementary Table 1). Using the variants identified in 1000G, we examined NCF1-CGD patients with 1 or 2 intact GTGT alleles indicating non-ΔGT variations. We considered variants rare or unique among 1000G and the human variation database, Genome Aggregation Database (gnomAD) (15), and predicted deleterious by the bioinformatic algorithm, Combined Annotation Dependent Depletion (16), using a threshold of CADD>20, identifying 6 variants (Figure 5, upper; Table 1). Each patient with 1 intact GTGT copy had a single mutation, while P1, with 2 GTGT alleles, carried 2 separate mutations; each variant had a frequency approximating 1/6, suggesting that patients were heterozygous for the variants. P1 had two variants, c.72+3G>A at the start of intron 1 and c.125G>A (p.R42Q) within exon 2, both previously reported (14). P2 also carried a previously reported mutation, c.574G>A (p.G192S), which occurs at the last base of exon 6 leading to impaired splicing of the exon (14). P3, P4, and P5 carried novel variants: c.892_905+11del spanning the exon 9/intron 9 boundary, c.839T>C (p.L280P), and c.107C>T (p.S36L), respectively. Three of the variants, c.107C>T, c.125G>A, and c.574G>A, are present in gnomAD at low frequency (~1/100,000) however only c.107C>T occurs in 1000G (n=2). While it is not always possible to phase variants with short-read sequencing, c.107C>T is within 35 bases of ΔGT; using the Integrative Genomics Viewer (17) (IGV), a platform allowing visualization of high throughput sequencing alignments, we determined this variant was allelic with ΔGT (Supplementary Figure 5), indicating the variant was not causative of CGD.
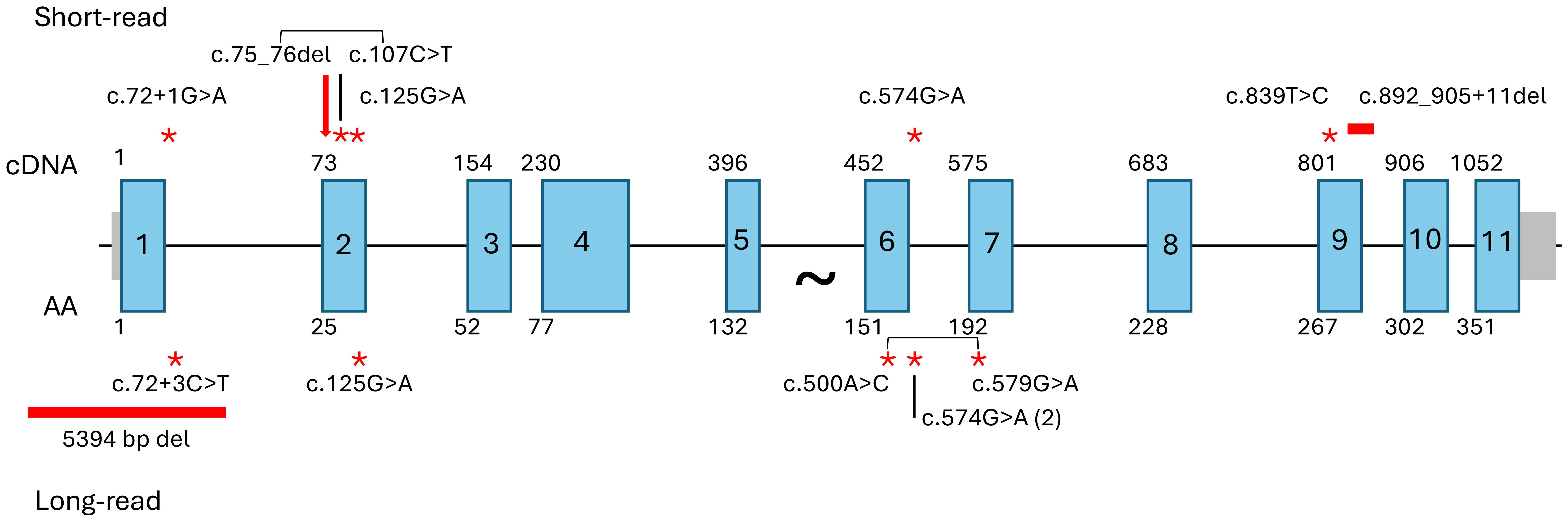
Figure 5. Identified NCF1 mutations. Mutations identified after pseudogene masking of short-read WES/WGS (upper) or long-read WGS (lower). Large arrow indicates ΔGT, point mutations noted by red asterisks, deletions by horizontal red bars. Square brackets indicate allelic variants observed in P5 (short-read) and P8 (long-read).
Long-read sequencing
Since we were unable to identify a second mutation in the P5/P6 family, we performed Oxford Nanopore long-read sequencing. With an average read length of 25kb, we confidently spanned the full NCF1 locus and were able to phase variants across the region. In P6, the niece of P5, we identified a 5394 bp deletion fully encompassing exon 1 and part of intron 1 (chr7:74,770,624-74,776,017), (Supplementary Figure 6). Examination of sequencing reads in IGV revealed the large deletion was not allelic with ΔGT (Supplementary Figure 6). Using primers spanning the deleted region, the presence of the 5394 bp deletion was confirmed in P5 and the father of P6 (P6-F, an obligate carrier) (Supplementary Figure 7A). Additionally, the c.107C>T variant in P5, allelic with ΔGT in P5, was not present in P6 suggesting a commonly inherited large deletion within this family and different ΔGT alleles present in P5 and P6. (Supplementary Figure 7B). It is notable that the deceased brother of P5 (uncle of P6) was diagnosed with CGD after Mycobacterium fortuitum infection at age 27 (18).
Two additional NCF1-CGD patients were diagnosed using long read sequencing. P7 carried one variant occurring at the last base of exon 6, (c.574G>A; p.G192S/splice) which was non-allelic with ΔGT. P8, with two copies of intact GTGT, had three damaging variants identified, c.72+3G>T at the start of intron 1, c.500A>C encoding p.Y167S, and c.579G>A encoding p.W193*. Examination of reads in IGV revealed that c.500A>C was allelic with the c.579G>A mutation but not the splice mutation (Supplementary Figure 8). Lastly, three presumed carriers of non-ΔGT mutations were sequenced. In two cases, a single, known pathogenic mutation was identified which was allelic with intact GTGT; P9, mother of P1, carried one copy of c.125G>A (p.R42Q), while P10 carried c.574G>A (p.G192S/splice). No mutation was identified in P11, the father of an NCF1-CGD patient. It is possible the patient carries a de novo mutation on the paternal allele or P11 is a germline mosaic.
Discussion
While chronic granulomatous disease has been diagnosed in the laboratory for more than 60 years, providing genetic diagnosis for p47phox-deficient patients requires specialized techniques performed in select laboratories. We have developed the ability to identify NCF1 mutations using a bioinformatic pipeline on existing whole exome and whole genome sequence data. By masking the pseudogene sequences and aligning all the reads to NCF1, we were able to identify variants within the NCF1/NCF1B/NCF1C locus. Using variant vs total read depth provided a variant allele frequency (AltAB) for known variants present in NCF1B or NCF1C or both (ΨNCF1). Normalizing AltAB in individual patient data to expected variant frequency for these known variants, we demonstrated full or partial replacement of NCF1 by a pseudogene in 39/42 patients with one patient carrying two non-ΔGT mutations unrelated to pseudogene sequences.
Our data are consistent with previous reports of recombination within the NCF1/NCF1B/NCF1C locus, confirming that ΔGT occurs by crossover of NCF1C/NCF1B into the NCF1 locus (8). Most frequently, the entire NCF1 locus is replaced, however crossovers between exons 2 and 4 or within intron 5 are detected as previously reported (19). The higher observed frequency of NCF1 replacement by NCF1C may be due to the NCF1/NCF1B/NCF1C locus organization with NCF1C in closer proximity and in reverse orientation to NCF1 enabling DNA hairpin loop formation and occurrence of crossover events. In many cases of autosomal recessive disease, founder mutations are prominent. While founder mutations may be present in some communities, given the variety of alleles present, it is likely the locus continues to undergo recombination among the NCF1/NCF1B/NCF1C alleles. This is supported by the presence of multiple different crossover loci identified here and in the literature (19).
Patients suspected to have CGD are first assessed using assays to quantify the ability of granulocytes to produce reactive oxygen species including DHR or NBT assays. These may be performed by various reference laboratories. Additional testing includes flow cytometric or immunoblotting for NADPH oxidase components (gp91phox, p22phox, p47phox, p67phox, and p40phox, EROS). In the setting of clinical suspicion plus abnormal functional testing, sequencing may confirm a genetic diagnosis, allowing screening of family members for disease or carrier status. To date, the diagnosis of NCF1/p47phox deficiency has been limited to functional testing in affected individuals. There are a handful of laboratories utilizing specialized techniques to enumerate ΔGT copies including Gene-scan (5), droplet digital PCR (ddPCR) (2), and restriction fragment length polymorphism (9), each of which may provide genetic diagnosis for ΔGT. Sequencing of NCF1 has been reported using primers anchoring on the exon 2 GTGT, allowing identification of non-ΔGT mutations (6, 20, 21), but this too must be performed in a specialized laboratory. As high-throughput sequencing becomes commonplace, the ability to determine NCF1 variants from high-throughput sequencing permits recognition of NCF1-CGD patients, regardless of clinical presentation. Incorporating our bioinformatic approach would enable identification of individuals presenting later in life, with colitis or Crohn’s disease, or those with previously unappreciated infections, and not limit diagnosis to those children suspected of having a primary immune deficiency.
With 80% of NCF1-CGD patients homozygous for ΔGT, gene correction has become an attractive therapeutic target. Since our data reveal the full replacement of NCF1 by pseudogene in the majority of NCF1-CGD patients, targeted correction of ΔGT would correct a pseudogene resulting in pseudogene-derived protein expression. Early studies using zinc-finger nucleases demonstrated pseudogene correction was sufficient to restore both p47phox expression and superoxide production (22). Characterizing pseudogene-derived p47phox function, with the associated amino acid differences from NCF1-derived p47phox, is an important consideration as gene-correction trials are pursued. Correction of one or both pseudogenes would likely result in a protein containing p.R90H, reported to cause an early-onset interferonopathy (23), or a lupus-like disease in mice (24). The 12 healthy individuals from 1000G, who have only p.R90H, suggest additional factors may play a role in the immune dysregulation reported for this variant. Additionally, pseudogene correction that restores myeloid cell function may also benefit patients with non-ΔGT mutations.
Here we have developed a technique using standard, high throughput sequencing to establish the genetic diagnosis of NCF1/p47phox CGD. Using this bioinformatic pipeline does not require specialized instrumentation, techniques, or the need for resequencing and may be performed on historic high-throughput sequence data. It is important to note that, depending on the sequencing platform used, this method may not establish whether the mutation occurs within NCF1 or one of the pseudogenes. Long-read sequencing spanning the full length of NCF1 allows phasing of identified variants with ΔGT or GTGT at the start of exon 2 and other pseudogene-specific SNPs; this is not possible with short-read sequencing. This approach should always be accompanied by functional assays demonstrating abnormal neutrophil respiratory burst and loss of p47phox protein. Additionally, the diagnosis of NCF1-CGD in older children and adults is not uncommon; adult cases have been diagnosed presenting with pneumonia (25–28) or in the setting of colitis (29, 30). These cases suggest a broader use for the pipeline beyond pediatric immune deficiency patients including individuals with recurrent infections, Aspergillus fumigatus pneumonia, Crohn’s disease, or other forms of colitis. Providing genetic and clinical diagnoses in these settings allows proper antimicrobial treatment and ongoing prophylaxis for the patients and the ability to screen at-risk family members for NCF1 mutation status. This method should be modifiable for other gene/pseudogene combinations which inhibit standard sequencing diagnosis such as IKBKG/NEMO deficiency, associated with immune deficiency with or without ectodermal dysplasia.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Institutional Review Board, National Institutes of Health. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
APH: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing. EK: Formal analysis, Methodology, Software, Writing – review & editing. JL: Formal analysis, Methodology, Software, Writing – review & editing. TJP: Formal analysis, Writing – review & editing. KL: Investigation, Writing – review & editing. KRM-B: Investigation, Writing – review & editing. DLP: Investigation, Writing – review & editing. JD: Writing – review & editing. DLF: Investigation, Writing – review & editing. CSZ: Writing – review & editing. JIG: Writing – review & editing. HLM: Writing – review & editing. SMH: Project administration, Resources, Supervision, Writing – review & editing. DBK: Conceptualization, Data curation, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded in whole or in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health and with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E.
Acknowledgments
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Conflict of interest
Authors KL, KRM-B, DLP, DLF, and DBK were employed by Leidos Biomedical Research, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1640496/full#supplementary-material
References
1. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. (2010) 363:2600–10. doi: 10.1056/NEJMoa1007097
2. Kuhns DB, Hsu AP, Sun D, Lau K, Fink D, Griffith P, et al. NCF1 (p47phox)-deficient chronic granulomatous disease: comprehensive genetic and flow cytometric analysis. Blood Adv. (2019) 3:136–47. doi: 10.1182/bloodadvances.2018023184
3. Bakri FG, Martel C, Khuri-Bulos N, Mahafzah A, El-Khateeb MS, Al-Wahadneh AM, et al. First report of clinical, functional, and molecular investigation of chronic granulomatous disease in nine Jordanian families. J Clin Immunol. (2009) 29:215–30. doi: 10.1007/s10875-008-9243-y
4. Fattahi F, Badalzadeh M, Dedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. (2011) 31:792–801. doi: 10.1007/s10875-011-9567-x
5. Dekker J, de Boer M, and Roos D. Gene-scan method for the recognition of carriers and patients with p47(phox)-deficient autosomal recessive chronic granulomatous disease. Exp Hematol. (2001) 29:1319–25. doi: 10.1016/S0301-472X(01)00731-7
6. Roos D, de Boer M, Koker MY, Dekker J, Singh-Gupta V, Ahlin A, et al. Chronic granulomatous disease caused by mutations other than the common GT deletion in NCF1, the gene encoding the p47phox component of the phagocyte NADPH oxidase. Hum Mutat. (2006) 27:1218–29. doi: 10.1002/humu.20413
7. Casimir CM, Bu-Ghanim HN, Rodaway AR, Bentley DL, Rowe P, Segal AW, et al. Autosomal recessive chronic granulomatous disease caused by deletion at a dinucleotide repeat. Proc Natl Acad Sci U S A. (1991) 88:2753–7. doi: 10.1073/pnas.88.7.2753
8. Roesler J, Curnutte JT, Rae J, Barrett D, Patino P, Chanock SJ, et al. Recombination events between the p47-phox gene and its highly homologous pseudogenes are the main cause of autosomal recessive chronic granulomatous disease. Blood. (2000) 95:2150–6. doi: 10.1182/blood.V95.6.2150
9. Wrona D, Siler U, and Reichenbach J. Novel Diagnostic tool for p47phox-deficient chronic granulomatous disease patient and carrier detection. Mol Ther Methods Clin Dev. (2019) 13:274–8. doi: 10.1016/j.omtm.2019.02.001
10. Quinlan AR and Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. (2010) 26:841–2. doi: 10.1093/bioinformatics/btq033
11. Boisson B, Honda Y, Ajiro M, Bustamante J, Bendavid M, Gennery AR, et al. Rescue of recurrent deep intronic mutation underlying cell type-dependent quantitative NEMO deficiency. J Clin Invest. (2019) 129:583–97. doi: 10.1172/JCI124011
12. Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discover to tens of thousands of samples. BioRxiv. (2017), 201178. doi: 10.1101/201178
13. Almeida de Jesus A, Lin B, Karlins E, Kahle D, Rastegar A, Mitchell J, et al. Validation of bioinformatics pipeline to detect NEMO-deleted exon 5 autoinflammatory syndrome (NEMO-NDAS) and preliminary clinical and immunologic characterization [abstract. Arthritis Rheumatol. (2021) 73. Available online at: https://acrabstracts.org/abstract/validation-of-bioinformatics-pipeline-to-detect-nemo-deleted-exon-5-autoinflammatory-syndrome-nemo-ndas-and-preliminary-clinical-and-immunologic-characterization/.
14. Roos D, van Leeuwen K, Hsu AP, Priel DL, Begtrup A, Brandon R, et al. Hematologically important mutations: The autosomal forms of chronic granulomatous disease (third update). Blood Cells Mol Dis. (2021) 92:102596. doi: 10.1016/j.bcmd.2021.102596
15. Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, et al. A genomic mutational constraint mamp using variation in 76,156 human genomes. Nature. (7993) 625:92–100. doi: 10.1038/s41586-023-06045-0
16. Schubach M, Maass T, Nazaretyan L, Röner S, and Kircher M. CADD v1.7: Using protein language models, regulator CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. (2024) 52:D1143–54. doi: 10.1093/nar/gkad989
17. Thorvaldsdottir H, Robinson JT, and Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. (2013) 14:178–92. doi: 10.1093/bib/bbs017
18. Chusid MJ, Parrillo JE, and Fauci AS. Chronic granulomatous disease. Diagnosis in a 27-year-old man with Mycobacterium fortuitum. JAMA. (1975) 233:1295–6. doi: 10.1001/jama.233.12.1295
19. Hayrapetyan A, Dencher PC, van Leeuwen K, de Boer M, and Roos D. Different unequal cross-over events between NCF1 and its pseudogenes in autosomal p47(phox)-deficient chronic granulomatous disease. Biochim Biophys Acta. (2013) 1832:1662–72. doi: 10.1016/j.bbadis.2013.05.001
20. Noack D, Rae J, Cross AR, Ellis BA, Newburger PE, Curnutte JT, et al. Autosomal recessive chronic granulomatous disease caused by defects in NCF-1, the gene encoding the phagocyte p47-phox: mutations not arising in the NCF-1 pseudogenes. Blood. (2001) 97:305–11. doi: 10.1182/blood.V97.1.305
21. Bakri FG, Mollin M, Beaumel S, Vigne B, Roux-Buisson N, Al-Wahadneh AM, et al. Second report of chronic granulomatous disease in Jordan: Clinical and genetic description of 31 patients from 21 different families, including families from Lybia and Iraq. Front Immunol. (2021) 12:639226. doi: 10.3389/fimmu.2021.639226
22. Merling RK, Kuhns DB, Sweeney CL, Wu X, Burkett S, Chu J, et al. Gene-edited pseudogene resurrection corrects p47phox-deficient chronic granulomatous disease. Blood Adv. (2016) 1:270–8. doi: 10.1182/bloodadvances.2016001214
23. Schnappauf O, Heale L, Dissanayake D, Tsai WL, Gadina M, Leto TL, et al. Homozygous variant p.Arg90His in NCF1 is associated with early-onset interferonopathy: a case report. Pediatr Rheumatol Online J. (2021) 19:54. doi: 10.1186/s12969-021-00536-y
24. Meng Y, Ma J, Yao C, Ye Z, Ding H, Liu C, et al. The NCF1 variant p.R90H aggravates autoimmunity by facilitating the activation of plasmacytoid dendritic cells. J Clin Invest. (2002) 132:e153619. doi: 10.1172/JCI153619
25. Siddiqui S, Anderson VL, Hilligoss DM, Abinun M, Kuijpers TW, Masur H, et al. Fulminant mulch pneumonitis: an emergency presentation of chronic granulomatous disease. Clin Infect Dis. (2007) 45:673–81. doi: 10.1086/520985
26. Williams D, Kadaria D, Sodhi A, Fox R, Williams G, Threlkeld S, et al. Chronic granulomatous disease presenting as. Aspergillus fumigatus pneumonia in a previously healthy young woman. Am J Case Rep. (2017) 18:351–4. doi: 10.12659/AJCR.902764
27. Schwenkenbecher P, Neyazi A, Donnerstag F, Ringshausen FC, Jacobs R, Stoll M, et al. Chronic granulomatous disease first diagnosed in adulthood presenting with spinal cord infection. Front Immunol. (2018) 9:1258. doi: 10.3389/fimmu.2018.01258
28. Marois L, Drouin D, Leduc C, Fernandez I, Manganas H, Gosse G, et al. Chronic granulomatous disease presenting at age 52 with fulminant mulch pneumonitis. J Allergy Clin Immunol Glob. (2022) 1:322–4. doi: 10.1016/j.jacig.2022.06.004
29. Ramanuja S, Wolf KM, and Sadat MA. Newly diagnosed chronic granulomatous disease in a 53-year-old woman with Crohn disease. Ann Allergy Asthma Immunol. (2005) 95:204–9. doi: 10.1016/S1081-1206(10)61212-4
Keywords: NCF1, CGD, chronic granulomatous disease (CGD), NGS, ONT long read sequencing, pseudogene, genetic diagnosis
Citation: Hsu AP, Karlins E, Lack J, Pepper TJ, Lau K, Marshall-Batty KR, Long Priel D, Davis J, Fink DL, Zerbe CS, Gallin JI, Malech HL, Holland SM and Kuhns DB (2025) Reliable genetic diagnosis of NCF1 (p47phox)-deficient chronic granulomatous disease using high-throughput sequencing. Front. Immunol. 16:1640496. doi: 10.3389/fimmu.2025.1640496
Received: 03 June 2025; Accepted: 24 July 2025;
Published: 18 August 2025.
Edited by:
Andrew R Gennery, Newcastle University, United KingdomReviewed by:
Kerstin Felgentreff, Ulm University Medical Center, GermanyConor John O’Donovan, University of Bristol, United Kingdom
Copyright © 2025 Hsu, Karlins, Lack, Pepper, Lau, Marshall-Batty, Long Priel, Davis, Fink, Zerbe, Gallin, Malech, Holland and Kuhns. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy P. Hsu, dHdpbnNAbWFpbC5uaWguZ292