 Xiangsong Cheng1†Shu Chen2†Yilong Fu3†Runze Jiang4Yanlong Jing2Bizhu Zhao3Dong Guo3
Xiangsong Cheng1†Shu Chen2†Yilong Fu3†Runze Jiang4Yanlong Jing2Bizhu Zhao3Dong Guo3 Liangyu Wang3
Liangyu Wang3 Zi Ye5*
Zi Ye5* Yumeng Li4*Xianliang Chen1*
Yumeng Li4*Xianliang Chen1*- 1Department of Respiratory Medicine, Heart Center, Henan Provincial People’s Hospital, Central China Fuwai Hospital of Zhengzhou University, Fuwai Central China Cardiovascular Hospital & Central China Branch of National Center for Cardiovascular Diseases, Zhengzhou, Henan, China
- 2School of Basic Medical Sciences, Xinxiang Medical University, Xinxiang, Henan, China
- 3Clinical Medicine, The First Clinical School of Zhengzhou University, Zhengzhou, Henan, China
- 4Shandong University of Traditional Chinese Medicine, Jinan, Shandong, China
- 5Department of Scientific Research, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
Background: Non-small cell lung cancer (NSCLC) was a major cause of cancer-related mortality globally. Despite advancements in immunotherapy and targeted therapies, clinical outcomes were still limited by tumor heterogeneity and treatment resistance. The transcription factor (TF) FOS, a key component of the AP-1 complex, was linked to tumor progression and therapy resistance in various cancers, but its precise mechanisms remained unclear, and its role in lung adenocarcinoma (LUAD) was unknown. We investigated the tumor microenvironment (TME) of LUAD using single-cell RNA sequencing (scRNA-seq) to identify potential therapeutic vulnerabilities and FOS-driven mechanisms.
Methods: We identified fourteen cell types by analyzing scRNA-seq data from LUAD samples (GSE164789) using Seurat (v4.4.0) and Harmony for batch correction. InferCNV was utilized to characterize the tumor cell subtypes after they were clustered using marker genes. CytoTRACE and Monocle were used to create pseudotime trajectories in order to map differentiation states. CellChat revealed intercellular communication networks, while SCENIC identified TF regulatory modules. The CCK-8, Edu, Transwell, and wound healing assays showed that FOS knockdown functionally validated A549 and NCI-H1975 cells. Furthermore, a prognostic model was developed.
Results: We discovered that invasive LUAD was dominated by a highly stem-like C0 MAFF+ tumor cell subtype that produced chemokines and activated lipid metabolism. These cells stimulated immunosuppression and tumor-associated macrophage (TAM) differentiation by interacting with macrophages via MIF-(CD74+CD44) signaling. Experiments using FOS knockdown demonstrated its role in maintaining invasion, migration, and proliferation. Using the MTRS model, patients were categorized into high- and low-risk cohorts, high-risk patients exhibited unique drug sensitivities. Immunoprofile analysis revealed higher M1 macrophages in high-risk patients, suggesting that FOS inhibition could repolarize TAMs and enhance immunotherapies.
Conclusion: Our studies show that FOS is a main regulator of C0 MAFF+ TCs in LUAD, polarizing macrophages via MIF and rewiring lipid metabolism to support cancer. The MTRS model offers clinical value for risk assessment even if FOS inhibition shows promise as a therapeutic approach to raise immunotherapy efficacy. Targeting the FOS could cause TME immunosuppression to be disrupted, thus LUAD presents a fresh precision oncology approach.
Introduction
Across the world, the incidence of lung cancer gradually climbed and it ultimately became the foremost reason for cancer fatalities. In addition, smoking served as a major factor that heightened the risk of developing lung cancer. Chronic pulmonary diseases, pulmonary infections, occupational and environmental exposures, and lifestyle factors were all considered non-tobacco risk factors (1). Roughly one-third to one-half of lung cancer cases experienced dyspnea, which could have resulted from direct malignancy in the airways or from involvement of lung parenchyma or pleura. Patients were also at risk of developing pulmonary embolism, pneumothorax, pleural effusion, and/or pericardial effusion. Other less frequent symptoms included hoarseness brought on by involvement of the recurrent laryngeal nerve and chest pain from local tumor infiltration (2).
Lung adenocarcinoma (LUAD) was the most prevalent histological subtype of non-small cell lung cancer (NSCLC), which accounted for about 85% of patient diagnoses (3). In LUAD, it was further categorized into adenocarcinoma in situ (AIS), minimally invasive adenocarcinoma (MIA) and invasive adenocarcinoma (IAC). For AIS or MIA, it was defined that if complete resection was performed, these patients could have a disease-specific survival rate of close to 100% (4). Furthermore, in IAC, a strong interaction induced by TGF-β signaling between cancer cells and the tumor microenvironment (TME) was identified, which was not observed in AIS and MIA, indicating that IAC represented a more malignant phenotype (5).
Surgical treatment was most suitable for the initial phase of lung cancer and was regarded as the best treatment approach (6). Nevertheless, the majority of lung cancer patients were generally detected only when the disease had already advanced to a later stage, which may be linked to a poor prognosis. Clinicians also faced difficulties due to treatment options and prognostic evaluation limitations (7). The substantial problem of drug resistance, whether in low-toxicity targeted therapies or conventional chemotherapy, was one of the causes of the high death rate linked to NSCLC (8). The EGFR was found to be a major gene implicated in lung cancer, since mutations were detected in more than 40% of adenocarcinomas. Three drugs were available for the treatment of EGFR-mutant cancers. In 2007, researchers discovered a second driver gene that was present in 5-7% of adenocarcinomas. This gene, known as ALK, encoded a poorly understood signaling protein that occasionally underwent gene rearrangements, causing persistent activation of the protein. However, the benefits of these targeted therapies were often temporary, with most tumors developing resistance after approximately one year of remission (9). Furthermore, because early-stage LUAD was usually asymptomatic, the diagnosis was frequently delayed, resulting in late-stage diagnoses for the majority of patients (10). Given that a 65% survival rate over five years, only 30% of patients received a stage I cancer diagnosis, in patients with advanced stages, this rate dropped to 5% or 6% (11). The overall survival rate for LUAD patients remained low despite improvements in surgical resection, chemotherapy, radiotherapy, and molecular targeted therapy (12). Immunotherapy, including LUAD, has emerged as a recognized cancer treatment approach in recent years (13). However, because immunotherapy suppressed immune activity in specific TME and caused resistance and adverse reactions, merely a minor fraction of the diseased gained benefits, with individual differences and tumor heterogeneity limiting its effectiveness (14). Due to the heterogeneity of LUAD, developing effective personalized therapies continued to pose a significant challenge (15).
Since FOS was identified among the first viral genes, it encoded a leucine zipper protein that could associate with JUN family members by dimerization. Through this process, the AP-1 TF complex was formed, which in turn had a significant impact on tumor cell growth, differentiation, survival, and how cells responded to DNA damage (16, 17). This AP-1 complex promoted tumor development by directly transcriptionally repressing the p53 served as a tumor suppressor (18). Existing studies indicated that FOS was potentially utilized as a core genetic target in the therapeutic approach to LUAD (19). FOS was shown to disrupt cell polarity and induce epithelial-mesenchymal transition (EMT) in breast cancer. Consequently, this process enhanced the ability of breast cancer cells to invade and metastasize (20). Within tongue carcinoma, more FOS was bound and expressed the more serious the tongue lesions were (21). Given its proven functions in other malignancies, FOS may also be essential for the evolution of LUAD.
A potent technique for researching cell biology at a never-before-seen level of resolution is single-cell RNA sequencing (scRNA-seq). It allowed scientists not only to analyze the heterogeneity of cells but also to detect rare, significant cell types while simultaneously exploring the interactions and communication occurring between different cells. Its wide range of uses included both fundamental and applied research fields (22). In order to clarify the cellular heterogeneity and features of the TME, we carried out a scRNA-seq investigation on LUAD cells. New therapeutic ideas developed from this research contributed to improve patient survival rates and prognosis. This investigation aims at investigating the function of FOS in LUAD with an eye toward the requirement of looking at the molecular pathways supporting treatment resistance and tailored therapeutic methods.
Materials and methods
Origination of data
To investigate the TME of LUAD, we analyzed scRNA-seq data (23, 24) obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) (GSE164789). In addition to analyzing gene expression patterns, the study combined clinical information and mutation data, which together provided a more comprehensive foundation for the analysis (25). Since the data were publicly accessible, ethical approval was not required.
Processed and visualized raw data
The Seurat package (v4.4.0), along with R software (v4.3.3), was employed to process the raw gene expression data, thereby facilitating robust analysis (26). The DoubletFinder package (v2.0.3) was first employed to effectively eliminate possible doublet cells while also filtering out cells of low quality. To eliminate low-quality cells, we applied stringent filtering criteria: cells with nFeature (number of detected genes) outside the range of 300-6,000 or nCount (total number of counts) beyond 500-75,000 were excluded. Eliminated also were cells with mitochondrial gene expression more than 25% or red blood cell gene expression more than 5%.
The “Normalize Data” function in Seurat helped us to normalize the data, subsequently, the “Find Variable Features” tool found 2,000 extremely highly variable genes (27). The “ScaleData” tool helped to further standardize gene expression counts so suited for principal component analysis. Harmony R package (v1.2.0) lower sample batch effects (28). Moreover, the “Cell Cycle Scoring” tool indicated cell cycle phases to ensure appropriate characterizing of cellular states (29). For dimensionality reduction, clustering analyses were performed and gene expression patterns were visualized using UMAP (30, 31).
Classification of cell subtypes
Cell subtypes were determined by performing clustering analysis in Seurat, utilizing the “FindNeighbors” and subsequently the “FindClusters” functions (32, 33). The diverse cell subtypes within the TME were accurately classified because clusters were annotated according to the average expression levels of established marker genes.
Assessment of cell stemness
Stemness of cells was evaluated by computing gene set activity scores from scRNA-seq data using the AUCell method. Gene sets might be ranked based on degree of enrichment inside individual cells. This approach defined the variations in tumor cell subgroups and their stemness characteristics.
Trajectory analysis of tumor cell subtypes
We used CytoTRACE, a computational tool for predicting developmental potential, to esitmate the differentiation status of tumor cell subtypes (34). Pseudotime trajectories were created from Monocle (v2.24.1), while lineage architecture was deduced from Slingshot (v2.8.0), which used a cluster-based minimal spanning tree (MST) (35). Smooth trajectory curves produced by the “getLineages” and “getCurves” let differentiation paths and branching events be seen.
Enrichment studies of cellular subtypes
Using “FindAllMarkers,” differentially expressed genes (DEGs) were found in Seurat. We investigated their biological relevance using Genomic Variant Analysis (GSVA) and Gene Set Enrichment Analysis (GSEA). Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), and ClusterProfiler (v4.8.2) derived functional annotations (36, 37). Using an adjusted P-value threshold of 0.05, we detected a range of GO terms that showed significant enrichment, thereby helping to elucidate the regulatory processes associated with various tumor cell subtypes (38, 39).
Analysis of interactions among cells
The CellChat R package (v1.6.1) was used to analyze cellular communications within the TME (40). The “IdentifyCommunicationPatterns” function measured discrete communication patterns, and the variations in the intensity of interactions between cells were analyzed using the “netVisual diffInteraction” (41). This analysis revealed key signaling pathways and ligand-receptor pairs mediating cellular crosstalk in LUAD. To visually examine the signals that enter and exit every cell, we utilized scatter plots, heatmaps, and a range of other visualization techniques. Moreover, we considered associations among different cell types to be meaningful when the P value was less than 0.05.
Single-cell regulatory network inference and clustering investigation
We built clusters of TCs and single-cell regulatory networks using Python (v3.9.19) and the pySCENIC library (v0.12.1) (42). Using data on rankings of human gene motif from (https://resources.aertslab.org/cistarget/), we determined the top five TFs displaying the most notable expression changes.
Established and assessed a prognostic prediction model
The central goal of this investigation was to determine how effectively specific genes associated with various LUAD subtypes could predict patient survival outcomes. After identifying the most significant prognostic genes, we demonstrated that they served as strong predictors for constructing reliable prognostic models (43). This identification process was carried out through both univariate and multivariate Cox proportional hazards analyses (44). Thereafter, we employed a risk evaluation technique, in which the final risk value was obtained by adding up the multiplied values of gene expression amounts and their matching coefficients: . Furthermore, we derived optimal cut-off values using the “surv_cutpoint” function, which enabled a comparative analysis of prognostic differences among patient subgroups. Survival analysis was subsequently conducted by means of the “Survival” package in R (v4.3.3), and survival curves were also constructed using the “ggsurvplot” function so as to examine the predictive accuracy of our risk model (45). In addition, we assessed the model’s reliability by constructing ROC curves (46) with the package of “timeROC” (v0.4.0), thus offering a thorough assessment of both the model’s accuracy and its calibration.
Analysis of immune microenvironment
To comprehensively analyze the immune landscape, we first used the CIBERSORT R software package (v0.1.0) to estimate immune cell scores for each patient (47, 48). Subsequently, we investigated the infiltration of immune cells in detail and also evaluated the differential expression of immune checkpoint-related genes (49). We further examined associations among risk scores, immune cell populations, and genes included in the model. Additionally, we made use of the Tumor Immune Dysfunction and Rejection (TIDE) platform (http://tide.dfci.harvard.edu) to predict patient responses to tumor immunotherapy.
Identification of malignant cells by inferCNV
The inferCNV R software package (v1.16.0, https://github.com/broadinstitute/inferCNV) served as our primary tool for CNV inference, enabling us to characterize the CNV patterns within multiple cell subtypes using the inferCNV algorithm. We assessed both relative gene expression and chromosome location data to infer CNV status across individual cells (50).
Analysis of drug sensitivity
To strengthen the clinical significance of our results regarding drug applications, we evaluated the sensitivity of multiple agents. Specifically, we employed the “pRRophetic” package (v0.5) to estimate the half-maximal inhibitory concentration (IC50) for every individual case and subsequently compared drug sensitivities across the high-risk and low-risk groups.
Cellular culture
The A549 cell strain was cultured in F-12K medium under standardized conditions, which included a temperature of 37 °C, 5% CO2, and 95% humidity. The medium was fortified by incorporating 10% fetal calf serum along with 1% antimicrobials. Likewise, the NCI-H1975 cell strains were routinely preserved in RPMI-1640 medium with similar environmental conditions, and this medium also contained 10% fetal calf serum as well as 1% antimicrobials to ensure the cells attained their best growth rates.
Transfection of the cells
A decrease in FOS expression was observed, which was partly a result of RNA synthesized by GenePharma (Suzhou, China). After introducing the FOS-targeting siRNAs (siFOS-1 and siFOS-2) combined with a control siRNA (si-NC), cells were distributed into 6- well chambers at a coverage level of 50%. Transfection was carried out in accordance with the instructions from the manufacturer, utilizing Lipofectamine 3000RNAiMAX (Invitrogen, USA). Each siRNA (RIbbio, China) was individually introduced into the cells.
Western blotting
Following achievement of 70% confluence in transfected cells, lysis was conducted with RIPA buffer. The lysates underwent centrifugation at 12,000 rpm for 15 minutes to clarify the extracts before being processed by SDS-PAGE. The separated proteins were then transferred onto a PVDF membrane, which was blocked at room temperature for 1.5 hours with 5% calf serum albumin. The membrane underwent incubation throughout the night at 4°C with an anti-FOS antibody, followed by a one-hour incubation with a secondary antibody. Finally, the presence of FOS protein was revealed using a chemiluminescent Western blot substrate.
Quantitative real-time polymerase chain reaction
To extract RNA, cell lysis was achieved with Trizol reagent. All water sources, laboratory instruments, and workspaces were maintained RNase-free throughout the protocol to ensure RNA stability. Once RNA was isolated, the PrimeScript™ reagent kit was used for reverse transcription. Finally, quantitative real-time PCR was conducted, using SYBR Green master mix as the fluorescent indicator for the detection of amplification.
Assay of the viability of the cells
To evaluate the survival of A549 and NCI-H1975 cells following transfection, the Cell Counting Kit-8 (CCK-8) method was applied (51). The cells were plated into 96-well chambers at 5 × 10³ cells per well and, following this, were left to adhere for 24 hours. Afterwards, every well gained 10 µL of CCK-8 reagent (A311-01, Vazyme), followed by an additional 2-hour incubation at 37°C in the absence of light. Light absorption at 450 nm was recorded each day from the first to the fourth day by means of a microtiter reader (A33978, Thermo). The collected light density data were averaged and graphed to illustrate changes in cell viability over the time course.
Experiments of 5-Ethynyl-2’-deoxyuridine proliferation
After the transfection procedure, A549 and NCI-H1975 cells were plated into 6-well chambers at 5 × 10³ cells per well. Cells were allowed to incubate for 24 hours at ambient temperature before adding the EdU working solution for another 2-hour period. Being rinsed two times with PBS, the cells were fixed in 4% paraformaldehyde for a duration of 15 minutes. After fixation, permeabilization was performed, and the cells were quenched for 15 minutes in a solution containing 0.5% Triton X-100 and 2 mg/ml glycine. Following this, 1 ml of Apollo solution and 1 ml of Hoechst stain were applied, and the cells were incubated for 30 minutes. Finally, fluorescence microscopy was employed at the end to image the cells and evaluate their proliferation.
Transwell assay
Initially, cells were deprived of serum for 24 hours by incubating them in a serum-free medium. Following this, the cellular suspension was blended with Matrigel (BD Biosciences, USA) and the upper well of the Costar plate was loaded with the cell suspension, while the lower well received serum-containing medium to form a chemotactic gradient. Then cells were allowed to migrate and invade in a constant-temperature apparatus for a duration of 48 hours. Afterwards, 4% paraformaldehyde solution served to fix the cells, and crystal violet staining was employed to evaluate their invasion capacity visually.
Wound healing assay
Cells with stable transfection were cultured in 6- well chambers and incubated at 37°C within a moist environment supplemented with 5% CO2 until they became confluent. Sterile 200 µL pipette tips were then used to create straight scratches in the cell monolayer. The wells were carefully washed with PBS to remove floating cells and debris. Following this, the cells were incubated with serum-free medium to facilitate migration. Photographs were captured at both the beginning (0 hours) and after 48 hours, and scratch widths were measured using Image-J for subsequent analysis.
Statistical procedures
The datasets were subjected to analysis using R (v4.3.3) and Python (v3.9.19). For comparisons between groups, Wilcoxon’s test and Pearson’s correlation coefficient were applied (52). We interpreted the significance at the statistical level following the criteria listed below: *P< 0.05, **P< 0.01, ***P< 0.001, and ****P< 0.0001, “ns” was used to denote results without statistical significance. These analytical methods and cutoffs were adopted to confirm the robustness of the findings.
Results
Single cell landscape of LUAD
To explore the ECs, we analyzed their CNVs using inferCNV with ECs as a reference (Supplementary Figure 1). We examined the obtained dataset to explore the single-cell profile present in the LUAD environment. Figure 1 illustrated our procedure. By examining five localized adenocarcinomas and twenty-six infiltrating adenocarcinomas from GSE164789, we employed dimensionality reduction clustering with UMAP plots. This analysis initially exhibited how thirty-one individual samples were distributed and subsequently identified fourteen cell types: T and NK cells, epithelial cells (EPCs), macrophages, plasma cells, mesenchymal cells (MCs), endothelial cells (ECs), monocytes, fibroblasts, B cells, conventional dendritic cells type 2 (cDC2), proliferating cells, conventional dendritic cells type 1 (cDC1), myofibroblasts, and plasmacytoid dendritic cells (pDCs). Furthermore, we presented the distribution of cells within three groups (AIS, IAC, MIA) and across three different phases of the cell cycle (G1, G2/M, S) (Figure 2A). We examined the expression profiles of nCount RNA, nFeature RNA, cell stemness AUC, and pMT among the fourteen cell types and three different groups (Figures 2B, C). Additionally, Figure 2D illustrated the cellular distribution of the dataset. Volcano plots revealed the presence of different genes among these cell types (Figure 2E). In EPCs, TCP11L2, POM121C, HYAL2, HBP1, and SYNE2 were found to be upregulated, while BCOR, LHFPL6, IKZF2, SRPK1, and SRSF8 exhibited downregulation. In macrophages, EIF5AL1, ZNF468, AGO1, USP25, and TMEM165 showed upregulation, whereas UPRT, PDHX, MTFR1L, TRIOBP, and TMEM141 showed downregulation. We also provided the top five marker genes corresponding to the fourteen cell types, as depicted in Figure 2F. Through investigating the ratios of individual cell types in the three groups alongside cell cycle data, we found that each cell type distributed across separate phases within the cell cycle, with EPCs and macrophages constituting the main components of the IAC group, predominantly located in the differentiated G1 phase (Figure 2G).
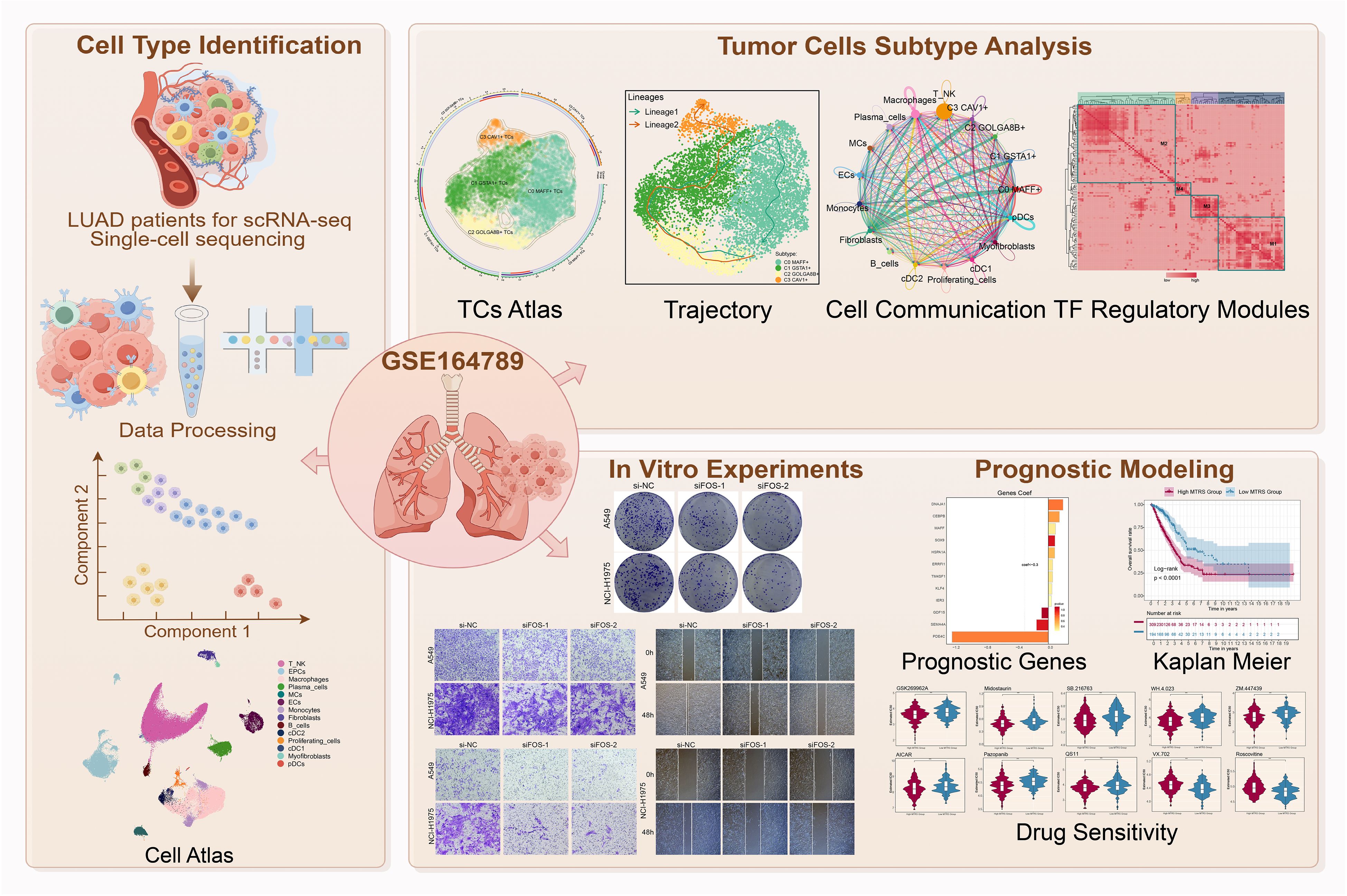
Figure 1. Graphical abstract. Workflow demonstrated LUAD single-cell RNA sequencing analysis of the GSE 164789 dataset.
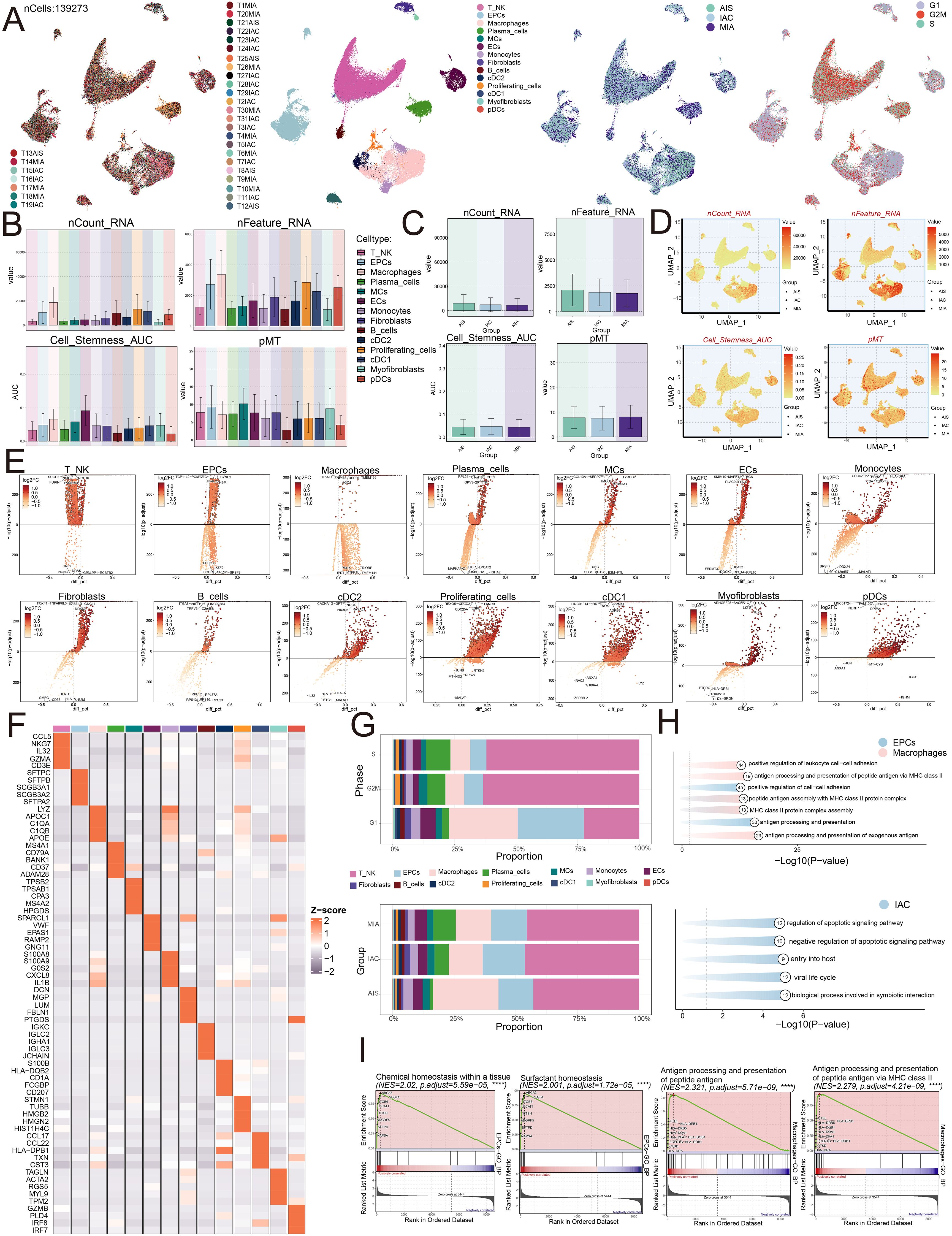
Figure 2. Single-cell profiling of LUAD. (A) The UMAP plots mapped out the distribution of thirty-one samples, and additionally portrayed the allocation of fourteen unique cell types—including T and NK cells, EPCs, Macrophages, Plasma cells, MCs, ECs, Monocytes, Fibroblasts, B cells, cDC2, Proliferating cells, cDC1, Myofibroblasts, and pDCs—across the total cell population. In addition, these plots provided a visual of the group distributions and various cell cycle phases (from left to right). (B) The bar graphs represented the levels of nCount RNA, nFeature RNA, cell stemness AUC, and pMT for each of the fourteen cell types. (C) Bar graphs were utilized to display the expression analyses of three groups in terms of nCount RNA, nFeature RNA, cell stemness AUC, and pMT. (D) The UMAP plots depicted the distribution of nCount RNA, nFeature RNA, cell stemness AUC, and pMT, with each group identified by different symbols. (E) Volcano plots were used to present the five most significantly upregulated and downregulated genes across the fourteen distinct cell types. (F) The heatmap presented the distribution of the top five marker gene expressions among diverse cell populations. (G) The distribution of cell types across various phases and groups was visualized using stacked bar graphs. (H) Enrichment analysis was visualized for EPCs, Macrophages, and the IAC group. (I) GSEA enrichment analysis revealed the upregulated pathways in EPCs and Macrophages.
We then conducted functional enrichment analysis. The analysis suggested that IAC group displayed increased activity in pathways connected to the regulation of apoptotic signaling, negative regulation of apoptotic signaling, entry into the host, viral life cycle, and biological processes involved in symbiotic interaction. EPCs indicated enrichment within pathways related to positive regulation of cell-cell adhesion and antigen processing and presentation, while macrophages were enriched in pathways associated with positive regulation of leukocyte cell-cell adhesion, antigen processing and presentation of peptide antigens via MHC class II, peptide antigen assembly with MHC class II protein complex, assembly of MHC class II protein complexes, and antigen processing and presentation of exogenous antigens (Figure 2H). Furthermore, EPCs demonstrated upregulation in pathways related to chemical homeostasis within tissues and surfactant homeostasis, whereas macrophages displayed significant upregulation in antigen processing and presentation of peptide antigens and antigen processing and presentation of peptide antigens via MHC class II (Figure 2I).
Visualization analysis of LUAD tumor cell subtypes
The involvement of the TME in tumorigenesis became widely recognized, as it harbored TCs that, through their interactions with other cells via the circulatory and lymphatic networks, played an essential part in both the emergence and advancement of cancer (53). We characterized the significance of TCs in the TME. Four subtypes of LUAD TCs were classified according to the expression of marker genes: C0 MAFF+ TCs, C1 GSTA1+ TCs, C2 GOLGA8B+ TC, and C3 CAV1+ TCs, and demonstrated the nCount RNA, nFeature RNA, cell stemness AUC, and pMT for each subtype (Figure 3A). Subsequently, we examined the primary distribution of TCs across different groups using UMAP plots (Figure 3B). Bubble plots illustrated the marker genes associated with various groups and tumor cell subtypes (Figures 3C, D). In MIA, we found that ATP5E, RPL11, SPINK5, MALAT1, and XIST were highly expressed. In AIS, MALAT1, WSB1, VMP1, EMP2, and NEAT1 exhibited high expression levels. In IAC, ATF3, AREG, C16orf89, ATP5E, and RPL11 showed elevated expression. The C0 subtype exhibited high expression of CXCL8, CXCL2, RRAD, ATF3, and AREG. The C1 subtype showed elevated levels of RPL11, ATP5E, GSTA1, C16orf89, and SPINK5. The C2 subtype had increased expression of NEAT1, MALAT1, VMP1, WSB1, and XIST. The C3 subtype displayed high expression of EMP2, CLIC5, CAV2, CAV1, and AGER. Figure 3E visualized these different genes. We found that JUN, ATF3, IRF1, IER5, and MAFF were upregulated in the C0 subtype. RPS1, RPL11, RPS14, RPS18, and RPL37 were upregulated in the C1 subtype. MALAT1, NEAT1, WSB1, MT-ND3, and VMP1 showed increased expression in the C2 subtype. CAV1, CAV2, AGER, EMP2, and NCKAP5 were upregulated in the C3 subtype. We subsequently analyzed how the top five marker genes were differentially expressed (Figure 3F).
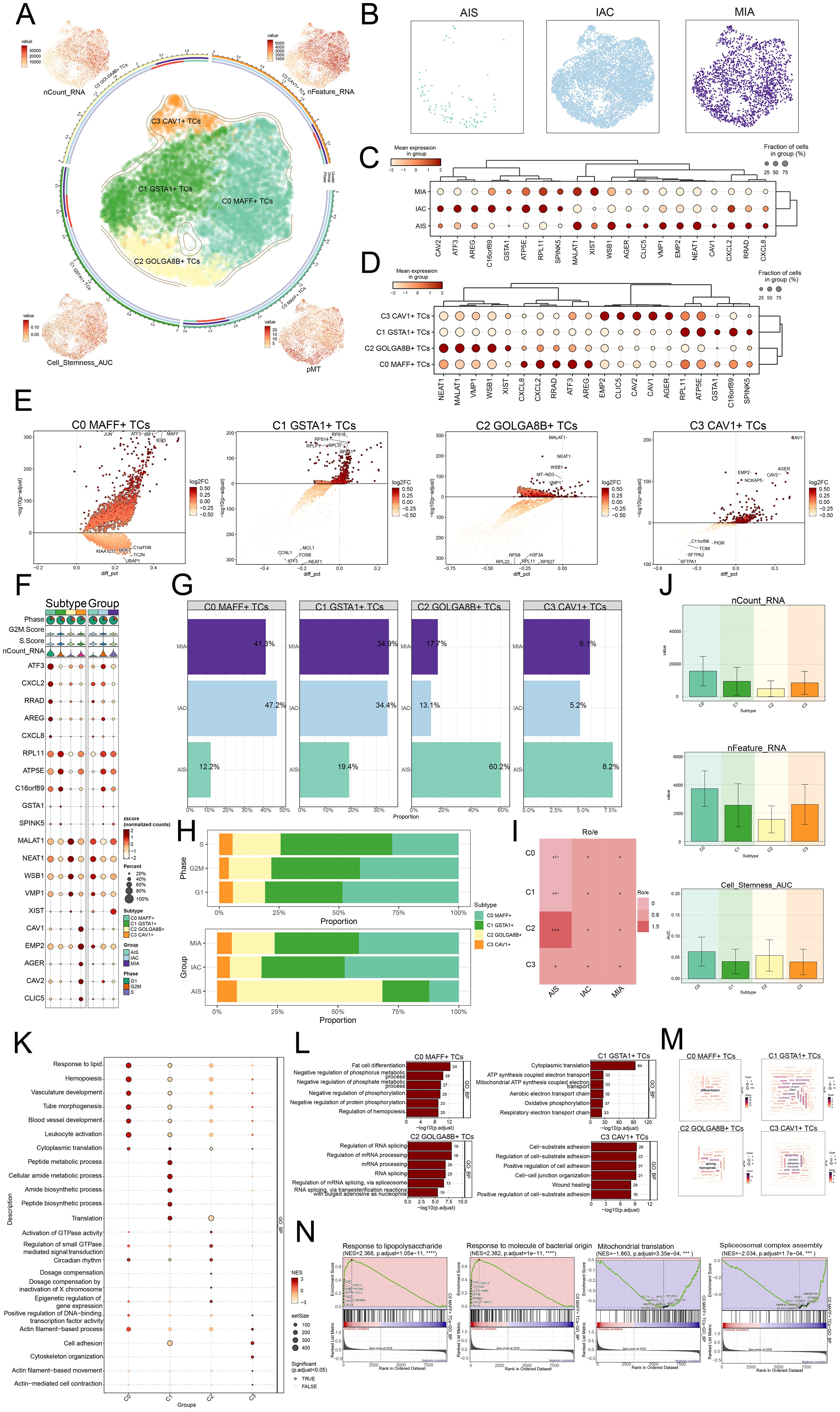
Figure 3. MAFF+ TCs specifically expressed in malignant EPCs and enrichment analysis in TCs of LUAD. (A) The arrangement of four distinct tumor cell subtypes in LUAD was shown using a circular plot, with boundary curves outlining each subtype. The total cell numbers within each class were represented on the outer axis using a logarithmic scale. UMAP plots placed at each of the four corners, moving clockwise from the top left, visualized the distribution of nCount RNA, nFeature RNA, pMT, and cell stemness AUC among all TCs (B) The UMAP plots also revealed how TCs were distributed among AIS, IAC, and MIA. (C) The bubble plot displayed the marker gene expression patterns within three groups. (D) The bubble plot presented the marker gene profiles in the four tumor cell subtypes, with bubble diameter corresponding to the percentage of cells with expression and color reflecting the normalized measurement. (E) Volcano plots displayed the most significantly upregulated and downregulated genes in each of the four subtypes. (F) For each subtype, the mean values of the top five differentially expressed genes were determined were also depicted using bubble plots, where bubble size and color denoted expression percentage and data normalization, respectively. (G) Bar graphs visualized the percentages of different groups within each tumor cell subtype. (H) Stacked bar graphs were also used to visualize the allocation of these subtypes within different phases and groups. (I) The group preference for each subtype was evaluated with the Ro/e score. (J) Bar graphs presented the expression levels of nCount RNA, nFeature RNA, and cell stemness AUC across the subtypes. (K) The GSEA results for each subtype were visualized in a bubble plot. (L) Bar graphs provided insights into biological processes across the subtypes. (M) Biological processes associated with each subtype were shown via word cloud graphs. (N) GSEA was used to analyze both positively and negatively enriched pathways in C0 MAFF+ TCs.
Moreover, ratios within separate groups, phases of development, and specific cell subtypes were determined, discovering that the C0 subtype represented the highest proportion within the IAC, accounting for up to 47.2% (Figures 3G-I). Therefore, the heterogeneity of the IAC group might be related to the C0 subtype.
Next, we presented the results of nCount RNA, nFeature RNA, and cell stemness AUC for different subtypes using bar graphs (Figure 3J). The findings suggested that, in these aspects, C0 demonstrated elevated levels of expression in these metrics relative to the other subtypes. Consequently, we inferred that the C0 subtype likely corresponded to a higher degree of malignancy.
Analysis of enrichment in LUAD TCs
To explore the biological functions of TCs in LUAD, we conducted GSEA analysis across different subtypes. We observed that the C0 subtype was primarily enriched in the response to lipid (Figure 3K). Subsequently, we analyzed the biological processes associated with the four subtypes and the C0 subtype was primarily identified as being enriched in fat cell differentiation, the C1 subtype in cytoplasmic translation, the C2 subtype in regulation of RNA splicing, and the C3 subtype in cell-substrate adhesion (Figure 3L). With regard to the biological processes, we found that the C0 subtype was associated with differentiation (Figure 3M). While showing considerable negative regulation in pathways linked to mitochondrial translation and spliceosomal complex assembly, the GSEA results also revealed that the C0 subtype displayed a significant positive regulation in pathways related with response to lipopolysaccharide and response to molecules of bacterial origin (Figure 3N). C0 subtype linked responses to lipid and molecules of bacterial origin, fat cell differentiation, and hemopoiesis. These results greatly affected the viability and growth of TCs, so facilitating the advancement of LUAD.
Pseudotime analysis revealed the heterogeneity of stemness and developmental stages among LUAD TCs subtypes
In our investigation, we examined the expression of genes connected to stemness in the different tumor cell subtypes and determined that the expression levels of ATF3, CXCL2, RRAD, AREG, and CXCL8 were significantly higher in the C0 subtype (Figure 4A). UMAP visualization revealed how these genes were distributed (Figure 4B). The bar graphs indicated that the expression profiles of ATF3, CXCL2, RRAD, AREG, and CXCL8 in the C0 subtype were expressed at greater levels than in the remaining tumor cell subtypes (Figure 4C).
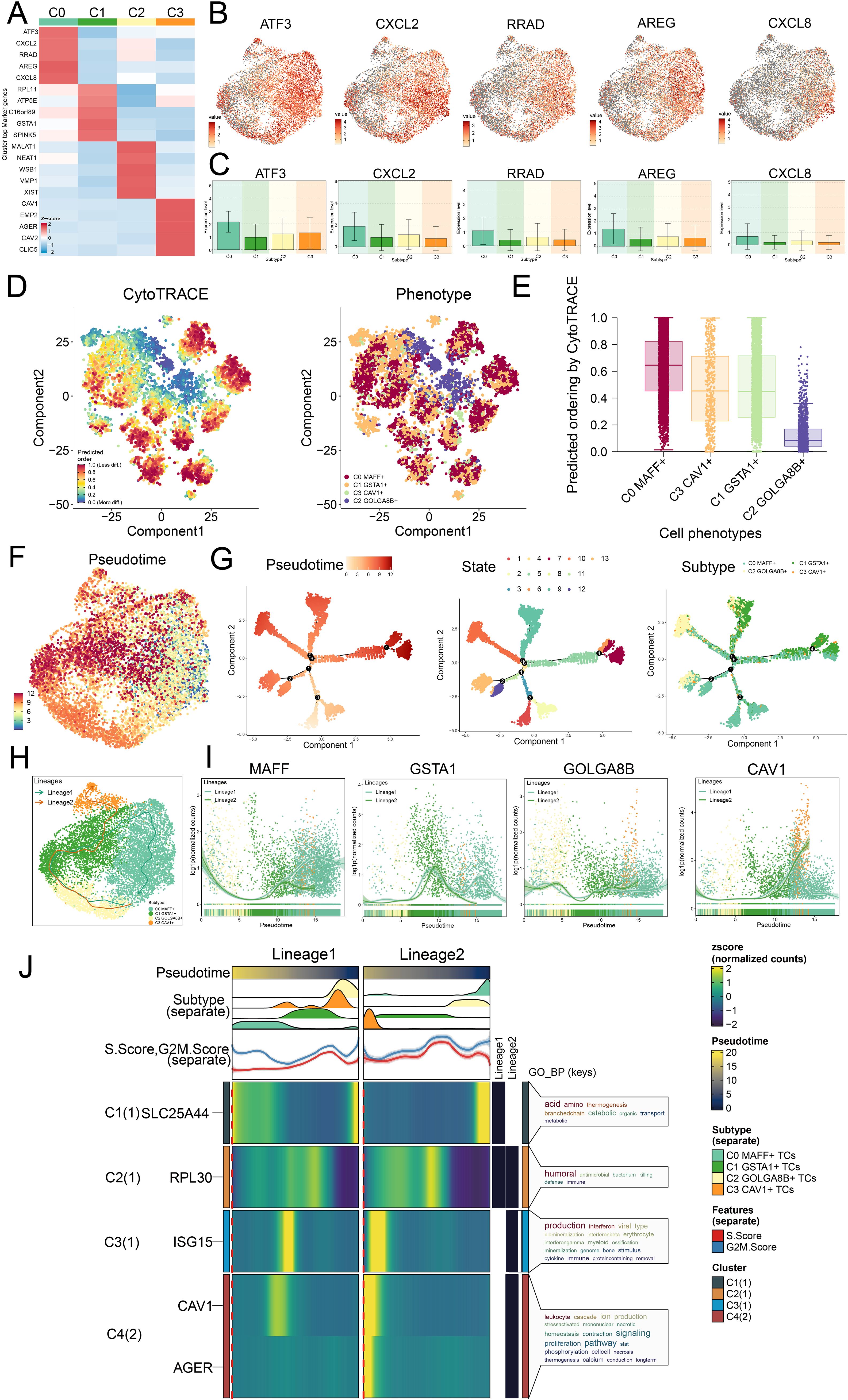
Figure 4. Trajectory analysis on the different tumor cell subtypes. (A) Z-scores for stemness-related marker genes across the four subtypes were displayed in a heatmap. (B) UMAP plots further mapped the distribution of five cell stemness genes among all TCs. (C) Bar graphs illustrated the expression profiles of these five pivotal genes across the four subtypes. (D) Left panel depicted the predicted order distribution within TCs as determined by CytoTRACE, with color gradients reflecting the degree of cell stemness. Right panel illustrated the allocation of tumor cell subtypes, each assigned a specific color. (E) To rank the stemness among tumor cell subtypes, CytoTRACE analysis was implemented. (F) A trajectory analysis was then conducted to map the developmental progression of tumor cell subtypes. (G) The differentiation trajectory was color-coded according to pseudotime (left), state (middle), and subtype (right), thereby offering comprehensive insight. (H) UMAP plot revealed the trajectory of the differentiation among the four identified subtypes, with C0 MAFF+ TCs positioned at the endpoint of lineage 1, solid lines traced the trajectories, and arrows denoted the progression from naive to mature. (I) Dynamic trend plots revealed the fluctuations in expression for the four marker genes analyzed. (J) Heatmaps displayed the GO enrichment pathways active during tumor cell differentiation, and the top bar graphs annotated both pseudotime and tumor cell subtype. Ridgeline plots outlined the density of subtype distribution across pseudotime, while trajectory plots presented the variation of S.Score (red) and G2/M. Score (blue) as pseudotime progressed.
In seeking to understand how LUAD tumor cell subtypes arose and developed, we investigated both their lineage relationships and differentiation states. In Figures 4D-G, we analyzed the results of trajectory and CytoTRACE. CytoTRACE analysis indicated that C0 MAFF+ TCs exhibited a higher degree of stemness (Figure 4E). Stemness was closely related to cellular differentiation, higher levels of stemness correspond to lower degrees of differentiation, and TCs in the latter stages of differentiation often possessed greater stemness.
In order to define how the tumor cell subtypes differentiated, we utilized Slingshot analysis studied their developmental trajectories, and further presented the results through UMAP visualization. We first presented two main differentiation trajectories for the four tumor cell subtypes: lineage 1: C2→C1→C0 and lineage 2: C2→C1→C3. The differences between the two trajectories were primarily observed in later stages, where C2 GOLGA8B+ TCs were located at the initial stages of differentiation in both lineage 1 and lineage 2, and C0 MAFF+ TCs were found at the final stage of lineage 1 (Figure 4H). Additionally, we analyzed the dynamic trends of marker gene expression among the four subtypes, the high expression of MAFF+ was predominantly observed at later stages (Figure 4I), further validating our Monocle analysis findings.
We ultimately conducted analysis of GO-BP enrichment on the subtypes to verify the biological processes pertinent to both lineages (Figure 4J). The dynamic timing approach demonstrated how gene expression in TCs shifted along the two pseudotime trajectories.
The analysis of intercellular communication along with visualization of the MIF signaling pathway
In order to better comprehend the intricacies of cellular responses, we sought to investigate the networks and intercellular connections that underpin ligand-receptor signaling, which allowed us to depict the interactions among different cell types. By employing analysis of CellChat, we established a comprehensive network of cell-cell interactions covering the majority of cell types, such as C0 MAFF+ TCs, C1 GSTA1+ TCs, C2 GOLGA8B+ TCs, C3 CAV1+ TCs, T and NK cells, macrophages, plasma cells, MCs, ECs, monocytes, fibroblasts, B cells, cDC2, proliferating cells, cDC1, myofibroblasts and pDCs (Figure 5A). We found that in the outgoing signaling pattern, C0 MAFF+ TCs predominantly exhibited pattern 1, while JAM, CADM, GDF, OCLN and UGRP1 displayed high expression levels within this pattern. Correspondingly, in the incoming signaling pattern, C0 MAFF+ TCs also primarily represented pattern 1, where SCT, CDH, OCLN, DESMOSOME, and TWEAK showed elevated expression levels (Figure 5B). The heatmaps illustrated the communication patterns and signaling pathways of each tumor cell subtype (Figure 5C). We found that the degree as well as the count of connections involving C0 MAFF+ TCs and macrophages were more significant in both afferent and efferent signals, so it could be inferred that there was a strong interaction between C0 MAFF+ TCs and macrophages (Figures 5D, E).
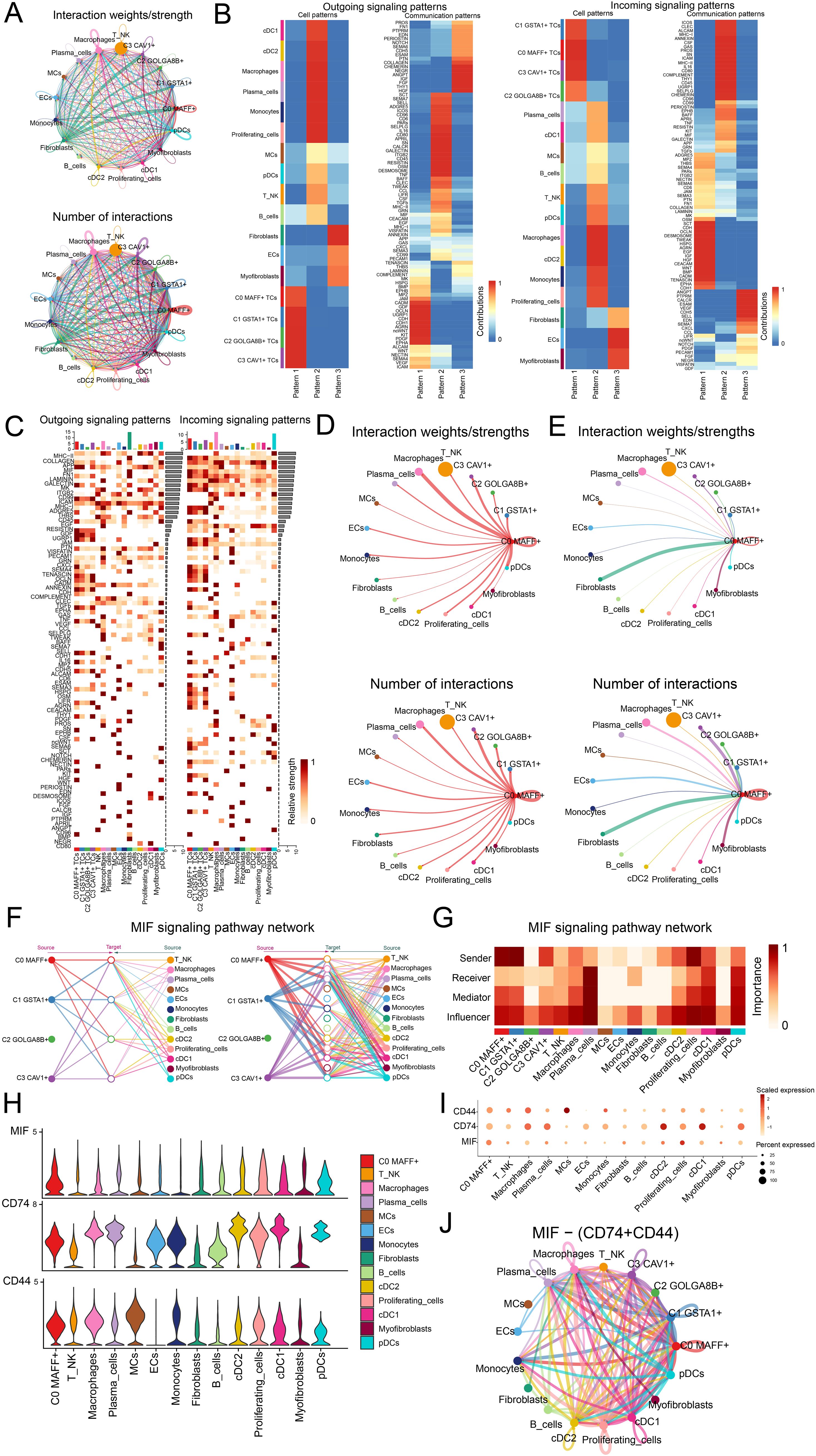
Figure 5. Analysis of the regulatory networks governing genes in C0 MAFF+ TCs. (A) The circle diagrams provided a summary of both the number and intensity of interactions observed between the four tumor cell subtypes and thirteen different cell types, thus shedding light on their interconnections. (B) Separate heatmaps showed the outgoing (left) and incoming (right) signaling effects of the four tumor cell subtypes and the thirteen cell types, as well as the specific input from various proteins within three distinct cell communication patterns. (C) Bar graphs were employed to evaluate and compare the outgoing and incoming signaling strengths among four tumor cell subtypes and thirteen types of cells. Meanwhile, the heatmaps provided a visualization of the intensity with which these proteins, implicated in cell communication, were received among the different groups. (D, E) These circle diagrams further illustrated the strength (top) and number (bottom) of interactions where C0 MAFF+ TCs acted as either the source or the target in relation to other cell types. (F) The hierarchical graph depicted the network of interactions between C0 MAFF+ TCs and other cell types within the MIF signaling pathway. (G) A further heatmap presented the centrality scores for the MIF signaling pathway. (H, I) Both the violin plot and bubble plot demonstrated that C0 MAFF+ TCs and macrophages potentially interacted through the ligand MIF and the receptors CD74 and CD44. (J) Finally, the circle diagram outlined the communication network of MIF-(CD74+CD44) ligand-receptor interactions with TCs receiving the signals.
To present the results more intuitively, we employed a hierarchical graph to depict the relationship between C0 MAFF+ TCs and macrophages. The results demonstrated that the primary interactions between C0 MAFF+ TCs and macrophages occurred through paracrine and autocrine signaling, leading to intense communication (Figure 5F). Subsequently, through network centrality analysis of the MIF signaling pathway, we examined the roles of C0 MAFF+ TCs and macrophages within this pathway. The findings revealed that C0 MAFF+ TCs acted as senders, mediators and influencers, whereas macrophages primarily functioned as influencers, senders and mediators, which could have been linked to the conversion of regular macrophages into tumor-associated macrophages (TAMs) (Figure 5G). We compared the receptor-ligand interactions between C0 MAFF+ TCs and other cell types, allowing us to infer that the MIF ligand from C0 MAFF+ TCs interacted with the CD74-CD44 receptors on macrophages (Figures 5H, I). Further supporting our conclusions was a circular diagram showing the interaction between C0 MAFF+ TCs and macrophages within the MIF-(CD74+CD44) signaling pathway (Figure 5J).
Overall, our work exposed the interactions between C0 MAFF+ TCs and macrophages in LUAD, stressing their possible relationship with the change of macrophages to TAMs, so fostering the development of LUAD.
TFs directed the tumorigenic mechanisms in C0 MAFF+ TCs
TFs governed gene expression through their ability to bind specific consensus sequences located in the local chromatin setting (54). TFs were typically considered to comprise two main domains: a DNA-binding domain and a functional domain. Additionally, these domains were seen as independent and separable, with the DNA-binding domain responsible for gene targeting and the functional domain for facilitating transcriptional regulation (55).
We employed the SCENIC method to perform two-dimensional clustering of TCs from LUAD with reference to various subtypes and groups (Figure 6A). Afterwards, we generated a matrix for the connection specificity index based on the similarity of AUCell scores, which categorized LUAD TCs into four regulatory modules: M1, M2, M3, and M4 (Figure 6B). A visualization analysis of these modules was conducted, revealing that C0 MAFF+ TCs exhibited increased expression in the M1 module compared to other subtypes. Furthermore, UMAP visualizations were employed to examine how each tumor cell subtype was distributed among the various modules, indicating that C0 MAFF+ TCs were predominantly located in the M1 module (Figures 6C, D).
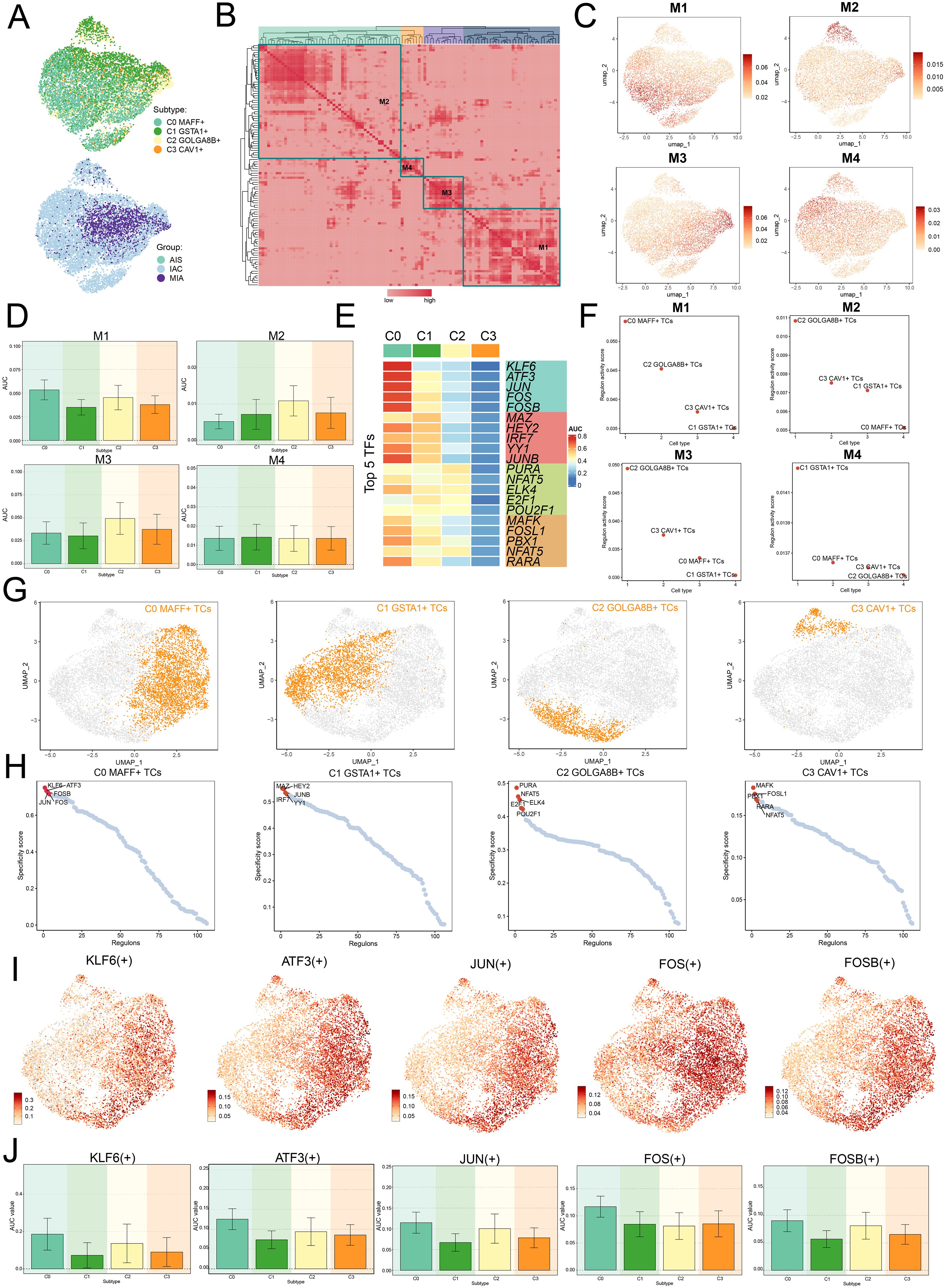
Figure 6. Clustering analysis of TFs and identification of the top five TFs in C0 MAFF+ TCs. (A) UMAP plots visualized all TCs, applying colors determined by regulatory module activity scores and categorizing them by both cell subtypes and group classification. (B) Identification of four regulatory modules among various tumor cell subtypes was visualized in the heatmap, relying on SCENIC modules and AUCell similarity assessment. (C) UMAP plots further illustrated the unique expression signatures of TFs within the four modules of TCs. (D) Bar graphs depicted the AUC values for the four tumor cell subtypes across modules M1, M2, M3, and M4. (E) The heatmap highlighted the top five TFs in each of the four tumor cell subtypes. (F) Scatter plots ranked the regulatory activity scores of TFs for various tumor cell subtypes in all four modules. (G, H) UMAP plots visualized the spatial distribution of each tumor cell, while scatter plots provided the ranking of TFs specificity scores for the top five TFs in every subtype of tumor cell. (I, J) UMAP plots exhibited the distribution of selected TFs, and the bar graphs reported the AUC values of the top five TFs in C0 MAFF+ TCs across different tumor cell subtypes.
Afterwards, we evaluated the top five TFs for each distinct subtype of TCs, with a particular focus on their specificity scores across the various modules. Notably, in M1, C0 MAFF+ TCs displayed the highest regulatory activity score, supporting our earlier conclusions (Figures 6E, F). Furthermore, we ranked the TFs for each subtype and looked at the distribution of various subtypes (Figures 6G, H). In C0 MAFF+ TCs, we examined the distribution and expression levels of five important TFs (KLF6, ATF3, JUN, FOS, and FOSB) in various subtypes. The findings showed that FOS expression was considerably elevated in C0 MAFF+ TCs relative to those in other subtypes (Figures 6I, J). It is still unknown, though, exactly how FOS affects LUAD. Consequently, validating the role of FOS in LUAD cells through in vitro functional studies became a principal focus.
In vitro validation through experimental approaches
We explored the role of FOS in LUAD by performing in vitro assays using A549 and NCI-H1975 cell lines. First, we inhibited FOS and analyzed the related mRNA and expressions of protein at baseline and post-knockdown. Compared to the control group, both cell lines showed substantially decreased mRNA and protein expression (Figure 7A). Furthermore, cell viability was greatly diminished after FOS inhibition (Figure 7B). We subsequently examined the potential for TCs to proliferate. A significant decrease in cell counts was observed in the colony development assay as a result of the FOS suppression (Figures 7C, D). In the EDU experiment, we noted a decrease in colony density following the knockdown, data analysis indicated that siFOS impeded proliferation (Figures 7E, F). To evaluate cell migratory and invasive behaviors, we performed assays of transwell and wound healing. Transwell assays depicted that siFOS TCs showed fewer migrated cells and lower cell density compared to the si-NC group, demonstrating that the knockdown of FOS impaired their migration and invasion potential (Figures 7G-I). The wound healing assays illustrated that siFOS inhibited wound healing of TCs in both A549 and NCI-H1975 cell lines (Figures 7J, K).
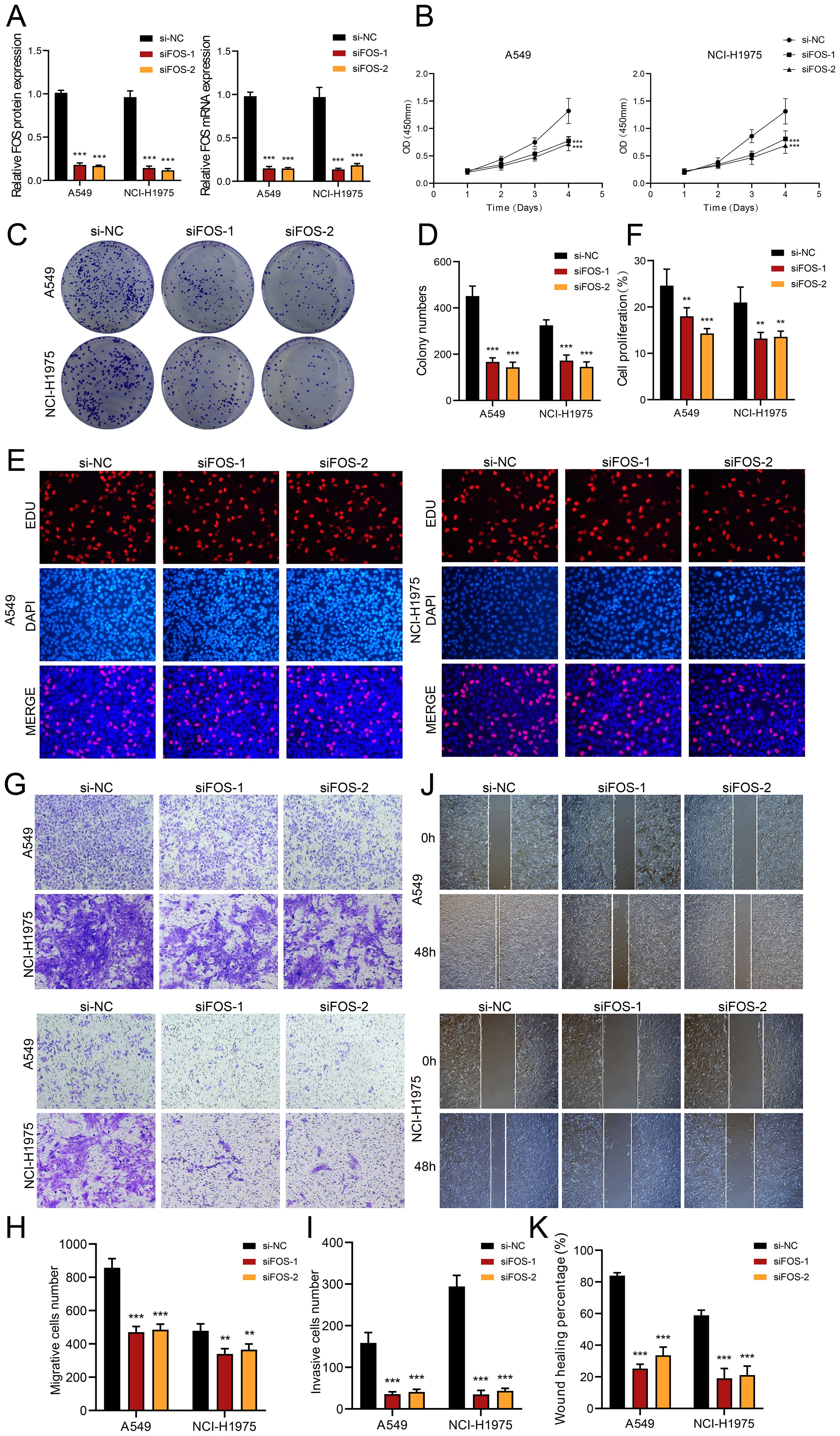
Figure 7. In vitro assays demonstrated the consequences of FOS knockdown. (A) The bar graphs illustrated the levels of genetically encoded proteins (left panel) and gene mRNA (right panel) in the si-NC, siFOS-1, and siFOS-2 groups within the A549 and NCI-H1975 cells. A notable decline in both mRNA and protein expression followed the reduction of FOS. (B) The line graph showed that all groups exhibited sustained growth in both cell lines as time progressed. (C, D) Colony formation assays indicated that the colony numbers dropped markedly after FOS was knocked down, while relevant bar graphs quantitatively displayed colony numbers for all groups in both cell types. (E, F) The assay using EDU staining indicated that proliferation was notably inhibited after FOS knockdown, as further validated by the bar graphs. (G-I) Transwell assays demonstrated that suppressing FOS expression led to decreased migration and invasion capacities in both A549 and NCI-H1975 cells. (J, K) The cell wound healing assays assessed migration ability in C0 MAFF+ TCs after FOS knockdown, which led to a statistically significant reduction in wound healing, as illustrated by the respective bar graphs. **P<0.01 and ***P<0.001.
Collectively, the results suggested that reducing FOS expression impaired cell activity and significantly decreased the migratory, invasive, and proliferative abilities of TCs. This underlined the important tumor-promoting action of FOS, which was essential in the evolution of TCs. Therefore, a main focus of LUAD treatment is FOS inhibition since it might increase patient survival and prognosis.
Developed a prognostic model for LUAD
We utilized univariate Cox regression analysis to explore prognostic indicators in patients and identified twenty-five genes that exhibited prognosis-related associations. We could observe the HR<1 of SEMA4A, PDE4C and GDF15, while the HR values of the remaining genes were >1 (Figure 8A). To reduce potential multicollinearity across genes, we further refined the list of prognosis-related genes (Figure 8B). Following this, we performed multivariate Cox regression analysis on twelve selected genes to determine their respective genetic risk coefficients (Figures 8C, D). Additionally, our analysis of the curve chart and scatter plot demonstrated that the low MAFF+ tumor risk score (MTRS) group exhibited lower risk scores and better survival outcomes compared to the high MTRS group, which was linked to poorer prognosis (Figure 8E). Heatmap further illustrated the distinct prognostic gene expression patterns between these groups, with SEMA4A, PDE4C, and GDF15 more strongly expressed in the low STRS group, suggesting their potential positive impact on prognosis (Figure 8F). Moreover, the ROC curves and corresponding AUC values at 1-year, 3-years, and 5-years were 0.73, 0.68 and 0.63 respectively, highlighting the strong predictive power of the model (Figure 8G). Kaplan-Meier survival analysis further corroborated the worse prognosis observed in the high MTRS group (Figure 8H). In summary, our analysis revealed a negative association between the risk score and survival, supporting the observation that higher risk scores were linked to shorter survival periods (Figure 8I).
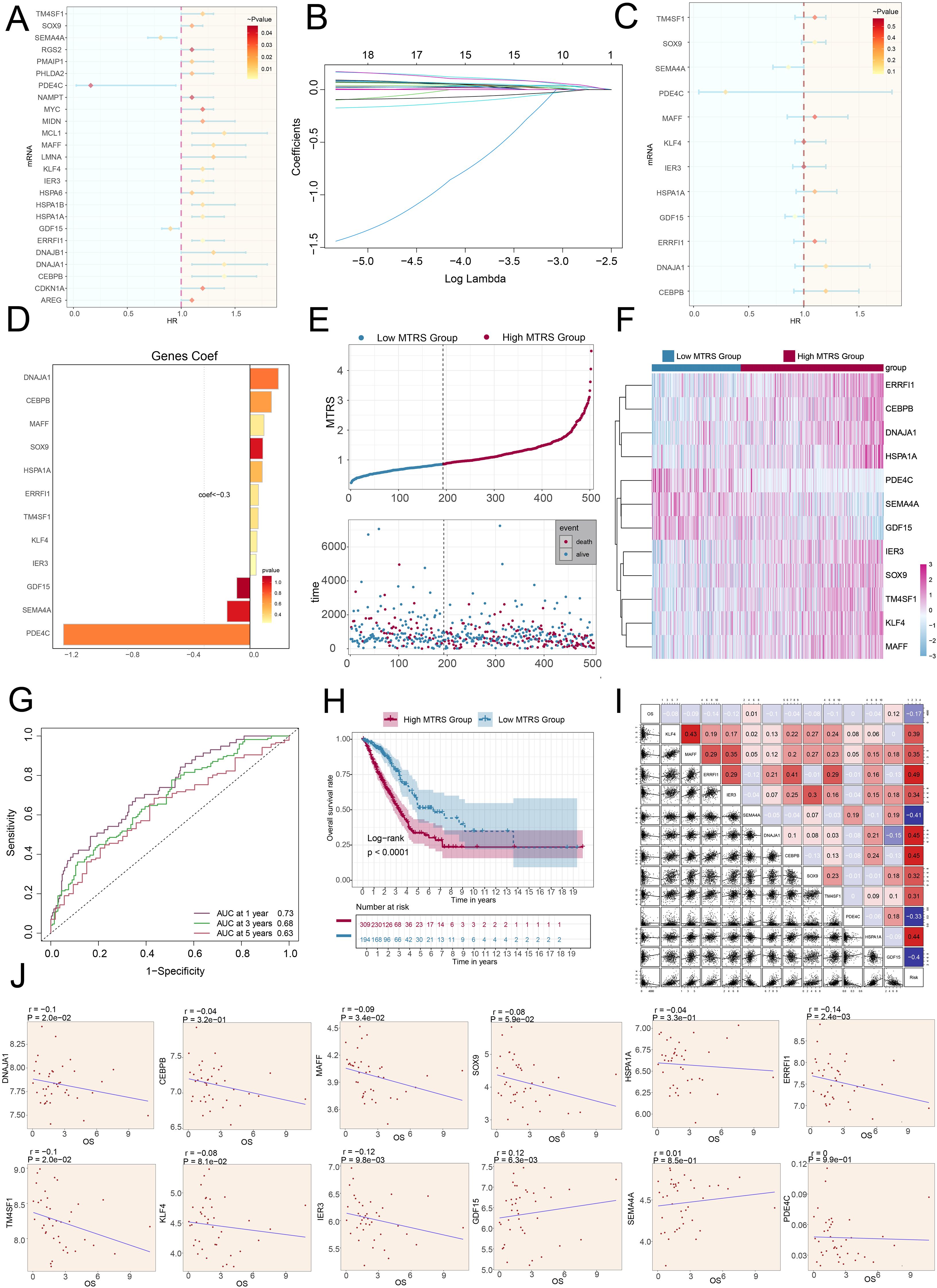
Figure 8. The construction and validation of the model of MTRS. (A) The forest plot represented the twenty-five most prognostically significant genes identified by univariate Cox analysis (HR<1 indicated protective factors, whereas HR>1 indicated risk factors.). (B) Each plotted line indicated the coefficients attributed to the variables, thereby selecting those with significant prognostic implications. (C) The forest plot further presented the top twelve prognostic genes, as determined by multivariate Cox regression. (D) The bar graph illustrated the correlation coefficients for these twelve selected genes. (E) The curve chart contrasted risk scores between low and high MTRS patient groups, and the scatter plot visualized survival status, marking survival with blue and death with red. (F) The heatmap illustrated the expression profiles of twelve risk genes across the two MTRS subgroups. (G) ROC curve analysis and its corresponding AUC values assessed the accuracy in predicting survival outcomes in patients. (H) The Kaplan-Meier method was applied to evaluate differences in survival between patients with high and low MTRS. (I) Both heatmaps and scatter plots visualized correlations among prognostic genes, overall survival, and genes used in constructing the model. (J) Scatter plots presented the twelve genes that were associated with overall survival.
Further examination of the expression levels of twelve prognostic genes in the high MTRS group and low MTRS group, the scatter plots showed that GDF15 and SEMA4A were beneficial for patient prognosis, while DNAJA1, CEBPB, MAFF, SOX9, HSPA1A, ERRFI1, TM4SF1, KLF4, and IER3were associated with poor patient prognosis (Figure 8J, Supplementary Figures 2A, B).
Analyses included the investigation of immunoinfiltration, enrichment assessment, and examination of drug sensitivity
The study compared gene expression levels and underlying biological processes across groups with high and low values, we made use of various visualization and enrichment analysis techniques. We first illustrated the estimated proportions of cell populations in both groups using a stacked bar graph (Figures 9A, B). Additionally, we utilized a bubble plot to illustrate the correlations among immune checkpoints, prognostic genes, overall survival, as well as the risk, revealing that KLF4 generally exhibited a positive correlation with immune checkpoint-associated genes, while HSPA1A showed a negative relationship (Figure 9C). Upon additional examination, we observed that higher risk scores were correlated with increased macrophage presence (Figure 9D). Moreover, the stromal and estimate appeared higher in the high MTRS group, while the immune score was lower compared to the low MTRS group (Figure 9E). Through box plot analysis, we discovered that the majority of immune checkpoint genes had considerably increase expression in the high MTRS group as opposed to the low group (Figure 9F).
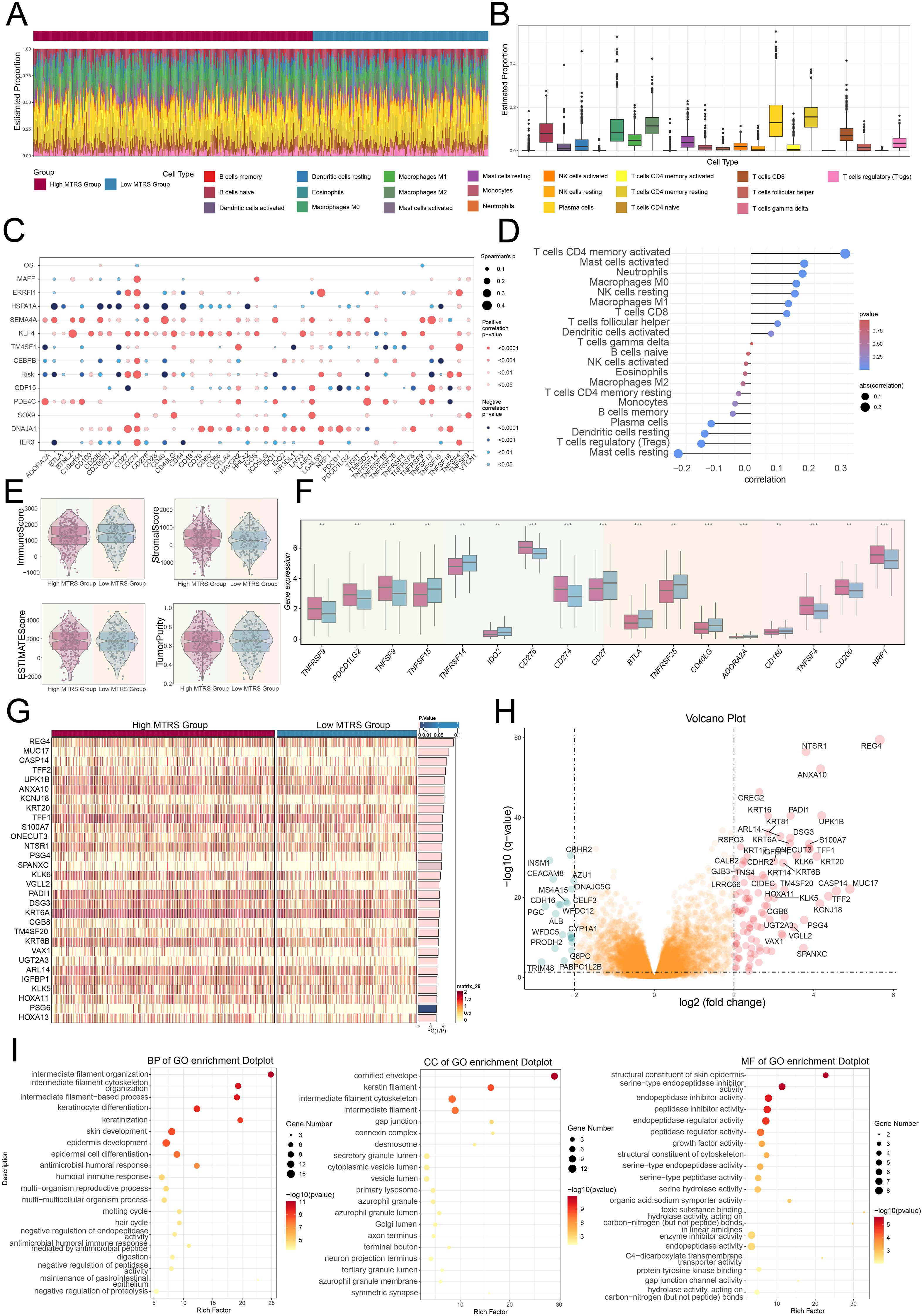
Figure 9. Immunoinfiltration differences and enrichment analysis. (A, B) Stacked bar graph, along with the box plot, demonstrated the calculated distributed twenty-two immune cell types across various risk score groups. (C) Bubble plot illustrated the strength of correlation that existed between risk genes and immune checkpoints. (D) Lollipop plot reflected the correlations between multiple immune-related pathways and risk scores, where bubble diameter indicated the magnitude of association and color signified the statistical significance (p-value). (E) The violin plots showed comparisons of immune score, stromal score, estimate score, and tumor purity between the high and low MTRS groups. (F) The box plot revealed significant differences in immune checkpoint expression when comparing high MTRS group to low MTRS group. (G) The heatmap showed that gene expression patterns varied distinctly between the two MTRS groups. (H) The volcano plot provided a visual summary of expression changes among DEGs. (I) The dot plots presented the GO enrichment results for BP, CC, and MF categories, respectively. **P< 0.01 and ***P< 0.001..
To shed light on the differences between the high and low scoring cohorts, we examined genes that were differentially expressed (Figure 9G). The volcano plot further depicted the trends of gene upregulation and downregulation among these DEGs (Figure 9H). Following this step, enrichment analysis was undertaken to explore the biological processes associated with the identified genes. We initially performed GO enrichment analysis, which identified significantly enriched terms in biological processes (BP), cellular component (CC), and molecular functions (MF) (Figure 9I). In terms of GO-BP, the genes were prominently enriched in pathways involved the organization of intermediate filaments, the structuring of the intermediate filament cytoskeleton, processes based on intermediate filaments, and the differentiation of keratinocytes. The cornified envelope, keratin filaments, intermediate filament cytoskeleton, and intermediate filaments were identified as the main sites of enrichment in terms of GO-CC. GO-MF analysis revealed enrichment included structural constituent of the skin epidermis as well as serine-type endopeptidase inhibitor, endopeptidase inhibitor, and peptidase inhibitor activities.
Afterwards, the heatmap offered a clear visual comparison that further supported our earlier findings (Figure 10A). Additionally, we performed KEGG pathway analysis, which highlighted enrichment in pathways such as the estrogen signaling pathway, staphylococcus aureus infection, steroid hormone biosynthesis, and retinol metabolism (Figure 10B). To better elucidate the functionality and enrichment of pathway associated with LUAD, further investigations were carried out, we also conducted GSEA analysis (Figure 10C). These results demonstrated positive enrichment trends in keratinization, differentiation of keratinocytes, development of the skin, and the epidermis underwent development. Conversely, negative enrichment trends were observed for positive regulation of myoblast fusion, peptide antigen assembly with MHC protein complex, metal ion export, and DNA replication-dependent chromatin organization.
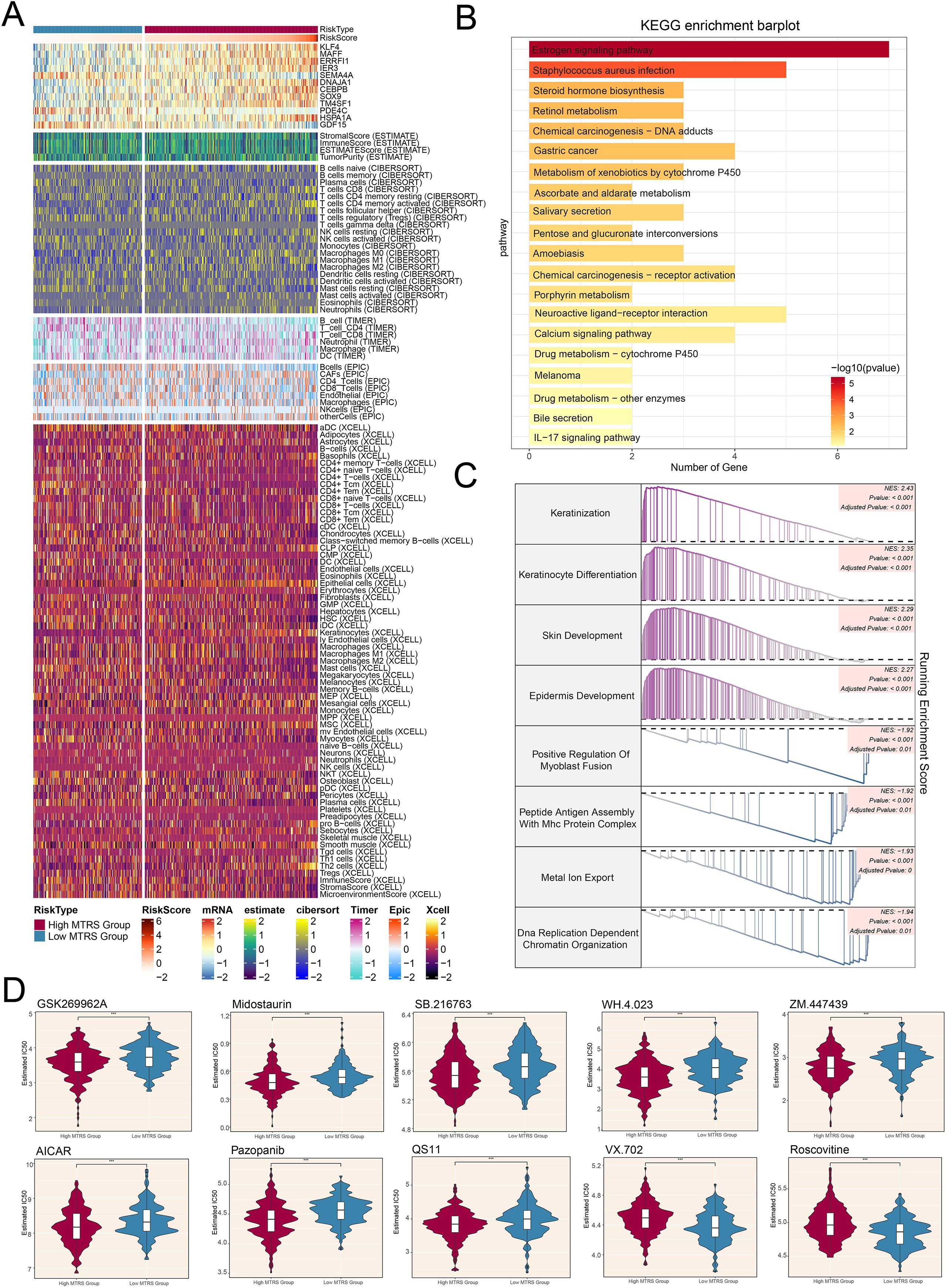
Figure 10. Differences in immune cell infiltration, results of enrichment analysis, and findings from drug sensitivity assessments across various risk groups. (A) Heatmap illustrated how risk scores of different immune cell types varied between the high and low MTRS groups. (B) The KEGG enrichment bar plot reported the twenty most significantly enriched pathways. (C) The GSEA enrichment analysis thoroughly summarized the gene sets that distinguished the high MTRS group from the low MTRS group. (D) Violin plots highlighted marked disparities in the IC50 values for different chemotherapeutic agents between patients belonging to the high and low MTRS categories. ***P<0.001..
In conclusion, the results of our drug sensitivity analysis indicated that the high MTRS group showed heightened sensitivity to chemotherapeutic agents (Figure 10D), including GSK269962A, Midostaurin, SB.216763, WH.4.023, ZM.447439, AICAR, Pazopanib, and QS11. Furthermore, it was determined that the low MTRS group exhibited decreased IC50 values for VX.702 and Roscovitine compared to the high MTRS group. This finding suggested that VX.702 and Roscovitine may have more favorable therapeutic effects for low MTRS group patients.
Discussion
Lung cancer is the most prevalent malignant tumor in both China and the world, with the highest incidence and mortality rates. Among its various pathological types, LUAD is the most common, accounting for approximately 40% of lung cancer cases (3). The anti-tumor efficacy in LUAD patients is often limited by chemotherapy resistance and evasion of apoptosis, leading to tumor recurrence and poor prognosis (56, 57). As a result, the survival duration for LUAD patients has been notably poor. Therefore, it became urgent to develop personalized treatments based on the progression of LUAD, necessitating a comprehensive investigation of the tumor’s heterogeneity to improve patient outcomes and identify potential therapeutic targets. Through scRNA-seq analysis, the characteristics related to both cells and molecules in LUAD tissue were studied, revealing fourteen distinct cell types. Temporal phase, sample origin, and pathway enrichment analyses were conducted for the different cell subtypes. The IAC group primarily contained EPCs and macrophages, suggesting that these cell types may play a key role in the development of malignant tumors. Tumors were composed of various cell types and components that competed with the normal microenvironment. Consequently, the interactions among these cell types might have facilitated the tumor’s aggressiveness and resistance to treatment (58).
Enrichment analysis revealed that EPCs exhibited positive enrichment in pathways associated with chemical homeostasis within a tissue and surfactant homeostasis. Pulmonary surfactant, a complex of lipids and proteins, served a key function in decreasing the interfacial tension at the alveolar air-liquid interface, and its production is regulated during development (59). Genes related to pulmonary surfactant have been regarded as excellent candidate genes for lung cancer risk due to their high expression levels in the lungs and their role in maintaining alveolar integrity (60). Studies demonstrated that the disruption of pulmonary surfactant homeostasis may be linked to the onset of lung cancer, with abnormalities in the genes and cellular processes related to surfactant maturation and function identified as risk factors for lung cancer (61–64). During the process of self-evolution, cancer cells give rise to a class of tumor stem cells characterized by self-renewal, metastatic spread, and treatment resistance, functioning as a major driver in cancer initiation and subsequent growth (65).
Accordingly, we conducted a study focused on TCs within LUAD. Marker gene profiles were used to assign tumor tissues to one of four subtypes in the initial step. Notably, it was observed that TCs with increased MAFF expression were mainly derived from the IAC group. CXCL8 was initially identified as an effective chemoattractant and activator of polymorphonuclear leukocytes. It also influenced cancer cell proliferation, migration, tumor angiogenesis, and metastasis, being expressed in various cancer cell types (66). CXCL2, a small secretory member of the CXC chemokine family (67), played a critical role in maintaining macrophage infiltration induced by cancer (68). RRAD was associated with multiple cancer types and significantly contributed to regulating aerobic glycolysis and cellular invasion and metastasis (69). ATF3 having a significant impact on regulating metabolism, immunity, and tumorigenesis (70). AREG promoted cellular proliferation in various tumors and inhibited apoptosis while a vital contribution was made in the extracellular matrix environment or bloodstream (71). The synergy among these genes effectively demonstrated the C0 MAFF+ tumor cell subtype’s role in promoting tumorigenesis. Furthermore, the C0 MAFF+ tumor cell subtype displayed elevated levels of nCount RNA, nFeature RNA, and stemness expression, indicating a higher degree of malignancy and differentiation potential for these cells. All things considered, there was a complex interaction between C0 MAFF+ TCs and the development of LUAD.
We carried out a number of cell subtype enrichment analyses in order to elucidate the association between cancer and C0 MAFF+ tumor cell subtype. The biological processes associated with the response to lipid and fat cell differentiation were the main ones in which C0 MAFF+ TCs participated. The metabolic activity of cancer cells often exhibited specific alterations, with this metabolic reprogramming supporting an increase in metabolic intermediates necessary for the synthesis of proteins, nucleic acids, and lipids, which are prerequisites for the rapid proliferation of cancer cells. The increased rate of lipid synthesis in tumor tissues has long been recognized as a significant aspect of the metabolic rewiring of transformed cells (72). Abnormal lipid synthesis and extracellular lipid uptake serve as beneficial modifications to meet the demands of uncontrolled cancer cell proliferation (73).
To rephrase, LUAD TCs tended to grow faster when supported by lipids, which was also linked to a higher degree of malignancy. The metabolic interactions between TCs and adipocytes induced the mesenchymal transformation of adipocytes and contributed to the reconfiguration of the stroma into a microenvironment more conducive to tumors. Regions of tumor-adipocyte interactions exhibited high densities of angiogenesis, indicating that TCs were in a more active state due to the nutrient-rich environment in these areas (74). To summarize, processes like lipid metabolism and adipocyte differentiation were key contributors to tumor formation and advancement, potentially promoting the rapid growth and spread of TCs and heightening tumor malignancy. In short, intervening in factors such as lipid metabolism or fat cell differentiation might have slowed the progression of LUAD.
The pseudotime analysis revealed that the C0 subtype was located at the terminal end of lineage 1, and the C0-associated gene MAFF was highly expressed primarily in the later stages, indicating a more advanced developmental state and a mature phase of differentiation. The high level of cellular stemness suggested a low differentiation status for C0 subtype, which correlated with higher malignancy and stronger resistance to chemotherapy and radiotherapy. This, in turn, leads to poor prognosis and a higher likelihood of recurrence in patients.
To further investigate the interactions between C0 MAFF+ TCs and other cells, CellChat identified that C0 MAFF+ TCs act on macrophages via the MIF-(CD74+CD44) signaling pathway. MIF, known as Macrophage Migration Inhibitory Factor, was a cytokine expressed in various cell types, including HCs, EPCs, ECs, MCs, and NCs. Altered expression of MIF was linked to various diseases, extending from inflammatory illnesses to organ abnormalities and different forms of malignancy (75). Enhanced expression of MIF in tumor tissues suggested a potential oncogenic role of macrophages (76). Studies have shown that the MIF-CD74 signaling pathway promotes tumor cell proliferation (77). It served as a crucial cytokine in the context of both tumorigenesis and inflammation, MIF triggers MAPK and PI3K signaling pathways by binding to the CD74 receptor (78). Activation of MAPK and PI3K pathways by MIF regulated fundamental cellular functions related to proliferation, differentiation, apoptosis, cell survival, and cancer development (79). In normal lung tissue, MIF mRNA and protein were expressed in bronchial epithelium, alveolar epithelium, vascular smooth muscle, and alveolar macrophages. In LUAD tumor tissues, levels of MIF mRNA and protein were significantly higher than those in normal alveolar EPCs, with elevated MIF mRNA levels in both TCs and premalignant states in LUAD (80). These findings indicated that high expression of MIF promotes tumor growth and metastasis in LUAD and created a TME conducive to tumor development. C0 MAFF+ TCs might promote the transformation of normal macrophages into TAMs by acting on the CD74-CD44 receptor of macrophages via the MIF ligand, thus, it blocked the natural immune response that targeted TCs. TAMs directly influenced tumor cell communication by transferring substances such as some non-coding RNAs through exosomes, which affected TCs (81). Additionally, TAMs induced immune checkpoint inhibition of T cells by upregulating PD-L1 expression and recruited Tregs via CCL22 to further suppress anti-tumor immune responses (82). TAMs primarily exhibited M2-like tumor-promoting effects within the TME and regulated various malignant processes, including angiogenesis, immunosuppression, and tumor metastasis (83). Therefore, targeting M2-like TAMs to deplete them in the TME or reversing M2-like TAMs to an M1-like phenotype directly enhanced their cytotoxicity and indirectly stimulated cytotoxic T cells to eliminate TCs, representing a potential strategy for anti-tumor immunotherapy (84, 85). Thus, inhibiting MIF within TCs emerged as a promising avenue for therapeutic intervention.
For a deeper understanding of the oncogenic mechanisms of the C0 MAFF+ tumor cell subtype, we analyzed the TFs within this subtype and identified the top five active TFs: KLF6, ATF3, JUN, FOS, and FOSB.
Members of the KLF protein family play vital roles in regulating key biological activities such as cell growth, specialization, metabolic regulation, programmed cell death, and inflammations. Aberrant KLF function can disturb cellular equilibrium and has been implicated in the pathogenesis of various diseases. KLF6 was implicated in cancer, inflammatory diseases, and cardiovascular disorders (86). As a TF induced by stress, ATF3 was vital for modulating metabolism, immune responses, and tumorigenesis. ATF3 expression was promoted by several extracellular stimuli, among them endoplasmic reticulum stress, cytokines, and chemokines. Additionally, ATF3 served as a major regulator of metabolic homeostasis (70). JUN belonged to the most widely analyzed elements of the AP-1 complex and was engaged in numerous activities, including cell division, programmed cell death, survival, cancer development, and tissue formation. Early studies identified JUN as a basic leucine zipper TF that functioned as a homo- or heterodimer to bind DNA and regulated gene transcription. Subsequent research demonstrated that extracellular signals can induce post-translational modifications of JUN, leading to altered transcriptional activity and target gene expression (87). Members of the FOS protein family could be divided into two groups: transforming (c-Fos and FosB) and non-transforming (Fra-1 and Fra-2) proteins (88). FOS encoded leucine zipper proteins that dimerize with proteins from the JUN family to form the TF complex AP-1, which played a key role in tumor cell growth, differentiation, survival, and DNA damage response (16, 17). Notably, overexpression of FOS could promote drug resistance and enhance EMT (89, 90). FOSB, also known as FBJ murine osteosarcoma viral oncogene homolog B, was a member of the FOS TF family (91). FOSB was an oncogene present in various tumors that promoted angiogenesis and regulated genes associated with drug sensitivity and invasive activity (92, 93). In summary, these findings provided innovative perspectives for future immunological interventions in LUAD.
To improve patient survival rates, enhance their quality of life, and prolong longevity, our study constructed a prognostic risk prediction model for LUAD by employing the top twelve marker genes of C0 MAFF+ TCs. We observed that the high MTRS group exhibited higher risk scores, indicating increased mortality rates and poorer prognoses. The results suggested that FOS contributed to the progression of LUAD and was generally correlated with adverse outcomes in cancer patients.
In vitro assays demonstrated that knocking down FOS greatly diminished the abilities of TCs to proliferate, migrate, and invade. Overexpression of FOS had been shown to promote drug-resistant phenotypes (89). Therefore, considering FOS targeting as a viable strategy to hinder these carcinogenic processes and improve treatment outcomes in LUAD patients appeared promising. Future studies could explore the combination of FOS-targeted therapies with existing treatments to enhance the efficacy of LUAD immunotherapy. Moreover, analysis of immune cell infiltration demonstrated that the high MTRS group exhibited significantly increased stromal scores, estimate scores, and levels of macrophage infiltration in comparison to the low MTRS group. Further examination of the associations in the relationship of MTRS to immune infiltration showed that MTRS was significantly and positively correlated with activated CD4 memory T cells, activated mast cells, and M1 macrophages, whereas it was negatively associated with M2 macrophages and resting mast cells. In most cases, macrophages were divided into two principal subtypes: the classically activated M1 and the alternatively activated M2 (94). M1 macrophages played a role in mediating resistance against tumors. In contrast, various forms of M2 macrophages were present in established tumors and promoted progression, tissue repair, remodeling, and exhibited immunoregulatory functions (95). The higher levels of macrophage infiltration in the high MTRS group favored the targeting of macrophages in LUAD patients, facilitating the occurrence of normal anti-tumor effects. Targeting M2 macrophages might be a feasible therapeutic approach, and enhancing M1 macrophages could further improve patient prognoses.
In our continued efforts to elucidate the function of macrophage phenotype control in anti-cancer therapy, with emphasis on promoting the transition from M2 to M1 macrophages, analysis indicated that STING agonists promoted the local production of anti-angiogenic factors and normalized tumor-associated vasculature (96), suggesting that reprogramming macrophages into anti-tumor states was promising. Additionally, previous research showed that tetrahedral DNA nanostructures actively entered macrophages to enhance M1 polarization (97), while mitochondrial DNA induced macrophage recruitment and M2 polarization through the TLR9 pathway, inhibiting this pathway reversed mitochondrial DNA-mediated M2 macrophage polarization (98). These approaches effectively suppressed M2 polarization and promoted M1 activation, highlighting the potential benefits of combining immunomodulatory therapies in LUAD. In our study, the group with elevated MTRS showed notably worse prognoses in comparison to the low MTRS group, therefore, therapeutic strategies targeting the polarization of TAMs from the M2 to the M1 might have offered significant benefits Furthermore, targeting FOS might enhance the immunotherapeutic response in patients with either high or low MTRS. Therefore, FOS could serve as a novel target for improving the reprogramming of macrophages. Later studies might have assessed the combination of FOS-targeted therapies with existing treatments to improve macrophage-based cancer immunotherapy outcomes.
Furthermore, we analyzed the samples for their sensitivity to drugs across different risk score groups to uncover potential differences, which also contributed to the development of personalized treatment strategies. Our study revealed that GSK269962A, Midostaurin, SB.216763, WH.4.023, ZM.447439, AICAR, Pazopanib, and QS11 demonstrated stronger efficacy in patients within the high MTRS group.
AICAR was a pharmacological precursor in purine nucleotide biosynthesis with antitumor properties (99). AICAR has long been one of the most commonly used pharmacological regulators of AMPK activity, and most early studies on the role of AMPK in metabolic regulation and cancer pathogenesis were entirely based on the use of AICAR as an AMPK activator (100). It was found that patients belonging to the high MTRS category exhibited higher sensitivity to AICAR. Studies have indicated that GSK269962A was a selective ROCK1 inhibitor (101), which can inhibit tumor growth (102).
Midostaurin was a multitarget kinase inhibitor initially developed as a protein kinase C inhibitor for the treatment of solid tumors (103). SB.216763, a drug that functioned as a Wnt signaling pathway suppressor, was shown to suppress the proliferation, migration, and invasion of TCs (104). WH.4.023 was a chemotherapy drug that significantly targeted the proto-oncogene tyrosine-protein kinase SRC and the tyrosine-protein kinase ABL1 (105, 106). ZM.447439 was an Aurora selective ATP-competitive inhibitor that could disrupt spindle integrity checkpoints and chromosome segregation, making it useful for more selective cancer treatment (107). Pazopanib was an orally administered small-molecule multitarget kinase inhibitor that inhibited angiogenesis induced by vascular endothelial growth factor and basic fibroblast growth factor, and could be used as a treatment for tumors (108). QS11 influenced protein transport by inhibiting the GTPase-activating protein ARFGAP1, thereby co-activating the Wnt/β-catenin signaling pathway. When overexpressed in breast cancer cells, it inhibited cell migration, indicating that QS11 might have potential applications in tumor regulation (109). Our findings highlighted the differences in drug sensitivity among LUAD patients, such findings emphasized the importance of conducting additional studies to better understand how they functioned, enhanced efficacy, and developed personalized treatment strategies to improve patient prognosis.
In our research, although we performed a comprehensive evaluation of LUAD and meticulously selected our study population, we recognized several shortcomings. Firstly, given the restricted sample size, it is possible that this study encountered errors in correlating the examined samples with the target genes, thereby possibly influencing the accuracy of the analysis. Furthermore, the limited number of samples might have resulted in imprecisions that could challenge the reliability of the conclusions derived from the research. Secondly, LUAD comprised multiple subtypes. Yet, considering the high prevalence of LUAD in lung cancer, our investigation’s specificity was possibly limited, yielding more generalized outcomes since different LUAD subtypes could possess unique features and respond differently to treatments. Finally, our experimental assessment of the interplay between TCs and macrophages was not as detailed as desired. TAMs and other TME cellular components co-evolved with TCs, so fostering tumor progression and resistance and contributing to tumor heterogeneity. Future studies should thus concentrate more on these interactions and their effect on tumor growth. We intended to start research on FOS and investigate cell-targeted therapy approaches against LUAD in next projects. Clarifying the FOS mechanisms in LUAD would help us to create fresh therapeutic approaches to raise patient prognosis. We also sought to investigate how different TME-targeting techniques might be combined into a coherent approach to improve efficacy and lower side effects, so offering more treatment choices for LUAD patients.
Conclusion
Driving lipid metabolic reprogramming and immunosuppression through MIF-(CD74+CD44)-mediated macrophage polarization, our work finds FOS as a master regulator of the malignant C0 MAFF+ tumor cell subtype in LUAD. The MTRS prognostic model stratifies patients rather well, high-risk cases show increased chemosensitivity to AICAR and Midostaurin. In vitro, functionally FOS knockdown suppressed proliferation, migration, and invasion, so highlighting its therapeutic potential. These results suggest FOS inhibition as a dual approach to target immune evasion and tumor-intrinsic malignancy so improving LUAD treatment. To maximize clinical benefit, future research should look at FOS-directed treatments in concert with immune checkpoint inhibitors.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
XSC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing – original draft, Writing – review & editing. SC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft. YF: Conceptualization, Data curation, Methodology, Project administration, Resources, Supervision, Writing – original draft. RJ: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. YJ: Data curation, Formal analysis, Investigation, Supervision, Writing – original draft. BZ: Data curation, Formal analysis, Methodology, Resources, Supervision, Writing – original draft. DG: Data curation, Formal analysis, Investigation, Resources, Writing – original draft. LW: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. ZY: Conceptualization, Data curation, Resources, Visualization, Writing – original draft. YL: Data curation, Formal analysis, Methodology, Project administration, Validation, Writing – original draft. XLC: Formal analysis, Funding acquisition, Resources, Validation, Visualization, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
We prepared our Graphical Abstract with the help of Figdraw, referencing Image ID: SAPPW80b35. Figdraw’s contribution to this publication was sincerely appreciated.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1649147/full#supplementary-material
Supplementary Figure 1 | InferCNV analysis. (A) The prediction of CNVs was performed using scRNA-seq data sourced from ECs, and orange was used to indicate amplification, whereas blue signified deletion.
Supplementary Figure 2 | The analysis and expression levels of prognostic genes. (A, B) The Kaplan-Meier survival analysis illustrated which prognostic genes were associated with C0 MAFF+ TCs, while the gene expression profiles were further examined in both high and low MTRS groups.
References
1. Bade BC and Dela Cruz CS. Lung cancer 2020. Clin Chest Med. (2020) 41:1–24. doi: 10.1016/j.ccm.2019.10.001
2. Nasim F, Sabath BF, and Eapen GA. Lung cancer. Med Clin North Am. (2019) 103:463–73. doi: 10.1016/j.mcna.2018.12.006
3. Herbst RS, Morgensztern D, and Boshoff C. The biology and management of non-small cell lung cancer. Nature. (2018) 553:446–54. doi: 10.1038/nature25183
4. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. (2011) 6:244–85. doi: 10.1097/JTO.0b013e318206a221
5. Zhu J, Fan Y, Xiong Y, Wang W, Chen J, Xia Y, et al. Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics. Exp Mol Med. (2022) 54:2060–76. doi: 10.1038/s12276-022-00896-9
6. Hoy H, Lynch T, and Beck M. Surgical treatment of lung cancer. Crit Care Nurs Clin North Am. (2019) 31:303–13. doi: 10.1016/j.cnc.2019.05.002
7. Pei Q, Luo Y, Chen Y, Li J, Xie D, and Ye T. Artificial intelligence in clinical applications for lung cancer: diagnosis, treatment and prognosis. Clin Chem Lab Med. (2022) 60:1974–83. doi: 10.1515/cclm-2022-0291
8. Ting C, Lee K, Wu S, Feng P, Chan Y, Chen Y, et al. FOSB(-)PCDHB13 axis disrupts the microtubule network in non-small cell lung cancer. Cancers (Basel). (2019) 11:1–21. doi: 10.3390/cancers11010107
9. Eisenstein M. Personalized medicine: Special treatment. Nature. (2014) 513:S8–09. doi: 10.1038/513S8a
10. Ji X, Zhu R, Gao C, Xie H, Gong X, and Luo J. Hypoxia-derived exosomes promote lung adenocarcinoma by regulating HS3ST1-GPC4-mediated glycolysis. Cancers (Basel). (2024) 16:1–19. doi: 10.3390/cancers16040695
11. Siegel RL, Miller KD, Fuchs HE, and Jemal A. Cancer statistics, 2022. CA Cancer J Clin. (2022) 72:7–33. doi: 10.3322/caac.21708
12. Xu Y, Lv D, Yan C, Su H, Zhang X, Shi Y, et al. METTL3 promotes lung adenocarcinoma tumor growth and inhibits ferroptosis by stabilizing SLC7A11 m(6)A modification. Cancer Cell Int. (2022) 22:11. doi: 10.1186/s12935-021-02433-6
13. Kita T, Araya T, Sakai T, Uchida Y, Matsuoka H, and Kasahara K. Nivolumab-induced polymyalgia rheumatica in a patient with lung adenocarcinoma. Am J Med Sci. (2021) 362:321–23. doi: 10.1016/j.amjms.2021.04.010
14. Niknafs N, Balan A, Cherry C, Hummelink K, Monkhorst K, Shao XM, et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat Med. (2023) 29:440–49. doi: 10.1038/s41591-022-02163-w
15. Yoshizawa A, Motoi N, Riely GJ, Sima CS, Gerald WL, Kris MG, et al. Impact of proposed IASLC/ATS/ERS classification of lung adenocarcinoma: prognostic subgroups and implications for further revision of staging based on analysis of 514 stage I cases. Mod Pathol. (2011) 24:653–64. doi: 10.1038/modpathol.2010.232
16. Lam SW, Cleven AHG, Kroon HM, Briaire-de Bruijn IH, Szuhai K, and Bovee JVMG. Utility of FOS as diagnostic marker for osteoid osteoma and osteoblastoma. Virchows Arch. (2020) 476:455–63. doi: 10.1007/s00428-019-02684-9
17. van IJzendoorn DGP, Forghany Z, Liebelt F, Vertegaal AC, Jochemsen AG, Bovee JVMG, et al. Functional analyses of a human vascular tumor FOS variant identify a novel degradation mechanism and a link to tumorigenesis. J Biol Chem. (2017) 292:21282–90. doi: 10.1074/jbc.C117.815845
18. Guinea-Viniegra J, Zenz R, Scheuch H, Jimenez M, Bakiri L, Petzelbauer P, et al. Differentiation-induced skin cancer suppression by FOS, p53, and TACE/ADAM17. J Clin Invest. (2012) 122:2898–910. doi: 10.1172/JCI63103
19. Ni KW and Sun GZ. The identification of key biomarkers in patients with lung adenocarcinoma based on bioinformatics. Math Biosci Eng. (2019) 16:7671–87. doi: 10.3934/mbe.2019384
20. Fialka I, Schwarz H, Reichmann E, Oft M, Busslinger M, and Beug H. The estrogen-dependent c-JunER protein causes a reversible loss of mammary epithelial cell polarity involving a destabilization of adherens junctions. J Cell Biol. (1996) 132:1115–32. doi: 10.1083/jcb.132.6.1115
21. Gupta S, Kumar P, Kaur H, Sharma N, Saluja D, Bharti AC, et al. Selective participation of c-Jun with Fra-2/c-Fos promotes aggressive tumor phenotypes and poor prognosis in tongue cancer. Sci Rep. (2015) 5:16811. doi: 10.1038/srep16811
22. Cheng C, Chen W, Jin H, and Chen X. A review of single-cell RNA-seq annotation, integration, and cell-cell communication. Cells. (2023) 12:1–39. doi: 10.3390/cells12151970
23. Luo Z, Wan R, Qiu J, Chen C, Chen Y, Zhao Z, et al. Multi-omics profiling of a self-assembling bioactive hydrogel for immunomodulation and myogenesis in volumetric muscle loss. Chem. (2025) 11. doi: 10.1016/j.chempr.2025.102645
24. Du H, Li S, Lu J, Tang L, Jiang X, He X, et al. Single-cell RNA-seq and bulk-seq identify RAB17 as a potential regulator of angiogenesis by human dermal microvascular endothelial cells in diabetic foot ulcers. Burns Trauma. (2023) 11:tkad020. doi: 10.1093/burnst/tkad020
25. Zhao Z, Cai H, Nie W, Wang X, Zhao Z, Zhao F, et al. Ectopic expression of GDF15 in cancer-associated fibroblasts enhances melanoma immunosuppression via the GFRAL/RET cascade. J Immunother Cancer. (2025) 13:e011036. doi: 10.1136/jitc-2024-011036
26. Nie W, Zhao Z, Xiahou Z, Zhang J, Liu Y, Wang Y, et al. Single-cell RNA sequencing reveals the potential role of Postn(+) fibroblasts in promoting the progression of myocardial fibrosis after myocardial infarction. Sci Rep. (2025) 15:22390. doi: 10.1038/s41598-025-04990-6
27. Lin Z, Wang F, Yin R, Li S, Bai Y, Zhang B, et al. Single-cell RNA sequencing and immune microenvironment analysis reveal PLOD2-driven Malignant transformation in cervical cancer. Front Immunol. (2024) 15:1522655. doi: 10.3389/fimmu.2024.1522655
28. Lin Z, Zhou Y, Liu Z, Nie W, Cao H, Li S, et al. Deciphering the tumor immune microenvironment: single-cell and spatial transcriptomic insights into cervical cancer fibroblasts. J Exp Clin Cancer Res. (2025) 44:194. doi: 10.1186/s13046-025-03432-5
29. Ge Q, Zhao Z, Li X, Yang F, Zhang M, Hao Z, et al. Deciphering the suppressive immune microenvironment of prostate cancer based on CD4+ regulatory T cells: Implications for prognosis and therapy prediction. Clin Transl Med. (2024) 14:e1552. doi: 10.1002/ctm2.1552
30. Becht E, McInnes L, Healy J, Dutertre C, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. (2018) 30:38–44. doi: 10.1038/nbt.4314
31. Diaz-Papkovich A, Anderson-Trocme L, and Gravel S. A review of UMAP in population genetics. J Hum Genet. (2021) 66:85–91. doi: 10.1038/s10038-020-00851-4
32. Ye B, Fan J, Xue L, Zhuang Y, Luo P, Jiang A, et al. iMLGAM: Integrated Machine Learning and Genetic Algorithm-driven Multiomics analysis for pan-cancer immunotherapy response prediction. Imeta. (2025) 4:e70011. doi: 10.1002/imt2.70011
33. Zhang N, Zhang H, Liu Z, Dai Z, Wu W, Zhou R, et al. An artificial intelligence network-guided signature for predicting outcome and immunotherapy response in lung adenocarcinoma patients based on 26 machine learning algorithms. Cell Prolif. (2023) 56:e13409. doi: 10.1111/cpr.13409
34. Fan G, Xie T, Li L, Tang L, Han X, and Shi Y. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. (2024) 8:75. doi: 10.1038/s41698-024-00564-3
35. Zhao Z, Zhao Z, Lin Z, Fan L, Xiahou Z, Dong Y, et al. Decoding multiple myeloma: single-cell insights into tumor heterogeneity, immune dynamics, and disease progression. Front Immunol. (2025) 16:1584350. doi: 10.3389/fimmu.2025.1584350
36. He Y, Luo Z, Nie X, Du Y, Sun R, Sun J, et al. An injectable multi-functional composite bioactive hydrogel for bone regeneration via immunoregulatory and osteogenesis effects. Adv Compos Hybrid Mater. (2025) 8:128. doi: 10.1007/s42114-025-01213-4
37. Luo S, Wang L, Xiao Y, Cao C, Liu Q, and Zhou Y. Single-cell RNA-sequencing integration analysis revealed immune cell heterogeneity in five human autoimmune diseases. Bio Integration. (2023) 4:145–59. doi: 10.15212/bioi-2023-0012
38. Li X, Lin Z, Chen S, Ni S, Lin G, Wang W, et al. Tiaogeng decoction improves mild cognitive impairment in menopausal APP/PS1 mice through the ERs/NF-kappa b/AQP1 signaling pathway. Phytomedicine. (2025) 138:156391. doi: 10.1016/j.phymed.2025.156391
39. Lin Z, Fan W, Yu X, Liu J, and Liu P. Research into the mechanism of intervention of SanQi in endometriosis based on network pharmacology and molecular docking technology. Med (Baltimore). (2022) 101:e30021. doi: 10.1097/MD.0000000000030021
40. Ye B, Wang Q, Zhu X, Zeng L, Luo H, Xiong Y, et al. Single-cell RNA sequencing identifies a novel proliferation cell type affecting clinical outcome of pancreatic ductal adenocarcinoma. Front Oncol. (2023) 13:1236435. doi: 10.3389/fonc.2023.1236435
41. Sun Y, Nie W, Xiahou Z, Wang X, Liu W, Liu Z, et al. Integrative single-cell and spatial transcriptomics uncover ELK4-mediated mechanisms in NDUFAB1+ tumor cells driving gastric cancer progression, metabolic reprogramming, and immune evasion. Front Immunol. (2025) 16. doi: 10.3389/fimmu.2025.1591123
42. Sun L, Shao W, Lin Z, Lin J, Zhao F, and Yu J. Single-cell RNA sequencing explored potential therapeutic targets by revealing the tumor microenvironment of neuroblastoma and its expression in cell death. Discov Oncol. (2024) 15:409. doi: 10.1007/s12672-024-01286-5
43. Huang X, Ma J, Wei Y, Chen H, and Chu W. Identification of biomarkers associated with diagnosis of postmenopausal osteoporosis patients based on bioinformatics and machine learning. Front Genet. (2023) 14:1198417. doi: 10.3389/fgene.2023.1198417
44. Simon N, Friedman J, Hastie T, and Tibshirani R. Regularization paths for cox’s proportional hazards model via coordinate descent. J Stat Softw. (2011) 39:1–13. doi: 10.18637/jss.v039.i05
45. Wan R, Pan L, Wang Q, Shen G, Guo R, Qin Y, et al. Decoding gastric cancer: machine learning insights into the significance of COMMDs family in immunotherapy and diagnosis. J Cancer. (2024) 15:3580–95. doi: 10.7150/jca.94360
46. Blanche P, Dartigues J, and Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. (2013) 32:5381–97. doi: 10.1002/sim.5958
47. Zhou H, Jing S, Liu Y, Wang X, Duan X, Xiong W, et al. Identifying the key genes of Epstein-Barr virus-regulated tumour immune microenvironment of gastric carcinomas. Cell Prolif. (2023) 56:e13373. doi: 10.1111/cpr.13373
48. Zhang D, Wang M, Huang X, Wang L, Liu Y, Zhou S, et al. GLS as a diagnostic biomarker in breast cancer: in-silico, in-situ, and in-vitro insights. Front Oncol. (2023) 13:1220038. doi: 10.3389/fonc.2023.1220038
49. Fang Y, Kong Y, Rong G, Luo Q, Liao W, and Zeng D. Systematic investigation of tumor microenvironment and antitumor immunity with IOBR. Med Res. (2025) 1:136–40. doi: 10.1002/mdr2.70001
50. Zheng M, Hu Y, Gou R, Nie X, Li X, Liu J, et al. Identification three LncRNA prognostic signature of ovarian cancer based on genome-wide copy number variation. BioMed Pharmacother. (2020) 124:109810. doi: 10.1016/j.biopha.2019.109810
51. Feng X, Luo Z, Zhang W, Wan R, Chen Y, Li F, et al. Zn-DHM nanozymes enhance muscle regeneration through ROS scavenging and macrophage polarization in volumetric muscle loss revealed by single-cell profiling. Adv Funct Mater. (2025), 2506476. doi: 10.1002/adfm.202506476
52. Zhang H, Zhang G, Xu P, Yu F, Li L, Huang R, et al. Optimized dynamic network biomarker deciphers a high-resolution heterogeneity within thyroid cancer molecular subtypes. Med Res. (2025) 1:10–31. doi: 10.1002/mdr2.70004
54. Garcia DA, Johnson TA, Presman DM, Fettweis G, Wagh K, Rinaldi L, et al. An intrinsically disordered region-mediated confinement state contributes to the dynamics and function of transcription factors. Mol Cell. (2021) 81:1484–98. doi: 10.1016/j.molcel.2021.01.013
55. Burdach J, Funnell APW, Mak KS, Artuz CM, Wienert B, Lim WF, et al. Regions outside the DNA-binding domain are critical for proper in vivo specificity of an archetypal zinc finger transcription factor. Nucleic Acids Res. (2014) 42:276–89. doi: 10.1093/nar/gkt895
56. Zhang Y, Du H, Li Y, Yuan Y, Chen B, and Sun S. Elevated TRIM23 expression predicts cisplatin resistance in lung adenocarcinoma. Cancer Sci. (2020) 111:637–46. doi: 10.1111/cas.14226
57. Arbour KC and Riely GJ. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: A review. Jama. (2019) 322:764–74. doi: 10.1001/jama.2019.11058
58. Bissell MJ and Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. (2011) 17:320–29. doi: 10.1038/nm.2328
59. Bry K, Lappalainen U, and Hallman M. Cytokines and production of surfactant components. Semin Perinatol. (1996) 20:194–205. doi: 10.1016/s0146-0005(96)80048-6
60. Shiels MS, Chaturvedi AK, Katki HA, Gochuico BR, Caporaso NE, and Engels EA. Circulating markers of interstitial lung disease and subsequent risk of lung cancer. Cancer Epidemiol Biomarkers Prev. (2011) 20:2262–72. doi: 10.1158/1055-9965.EPI-11-0326
61. Frishman WH, Charlap S, Kimmel B, Goldberger J, Phillippides G, and Klein N. Calcium-channel blockers for combined angina pectoris and systemic hypertension. Am J Cardiol. (1986) 57:22D–9D. doi: 10.1016/0002-9149(86)90801-5
62. Taguchi A, Hanash S, Rundle A, McKeague IW, Tang D, Darakjy S, et al. Circulating pro-surfactant protein B as a risk biomarker for lung cancer. Cancer Epidemiol Biomarkers Prev. (2013) 22:1756–61. doi: 10.1158/1055-9965.EPI-13-0251
63. Sin DD, Tammemagi CM, Lam S, Barnett MJ, Duan X, Tam A, et al. Pro-surfactant protein B as a biomarker for lung cancer prediction. J Clin Oncol. (2013) 31:4536–43. doi: 10.1200/JCO.2013.50.6105
64. Wikoff WR, Hanash S, DeFelice B, Miyamoto S, Barnett M, Zhao Y, et al. Diacetylspermine is a novel prediagnostic serum biomarker for non-small-cell lung cancer and has additive performance with pro-surfactant protein B. J Clin Oncol. (2015) 33:3880–86. doi: 10.1200/JCO.2015.61.7779
65. Najafi M, Mortezaee K, and Ahadi R. Cancer stem cell (a)symmetry & plasticity: Tumorigenesis and therapy relevance. Life Sci. (2019) 231:116520. doi: 10.1016/j.lfs.2019.05.076
66. Kotyza J. Interleukin-8 (CXCL8) in tumor associated non-vascular extracellular fluids: its diagnostic and prognostic values. A review. Int J Biol Markers. (2012) 27:169–78. doi: 10.5301/JBM.2012.9261
67. Ding J, Xu K, Zhang J, Lin B, Wang Y, Yin S, et al. Overexpression of CXCL2 inhibits cell proliferation and promotes apoptosis in hepatocellular carcinoma. BMB Rep. (2018) 51:630–35. doi: 10.5483/BMBRep.2018.51.12.140
68. Zhang Y, Sang R, Bao J, Jiang Z, Qian D, Zhou Y, et al. Schwann cell-derived CXCL2 contributes to cancer pain by modulating macrophage infiltration in a mouse breast cancer model. Brain Behav Immun. (2023) 109:308–20. doi: 10.1016/j.bbi.2023.02.004
69. Shang R, Wang J, Sun W, Dai B, Ruan B, Zhang Z, et al. RRAD inhibits aerobic glycolysis, invasion, and migration and is associated with poor prognosis in hepatocellular carcinoma. Tumour Biol. (2016) 37:5097–105. doi: 10.1007/s13277-015-4329-7
70. Ku H and Cheng C. Master regulator activating transcription factor 3 (ATF3) in metabolic homeostasis and cancer. Front Endocrinol (Lausanne). (2020) 11:556. doi: 10.3389/fendo.2020.00556
71. Weissferdt A, Lin H, Woods D, Tang X, Fujimoto J, Wistuba II, et al. HER family receptor and ligand status in thymic carcinoma. Lung Cancer. (2012) 77:515–21. doi: 10.1016/j.lungcan.2012.05.108
72. Baenke F, Peck B, Miess H, and Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. (2013) 6:1353–63. doi: 10.1242/dmm.011338
73. Rossi Sebastiano M and Konstantinidou G. Targeting long chain acyl-coA synthetases for cancer therapy. Int J Mol Sci. (2019) 20:1–16. doi: 10.3390/ijms20153624
74. Zhu Q, Zhu Y, Hepler C, Zhang Q, Park J, Gliniak C, et al. Adipocyte mesenchymal transition contributes to mammary tumor progression. Cell Rep. (2022) 40:111362. doi: 10.1016/j.celrep.2022.111362
75. Jankauskas SS, Wong DWL, Bucala R, Djudjaj S, and Boor P. Evolving complexity of MIF signaling. Cell Signal. (2019) 57:76–88. doi: 10.1016/j.cellsig.2019.01.006
76. Zhang H, Dai J, Mu Q, Zhao X, Lin Z, Wang K, et al. Macrophage heterogeneity and oncogenic mechanisms in lung adenocarcinoma: insights from scRNA-seq analysis and predictive modeling. Front Immunol. (2024) 15:1491872. doi: 10.3389/fimmu.2024.1491872
77. D’Amato-Brito C, Cipriano D, Colin DJ, Germain S, Seimbille Y, Robert JH, et al. Role of MIF/CD74 signaling pathway in the development of pleural mesothelioma. Oncotarget. (2016) 7:11512–25. doi: 10.18632/oncotarget.7314
78. Osipyan A, Chen D, and Dekker FJ. Epigenetic regulation in macrophage migration inhibitory factor (MIF)-mediated signaling in cancer and inflammation. Drug Discov Today. (2021) 26:1728–34. doi: 10.1016/j.drudis.2021.03.012
79. Koh HM and Kim DC. Prognostic significance of macrophage migration inhibitory factor expression in cancer patients: A systematic review and meta-analysis. Med (Baltimore). (2020) 99:e21575. doi: 10.1097/MD.0000000000021575
80. Kamimura A, Kamachi M, Nishihira J, Ogura S, Isobe H, Dosaka-Akita H, et al. Intracellular distribution of macrophage migration inhibitory factor predicts the prognosis of patients with adenocarcinoma of the lung. Cancer. (2000) 89:334–41. doi: 10.1002/1097-0142(20000715)89:2<334::AID-CNCR18>3.0.CO;2-N
81. Yang M, Chen J, Su F, Yu B, Su F, Lin L, et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer. (2011) 10:117. doi: 10.1186/1476-4598-10-117
82. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PloS One. (2011) 6:e19495. doi: 10.1371/journal.pone.0019495
83. Gao J, Liang Y, and Wang L. Shaping polarization of tumor-associated macrophages in cancer immunotherapy. Front Immunol. (2022) 13:888713. doi: 10.3389/fimmu.2022.888713
84. Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. (2018) 9:873. doi: 10.1038/s41467-018-03225-9
85. Pathria P, Louis TL, and Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. (2019) 40:310–27. doi: 10.1016/j.it.2019.02.003
86. Syafruddin SE, Mohtar MA, Wan Mohamad Nazarie WF, and Low TY. Two sides of the same coin: the roles of KLF6 in physiology and pathophysiology. Biomolecules. (2020) 10:1–22. doi: 10.3390/biom10101378
87. Meng Q and Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell. (2011) 2:889–98. doi: 10.1007/s13238-011-1113-3
88. Tulchinsky E. Fos family members: regulation, structure and role in oncogenic transformation. Histol Histopathol. (2000) 15:921–28. doi: 10.14670/HH-15.921
89. Zhao Y, Luo A, Li S, Zhang W, Chen H, Li Y, et al. Inhibitor of differentiation/DNA binding 1 (ID1) inhibits etoposide-induced apoptosis in a c-jun/c-fos-dependent manner. J Biol Chem. (2016) 291:6831–42. doi: 10.1074/jbc.M115.704361
90. Muhammad N, Bhattacharya S, Steele R, Phillips N, and Ray RB. Involvement of c-fos in the promotion of cancer stem-like cell properties in head and neck squamous cell carcinoma. Clin Cancer Res. (2017) 23:3120–28. doi: 10.1158/1078-0432.CCR-16-2811
91. Nestler EJ. ▵FosB: a transcriptional regulator of stress and antidepressant responses. Eur J Pharmacol. (2015) 753:66–72. doi: 10.1016/j.ejphar.2014.10.034
92. Shahzad MMK, Arevalo JM, Armaiz-Pena GN, Lu C, Stone RL, Moreno-Smith M, et al. Stress effects on FosB- and interleukin-8 (IL8)-driven ovarian cancer growth and metastasis. J Biol Chem. (2010) 285:35462–70. doi: 10.1074/jbc.M110.109579
93. Na H, Ryu J, and Kim K. Knockout of FosB gene changes drug sensitivity and invasion activity via the regulation of Bcl-2, E-cadherin, beta-catenin, and vimentin expression. Biochem Biophys Res Commun. (2021) 567:131–37. doi: 10.1016/j.bbrc.2021.06.031
94. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili S, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
95. Mantovani A, Sica A, and Locati M. Macrophage polarization comes of age. Immunity. (2005) 23:344–46. doi: 10.1016/j.immuni.2005.10.001
96. Chelvanambi M, Fecek RJ, Taylor JL, and Storkus WJ. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J Immunother Cancer. (2021) 9:e001906. doi: 10.1136/jitc-2020-001906
97. Liang S, Li J, Zou Z, Mao M, Ming S, Lin F, et al. Tetrahedral DNA nanostructures synergize with MnO(2) to enhance antitumor immunity via promoting STING activation and M1 polarization. Acta Pharm Sin B. (2022) 12:2494–505. doi: 10.1016/j.apsb.2021.12.010
98. Yang Q, Cui M, Wang J, Zhao Y, Yin W, Liao Z, et al. Circulating mitochondrial DNA promotes M2 polarization of tumor associated macrophages and HCC resistance to sorafenib. Cell Death Dis. (2025) 16:153. doi: 10.1038/s41419-025-07473-8
99. Philippe C, Pinson B, Dompierre J, Pantesco V, Viollet B, Daignan-Fornier B, et al. AICAR antiproliferative properties involve the AMPK-independent activation of the tumor suppressors LATS 1 and 2. Neoplasia. (2018) 20:555–62. doi: 10.1016/j.neo.2018.03.006
100. Visnjic D, Lalic H, Dembitz V, Tomic B, and Smoljo T. AICAr, a widely used AMPK activator with important AMPK-independent effects: A systematic review. Cells. (2021) 10:1–17. doi: 10.3390/cells10051095
101. Pan T, Wang S, Feng H, Xu J, Zhang M, Yao Y, et al. Preclinical evaluation of the ROCK1 inhibitor, GSK269962A, in acute myeloid leukemia. Front Pharmacol. (2022) 13:1064470. doi: 10.3389/fphar.2022.1064470
102. Vogel CJ, Smit MA, Maddalo G, Possik PA, Sparidans RW, van der Burg SH, et al. Cooperative induction of apoptosis in NRAS mutant melanoma by inhibition of MEK and ROCK. Pigment Cell Melanoma Res. (2015) 28:307–17. doi: 10.1111/pcmr.12364
103. Propper DJ, McDonald AC, Man A, Thavasu P, Balkwill F, Braybrooke JP, et al. Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C. J Clin Oncol. (2001) 19:1485–92. doi: 10.1200/JCO.2001.19.5.1485
104. Zeng J, Liu X, Li X, Zheng Y, Liu B, and Xiao Y. Daucosterol inhibits the proliferation, migration, and invasion of hepatocellular carcinoma cells via wnt/beta-catenin signaling. Molecules. (2017) 22:1–10. doi: 10.3390/molecules22060862
105. Kafita D, Daka V, Nkhoma P, Zulu M, Zulu E, Tembo R, et al. High ELF4 expression in human cancers is associated with worse disease outcomes and increased resistance to anticancer drugs. PloS One. (2021) 16:e0248984. doi: 10.1371/journal.pone.0248984
106. Zhou M, Wang B, Liang R, and Luan X. Role of lysine acetylation-related genes in the diagnosis and prognosis of glioma. Sci Rep. (2025) 15:20762. doi: 10.1038/s41598-025-07738-4
107. Long Z, Xu J, Yan M, Zhang J, Guan Z, Xu D, et al. ZM 447439 inhibition of aurora kinase induces Hep2 cancer cell apoptosis in three-dimensional culture. Cell Cycle. (2008) 7:1473–79. doi: 10.4161/cc.7.10.5949
108. Miyamoto S, Kakutani S, Sato Y, Hanashi A, Kinoshita Y, and Ishikawa A. Drug review: pazopanib. Jpn J Clin Oncol. (2018) 48:503–13. doi: 10.1093/jjco/hyy053
Keywords: lung adenocarcinoma, MAFF, tumor microenvironment, FOS, immunotherapy, drug sensitivity
Citation: Cheng X, Chen S, Fu Y, Jiang R, Jing Y, Zhao B, Guo D, Wang L, Ye Z, Li Y and Chen X (2025) Targeted and personalized immunotherapy in lung adenocarcinoma: single-cell RNA sequencing of MAFF+ tumor cells and the therapeutic potential of FOS. Front. Immunol. 16:1649147. doi: 10.3389/fimmu.2025.1649147
Received: 18 June 2025; Accepted: 08 August 2025;
Published: 27 August 2025.
Edited by:
Pengpeng Zhang, Tianjin Medical University Cancer Institute and Hospital, ChinaReviewed by:
Sujit Nair, Phytoveda Pvt. Ltd, IndiaLuo Qiang, Children’s Hospital of Chongqing Medical University, China
Wei Pan, Foshan Women and Children Hospital, China
Copyright © 2025 Cheng, Chen, Fu, Jiang, Jing, Zhao, Guo, Wang, Ye, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zi Ye, eWV6aTAzMjZhQDE2My5jb20=; Yumeng Li, eWluZ3poaWxpZmVuZzE5OTVAMTYzLmNvbQ==; Xianliang Chen, MTM1OTg4MDgwMzVAMTI2LmNvbQ==
†These authors have contributed equally to this work