 Maine Luellah Demaret Bardou1*
Maine Luellah Demaret Bardou1* Rosemeire Navickas Constantino-Silva1
Rosemeire Navickas Constantino-Silva1 Maria Luiza Oliva Alonso2
Maria Luiza Oliva Alonso2 Ana Júlia Ribeiro Teixeira3
Ana Júlia Ribeiro Teixeira3 Pedro Francisco Giavina-Bianchi3
Pedro Francisco Giavina-Bianchi3 Eli Mansour4
Eli Mansour4 João Bosco Pesquero5
João Bosco Pesquero5 Solange Oliveira Rodrigues Valle2
Solange Oliveira Rodrigues Valle2 Anete Sevciovic Grumach1
Anete Sevciovic Grumach1- 1Clinical Immunology, Centro Universitario Faculdade de Medicina ABC, Santo Andre, SP, Brazil
- 2Department of Clinical Medicine, Immunology Service, Hospital Universitario Clementino Fraga Filho, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 3Division of Allergy and Clinical Immunology, University of São Paulo, São Paulo, SP, Brazil
- 4Allergy and Immunology, Department of Internal Medicine, School of Medical Sciences, University of Campinas, Campinas, Brazil
- 5Department of Biophysics, Federal University of São Paulo, São Paulo, SP, Brazil
Introduction: Hereditary Angioedema (HAE) is a rare genetic disease characterized by recurrent episodes of edema and classified into HAE with C1 inhibitor deficiency (HAE-C1INH types 1 and 2) and HAE with normal C1INH (HAE-nC1INH). This study evaluates the function of C1 inhibitor (fC1INH) in patients with suspected HAE using several laboratory methods: dried blood spot (DBS), chromogenic assay, and ELISA with FXIIa and PKa (plasma kallikrein). The comparative approach aims to improve early detection and understanding of C1INH dysfunction in all HAE subtypes to reflect real-world diagnostic scenarios.
Methods: We assessed the diagnostic performance of four fC1INH assays in a cohort of 148 HAE patients: 84 with HAE-C1INH (72 type 1 and 12 type 2) and 64 with HAE-nC1INH (53 HAE-FXII and 11 HAE-UNK). The gold-standard chromogenic assay and the two substrate-specific ELISAs (PKa and FXIIa) were compared to a novel DBS-based LC-MS/MS assay using endogenous C1s activity. For all fC1INH assays, values >50% were considered within the normal range.
Results: In HAE-C1INH, the DBS assay showed the highest specificity (type 1: 98.6%, type 2: 100%) and 100% sensitivity for both subtypes. ELISA-FXIIa also performed well (specificity: 97.2% and 91.7%). In contrast, ELISA-PKa and the chromogenic assay showed reduced specificity in type 2 (25% and 66.7%, respectively). Among patients with HAE-FXII, fC1INH levels were reduced by 36.5% by ELISA-FXIIa (19/52), 19.1% by DBS (9/47), and 3.8% by ELISA-PKa (2/52), and no alterations were detected by the chromogenic assay. Some of the changes seen in other tests may be partly related to pregnancy in a few patients. In the HAE-UNK group, all 11 patients had fC1INH >50% in all methods.
Conclusion: DBS-based LC-MS/MS and ELISA-FXIIa offer promising accuracy and broader applicability for early diagnosis of HAE types 1 and 2. The use of novel substrates and the inclusion of a clinically realistic cohort may enhance the translational relevance of these findings.
1 Introduction
Hereditary Angioedema (HAE) is a rare, potentially life-threatening genetic disorder characterized by recurrent, unpredictable episodes of subcutaneous and submucosal edema. The most commonly affected sites include the face, extremities, gastrointestinal tract, genitalia, and upper airways. Misdiagnosis or delayed recognition may result in unnecessary surgical interventions, avoidable morbidity, and, in some cases, airway compromise due to laryngeal edema (1–3).
HAE is classified based on the presence or absence of C1 inhibitor (C1INH) deficiency. HAE-C1INH results from either a quantitative reduction (type 1) or a dysfunctional protein with normal or elevated levels (type 2) (2). In contrast, HAE with normal C1INH (HAE-nC1INH) includes patients with normal antigenic and functional C1INH levels (fC1INH), and in a subset of these cases, pathogenic variants have been identified in genes already known to be associated with the disease.
C1INH is a serine protease inhibitor that regulates the complement, coagulation, fibrinolytic, kinin-kallikrein, and contact systems by inhibiting proteases such as plasma kallikrein (PKa), factor XI (FXI), and factor XII (FXII) (5, 6). C1INH deficiency leads to excessive bradykinin production through uncontrolled PKa-mediated cleavage of high-molecular-weight kininogen (HK), resulting in increased vascular permeability (7, 8).
More than 800 variants in the SERPING1 gene have been identified in patients with C1INH deficiency (HAE-C1INH) (https://databases.lovd.nl/shared/variants/SERPING1) (9, 10). It is the most prevalent HAE type, with an estimated frequency from 1:50,000 to 1:100,000 (11). HAE-C1INH-Type1 accounts for approximately 85% of cases and is characterized by reduced antigenic and functional levels of C1INH. In HAE-C1INH-Type2, C1INH levels are normal or elevated, but function is impaired. In both subtypes, fC1INH levels are typically <50%, making it a reliable diagnostic biomarker (2). Complement component C4 is usually decreased; however, its diagnostic value is limited due to suboptimal sensitivity and specificity (12–14). While SERPING1 sequencing may be informative, particularly in early or prenatal contexts, C1INH antigen and function assays are cost-effective and remain first-line diagnostic tools (3, 15, 16).
Diagnosis of HAE-nC1INH relies on molecular testing. The most extensively studied subtype HAE-FXII is caused by variants in the F12 gene, which increase susceptibility to cleavage by plasma kallikrein and promote excessive bradykinin production (4, 17). Several other rare genetic variants have been identified, including mutations in plasminogen (HAE-PLG) (18), angiopoietin 1 (HAE-ANGPT1) (19), kininogen1 (HAE-KNG1) (20), myoferlin (HAE-MYOF) (21), heparan sulfate 3-O-sulfotransferase 6 (HAE-HS3ST6) (22), carboxypeptidase N (HAE-CPN1) (23), and disabled homologous interacting protein 2 (HAE-DAB2IP) (24). Patients with no identifiable mutations are termed HAE-UNK and typically present with a family history, lack of response to antihistamines or omalizumab, and positive response to HAE-specific treatments (3, 4, 25).
All suspected cases of HAE should be assessed for C1INH antigen, function, and C4 levels (3, 16, 26). However, logistical barriers persist. Complement proteins are thermolabile and require prompt aliquoting and storage at −80°C for accurate results (2, 27, 28). Access to specialized laboratories remains limited, and shipping samples on dry ice incurs high costs (29). Recent innovations include dried blood spot (DBS) sampling coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS), which has shown high sensitivity and practicality for remote testing (30–33). In this study, we aimed to evaluate different subtypes of HAE using diverse methodologies for assessing fC1INH, with a focus on comparing the sensitivity and specificity of these diagnostic approaches.
2 Methods
2.1 Study design and setting
This multicenter, cross-sectional study was coordinated by the Centro Universitário FMABC (CEUFMABC) in Santo André, Brazil, an internationally accredited ACARE (Angioedema Center of Reference and Excellence). To ensure a robust and representative cohort, patient recruitment and sample collection were conducted in collaboration with reference centers from the Brazilian Group for the Study of Hereditary Angioedema (GEBRAEH), including the University of Campinas, the Federal University of Rio de Janeiro (Hospital Clementino Fraga Filho), and the University of São Paulo. Standardized protocols were implemented across all participating centers, covering sample collection timing, tube type, centrifugation parameters, storage conditions, and transportation logistics. All samples were centrally processed at CEUFMABC.
2.2 Study participants and diagnostic workflow
This study was approved by the Brazilian National Research Ethics Commission (CAAE: 41812720010010082), and written informed consent was obtained from all participants and/or their legal guardians.
We enrolled individuals aged over 1 year who met at least one of the following criteria: 1) a confirmed diagnosis of HAE, 2) presence of suggestive symptoms without a definitive diagnosis, or 3) being an asymptomatic first-degree relative of an index case. Diagnostic classification was based on clinical history, family pedigree, and biochemical evaluation, including C1 inhibitor level (C1INHq), fC1INH, and complement C4.
Exclusion criteria comprised comorbidities known to interfere with complement activity—such as hepatic or renal disease, autoimmune disorders, chronic infections, or hematologic malignancies. Additionally, individuals ultimately diagnosed with mast cell-mediated angioedema during follow-up were excluded from the study.
In patients with suspected HAE and normal C1INH levels and function (HAE-nC1INH), genetic testing with Sanger sequencing of exon 9 of the FXII gene was performed on an ABI 3500 Genetic Analyzer (Applied Biosystems) at the JB Pesquero Molecular Genetics Laboratory, a facility specialized in genomic diagnostics for HAE. When no variant was identified, whole-exome sequencing was conducted to investigate additional known pathogenic variants.
2.3 Sample collection and processing
Peripheral blood was collected into sodium citrate, EDTA, and serum tubes. Plasma and serum were separated by centrifugation at 1,207×g for 10 min at 4°C and stored at −80°C. For DBS analysis, 50 µL of EDTA-anticoagulated whole blood was applied to filter paper, dried at room temperature for ≥3 h, and stored at −20°C until shipment to Revvity Omics (USA) for LC-MS/MS analysis.
2.4 Complement quantification and functional C1INH assays
2.4.1 Antigenic quantification of C1INH and C4
C1INHq and C4 were determined by radial immunodiffusion (Binding Site, Birmingham, United Kingdom). Reference values were 19.5–34.5 mg/dL for C1INHq and 20–40 mg/dL for C4.
2.4.2 Functional assay: chromogenic method
Functional C1INH activity was assessed in citrated plasma using the Technochrom® C1INH kit (Technoclone, Vienna, Austria), which measures the ability of C1INH to inhibit cleavage of the synthetic substrate Z-Lys-SBzl·HCl by C1s. The reaction releases free thiol, which reacts with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB, Ellman’s reagent), forming a yellow compound quantified spectrophotometrically at 412 nm. Lower absorbance indicates higher C1INH activity. Values above 50% were considered within the normal range.
2.4.3 Functional assay: DBS LC-MS/MS
This method was developed and validated by Revvity Omics (CLIA-certified for high-complexity testing) to quantify cbz-Lys, the cleavage product generated by free C1s. C1INH extracted from DBS cards was incubated with an excess of C1s, and the resulting cleavage products were quantified by LC-MS/MS. Although the manufacturer’s reference value is >62.8%, a cutoff of >50% was adopted based on previously published studies (3, 16).
2.4.4 Functional assay: ELISA with FXIIa and PKa
Two in-house ELISAs were performed as previously described by Joseph et al. (34) to assess functional C1INH activity using biotinylated activated factor XII (FXIIa) and PKa as substrates. Nunc Maxisorp 96-well plates were coated with streptavidin (5 µg/mL) in carbonate/bicarbonate buffer (pH 9.6) and incubated overnight at 4°C. After blocking with PBS containing 1.5% BSA for 2 h at room temperature and washing with PBS-Tween, 50 µL of binding buffer, 25 µL of C1INH standards (R&D Systems, Minneapolis, MN, United States; 5, 2.5, 1.25, 0.63, 0.31, 0.16, and 0 µg/mL), and diluted plasma samples (1:100) were added, followed by 25 µL of biotinylated FXIIa or PKa (Enzyme Research, South Bend, Indiana, United States). The plate was incubated at 37°C for 1 h. After additional washes, HRP-conjugated anti-C1INH antibody (1:10,000; Thermo Fisher, Waltham, MA, United States) was added and incubated for 1 h. The reaction was developed with chromogenic substrate and stopped after 5–10 min. Absorbance was read at 450 nm. Values above 50% were considered within the normal range. General-use reagents (PBS, BSA, buffers, and streptavidin) were purchased from Sigma (Burlington, MA, United States).
2.5 Cohort description and diagnostic grouping
A total of 187 biological samples were collected from individuals, including patients with a confirmed diagnosis of HAE, symptomatic and asymptomatic relatives of HAE patients, and individuals with recurrent angioedema of unknown etiology. Twenty-eight participants were excluded from the final analysis due to either negative diagnostic results among relatives (n = 15) or the inability to obtain confirmatory samples (n = 13). In addition, 11 samples from patients initially suspected of having HAE were excluded following diagnostic reclassification as mast cell-mediated angioedema, based on established clinical and laboratory criteria.
The final study cohort of 148 included patients with HAE-C1INH (type 1, n = 72; type 2, n = 12) and HAE-nC1INH, comprising HAE-FXII (n = 53) and HAE-UNK (n = 11). Regarding control samples, the number varied according to the method employed: DBS (n = 27, including 15 from relatives with negative results), chromogenic assay (n = 14, all from relatives with negative results), PKa (n = 64), and FXIIa (n = 61) (Supplementary Table S1).
Of the 72 individuals with HAE-C1INH-Type1, 51 were female patients (70.8%), with a mean age of 39.8 years (range: 3–79) (Supplementary Table S2). Among the 12 patients with HAE-C1INH-Type2, 7 were female subjects (58.3%), with a mean age of 35.5 years (range: 11–56); one woman was experiencing an AE attack at the time of sample collection (Supplementary Table S3).
An additional 64 patients were diagnosed with HAE-nC1INH, including 53 with a pathogenic variant in the F12 gene (HAE-FXII) and 11 without an identifiable mutation (HAE-UNK). Sixteen newly confirmed diagnoses were made within the HAE-FXII group. The majority of HAE-FXII patients were women (37 out of 53; 69.8%), with a mean age of 40 years (range: 11–56), of whom 3 were pregnant at the time of sample collection (Supplementary Table S4). All 11 patients in the HAE-UNK group were women (100%), with a mean age of 39.9 years (range: 24–68). These patients had a positive family history of angioedema, comprising three distinct families: seven individuals from family 57, two from family 58, and two from family 59. All responded to HAE-specific treatments but not to therapies targeting mast cell-mediated angioedema. Whole-exome sequencing revealed no known pathogenic variants in this group, except for a variant of uncertain significance (VUS) in the ANGPT1 gene, identified in a mother and daughter from a family with multiple affected members for whom further genetic analysis was not possible (Supplementary Table S5).
3 Results
Median C4 and C1INHq levels were markedly reduced in patients with HAE-C1INH-Type1, with values of 10.1 mg/dL (range: 6.0–27.9) and 8.88 mg/dL (range: 3.8–14.9), respectively (Figures 1, 2). Reference values were 19.5–34.5 mg/dL for C1INHq and 20–40 mg/dL for C4. Notably, five individuals (unrelated adults aged 28 to 72 years) had C4 values within the elevated range (Supplementary Table S1). Patients with HAE-C1INH-Type2 showed similarly low C4 levels (median: 10.1 mg/dL; range: 5.0–17.1), while C1INHq levels were elevated (median: 31.8 mg/dL; range: 10.8–70.0), consistent with the known biochemical profile of this subtype. One patient experiencing an AE attack at the time of sample collection had reduced C1INHq levels. Additionally, one child in this group also had reduced C1INHq levels (Figures 1, 2).
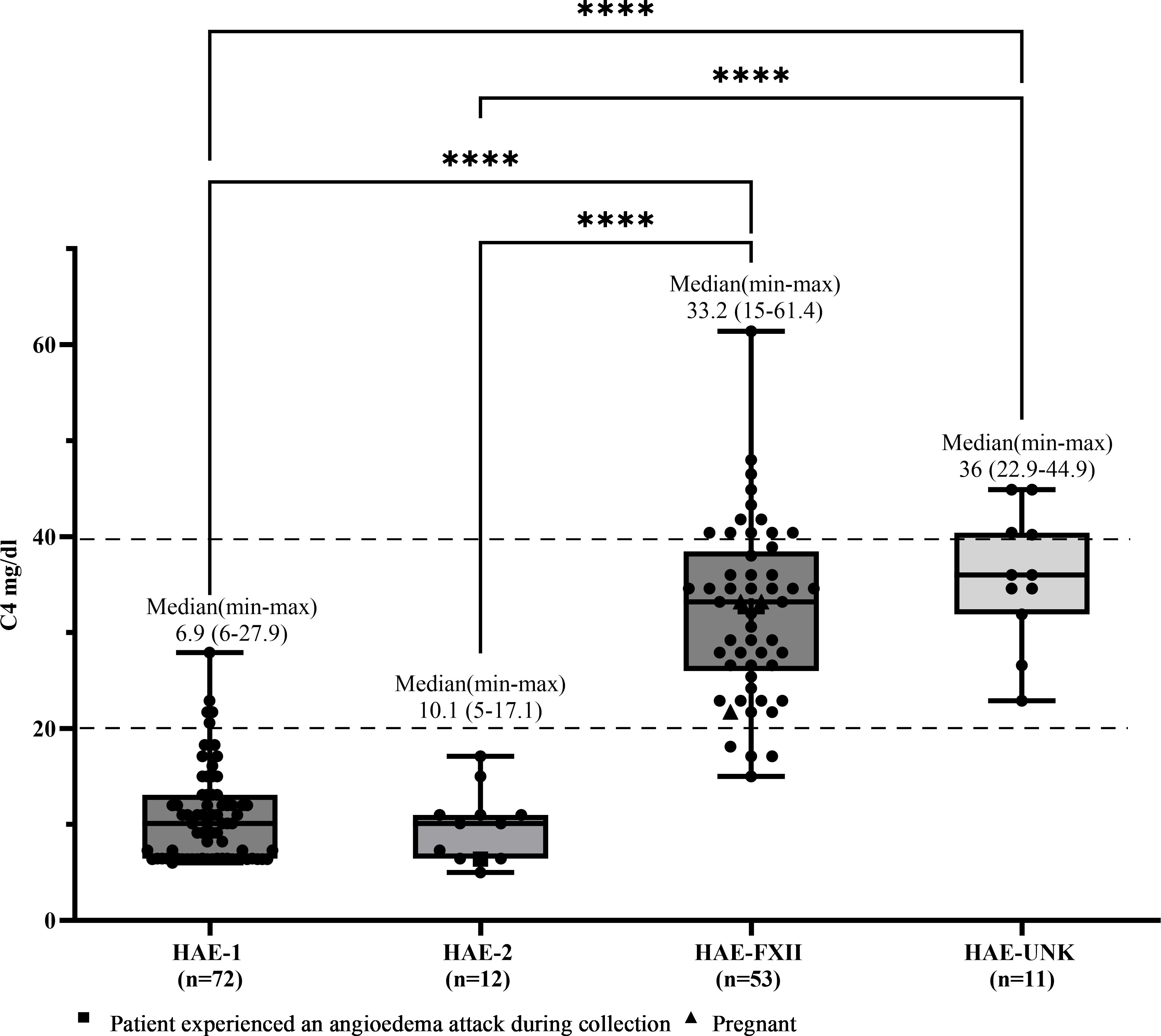
Figure 1. C4 levels according to types of hereditary angioedema. Box-and-whisker plots represent serum C4 concentrations (mg/dL) in patients with hereditary angioedema (HAE) due to C1INH deficiency type 1 (HAE-1) and HAE due to C1INH deficiency type 2 (HAE-2), as well as HAE with normal C1 inhibitor levels, subdivided into HAE with factor XII mutation (HAE-FXII) and HAE of unknown mutation (HAE-UNK). The normal reference interval for serum C4 is indicated by dashed lines. Boxes represent the interquartile range (IQR); the horizontal line within each box indicates the median, and whiskers extend from the minimum to the maximum values. Individual data points are overlaid. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn's multiple comparisons test. (****p <0.0001).
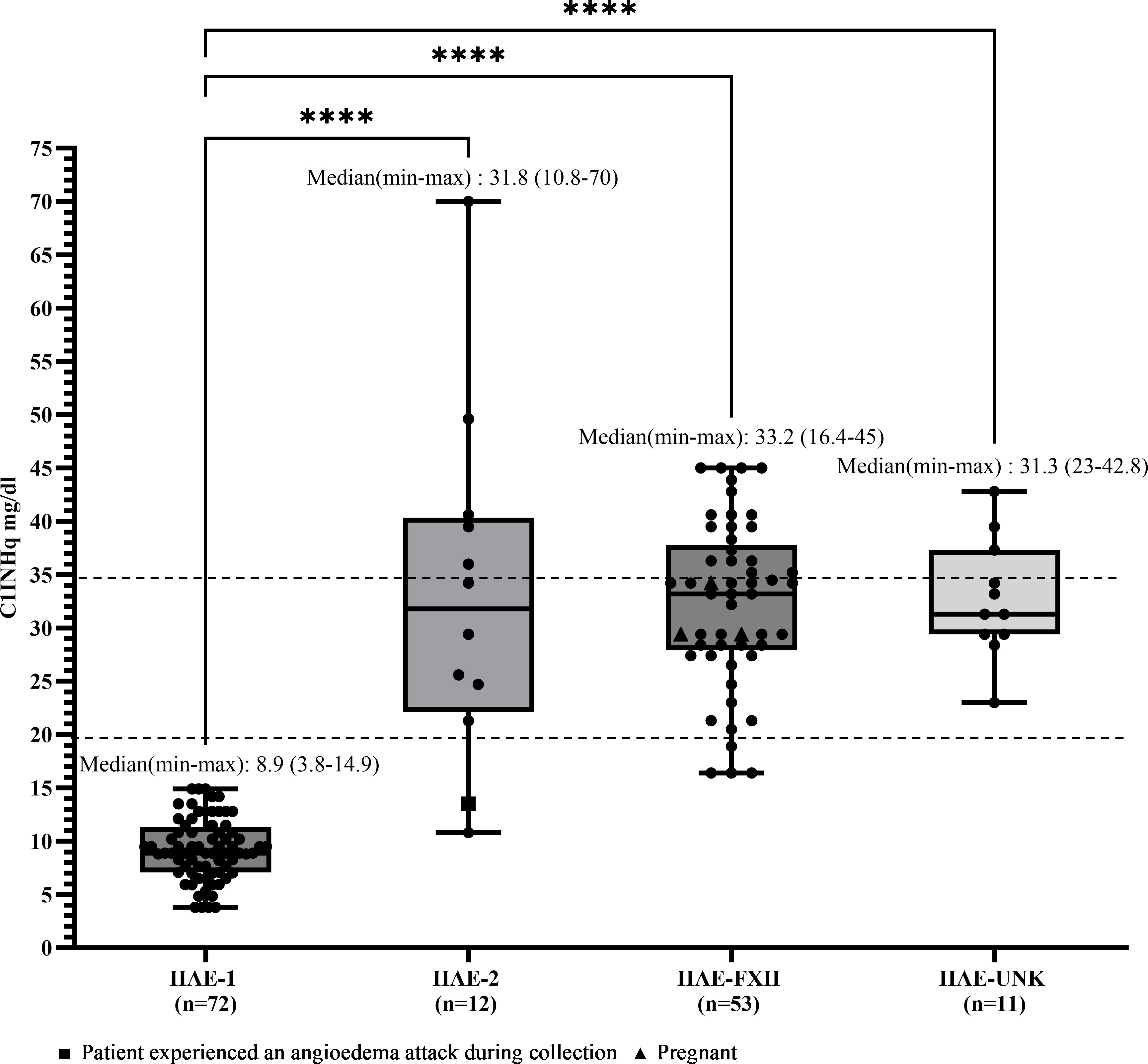
Figure 2. C1INHq levels according to HAE diagnosis. Box-and-whisker plots represent C1 inhibitor level concentrations (CIINIIq, mg/dL) in patients with hereditary angioedema (HAE) due to CIINH deficiency type 1 (HAE-1) and HAE due to C1INH deficiency type 2 (HAE-2), as well as HAE with normal C1 inhibitor levels, subdivided into HAE with factor XII mutation (HAE-FXII) and HAE of unknown mutation (HAE-UNK). The normal reference interval for CIINHq levels is indicated by dashed lines. Boxes represent the interquartile range (IQR); the horizontal line within each box indicates the median, and whiskers extend from the minimum to the maximum values. Individual data points are overlaid. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn's multiple comparisons test. (****p<0.0001).
In patients with HAE-FXII, both C4 and C1INHq levels were within the normal range (median for both: 33.2 mg/dL), although one young adult had a decreased C4 level of 15 mg/dL. In the HAE-UNK group, median C4 and C1INHq levels were 36.0 mg/dL (range: 22.9–44.9) and 31.3 mg/dL (range: 23.0–42.8), respectively (Figures 1, 2).
Sensitivity was calculated as the proportion of true positive (TP) results among patients with HAE-C1INH type 1, using a cutoff of 50% for fC1INH levels. As all patients with HAE-C1INH type 1 are expected to have fC1INH activity below 50%, values ≤50% were considered positive (i.e., abnormal). The number of true positives (TP) for each method was 71/72 for the DBS assay, 45/59 for the chromogenic assay, 55/72 for the ELISA PKa method, and 69/71 for the ELISA FXIIa method. Sensitivity was expressed as TP/(TP + FN), with false negatives (FN) defined as patients with a confirmed diagnosis of HAE-C1INH type 1 who unexpectedly showed fC1INH values above 50% and were therefore misclassified as normal. Among the functional assays, the DBS method showed the highest sensitivity for detecting fC1INH deficiency in HAE-C1INH type 1 patients (71/72; 98.6%), followed by ELISA-FXIIa (69/71; 97.2%), the chromogenic assay (45/59; 76.3%), and ELISA-PKa (55/72; 76.4%). Median fC1INH values (range) were as follows: DBS, 0% (0–78.4); chromogenic, 39.8% (2–102); ELISA-PKa, 40% (0–94); and ELISA-FXIIa, 14% (0–75) (Figure 3).
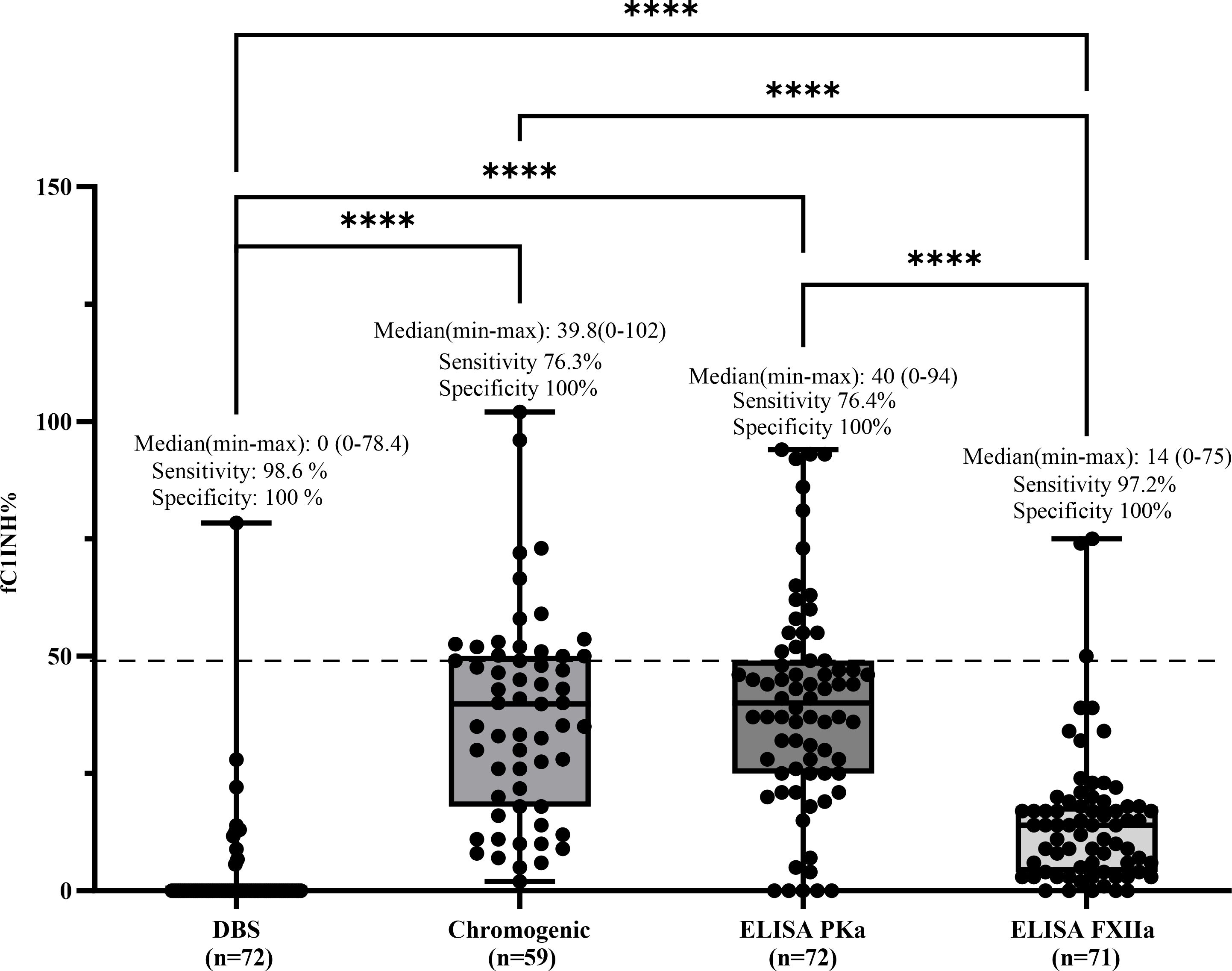
Figure 3. Comparison of fC1INH % values by different methods in HAE-C1INH-Type1. Box-and-whisker plots represent functional C1 inhibitor (fC1INH%) levels in patients with hereditary angioedema due to C1INH deficiency type 1 (HAE-C1INH-Type1), assessed using four different methods: dried blood spot (DBS), chromogenic assay, and ELISAs employing kallikrein (PKa) or activated factor XII (FXIIa) as substrates. The dashed line indicates the lower reference limit for normal fC1INH activity (50%). Boxes represent the interquartile range (IQR); the horizontal line within each box indicates the median, and whiskers extend from minimum to maximum values. Individual data points are overlaid. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn's multiple comparisons test. (****p < 0.0001). Sensitivity was calculated by identifying true positive results defined as fC1INH activity below 50% in patients with confirmed HAE-1. Specificity was assessed using samples from individuals without C1INH deficiency, where true negatives were defined as fC1INH activity ≥50%.
For patients with HAE-C1INH type 2, sensitivity was calculated using the same rationale, since these patients also present with functional deficiency of C1 inhibitor, despite having normal or elevated antigenic levels. Therefore, all values ≤50% were considered true positives. Any result >50% was interpreted as a false negative, representing an inappropriately normal result in a patient with a confirmed diagnosis. Based on these criteria, the DBS method correctly identified all 12 patients with values ≤50%, resulting in a sensitivity of 100%. ELISA-FXIIa detected 11 out of 12 cases (sensitivity of 91.7%), the chromogenic method identified 6 out of 9 patients (66.7%), and ELISA-PKa detected only 3 out of 12 (25%). Median values (range) in this group were as follows: DBS, 0% (0–22.78); chromogenic, 38% (0–66); ELISA-PKa, 57% (38–143); and ELISA-FXIIa, 8% (0–95) (Figure 4).
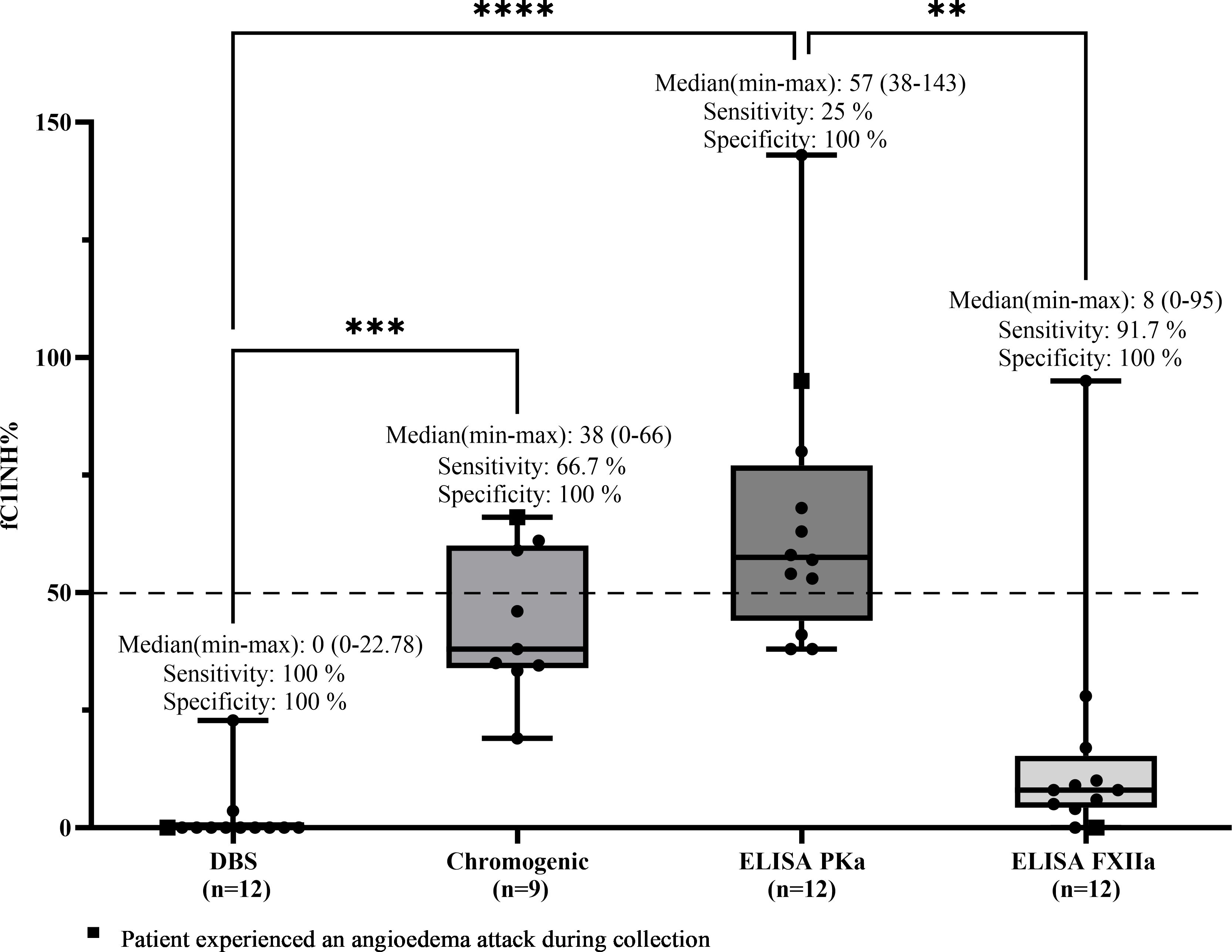
Figure 4. Comparison of fC1INH % values by different methods in HAE-C1INH-Type2. Box-and-whisker plots represent functional C1 inhibitor (fC1INH%) levels in patients with hereditary angioedema due to C1INH deficiency type 2 (HAE-C1INH-Type2), assessed using four different methods: dried blood spot (DBS), chromogenic assay, and ELISAs employing kallikrein (PKa) or activated factor XII (FXIIa) as substrates. The dashed line indicates the lower reference limit for normal fC1INH activity (50%). Boxes represent the interquartile range (IQR); the horizontal line within each box indicates the median, and whiskers extend from minimum to maximum values. Individual data points are overlaid. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn's multiple comparisons test (**p < 0.01; ***p < 0.001; ****p < 0.0001); Sensitivity was calculated by identifying true positive results-defined as fC1INH activity below 50% in patients with confirmed HAE-1. Specificity was assessed using samples from individuals without C1INH deficiency, where true negatives were defined as fC1INH activity ≥50%.
In contrast, patients with HAE-FXII and HAE of unknown cause (HAE-UNK) are expected to have normal fC1INH function. For these groups, values below 50% were not expected and were therefore interpreted as altered or falsely abnormal results. In these cases, the prevalence of altered results was calculated, defined as the proportion of individuals with fC1INH values ≤50%. In the HAE-FXII group (n = 47), the DBS method showed altered values in 9 patients (19.1%, including all 3 pregnant women in the study), the chromogenic method in none (0%), ELISA-PKa in 2 out of 52 patients (3.8%, both pregnant), and ELISA-FXIIa in 19 out of 52 patients (36.5%, including 2 pregnant women). Median fC1INH values (range) were as follows: DBS, 74% (0–126); chromogenic, 120% (69–152); ELISA-PKa, 132.9% (0–433); and ELISA-FXIIa, 55.6% (0–95) (Figure 5).
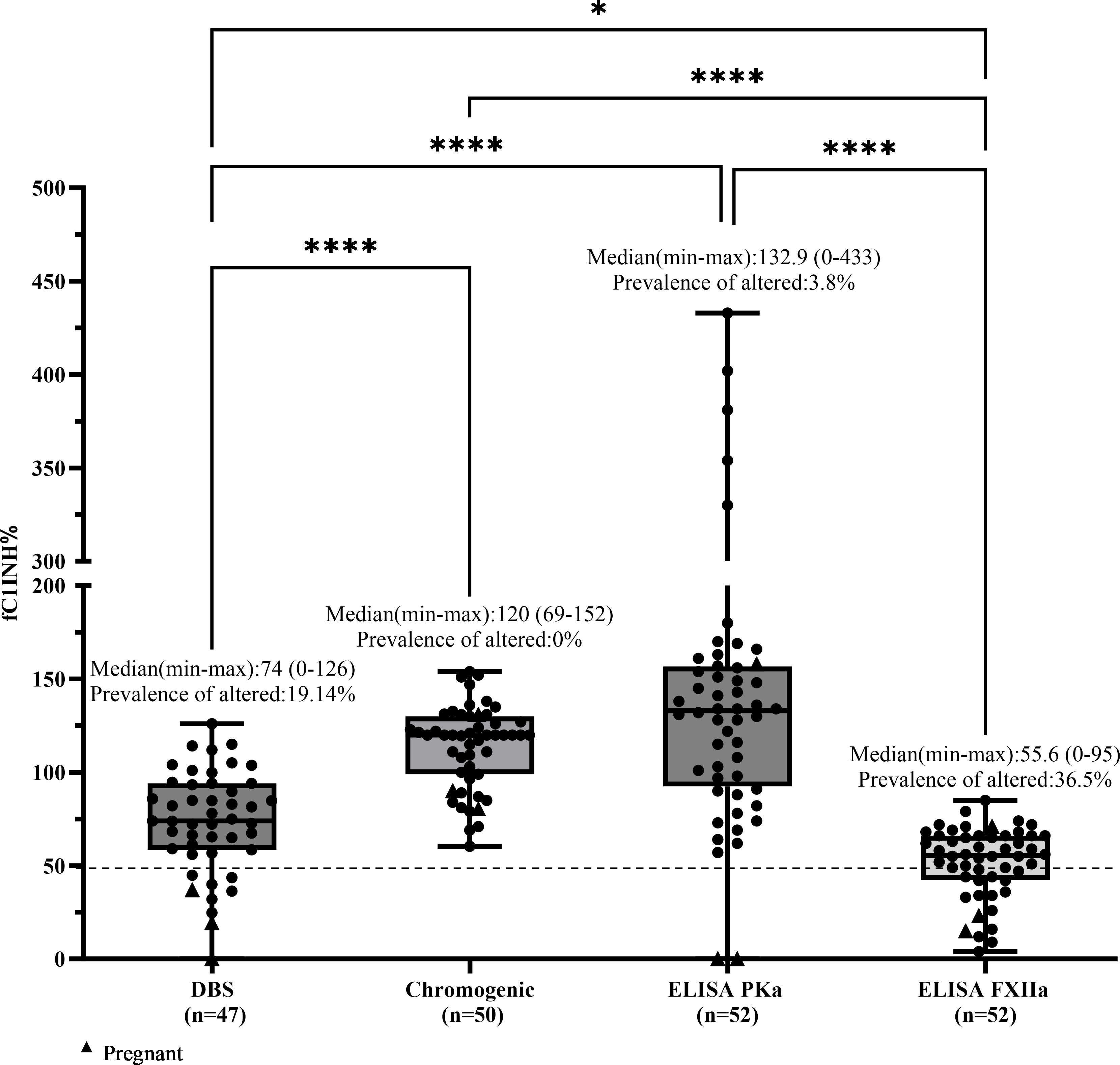
Figure 5. Comparison of fC1INH % values by different methods in HAE-FXII. Box-and-whisker plots depict functional C1 inhibitor (fC1INH%) levels in patients with HAE with normal CIINH due to a Factor XII variant (HAE-FXII), assessed using four different methods: dried blood spot (DBS), chromogenic assay, and ELISAs employing kallikrein (PKa) or activated factor XII (FXIIa) as substrates. The dashed line represents the lower reference limit for normal fC1INH activity (50%). Boxes indicate the interquartile range (IQR); the horizontal line within each box shows the median, and whiskers extend from the minimum to maximum values. Individual data points are overlaid. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn's multiple comparisons test (*p < 0.05; ****p < 0.0001).
In the HAE-UNK group (n = 11), all patients showed fC1INH levels above 50% across all methods, resulting in a prevalence of altered values of 0% for all assays. Median values (range) were as follows: DBS, 109.6% (62–131); chromogenic, 120% (69–152); ELISA-PKa, 79% (62.4–409); and ELISA-FXIIa, 67% (50–102) (Figure 6).
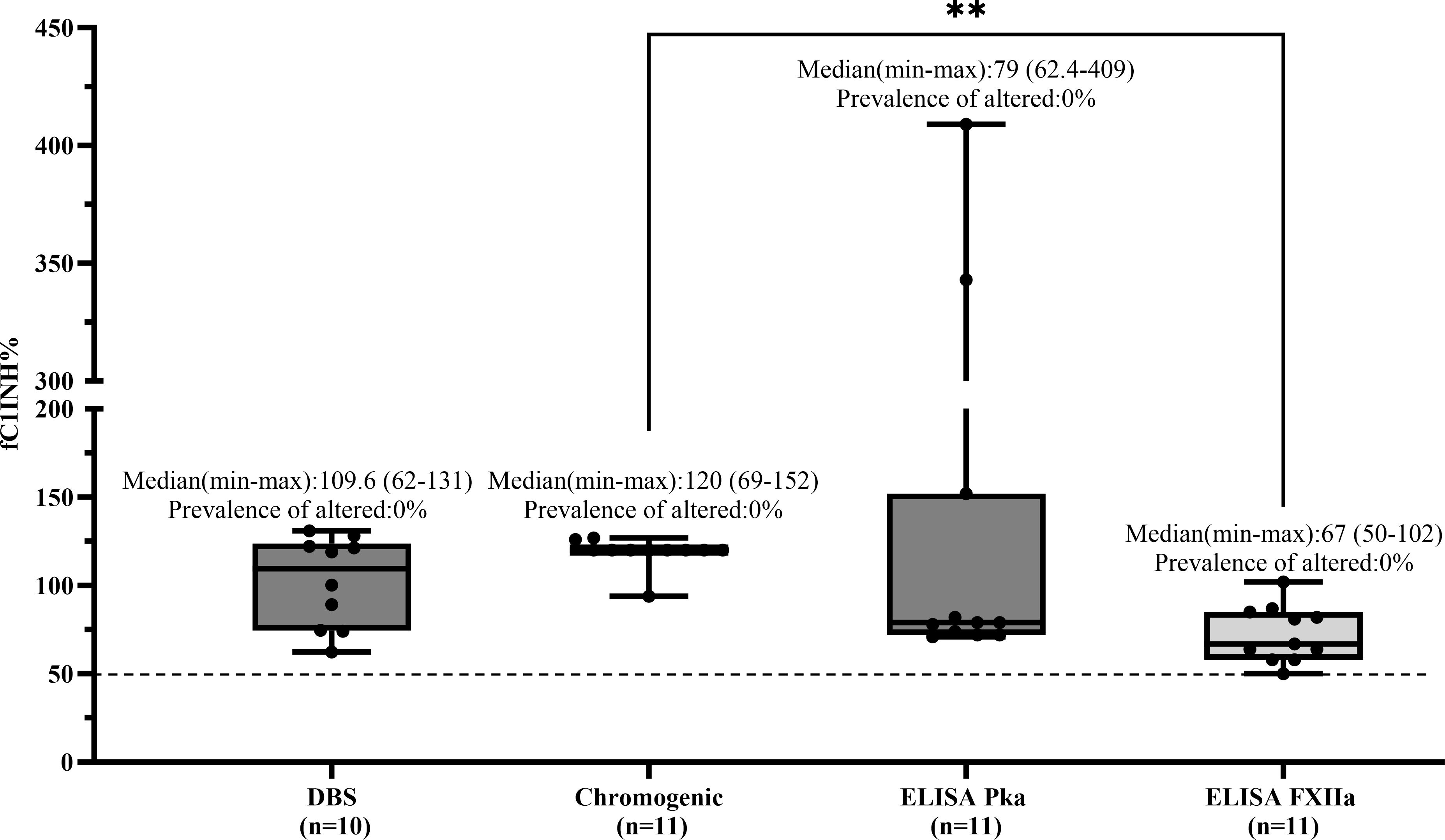
Figure 6. Comparison of fC1INH % values by different methods in HAE-UNK. Box-and-whisker plots depict functional C1 inhibitor (fC1INH%) levels in patients with HAE with normal C1INH with unknown mutation (HAE-UNK), assessed using four different methods: dried blood spot (DBS), chromogenic assay, and ELISAs employing kallikrein (PKa) or activated factor XII (FXIIa) as substrates. The dashed line represents the lower reference limit for normal fC1INH activity (50%). Boxes indicate the interquartile range (IQR); the horizontal line within each box shows the median, and whiskers extend from the minimum to maximum values. Individual data points are overlaid. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn's multiple comparisons test (**p < 0.01).
Specificity was defined as the proportion of true negative results among healthy controls. All control individuals had fC1INH levels >50% across all methods evaluated and were therefore classified as true negatives (TN). As no false positives (FP) were observed, specificity was calculated as TN/(TN + FP), resulting in 100% for all assays tested.
4 Discussion
HAE remains underrecognized globally, especially in settings with limited access to specialized laboratory testing (29, 35). Although clinical criteria are well established, biochemical confirmation remains critical for accurate subtype differentiation and therapeutic decision-making. Standard diagnostics rely on the evaluation of C4 and C1INH antigenic and functional C1INH levels. However, these markers are variably affected by pre-analytical factors such as sample handling and storage, which may reduce the reliability of results (27, 28, 36, 37).
This study presents novel comparative methods of evaluation of fC1INH performed in a population of patients that closely mirrors real-world clinical scenarios. Unlike many previous studies that stratify patients only after the complete diagnostic workup, this work reflects the practical reality: all patients with suspected HAE initially undergo functional C1INH testing, regardless of their final classification (HAE-C1INH or HAE-nC1INH). This approach enhances the applicability and relevance of the findings to early diagnostic pathways. A key methodological innovation of this study lies in the head-to-head comparison of multiple fC1INH assays, DBS, chromogenic assay, and ELISA, using distinct substrates. While both DBS and chromogenic assays utilized C1s, in line with standard diagnostic protocols, the in-house ELISA developed specifically for this research employed novel substrates: activated factor XII (FXIIa) and plasma kallikrein (PKa) (34). These components target the kinin-kallikrein system (KKS), offering a unique perspective on C1INH functionality. This innovative use of FXIIa and PKa not only broadens the understanding of KKS regulation but also suggests the potential for greater sensitivity in detecting functional abnormalities, particularly in diagnostically challenging cases.
In HAE-C1INH-Type1, patients are expected to have uniformly reduced levels of C4, C1INHq, and fC1INH. Our findings largely confirm this biochemical pattern: 72 patients classified as type 1 exhibited low median values across all markers, and the majority had fC1INH <50% by DBS, ELISA-FXIIa, chromogenic, and ELISA-PKa. However, exceptions were noted: five type 1 patients (one child and four adults) had C4 within or above the reference range, underscoring the limitations of relying on a single marker (3, 12).
In HAE-C1INH-Type2, the classic profile includes reduced C4, normal or elevated C1INHq, and diminished function. Our data support this: the median C1INHq was elevated with a normal range extending up to 70 mg/dL, while C4 remained low. Functionally, only DBS (100%) and ELISA-FXIIa (91.7%) reliably detected reduced fC1INH, while the chromogenic assay (66.7%) and ELISA-PKa (25%) showed low or very low sensitivity.
Notably, while the chromogenic assay remains the conventional reference, our data confirm its limited sensitivity for HAE-C1INH-Type2 and variable performance in type 1. This may reflect assay vulnerability to pre-analytical influences, as described in the literature (37–39). Our results suggest that reliance on this assay alone may miss a substantial proportion of affected individuals, particularly in the presence of borderline or fluctuating levels.
In contrast, the DBS LC-MS/MS method and ELISA-FXIIa assay demonstrated superior diagnostic performance across both subtypes. DBS detected nearly all functional deficiencies in types 1 and 2, offering advantages of simplified logistics and greater reproducibility, even in decentralized settings. While the DBS approach offers logistical benefits such as minimal sample volume, ambient stability, and remote usability, its implementation is still constrained by the need for LC-MS/MS infrastructure, a high-complexity technology still unavailable in many laboratories and countries, particularly in low-resource settings (30–33). ELISA-FXIIa also showed high accuracy, though its in-house nature and lack of commercial availability remain barriers to broad implementation. However, these two assays were less specific in the HAE-FXII group, where fC1INH levels are expected to be normal. Apparent reductions in fC1INH were observed in 19.1% of cases by DBS and in 36.5% by ELISA-FXIIa, particularly among pregnant women, likely reflecting expected physiological changes during pregnancy, such as hormonal modulation or increased C1INH consumption. These findings underscore the high sensitivity of the assays in detecting altered function; however, no clear explanation has been established for other isolated reductions observed in non-pregnant individuals (17, 40).
In patients with HAE-UNK, all assays resulted in normal fC1INH levels, and the group showed biochemical profiles consistent with preserved complement function, reinforcing the need for genetic exploration and assessment of treatment response for diagnosis (3, 4).
Finally, our findings reinforce the limited utility of C4 as a standalone screening marker. Although historically used in the initial evaluation of HAE, its sensitivity and specificity are suboptimal. Notably, several genetically confirmed cases exhibited normal C4 levels, highlighting that a normal C4 result does not exclude the diagnosis of HAE. Therefore, C4 measurement should be repeated during symptomatic episodes to improve diagnostic accuracy (12).
The strengths of this study include its multicenter design, a large and well-characterized cohort with confirmed genetic diagnoses, standardized sample processing, and comparative analysis of four distinct functional assays. The inclusion of controls provided an internal benchmark for specificity and variability.
In summary, this work supports the integration of functional assays—especially DBS and ELISA-FXIIa—into diagnostic workflows and cautions against overreliance on C4 or chromogenic testing alone. A combination of clinical evaluation, comprehensive complement testing, and genetic confirmation remains essential for accurate diagnosis and optimal care.
5 Conclusion
This study demonstrates that both the DBS LC-MS/MS method and the in-house ELISA-FXIIa assay offer high diagnostic accuracy for HAE-C1INH, where conventional assays such as the chromogenic method may lack sensitivity. DBS stands out for its practical advantages, such as simple collection, low sample volume, and ambient stability, which make it particularly suited for use in remote or resource-limited settings. While ELISA-FXIIa also showed strong performance, its broader clinical adoption is constrained by limited availability and standardization.
In clinical practice, DBS may serve as a valuable frontline tool for the biochemical confirmation of HAE-C1INH, complementing existing protocols. For patients with HAE-nC1INH, genetic testing remains essential for diagnosis. Future research should prioritize the validation of these functional assays in larger, diverse populations and explore the integration of emerging biomarkers to further improve diagnostic accuracy across all HAE subtypes.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by the Brazilian National Research Ethics Commission (CAAE: 41812720010010082). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
MB: Conceptualization, Formal Analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. RC-S: Investigation, Methodology, Validation, Writing – review & editing. MA: Writing – review & editing. AT: Writing – review & editing. PG-B: Writing – review & editing. EM: Writing – review & editing. JP: Writing – review & editing. SV: Writing – review & editing. AG: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. MLDB received a Researcher-Initiative Grant from LASID (sponsored by Takeda) for the period 2022–2023. The sponsors had no influence on the design of the study, nor on the collection, analysis, or interpretation of the data. ASG holds a CNPq Research Productivity Fellowship (grant number 309824/2021-4), and this study was supported by an Investigator-Initiated Research Grant (IISR-2020-103263) from Shire Pharmaceuticals, now part of the Takeda group of companies.
Acknowledgments
The authors express their heartfelt gratitude to the patients and their families for their trust, generosity, and participation, which were essential to the success of this study. We also thank the collaborating centers for their invaluable support and the dedicated team at FMABC, whose commitment and expertise were fundamental throughout every stage of the research.
Conflict of interest
MB received a Researcher-Initiative Grant from LASID, sponsored by Shire/Takeda, for independent research support unrelated to the execution or content of this study. AG holds a CNPq Research Productivity Fellowship grant number 309824/2021-4, and this study was supported by an Investigator-Initiated Research Grant IISR-2020-103263 from Shire Pharmaceuticals, now part of the Takeda group of companies. AG has served as a speaker and/or consultant for Takeda, CSL Behring, Pharvaris, KalVista, Exeltis, Pint-Pharma, Biomarin, Binding Site, Multicare, and Astra. MA, PG-B, and SV received financial support and/or honoraria from Takeda and CSL Behring. EM has served as a speaker and consultant for Takeda, CSL Behring, Novartis, GSK, Pint Pharma, and Sanofi.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. In accordance with Frontiers’ editorial policies, we declare that OpenAI’s ChatGPT was used solely to assist in improving the English language of the manuscript (grammar, clarity, and style). The tool was not used for generating content, interpreting data, or drawing scientific conclusions. All intellectual and scientific contributions remain the sole responsibility of the authors, who reviewed and approved the final version of the text.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1654078/full#supplementary-material
Abbreviations
AE, angioedema; C1INH, C1 inhibitor; C1INHq, C1 inhibitor level; DBS, dried blood spot; fC1INH, functional C1 inhibitor; HAE, hereditary angioedema; HAE-C1INH, HAE due to C1INH deficiency; HAE-C1INH-Type1, HAE due to C1INH deficiency type 1; HAE-C1INH-Type2, HAE due to C1INH deficiency type 2; HAE-nC1INH, HAE with normal C1INH; HAE-FXII, HAE with factor XII variant; HAE-ANGPT1, HAE with angiopoietin 1 variant; HAE-UNK, HAE with unknown mutation; cHK, cleaved high-molecular-weight kininogen; FXI, coagulation factor XI (Rosenthal factor); FXII, coagulation factor XII (Hageman factor); FXIIa, activated factor XII; KKS, kallikrein-kinin system; LC-MS/MS, liquid chromatography-tandem mass spectrometry; PKa, plasma kallikrein
References
1. Bork K, Meng G, Staubach P, and Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. (2006) 119:267–74. doi: 10.1016/j.amjmed.2005.09.064
2. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy.(2014) 69:602–16. doi: 10.1111/all.12380
3. Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema - The 2021 revision and update. World Allergy Organ J. (2022) 15:100627. doi: 10.1016/j.waojou.2022.100627
4. Zuraw BL, Bork K, Bouillet L, Christiansen SC, Farkas H, Germenis AE, et al. Hereditary angioedema with normal C1 inhibitor: an updated international consensus paper on diagnosis, pathophysiology, and treatment. Clin Rev Allergy Immunol. (2025) 68:24. doi: 10.1007/s12016-025-09027-4
5. Sainz IM, Pixley RA, and Colman RW. Fifty years of research on the plasma kallikrein-kinin system: from protein structure and function to cell biology and in-vivo pathophysiology. Thromb Haemost.(2007) 98:77–83. doi: 10.1160/TH07-04-0250
6. Kaplan AP and Joseph K. Complement, kinins, and hereditary angioedema: mechanisms of plasma instability when C1 inhibitor is absent. Clin Rev Allergy Immunol. (2016) 51:207–15. doi: 10.1007/s12016-016-8555-6
7. Schapira M, Silver LD, Scott CF, Schmaier AH, Prograis LJ Jr., Curd JG, et al. Prekallikrein activation and high-molecular-weight kininogen consumption in hereditary angioedema. N Engl J Med. (1983) 308:1050–3. doi: 10.1056/NEJM198305053081802
8. López Lera A. Pathophysiology and underlying mechanisms in hereditary angioedema. Balkan Med J. (2021) 38:82–8. doi: 10.4274/balkanmedj.galenos.2020.2020.10.166
9. Ponard D, Gaboriaud C, Charignon D, Ghannam A, Wagenaar-Bos IGA, Roem D, et al. SERPING1 mutation update: Mutation spectrum and C1 Inhibitor phenotypes. Hum Mutat. (2020) 41:38–57. doi: 10.1002/humu.23917
10. Germenis AE, Rijavec M, and Veronez CL. Leveraging genetics for hereditary angioedema: A road map to precision medicine. Clin Rev Allergy Immunol. (2021) 60:416–28. doi: 10.1007/s12016-021-08836-7
11. Lumry WR and Settipane RA. Hereditary angioedema: Epidemiology and burden of disease. Allergy Asthma Proc. (2020) 41:S08–s13. doi: 10.2500/aap.2020.41.200050
12. Tarzi MD, Hickey A, Förster T, Mohammadi M, and Longhurst HJ. An evaluation of tests used for the diagnosis and monitoring of C1 inhibitor deficiency: normal serum C4 does not exclude hereditary angio-oedema. Clin Exp Immunol. (2007) 149:513–6. doi: 10.1111/j.1365-2249.2007.03438.x
13. Aabom A, Bygum A, and Koch C. Complement factor C4 activation in patients with hereditary angioedema. Clin Biochem. (2017) 50:816–21. doi: 10.1016/j.clinbiochem.2017.04.007
14. Karim Y, Griffiths H, and Deacock S. Normal complement C4 values do not exclude hereditary angioedema. J Clin Pathol. (2004) 57:213–4. doi: 10.1136/jcp.2003.12021
15. Pedrosa M, Phillips-Angles E, López-Lera A, López-Trascasa M, and Caballero T. Complement study versus CINH gene testing for the diagnosis of type I hereditary angioedema in children. J Clin Immunol. (2016) 36:16–8. doi: 10.1007/s10875-015-0222-9
16. Betschel S, Badiou J, Binkley K, Borici-Mazi R, Hébert J, Kanani A, et al. The international/canadian hereditary angioedema guideline. Allergy Asthma Clin Immunol. (2019) 15:72. doi: 10.1186/s13223-019-0376-8
17. Ivanov I, Matafonov A, Sun MF, Mohammed BM, Cheng Q, Dickeson SK, et al. A mechanism for hereditary angioedema with normal C1 inhibitor: an inhibitory regulatory role for the factor XII heavy chain. Blood.(2019) 133:1152–63. doi: 10.1182/blood-2018-06-860270
18. Bork K, Wulff K, Steinmüller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy.(2018) 73:442–50. doi: 10.1111/all.13623
19. Bafunno V, Firinu D, D’Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. (2018) 141:1009–17. doi: 10.1016/j.jaci.2017.05.020
20. Bork K, Wulff K, Rossmann H, Steinmüller-Magin L, Braenne I, Witzke G, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy.(2019) 74:2479–81. doi: 10.1111/all.13869
21. Ariano A, D’Apolito M, Bova M, Bellanti F, Loffredo S, D’Andrea G, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy.(2020) 75:2989–92. doi: 10.1111/all.14454
22. Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. (2021) 148:1041–8. doi: 10.1016/j.jaci.2021.01.011
23. Vincent D, Parsopoulou F, Martin L, Gaboriaud C, Demongeot J, Loules G, et al. Hereditary angioedema with normal C1 inhibitor associated with carboxypeptidase N deficiency. J Allergy Clin Immunol Glob. (2024) 3:100223. doi: 10.1016/j.jacig.2024.100223
24. D’Apolito M, Santacroce R, Vazquez DO, Cordisco G, Fantini CA, D’Andrea G, et al. DAB2IP associates with hereditary angioedema: Insights into the role of VEGF signaling in HAE pathophysiology. J Allergy Clin Immunol. (2024) 154(3):698–706. doi: 10.1016/j.jaci.2024.05.017
25. Reshef A, Buttgereit T, Betschel SD, Caballero T, Farkas H, Grumach AS, et al. Definition, acronyms, nomenclature, and classification of angioedema (DANCE): AAAAI, ACAAI, ACARE, and APAAACI DANCE consensus. J Allergy Clin Immunol. (2024) 154:398–411.e1. doi: 10.1016/j.jaci.2024.03.024
26. Busse PJ, Christiansen SC, Riedl MA, Banerji A, Bernstein JA, Castaldo AJ, et al. US HAEA medical advisory board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract. (2021) 9:132–50.e3. doi: 10.1016/j.jaip.2020.08.046
27. Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, Bork K, Bucher C, Bygum A, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods. (2008) 338:14–20. doi: 10.1016/j.jim.2008.06.004
28. Grumach AS and Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. (2014) 61:110–7. doi: 10.1016/j.molimm.2014.06.030
29. Veronez CL, Mendes AR, Leite CS, Gomes CP, Grumach AS, Pesquero JB, et al. The panorama of primary angioedema in the Brazilian population. J Allergy Clin Immunol Pract. (2021) 9:2293–304.e5. doi: 10.1016/j.jaip.2020.11.039
30. Lai Y, Zhang G, Zhou Z, Inhaber N, Bernstein JA, Chockalingam PS, et al. A novel functional C1 inhibitor activity assay in dried blood spot for diagnosis of Hereditary angioedema. Clin Chim Acta. (2020) 504:155–62. doi: 10.1016/j.cca.2020.02.010
31. Lai Y, Zhang G, Inhaber N, Bernstein JA, Cwik M, Zhou Z, et al. A robust multiplexed assay to quantify C1-inhibitor, C1q, and C4 proteins for in vitro diagnosis of hereditary angioedema from dried blood spot. J Pharm BioMed Anal. (2021) 195:113844. doi: 10.1016/j.jpba.2020.113844
32. Wong JCY, Lam DLY, Yim JSH, Lee E, Shi W, Chiang V, et al. Validating and utilizing dried blood spots for family screening: Screening Programme Providing Outreach for Testing Hereditary Angioedema (SPPOT-HAE). J Allergy Clin Immunology: Global.(2025) 4:100381. doi: 10.1016/j.jacig.2024.100381
33. Bernstein JA, Cheng J, Pisani T, Sexton D, Whitaker RE, Estepan DN, et al. Clinical validity of dried blood spot assay for the measurement of functional C1 inhibitor in hereditary angioedema. J Allergy Clin Immunology: Global. (2025) 4(2):100401. doi: 10.1016/j.jacig.2025.100401
34. Joseph K, Bains S, Tholanikunnel BG, Bygum A, Aabom A, Koch C, et al. A novel assay to diagnose hereditary angioedema utilizing inhibition of bradykinin-forming enzymes. Allergy.(2015) 70:115–9. doi: 10.1111/all.12520
35. Faradiba S, Marilyn UP, Eduardo S, Regina DG, Maria G, Alvaro C, et al. O atendimento médico de pacientes com doenças imunoalérgicas no Brasil: reflexoes e propostas para a melhoria. Arq Asma Alerg Imunol.(2017) 2:327–34. doi: 10.5935/2526-5393.20180040
36. Germenis AE and Cicardi M. Driving towards Precision Medicine for angioedema without wheals. J Autoimmun. (2019) 104:102312. doi: 10.1016/j.jaut.2019.102312
37. Grumach AS, Riedl MA, Cheng L, Jain S, Nova Estepan D, and Zanichelli A. Hereditary angioedema diagnosis: Reflecting on the past, envisioning the future. World Allergy Organ J. (2025) 18(6):101060. doi: 10.1016/j.waojou.2025.101060
38. Gompels MM and Lock RJ. Laboratory testing for C1 inhibitor deficiency: a comparison of two approaches to C1 inhibitor function. Ann Clin Biochem. (2007) 44(Pt 1):75–8. doi: 10.1258/000456307779596020
39. Charest-Morin X, Betschel S, Borici-Mazi R, Kanani A, Lacuesta G, Rivard G-É, et al. The diagnosis of hereditary angioedema with C1 inhibitor deficiency: a survey of Canadian physicians and laboratories. Allergy Asthma Clin Immunol. (2018) 14:83. doi: 10.1186/s13223-018-0307-0
Keywords: hereditary angioedema, C1 inhibitor, factor XII, complement C4, biomarker, diagnosis
Citation: Bardou MLD, Constantino-Silva RN, Alonso MLO, Teixeira AJR, Giavina-Bianchi PF, Mansour E, Pesquero JB, Valle SOR and Grumach AS (2025) Evaluating functional C1INH with multiple laboratory methods across Hereditary Angioedema types. Front. Immunol. 16:1654078. doi: 10.3389/fimmu.2025.1654078
Received: 25 June 2025; Accepted: 04 August 2025;
Published: 26 August 2025.
Edited by:
Anthony Dorr, Barts Health NHS Trust, United KingdomReviewed by:
Maria Bova, University of Naples Federico II, ItalyAndrea Balduit, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), Italy
Copyright © 2025 Bardou, Constantino-Silva, Alonso, Teixeira, Giavina-Bianchi, Mansour, Pesquero, Valle and Grumach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maine Luellah Demaret Bardou, bWFpbmVfYmFyZG91QGhvdG1haWwuY29t