 Natasha Palmer
Natasha Palmer Salim Khakoo
Salim Khakoo Tilman Sanchez-Elsner
Tilman Sanchez-Elsner Andres F. Vallejo
Andres F. Vallejo- School of Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
The tumour microenvironment (TME) is a complex and dynamic environment containing diverse cellular, stromal and soluble factors, that collectively influence cancer progression, immune evasion and therapeutic resistance. Among the immune components of the TME, macrophages and natural killer (NK) cells are key players, whose interactions, particularly their crosstalk, critically shape anti-tumour immunity. The macrophage–NK cell interplay can either promote or suppress immune responses depending on the context, representing both a challenge and a therapeutic opportunity. NK cells are key effectors capable of recognising and eliminating malignant cells without prior sensitisation, whereas macrophages exhibit remarkable plasticity, functioning as either promoters or suppressors of tumour immunity depending on their activation state. This review focuses on current strategies to harness macrophages in cancer therapy, including phenotype repolarisation, selective depletion, and disruption or enhancement of the macrophage-NK cell crosstalk to enhance NK cell-mediated tumour surveillance. Finally, we highlight emerging technologies, such as single-cell RNA sequencing, spatial transcriptomics, and proteomics, as powerful tools to elucidate the dynamic interplay between macrophages and NK cells and inform the next generation of immunotherapeutic interventions.
1 Macrophages: versatile regulators of tissue homeostasis and immunity
Macrophages are innate immune cells with essential roles across homeostasis, inflammation, and cancer. They originate from multiple developmental pathways, including embryonic progenitors such as yolk sac and foetal liver precursors, which give rise to long-lived tissue-resident populations like microglia in the brain, Kupffer cells in the liver, and alveolar macrophages in the lung (1, 2). These populations are maintained independently of circulating monocytes throughout adult life (3). By contrast, macrophages associated with inflammation or pathology are typically derived from adult haematopoietic stem cells via monocytes, which are recruited to tissues in response to chemotactic signals and differentiate in situ (4–6).
Across tissues, macrophages carry out a core set of functions, including the phagocytosis of pathogens, apoptotic cells, and debris; the presentation of antigens via Major Histocompatibility Complex (MHC) molecules to T cells; and the secretion of cytokines and chemokines that modulate both innate and adaptive immune responses (7). However, their phenotype and function are shaped by the local tissue microenvironment and inflammatory cues, giving rise to considerable heterogeneity.
In vitro models have classically categorised macrophage polarisation into ‘M1’ and ‘M2’ phenotypic states, pro-inflammatory and anti-inflammatory respectively, based on stimulation with microbial products (e.g., LPS, IFN-γ) (8) or anti-inflammatory cytokines (e.g., IL-4, IL-13) (9). While this M1/M2 paradigm has provided a useful conceptual framework, it does not adequately reflect the complexity of heterogeneity of macrophage phenotypes observed in vivo, particularly within pathological settings such as the tumour microenvironment (TME). Tumour-associated macrophages (TAMs), for instance, do not conform neatly to M1 or M2 phenotypes but rather exhibit a spectrum of activation states that can simultaneously support immunosuppression, tissue remodelling, and tumour progression (10, 11). Unravelling this functional and phenotypic diversity remains a major focus in immunology, with implications for both fundamental biology and therapeutic targeting.
1.1 Activating and inhibitory receptors: regulating macrophage activation
Macrophage activation is regulated by a balance of stimulatory and inhibitory signals that enable these plastic cells to dynamically adapt to their environment. Among the main activating pathways are pattern recognition receptors (PRRs), notably Toll-like receptors (TLRs). TLRs recognise conserved pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), initiating downstream signalling cascades such as NF-κB activation. This leads to the production of pro-inflammatory cytokines, including TNF-α, IL-6 and IL-12 (12, 13). Co-stimulatory receptors like CD40 also promote macrophage activation, particularly through interaction with CD40L on CD4+ T cells, resulting in similar cytokine production (14–17). Triggering Receptor Expressed on Myeloid Cells-1 (TREM-1) further amplifies inflammatory responses by synergising with TLR signalling (18), whereas TREM-2 is associated with a regulatory phenotype, supporting phagocytosis and tissue remodelling (19).
Conversely, macrophage activation is tightly controlled by inhibitory receptors that prevent excessive tissue damage and maintain immune homeostasis. The checkpoint receptor programmed death-1 (PD-1) and its ligand PD-L1 modulate macrophage function by dampening inflammatory responses (20). The scavenger receptor Macrophage Receptor with Collagenous Structure (MARCO), also plays a immunoregulatory role by mediating the clearance of apoptotic cells and microbial components (21, 22). Additional regulatory pathways include the CD47–SIRPα axis, which inhibits macrophage phagocytosis and is frequently exploited by cancer cells to avoid clearance (23).
2 Natural killer cells: key players in immune surveillance and tumour defence
Natural killer (NK) cells are critical effectors of the innate immune system, capable of detecting and eliminating transformed or infected cells. These cytotoxic lymphocytes develop in the bone marrow and undergo maturation in secondary lymphoid organs, such as the spleen, lymph nodes and tonsils (24). Once in circulation, where they comprise 5-15% of peripheral blood lymphocytes, NK cells traffic to diverse tissues, including lymphoid and non-lymphoid sites like the liver and lungs (25). NK cell populations are broadly divided into two functional subsets: the cytotoxic CD56dim subset, which predominates in peripheral blood and the spleen, and the CD56bright subset, enriched in secondary lymphoid tissues and characterised by robust cytokine production, including IFN-γ, TNF-α, and GM-CSF (26–28). CD56bright NK cells also secrete chemokines such as CCL3–5 and CXCL8 (24, 29), contributing to immune cell recruitment and orchestration of early immune responses.
1.2 Activating and inhibitory receptors: coordinating NK cell responses
NK cell function is intricately regulated by a balance of activating and inhibitory germline-encoded receptors. Activating receptors, including NKG2D, DNAM-1, and natural cytotoxicity receptors (NCRs), recognise ligands that are upregulated on stressed, infected, or malignant cells (30–32). Conversely, inhibitory receptors, such as those within the killer immunoglobulin-like receptor (KIR) family and the NKG2A–CD94 heterodimer, monitor the expression of self-MHC class I molecules, preventing autoreactivity (33–35). This delicate receptor balance ensures that NK cells maintain tolerance to healthy cells while retaining the ability to target cells that have downregulated MHC class I expression, a common immune evasion mechanism in tumour cells. Thus, NK cells provide a crucial layer of immune surveillance that operates independently of T cell recognition (36).
The activation of NK cells typically requires the engagement of multiple activating receptors; however, CD16 (FcγRIIIa), the receptor responsible for mediating antibody-dependent cellular cytotoxicity (ADCC), can induce cytotoxic responses in the absence of other activating signals (37, 38). The KIR family, which includes both activating and inhibitory isoforms, plays a critical role in NK cell education, ensuring functional licensing and the development of tolerance to self (39). This process enables NK cells to discern between healthy and aberrant cells, with the clonal distribution of activating and inhibitory receptors across the NK cell repertoire allowing for context-dependent responses to both tumorigenic and infectious cells (40).
3 The role of macrophage-NK cell communication in immune defence
Macrophages and NK cells influence each other through dynamic, bidirectional interactions that shape immune responses (Table 1), and often these interactions are dependent on their anatomical context. Macrophages are largely tissue-resident and adapt to the specific microenvironment of their organ (49). While NK cells are traditionally described as circulating between blood and lymphoid tissues, they can be recruited to sites of inflammation or tumours (50–52), and subsets of tissue-resident NK cells have been identified in organs such as the liver and uterus during pregnancy, where they exhibit distinct phenotypic and functional properties (53, 54). In peripheral tissues, including the liver and lung, macrophages can activate NK cells through direct cell-to-cell contact and the secretion of soluble mediators. Notably, co-culture studies have demonstrated that blocking activating receptors such as DNAM-1 or 2B4, or neutralising IL-18, leads to reduced NK cell-derived IFN-γ (41), underscoring the importance of both receptor–ligand interactions and cytokine signalling in this axis. In addition, macrophage-derived IL-1β and type I interferons, particularly IFN-β, have been shown to upregulate the expression of activating NK cell receptors including NKp44 and NKG2D, thereby enhancing IFN-γ production (43).
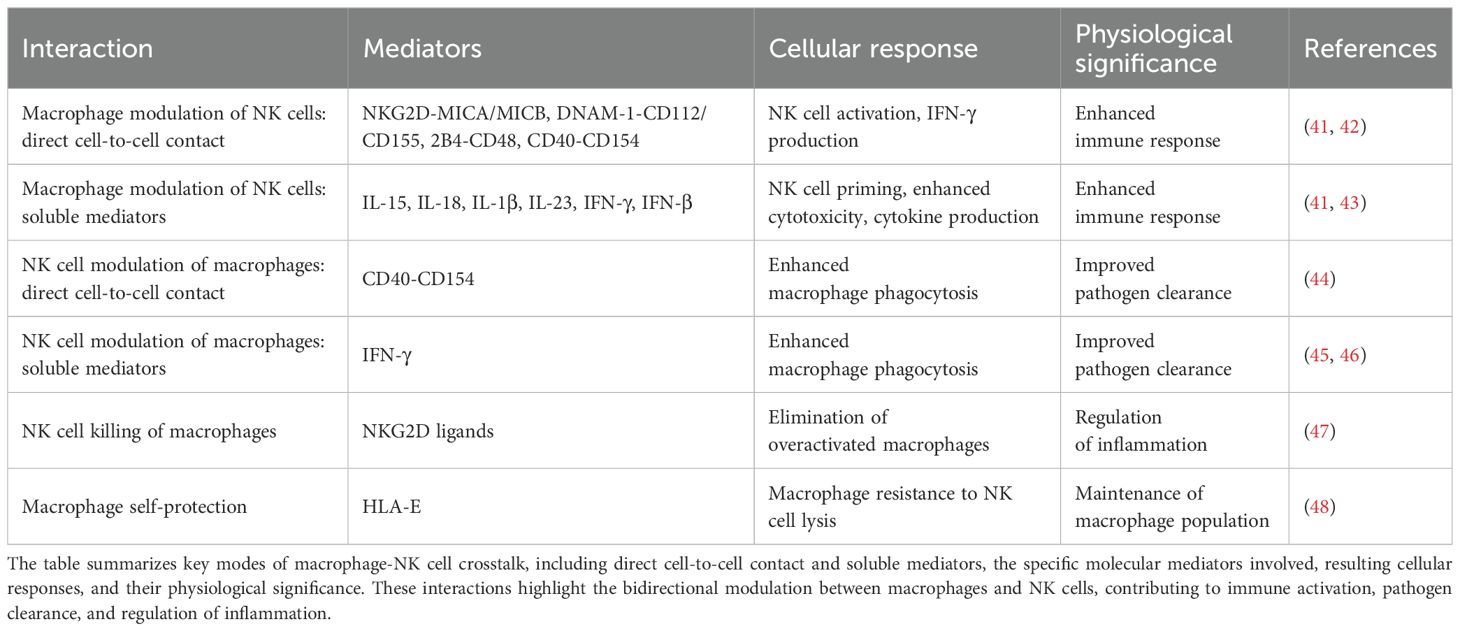
Table 1. Key interactions between macrophages and NK cells.
The functional role of NKG2D-mediated signalling has been particularly well characterised in uterine NK cells, where recognition of macrophage-expressed MICA drives robust IFN-γ responses (42). Conversely, NK cells can reciprocally activate macrophages. Engagement of CD40 on macrophages by CD154 expressed on NK cells induces the production of pro-inflammatory cytokines (55, 56), and macrophages from CD40-deficient mice exhibit impaired phagocytic activity in the presence of NK cells (44). Additionally, NK cell-derived IFN-γ can reprogramme immunosuppressive macrophages towards a more immunostimulatory phenotype (45), characterised by enhanced secretion of IL-12, TNF-α, and CXCL chemokines (57). This reciprocal activation is further amplified by a positive cytokine feedback loop: macrophage-derived IL-12, IL-15, and IL-18 activates NK cells, which in turn produce IFN-γ, TNF-α, and GM-CSF that further stimulate macrophage function and inflammatory cytokine production (46, 58, 59).
Importantly, this cooperative relationship is tempered by regulatory mechanisms that prevent excessive inflammation and preserve tissue integrity. For instance, type I interferons and Toll-like receptor (TLR) agonists can upregulate NKG2D ligands (such as MICA and ULBP1–3) on macrophages (42, 47), promoting their recognition and lysis by NK cells via NKG2D (47). However, inhibitory signalling through NKG2A, which recognises HLA-E on macrophages, restrains NK cell-mediated cytotoxicity (48). Macrophages activated by inflammatory stimuli express higher levels of HLA-E, providing increased protection against NK cell killing (48). Additionally, blockade of activating NK cell receptors, including NKp46 and DNAM-1, reduces macrophage susceptibility to NK cell-mediated cytotoxicity (60).
Together, these observations emphasise a finely tuned balance between NK cell-mediated activation and restraint, ensuring sufficient immune surveillance without depleting critical macrophage populations. In the context of cancer, selectively modulating this axis, such as through temporal regulation of NKG2D ligand expression, may offer a strategy to eliminate immunosuppressive tumour-associated macrophages while preserving homeostatic ones in healthy tissues.
4 Macrophage-NK cell crosstalk in the tumour microenvironment: balancing immunity and tolerance
TAMs and NK cells are key components of the TME, each contributing in distinct and often opposing ways, with their dynamic crosstalk shaping the TME (Figure 1). TAMs are typically the most abundant immune population within the TME, comprising up to 50% of infiltrating immune cells and contributing significantly to tumour structure and immunoregulation (61, 62). While they share some functional overlap with myeloid-derived suppressor cells (MDSCs), such as the ability to dampen anti-tumour immunity, TAMs are generally more differentiated and tissue-resident, whereas MDSCs represent a heterogeneous population of immature myeloid cells that expand during cancer and other chronic inflammatory conditions (63).
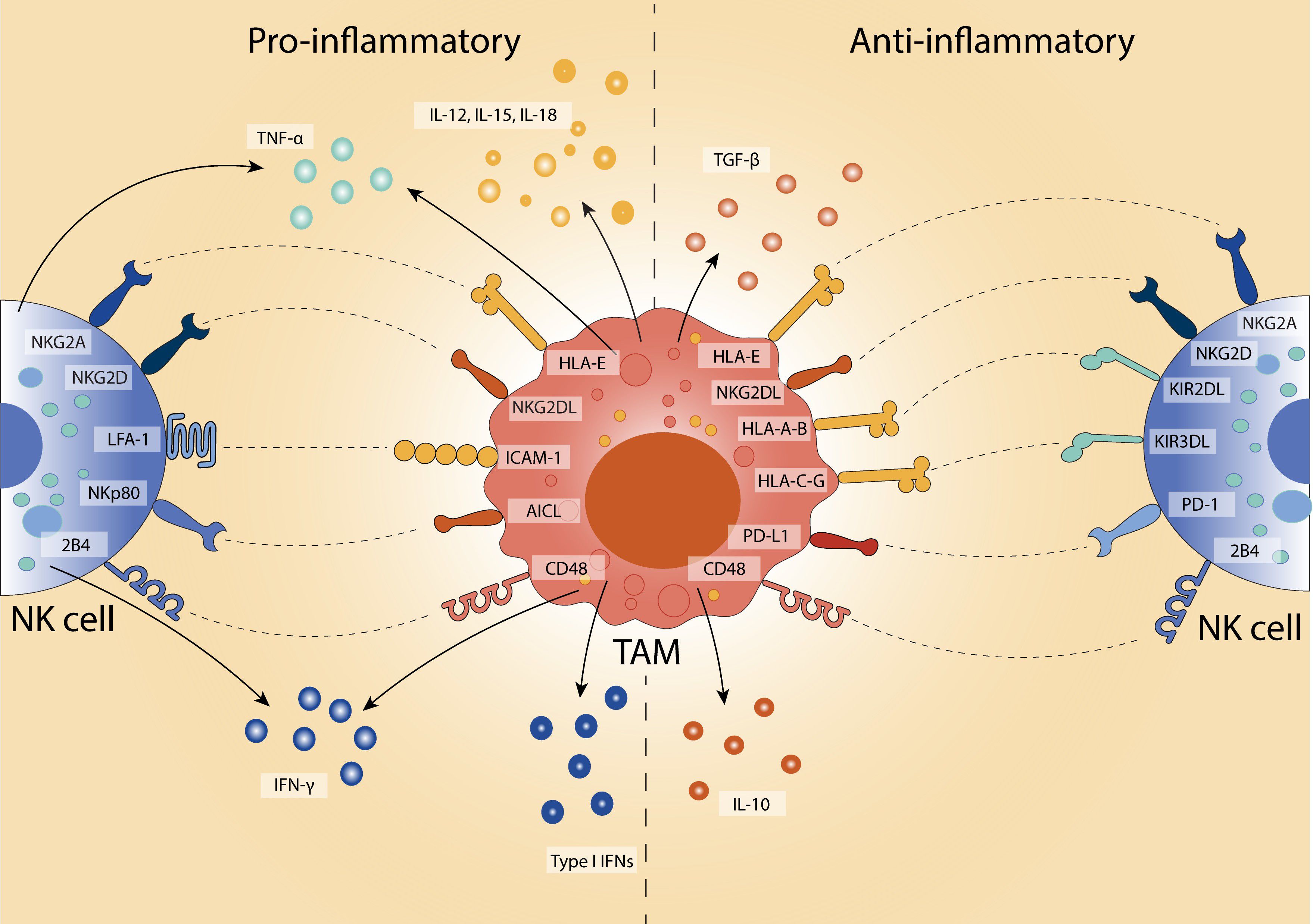
Figure 1. Polarised TAM-NK cell interactions in the TME. TAMs modulate NK cell activity depending on their polarisation state. Pro-inflammatory TAMs (left) promote anti-tumour responses by secreting IL-12, IL-15, IL-18, and TNF-α, and enhancing NK cell activation and IFN-γ production through engagement of activating receptors (e.g., NKG2D, NKp80, LFA-1). In contrast, anti-inflammatory TAMs (right) facilitate immune suppression via IL-10 and TGF-β, and upregulating inhibitory ligands (e.g., PD-L1, HLA-E, NKG2DL) that engage inhibitory NK cell receptors (e.g., PD-1, NKG2A, KIRs). This bidirectional crosstalk shapes the tumour microenvironment by either enhancing or suppressing NK cell cytotoxicity.
In early-stage tumours, macrophages may exhibit immunostimulatory phenotypes and exert tumoricidal functions (64), partly through nitric oxide production (65), and have been associated with improved clinical outcomes in colorectal (66), lung (67), ovarian (68), and breast cancers (69). However, as tumours progress, the local cytokine milieu shifts to favour anti-inflammatory cues, such as IL-10 and CSF1, driving the recruitment of monocytes and their subsequent differentiation into immunosuppressive TAMs (68, 70). These TAMs often adopt a phenotype resembling anti-inflammatory macrophages and are associated with poor prognosis across a wide spectrum of malignancies, including pancreatic, breast, endometrial, and brain cancers, as well as lymphomas and melanomas (7).
In contrast, NK cells are potent effectors of anti-tumour immunity. Their ability to directly lyse malignant cells and secrete pro-inflammatory cytokines such as IFN-γ renders them crucial for early tumour surveillance. Preclinical models demonstrate that prolonged NK cell depletion accelerates tumour progression (71–73), while clinical studies link reduced NK cell infiltration or impaired function with increased metastatic potential and recurrence, particularly in colorectal, head and neck, and pharyngeal cancers (74–77). Despite robust in vitro cytotoxicity against tumour targets such as melanoma, adoptive NK cell transfer has yielded limited clinical efficacy, often due to suppressive factors within the TME, including downregulation of activating receptors such as NKG2D (78).
4.1 Soluble mediators of communication: cytokine-driven modulation
Macrophage-NK cell communication within the TME is shaped by both soluble mediators and direct cell-cell interactions. Although these cells often reside in close proximity in the TME, cytokines such as TAM- and tumour-derived IL-10 still exert broad immunosuppressive effects, dampening both macrophage and NK cell effector functions (46, 79). Interestingly, in vitro addition of IL-15 can restore NK cell cytotoxicity in the presence of IL-10, suggesting that IL-10’s suppressive effects may be context-dependent (80). TGF-β, a prominent immunoregulatory cytokine secreted by both TAMs and tumour cells, suppresses NK cell activation by downregulating NKG2D and NKp30 (81), while concurrently promoting macrophage polarisation toward an immunosuppressive phenotype and reducing pro-inflammatory cytokine output (82). In gastric cancer, macrophage-derived TGF-β induces a marked functional impairment in NK cells, characterised by reduced IFN-γ production and diminished expression of NKp30, NKp46, and 2B4 (83). Notably, this suppression can be partially reversed by exogenous IL-15 (84).
NK cells also exert feedback effects on macrophages. In prostate cancer, NK cell-derived IL-8 recruits macrophages and skews their polarisation toward a tumour-promoting phenotype (85). Similarly, activated NK cells release cytotoxic granules containing effector molecules such as granzymes which can act on macrophages. For example, granzyme A can stimulate macrophages to produce pro-inflammatory cytokines including IL-6, IL-8, IL-1β and TNF-α (86–88). Conversely, macrophages, particularly those with anti-tumour activity, are a major source of type I interferons (IFN-α and IFN-β), which are essential for NK cell development and activation (89). These interferons reduce TAM frequency, promote inflammatory polarisation, and induce chemokines such as CXCL10 and CXCL11, which recruit NK cells via CXCR3 (90–94). IFN-β also enhances NK cell cytotoxic potential by upregulating NKG2D and inducing IL-15, a cytokine critical for both macrophage activation and NK cell survival, proliferation, and function (43). TAMs within the TME frequently produce IL-12, IL-15, and IL-18, a group of cytokines that work together to sustain NK cell activity (44, 48, 95, 96).
TNF-α, produced by both macrophages and NK cells, reinforces this axis by suppressing anti-inflammation-associated gene expression and enhancing IL-15 signalling pathways in NK cells (97, 98). Via TNFR1 and TNFR2, TNF-α activates pro-inflammatory signalling cascades that can culminate in tumour cell death (99, 100). Importantly, the outcome of macrophage–NK cell crosstalk depends on the prevailing balance of stimulatory versus inhibitory cytokines and on the spatial and temporal context of their interaction. Therapeutic strategies must therefore account for the heterogeneity of cytokine networks and macrophage polarisation states within individual tumours.
4.2 Receptor-ligand interactions: immune synapses in tumour immunity
In addition to soluble factors, direct contact between macrophages and NK cells is also important in shaping immune responses. NK cell activity is modulated through a network of activating and inhibitory receptors that respond to ligand expression on both tumour cells and myeloid populations. For instance, NKp46-deficient mice exhibit impaired tumour control in lymphoma and melanoma models (101–104), while reduced NKp30 and NKp46 expression in patients with leukaemia or cervical cancer correlates with poorer outcomes (105–107). Conversely, high expression of DNAM-1 enhances NK cell responses against both haematological malignancies and solid tumours (108).
Within the TME, tumour-derived CSF1 induces macrophage expression of NKG2D ligands, including MICA/B and ULBPs, thereby promoting NK cell activation (109). However, chronic NKG2D stimulation, as observed in acute myeloid leukaemia and hepatocellular carcinoma (HCC), can lead to NK cell exhaustion (110–112). Similarly, the 2B4-CD48 axis supports NK cell activation and IFN-γ production (41, 47, 113), but persistent engagement in the context of HCC drives functional impairment (114). Other macrophage-expressed ligands, such as AICL and ICAM-1, engage NKp80 and LFA-1 on NK cells, respectively, facilitating activation and migration (115–117). Notably, dectin-1, a C-type lectin receptor expressed by macrophages, recognises tumour-specific glycan structures and initiates signalling pathways that enhance NK cell cytotoxicity (118).
These receptor-ligand interactions act as important checkpoints that help control anti-tumour immune responses. However, because these signals are highly dynamic and influenced by the suppressive nature of the TME, therapeutic strategies need to be carefully tailored. New technologies, such as co-culture systems, single-cell transcriptomics, and spatial profiling, will be key to mapping these interactions within the TME and uncovering precise targets for immune-based therapies.
5 Harnessing macrophages: innovative approaches to target cancer
While T cells have been the main target of cancer immunotherapy for years, focusing on TAMs offers distinct advantages. Unlike T cells, which face challenges such as exhaustion, antigen escape, and TME infiltration (119–122), macrophages are already abundant and well-established within tumours (61, 62). TAMs play a pivotal role in shaping the TME by promoting tumour growth, suppressing T cell function, supporting angiogenesis, and remodelling the extracellular matrix (123–125). Additionally, they are key regulators of other immune populations (126–129); within the TME, the immunosuppressive interplay between TAMs and NK cells can further dampen effective anti-tumour responses. As they are less reliant on tumour-specific antigens, therapies targeting TAMs may also be less susceptible to immune evasion and more broadly applicable (130, 131).
Previous reviews have discussed immunotherapeutic strategies targeting TAMs (46, 132), yet few studies have addressed the therapeutic potential of modulating TAMs specifically to augment NK cell function. This remains a significant and underexplored area, with emerging evidence suggesting that TAM modulation can shape NK cell recruitment, activation and cytotoxicity. A summary of therapeutic approaches under clinical and preclinical investigation is outlined in Table 2. Current therapeutic strategies focus on three main approaches: reprogramming macrophage phenotype, depleting immunosuppressive subsets, and modulating TAM–NK cell interactions to restore cytotoxic activity.
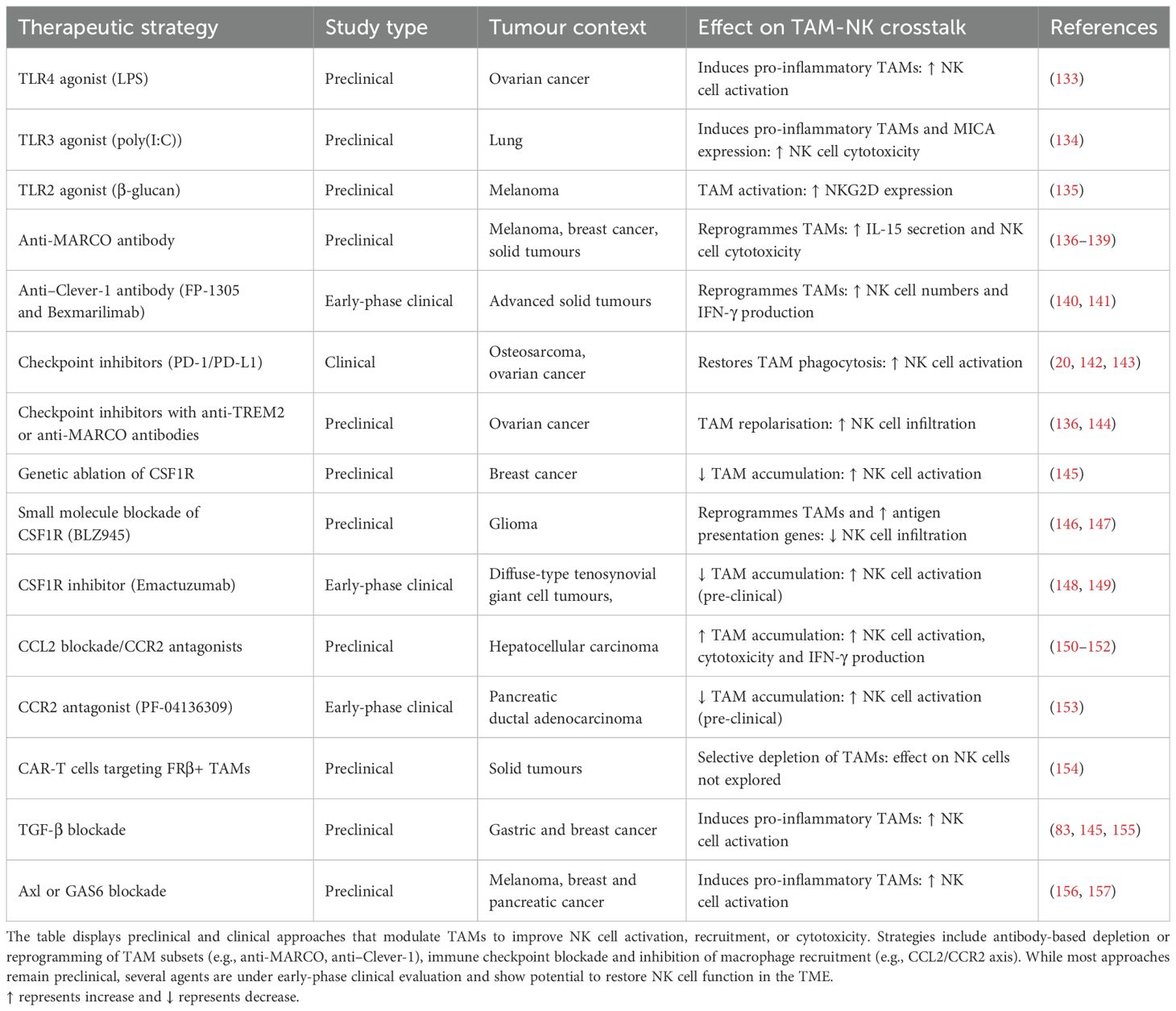
Table 2. Cancer therapeutic strategies targeting TAMs to enhance NK cell effector function.
5.1 Repolarising TAMs: shifting macrophages to anti-tumour action
One strategy to increase the anti-tumour response involves reprogramming TAMs from a tumour-promoting state toward a more pro-inflammatory, tumour suppressive phenotype, thereby enhancing NK cell recruitment and effector function (Figure 2). In vitro, macrophages described as ‘M2-like’ can be repolarised to an ‘M1-like’ phenotype, promoting IFN-γ secretion and restoring cytotoxicity in resting NK cells against multiple tumour targets, achieving activity comparable to IL-12-conditioned NK cells (41, 48, 135). Repolarised macrophages also secrete increased levels of IL-12, IL-18, and TNF-α (41), and upregulate NK cell-activating receptors such as NKp30, NKp44, NKp46, and NKG2D (47, 135). Notably, these repolarised macrophages exhibit resistance to NK cell-mediated cytotoxicity (48), suggesting a cooperative rather than antagonistic relationship between the two cell types.
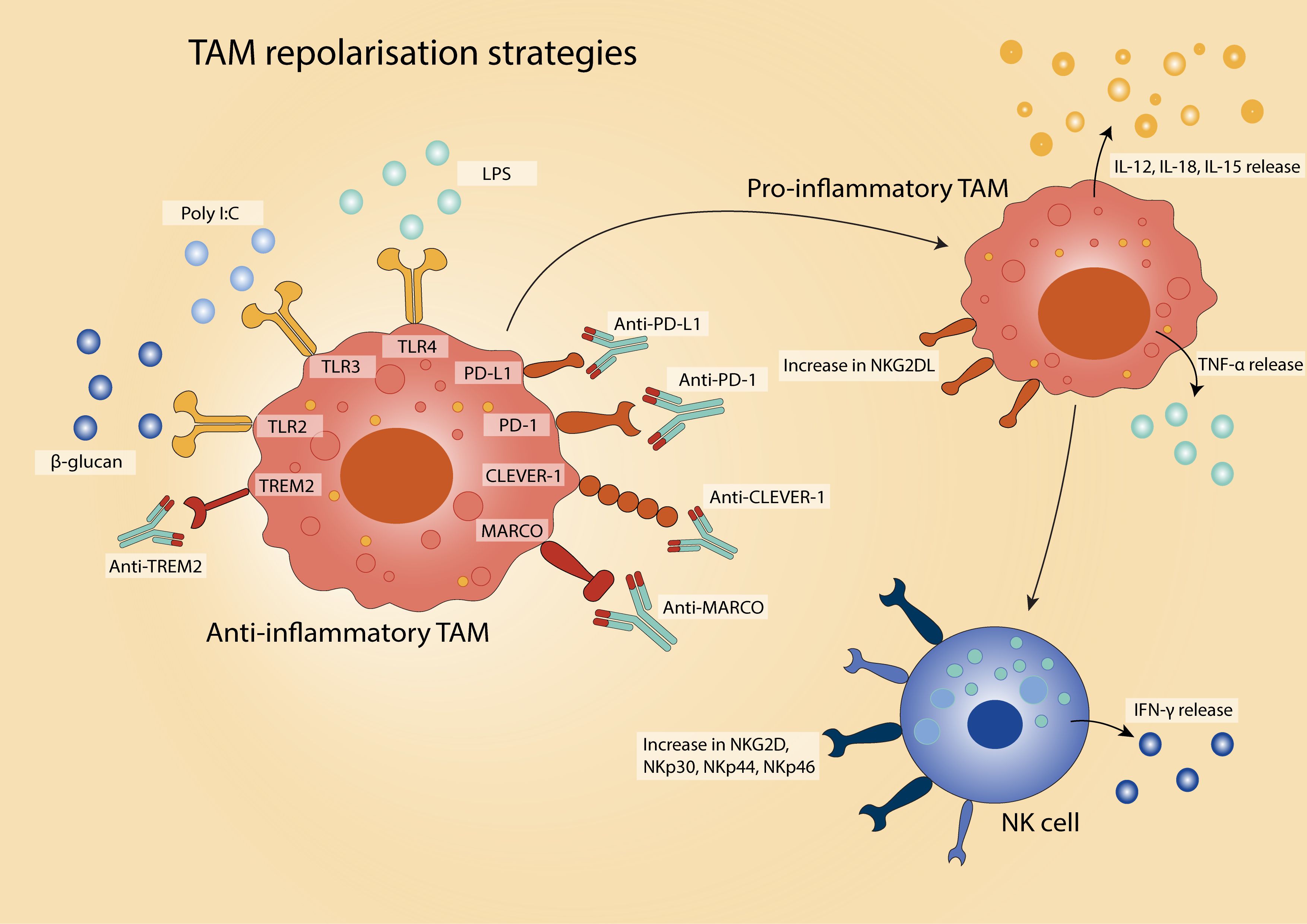
Figure 2. Strategies for TAM repolarisation in the TME. There are various approaches to reprogramme TAMs from an anti-inflammatory, pro-tumour phenotype to a pro-inflammatory, anti-tumour state. Therapeutic strategies include activation of pro-inflammatory-inducing pathways via agents targeting Toll-like receptors (TLRs), and immune checkpoint blockade (e.g., anti-PD-1/PD-L1) to relieve TAM-mediated immunosuppression. The reprogrammed TAMs exhibit enhanced phagocytic activity, antigen presentation, and pro-inflammatory cytokine secretion, contributing to improved anti-tumour immunity.
Toll-like receptor (TLR) agonists represent a widely studied class of agents capable of driving this phenotypic shift. For instance, stimulation with lipopolysaccharide (LPS, TLR4 agonist) enhances the ability of TAMs isolated from ovarian cancer to activate NK cells and promote tumour cell lysis (133). Similarly, poly(I:C) (a synthetic TLR3 ligand) induces pro-inflammatory phenotype polarisation of alveolar macrophages in lung cancer, resulting in heightened NK cell cytotoxicity and suppression of metastatic growth (134). In uterine macrophages, poly(I:C) also upregulates the NKG2D ligand MICA, enabling robust NK cell activation via NKG2D engagement (42). In melanoma, β-glucan (a TLR2 agonist) increases NKG2D expression on NK cells and enhances tumour control in a manner dependent on NK cell presence (135).
Beyond TLR-based interventions, antibody-mediated targeting of TAM-associated surface receptors offers an additional strategy to reprogramme macrophage function and restore NK cell responsiveness. In melanoma, TAMs expressing the scavenger receptor MARCO are found near NK cells (136, 137). Antibody blockade of MARCO reprogrammes TAMs toward an immunostimulatory phenotype, enhances IL-15 secretion, and increases NK cell infiltration and cytotoxicity (136, 138). In preclinical breast cancer models, anti-MARCO treatment also reduces tumour burden and metastatic dissemination (137). A similar effect has been observed with antibodies targeting Clever-1 (also known as stabilin-1), which boosts NK cell numbers and IFN-γ production in patients with advanced-stage solid tumours (140). Treatment with the anti–Clever-1 antibody bexmarilimab in patients with solid tumours has similarly been associated with increased IFN-γ and NK cell activation markers (141).
Immune checkpoint blockade has also emerged as a strategy to reprogramme TAMs. TAMs express both PD-1 and PD-L1, and blockade of this axis not only restores tumour cell phagocytosis but also enhances NK cell activation (20). Macrophage-targeted anti–PD-L1 therapy has been shown to increase IFN-γ production by NK cells (142), while anti–PD-1 treatment in osteosarcoma models expands the anti-tumour macrophage population (143). Importantly, combining immune checkpoint inhibitors with TAM-targeting agents such as anti-MARCO or anti-TREM2 antibodies enhances therapeutic efficacy in preclinical models, including ovarian cancer (136, 144).
5.2 Depleting TAMs: targeting the macrophages that aid tumour growth
The frequency and distribution of TAMs strongly correlate with poor prognosis across multiple malignancies, including breast, prostate, ovarian and lung cancers (158–164). In preclinical breast cancer models, macrophage depletion significantly delays tumour progression, positioning TAMs as a promising therapeutic target (165). However, efforts to deplete macrophages (Figure 3), have revealed the need for refined approaches that distinguish between immunosuppressive and immunostimulatory subsets.
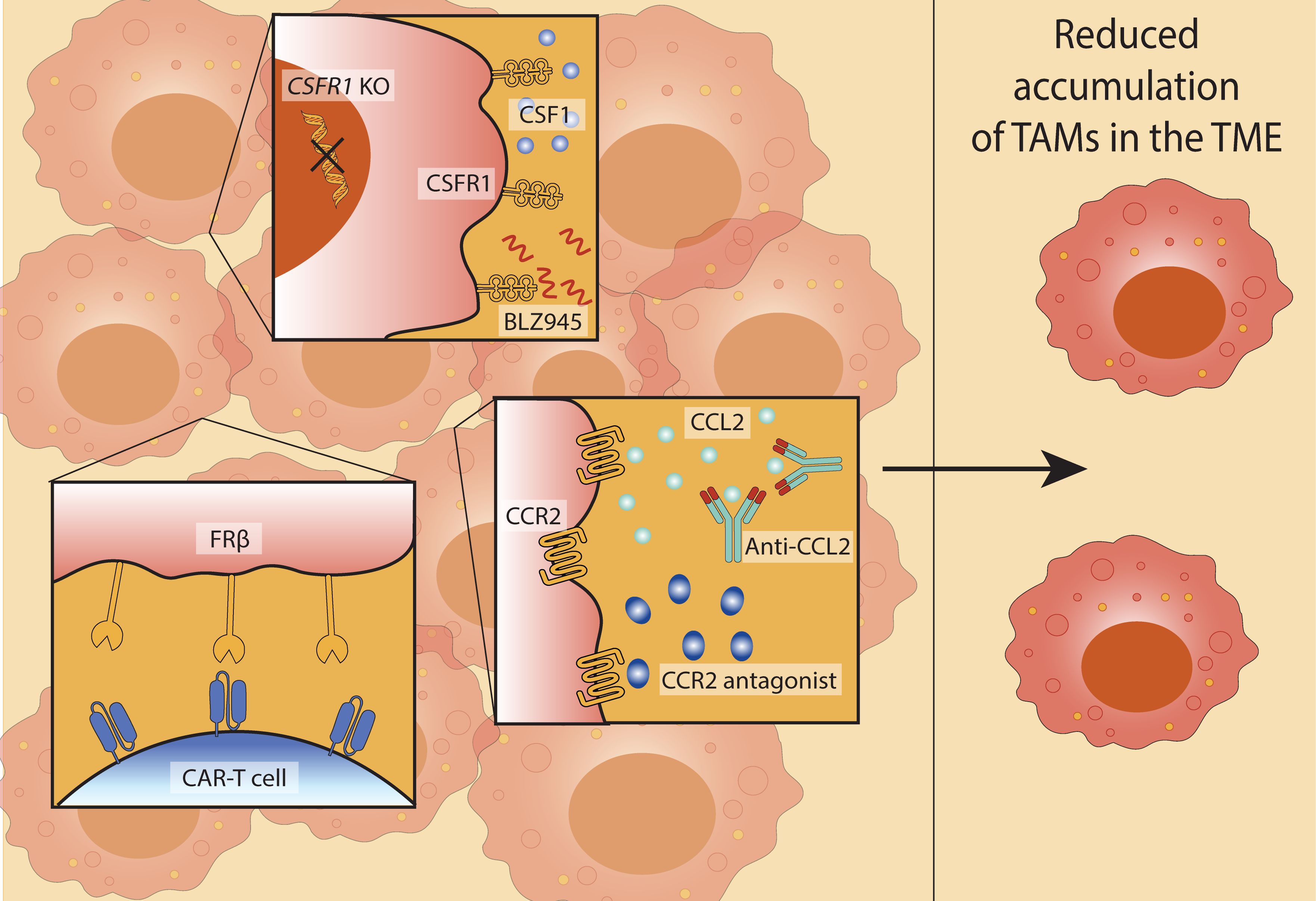
Figure 3. Therapeutic strategies to reduce TAM accumulation in the TME. Approaches to target TAM recruitment and survival to limit their accumulation in the TME include: inhibition of the colony-stimulating factor 1 receptor (CSF1R) pathway (through genetic knockout (CSF1R KO) or pharmacological inhibition using CSF1R inhibitor BLZ945) (top); disruption of the CCL2-CCR2 axis using anti-CCL2 antibodies or CCR2 antagonists (centre); chimeric antigen receptor (CAR)-T cells engineered to recognise the TAM marker folate receptor β (FRβ) (bottom). Collectively, these strategies contribute to reduced TAM accumulation in the TME, potentially enhancing anti-tumour immunity.
The colony-stimulating factor 1 (CSF1)-CSF1 receptor (CSF1R) axis represents a key regulatory pathway for macrophage survival and differentiation (166). Tumour cells, macrophages, and other stromal components secrete CSF1, sustaining TAM viability within the TME (167, 168). Genetic ablation of CSF1R in murine breast cancer models reduces TAM accumulation and promotes NK cell activation, with adoptive NK cell transfer further enhancing tumour control (145). Blockade of CSF1R using small-molecule inhibitors, such as BLZ945, has shown efficacy in glioma, where treatment led to TAMs with increased expression of antigen presentation genes (146) and downregulation of pro-tumour macrophage markers (147). Notably, these latter effects appear to reflect macrophage repolarisation rather than depletion.
Another strategy to limit TAM accumulation involves targeting monocyte recruitment through the CCL2–CCR2 chemokine axis. Tumour-derived CCL2 facilitates the recruitment of CCR2-expressing monocytes, which subsequently differentiate into TAMs. Inhibition of this pathway, via anti-CCL2 antibodies or CCR2 antagonists, reduces TAM numbers and restores NK cell effector function (150–152). In hepatocellular carcinoma models, CCL2 blockade enhances NK cell activation, IFN-γ production, and cytotoxicity, supporting the therapeutic value of this approach (150).
Nevertheless, blockade of monocyte recruitment can trigger compensatory mechanisms, such as increased neutrophil infiltration, which may counteract therapeutic benefits (62). One strategy to circumvent this is to selectively target TAM-associated receptors. For instance, chimeric antigen receptor (CAR)-T cells engineered to recognise folate receptor beta (FRβ), a marker enriched on immunosuppressive TAMs, successfully deplete this population, promote pro-inflammatory polarisation, and suppress tumour growth in preclinical models (154).
5.3 Modulation of TAM-NK cell crosstalk: shaping the immune communication to combat cancer
Rather than indiscriminately depleting macrophages, a more refined therapeutic strategy involves selectively modulating the bidirectional interactions between TAMs and NK cells within the TME. By either amplifying beneficial communication or disrupting suppressive crosstalk, it may be possible to restore NK cell function while preserving the immune-regulatory roles of macrophages critical for tissue integrity (Figure 4).
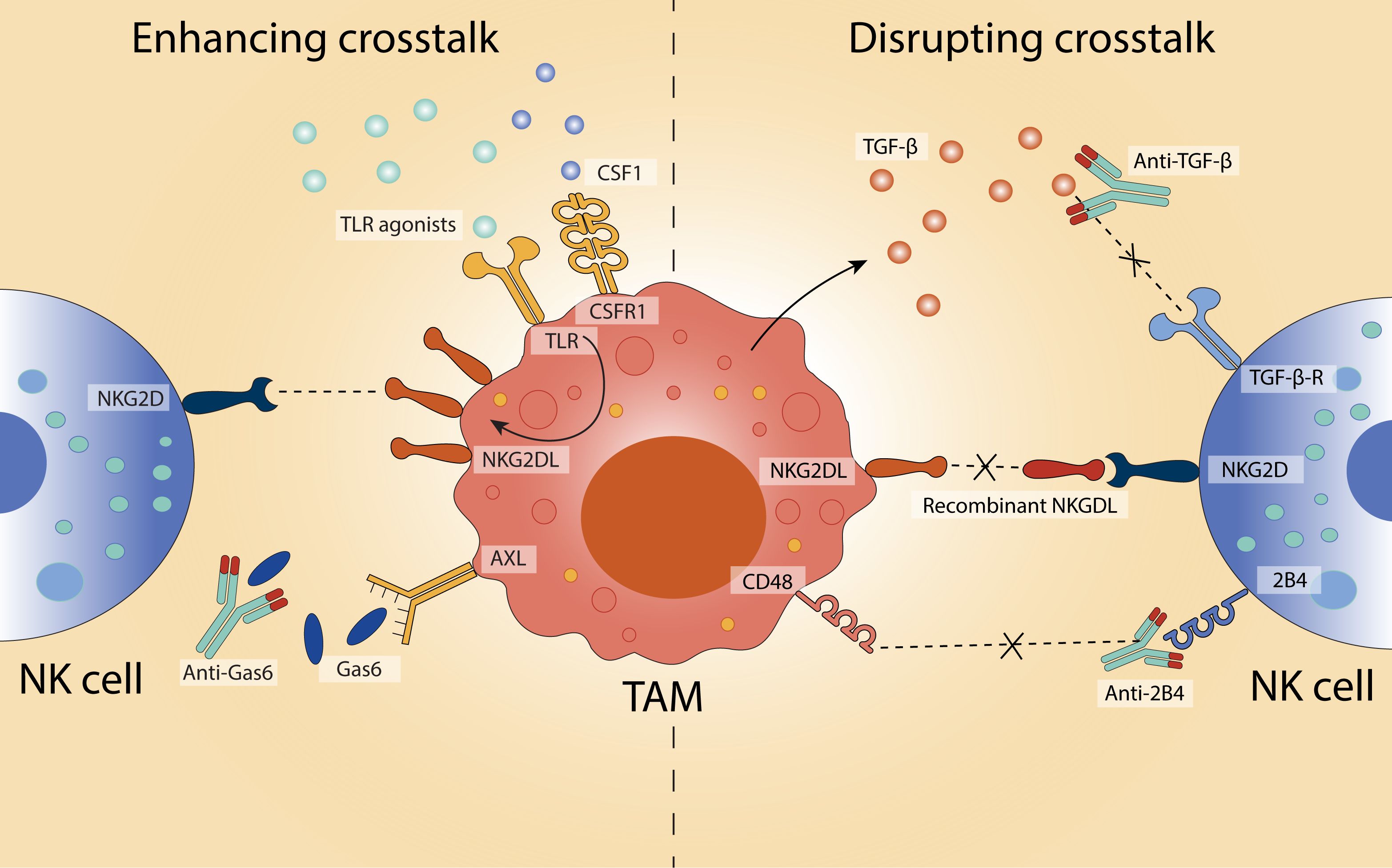
Figure 4. Modulating TAM–NK cell crosstalk to influence anti-tumour immunity. Strategies to either enhance or disrupt the interaction between TAMs and NK cells in the TME aim to restore or enhance NK cell cytotoxicity and contribute to anti-tumour responses. On the left, enhancing crosstalk is achieved through TLR agonists and CSFR1 stimulation, which upregulate NKG2D ligands (NKG2DL) on TAMs and promote NK cell activation. Inhibition of Gas6-AXL signalling using anti-Gas6 antibodies further supports immune activation. On the right, disruption of immunosuppressive crosstalk is demonstrated via blockade of TGF-β signalling (using anti-TGF-β antibodies) and interference with inhibitory CD48-2B4 interactions (via anti-2B4 antibodies). Additionally, recombinant NKG2DL can be used to inhibit NKG2D-mediated suppression.
One approach to enhance NK cell function is to increase expression of activating ligands on macrophages, particularly those that engage the NK cell receptor NKG2D. In mice, peritoneal macrophages stimulated with TLR agonists, including LPS (TLR4), Pam3CSK4 (TLR2), zymosan (TLR2/6), and poly(I:C) (TLR3), upregulate the NKG2D ligand RAE-1 (169). Tumour-derived CSF1 similarly drives RAE-1 expression on TAMs (109). Poly(I:C) treatment also induces the expression of other murine NKG2D ligands, such as H60 and MULT-1, while promoting secretion of type I interferons and cytokines critical for NK cell activation, including IFN-β, IL-12, IL-15, and IL-18 (48). Neutralisation of IFN-β or IL-15 diminishes NKG2D expression and NK cell cytotoxicity, demonstrating the centrality of this axis to immune activation (48). In human monocytes, analogous ligand upregulation can be achieved through IFN-α or TLR4 stimulation (170). LPS increases surface MICA and ULBP1–3 expression and enhances NK cell-derived IFN-γ in a MICA-NKG2D-dependent manner (170). However, prolonged or high-dose LPS exposure can paradoxically render macrophages more susceptible to NK cell-mediated lysis via NKG2D recognition, highlighting the delicate balance required in therapeutic modulation (47).
While the induction of activating ligands on macrophages can bolster NK cell function, persistent ligand expression can drive NK cell exhaustion. In both murine and human models, sustained exposure to RAE-1δ+ or MICAhigh TAMs results in reduced NKG2D expression and diminished cytotoxic capacity (109, 171). In melanoma models, recombinant NKG2D ligands have been shown to partially restore NK cell responsiveness (109). Similar effects are observed with the CD48–2B4 axis: TAMs expressing CD48 transiently activate NK cells, but prolonged stimulation leads to functional exhaustion (114). In hepatocellular carcinoma, blockade of 2B4 reverses this dysfunction and restores NK cell IFN-γ production (114).
Disruption of suppressive cytokine signalling represents another avenue to restore NK cell cytotoxicity. TGF-β, a canonical immunosuppressive cytokine secreted by both tumour cells and TAMs, impairs NK cell function by downregulating activating receptors such as NKG2D and NKp30 (81). In human gastric cancer and murine breast cancer models, TGF-β blockade restores NK cell activity and augments anti-tumour responses (83, 145, 155). Targeting macrophage-intrinsic suppressive pathways is also showing promise. The receptor tyrosine kinase AXL, often upregulated on TAMs in breast, ovarian, renal, and lung cancers, correlates with poor clinical outcomes and skews macrophages toward an immunosuppressive phenotype (172). In leukaemia, AXL is induced by tumour-derived GAS6, which also acts on NK cells to reduce NKG2D expression and impair cytotoxicity (173, 174). Blockade of either AXL or GAS6 enhances NK cell activation, reduces metastasis, and promotes tumour control in breast cancer, melanoma, and pancreatic cancer models (156, 157).
Immune checkpoint inhibition remains a promising approach in cancer immunotherapy. While there is limited direct evidence of checkpoint molecule-mediated interactions between TAMs and NK cells, therapeutic strategies targeting these checkpoints often modulate the activity of both cell types, suggesting underlying immune crosstalk that could be targeted. For example, V-domain Ig suppressor of T cell activation (VISTA) is highly expressed on TAMs, and its blockade improves survival in murine models of leukaemia and lymphoma (175). Notably, although NK cells do not express VISTA, anti-VISTA antibodies have demonstrated increased NK cell maturation and activation (176, 177), implying that indirect modulation through other immune populations, possibly including macrophages, is at play.
Lymphocyte activation gene-3 (LAG-3), an inhibitory receptor expressed on NK cells, is associated with reduced IFN-γ production and proliferation (178). Blockade of LAG-3 has been effective in restoring NK cell function in chronic lymphocytic leukaemia, including increased production of IFN-γ and IL-12 (179). Although there is limited evidence for direct interaction between TAMs and NK cells via LAG-3, LAG-3+ T cells can bind MHC class II on macrophages (180), and soluble LAG-3 binding to MHC class II on macrophages inhibits monocyte-to-macrophage differentiation (181).
T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) is expressed on both TAMs and NK cells (182, 183). In vitro, TIM-3 blockade enhances NK cell cytotoxicity against cancer cell lines and primary multiple myeloma cells, accompanied by increased IFN-γ production; in vivo, this corresponds to reduced tumour growth (184, 185). In macrophages, blocking TIM-3 inhibits polarisation toward an immunosuppressive phenotype in glioblastoma (186). Blockade of T cell immunoreceptor with Ig and ITIM domains (TIGIT), also present on both NK cells and TAMs, enhances NK cell cytotoxicity against melanoma cells in vitro and reduces metastatic growth in murine melanoma models (187). In TAMs, TIGIT supports an immunosuppressive phenotype (188), but inhibition can reprogramme these cells toward a pro-inflammatory, anti-tumour state (189).
5.4 Strengths, limitations, and translational challenges of harnessing macrophages
Several strategies to manipulate TAMs with the aim of enhancing NK cell function have emerged, including approaches to repolarise their phenotype, deplete suppressive subsets, and enhance TAM-NK cell crosstalk. Among these, antibody-based therapies targeting specific surface markers on pro-tumoural TAMs represent some of the most promising and clinically advanced avenues. Anti-MARCO antibodies have shown tumour-reducing effects across multiple models (136–138), although MARCO is also expressed on non-tumour macrophages (21), highlighting the need for tissue-specific profiling and validation. Anti-Clever-1 antibodies offer more selectivity by targeting immunosuppressive TAMs while sparing homeostatic populations (190), though their broader applicability across tumour types and their impact on NK cells remain underexplored. CAR-T strategies directed at TAM markers such as FRβ also offer a degree of specificity (154), yet their effects on NK cells, as well as their persistence, trafficking, and safety profiles, remain largely uncharacterised.
While several macrophage-targeted therapies, including anti-MARCO and anti-Clever-1 antibodies, have entered early-phase clinical trials, few include NK cell-specific endpoints. Most focus on T cell responses or cytokine outputs, neglecting NK-relevant metrics such as CD107a expression or intratumoural infiltration. For instance, clinical trials assessing CSF1R inhibition (e.g., NCT02526017) or checkpoint blockade (e.g., NCT02817633) do not evaluate NK functional markers (191, 192). Many of the strategies outlined in Table 2 remain preclinical, with toxicity, tumour-type specificity, and durability yet to be addressed.
Compared to T cell-based approaches, TAM-targeted therapies offer distinct advantages. Their antigen-independent mechanism avoids issues of antigen loss or MHC downregulation (193, 194). TAM repolarisation allows in situ immune reprogramming, potentially overcoming the trafficking barriers faced by adoptively transferred T cells (195). Repolarised TAMs can also secrete IL-15 and IL-12, enhancing NK cytotoxicity without the systemic toxicity of exogenous cytokines (41). Furthermore, because activation is localised, repolarisation carries a lower risk of cytokine release syndrome compared to CAR-T therapies (196).
However, macrophage plasticity poses a significant barrier to durable responses; macrophages are highly responsive to local cues, including the immunosuppressive signals of the TME, and therefore may not maintain an anti-tumour phenotype over time (197–199). Additionally, unlike memory-forming T cells, macrophages do not clonally expand (1, 3). Repolarising agents like TLR agonists [e.g., LPS, poly(I:C)] can upregulate NK-activating ligands but often lack tumour specificity, leading to off-target inflammation (200–203). Even more tolerable agents like β-glucan or inhibitors of broadly expressed molecules (e.g., Axl, TGF-β) may cause systemic toxicity without targeted delivery strategies (204–206).
TAM depletion strategies offer a different approach, aiming to remove suppressive macrophages and reduce inhibitory signalling toward NK cells. However, these are often not selective enough, for example CSF1R blockade can deplete supportive myeloid cells that produce IL-15 and IL-18, cytokines that are important for NK survival (207). In lung cancer models, CSF1R inhibitor BLZ945 impaired NK infiltration and increased metastasis (207). Depleting CD206+ TAMs similarly disrupted NK recruitment, indicating that not all TAMs are suppressive (208). Thus, complete depletion may inadvertently remove macrophages that support NK-mediated immunity.
Moreover, TAM populations may exhibit resistance to CSF1R inhibition (209) or are replenished via alternative recruitment (e.g.,CCR5) (210), while compensatory upregulation of PD-L1 or other inhibitory molecules may undermine efficacy (167). Although depletion strategies bypass the need for antigen specificity, they carry a higher risk of disrupting tissue-resident macrophages involved in homeostasis (208). The functional heterogeneity of TAMs demands greater precision in distinguishing suppressive subsets from those with beneficial roles (211).
Enhancing TAM–NK cell crosstalk offers a mechanistically attractive alternative. Instead of depleting or repolarising macrophages, this strategy modulates communication pathways to restore NK cell activity. This approach may be especially effective in NK-sensitive tumours where T cell responses are limited (212). However, it requires the presence of functional NK cells, limiting efficacy in tumours with low infiltration unless paired with adoptive NK cell transfer (213, 214).
Each TAM-targeting approach has trade-offs. Repolarisation preserves beneficial functions, but is vulnerable to reversal due to plasticity. Depletion removes immunosuppressive signalling, but risks harming supportive macrophages. Crosstalk enhancement provides targeted immune recalibration with minimal disruption, but depends on the presence of responsive NK cells. Of these, crosstalk modulation may strike the most effective balance: restoring NK function while maintaining macrophage-mediated tissue integrity. Ultimately, advancing TAM-targeted therapies will require a more nuanced understanding of macrophage-NK cell dynamics. Emerging technologies such as single-cell RNA sequencing, spatial transcriptomics, and proteomics offer powerful tools to explore this complexity.
6 Resolving the spatiotemporal crosstalk between macrophages and NK cells through single-cell and spatial multi-omics
While macrophage and NK cell interactions have been studied using flow cytometry, bulk RNA-sequencing, and co-culture models, these methods fall short in capturing spatial context, heterogeneity, and post-translational modifications. Emerging high-dimensional approaches such as spatial transcriptomics (ST), single-cell RNA sequencing (scRNA-seq), and proteomics, promise to overcome these limitations, offering a systems-level perspective on immune cell interplay.
6.1 Single-cell sequencing: unravelling cellular heterogeneity
Single-cell RNA sequencing (scRNA-seq) has transformed our understanding of immune cell heterogeneity and function by enabling transcriptomic profiling at single-cell resolution. This technology is particularly valuable for dissecting the diverse phenotypic states of macrophages within the tumour microenvironment (TME), a complexity underscored by findings such as the enhanced ability of spleen macrophages, compared to their lung counterparts, to potentiate NK cell cytotoxicity (215). For instance, scRNA-seq identified a neuron-like TAM subset in lung adenocarcinoma that promotes tumoural neurogenesis (216). Similarly, scRNA-seq has provided insights into the regulation of tumour-infiltrating NK cells, revealing that inhibition of the transcription factor HIF-1α can enhance cytotoxicity, suggesting novel therapeutic avenues (217). Integrative scRNA-seq analyses on NK cells from over 700 patients with 24 types of cancer shows heterogeneity in NK cell composition in a tumour-type-specific manner and importantly, also identified a population of tumour-associated NK cells that show impaired anti-tumour functions (218).
Beyond cellular profiling, integrating ligand-receptor interaction frameworks such as CellPhoneDB (219) and NicheNet (220) with scRNA-seq data has enabled the prediction of intercellular communication networks. In ovarian carcinoma, this approach revealed robust crosstalk, mediated by CXCL and CCL chemokines, between anti-tumour TAM subsets and cytotoxic NK cells (221). Moving forward, combining scRNA-seq with computational inference of cell-cell interactions holds significant promise for uncovering regulatory mechanisms such as cytokine feedback loops, immune checkpoint modulation, and metabolic coordination within the TME.
6.2 Spatial transcriptomics: adding the context of location
While scRNA-seq provides powerful insights into cellular heterogeneity, it lacks spatial resolution. Spatial transcriptomics (ST) addresses this limitation by mapping gene expression directly onto intact tissue sections, preserving the native architecture and cellular context. This spatial dimension is particularly crucial for elucidating cell–cell interactions within complex environments such as the TME.
ST has proven instrumental in characterising macrophage infiltration patterns in non-small cell lung cancer (NSCLC). In patient samples from anti-PD-1/PD-L1 immunotherapy trials, spatial profiling revealed the distribution of CD163+ macrophages across tumour and stromal areas, with high densities correlating with poor clinical outcomes (222). These findings underscore the multifaceted roles of TAMs in modulating immune responses and contributing to therapeutic resistance. In pancreatic adenocarcinoma, integration of scRNA-seq with ST enabled the identification of an anti-tumour macrophage population marked by IRF7 activity, which limited tumour progression through lipid metabolism-dependent mechanisms (223). More broadly, multi-modal spatial analyses have revealed that TAMs are not homogeneously distributed across the TME. Rather, distinct TAM subsets occupy defined niches: pro-inflammatory TAMs are enriched in tumour cores, whereas anti-inflammatory TAMs preferentially localise to invasive margins in gastric and pancreatic cancers (224, 225). Moreover, TAM function appears spatially encoded - those residing in perivascular or hypoxic regions exhibit immunosuppressive phenotypes, while macrophages at the invasive front can display anti-tumour activity (225). These spatially resolved phenotypes suggest that local tissue architecture and microenvironmental cues shape macrophage polarisation and function.
Importantly, recent spatial transcriptomic studies have begun to map TAM and NK cell crosstalk within the TME, revealing key mechanisms of immune exclusion and dysfunction (Figure 5). In NSCLC, TREM2+ macrophages are highly enriched in tumour cores, where they physically and functionally restrict NK cell infiltration; antibody-mediated TREM2 blockade reactivates NK cells, highlighting a targetable axis of suppression (227). Similarly, in adenocarcinoma and squamous-cell carcinoma, clusters of anti-inflammatory macrophages form immunosuppressive hotspots, inversely correlating with NK cell abundance (226). These spatially resolved TAM-NK interactions further support the notion that immune spatial context may influence response to immunotherapy.
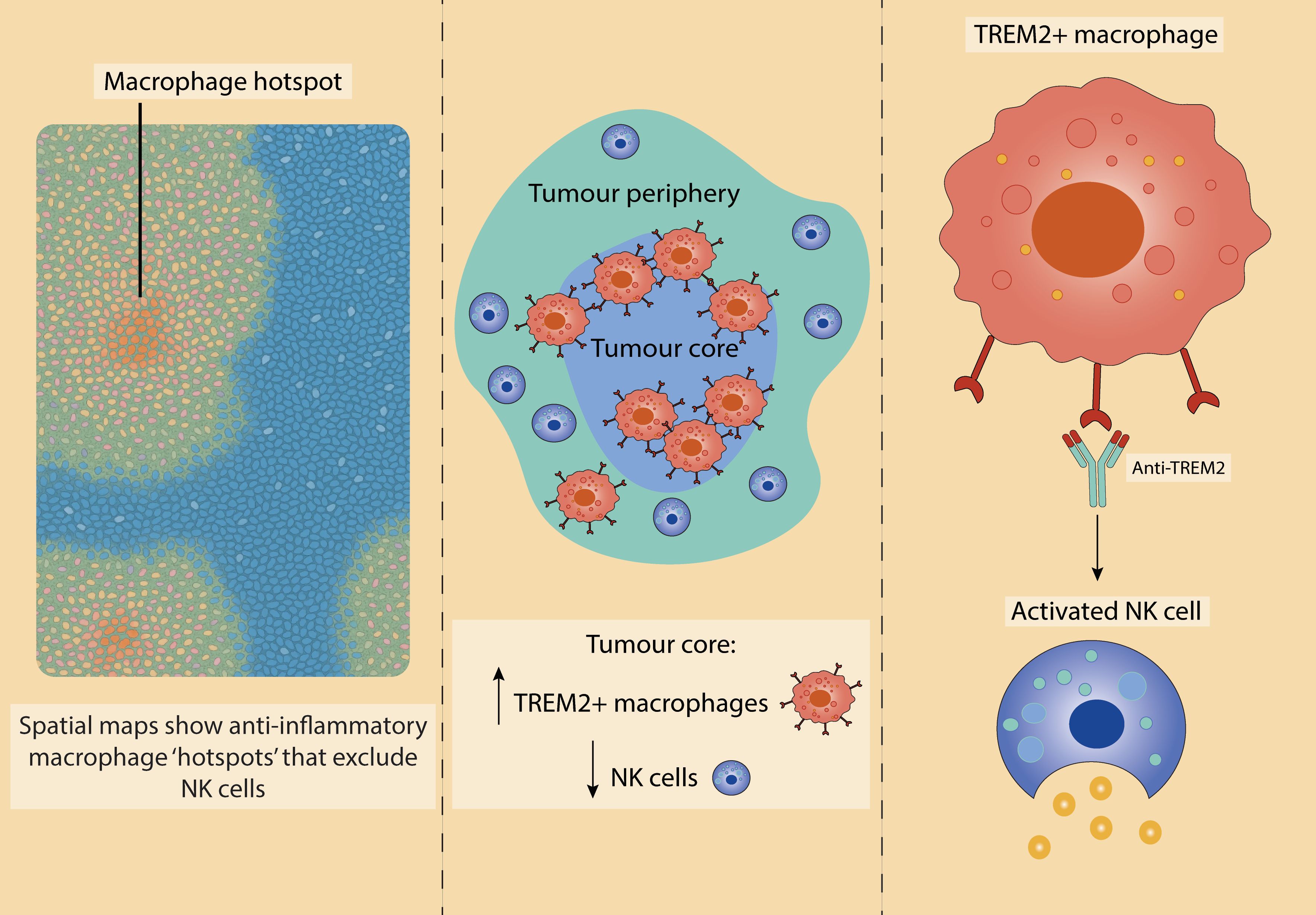
Figure 5. Spatial transcriptomic insights into TAM-NK cell crosstalk in the TME. Recent spatial transcriptomic analyses reveal altered distributions of TAMs and NK cells across tumour regions. Left: Anti-inflammatory macrophage-rich niches predominate in the TME of both adenocarcinoma and squamous cell carcinoma, with a corresponding reduction in NK cell infiltration and cytotoxic gene expression. Based on work by De Zuani et al. (226). Middle: In non-small cell lung cancer, TREM2+ TAMs are enriched within the tumour core, acting as a barrier to NK cell infiltration and promoting an immunosuppressive landscape. Right: Therapeutic blockade of TREM2 reverses this exclusion, enhancing NK cell activation. Middle and right panels based on findings from Park et al. (227).
Future studies examining macrophage–NK cell crosstalk will benefit from ST’s ability to reveal sites of cellular co-localisation, spatially restricted cytokine gradients, and the organisation of functional niches. Combining ST with scRNA-seq facilitates high-resolution mapping of cellular diversity within the TME. One notable advancement is Zman-seq, a dynamic single-cell technology that captures transcriptomic changes over time (228). In glioblastoma, Zman-seq uncovered a rapid sequence of immunological events: NK cells acquired a dysfunctional phenotype within 24 hours, driven by TGFβ1 signalling, followed by the differentiation of infiltrating monocytes into immunosuppressive TAMs within 36–48 hours, characterised by upregulation of suppressive myeloid checkpoints (228). These findings point to a critical early window for therapeutic intervention to preserve immune cell function.
Further integration of ST with high-dimensional imaging techniques, such as co-detection by indexing (CODEX) (229) or multiplexed ion beam imaging (MIBI) (230), will allow transcriptomic data to be overlaid with protein-level information. This multimodal approach will enhance validation of spatial signatures and enable a more comprehensive characterisation of immune cell states and interactions within the TME. However, current ST platforms still face major limitations. Researchers must often choose between single-cell resolution and full transcriptome coverage, meaning high resolution and comprehensive data is hard to achieve at the same time. Even at single-cell resolution, the number of genes reliably captured per cell is often too low to support detailed analyses, especially when compared to liquid-based single-cell RNA-seq (231). In addition, important parts of the TME, such as the extracellular matrix, are still not well captured by most current technologies, leaving major gaps in how we understand tumour structure and cell communication.
6.3 Proteomics: capturing functional states and post-translational dynamics
In the context of tumours, it is proteins, not RNA, that ultimately drive cell behaviour. While transcriptomic data offers important insights, gene expression levels often do not correlate with protein abundance or activity. Proteins interact directly, undergo post-translation modifications, and include secreted factors that mediate communication between cells – critical factors that are missed by RNA-based methods. This underscores the importance of integrating proteomic data to gain a more accurate and comprehensive understanding of cellular function and tumour biology.
Proteomic analyses have shed light on the heterogeneity and function of macrophage subsets. In models of liver fibrosis, distinct populations, including embryo-derived liver-resident Kupffer cells (EmKCs) and monocyte-derived Kupffer cells (MoKCs), were delineated based on unique proteomic signatures (232). In melanoma, proteomic profiling of TAMs revealed a shift toward enhanced cholesterol metabolism and reduced immune activation during tumour progression (233). Similar approaches have elucidated diversity within the NK cell compartment. For example, proteomics has distinguished memory-like NK cells from naïve populations by differential expression of key regulatory proteins (234). Furthermore, NK cell-derived extracellular vesicles, characterised through proteomic profiling, have been shown to contain effector molecules such as Fas ligand, TRAIL, NKG2D, and β-actin, which collectively contribute to their anti-tumour function (235).
Looking ahead, mass spectrometry-based proteomics and targeted platforms such as cytometry by time-of-flight (CyTOF) will be instrumental in dissecting macrophage-NK cell interactions. These approaches enable the detection of activation markers, secreted cytokines, and metabolic enzymes, providing functional insights beyond transcriptomic profiling. When integrated with scRNA-seq and ST, proteomics enables a multidimensional view of immune cell states and interactions - essential for understanding the dynamic regulation of immunity within the TME and informing next-generation immunotherapies.
While scRNA-seq, ST, and proteomics each provide distinct insights into macrophage and NK cell biology, combining these three technologies will allow macrophage-NK cell crosstalk to be studied at a cellular, spatial and functional level. For instance, a study in gastric cancer using integrated spatial multi-omics has shown that the TME has distinct, tissue-specific metabolism signatures (236). In glioblastoma, integrative spatial analysis showed cell organisation is associated with hypoxic cancer cells, with a distinct macrophage state marked by pro-inflammatory cytokine expression being identified (237). Combining scRNA-seq, ST, and proteomics provides a layered view of the TME, allowing transcriptional states, spatial relationships, and protein activity to be mapped together. This approach is well-suited to dissecting macrophage-NK cell interactions, revealing functional states and immunoregulatory niches that shape their crosstalk.
7 Concluding remarks
Harnessing the interplay between macrophages and NK cells within the TME represents a promising strategy to heighten innate immunity and drive durable anti-tumour responses. Unlike conventional T cell–focused therapies, targeting the mechanisms that govern macrophage-NK cell communication has the potential to initiate more coordinated, tissue-integrated immune activation capable of overcoming the immunosuppressive barriers within the TME. TAMs are abundant and actively shape the immune landscape, making them more accessible and impactful therapeutic targets compared to T cells, which often face challenges such as exhaustion, antigen escape, and poor infiltration.
However, current TAM-targeting strategies have limitations. Approaches aimed at repolarising macrophages toward a simplified “M1-like” phenotype underestimate the complexity and plasticity of TAMs, whose states rapidly shift in response to local cues, reducing the durability of such interventions. Depletion strategies lack selectivity, risking the loss of macrophage subsets essential for supporting NK cell function and maintaining tissue homeostasis. Given these challenges, the most promising path forward lies in directly modulating TAM-NK cell crosstalk through manipulating cell communication to restore NK cell cytotoxicity and promote anti-tumour activity. So far this has been achieved through upregulating NKG2D ligands on macrophages or blocking suppressive TGF-β secretion.
However, to fully exploit this therapeutic potential, future research must address several key areas. Mapping the spatial and temporal dynamics of macrophage-NK cell interactions using advanced technologies such as spatial transcriptomics and proteomics will provide detailed insights into their behaviour within the TME. It is also essential to identify selective strategies that target immunosuppressive TAM subsets, while preserving macrophage populations that support NK cell function. Understanding the mechanisms underlying NK cell dysfunction driven by chronic TAM engagement will guide the development of approaches to restore NK cytotoxic capacity. Furthermore, establishing physiologically relevant model systems, including tumour organoids and patient-derived co-cultures, will better enable preclinical testing of combination therapies. Finally, integrating TAM-NK cell-targeting strategies with existing immuno-oncology approaches, such as checkpoint blockade and adoptive cell therapies, may amplify therapeutic outcomes and broaden patient responsiveness. Collectively, these directions will deepen our understanding of innate immune crosstalk in cancer and provide a foundation for the development of next-generation immunotherapies.
Author contributions
NP: Writing – review & editing, Conceptualization, Writing – original draft, Visualization. SK: Supervision, Writing – review & editing. TS-E: Supervision, Writing – review & editing. AV: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. NP is supported by funding from the Medical Research Council (DTP award MR/N014308/1). SK is supported by CRUK Cancer Research UK through the HUNTER HCC expediter network award (C9380/A18084).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
2. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
3. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
4. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
5. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate mapping via ms4a3-expression history traces monocyte-derived cells. Cell. (2019) 178:1509–25. doi: 10.1016/j.cell.2019.08.009
6. Gutknecht MF and Bouton AH. Functional significance of mononuclear phagocyte populations generated through adult hematopoiesis. J Leukoc Biol. (2014) 96:969–80. doi: 10.1189/jlb.1RI0414-195R
7. Hirayama D, Iida T, and Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci. (2017) 19:92. doi: 10.3390/ijms19010092
8. Ruytinx P, Proost P, Van Damme J, and Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol. (2018) 9:1930. doi: 10.3389/fimmu.2018.01930
9. Abdelaziz MH, Abdelwahab SF, Wan J, Cai W, Huixuan W, Jianjun C, et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J Transl Med. (2020) 18:58. doi: 10.1186/s12967-020-02251-w
10. Vitale I, Manic G, Coussens LM, Kroemer G, and Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
11. Mills CD, Kincaid K, Alt JM, Heilman MJ, and Hill AM. -1/M-2 macrophages and the th1/th2 paradigm1. M J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
12. Ernst O, Vayttaden SJ, and Fraser IDC. Measurement of NF-κB activation in TLR-activated macrophages. Methods Mol Biol Clifton NJ. (2018) 1714:67–78. doi: 10.1007/978-1-4939-7519-8_5
13. Kawai T and Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
14. Stout RD, Suttles J, Xu J, Grewal IS, and Flavell RA. Impaired T cell-mediated macrophage activation in CD40 ligand-deficient mice. J Immunol. (1996) 156:8–11. doi: 10.4049/jimmunol.156.1.8
15. Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl LM, and Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages1. J Immunol. (2005) 174:1081–90. doi: 10.4049/jimmunol.174.2.1081
16. Kornbluth RS, Kee K, and Richman DD. CD40 ligand (CD154) stimulation of macrophages to produce HIV-1-suppressive β-chemokines. Proc Natl Acad Sci. (1998) 95:5205–10. doi: 10.1073/pnas.95.9.5205
17. Shu U, Kiniwa M, Wu CY, Maliszewski C, Vezzio N, Hakimi J, et al. Activated T cells induce interleukin-12 production by monocytes via CD40-CD40 ligand interaction. Eur J Immunol. (1995) 25:1125–8. doi: 10.1002/eji.1830250442
18. Bleharski JR, Kiessler V, Buonsanti C, Sieling PA, Stenger S, Colonna M, et al. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response1. J Immunol. (2003) 170:3812–8. doi: 10.4049/jimmunol.170.7.3812
19. Telemaco Contreras Colmenares M, de Oliveira Matos A, Henrique dos Santos Dantas P, Rodrigues do Carmo Neto J, Silva-Sales M, and Sales-Campos H. Unveiling the impact of TREM-2+ Macrophages in metabolic disorders. Cell Immunol. (2024), 405–6:104882. doi: 10.1016/j.cellimm.2024.104882
20. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
21. Kanno S, Hirano S, Sakamoto T, Furuyama A, Takase H, Kato H, et al. Scavenger receptor MARCO contributes to cellular internalization of exosomes by dynamin-dependent endocytosis and macropinocytosis. Sci Rep. (2020) 10:21795. doi: 10.1038/s41598-020-78464-2
22. Xing Q, Feng Y, Sun H, Yang S, Sun T, Guo X, et al. Scavenger receptor MARCO contributes to macrophage phagocytosis and clearance of tumor cells. Exp Cell Res. (2021) 408:112862. doi: 10.1016/j.yexcr.2021.112862
23. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-hodgkin lymphoma. Cell. (2010) 142:699–713. doi: 10.1016/j.cell.2010.07.044
24. Abel AM, Yang C, Thakar MS, and Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
25. Liu S, Galat V, Galat4 Y, Lee YKA, Wainwright D, and Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol OncolJ Hematol Oncol. (2021) 14:7. doi: 10.1186/s13045-020-01014-w
26. Zu S, Lu Y, Xing R, Chen X, and Zhang L. Changes in subset distribution and impaired function of circulating natural killer cells in patients with colorectal cancer. Sci Rep. (2024) 14:12188. doi: 10.1038/s41598-024-63103-x
27. Weber S, Menees KB, Park J, Agin-Liebes J, Lin CC, Alcalay RN, et al. Distinctive CD56dim NK subset profiles and increased NKG2D expression in blood NK cells of Parkinson’s disease patients. NPJ Park Dis. (2024) 10:36. doi: 10.1038/s41531-024-00652-y
28. Saito S, Nishikawa K, Morii T, Enomoto M, Narita N, Motoyoshi K, et al. Cytokine production by CD16-CD56bright natural killer cells in the human early pregnancy decidua. Int Immunol. (1993) 5:559–63. doi: 10.1093/intimm/5.5.559
29. Walzer T, Dalod M, Robbins SH, Zitvogel L, and Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force. Blood. (2005) 106:2252–8. doi: 10.1182/blood-2005-03-1154
30. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR–dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. (2009) 113:3503–11. doi: 10.1182/blood-2008-08-173914
31. Rölle A, Mousavi-Jazi M, Eriksson M, Odeberg J, Söderberg-Nauclér C, Cosman D, et al. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein1. J Immunol. (2003) 171:902–8. doi: 10.4049/jimmunol.171.2.902
32. Siewiera J, Gouilly J, Hocine HR, Cartron G, Levy C, Al-Daccak R, et al. Natural cytotoxicity receptor splice variants orchestrate the distinct functions of human natural killer cell subtypes. Nat Commun. (2015) 6:10183. doi: 10.1038/ncomms10183
33. Sönmez C, Wölfer J, Holling M, Brokinkel B, Stummer W, Wiendl H, et al. Blockade of inhibitory killer cell immunoglobulin-like receptors and IL-2 triggering reverses the functional hypoactivity of tumor-derived NK-cells in glioblastomas. Sci Rep. (2022) 12:6769. doi: 10.1038/s41598-022-10680-4
34. Campbell KS and Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology. (2011) 132:315–25. doi: 10.1111/j.1365-2567.2010.03398.x
35. Brooks AG, Posch PE, Scorzelli CJ, Borrego F, and Coligan JE. NKG2A complexed with CD94 defines a novel inhibitory natural killer cell receptor. J Exp Med. (1997) 185:795–800. doi: 10.1084/jem.185.4.795
36. Garcia-Lora A, Algarra I, and Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. (2003) 195:346–55. doi: 10.1002/jcp.10290
37. Bryceson YT, March ME, Ljunggren HG, and Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. (2006) 107:159–66. doi: 10.1182/blood-2005-04-1351
38. Lee HR, Son CH, Koh EK, Bae JH, Kang CD, Yang K, et al. Expansion of cytotoxic natural killer cells using irradiated autologous peripheral blood mononuclear cells and anti-CD16 antibody. Sci Rep. (2017) 7:11075. doi: 10.1038/s41598-017-09259-1
39. Sekine T, Marin D, Cao K, Li L, Mehta P, Shaim H, et al. Specific combinations of donor and recipient KIR-HLA genotypes predict for large differences in outcome after cord blood transplantation. Blood. (2016) 128:297–312. doi: 10.1182/blood-2016-03-706317
40. Béziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Björklund AT, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood. (2013) 121:2678–88. doi: 10.1182/blood-2012-10-459545
41. Bellora F, Castriconi R, Dondero A, Reggiardo G, Moretta L, Mantovani A, et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc Natl Acad Sci. (2010) 107:21659–64. doi: 10.1073/pnas.1007654108
42. Basu S, Eriksson M, Pioli PA, Conejo-Garcia J, Mselle TF, Yamamoto S, et al. Human Uterine NK Cells Interact with Uterine Macrophages via NKG2D upon Stimulation with PAMPs. Am J Reprod Immunol. (2009) 61:52–61. doi: 10.1111/j.1600-0897.2008.00661.x
43. Mattiola I, Pesant M, Tentorio PF, Molgora M, Marcenaro E, Lugli E, et al. Priming of human resting NK cells by autologous M1 macrophages via the engagement of IL-1β, IFN-β, and IL-15 pathways. J Immunol. (2015) 195:2818–28. doi: 10.4049/jimmunol.1500325
44. Scott M, Hoth J, Stagner M, Gardner S, Peyton J, and Cheadle W. CD40–CD154 interactions between macrophages and natural killer cells during sepsis are critical for macrophage activation and are not interferon gamma dependent. Clin Exp Immunol. (2004) 137:469–77. doi: 10.1111/j.1365-2249.2004.02547.x
45. Wu C, Xue Y, Wang P, Lin L, Liu Q, Li N, et al. IFN-γ Primes Macrophage Activation by Increasing Phosphatase and Tensin Homolog via Downregulation of miR-3473b. J Immunol. (2014) 193:3036–44. doi: 10.4049/jimmunol.1302379
46. Zhou J, Zhang S, and Guo C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int Immunopharmacol. (2021) 101:108374. doi: 10.1016/j.intimp.2021.108374
47. Nedvetzki S, Sowinski S, Eagle RA, Harris J, Vély F, Pende D, et al. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood. (2007) 109:3776–85. doi: 10.1182/blood-2006-10-052977
48. Zhou Z, Zhang C, Zhang J, and Tian Z. Macrophages help NK cells to attack tumor cells by stimulatory NKG2D ligand but protect themselves from NK killing by inhibitory ligand qa-1. PloS One. (2012) 7:e36928. doi: 10.1371/journal.pone.0036928
49. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
50. he RG, qing LY, Tian L, Zhang T, mei YD, Yu J, et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct Target Ther. (2022) 7:205. doi: 10.1038/s41392-022-01058-z
51. Glas R, Franksson L, Une C, Eloranta ML, Öhlén C, Örn A, et al. Recruitment and activation of natural killer (Nk) cells in vivo determined by the target cell phenotype: an adaptive component of nk cell–mediated responses. J Exp Med. (2000) 191:129–38. doi: 10.1084/jem.191.1.129
52. Shingu K, Helfritz A, Kuhlmann S, Zielinska-Skowronek M, Jacobs R, Schmidt RE, et al. Kinetics of the early recruitment of leukocyte subsets at the sites of tumor cells in the lungs: Natural killer (NK) cells rapidly attract monocytes but not lymphocytes in the surveillance of micrometastasis. Int J Cancer. (2002) 99:74–81. doi: 10.1002/ijc.10279
53. Moffett A and Colucci F. Uterine NK cells: active regulators at the maternal-fetal interface. J Clin Invest. (2014) 124:1872–9. doi: 10.1172/JCI68107
54. Peng H and Sun R. Liver-resident NK cells and their potential functions. Cell Mol Immunol. (2017) 14:890–4. doi: 10.1038/cmi.2017.72
55. Andrade RM, Portillo JAC, Wessendarp M, and Subauste CS. CD40 signaling in macrophages induces activity against an intracellular pathogen independently of gamma interferon and reactive nitrogen intermediates. Infect Immun. (2005) 73:3115–23. doi: 10.1128/IAI.73.5.3115-3123.2005
56. Carbone E, Ruggiero G, Terrazzano G, Palomba C, Manzo C, Fontana S, et al. A new mechanism of NK cell cytotoxicity activation: the CD40–CD40 ligand interaction. J Exp Med. (1997) 185:2053–60. doi: 10.1084/jem.185.12.2053
57. Müller E, Christopoulos PF, Halder S, Lunde A, Beraki K, Speth M, et al. Toll-like receptor ligands and interferon-γ Synergize for induction of antitumor M1 macrophages. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.01383
58. de Groen RA, Boltjes A, Hou J, Liu BS, McPhee F, Friborg J, et al. IFN-λ-mediated IL-12 production in macrophages induces IFN-γ production in human NK cells. Eur J Immunol. (2015) 45:250–9. doi: 10.1002/eji.201444903
59. Bloemendaal FM, Koelink PJ, van Schie KA, Rispens T, Peters CP, Buskens CJ, et al. TNF-anti-TNF immune complexes inhibit IL-12/IL-23 secretion by inflammatory macrophages via an fc-dependent mechanism. J Crohns Colitis. (2018) 12:1122–30. doi: 10.1093/ecco-jcc/jjy075
60. Michel T, Hentges F, and Zimmer J. Consequences of the crosstalk between monocytes/macrophages and natural killer cells. Front Immunol. (2012) 3:403. doi: 10.3389/fimmu.2012.00403
61. Vinogradov S, Warren G, and Wei X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomed. (2014) 9:695–707. doi: 10.2217/nnm.14.13
62. Mantovani A, Allavena P, Marchesi F, and Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
63. Ugel S, De Sanctis F, Mandruzzato S, and Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. (2015) 125:3365–76. doi: 10.1172/JCI80006
64. Boutilier AJ and Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. (2021) 22:6995. doi: 10.3390/ijms22136995
65. Duan Z and Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther. (2021) 6:1–21. doi: 10.1038/s41392-021-00506-6
66. Forssell J, Öberg Å, Henriksson ML, Stenling R, Jung A, and Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. (2007) 13:1472–9. doi: 10.1158/1078-0432.CCR-06-2073
67. Ma J, Liu L, Che G, Yu N, Dai F, and You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer. (2010) 10:112. doi: 10.1186/1471-2407-10-112
68. Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. (2014) 7:19. doi: 10.1186/1757-2215-7-19
69. Guo L, Cheng X, Chen H, Chen C, Xie S, Zhao M, et al. Induction of breast cancer stem cells by M1 macrophages through Lin-28B-let-7-HMGA2 axis. Cancer Lett. (2019) 452:213–25. doi: 10.1016/j.canlet.2019.03.032
70. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. (2004) 4:71–8. doi: 10.1038/nrc1256
71. Dithmar SA, Rusciano DA, Armstrong CA, Lynn MJ, and Grossniklaus HE. Depletion of NK cell activity results in growth of hepatic micrometastases in a murine ocular melanoma model. Curr Eye Res. (1999) 19:426–31. doi: 10.1076/ceyr.19.5.426.5294
72. Habu S, Fukui H, Shimamura K, Kasai M, Nagai Y, Okumura K, et al. In vivo effects of anti-asialo GM1. I. Reduction of NK activity and enhancement of transplanted tumor growth in nude mice. J Immunol Baltim Md 1950. (1981) 127:34–8.
73. Frese-Schaper M, Keil A, Yagita H, Steiner SK, Falk W, Schmid RA, et al. Influence of natural killer cells and perforin-mediated cytolysis on the development of chemically induced lung cancer in A/J mice. Cancer Immunol Immunother CII. (2014) 63:571–80. doi: 10.1007/s00262-014-1535-x
74. Tartter PI, Steinberg B, Barron DM, and Martinelli G. The prognostic significance of natural killer cytotoxicity in patients with colorectal cancer. Arch Surg. (1987) 122:1264–8. doi: 10.1001/archsurg.1987.01400230050009
75. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. (2011) 17:678–89. doi: 10.1158/1078-0432.CCR-10-2173
76. Schantz SP and Ordonez NG. Quantitation of natural killer cell function and risk of metastatic poorly differentiated head and neck cancer. Nat Immun Cell Growth Regul. (1991) 10:278–88.
77. Schantz SP, Savage HE, Racz T, Taylor DL, and Sacks PG. Natural killer cells and metastases from pharyngeal carcinoma. Am J Surg. (1989) 158:361–6. doi: 10.1016/0002-9610(89)90134-7
78. Parkhurst MR, Riley JP, Dudley ME, and Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res Off J Am Assoc Cancer Res. (2011) 17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347
79. Schierloh P, Alemán M, Yokobori N, Alves L, Roldán N, Abbate E, et al. NK cell activity in tuberculosis is associated with impaired CD11a and ICAM-1 expression: a regulatory role of monocytes in NK activation. Immunology. (2005) 116:541–52. doi: 10.1111/j.1365-2567.2005.02259.x
80. Park JY, Lee SH, Yoon SR, Park YJ, Jung H, Kim TD, et al. IL-15-induced IL-10 increases the cytolytic activity of human natural killer cells. Mol Cells. (2011) 32:265–72. doi: 10.1007/s10059-011-1057-8
81. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
82. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
83. Peng LS, Zhang JY, Teng YS, Zhao YL, Wang TT, Mao FY, et al. Tumor-associated monocytes/macrophages impair NK-cell function via TGFβ1 in human gastric cancer. Cancer Immunol Res. (2017) 5:248–56. doi: 10.1158/2326-6066.CIR-16-0152
84. Dean I, Lee CYC, Tuong ZK, Li Z, Tibbitt CA, Willis C, et al. Rapid functional impairment of natural killer cells following tumor entry limits anti-tumor immunity. Nat Commun. (2024) 15:683. doi: 10.1038/s41467-024-44789-z
85. Gallazzi M, Baci D, Mortara L, Bosi A, Buono G, Naselli A, et al. Prostate cancer peripheral blood NK cells show enhanced CD9, CD49a, CXCR4, CXCL8, MMP-9 production and secrete monocyte-recruiting and polarizing factors. Front Immunol. (2020) 11:586126. doi: 10.3389/fimmu.2020.586126
86. Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, et al. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity. (2008) 29:720–33. doi: 10.1016/j.immuni.2008.08.014
87. Garzón-Tituaña M, Sierra-Monzón JL, Comas L, Santiago L, Khaliulina-Ushakova T, Uranga-Murillo I, et al. Granzyme A inhibition reduces inflammation and increases survival during abdominal sepsis. Theranostics. (2021) 11:3781–95. doi: 10.7150/thno.49288
88. Wensink AC, Kok HM, Meeldijk J, Fermie J, Froelich CJ, Hack CE, et al. Granzymes A and K differentially potentiate LPS-induced cytokine response. Cell Death Discov. (2016) 2:16084. doi: 10.1038/cddiscovery.2016.84
89. Pannetier D, Faure C, Georges-Courbot MC, Deubel V, and Baize S. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to mopeia virus infection. J Virol. (2004) 78:10516–24. doi: 10.1128/JVI.78.19.10516-10524.2004
90. Xie C, Liu C, Wu B, Lin Y, Ma T, Xiong H, et al. Effects of IRF1 and IFN-β interaction on the M1 polarization of macrophages and its antitumor function. Int J Mol Med. (2016) 38:148–60. doi: 10.3892/ijmm.2016.2583
91. Liu Q, Li J, Zong Q, Duan Z, Liu F, Duan W, et al. Interferon-induced polarization of M1 macrophages mediates antiviral activity against the hepatitis B virus via the hepcidin-ferroportin axis. Int Immunopharmacol. (2024) 134:112219. doi: 10.1016/j.intimp.2024.112219
92. Makuch E, Jasyk I, Kula A, Lipiński T, and Siednienko J. IFNβ-induced CXCL10 chemokine expression is regulated by pellino3 ligase in monocytes and macrophages. Int J Mol Sci. (2022) 23:14915. doi: 10.3390/ijms232314915
93. Zhang F, Mears JR, Shakib L, Beynor JI, Shanaj S, Korsunsky I, et al. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med. (2021) 13:64. doi: 10.1186/s13073-021-00881-3
94. Fujita H, Asahina A, Tada Y, Fujiwara H, and Tamaki K. Type I interferons inhibit maturation and activation of mouse langerhans cells. J Invest Dermatol. (2005) 125:126–33. doi: 10.1111/j.0022-202X.2005.23803.x
95. Watkins SK, Li B, Richardson KS, Head K, Egilmez NK, Zeng Q, et al. Rapid release of cytoplasmic IL-15 from tumor associated macrophages is an initial and critical event in IL-12 initiated tumor regression. Eur J Immunol. (2009) 39:2126–35. doi: 10.1002/eji.200839010
96. Lusty E, Poznanski SM, Kwofie K, Mandur TS, Lee DA, Richards CD, et al. IL-18/IL-15/IL-12 synergy induces elevated and prolonged IFN-γ production by ex vivo expanded NK cells which is not due to enhanced STAT4 activation. Mol Immunol. (2017) 88:138–47. doi: 10.1016/j.molimm.2017.06.025
97. Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep. (2015) 12:1902–14. doi: 10.1016/j.celrep.2015.08.033
98. Lee J, Lee SH, Shin N, Jeong M, Kim MS, Kim MJ, et al. Tumor necrosis factor-α enhances IL-15-induced natural killer cell differentiation. Biochem Biophys Res Commun. (2009) 386:718–23. doi: 10.1016/j.bbrc.2009.06.120
99. McCulloch TR, Rossi GR, Alim L, Lam PY, Wong JKM, Coleborn E, et al. Dichotomous outcomes of TNFR1 and TNFR2 signaling in NK cell-mediated immune responses during inflammation. Nat Commun. (2024) 15:9871. doi: 10.1038/s41467-024-54232-y
100. Khan AUH, Ali AK, Marr B, Jo D, Ahmadvand S, Fong-McMaster C, et al. The TNFα/TNFR2 axis mediates natural killer cell proliferation by promoting aerobic glycolysis. Cell Mol Immunol. (2023) 20:1140–55. doi: 10.1038/s41423-023-01071-4
101. Merzoug LB, Marie S, Satoh-Takayama N, Lesjean S, Albanesi M, Luche H, et al. Conditional ablation of NKp46+ cells using a novel Ncr1greenCre mouse strain: NK cells are essential for protection against pulmonary B16 metastases. Eur J Immunol. (2014) 44:3380–91. doi: 10.1002/eji.201444643
102. Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, et al. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol. (2012) 188:2509–15. doi: 10.4049/jimmunol.1102461
103. Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. (2009) 119:1251–63. doi: 10.1172/JCI36022
104. Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, and Mandelboim O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR11. J Immunol. (2009) 182:2221–30. doi: 10.4049/jimmunol.0801878
105. Hadadi L, Hafezi M, Amirzargar AA, Sharifian RA, Abediankenari S, and Asgarian-Omran H. Dysregulated expression of tim-3 and NKp30 receptors on NK cells of patients with chronic lymphocytic leukemia. Oncol Res Treat. (2019) 42:202–8. doi: 10.1159/000497208
106. Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood. (2002) 99:3661–7. doi: 10.1182/blood.v99.10.3661
107. Chretien AS, Fauriat C, Orlanducci F, Rey J, Borg GB, Gautherot E, et al. NKp30 expression is a prognostic immune biomarker for stratification of patients with intermediate-risk acute myeloid leukemia. Oncotarget. (2017) 8:49548–63. doi: 10.18632/oncotarget.17747
108. Cifaldi L, Melaiu O, Giovannoni R, Benvenuto M, Focaccetti C, Nardozi D, et al. DNAM-1 chimeric receptor-engineered NK cells: a new frontier for CAR-NK cell-based immunotherapy. Front Immunol. (2023) 14:1197053. doi: 10.3389/fimmu.2023.1197053
109. Thompson TW, Jackson BT, Li PJ, Wang J, Kim AB, Huang KTH, et al. Tumor-derived CSF-1 induces the NKG2D ligand RAE-1δ on tumor-infiltrating macrophages. Yokoyama WM editor. eLife. (2018) 7:e32919. doi: 10.7554/eLife.32919.022
110. Siemaszko J, Marzec-Przyszlak A, and Bogunia-Kubik K. NKG2D natural killer cell receptor—A short description and potential clinical applications. Cells. (2021) 10:1420. doi: 10.3390/cells10061420
111. Wang W, Gao L, and Ma YG. Effect of NKG2D in eliminating hematological Malignant cell lines by natural killer cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2012) 20:296–9.
112. Sallman DA, Brayer J, Sagatys EM, Lonez C, Breman E, Agaugué S, et al. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica. (2018) 103:e424–6. doi: 10.3324/haematol.2017.186742
113. Romo N, Magri G, Muntasell A, Heredia G, Baía D, Angulo A, et al. Natural killer cell-mediated response to human cytomegalovirus-infected macrophages is modulated by their functional polarization. J Leukoc Biol. (2011) 90:717–26. doi: 10.1189/jlb.0311171
114. Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D, et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatol Baltim Md. (2013) 57:1107–16. doi: 10.1002/hep.26192
115. Welte S, Kuttruff S, Waldhauer I, and Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat Immunol. (2006) 7:1334–42. doi: 10.1038/ni1402
116. Gao N, Wang C, Yu Y, Xie L, Xing Y, Zhang Y, et al. LFA-1/ICAM-1 promotes NK cell cytotoxicity associated with the pathogenesis of ocular toxoplasmosis in murine model. PloS Negl Trop Dis. (2022) 16:e0010848. doi: 10.1371/journal.pntd.0010848
117. Hooda V and Sharma A. Interactions of NK cells and macrophages: from infections to cancer therapeutics. Immunology. (2025) 174:287–95. doi: 10.1111/imm.13886
118. Chiba S, Ikushima H, Ueki H, Yanai H, Kimura Y, Hangai S, et al. editor. eLife. (2014) 3:e04177. doi: 10.7554/eLife.04177
119. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
120. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
121. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
122. Li H, leum van der AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. (2019) 176:775–89. doi: 10.1016/j.cell.2018.11.043
123. Mitsudomi T and Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. (2010) 277:301–8. doi: 10.1111/j.1742-4658.2009.07448.x
124. Liu C, Zhang W, Wang J, Si T, and Xing W. Tumor-associated macrophage-derived transforming growth factor-β promotes colorectal cancer progression through HIF1-TRIB3 signaling. Cancer Sci. (2021) 112:4198–207. doi: 10.1111/cas.15101
125. Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, et al. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res. (2011) 17:7230–9. doi: 10.1158/1078-0432.CCR-11-1354
126. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. (2014) 33:2423–31. doi: 10.1038/onc.2013.191
127. Cat B, Stuhlmann D, Steinbrenner H, Alili L, Holtkötter O, Sies H, et al. Enhancement of tumor invasion depends on transdifferentiation of skin fibroblasts mediated by reactive oxygen species. J Cell Sci. (2006) 119:2727–38. doi: 10.1242/jcs.03011
128. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. (2021) 595:578–84. doi: 10.1038/s41586-021-03651-8
129. Lievense LA, Cornelissen R, Bezemer K, Kaijen-Lambers MEH, Hegmans JPJJ, and Aerts JGJV. Pleural effusion of patients with Malignant mesothelioma induces macrophage-mediated T cell suppression. J Thorac Oncol. (2016) 11:1755–64. doi: 10.1016/j.jtho.2016.06.021
130. Muraoka D, Seo N, Hayashi T, Tahara Y, Fujii K, Tawara I, et al. Antigen delivery targeted to tumor-associated macrophages overcomes tumor immune resistance. J Clin Invest. (2019) 129:1278–94. doi: 10.1172/JCI97642
131. Tang X, Mo C, Wang Y, Wei D, and Xiao H. Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology. (2013) 138:93–104. doi: 10.1111/imm.12023
132. Cassetta L and Kitamura T. Macrophage targeting: opening new possibilities for cancer immunotherapy. Immunology. (2018) 155:285–93. doi: 10.1111/imm.12976
133. Bellora F, Castriconi R, Dondero A, Pessino A, Nencioni A, Liggieri G, et al. TLR activation of tumor-associated macrophages from ovarian cancer patients triggers cytolytic activity of NK cells. Eur J Immunol. (2014) 44:1814–22. doi: 10.1002/eji.201344130
134. Sommariva M, Le Noci V, Storti C, Bianchi F, Tagliabue E, Balsari A, et al. Activation of NK cell cytotoxicity by aerosolized CpG-ODN/poly(I:C) against lung melanoma metastases is mediated by alveolar macrophages. Cell Immunol. (2017) 313:52–8. doi: 10.1016/j.cellimm.2017.01.004
135. Zhu Z, He L, Bai Y, Xia L, Sun X, and Qi C. Yeast β-glucan modulates macrophages and improves antitumor NK-cell responses in cancer. Clin Exp Immunol. (2023) 214:50–60. doi: 10.1093/cei/uxad080
136. Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjärvi S, Oliynyk G, et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci U S A. (2020) 117:32005–16. doi: 10.1073/pnas.2015343117
137. Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
138. Haeggblom L, Dash S, Vayn Y, Rudolph J, Palmer R, Yang X, et al. Therapeutic targeting of MARCO with PY265 antibody promotes myeloid cell reprogramming and unleashes anti-tumor immunity. J Immunother Cancer. (2022) 10:1342. doi: 10.1136/jitc-2022-sitc2022.1342
139. Takahashi H, Perez-Villarroel P, Falahat R, and Mulé JJ. Targeting MARCO in combination with anti-CTLA-4 leads to enhanced melanoma regression and immune cell infiltration via macrophage reprogramming. J Immunother Cancer. (2025) 13:e011030. doi: 10.1136/jitc-2024-011030
140. Virtakoivu R, Rannikko JH, Viitala M, Vaura F, Takeda A, Lönnberg T, et al. Systemic blockade of clever-1 elicits lymphocyte activation alongside checkpoint molecule downregulation in patients with solid tumors: results from a phase I/II clinical trial. Clin Cancer Res. (2021) 27:4205–20. doi: 10.1158/1078-0432.CCR-20-4862
141. Rannikko JH, Verlingue L, de Miguel M, Pasanen A, Robbrecht D, Skytta T, et al. Bexmarilimab-induced macrophage activation leads to treatment benefit in solid tumors: The phase I/II first-in-human MATINS trial. Cell Rep Med. (2023) 4. doi: 10.1101/2023.04.17.23288693
142. Ehlers FAI, Mahaweni NM, van de Waterweg Berends A, Saya T, Bos GMJ, and Wieten L. Exploring the potential of combining IL-2-activated NK cells with an anti-PDL1 monoclonal antibody to target multiple myeloma-associated macrophages. Cancer Immunol Immunother. (2023) 72:1789–801. doi: 10.1007/s00262-022-03365-4
143. Dhupkar P, Gordon N, Stewart J, and Kleinerman ES. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. (2018) 7:2654–64. doi: 10.1002/cam4.1518
144. Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, Lee T, et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. (2021) 37:109844. doi: 10.1016/j.celrep.2021.109844
145. Brownlie D, Doughty-Shenton D YH, Soong D, Nixon C, Carragher N O, Carlin L M, et al. Metastasis-associated macrophages constrain antitumor capability of natural killer cells in the metastatic site at least partially by membrane bound transforming growth factor β. J Immunother Cancer. (2021) 9:e001740. doi: 10.1136/jitc-2020-001740
146. Pfirschke C, Zilionis R, Engblom C, Messemaker M, Zou AE, Rickelt S, et al. Macrophage-targeted therapy unlocks antitumoral cross-talk between IFNγ-secreting lymphocytes and IL12-producing dendritic cells. Cancer Immunol Res. (2022) 10:40–55. doi: 10.1158/2326-6066.CIR-21-0326
147. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
148. Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D’Angelo SP, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann Oncol. (2019) 30:1381–92. doi: 10.1093/annonc/mdz163
149. Cassier PA, Italiano A, Gomez-Roca CA, Tourneau CL, Toulmonde M, Cannarile MA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. (2015) 16:949–56. doi: 10.1016/S1470-2045(15)00132-1
150. Teng KY, Han J, Zhang X, Hsu SH, He S, Wani NA, et al. Blocking the CCL2-CCR2 axis using CCL2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol Cancer Ther. (2017) 16:312–22. doi: 10.1158/1535-7163.MCT-16-0124
151. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
152. Yao W, Ba Q, Li X, Li H, Zhang S, Yuan Y, et al. A natural CCR2 antagonist relieves tumor-associated macrophage-mediated immunosuppression to produce a therapeutic effect for liver cancer. eBioMedicine. (2017) 22:58–67. doi: 10.1016/j.ebiom.2017.07.014
153. Noel M, O’Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. (2020) 38:800–11. doi: 10.1007/s10637-019-00830-3
154. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. (2021) 12:877. doi: 10.1038/s41467-021-20893-2
155. Krneta T, Gillgrass A, Poznanski S, Chew M, Lee AJ, Kolb M, et al. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J Leukoc Biol. (2017) 101:285–95. doi: 10.1189/jlb.3A1215-552R
156. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. (2014) 507:508–12. doi: 10.1038/nature12998
157. Ireland L, Luckett T, Schmid MC, and Mielgo A. Blockade of stromal gas6 alters cancer cell plasticity, activates NK cells, and inhibits pancreatic cancer metastasis. Front Immunol. (2020) 11:297. doi: 10.3389/fimmu.2020.00297
158. Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas1. Clin Cancer Res. (1999) 5:1107–13.
159. Hanada T, Nakagawa M, Emoto A, Nomura T, Nasu N, and Nomura Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int J Urol. (2000) 7:263–9. doi: 10.1046/j.1442-2042.2000.00190.x
160. Fujimoto J, Sakaguchi H, Aoki I, and Tamaya T. Clinical implications of expression of interleukin 8 related to angiogenesis in uterine cervical cancers. Cancer Res. (2000) 60:2632–5.
161. Salvesen HB and Akslen LA. Significance of tumour-associated macrophages, vascular endothelial growth factor and thrombospondin-1 expression for tumour angiogenesis and prognosis in endometrial carcinomas. Int J Cancer. (1999) 84:538–43. doi: 10.1002/(SICI)1097-0215(19991022)84:5<538::AID-IJC17>3.0.CO;2-B
162. Volodko N, Reiner A, Rudas M, and Jakesz R. Tumour-associated macrophages in breast cancer and their prognostic correlations. Breast. (1998) 7:99–105. doi: 10.1016/S0960-9776(98)90065-0
163. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, and Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma1. Cancer Res. (1996) 56:4625–9.
164. Bingle L, Brown NJ, and Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. (2002) 196:254–65. doi: 10.1002/path.1027
165. Lin EY, Nguyen AV, Russell RG, and Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to Malignancy. J Exp Med. (2001) 193:727–40. doi: 10.1084/jem.193.6.727
166. Yu W, Chen J, Xiong Y, Pixley FJ, Yeung YG, and Stanley ER. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J Biol Chem. (2012) 287:13694–704. doi: 10.1074/jbc.M112.355610
167. Magkouta SF, Vaitsi PC, Pappas AG, Iliopoulou M, Kosti CN, Psarra K, et al. CSF1/CSF1R axis blockade limits mesothelioma and enhances efficiency of anti-PDL1 immunotherapy. Cancers. (2021) 13:2546. doi: 10.3390/cancers13112546
168. Ding J, Guo C, Hu P, Chen J, Liu Q, Wu X, et al. CSF1 is involved in breast cancer progression through inducing monocyte differentiation and homing. Int J Oncol. (2016) 49:2064–74. doi: 10.3892/ijo.2016.3680
169. Hamerman JA, Ogasawara K, and Lanier LL. Cutting edge: toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor 1. J Immunol. (2004) 172:2001–5. doi: 10.4049/jimmunol.172.4.2001
170. Kloss M, Decker P, Baltz KM, Baessler T, Jung G, Rammensee HG, et al. Interaction of monocytes with NK cells upon toll-like receptor-induced expression of the NKG2D ligand MICA1. J Immunol. (2008) 181:6711–9. doi: 10.4049/jimmunol.181.10.6711
171. Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. (2015) 348:136–9. doi: 10.1126/science.1258867
172. Kim J, Nam G, Shin YK, Vilaplana-Lopera N, Jeung HC, Moon EJ, et al. Targeting AXL using the AVB-500 soluble receptor and through genetic knockdown inhibits bile duct cancer growth and metastasis. Cancers. (2023) 15:1882. doi: 10.3390/cancers15061882
173. Tirado-Gonzalez I, Descot A, Soetopo D, Nevmerzhitskaya A, Schäffer A, Kur IM, et al. AXL inhibition in macrophages stimulates host-versus-leukemia immunity and eradicates naïve and treatment-resistant leukemia. Cancer Discov. (2021) 11:2924–43. doi: 10.1158/2159-8290.CD-20-1378
174. Chirino LM, Kumar S, Okumura M, Sterner DE, Mattern M, Butt TR, et al. TAM receptors attenuate murine NK-cell responses via E3 ubiquitin ligase Cbl-b. Eur J Immunol. (2020) 50:48–55. doi: 10.1002/eji.201948204
175. Zhong C, Wang L, Liu Y, Wang X, Xia Z, Li Y, et al. PSGL-1 is a phagocytosis checkpoint that enables tumor escape from macrophage clearance. Sci Immunol. (2025) 10:eadn4302. doi: 10.1126/sciimmunol.adn4302
176. Noelle R, Johnson M, Rodon J, Zauderer M, Lewis L, Severgnini M, et al. 761 Pharmacokinetic and pharmacodynamic data from a phase 1 study of CI-8993 Anti-VISTA antibody in patients with advanced solid tumors. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0761
177. Guillaudeux T, Lustig K, Frazier E, Kabi N, Katz C, Peckham D, et al. 425 KVA12.1: an anti-VISTA monoclonal antibody with strong single agent anti-tumor activity and no evidence of cytokine mediated toxicity. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0425
178. Ge H, Guo N, Liu Y, Lang B, Yin X, Yu X, et al. The inhibitory receptor LAG3 affects NK cell IFN-γ production through glycolysis and the PSAT1/STAT1/IFNG pathway. mBio. (2025) 16:e0023025. doi: 10.1128/mbio.00230-25
179. Sordo-Bahamonde C, Lorenzo-Herrero S, González-Rodríguez AP, Payer ÁR, González-García E, López-Soto A, et al. LAG-3 blockade with relatlimab (BMS-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers. (2021) 13:2112. doi: 10.3390/cancers13092112
180. Bauché D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3+ Regulatory T cells restrain interleukin-23-producing CX3CR1+ Gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. (2018) 49:342–52. doi: 10.1016/j.immuni.2018.07.007
181. Buisson S and Triebel F. LAG-3 (CD223) reduces macrophage and dendritic cell differentiation from monocyte precursors. Immunology. (2005) 114:369–74. doi: 10.1111/j.1365-2567.2004.02087.x
182. Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. (2012) 119:3734–43. doi: 10.1182/blood-2011-11-392951
183. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
184. Hou H, Liu W, Wu S, Lu Y, Peng J, Zhu Y, et al. Tim-3 negatively mediates natural killer cell function in LPS-induced endotoxic shock. PloS One. (2014) 9:e110585. doi: 10.1371/journal.pone.0110585
185. Jiang W, Li F, Jiang Y, Li S, Liu X, Xu Y, et al. Tim-3 blockade elicits potent anti-multiple myeloma immunity of natural killer cells. Front Oncol. (2022) 12:739976. doi: 10.3389/fonc.2022.739976
186. Ni X, Wu W, Sun X, Ma J, Yu Z, He X, et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci Adv. (2022) 8:eabl5165. doi: 10.14791/btrt.2022.10.Suppl
187. Chauvin JM, Ka M, Pagliano O, Menna C, Ding Q, DeBlasio R, et al. IL15 stimulation with TIGIT blockade reverses CD155-mediated NK-cell dysfunction in melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. (2020) 26:5520–33. doi: 10.1158/1078-0432.CCR-20-0575
188. Chen X, Lu PH, Liu L, Fang ZM, Duan W, Liu ZL, et al. TIGIT negatively regulates inflammation by altering macrophage phenotype. Immunobiology. (2016) 221:48–55. doi: 10.1016/j.imbio.2015.08.003
189. Brauneck F, Fischer B, Witt M, Muschhammer J, Oelrich J, da Costa Avelar PH, et al. TIGIT blockade repolarizes AML-associated TIGIT+ M2 macrophages to an M1 phenotype and increases CD47-mediated phagocytosis. J Immunother Cancer. (2022) 10:e004794. doi: 10.1136/jitc-2022-004794
190. Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: A randomized clinical trial. JAMA. (2021) 325:632–44. doi: 10.1001/jama.2021.0202
191. Weiss SA, Djureinovic D, Jessel S, Krykbaeva I, Zhang L, Jilaveanu L, et al. A phase I study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non–small cell lung cancer resistant to anti-PD-1/PD-L1. Clin Cancer Res Off J Am Assoc Cancer Res. (2021) 27:4757–67. doi: 10.1158/1078-0432.CCR-21-0903
192. Falchook GS, Ribas A, Davar D, Eroglu Z, Wang JS, Luke JJ, et al. Phase 1 trial of TIM-3 inhibitor cobolimab monotherapy and in combination with PD-1 inhibitors nivolumab or dostarlimab (AMBER). J Clin Oncol. (2022) 40:2504–4. doi: 10.1200/JCO.2022.40.16_suppl.2504
193. DhatChinamoorthy K, Colbert JD, and Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
194. Xia Y, Rao L, Yao H, Wang Z, Ning P, and Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. (2020) 32:2002054. doi: 10.1002/adma.202002054
195. White LG, Goy HE, Rose AJ, and McLellan AD. Controlling cell trafficking: addressing failures in CAR T and NK cell therapy of solid tumours. Cancers. (2022) 14:978. doi: 10.3390/cancers14040978
196. Lei W, Xie M, Jiang Q, Xu N, Li P, Liang A, et al. Treatment-related adverse events of chimeric antigen receptor T-cell (CAR T) in clinical trials: A systematic review and meta-analysis. Cancers. (2021) 13:3912. doi: 10.3390/cancers13153912
197. Ricketts TD, Prieto-Dominguez N, Gowda PS, and Ubil E. Mechanisms of macrophage plasticity in the tumor environment: manipulating activation state to improve outcomes. Front Immunol. (2021) 12:642285. doi: 10.3389/fimmu.2021.642285
198. Li L and Tian Y. The role of metabolic reprogramming of tumor-associated macrophages in shaping the immunosuppressive tumor microenvironment. BioMed Pharmacother Biomedecine Pharmacother. (2023) 161:114504. doi: 10.1016/j.biopha.2023.114504
199. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
200. Nürnberger F, Ott D, Claßen R, Rummel C, Roth J, and Leisengang S. Systemic lipopolysaccharide challenge induces inflammatory changes in rat dorsal root ganglia: an ex vivo study. Int J Mol Sci. (2022) 23:13124. doi: 10.3390/ijms232113124
201. Juskewitch JE, Knudsen BE, Platt JL, Nath KA, Knutson KL, Brunn GJ, et al. LPS-induced murine systemic inflammation is driven by parenchymal cell activation and exclusively predicted by early MCP-1 plasma levels. Am J Pathol. (2012) 180:32–40. doi: 10.1016/j.ajpath.2011.10.001
202. Boukhvalova MS, Sotomayor TB, Point RC, Pletneva LM, Prince GA, and Blanco JCG. Activation of interferon response through toll-like receptor 3 impacts viral pathogenesis and pulmonary toll-like receptor expression during respiratory syncytial virus and influenza infections in the cotton rat sigmodon hispidus model. J Interferon Cytokine Res. (2010) 30:229–42. doi: 10.1089/jir.2009.0025
203. Field R, Campion S, Warren C, Murray C, and Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNα/β and IL-1β responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. (2010) 24:996–1007. doi: 10.1016/j.bbi.2010.04.004
204. Modak S, Kushner BH, Kramer K, Vickers A, Cheung IY, and Cheung NKV. Anti-GD2 antibody 3F8 and barley-derived (1 → 3),(1 → 4)-β-D-glucan. Oncoimmunology. (2013) 2:e23402. doi: 10.4161/onci.23402
205. Bhalla S, Fattah FJ, Ahn C, Williams J, Macchiaroli A, Padro J, et al. Phase 1 trial of bemcentinib (BGB324), a first-in-class, selective AXL inhibitor, with docetaxel in patients with previously treated advanced non-small cell lung cancer. Lung Cancer Amst Neth. (2023) 182:107291. doi: 10.1016/j.lungcan.2023.107291
206. Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. (2011) 39:916–24. doi: 10.1177/0192623311416259
207. Beffinger M, de Lara PT, Tugues S, Vermeer M, Montagnolo Y, Ohs I, et al. CSF1R-dependent myeloid cells are required for NK−mediated control of metastasis. JCI Insight. (2018) 3:e97792. doi: 10.1172/jci.insight.97792
208. Ray A, Hu KH, Kersten K, Courau T, Kuhn NF, Zaleta-Linares I, et al. Targeting CD206+ macrophages disrupts the establishment of a key antitumor immune axis. J Exp Med. (2024) 222:e20240957. doi: 10.1084/jem.20240957
209. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. (2016) 352:aad3018. doi: 10.1126/science.aad3018
210. Chen Z, Xie X, Jiang N, Li J, Shen L, and Zhang Y. CCR5 signaling promotes lipopolysaccharide-induced macrophage recruitment and alveolar developmental arrest. Cell Death Dis. (2021) 12:1–14. doi: 10.1038/s41419-021-03464-7
211. Chu X, Tian Y, and Lv C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol Cancer. (2024) 23:150. doi: 10.1186/s12943-024-02064-1
212. Wei C, Ma Y, Wang F, Liao Y, Chen Y, Zhao B, et al. Igniting hope for tumor immunotherapy: promoting the “Hot and cold” Tumor transition. Clin Med Insights Oncol. (2022) 16:11795549221120708. doi: 10.1177/11795549221120708
213. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
214. Iliopoulou EG, Kountourakis P, Karamouzis MV, Doufexis D, Ardavanis A, Baxevanis CN, et al. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol Immunother. (2010) 59:1781–9. doi: 10.1007/s00262-010-0904-3
215. Michel T, Poli A, Domingues O, Mauffray M, Thérésine M, Brons NHC, et al. Mouse lung and spleen natural killer cells have phenotypic and functional differences, in part influenced by macrophages. PloS One. (2012) 7:e51230. doi: 10.1371/journal.pone.0051230
216. Tang PCT, Chung JYF, Liao J, Chan MKK, Chan ASW, Cheng G, et al. Single-cell RNA sequencing uncovers a neuron-like macrophage subset associated with cancer pain. Sci Adv. (2022) 8:eabn5535. doi: 10.1126/sciadv.abn5535
217. Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Bühler L, et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1α Unleashes NK cell activity. Immunity. (2020) 52:1075–87. doi: 10.1016/j.immuni.2020.05.001
218. Tang F, Li J, Qi L, Liu D, Bo Y, Qin S, et al. A pan-cancer single-cell panorama of human natural killer cells. Cell. (2023) 186:4235–51. doi: 10.1016/j.cell.2023.07.034
219. Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. (2020) 15:1484–506. doi: 10.1038/s41596-020-0292-x
220. Browaeys R, Saelens W, and Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. (2020) 17:159–62. doi: 10.1038/s41592-019-0667-5
221. Meng L and Sun S. Single-cell RNA sequencing reveals the change in cytotoxic NK/T cells, epithelial cells and myeloid cells of the tumor microenvironment of high-grade serous ovarian carcinoma. Discov Oncol. (2024) 15:417. doi: 10.1007/s12672-024-01290-9
222. Larroquette M, Guegan JP, Besse B, Cousin S, Brunet M, Le Moulec S, et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J Immunother Cancer. (2022) 10:e003890. doi: 10.1136/jitc-2021-003890
223. Zhan T, Zou Y, Han Z, Tian X, Chen M, Liu J, et al. Single-cell sequencing combined with spatial transcriptomics reveals that the IRF7 gene in M1 macrophages inhibits the occurrence of pancreatic cancer by regulating lipid metabolism-related mechanisms. Clin Transl Med. (2024) 14:e1799. doi: 10.1002/ctm2.1799
224. Huang YK, Wang M, Sun Y, Di Costanzo N, Mitchell C, Achuthan A, et al. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat Commun. (2019) 10:3928. doi: 10.1038/s41467-019-11788-4
225. Rakina M, Larionova I, and Kzhyshkowska J. Macrophage diversity in human cancers: New insight provided by single-cell resolution and spatial context. Heliyon. (2024) 10. doi: 10.1016/j.heliyon.2024.e28332
226. De Zuani M, Xue H, Park JS, Dentro SC, Seferbekova Z, Tessier J, et al. Single-cell and spatial transcriptomics analysis of non-small cell lung cancer. Nat Commun. (2024) 15:4388. doi: 10.1038/s41467-024-48700-8
227. Park MD, Reyes-Torres I, LeBerichel J, Hamon P, LaMarche NM, Hegde S, et al. TREM2 macrophages drive NK cell paucity and dysfunction in lung cancer. Nat Immunol. (2023) 24:792–801. doi: 10.1038/s41590-023-01475-4
228. Kirschenbaum D, Xie K, Ingelfinger F, Katzenelenbogen Y, Abadie K, Look T, et al. Time-resolved single-cell transcriptomics defines immune trajectories in glioblastoma. Cell. (2024) 187:149–65. doi: 10.1016/j.cell.2023.11.032
229. Black S, Phillips D, Hickey JW, Kennedy-Darling J, Venkataraaman VG, Samusik N, et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat Protoc. (2021) 16:3802–35. doi: 10.1038/s41596-021-00556-8
230. Ptacek J, Locke D, Finck R, Cvijic ME, Li Z, Tarolli JG, et al. Multiplexed ion beam imaging (MIBI) for characterization of the tumor microenvironment across tumor types. Lab Invest. (2020) 100:1111–23. doi: 10.1038/s41374-020-0417-4
231. Choe K, Pak U, Pang Y, Hao W, and Yang X. Advances and challenges in spatial transcriptomics for developmental biology. Biomolecules. (2023) 13:156. doi: 10.3390/biom13010156
232. Yang W, Chen L, Zhang J, Qiu C, Hou W, Zhang X, et al. In-depth proteomic analysis reveals phenotypic diversity of macrophages in liver fibrosis. J Proteome Res. (2024) 23:5166–76. doi: 10.1021/acs.jproteome.4c00681
233. Liu Y, Liu D, Liu Y, Fu B, Ji S, Wang R, et al. Comprehensive proteomics analysis reveals dynamic phenotypes of tumor-associated macrophages and their precursor cells in tumor progression. J Proteome Res. (2024) 23:822–33. doi: 10.1021/acs.jproteome.3c00725
234. Arellano-Ballestero H, Zubiak A, Dally C, Orchard K, Alrubayyi A, Charalambous X, et al. Proteomic and phenotypic characteristics of memory-like natural killer cells for cancer immunotherapy. J Immunother Cancer. (2024) 12:e008717. doi: 10.1136/jitc-2023-008717
235. Choi JW, Lim S, Kang JH, Hwang SH, Hwang KC, Kim SW, et al. Proteome analysis of human natural killer cell derived extracellular vesicles for identification of anticancer effectors. Molecules. (2020) 25:5216. doi: 10.3390/molecules25215216
236. Sun C, Wang A, Zhou Y, Chen P, Wang X, Huang J, et al. Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat Commun. (2023) 14:2692. doi: 10.1038/s41467-023-38360-5
Keywords: macrophages, natural killer cells, immune crosstalk, cancer, immunotherapy
Citation: Palmer N, Khakoo S, Sanchez-Elsner T and Vallejo AF (2025) Enhancing natural killer cell anti-tumour activity through macrophage manipulation. Front. Immunol. 16:1656925. doi: 10.3389/fimmu.2025.1656925
Received: 30 June 2025; Accepted: 11 August 2025;
Published: 29 August 2025.
Edited by:
Italia Falcone, Regina Elena National Cancer Institute (IRCCS), ItalyReviewed by:
Cecilia Pesini Martín, University of Zaragoza, SpainAna Sami, Queen Mary University of London, United Kingdom
Abdalla Abdrabou, Chan Zuckerberg Biohub, United States
Copyright © 2025 Palmer, Khakoo, Sanchez-Elsner and Vallejo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salim Khakoo, Uy5JLktoYWtvb0Bzb3Rvbi5hYy51aw==; Andres F. Vallejo, YWZ2cDFmMTdAc290b24uYWMudWs=