 Feifei Wei1,2‡
Feifei Wei1,2‡ Hiroyuki Takeda3‡
Hiroyuki Takeda3‡ Koichi Azuma4‡
Koichi Azuma4‡ Yoshiro Nakahara5,6Yuka Igarashi2
Yoshiro Nakahara5,6Yuka Igarashi2 Kenta Murotani7Haruhiro Saito5
Kenta Murotani7Haruhiro Saito5 Shuji Murakami5Tetsuro Kondo5
Shuji Murakami5Tetsuro Kondo5 Taku Kouro1,2Hidetomo Himuro1,2
Taku Kouro1,2Hidetomo Himuro1,2 Kayoko Tsuji1,2Mitsuru Komahashi1,2,8Tatsuya Sawasaki9*†
Kayoko Tsuji1,2Mitsuru Komahashi1,2,8Tatsuya Sawasaki9*† Tetsuro Sasada1,2*†
Tetsuro Sasada1,2*†- 1Division of Cancer Immunotherapy, Kanagawa Cancer Center Research Institute, Yokohama, Kanagawa, Japan
- 2Cancer Vaccine and Immunotherapy Center, Kanagawa Cancer Center, Yokohama, Kanagawa, Japan
- 3Division of Proteo-Drug-Discovery Sciences, Proteo-Science Center, PIAS, Ehime University, Matsuyama, Ehime, Japan
- 4Department of Internal Medicine, Kurume University School of Medicine, Kurume, Fukuoka, Japan
- 5Department of Thoracic Oncology, Kanagawa Cancer Center, Yokohama, Kanagawa, Japan
- 6Department of Respiratory Medicine, Kitasato University School of Medicine, Sagamihara, Kanagawa, Japan
- 7Biostatistics Center, Kurume University School of Medicine, Kurume, Fukuoka, Japan
- 8Department of Pediatric Surgery, Nihon University School of Medicine, Tokyo, Japan
- 9Division of Cell-Free Sciences, Proteo-Science Center, PIAS, Ehime University, Matsuyama, Ehime, Japan
Introduction: Immune checkpoint inhibitors (ICIs) targeting the programmed death-1/ligand-1 (PD-1/PD-L1) axis have significantly improved treatment outcomes in non-small cell lung cancer (NSCLC); however, challenges remain owing to the limited durability of therapeutic responses and the occurrence of immune-related adverse events (irAEs). This study aimed to characterize dynamic changes in the circulating autoantibody (CAAB) profile during ICI treatment and explore their association with treatment outcomes in patients with NSCLC.
Methods: A panel of 59 CAABs showing substantial treatment-related changes was initially identified using AlphaScreen assays in a primary screening of five patients who developed ir-pneumonitis. These CAABs were subsequently profiled in paired pre-and post-treatment plasma samples obtained from 179 patients with NSCLC treated with anti-PD-1/PD-L1 therapy at two Japanese centers. Associations between CAAB dynamics and clinical parameters—including baseline characteristics, treatment regimens, and treatment outcomes (irAEs, ir-pneumonitis, response, progression-free survival [PFS], and overall survival [OS])—were evaluated using permutational multivariate analysis of variance and univariate binary logistic and Cox regression, elastic net regularization regression, and random forest regression.
Results: Using permutational multivariate analysis of variance and univariate binary logistic/Cox regression, we comprehensively assessed the global associations between CAAB dynamics and eight clinical parameters, including background factors (PD-L1 expression and treatment line), treatment regimens (chemotherapy exposure), and treatment outcomes (irAE occurrence, ir-pneumonitis development, RECIST-assessed response, PFS, and OS), indicating that chemotherapy exposure was the only significant and strong factor influencing CAAB dynamics. In patients receiving ICI monotherapy, univariate logistic or Cox regression analyses were performed to identify individual CAABs significantly associated with each outcome, highlighting both shared and distinct immunological features underlying different clinical endpoints. Through machine learning-based evaluation of the predictive potential of CAAB dynamics for five treatment outcomes across the overall cohort and six subgroups defined by three stratification variables, four optimized CAAB signatures with robust predictive performance for ICI treatment outcomes were established.
Conclusions: These findings suggest the involvement of distinct immune pathways in therapeutic benefits and toxicity. Collectively, our results provide mechanistic insights into ICI-induced humoral immune regulation, highlight the potential utility of CAABs as biomarkers to enhance benefit-to-risk assessment, and guide the development of personalized immunotherapy strategies for NSCLC.
1 Introduction
Although immune checkpoint inhibitors (ICIs) targeting the programmed death-1/ligand-1 (PD-1/PD-L1) axis have revolutionized the therapeutic paradigms for non-small cell lung cancer (NSCLC), two major clinical challenges remain. First, resistance limits the proportion of patients who are able to achieve a durable therapeutic response. Second, a spectrum of organ-specific inflammatory toxicities, known as immune-related adverse events (irAEs), further complicate treatment management (1, 2). These issues highlight the urgent need to elucidate the precise immunomodulatory mechanisms by which ICIs crosstalk with components involved in the host immune homeostasis. Comparative analysis of the dynamic alterations in the circulating autoantibody (CAAB) repertoire, which refers to the diversity and composition of autoantibodies present in peripheral blood, preceding and following ICI administration may yield novel mechanistic insights into treatment-induced immunomodulation. Thus, elucidating the CAAB repertoire may advance our understanding of the role of humoral immunity in both therapeutic efficacy and disruption of immune homeostasis.
Current evidence in cancer immunotherapy indicates that baseline autoantibody levels and treatment-induced antibody dynamics are associated with irAE development and therapeutic efficacy in ICI therapy (3–19). Notably, small-scale clinical studies have revealed that anti-PD-1 monotherapy induces patterns of circulating antibody/B-cell/plasmablast remodeling that are different those induced by anti-cytotoxic T-lymphocyte-associated antigen 4 (CTLA4)-containing ICI regimens, although the underlying mechanisms remain elusive (15, 20). Critical unknowns persist regarding (i) the dynamics of CAAB during PD-1/PD-L1 blockade in a large patient cohort and (ii) the existence and clinical relevance of associations between temporal autoantibody profile shifts and treatment options (monotherapy or combination therapy with chemotherapy), therapeutic response, irAE incidence, and severe complications such as pneumonitis.
In this study, we profiled 59 specific CAABs in pairs before and after treatment, using plasma samples from 179 patients who received anti-PD-1/PD-L1 therapy. By examining the antibody dynamics, we quantitatively assessed the associations between changes in the CAAB dynamic repertoire and clinical parameters, including baseline characteristics (PD-L1 expression and treatment line), treatment regimens (chemotherapy exposure), and treatment outcomes [irAE occurrence, ir-pneumonitis development, response, progression-free survival (PFS), and overall survival (OS)] (Figure 1A). These findings provide novel insights into the systemic immunomodulatory effects of the PD-1/PD-L1 blockade and potential autoantibody targets for disentangling therapeutic benefits from associated risks, thereby informing benefit-to-risk assessments and development of novel adjunctive strategies.
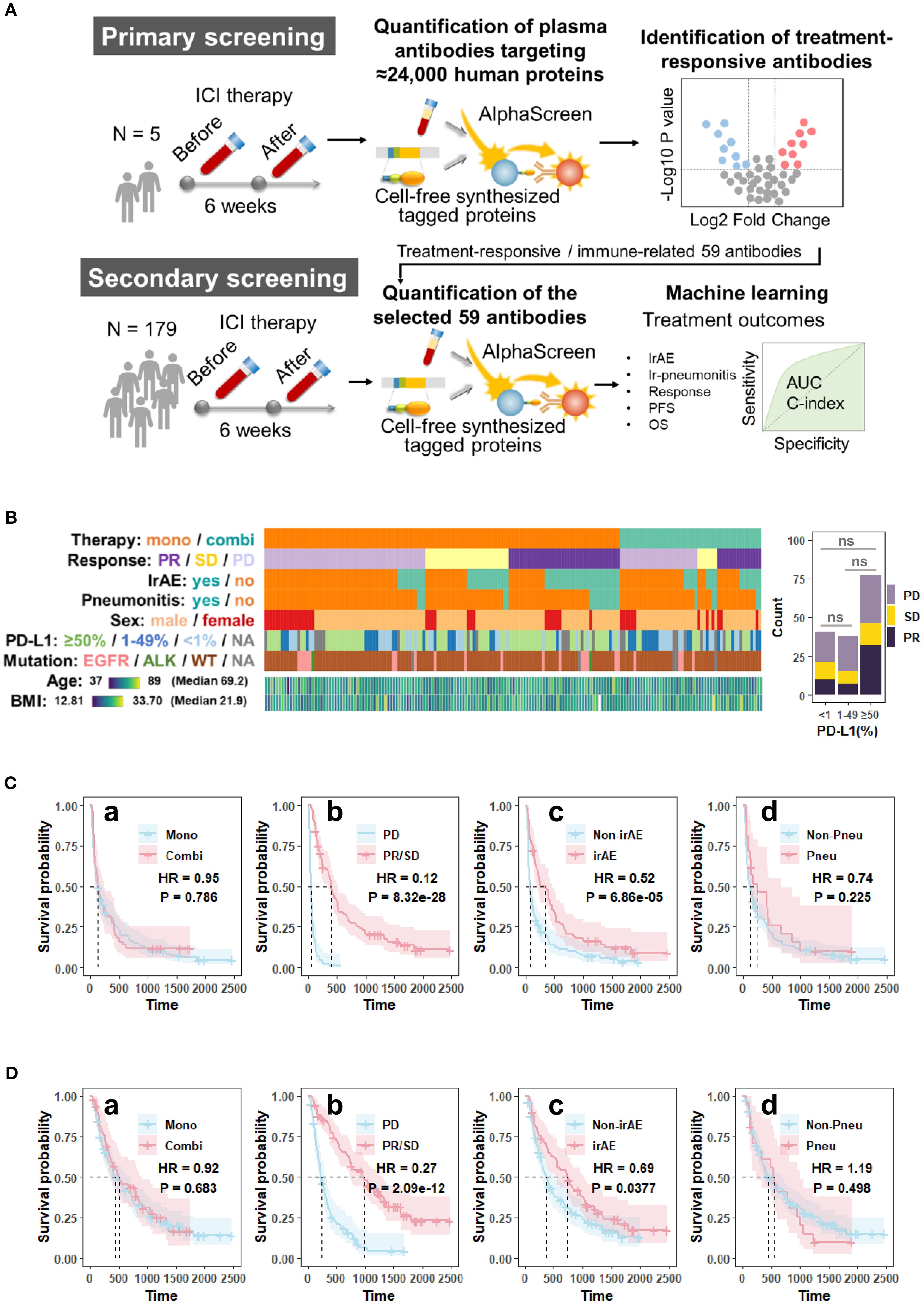
Figure 1. Overview of (A) the study design; (B) patient demographics, clinical characteristics, and the association between PD-L1 expression and treatment response; (C) Kaplan-Meier survival curves for progression-free survival (PFS); and (D) overall survival (OS) of the cohort (n = 179). The survival curves were generated using Cox regression based on different grouping criteria: a treatment option, b best clinical response, c irAE, and d ir-pneumonitis. ALK, anaplastic lymphoma kinase gene; AUC, area under the receiver operating characteristic curve; BMI, body mass index; Combi, combination therapy; EGFR, epidermal growth factor receptor gene; HR, hazard ratio in the Cox proportional hazards model; ICI, immune checkpoint inhibitor; irAE, immune-related adverse event; Mono, monotherapy; ns, not significant (p > 0.05, Chi-square test); OS, overall survival; P, p-value in the Cox proportional hazards model; PD, progressive disease; PD-L1, programmed death-ligand 1; PFS, progression-free; Pneu, ir-pneumonitis; survival; PR, partial response; SD, stable disease; WT, wild type.
2 Materials and methods
2.1 Patients and data collection
This study included patients diagnosed with advanced, recurrent, or metastatic NSCLC who received anti-PD-1 (nivolumab or pembrolizumab) or anti-PD-L1 therapy (atezolizumab), either as monotherapy or in combination with chemotherapy, at Kurume University Hospital (Kurume, Japan) and Kanagawa Cancer Center (Yokohama, Japan). The patient cohort partially overlapped with that of a previous study conducted by our group (21, 22). The cohort consisted of 179 patients enrolled between February 2016 and August 2019. The clinical course of the enrolled patients was followed until July 2024. The patient characteristics are summarized in Figures 1B–D, and Table 1. Tumor PD-L1 expression was assessed via immunohistochemical staining of paraffin-embedded tumor sections using anti-PD-L1 monoclonal antibodies (clone E1L3N: Cell Signaling Technology, Danvers, MA, USA and clone 22C3: Agilent Technologies/Dako, Carpinteria, CA, USA). For most patients, PD-L1 expression was evaluated in tumor specimens collected prior to first-line therapy. This analysis was also done in patients who received ICIs as second- or subsequent-line treatment. Clinical response was evaluated based on the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. IrAEs were defined according to the Common Terminology Criteria for Adverse Events (CTCAE) v5.0 grading scale. This study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Institutional Review Boards of Kurume University Hospital (approval numbers: 15210 and 19240) and Kanagawa Cancer Center (approval number: 28-85). Informed consent was obtained from all enrolled patients after the nature of the study and its possible consequences were explained.
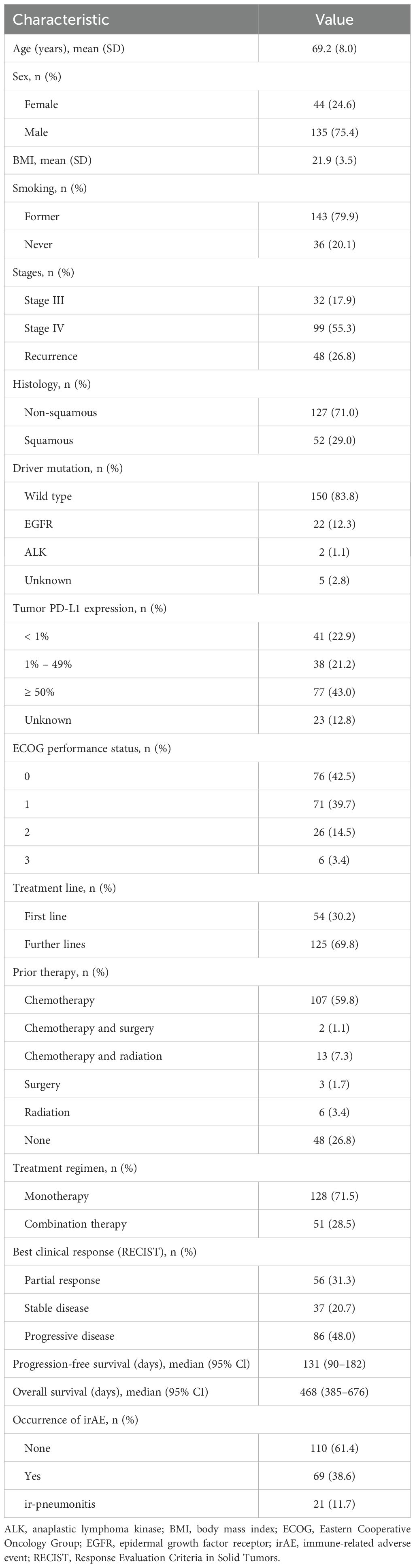
Table 1. Summary of patient characteristics in the study.
2.2 Plasma antibody profiling
Peripheral blood samples were collected in heparin-coated tubes before and 6 weeks after therapy initiation. Plasma was separated from whole blood via centrifugation and stored at −80 °C until analysis. Plasma autoantibodies were assessed via the AlphaScreen assay using two human protein arrays generated using a wheat germ cell-free protein production system (23, 24). The HuPEX protein array, containing 19,713 human proteins, was purchased from CellFree Science Co., Ltd. (Yokohama, Japan). The Ehime-Kazusa protein array, consisting of 4,144 human proteins, was prepared in-house using a cell-free protein synthesis reagent. Gene resources for the protein array were provided by the Kazusa DNA Research Institute (Kisarazu, Japan) (25). Each cDNA clone was subcloned into the pEU-E01-FLAG-GST-K1–02 vector (26). In vitro transcription and translation were performed using the WEPRO7240 Expression Kit (CellFree Science) as previously described (27, 28). Briefly, the DNA fragments coding for each protein were amplified via PCR and used as templates for in vitro transcription. The mRNA generated by in vitro transcription was used as a template for in vitro translation (total reaction mixture, 5 µL; components, 2.5 µL mRNA; 1.67 µL WEPRO 7240 wheat germ extract; 0.14 µL creatine kinase [20 mg/mL]; and 0.11 µL RNase inhibitor) was layered below 50 µL SUB-AMIX SGC substrate solution in a 384-well plate and incubated at 20°C for 18 h. The translated protein arrays were diluted 2-fold with AlphaScreen buffer (100 mM Tris-HCl, pH 8.0; 0.01% Tween 20; 1 mg/mL BSA), aliquoted into 384-well plates, flash-frozen in liquid nitrogen, and stored at −80°C.
The AlphaScreen assay was performed as previously described, with minor modifications (27). Specificity was achieved through two independent high-affinity binding steps (1): glutathione-coated donor beads that selectively bind GST-tagged recombinant antigens, and (2) Protein G–conjugated acceptor beads that specifically recognize human IgG, thereby minimizing nonspecific interactions. For primary screening, autoantibody reactivity was assessed after incubating 10 paired plasma samples from patients with NSCLC (five pre- and post-ICI treatment, respectively) with a library of 23,857 human proteins. Plasma samples were diluted 1:667 in AlphaScreen reaction buffer (100 mM Tris-HCl, pH 8.0, 0.01% [v/v] Tween 20, 0.1% [w/v] bovine serum albumin), and 20 µL diluted plasma was dispensed into each well of OptiPlate-384 plates (Revvity, Yokohama, Japan) using a Liquidator96 pipetting system. Next, 1 µL of each FLAG-GST tagged protein was transferred from 384-well stock plate to the reaction plate using JANUS automated dispensing workstation equipped with a NanoHead 384-channel microsyringe head (Revvity). Subsequently, 9 µL detection mixture (containing 0.06 µL AlphaScreen GSH Donor Beads [Revvity] and 0.06 µL Protein G-conjugated AlphaScreen acceptor beads in reaction buffer) was added to the reaction plates with a FlexDrop dispenser (27). After incubation at 25°C for 1 h in the dark, AlphaScreen signals were detected using an EnVision plate reader (Revvity).
For secondary screening, 358 paired plasma samples from 179 patients with NSCLC (pre- and post-treatment) were tested for the 59 selected proteins, each in quadruplicate. All reactions were performed on AlphaPlate-1536 plates (Revvity). Three microliters of protein dilution containing 0.05 µL FLAG-GST tagged protein was dispensed into an AlphaPlate 1536 plate (Revvity) via a Multidrop Combi nL (Thermo Fisher Scientific, Tokyo, Japan). All 358 plasma samples were diluted 1:40 and dispensed into a 384 well plate. Subsequently, 0.2 µL of the 1:40 diluted plasma from each well of these 384-well plates was dispensed into the 1536-well reaction plates via a JANUS workstation with a NanoHead. Finally, 1.8 µL detection mixture (containing 0.01 µL AlphaScreen GSH Donor Beads and 0.01 µL Protein G-conjugated AlphaScreen acceptor beads) was added to the reaction plates via a Multidrop Combi nL. After incubation at 25°C for 1 h in the dark, AlphaScreen signals were detected with an EnVision plate reader. The secondary screening minimized inter-plate variability by ensuring that paired plasma samples (pre- and post-ICI) from the same patient were analyzed on the same plate. To further minimize experimental variability, all assays were conducted on a single 1536-well plate uniformly coated with the same antigen. Plasma samples from all patients were dispensed in quadruplicate. This design enabled direct assessment of treatment-induced changes both within individual patients and across the cohort, while controlling for inter-plate variability.
2.3 Machine learning and statistical analysis
All analyses were performed using R (version 4.4.1; https://www.r-project.org). Univariate binary logistic and Cox proportional hazards regression analyses were performed using the glm function (R Basic package). P-values were derived using Wald z-tests and subsequently adjusted for multiple comparisons using the Benjamini–Hochberg false discovery rate (FDR) procedure. To mitigate overfitting in multivariate predictive modeling, we applied elastic net (EN) regularized regression and random forest (RF) regression for binary outcome variables (irAE, ir-pneumonitis, and treatment response), and Cox proportional hazards regression with elastic net regularization (EN-Cox) and random survival forest (RSF) for survival outcomes (PFS and OS) within a machine learning framework (21). The cohort was randomly stratified into training (80%) and validation (20%) groups. Model development and hyperparameter optimization were conducted using nested cross-validation (CV) exclusively on the training set. The final models were subsequently retrained on the entire training set and independently evaluated on the validation set, maintaining a strict separation between the development and evaluation datasets to prevent data leakage. For EN regression, hyperparameter tuning (α = 0.5 for balanced L1/L2 regularization) was performed using 10-fold CV via the cv.glmnet implementation (glmnet package), optimizing the regularization parameter λ to minimize classification error. The λ value yielding minimum CV error was selected for final model training, followed by external validation on the holdout set. For the RF regression, feature selection was performed via sequential variable inclusion based on importance rankings, with variable importance assessed using the mean Gini index (randomForest package). The optimal feature subset (important factors) was identified by maximizing predictive accuracy during stepwise variable addition. Model performance was assessed using leave-one-out cross-validation (LOO-CV) of the training set with discriminatory power quantified by the area under the receiver operating characteristic curve (AUC). All RF implementations utilized an ensemble of 1,000 decorrelated decision trees with the minimum node purity set to 1, following established methodologies in high-dimensional regression (29). Optimization of the model cutoff value was performed by maximizing Youden’s J statistic (sensitivity + specificity - 1), implemented through the coords function in R. Permutational multivariate analysis of variance (PERMANOVA) was performed using the Adonis function (vegan package). Nonparametric comparisons were performed using the Wilcoxon signed-rank test via the wilcox.test function. Survival analyses included Kaplan–Meier curve generation with log-rank testing, alongside Cox proportional hazards modeling, executed using the survival and survminer packages in R.
3 Results
3.1 Patient characteristics
As shown in Figure 1 and Table 1, of the 179 patients with NSCLC included in this study, 128 (71.5%) received anti-PD-1/PD-L1 monotherapy and 51 (28.5%) received combination therapy with chemotherapy. The best overall response was partial response (PR) in 56 patients (31.3%), stable disease (SD) in 37 patients (20.7%), and progressive disease (PD) in 86 patients (48.0%). IrAEs occurred in 69 patients (38.6%) and ir-pneumonitis was observed in 21 patients (11.7%). The median PFS in the cohort was 131 days and the median OS was 468 days. Figure 1C illustrates PFS across the clinical subgroups. No significant differences in PFS were observed between patients who received combination therapy and those who received monotherapy (hazard ratio [HR] = 0.95, p = 0.786). Patients in the responder group (PR + SD) demonstrated significantly prolonged PFS compared to those in the PD group (HR = 0.12, p < 0.001). Similarly, patients who developed irAEs had a longer PFS than those who did not (HR = 0.52, p < 0.001). However, patients with ir-pneumonitis did not show a significant difference in PFS compared to those without ir-pneumonitis (HR = 0.74, p = 0.225). Figure 1D shows OS data across the same subgroups. Consistent with the PFS results, the OS did not differ significantly between the ICI monotherapy and combination therapy (HR = 0.92, p = 0.683) groups. The responder group exhibited significantly improved OS compared with the PD group (HR = 0.27, p < 0.001). Additionally, patients with irAEs had a longer OS than those without irAEs (HR = 0.69, p = 0.0377). In contrast, patients who developed ir-pneumonitis showed no OS benefit from treatment (HR = 1.19, p = 0.498).
3.2 Circulating autoantibody titers exhibit a predominant decrease during PD-1/PD-L1 blockade
For primary screening, we used AlphaScreen to quantify plasma IgG antibodies against approximately 24,000 biotinylated proteins in pre- and post-treatment plasma from five patients with NSCLC who developed ir-pneumonitis during ICI treatment. Based on pre/post signal ratios, 37 antibodies showed significant changes (p < 0.05, Student’s t-test; fold changes < 0.7 or > 1.4). To offset the limited sample size, 22 additional antibodies were included due to notable fold changes and prior associations with immune-related pathways, cancer biology, or ICI responses reported in the literature. In total, 59 antibodies were selected for further analysis (Supplementary Table S1).
In secondary screening, these 59 candidates were quantified in pre- and post-treatment plasma from a formal cohort of 179 patients. A volcano plot (Figure 2A) illustrates changes in CAAB levels before and after anti-PD-1/PD-L1 therapy: 53 decreased (average log2-transformed fold-change < 0), 30 significantly (p < 0.05), including BASP1, ITGAE, CPB1, and CD200 autoantibodies. Six increased (average log2-transformed fold-change > 0), with two autoantibodies against CASP10 and SCGN reached statistical significance.
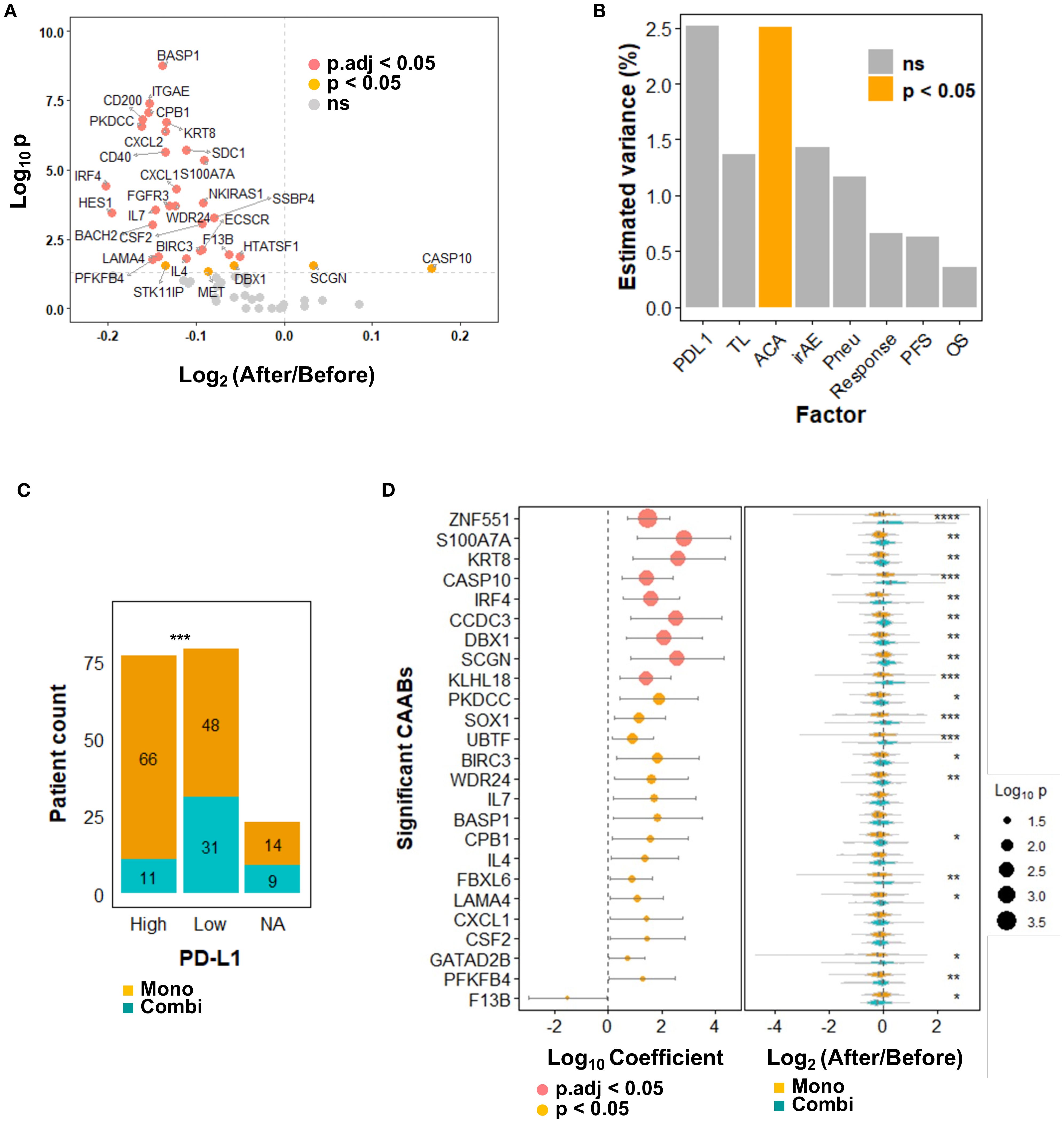
Figure 2. Dynamics of 59 protein-specific circulating autoantibodies (CAABs) before and after ICI therapy (n = 179). (A) Volcano plot illustrating differential antibody responses: Wilcoxon signed-rank test p-values; p.adj indicates false discovery rate (FDR) correction using the Benjamini-Hochberg method. (B) PERMANOVA analysis quantifying inter-individual variation in CAAB dynamics explained by background factors (PD-L1 expression and treatment line), treatment regimens (monotherapy vs. combination therapy), and treatment outcomes (irAE occurrence, ir-pneumonitis development, RECIST-assessed response, PFS, and OS). Chemotherapy exposure emerged as the only significant (p < 0.05) modulator of CAAB dynamics among ICI-related clinical parameters; no factor showed p.adj < 0.05. (C) Association between PD-L1 expression (≥ 50%: High; < 50%: Low) and treatment options in the present cohort: Chi-square test p-values; ***p < 0.001. (D) Univariate logistic regression evaluating chemotherapy-associated CAABs: Wald test p-values; all significant CAABs (p < 0.05) are displayed; p.adj indicates Benjamini-Hochberg-adjusted FDR. Violin plots demonstrate group-wise comparisons using Wilcoxon rank-sum test significance levels: ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05. ACA, anticancer agent; Combi, combination therapy; FDR, false discovery rate; ICI, immune checkpoint inhibitor; irAE, immune-related adverse event; Mono, monotherapy; NA, not available; ns, not significant; OS, overall survival; PERMANOVA, permutational multivariate analysis of variance; PFS, progression-free survival; Pneu, ir-pneumonitis; TL, treatment line.
3.3 Chemotherapy exposure as the predominant determinant of circulating autoantibody repertoire changes
We calculated the log2-transformed fold-change for each CAAB, designating this dataset as the dynamic repertoire. Clinical relevance of the CAAB dynamic repertoire by computing multivariate effect sizes for treatment regimen, treatment line, PD-L1 expression, occurrence of irAE and ir-pneumonitis, therapeutic response, PFS, and OS using PERMANOVA (30). As shown in Figure 2B, the CAAB dynamic repertoire exhibited a significant association only with chemotherapy exposure among all clinical factors assessed (highest R² = 2.5%, p < 0.05), indicating marked sensitivity to anticancer agent administration. Regarding clinical baseline characteristics, CAAB dynamics demonstrated a stronger effect size with PD-L1 expression than with the line of therapy, although without significance. In the present cohort, the therapeutic decisions (monotherapy versus combination therapy) were directly influenced by PD-L1 expression (Figure 2C) (31). Among the five treatment outcome measures—irAEs, ir-pneumonitis, RECIST-assessed response, PFS, and OS—the effect size of the association with CAAB dynamics decreased in the following order: irAEs, ir-pneumonitis, response, PFS, and OS, although none reached statistical significance (Figure 2B). To further elucidate which CAABs exhibited the strongest association with concomitant anticancer agent administration during ICI therapy, we performed univariate logistic regression analyses stratified by anticancer agent use. CAABs with a significance threshold of p < 0.05 in the regression analyses are summarized in Figure 2D. Nine CAABs retained statistical significance after FDR correction: antibodies against ZNF551, S100A7A, KRT8, CASP10, IRF4, CCDC3, DBX1, SCGN, and KLHL18. Several other CAABs exhibited notable associations, including PKDCC, SOX1, UBTF, BIRC3, WDR24, IL7, BASP1, CPB1, IL4, FBXL6, LAMA4, CXCL1, CSF2, GATAD2B, PFKFB4, and F13B. Plasma levels of these autoantibodies were significantly modulated by anticancer agent coadministration. Violin plots (Figure 2D) illustrate distinct expression patterns. ZNF551-targeting CAABs decreased after monotherapy but increased with combination therapy. Similarly, S100A7A and KRT8 CAABs decreased with monotherapy, but remained stable before and after combination therapy. CASP10 autoantibody levels increased in both groups, albeit less markedly in monotherapy.
3.4 Circulating autoantibody dynamics associated with treatment outcomes under anti-PD-1/PD-L1 monotherapy
Through the above analyses, we identified that the treatment regimen was the primary significant factor influencing the dynamic repertoire of CAAB during ICI therapy. Given the non-negligible influence of anticancer agents, we further examined the association between CAAB fluctuation patterns and treatment outcomes in a subgroup of patients receiving ICI monotherapy. To identify the autoantibodies specifically associated with each treatment outcome (irAEs, ir-pneumonitis, treatment response, PFS, and OS), we performed individual univariate logistic/Cox regression analyses for each factor. Figure 3A shows that in patients receiving ICI monotherapy, CAABs targeting DBX1, BIRC3, BASP1, STAT4, and SPATC1L were significantly associated with the occurrence of irAEs. The irAE-positive subgroup exhibited more pronounced post-treatment reductions in autoantibody titers than the controls. Figure 3B presents the univariate regression analyses of pneumonitis-associated CAABs, revealing significant correlations for autoantibodies targeting CXCL2, ROPN1, SPATC1L, FURIN, and DBX1. Consistent with the irAE pattern, patients with ir-pneumonitis showed greater treatment-related decreases in CAABs, with CXCL2 showing the greatest reduction. Regarding the therapeutic efficacy, response-associated CAABs included autoantibodies against SNCA, CCDC3, ECSCR, STAT4, and BIRC3 (Figure 3C). The treatment responders displayed a significantly greater absolute reduction in CAAB levels after ICI administration. PFS- and OS-associated CAABs included autoantibodies against F13B, ECSCR, and CXCL1 (Figures 3D, E). The Venn diagram in Figure 3F illustrates both shared and unique CAABs across different treatment outcomes. Autoantibodies against BIRC3 and STAT4 were common to irAEs and treatment responses, whereas SPATC1L and DBX1 were associated with both irAEs and ir-pneumonitis. Treatment response was characterized by distinct autoantibodies targeting SNCA, CCDC3, and ECSCR, with ECSCR additionally shared with PFS. Autoantibodies against CXCL1 were specifically associated with OS, while ir-pneumonitis showed unique associations with CXCL2, ROPN1, and FURIN, highlighting the differential CAAB dynamics underlying these outcomes.
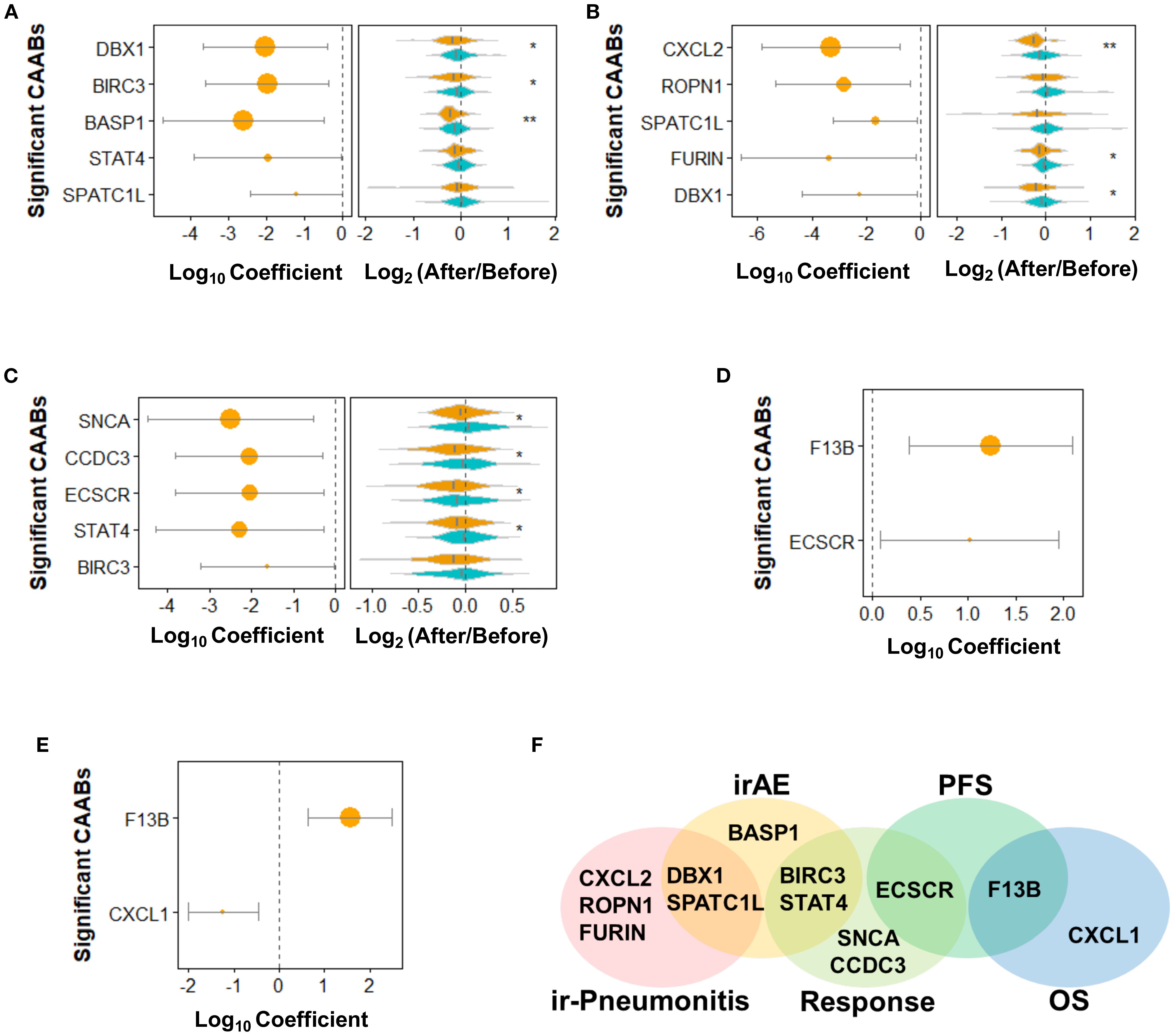
Figure 3. Dynamics of 59 protein-specific circulating autoantibodies (CAABs) during ICI therapy in patients receiving monotherapy (n = 128). Univariate logistic regression was used to evaluate (A) irAE-, (B) ir-pneumonitis-, and (C) RECIST-assessed response-associated CAABs; univariate Cox regression was used to evaluate (D) PFS- and (E) OS-associated CAABs. All significant CAABs (p < 0.05, Wald test) are displayed; no CAABs showed. p.adj < 0.05 after Benjamini-Hochberg FDR adjustment. Violin plots demonstrate group-wise comparisons using Wilcoxon rank-sum test significance levels: **p < 0.01; *p < 0.05. (F) Summary Venn diagram of CAABs significantly associated with irAE, ir-pneumonitis, response, PFS, and OS. FDR, false discovery rate; ICI, immune checkpoint inhibitor; irAE, immune-related adverse event.
3.5 Machine learning for prediction of treatment outcomes using circulating autoantibody dynamics
Next, we assessed the potential of CAAB as a predictive biomarker for treatment outcomes associated with anti-PD-1/PD-L1–based ICI therapy. The predictive performance of CAAB dynamics for five treatment outcomes across the overall cohort and six subgroups defined by three stratification variables is summarized in Table 2. As shown, models using the entire cohort without stratification demonstrated limited predictive accuracy. In contrast, stratified analyses revealed notable performance: for irAE prediction, the EN model in the high PD-L1 subgroup attained AUCs of 0.81 and 0.68 for the training and test sets, respectively (Figure 4); for ir-pneumonitis prediction, the RF model in the monotherapy subgroup achieved AUCs of 0.83 and 0.88 (Figure 5); for response prediction, the EN model in the combination therapy subgroup exhibited AUCs of 0.90 and 0.79; and in the first-line treatment subgroup, the RF model achieved AUCs of 0.79 and 0.89 (Figure 6). By contrast, the predictive capacity of CAAB dynamics for PFS and OS remained limited.
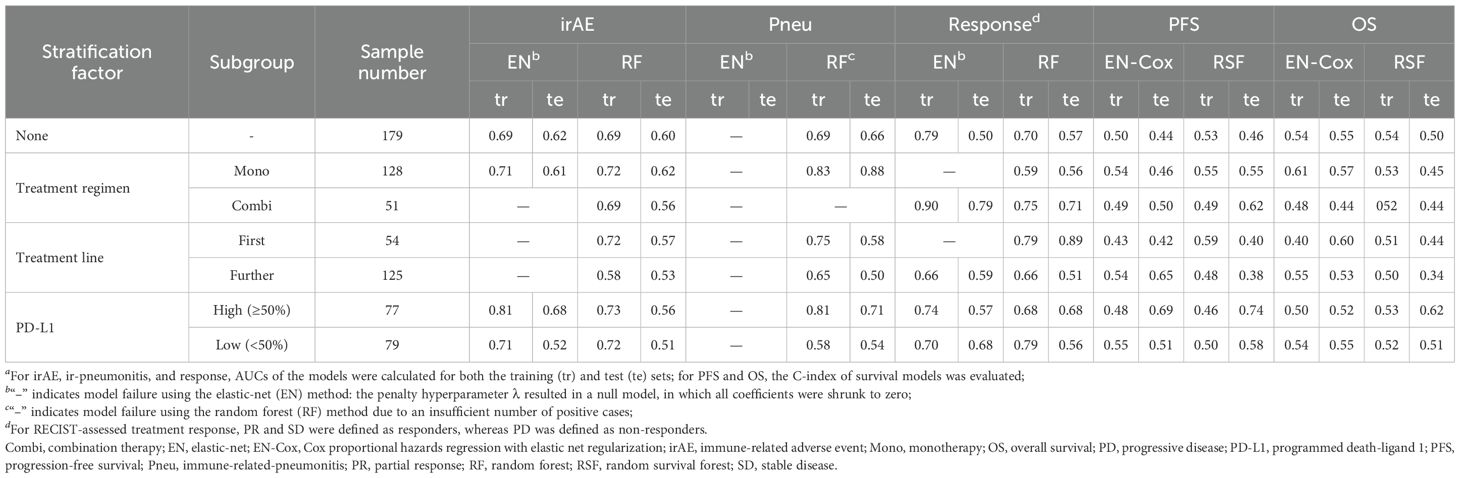
Table 2. Predictive performance of circulating autoantibody (CAAB) dynamics for five treatment outcomes overall and in six subgroups defined by three stratification conditions.a
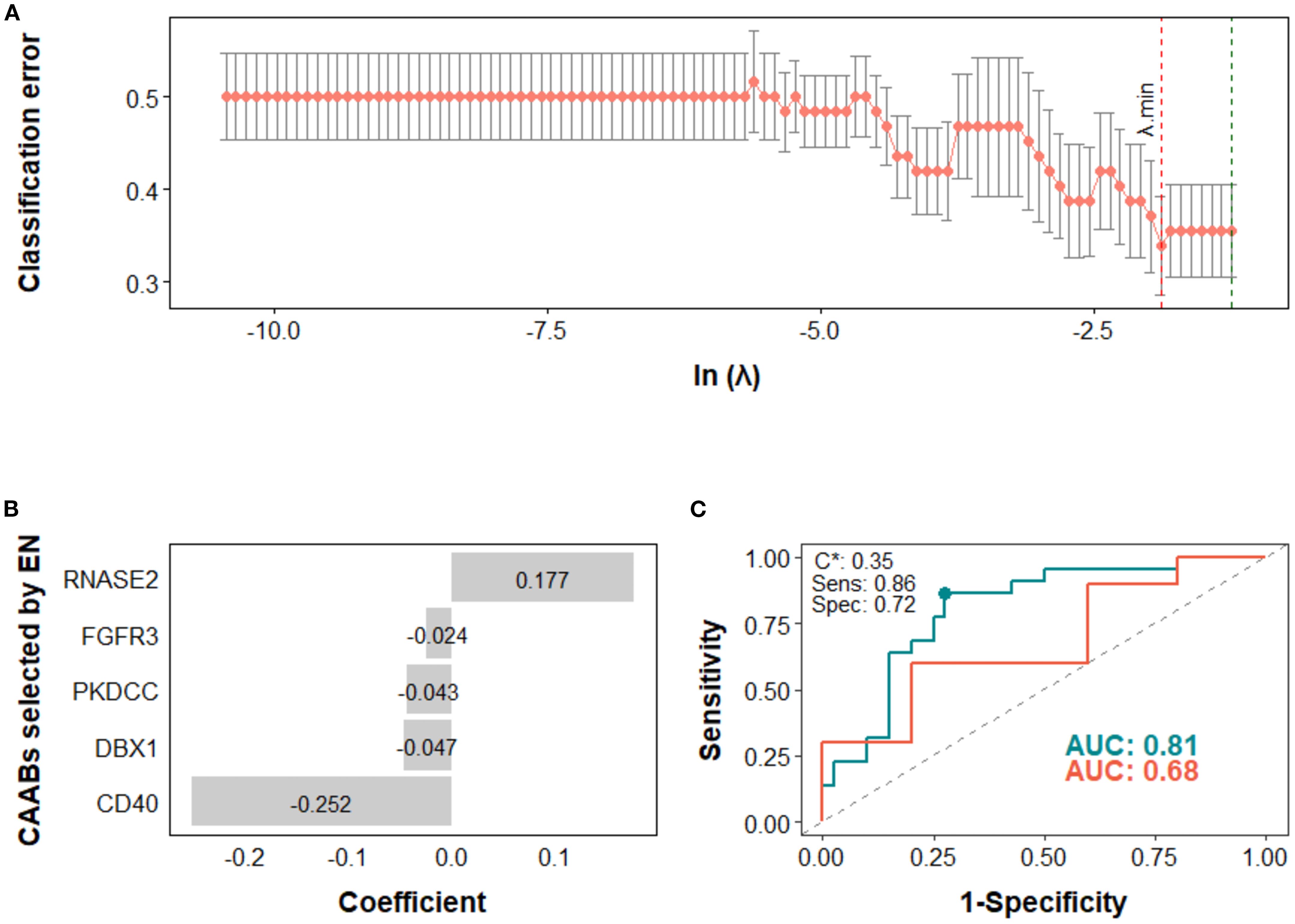
Figure 4. Predictive modeling of irAE occurrence using dynamic profiles of circulating autoantibodies (CAABs) in the high PD-L1 subgroup (n = 77). In the elastic-net (EN) regression-based models: (A) the penalty hyperparameter (λ) was determined through 10-fold cross-validation (CV) across a range of λ values; the optimal λ (λ.min) corresponding to the minimum CV classification error was selected. (B) CAABs selected by EN at λ.min. (C) ROC curves derived from the training and test sets, with AUCs of 0.81 and 0.68, respectively; the model threshold (C*) was optimized using the maximum Youden’s J statistic, yielding a sensitivity of 0.86 and specificity of 0.72. AUC, area under the receiver operating characteristic curve; CV, cross-validation; EN, elastic-net; ROC, receiver operating characteristic curve; Sens, sensitivity; Spec, specificity.
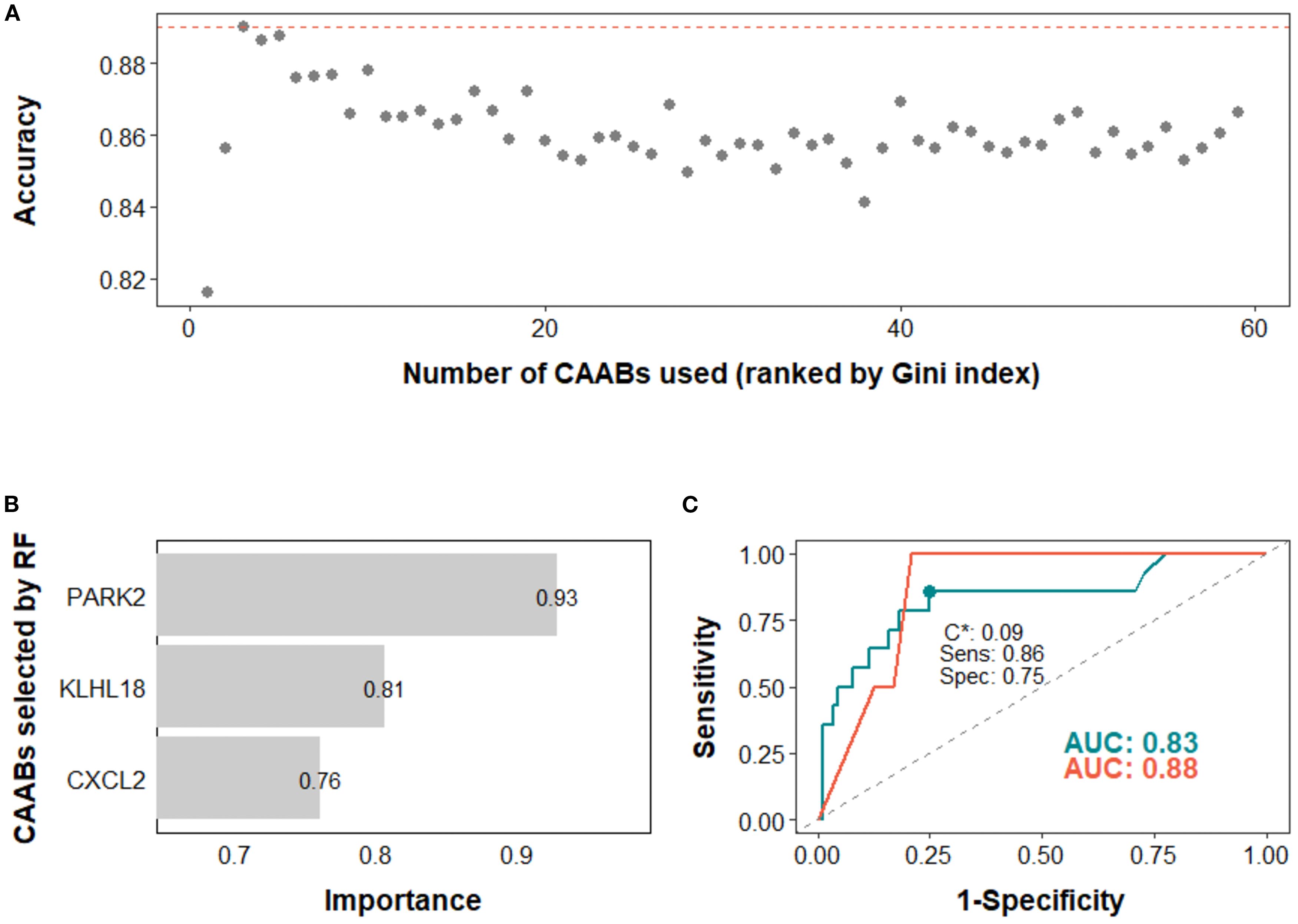
Figure 5. Predictive modeling of ir-pneumonitis occurrence using dynamic profiles of circulating autoantibodies (CAABs) in the monotherapy subgroup (n = 128). In the random forest (RF)-based models: (A) predictive accuracy during stepwise addition of CAABs according to their importance ranked by the Gini index; the RF model using the top three CAABs achieved the highest predictive accuracy. (B) CAABs selected by the RF model. (C) ROC curves derived from the training and test sets, with AUCs of 0.83 and 0.88, respectively; the model threshold (C*) was optimized using the maximum Youden’s J statistic, yielding a sensitivity of 0.86 and a specificity of 0.75. AUC, area under the receiver operating characteristic curve; RF, random forest; ROC, receiver operating characteristic curve; Sens, sensitivity; Spec, specificity.
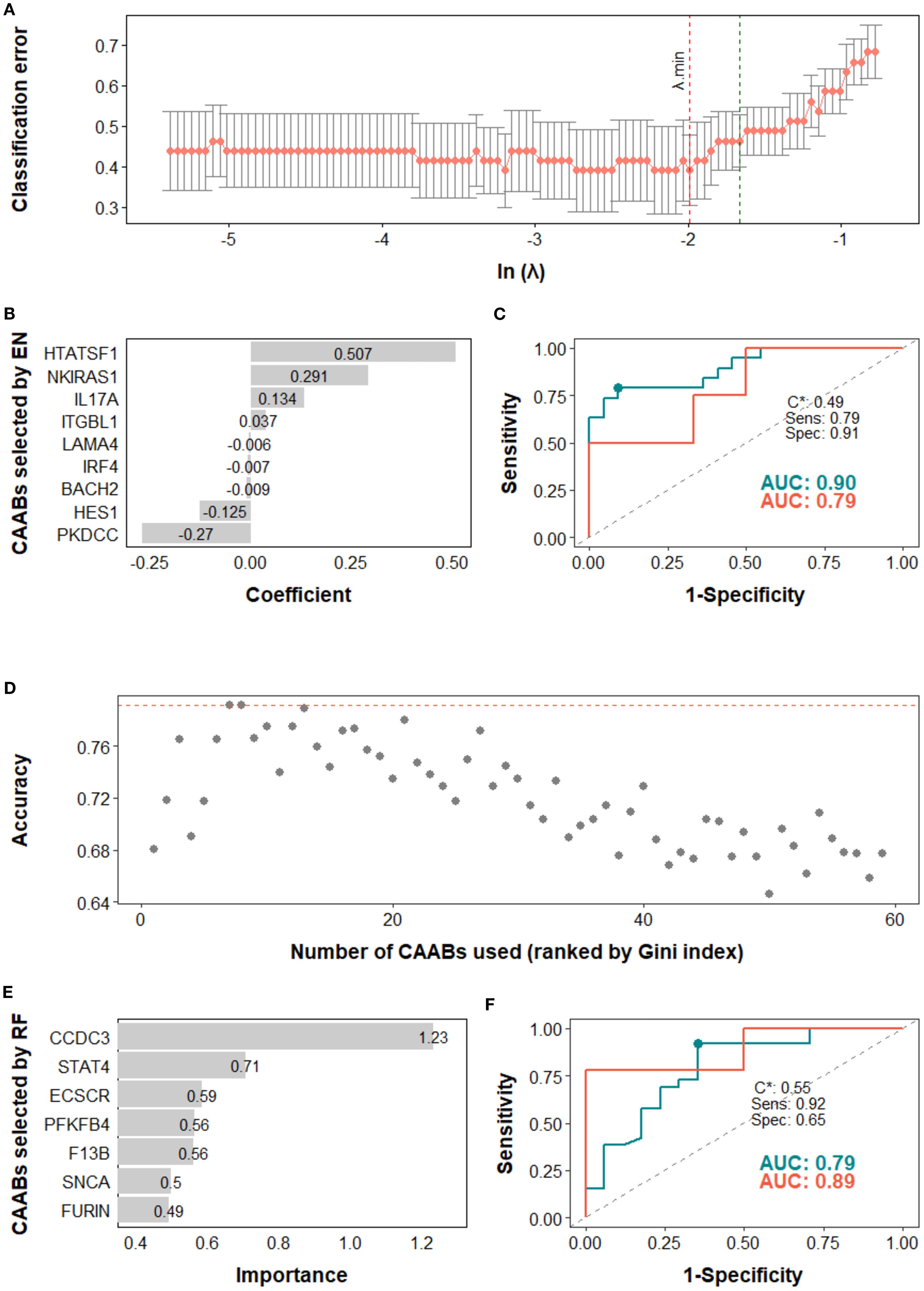
Figure 6. Predictive modeling of treatment response using dynamic profiles of circulating autoantibodies (CAABs) in the (A–C) combination therapy subgroup (n = 51) and (D–F) first line subgroup (n = 54). In the elastic-net (EN) regression-based models of combination therapy subgroup: (A) the penalty hyperparameter (λ) was determined through 10-fold cross-validation (CV) across a range of λ values; the optimal λ (λ.min) corresponding to the minimum CV classification error was selected. (B) CAABs selected by EN at λ.min. (C) ROC curves derived from the training and test sets, with AUCs of 0.90 and 0.79, respectively; the model threshold (C*) was optimized using the maximum Youden’s J statistic, yielding a sensitivity of 0.79 and specificity of 0.91. In the random forest (RF)-based models of first line subgroup: (D) predictive accuracy during stepwise addition of CAABs according to their importance ranked by the Gini index; the RF model using the top seven CAABs achieved the highest predictive accuracy. (E) CAABs selected by the RF model. (F) ROC curves derived from the training and test sets, with AUCs of 0.79 and 0.89, respectively; the model threshold (C*) was optimized using the maximum Youden’s J statistic, yielding a sensitivity of 0.92 and a specificity of 0.65. AUC, area under the receiver operating characteristic curve; CV, cross-validation; EN, elastic-net; RF, random forest; ROC, receiver operating characteristic curve; Sens, sensitivity; Spec, specificity.
In the high PD-L1 subgroup, the EN model selected five CAABs (RNASE2, FGFR3, PKDCC, DBX1, and CD40). The optimized cutoff value of 0.35 yielded a specificity of 0.86 and a sensitivity of 0.72 for predicting irAEs (Figure 4). the RF model with the top three features (PARK2, KLHL18, CXCL2) achieved the best performance, with a cutoff of 0.09 (specificity 0.86, sensitivity 0.75) for predicting ir-pneumonitis (Figure 5). For treatment response in the combination therapy subgroup, the EN model highlighted nine CAABs (HTATSF1, NKIRAS1, IL17A, ITGBL1, LAMA4, IRF4, BACH2, HES1, PKDCC), with a cutoff of 0.49 (specificity 0.79, sensitivity 0.91) (Figures 6A–C). In the first-line treatment subgroup, the RF model selected seven CAABs (CCDC3, STAT4, ECSCR, PFKFB4, F13B, SNCA, FURIN), with a cutoff of 0.55 (specificity 0.92, sensitivity 0.65) (Figures 6D–F). Both models demonstrated favorable predictive performance for treatment response within their respective subgroups.
These results indicate that patient background and treatment options distinctly shape CAAB dynamics, and that capturing subset-specific dynamic signatures may enable effective prediction of treatment outcomes across different clinical contexts or therapeutic regimens.
4 Discussion
Characterizing the dynamic changes in CAAB profiles during ICI therapy provides critical insights into the immunomodulatory effects of ICIs on host immunity. In this study, we initially identified 59 autoantibodies that exhibited substantial alterations during anti-PD-1/PD-L1 treatment in a primary screening of peripheral blood samples from five patients with NSCLC who developed ir-pneumonitis following ICI therapy. These candidate CAABs were subsequently quantified in plasma samples from an expanded cohort of 179 patients. PERMANOVA was applied to comprehensively assess the global associations between CAAB dynamics and eight clinical parameters, including background factors (PD-L1 expression and treatment line), treatment regimens (chemotherapy exposure), and treatment outcomes (irAE occurrence, ir-pneumonitis development, RECIST-assessed response, PFS, and OS). For patients receiving ICI monotherapy, univariate logistic or Cox regression analyses were performed to identify individual CAABs significantly associated with each treatment outcome factor. Finally, within a machine learning framework incorporating rigorous robustness evaluations, we examined the predictive potential of CAAB dynamics for five treatment outcomes in the overall cohort and across six subgroups defined by three stratification variables, and established four optimized CAAB signatures relevant to ICI treatment outcomes. Among all clinical factors assessed, the CAAB dynamic repertoire showed a significant association solely with chemotherapy exposure, highlighting its pronounced sensitivity to anticancer drug administration. Several mechanisms may explain this phenomenon. For example, cytotoxic anticancer agents can induce immunogenic cell death, such as apoptosis and necrosis, in both tumor and healthy cells, resulting in the release of self-antigens. This antigen release may enhance antigen presentation by antigen-presenting cells and promote a pro-inflammatory environment, ultimately contributing to the breakdown of immune tolerance and activation of autoreactive lymphocytes. In addition, some anticancer agents have been reported to reduce immunosuppressive cell populations, including regulatory T cells and myeloid-derived suppressor cells (MDSCs), thereby facilitating the production of autoantibodies (32). CAAB dynamics also demonstrated a relatively strong, albeit non-significant, correlation with PD-L1 expression. Considering the substantial influence of PD-L1 status on treatment options in the present study cohort, further investigations are warranted to clarify the potential association between PD-L1 expression and CAAB dynamics during ICI therapy.
In the monotherapy group, univariate logistic and Cox regression analysis revealed associations between CAAB dynamics and treatment outcomes during ICI therapy, highlighting both shared and distinct immunological features underlying different clinical endpoints. Although irAEs have been proposed as a potential clinical marker of ICI responsiveness, the exact nature and extent of this relationship remain incompletely understood (1, 33–36). This biological distinction underscores the importance of methodologically separating irAE-related signals from those linked to therapeutic efficacy, and highlighting the need for comprehensive benefit-to-risk assessment to guide clinical decision-making (37). Notably, CAABs against BIRC3 and STAT4 were associated with both treatment response and irAE occurrence, suggesting that the underlying immune mechanisms linked to these antibodies may contribute to therapeutic efficacy, while also driving adverse immune activation. In contrast, CAABs against SNCA, CCDC3, and ECSCR were significantly associated only with treatment response, indicating the possible existence of tumor-specific immune regulatory pathways independent of irAEs. These findings offer a novel perspective for disentangling treatment efficacy from immune-related toxicity, thereby supporting optimized benefit-to-risk assessments in clinical practice.
In addition, we observed that the majority of selected CAABs showed decreased plasma concentrations following ICI treatment. Previous studies have reported that blocking PD-1/PD-L1 enhances the maturation of PD-1-expressing T follicular helper (Tfh) cells on B cells, thereby augmenting antibody production against exogenous antigens (38, 39). However, clinical data suggest divergent effects depending on the ICI class. In a cohort of 39 patients, anti-CTLA-4 monotherapy or CTLA-4–containing combinations were associated with increased circulating plasmablasts (CD38+CD27+), whereas anti-PD-1 monotherapy was linked to reduced plasmablast numbers (20). Similarly, a study involving 48 patients reported that anti-CTLA-4 treatment generally led to increased CAAB levels, whereas anti-PD-L1 treatment tended to reduce them (15). More recently, statistical analyses of CAABs in 102 patients corroborated these trends (16). Although the mechanisms underlying these observations are poorly understood, these divergent outcomes may partly reflect the distinct sites and timing of immune modulation. CTLA-4 inhibition acts primarily during the priming phase within secondary lymphoid organs, promoting polyclonal B-cell activation, including autoreactive clones, and sustaining plasmablast output and survival (9, 20). In contrast, PD-1/PD-L1 blockade acts mainly within germinal centers and peripheral tissues, enhancing the quality of Tfh–mediated B-cell help rather than inducing global plasmablast expansion, and favoring transient expansion of high-affinity, antigen-specific clones (38). Such responses may wane once the antigenic stimulus declines. Another speculative mechanism is local immune-complex formation within tumor or inflamed tissues, which could promote antibody sequestration or consumption, thereby lowering circulating titers despite ongoing local humoral activity. However, these tissue-level antibody dynamics have not been directly demonstrated in the context of PD-1 versus CTLA-4 blockade, meriting further mechanistic investigation.
The best-performing model for predicting ir-pneumonitis was the RF model in the monotherapy subgroup. Identified CAABs targeting CXCL2, PARK2, and KLHL18 may contribute to pathogenic immune hyperactivation, thus warranting further investigation. Our previous findings indicated that decreased plasma CXCL2 levels after ICI treatment are associated with irAE occurrence in patients with NSCLC (40). CXCL2 promotes the recruitment of MDSCs via CXCR2-mediated signaling, thereby contributing to the establishment of an immunosuppressive microenvironment (41, 42). In contrast, reduced CXCL2 levels may reflect a shift toward a pro-inflammatory milieu. Among patients who developed ir-pneumonitis, we observed lower levels of CXCL2-specific CAABs. This reduction may reflect diminished antigen-driven antibody production or sequestration within CXCL2–antibody complexes, resulting in lower detectable plasma levels. Further work is required to test these hypotheses and clarify their relevance to irAE pathogenesis.
The overlapping associations of BIRC3 and STAT4 with both therapeutic efficacy and the occurrence of irAEs, together with the selective link of CXCL2 with pneumonitis, suggest the existence of a shared yet bi-directional immunoregulatory axis. BIRC3, a regulator of nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB)-dependent transcription, and STAT4, a mediator of IL-12-driven type 1 helper T cell (Th1) polarization and type II interferon (IFN-γ) production, can amplify systemic pro-inflammatory signaling that enhances anti-tumor immunity while heightening susceptibility to multi-organ irAEs (43, 44). In contrast, CXCL2 recruits CXCR2+ MDSCs, providing a counter-regulatory brake on inflammation, with pulmonary tissue particularly dependent on this chemokine axis (45). In our cohort, reductions in BIRC3- and STAT4-specific autoantibodies were associated both with enhanced therapeutic efficacy and increased irAEs. One plausible explanation is that early activation of NF-κB/Th1 programs transiently augments antigen presentation and autoantibody production, followed by antigen clearance or a shift toward cell-mediated immunity, leading to lower autoantibody titers despite sustained pathway activity. By contrast, decreases in CXCL2-specific autoantibodies were unrelated to efficacy but correlated with pneumonitis, consistent with the idea that disrupting the CXCL2–CXCR2 axis removes a local anti-inflammatory safeguard in the lung, creating a predisposition to organ-restricted toxicity without broadly influencing systemic anti-tumor immunity (45). These findings are hypothesis-generating and highlight the need for prospective validation through longitudinal protein measurements, immune cell phenotyping, and pathway-level analyses.
For clinical translation of the high-performance predictive models identified in this study, peripheral blood samples should be collected prior to and six weeks following the initiation of ICI therapy. Titers of the CAABs selected by each model are then quantified, fold changes calculated, and incorporated into the model to generate a predictive score. Patients with scores exceeding the model-specific optimal threshold would be classified as at risk for adverse events or as potential responders. Future studies aimed at optimizing blood sampling intervals may further improve the timeliness and accuracy of these predictions.
This study has several limitations. The initial screening phase was restricted to five patients due to practical and financial constraints, which inevitably narrows the scope of this analysis. Identifying approximately 60 CAABs from a proteome-wide panel encompassing over 20,000 potential targets posed a substantial methodological challenge. Given the limited sample size, the presence of variability and possibility of overlooking relevant antigens cannot be excluded. As a result, the present findings provide a partial snapshot of the broader treatment-induced alterations in the antibody repertoire. The specificity of the autoantibody detection platform and selection strategy precluded the use of suitable publicly available datasets, preventing evaluation of model performance across independent or multi-ethnic populations. To achieve a more comprehensive understanding of CAAB dynamics in the context of ICI therapy, future investigations should involve larger and more diverse patient populations and employ unbiased, high-throughput proteomic profiling strategies.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Institutional Review Boards of Kurume University Hospital and Kanagawa Cancer Center. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
FW: Conceptualization, Data curation, Formal analysis, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. HT: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. KA: Data curation, Formal analysis, Methodology, Project administration, Resources, Writing – review & editing. YN: Resources, Writing – review & editing. YI: Resources, Writing – review & editing. KM: Data curation, Formal analysis, Methodology, Writing – review & editing. HS: Resources, Writing – review & editing. SM: Resources, Writing – review & editing. TeK: Resources, Writing – review & editing. TaK: Resources, Writing – review & editing. HH: Resources, Writing – review & editing. KT: Resources, Writing – review & editing. MK: Resources, Writing – review & editing. TaS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Supervision, Writing – review & editing. TeS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Japan Society for the Promotion of Science (JSPS) Grant: Grant-in-Aid for Challenging Exploratory Research: 18K19490 (TeS) and Grant-in-Aid for Scientific Research (C): 22K08724 and 25K10526 (FW); AMED Grant Number JP19ae0101076 (TeS); The Cell Science Research Foundation Fellowship (FW). The establishment of the Ehime-Kazusa protein array and screening assay were partially supported by the grant from Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT). This research was partially supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP19am0101077, and Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP22ama121010 (TaS).
Acknowledgments
We would like to thank Norikazu Matsuo (Kurume University) for patient enrollment and data collection, as well as Junya Otake, Megumi Abe, Akiko Orikasa, and Makoto Wakatsuki (Kanagawa Cancer Center Research Institute) for their assistance with sample handling and data collection. The establishment of the Ehime-Kazusa protein array was performed by the collaboration with the Kazusa DNA Research Institute. Protein array screening was supported by the Division of Applied Protein Research Support, the Advanced Research Support Center (ADRES), Ehime University.
Conflict of interest
KA received personal fees from AstraZeneca, Bristol Myers Squibb, Chugai Pharmaceutical, MSD Oncology, and Ono Pharmaceutical. YN received honoraria from Ono, Takeda, Eli Lilly, Kyowa Kirin, Boehringer Ingelheim, AstraZeneca, and Bristol Myers Squibb, and research funds from Bristol Myers Squibb. HS received honoraria from Boehringer Ingelheim, Eli Lilly, Pfizer, AstraZeneca, Bristol Myers Squibb, Chugai, and Ono, and research funds from AstraZeneca, Bristol Myers Squibb, Chugai, and Ono. SM received honoraria from AstraZeneca, Chugai, Boehringer Ingelheim, Taiho, Pfizer, MSD, and Ono. TeK received honoraria from AstraZeneca, Daiichi-Sankyo, and Otsuka, and research funds from AstraZeneca, Chugai, and Taiho. TeS received research funds from Taiho and Ajinomoto.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1666030/full#supplementary-material
References
1. Das S and Johnson DB. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J Immunother Cancer. (2019) 7:306. doi: 10.1186/s40425-019-0805-8
2. Remon J, Passiglia F, Ahn MJ, Barlesi F, Forde PM, Garon EB, et al. Immune checkpoint inhibitors in thoracic Malignancies: review of the existing evidence by an iaslc expert panel and recommendations. J Thorac Oncol. (2020) 15:914–47. doi: 10.1016/j.jtho.2020.03.006
3. Ghosh N, Postow M, Zhu C, Jannat-Khah D, Li QZ, Vitone G, et al. Lower baseline autoantibody levels are associated with immune-related adverse events from immune checkpoint inhibition. J Immunother Cancer. (2022) 10:e004008. doi: 10.1136/jitc-2021-004008
4. Daban A, Gonnin C, Phan L, Saldmann A, Granier C, Lillo-Lelouet A, et al. Preexisting autoantibodies as predictor of immune related adverse events (Iraes) for advanced solid tumors treated with immune checkpoint inhibitors (Icis). Oncoimmunology. (2023) 12:2204754. doi: 10.1080/2162402X.2023.2204754
5. Alserawan L, Anguera G, Zamora Atenza C, Serra Lopez J, Martinez-Martinez L, Riudavets Melia M, et al. Association between changes in the patterns of antinuclear autoantibodies during immune checkpoint inhibition therapy and the development of severe immune related adverse events. Int J Mol Sci. (2022) 23:12641. doi: 10.3390/ijms232012641
6. Taylor J, Gandhi A, Gray E, and Zaenker P. Checkpoint inhibitor immune-related adverse events: A focused review on autoantibodies and B cells as biomarkers, advancements and future possibilities. Front Immunol. (2022) 13:991433. doi: 10.3389/fimmu.2022.991433
7. Gowen MF, Giles KM, Simpson D, Tchack J, Zhou H, Moran U, et al. Baseline antibody profiles predict toxicity in melanoma patients treated with immune checkpoint inhibitors. J Transl Med. (2018) 16:82. doi: 10.1186/s12967-018-1452-4
8. Toi Y, Sugawara S, Sugisaka J, Ono H, Kawashima Y, Aiba T, et al. Profiling preexisting antibodies in patients treated with anti-pd-1 therapy for advanced non-small cell lung cancer. JAMA Oncol. (2019) 5:376–83. doi: 10.1001/jamaoncol.2018.5860
9. de Moel EC, Rozeman EA, Kapiteijn EH, Verdegaal EME, Grummels A, Bakker JA, et al. Autoantibody development under treatment with immune-checkpoint inhibitors. Cancer Immunol Res. (2019) 7:6–11. doi: 10.1158/2326-6066.CIR-18-0245
10. Tahir SA, Gao J, Miura Y, Blando J, Tidwell RSS, Zhao H, et al. Autoimmune antibodies correlate with immune checkpoint therapy-induced toxicities. Proc Natl Acad Sci U.S.A. (2019) 116:22246–51. doi: 10.1073/pnas.1908079116
11. Tan Q, Wang D, Yang J, Xing P, Yang S, Li Y, et al. Autoantibody profiling identifies predictive biomarkers of response to anti-pd1 therapy in cancer patients. Theranostics. (2020) 10:6399–410. doi: 10.7150/thno.45816
12. Johannet P, Liu W, Fenyo D, Wind-Rotolo M, Krogsgaard M, Mehnert JM, et al. Baseline serum autoantibody signatures predict recurrence and toxicity in melanoma patients receiving adjuvant immune checkpoint blockade. Clin Cancer Res. (2022) 28:4121–30. doi: 10.1158/1078-0432.CCR-22-0404
13. Soussan S, Pupier G, Cremer I, Joubert PE, Sautes-Fridman C, Fridman WH, et al. Unraveling the complex interplay between anti-tumor immune response and autoimmunity mediated by B cells and autoantibodies in the era of anti-checkpoint monoclonal antibody therapies. Front Immunol. (2024) 15:1343020. doi: 10.3389/fimmu.2024.1343020
14. Borgers JSW, van Wesemael TJ, Gelderman KA, Rispens T, Verdegaal EME, Moes D, et al. Autoantibody-positivity before and seroconversion during treatment with anti-pd-1 is associated with immune-related adverse events in patients with melanoma. J Immunother Cancer. (2024) 12:e009215. doi: 10.1136/jitc-2024-009215
15. Mu-Mosley H, von Itzstein MS, Fattah F, Liu J, Zhu C, Xie Y, et al. Distinct autoantibody profiles across checkpoint inhibitor types and toxicities. Oncoimmunology. (2024) 13:2351255. doi: 10.1080/2162402X.2024.2351255
16. Dai Y, Aizenbud L, Qin K, Austin M, Jaycox JR, Cunningham J, et al. Humoral determinants of checkpoint immunotherapy. Nature. (2025) 644:527–36. doi: 10.1038/s41586-025-09188-4
17. Dai L, Tan Q, Li L, Lou N, Zheng C, Yang J, et al. High-throughput antigen microarray identifies longitudinal prognostic autoantibody for chemoimmunotherapy in advanced non-small cell lung cancer. Mol Cell Proteomics. (2024) 23:100749. doi: 10.1016/j.mcpro.2024.100749
18. Derbala MH, Hajjar J, Stephen B, Gurses SA, Kwiatkowski E, Budde P, et al. Autoantibody profiling to predict response to the anti-pd-1 therapy, pembrolizumab, in rare tumors. ESMO Open. (2025) 10:105518. doi: 10.1016/j.esmoop.2025.105518
19. Zheng N, Li Y, Peng Z, Tang Y, Liang Z, Wang H, et al. Five autoantibodies identified from immune complexes as breast cancer biomarkers. Front Immunol. (2025) 16:1640054. doi: 10.3389/fimmu.2025.1640054
20. Das R, Bar N, Ferreira M, Newman AM, Zhang L, Bailur JK, et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J Clin Invest. (2018) 128:715–20. doi: 10.1172/JCI96798
21. Wei F, Azuma K, Nakahara Y, Saito H, Matsuo N, Tagami T, et al. Machine learning for prediction of immunotherapeutic outcome in non-small-cell lung cancer based on circulating cytokine signatures. J Immunother Cancer. (2023) 11:e006788. doi: 10.1136/jitc-2023-006788
22. Nakahara Y, Kouro T, Motoyama S, Miura M, Fujita K, Igarashi Y, et al. Circulating il-6 and not its circulating signaling components sil-6r and sgp130 demonstrate clinical significance in nsclc patients treated with immune checkpoint inhibitors. Front Cell Dev Biol. (2023) 11:1324898. doi: 10.3389/fcell.2023.1324898
23. Nagayoshi Y, Nakamura M, Matsuoka K, Ohtsuka T, Mori Y, Kono H, et al. Profiling of autoantibodies in sera of pancreatic cancer patients. Ann Surg Oncol. (2014) 21 Suppl 3:S459–65. doi: 10.1245/s10434-014-3574-0
24. Onishi S, Adnan E, Ishizaki J, Miyazaki T, Tanaka Y, Matsumoto T, et al. Novel autoantigens associated with lupus nephritis. PloS One. (2015) 10:e0126564. doi: 10.1371/journal.pone.0126564
25. Gralla RJ, Clark RA, Kris MG, and Tyson LB. Methodology in anti-emetic trials. Eur J Cancer. (1991) 27 Suppl 1:S5–8.
26. Yamanaka S, Murai H, Saito D, Abe G, Tokunaga E, Iwasaki T, et al. Thalidomide and its metabolite 5-hydroxythalidomide induce teratogenicity via the cereblon neosubstrate plzf. EMBO J. (2021) 40:e105375. doi: 10.15252/embj.2020105375
27. Takeda H. Autoantibody profiling using human autoantigen protein array and alphascreen. Methods Mol Biol. (2024) 2766:107–28. doi: 10.1007/978-1-0716-3682-4_12
28. Nishiyama K, Maekawa M, Nakagita T, Nakayama J, Kiyoi T, Chosei M, et al. Cnksr1 serves as a scaffold to activate an egfr phosphatase via exclusive interaction with rhob-gtp. Life Sci Alliance. (2021) 4: e202101095. doi: 10.26508/lsa.202101095
29. Wei F, Kouro T, Nakamura Y, Ueda H, Iiizumi S, Hasegawa K, et al. Enhancing mass spectrometry-based tumor immunopeptide identification: machine learning filter leveraging hla binding affinity, aliphatic index and retention time deviation. Comput Struct Biotechnol J. (2024) 23:859–69. doi: 10.1016/j.csbj.2024.01.023
30. Chen L, Zhernakova DV, Kurilshikov A, Andreu-Sanchez S, Wang D, Augustijn HE, et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat Med. (2022) 28:2333–43. doi: 10.1038/s41591-022-02014-8
31. Wang Y, Safi M, Hirsch FR, Lu S, Peters S, Govindan R, et al. Immunotherapy for advanced-stage squamous cell lung cancer: the state of the art and outstanding questions. Nat Rev Clin Oncol. (2025) 22:200–14. doi: 10.1038/s41571-024-00979-8
32. Galluzzi L, Guilbaud E, Schmidt D, Kroemer G, and Marincola FM. Targeting immunogenic cell stress and death for cancer therapy. Nat Rev Drug Discov. (2024) 23:445–60. doi: 10.1038/s41573-024-00920-9
33. Rogado J, Sanchez-Torres JM, Romero-Laorden N, Ballesteros AI, Pacheco-Barcia V, Ramos-Levi A, et al. Immune-related adverse events predict the therapeutic efficacy of anti-pd-1 antibodies in cancer patients. Eur J Cancer. (2019) 109:21–7. doi: 10.1016/j.ejca.2018.10.014
34. Toi Y, Sugawara S, Kawashima Y, Aiba T, Kawana S, Saito R, et al. Association of immune-related adverse events with clinical benefit in patients with advanced non-small-cell lung cancer treated with nivolumab. Oncologist. (2018) 23:1358–65. doi: 10.1634/theoncologist.2017-0384
35. Grangeon M, Tomasini P, Chaleat S, Jeanson A, Souquet-Bressand M, Khobta N, et al. Association between immune-related adverse events and efficacy of immune checkpoint inhibitors in non-small-cell lung cancer. Clin Lung Cancer. (2019) 20:201–7. doi: 10.1016/j.cllc.2018.10.002
36. Maher VE, Fernandes LL, Weinstock C, Tang S, Agarwal S, Brave M, et al. Analysis of the association between adverse events and outcome in patients receiving a programmed death protein 1 or programmed death ligand 1 antibody. J Clin Oncol. (2019) 37:2730–7. doi: 10.1200/JCO.19.00318
37. Jing Y, Yang J, Johnson DB, Moslehi JJ, and Han L. Harnessing big data to characterize immune-related adverse events. Nat Rev Clin Oncol. (2022) 19:269–80. doi: 10.1038/s41571-021-00597-8
38. Herati RS, Knorr DA, Vella LA, Silva LV, Chilukuri L, Apostolidis SA, et al. Pd-1 directed immunotherapy alters tfh and humoral immune responses to seasonal influenza vaccine. Nat Immunol. (2022) 23:1183–92. doi: 10.1038/s41590-022-01274-3
39. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing siv-specific immunity in vivo by pd-1 blockade. Nature. (2009) 458:206–10. doi: 10.1038/nature07662
40. Matsuo N, Azuma K, Hattori S, Ohtake J, Kawahara A, Ishii H, et al. Association between soluble immune mediators and tumor responses in patients with nonsmall cell lung cancer treated with anti-pd-1 inhibitor. Int J Cancer. (2019) 144:1170–9. doi: 10.1002/ijc.31923
41. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of cxcr2-mediated mdsc tumor trafficking enhances anti-pd1 efficacy. Sci Transl Med. (2014) 6:237ra67. doi: 10.1126/scitranslmed.3007974
42. Bullock K and Richmond A. Suppressing mdsc recruitment to the tumor microenvironment by antagonizing cxcr2 to enhance the efficacy of immunotherapy. Cancers (Basel). (2021) 13:6293. doi: 10.3390/cancers13246293
43. Frazzi R. Birc3 and birc5: multi-faceted inhibitors in cancer. Cell Biosci. (2021) 11:8. doi: 10.1186/s13578-020-00521-0
44. Tolomeo M and Cascio A. Stat4 and stat6, their role in cellular and humoral immunity and in diverse human diseases. Int Rev Immunol. (2024) 43:394–418. doi: 10.1080/08830185.2024.2395274
Keywords: non-small cell lung cancer, immune checkpoint inhibitor, circulating autoantibody, immune-related adverse events, immune-related pneumonitis, treatment response, machine learning
Citation: Wei F, Takeda H, Azuma K, Nakahara Y, Igarashi Y, Murotani K, Saito H, Murakami S, Kondo T, Kouro T, Himuro H, Tsuji K, Komahashi M, Sawasaki T and Sasada T (2025) Machine learning–based insights into circulating autoantibody dynamics and treatment outcomes in patients with NSCLC receiving immune checkpoint inhibitors. Front. Immunol. 16:1666030. doi: 10.3389/fimmu.2025.1666030
Received: 15 July 2025; Accepted: 18 September 2025;
Published: 03 October 2025.
Edited by:
Cuiling Zheng, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Yingkun Xu, Shandong University, ChinaKecheng Huang, Huazhong University of Science and Technology, China
Copyright © 2025 Wei, Takeda, Azuma, Nakahara, Igarashi, Murotani, Saito, Murakami, Kondo, Kouro, Himuro, Tsuji, Komahashi, Sawasaki and Sasada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tetsuro Sasada, dHNhc2FkYUBrY2NoLmpw; Tatsuya Sawasaki, c2F3YXNha2kudGF0c3V5YS5tZkBlaGltZS11LmFjLmpw
†These authors share last authorship
‡These authors have contributed equally to this work and share first authorship