 Alexander Pine1
Alexander Pine1 Ayesha Butt1
Ayesha Butt1 Laura Andreoli2
Laura Andreoli2 Jason S. Knight3
Jason S. Knight3 Maria Gerosa4
Maria Gerosa4 Irene Cecchi5D. Ware Branch6Rosario Lopez-Pedrera7H. Michael Belmont8
Irene Cecchi5D. Ware Branch6Rosario Lopez-Pedrera7H. Michael Belmont8 Nina Kello9Michelle Petri10
Nina Kello9Michelle Petri10 Ricard Cervera11
Ricard Cervera11 Vittorio Pengo12
Vittorio Pengo12 Pier Luigi Meroni4Hannah Cohen13
Pier Luigi Meroni4Hannah Cohen13 Rohan Willis14Maria Laura Bertolccini15George Goshua1Sean Gu16
Rohan Willis14Maria Laura Bertolccini15George Goshua1Sean Gu16 John Hwa16Alfred I. Lee1
John Hwa16Alfred I. Lee1 Doruk Erkan17
Doruk Erkan17 Anish V. Sharda1*
Anish V. Sharda1*- 1Section of Hematology, Yale University School of Medicine, New Haven, CT, United States
- 2University of Brescia, Brescia, Italy
- 3University of Michigan, Ann Arbor, MI, United States
- 4University of Milan, Milan, Italy
- 5University of Turin, Turin, Italy
- 6University of Utah and Intermountain Healthcare, Salt Lake, UT, United States
- 7Maimonides Institute of Biomedical Research of Cordoba, Córdoba, Spain
- 8New York University Langone Medical Center, New York, NY, United States
- 9Northwell Health, New Hyde Park, NY, United States
- 10Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 11Department of Autoimmune Diseases, Hospital Clínic, Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), University of Barcelona, Barcelona, Catalonia, Spain
- 12University of Padova, Padova, Italy
- 13University College London, London, United Kingdom
- 14University of Texas Medical, Galveston, TX, United States
- 15St Thomas’ Hospital, London, United Kingdom
- 16Yale Cardiovascular Research Center, Yale University School of Medicine, New Haven, CT, United States
- 17Hospital for Special Surgery and Weill Cornell Medicine, New York, NY, United States
Introduction: Antiphospholipid syndrome (APS) is an autoimmune disease with thromboembolic and obstetric morbidity arising via a model of immunothrombosis. Individuals with APS may present with thrombotic (TAPS), obstetric (OAPS), or microvascular (MAPS) disease, while many have circulating antiphospholipid antibodies (aPL) without APS classification (NoAPS). Multiple pathophysiologic mechanisms have been proposed in APS, including activation by aPL of platelets, endothelial and immune cells, as well as complement and coagulation pathways; however, the pathophysiology of APS, particularly transition of clinical APS from aPL remains unclear.
Methods: Seeking to define the inflammatory signature of APS, we carried out an unbiased proteomic screen of persistently aPL-positive patients with different clinical phenotypes from the international APS Alliance for Clinical Trials and International Networking (ACTION) Registry and compared them to 10 healthy controls. 6398 unique proteins were estimated using an DNA aptamer-based assay. Subsequently, we validated our findings in 34 additional patients.
Results: Our data show that the mere presence of aPL confers a distinct thromboinflammatory signature characterized by the activation of coagulation, complement, innate and adaptive immune response pathways shared by all APS subtypes. Pathway enrichment analysis revealed increasing enrichment with rising statistical significance of thrombosis, complement, neutrophil and other innate and adaptive immune activation, as well as extracellular matrix (ECM) organization with increasing clinical severity, suggesting a model of progressive thromboinflammation in evolution of APS from NoAPS to TAPS and MAPS.
Conclusions: Our findings provide novel insights into the pathogenesis of APS and identify potential novel targets for diagnostic and therapeutic intervention in APS across its entire spectrum.
Introduction
Antiphospholipid syndrome (APS) is an autoimmune disease characterized by the presence of circulating antiphospholipid antibodies (aPL), such as anticardiolipin and anti-β2-glycoprotein 1 (anti-β2 GP1) antibodies, and associated with thromboembolic disease and obstetrical morbidity (1, 2). A striking feature of APS is the propensity to both arterial and venous thromboembolic disease, as well as thrombosis of the microcirculation (1, 2). The resulting thrombotic complications, particularly strokes, myocardial infarction, venous thromboembolism and microvascular thrombi of the skin, lungs, and kidneys, are associated with increased morbidity and mortality (1, 2). Obstetric morbidity in APS manifests as placental insufficiency, recurrent miscarriages, fetal demise, and preeclampsia/eclampsia (1). APS is sometimes associated with other autoimmune diseases, particularly systemic lupus erythematosus, but can occur as a primary syndrome (1, 2). The clinical spectrum of APS can range from individuals with circulating aPL but no thromboembolic/obstetric disease (NoAPS), to those with moderate-to-large vessel thrombosis (TAPS), pregnancy morbidity (OAPS), and/or microvascular disease (MAPS) (e.g., skin necrosis, aPL-nephropathy or diffuse alveolar hemorrhage) (1, 3). While the clinical manifestations of APS are well-recognized, molecular mechanisms driving antibody-induced thrombosis and obstetric morbidity remain incompletely understood. As a result, other than anticoagulation, therapeutic strategies in APS are currently limited.
Although the presence of aPL are necessary for the development of thrombosis, what precipitates thromboembolic disease remains unknown (2). As such, a ‘two-hit’ concept of APS has been proposed, which hypothesizes that vascular injury or inflammatory stimulus (second hit) triggers thrombosis in a generalized procoagulant state created by aPL (first hit) (4). The pathophysiologic mechanisms distinguishing different APS subtypes also remain unknown. aPL interact with phospholipid binding proteins present on cellular membranes (4–6). β2-glycoprotein I (β2 GP1) present on circulating cells such as platelets, monocytes, and endothelial cells is believed to be the primary target. Murine models of APS show that mice treated with anti-β2 GP1 antibodies become more susceptible to thrombosis upon vascular injury (7). Multiple broader pathophysiologic mechanisms have been proposed in APS, including activation of platelets, endothelial cells, monocytes and neutrophils, direct activation of coagulation, inhibition of natural anticoagulant systems, and complement activation (4, 5, 8–10). In vitro studies have indicated possible activation by aPL of toll-like receptors (TLR2, TLR4) on monocytes, neutrophils, platelets and endothelial cells, leading to thromboinflammation (11–14). However, it remains unclear which cells in the circulation, and what inflammatory pathways, are the primary targets of aPL (4).
Proteome profiling may aid in answering some of these questions and has become a valuable tool in understanding pathophysiology and facilitating biomarker discovery. Proteomics is especially valuable for determining the activity of transcriptomically quiescent cells, such as neutrophils and platelets. Plasma proteomics has been valuable in understanding inflammatory responses to infections such as COVID-19, rheumatologic diseases, diabetes, liver disease and Alzheimer’s dementia (15–18). Seeking to define the inflammatory signature of APS and to enable biomarker discovery, we carried out an unbiased proteomic screen of patients with different APS subtypes, and validated our findings in a second cohort. We found that the mere presence of aPL triggers multiple thromboinflammatory pathways characterized by activation of complement, coagulation, and both innate and adaptive immune systems. Furthermore, the clinical spectrum of APS, from NoAPS to MAPS/CAPS (catastrophic antiphospholipid syndrome), represents a progression of these thromboinflammatory pathways culminating in extracellular matrix (ECM) involvement and tissue inflammation.
Material and methods
Samples procurement and study approval
Plasma samples were obtained from APS ACTION registry, an international and collaborative clinical registry and repository of blood samples. All patients included in this study were persistently aPL-positive (measured on two occasions 12 weeks apart) and without systemic autoimmune rheumatic diseases (SARDs) (19). Samples were not obtained during the time of the event, but at the time of outpatient referral/enrollment. Samples were split into discovery (North American Centers) and validation (European Centers) cohorts (Figure 1, Table 1).
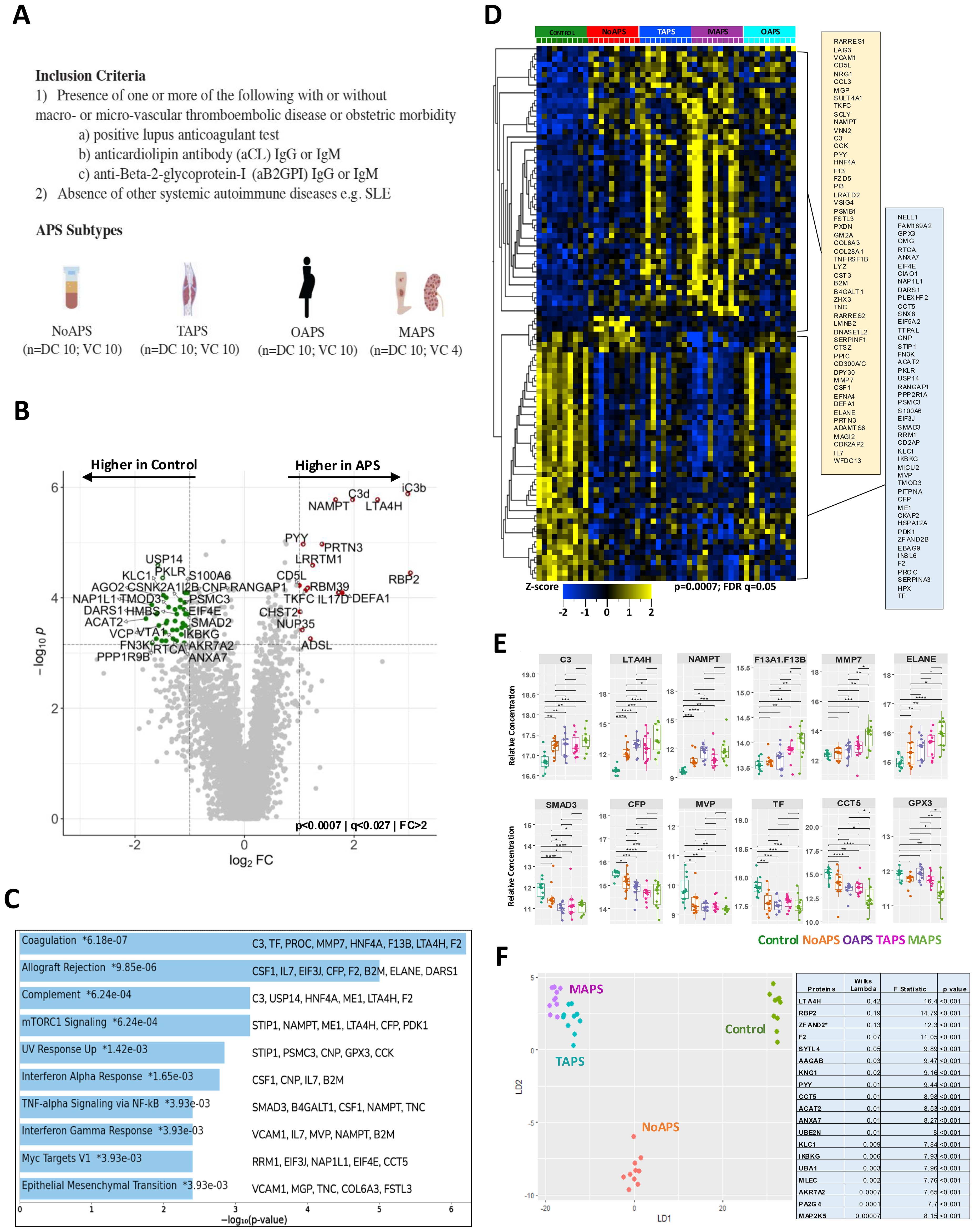
Figure 1. Presence of antiphospholipid antibodies confers a distinct proteomic signature shared by all APS subtypes. (A) Study cohorts and inclusion criteria. APS subtypes: NoAPS, aPL positivity without thromboembolism/obstetric morbidity; TAPS, history of thromboembolism, not meeting criteria for microvascular APS; OAPS, history of obstetric morbidity only; MAPS, biopsy-proven microvascular APS or history of definite catastrophic APS (61). n, sample size; DC, discovery cohort; VC, validation cohort. (B) Volcano plot of differentially abundant proteins between controls and all APS combined (p < 0.0007; q < 0.027; fold-change (FC)>2). All proteins are shown. Red dots, proteins higher in APS; green dots, proteins higher in controls. (C) Top 10 pathways enriched (KEGG 2021) in APS derived from protein list in B, along with the corresponding p-values. Asterisk (*) denotes that the term also has a adjusted p < 0.005. (E) Select differentially abundant proteins (C3, LTA4H, NAMPT, F13, MMP7, ELANE, SMAD3, CFP, MVP, TF, CCT5, GPX3); FDR-corrected t-test on log2-normalized data; ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05. (D) Z-score heatmap of differentially abundant proteins (rows; table on right) between different APS subtypes and controls (columns); ANOVA, p ≤ 0.0007, FDR q ≤ 0.05. (F) Unsupervised clustering of non-obstetric APS and controls by linear discriminant analysis (proteins tabulated on right together with Wilks lambda, F-statistic and p-value).
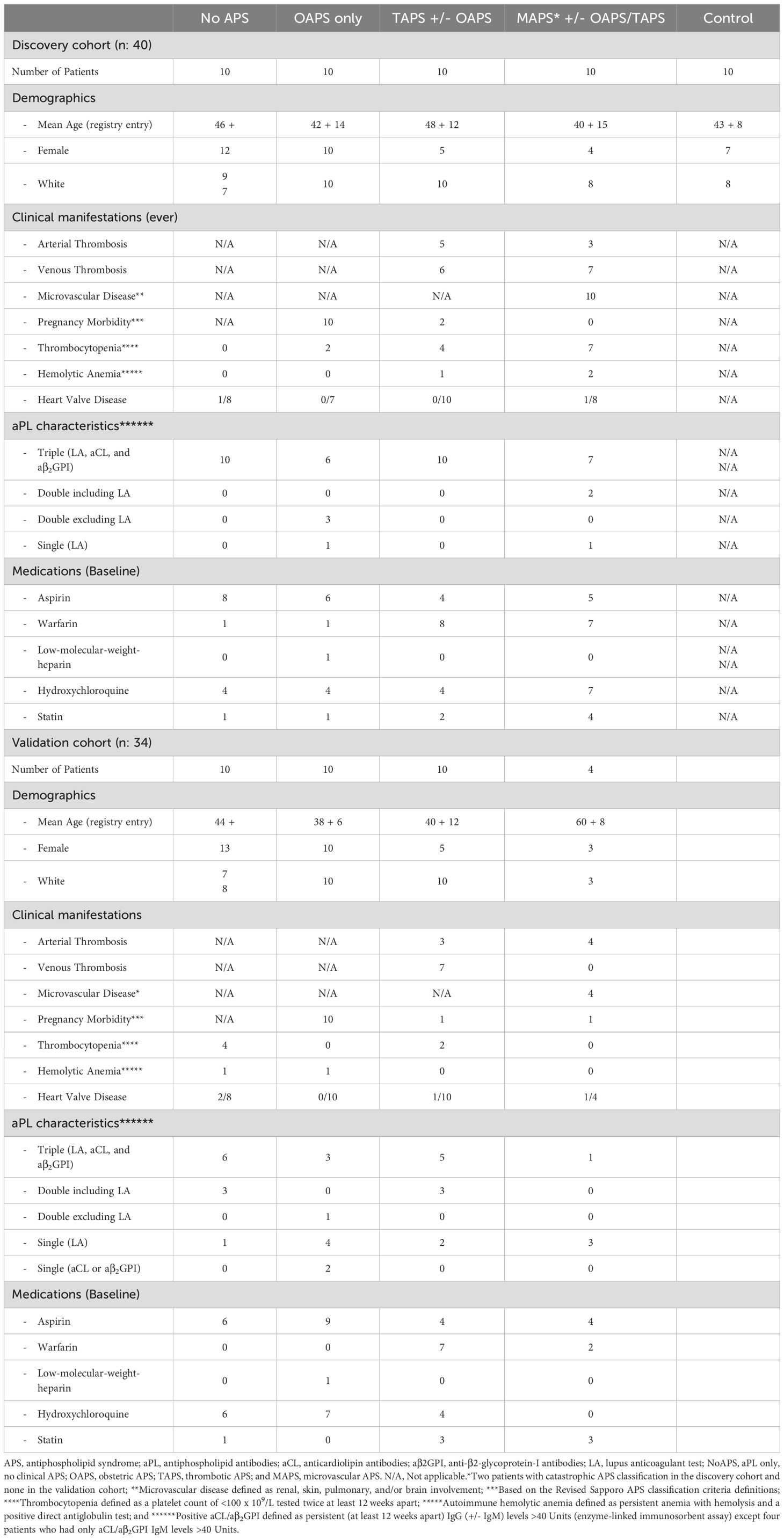
Table 1. Demographic, clinical, and laboratory characteristics of APS patients and controls.
Control subjects were healthy volunteers with no history of thromboembolism, SARD, cancer, or anticoagulant/antiplatelet medication use, enrolled from healthcare employees at hematology clinic at Yale New Haven Hospital (Table 1).
The protocol was approved by the Institutional Review Boards of all participating institutions. Informed consent was obtained from all participants. Blood samples were collected in EDTA tubes and processed within two hours.
SomaScan assay
We measured plasma proteins using SomaScan assay, a technology based on highly-specific single-stranded DNA aptamers, which quantifies relative concentrations of plasma proteins (20). The characteristics, sensitivity/specificity and reproducibility of SomaScan to human targets have been previously described (20–23). 6398 unique proteins were measured in the discovery cohort using SomaLogic 7K assay. Subsequently, 1500 proteins were measured in validation cohort using same platform. Median CV between two runs was 4.3% with 90% of analytes having a CV <8.7%.
Data analysis
All values were technically valid and above the limit-of-detection and there were no missing values (24). First, raw data was log2-normalized. To identify differentially abundant proteins between two groups we used t-test; in case of more than two comparators we used ANOVA. In each case, we applied Benjamini-Hochberg correction for multiple comparisons to control the false discovery rate (FDR) at least q < 0.1 or q < 0.5, thereby minimizing type I errors inherent in simultaneous testing of thousands of proteins. Protein abundance was considered different when the test of statistical significance was met at the specified p- and q-values. We used hierarchical clustering and z-score heat maps (normalized to a mean of 0, variance of 1) to visualize differential proteins, which were considered upregulated or downregulated if the difference was at least 1.5-fold. These analyses were carried out in Qlucore Omics Explorer (Lund, Sweden). Box, volcano, and radar plots were generated with ggplot2, EnhancedVolcano, and ggradar packages, respectively, in R/RStudio (R Core Team, Vienna, Austria/RStudio Team, Boston, MA).
For linear discriminant analysis (LDA), we implemented a two-stage feature selection approach to address the high-dimensional nature of the proteomics data (p >> n). First, we generated candidate differential proteins using the Wilcoxon rank-sum test with FDR adjustment at q<0.2, chosen as a dimension reduction strategy to retain potentially informative features while mitigating multicollinearity. Second, we applied stepwise forward variable selection using greedy Wilk’s Lambda criterion to select proteins based on their discriminatory capacity. The LDA procedure projects high-dimensional protein data onto 2–3 linear discriminants (depending on group number), with each subject’s position determined by linear discriminant scores to enable visualization and classification. This analysis was performed using MASS and klaR packages in R.
Functional enrichment analysis
We utilized gene annotation and analysis resources for functional enrichment analysis has been previously done (15, 16, 18). We specifically used Enrichr and Metascape suites for this analysis (25, 26).
Results
aPL confer a distinct plasma proteomic signature shared by all APS subtypes
We performed an unbiased plasma proteomic screen in 40 persistently aPL-positive patients: 10 each with NoAPS, TAPS, MAPS, or OAPS (Figure 1A). The median age was 48 years, with 70% females. Approximately 70% were ‘triple-positive’ (i.e., positive for lupus anticoagulant, anticardiolipin antibodies IgG/M ‗ 40U, and anti-β2 GP1 IgG/M ‗ 40U), none with a concomitant diagnosis of SARDs. The clinical characteristics of the cohort are described in Table 1. Ten additional healthy adults, without any history of thrombosis, SARD or cancer, were enrolled as controls.
We measured 6398 unique plasma proteins in all 50 samples by SomaLogic 7K assay (20). First, to characterize broadly plasma proteomic abnormalities in APS, we compared all APS subtypes combined versus controls. The volcano plot in Figure 1B highlights the differentially abundant proteins between the two groups. A comprehensive list of all measured plasma proteins, difference between control and APS, and relevant statistics are provided in Supplementary Table 1. The most significantly differential proteins have been discussed in greater detail below including in subsequent analyses.
Next, we carried out functional enrichment analysis from the list of differentially abundant proteins obtained above, to identify the implicated biological pathways (Figure 1C, Supplementary Table 2). The most significant pathways were Coagulation (C3, tissue factor, protein C, MMP7, HNF4A, LTAH4, and prothrombin), Allograft Rejection (CSF1, IL7, EIF3J, CFP, prothrombin, B2M, ELANE, DARS1) and Complement (C3, USP14, HNF4A, ME1, LTA4H, prothrombin). Additionally, alterations in Interferon, IL-6/JAK/STAT3, PI3K/AKT/mTOR, and ILT-STAT4 signaling pathways highlight the thromboinflammatory milieu that defines APS, including NoAPS.
Following this, we compared each of the four APS subtypes with healthy controls using ANOVA. This analysis revealed a proteomic signature shared by all APS subtypes, including NoAPS group, as compared with controls (Figure 1D). There was heterogeneity among APS patients, even within the same categories, but despite this, different APS subtypes were hierarchically clustered. The differentially abundant proteins are listed next to the heat map. Generally, the z-scores for the differentially abundant proteins altered in APS were more significant in TAPS, but particularly in MAPS subtype, suggesting that plasma thromboinflammatory abnormalities in APS reflect APS severity. OAPS appeared distinct, which was somewhat unexpected (27, 28). For subsequent analyses, OAPS was explored separately.
To study proteomic effects, if any, of treatments, we carried out analysis stratified on vitamin K antagonists (15 out of 20 on warfarin), hydroxychloroquine (9 out of 20 on hydroxychloroquine), statins (8 out of 20 on statins) and aspirin (9 out of 20 on aspirin) in TAPS and MAPS combined (Supplementary Figure S2). As expected, Vitamin K-dependent proteins were found to be lower in individuals on VKA (Supplementary Figures S2A, B), but proteomic abnormalities in hydroxychloroquine group could only be detected at very high false discovery rates (Supplementary Figures S2C, D). Individuals on statin had proteomic abnormalities that included higher insulin-like growth factor binding protein 1 (IGFBP1) and two members of sialic-acid binding immunoglobulin-like lectins (SIGLECs) (Supplementary Figures S2E, F). There were no significant abnormalities noted in aspirin group (Supplementary Figure S2G). None of the treatments affected proteomic abnormalities previously found to be associated with APS.
Several proteins displayed a positive trend across the phenotypes from NoAPS to MAPS (Figure 1E, top panel), while others showed a negative trend (Figure 1E, lower panel), suggesting an association with clinical severity of APS. Notably, tissue factor (TF) and complement factor P (CFP), a positive regulator of the alternative complement pathway, were lower in APS, particularly in MAPS. Patients with a ‘triple-positive’ aPL profile are known to be at risk for subsequent events. Our data suggests that NoAPS can be associated with a protein profile linked to thromboinflammation, which is shared across APS subtypes (29).
Next, we attempted unsupervised clustering using linear discriminant analysis to identify proteins that would classify individual samples to their phenotypes (NoAPS, TAPS, MAPS) and controls. Indeed, LDA distinctly separated NoAPS, TAPS and MAPS from controls (Figure 1F). The set of discriminant proteins with corresponding discriminatory metric (Wilk’s lambda) is shown in the table and discussed in a greater detail below; proteins with lower Wilk’s lambda are more discriminatory.
Proteomic abnormalities associated with aPL
The ‘two-hit’ model of APS posits that vascular injury or inflammation (“second hit”) triggers thromboembolism in a circulation previously primed by aPL (“first hit”) (5). Given the significant plasma proteomic changes in NoAPS cohort seen above (Figures 1B, C), we wanted to further characterize the thromboinflammatory milieu associated with this “first hit”. Therefore, we performed pairwise analysis between NoAPS and controls. The most significant differences were in C3, RBP2, LTA4H, NAMPT, IL17D, CCL5, and CXCL2 (high in NoAPS), as well as SYTL4, ARHGAP45, INPP5B and PRKCB (low in NoAPS) (Figure 2A). Hierarchical clustering of differentially abundant proteins obtained in this analysis also showed a clear distinction between NoAPS and controls (Supplementary Figure S1). Functional enrichment analysis again highlighted alterations in complement, coagulation, innate immunity, IL6 as well as TNF-alpha signaling pathways, among others (Figure 2B). This, we propose, is the thromboinflammatory signature of the “first hit” of APS. Interestingly, Human Phenotype Ontology identified multiple thromboembolism phenotypes using the set of proteins differentially abundant between NoAPS and controls (Supplementary Figure S1B). Pairwise comparisons of controls with TAPS or MAPS also identified similar protein sets, particularly C3, LTA4H, NAMPT, PRTN3, and RBP2, (Supplementary Figure S1C).
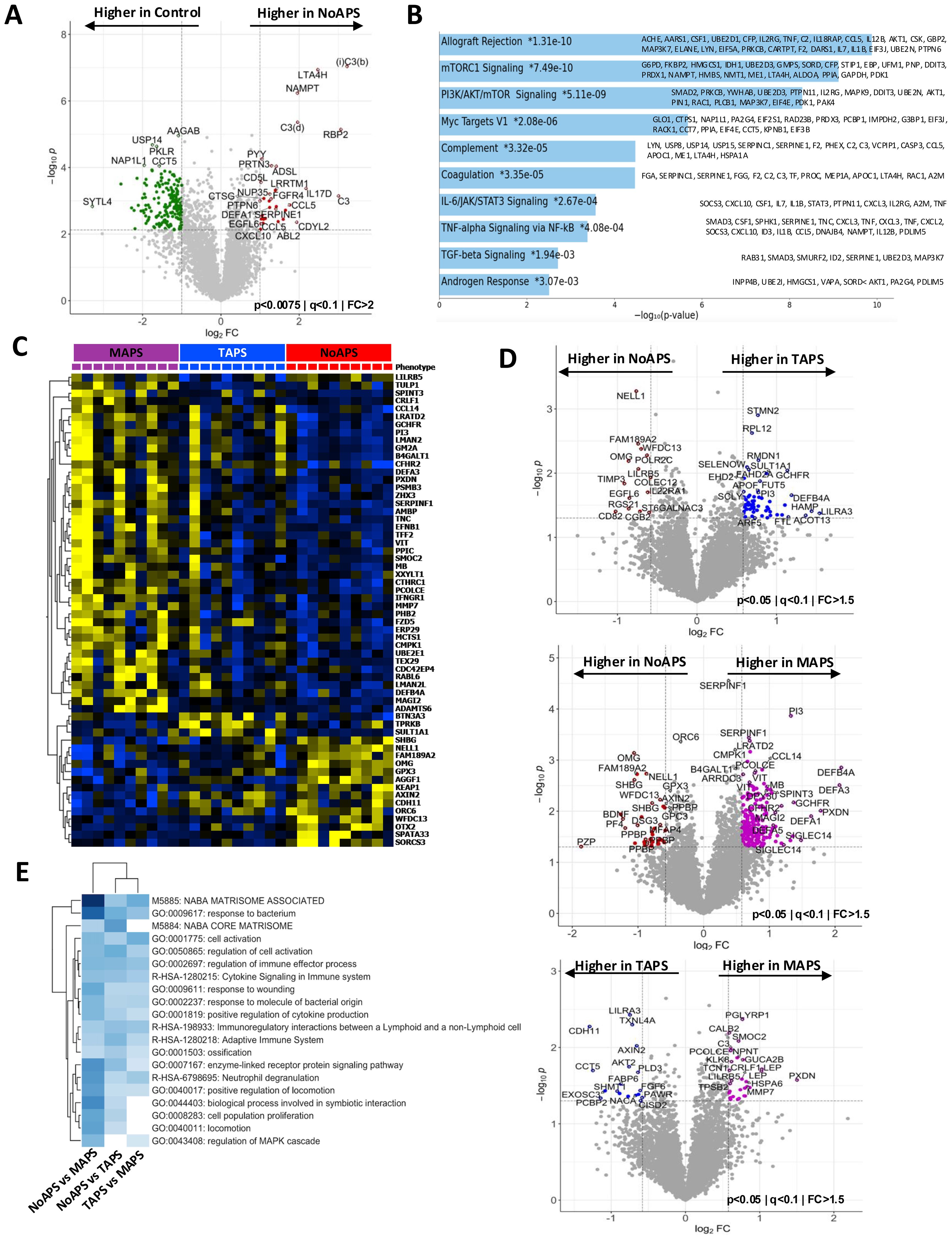
Figure 2. Plasma proteomic abnormalities associated with the ‘first-hit’; increasing thromboinflammation in TAPS and MAPS. (A) Volcano plot of differentially abundant proteins between NoAPS and controls (p < 0.0075; q < 0.1; fold-change (FC)>2). All proteins are shown. Red dots, proteins significantly higher in aPL; green dots, proteins significantly higher in controls. (B) Bar chart showing top 10 enriched terms (KEGG 2021) in the protein list generated by the pairwise analysis of NoAPS vs control in A, along with the corresponding p-values. Asterisk (*) denotes that the term also has a p < 0.005). (C) Z-score heatmap of differentially abundant proteins (rows; tabulated on right) between NoAPS, TAPS and MAPS (columns); ANOVA, p ≤ 0.0007, FDR q ≤ 0.1. (D) Volcano plots of differentially abundant proteins derived from three individual pairwise analyses between NoAPS and TAPS (top), NoAPS and MAPS (middle), and TAPS and MAPS (bottom) (p < 0.05; i<0.1; fold-change (FC)>1.5). All proteins are shown. Colored dots depicting differentially abundant proteins, directionality shown on the plots. (E) Heatmap showing the top 20 enriched terms derived from the protein sets (p < 0.05) obtained from the pairwise analyses between NoAPS and TAPS, NoAPS and MAPS and TAPS and MAPS in D.
C3 and other complement abnormalities are well established in APS and have also been targeted therapeutically (4). Leukotriene A4H (LTA4H), which was also one of the top hits in LDA (Figure 1E), although primarily synthesized by liver, is released by many circulating immune cells, including neutrophils, and there is evolving literature on the role of LTA4H in inflammatory diseases and preclinical data on therapeutic utility of LTA4H inhibitors (30, 31). Circulating NAMPT, acting as a damage associated membrane protein (DAMP), is a well-known regulator of inflammation specifically through activation of pattern recognition receptors toll-like receptors (TLRs) such as TLR4 (32, 33). Isoforms of RBP have also been identified as proinflammatory mediators in conditions such as type 2 diabetes mellitus (32, 33).
Our data clearly show that many of the inflammatory pathways recognized in the context of severe clinical APS subtypes, are already active in asymptomatic individuals with circulating aPL.
From laboratory APS to thromboembolic disease: proteomic abnormalities in thrombotic APS
Identification of pathophysiologic abnormalities associated with development of clinical APS from NoAPS would allow for risk stratification of APS but also potentially novel therapeutic biomarkers. We hypothesized that this would manifest through differences in proteomic profiles of varying APS subtypes and performed pairwise analyses between NoAPS and TAPS, NoAPS and MAPS, and TAPS and MAPS cohorts.
First, hierarchical clustering of differentially abundant proteins obtained from multigroup ANOVA showed a clear distinction across NoAPS, TAPS, and MAPS (Figure 2C). A trend between APS severity and plasma proteomic abnormality, in both the number of differentially abundant proteins and z scores, was again noted, with MAPS having the greatest number of alterations, while TAPS appearing intermediate. The volcano plots highlight the most significant differentially abundant proteins between NoAPS and TAPS, NoAPS and MAPS, and TAPS and MAPS (Figure 2D). Again, the number of differentially abundant proteins (defined by adjusted p-value <0.05) was highest in the NoAPS vs. MAPS (440), as compared with NoAPS vs. TAPS (225), and TAPS vs. MAPS (190), with many proteins being common (116 proteins) (Supplementary Table 3). This suggests that plasma proteomic alterations increase with APS severity.
Lastly, we performed functional enrichment analyses using Metascape suite, which allowed us to compare the three proteins sets obtained from the pairwise comparisons above (NoAPS vs MAPS, NoAPS vs TAPS and TAPS vs MAPS) (Figure 2E). Among the top 20 functional pathways identified and shared between the three groups, the most significant differences were identified in NoAPS vs. MAPS, based on the z scores. This analysis provides insights into the inflammatory responses in APS beyond that of laboratory APS. Although neutrophil degranulation, cell signaling and proliferation, cytokine signaling, and pathways related to innate and adaptive immune systems were activated, the most significant changes were seen in pathways connected to ECM organization. Increasing strength of alterations in coagulation pathway was also notable. In summary, our data reveals a pattern of progressive thromboinflammation with increasing clinical APS severity.
Microvascular antiphospholipid syndrome is characterized by increasing thromboinflammation and extracellular matrix reorganization
Organ or life-threatening multisystemic thromboembolic disease is a hallmark of MAPS/CAPS. Infections as well as individual level risk factors such as inherited complement factor abnormalities have been identified as provoking factors in some individuals, but what triggers these severe clinical manifestations of APS remains largely undefined (4, 34, 35). Therefore, we attempted to identify biologic pathways that distinguish MAPS from TAPS.
The pairwise comparison between TAPS and MAPS identified differentially abundant proteins that could distinctly cluster the two cohorts (Figure 2D, Supplementary Figure S1D). In Figure 3A, the relative mean protein levels of 50 top hits are shown as a radar plot; 34 proteins were higher in MAPS than TAPS. To further understand the functional nature of these proteomic changes, we created a network plot of biologic pathways identified from this protein list (adjusted p-value < 0.05) (Figure 3B). Many of the pathways identified, specifically intracellular signaling pathways, vesicular trafficking, protein phosphorylation, angiogenesis, and metabolism, extend beyond the abnormalities already noted in NoAPS and TAPS.
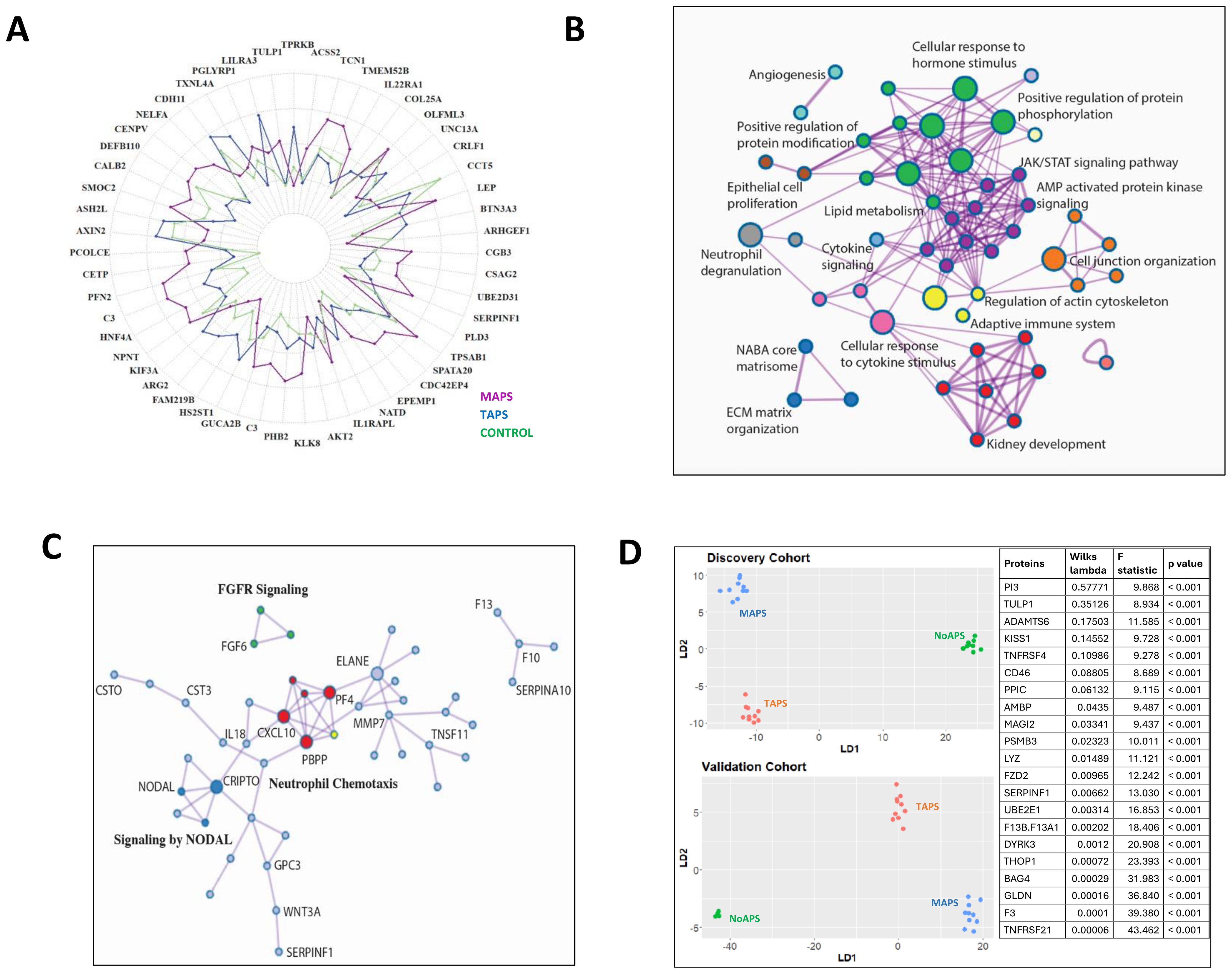
Figure 3. Thromboinflammation resulting in extracellular matrix reorganization in MAPS (A) Radial plot of the top 50 differentially abundant proteins comparing MAPS (purple) with TAPS (blue); relative protein abundance in controls (green) shown as baseline. (B) Network plot of enriched terms obtained from differential protein list from TAPS vs. MAPS comparison (Figure 2D). Terms with a similarity of >0.3 are connected by edges. Each node represents an enriched term and is colored by its cluster ID. The most significant of the enriched terms are labeled in the network plot. (C) Protein-protein interaction enrichment analysis of 85 proteins belonging to ECM-associated pathways complied from the pairwise analyses between aPL or TAPS and MAPS. Network contains proteins that form physical interactions with at least one other member in the list. Pathway enrichment analysis applied to the most densely connected network components. Most significant terms include neutrophil chemotaxis (red), FGFR signaling (green), and NODAL pathways (dark blue). (D) Linear discriminant analysis/unsupervised clustering of NoAPS, TAPS and MAPS in validation cohort (bottom plot) using protein set generated by linear discriminant analysis in discovery cohort (top plot) (outlined in the panel on right together with Wilks lambda, F-statistic and p-value).
Notably, ECM abnormalities and many kidney-associated pathways suggest increasing tissue inflammation in MAPS. The role of ECM in inflammation and thrombosis, is well established (36, 37). The proteins belonging to the ECM-related pathways, compiled from both TAPS vs. MAPS and NoAPS vs. MAPS analyses, included proteases (neutrophil elastase (ELANE), cathepsins G (CTSG), O (CTSO), and V (CTSV)), protease inhibitors (cystatins B (CYTB) and C (CTS3), serine proteinase inhibitors (SERPIN) B8, B9, A7, A10 and F1; peptidase inhibitor 3 (PI3) and PZP alpha-2-macroglobulin like (PZP)); metalloproteinases (matrix metallopeptidase 7 (MMP7), pappalysin 1 (PAPPA), ADAMTS4, 6 and 13), metalloproteinase inhibitor TIMP3, growth factors (fibroblast growth factors (FGF) 6, 9, and 20; platelet derived growth factor (PDGF); growth differentiation factors (GDF) 2 and 15; and angiopoietin 2 (ANGPT2)), glycans and glycan-binding receptors (glypicans (GPC) 1 and 5; C-type lectin receptors (CLEC) 3B, 4G and 9A; collectin subfamily member 12 (COLEC12)), and cytokines CXCL10, CCL16, interleukin 7 (IL7), IL17, IL19; and TNF superfamily members (TNFSF) 11 and 12. Protein-protein interaction enrichment analysis of these 85 ECM-associated proteins was performed; the accompanying plot highlights the most densely connected network components underscoring the role of ECM proteins in neutrophil chemotaxis. chemokine-mediated signaling, FGF activation and NODAL pathways in MAPS (Figure 3C). Overall, the involvement of ECM, metabolism, intracellular signaling and trafficking pathways, signify the depth of MAPS-associated inflammation.
Validation
We sought to validate these findings in an additional cohort comprising of 34 individuals (10 NoAPS, 10 TAPS, 4 MAPS, and 10 OAPS; Table 1). Other than the geographic location, there were no differences in the clinical characteristics between the two cohorts. For validation, we estimated the 1500 most differentially abundant proteins identified in the original analysis using the same assay (Supplementary Table 4). We were able to obtain hierarchical clustering of different APS subtypes in the validation cohort (Supplementary Figure S3).
Next, we wanted to see if a single set of discriminant proteins could cluster NoAPS, TAPS and MAPS in both the cohorts. For this, we first repeated linear discriminant analysis of the discovery cohort with the 1500 proteins measured in the validation cohort (Figure 3D top). Subsequently, we attempted clustering of the validation cohort by this protein set. Notably, the same set of discriminant proteins identified from the discovery cohort, clustered NoAPS, TAPS and MAPS in validation cohort (Figure 3D bottom). The proteins featured in this analysis (table on the right) included cytokines (TNFRSF21 and TNFRSF4), TNF receptor signal transducers (BAG4, DYRK3) coagulation factors (tissue factor, factor 13), proteases (lysozyme), protease inhibitors (PI3), and ECM regulators (ADAMTS6), that play a role in innate and adaptive immunity, thrombosis, ECM organization, and regulation of tissue inflammation.
Obstetric antiphospholipid syndrome
Although OAPS and NoAPS shared many of the proteomic alterations, individuals with a history of OAPS appeared distinct from TAPS and MAPS (Figure 1B). To evaluate plasma proteomic changes that characterize OAPS, first, we compared OAPS with controls. This analysis highlighted previously noted abnormalities shared by all APS subtypes, including C3, LTA4H, RBP2 and NAMPT (Supplementary Figure S4A). Next, we compared OAPS and TAPS. The volcano plot in Figure 4A shows the differentially abundant proteins between OAPS and TAPS, while Figure 4B highlights the functional enrichment analysis of these differential proteins. Restricting this analysis only to females with TAPS (n=7) did not alter the findings (Supplementary Figure S4B). Epithelial Mesenchymal Transition (SFRP4, FOXC2, LAMA2, TNC, VIM and FBLN5) was the most significantly altered term including proteins primarily higher in OAPS. Additionally, KRAS Signaling (EGF, GP1BA, PNMT, ITIH3, GDNF) was noted to be distinctive from previous analyses of TAPS and MAPS, also driven by abnormalities in OAPS.
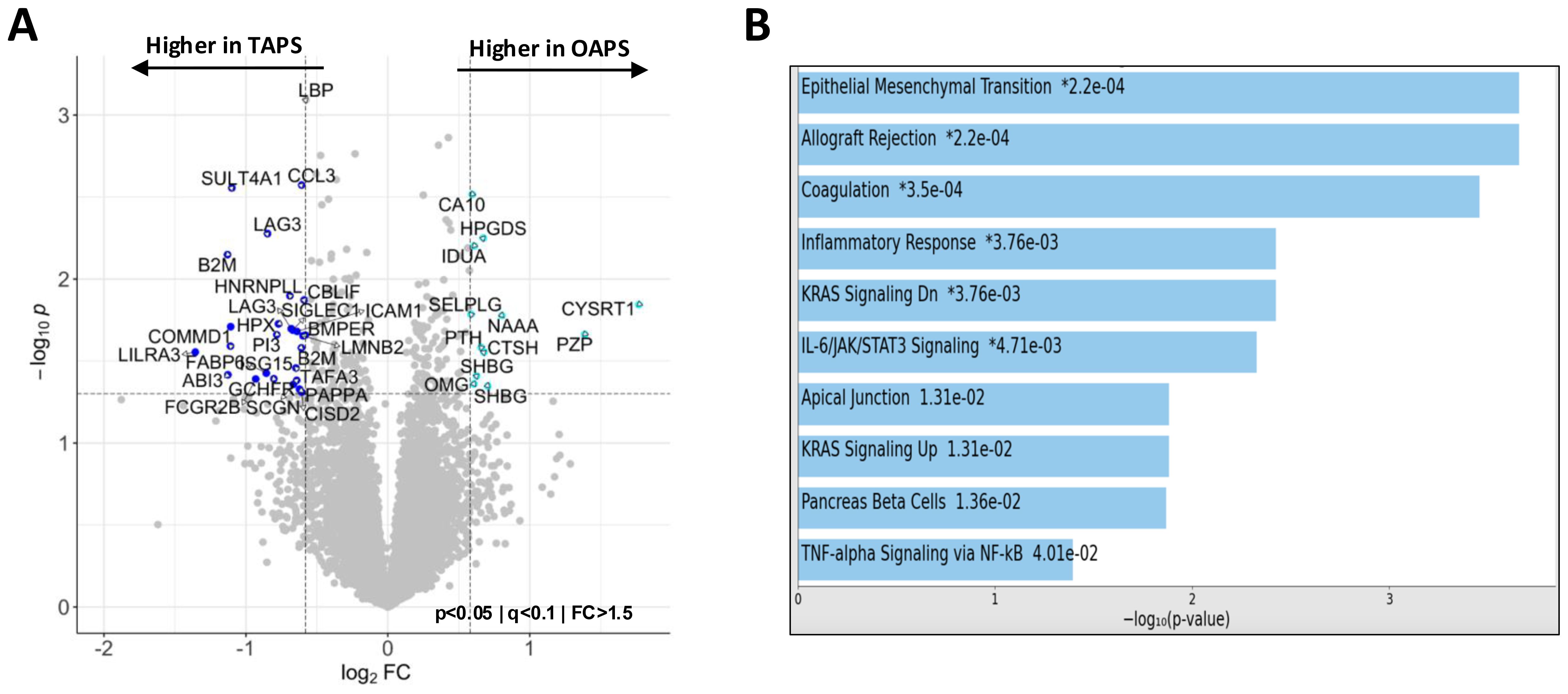
Figure 4. Plasma proteomics abnormalities in OAPS. (A) Volcano plot of differentially abundant proteins between TAPS and OAPS (p<0.05; q<0.1; fold change (FC)>1.5). (B) Bar chart showing the top 10 enriched terms (KEGG 2021 Human Library) in the protein list generated by the pairwise analysis of TAPS vs OAPS in A, along with the corresponding P values. Asterisk (*) denotes that the term also has a significant adjusted P value (<0.005).
Discussion
Understanding molecular mechanisms underlying APS could facilitate biomarker discovery for both diagnostic as well as therapeutic purposes. With this goal, we carried out comprehensive plasma proteomic profiling of patients with four different subtypes of primary APS (laboratory, thrombotic, obstetric, and microvascular/catastrophic) using a highly sensitive and specific single stranded-DNA aptamer-based assay (20, 21). Despite heterogeneity among APS patients, our data show that mere presence of circulating aPL induces multiple inflammatory pathways, particularly in individuals with ‘high-risk’ phenotype, a signature shared by all APS subtypes and consistent with the “first hit” of APS. Subsequently, the evolution of APS from laboratory to thrombotic and especially microvascular/catastrophic disease is characterized by increasing activation of coagulation and complement systems, native and adaptive immunity, culminating in ECM organization and tissue inflammation (Figure 5).
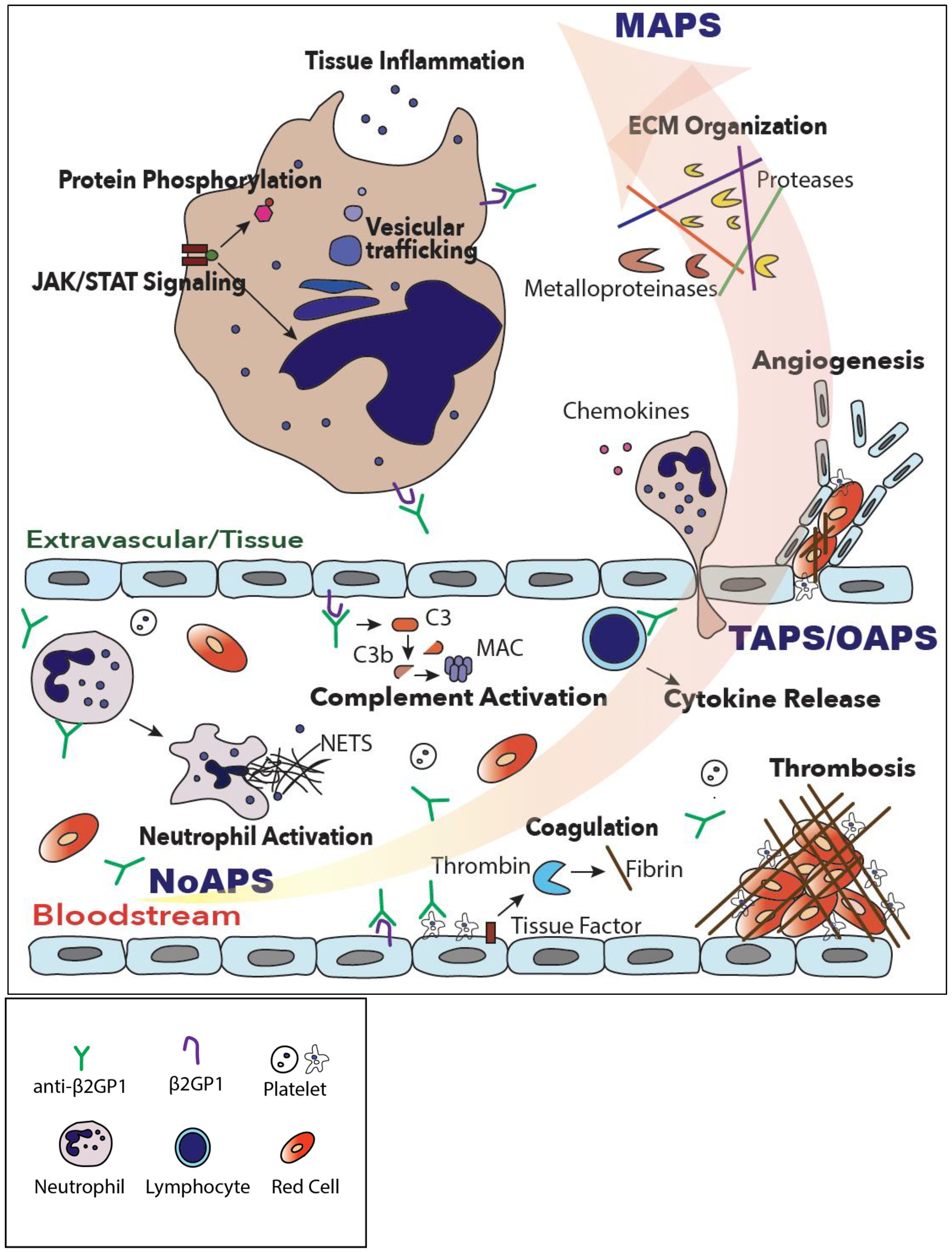
Figure 5. A model of progressive thromboinflammation in primary APS. While complement, coagulation and neutrophil activation are already evident in high-risk, ‘triple-positive’, laboratory APS (NoAPS), clinical subtypes of APS, OAPS, TAPS but particularly microvascular/catastrophic APS (MAPS), are characterized by severe thromboinflammation involving activation of intracellular signaling pathways, vesicular trafficking, protein phosphorylation, tissue inflammation and extracellular matrix organization.
Coagulation functional pathway abnormalities were identified as one of the principal proteomic abnormalities in APS. These proteins included procoagulants such as tissue factor, fibrinogen, factor XIII, as well as natural anticoagulants such as antithrombin, but also proteins related to pathobiology of coagulation such as complement proteins (C3), metalloproteinases (MMP7) and other serine protease inhibitors (SERPINE1 or PAI-1). Notably, we found reduce levels of plasma TF in APS, which could either reflect consumption and clearance of circulating soluble TF or elevated membrane binding. Similarly, CFP was lower in APS, which could reflect consumption or compensatory regulation of this positive regulator of complement pathway. We also identified abnormalities in factor II (prothrombin), and protein C, but these vitamin K-dependent proteins could have been affected by warfarin that some subjects were taking for anticoagulation. Additionally, complement proteins including C3 and complement regulators such as CFP, CD55, CD46, HNF4A, were differentially abundant in APS. Complement abnormalities have been well known in APS and have also been exploited therapeutically in treatment of MAPS (34). More importantly, our data provide evidence of progressive and overlapping activation of coagulation and complement pathways in development of thrombotic and microvascular forms of APS.
Some of the proteins that were highly significant and discriminatory in severe types of APS included LTA4H, NAMPT, TNFRSF21, TF, SULT4A1, which can potentially be used as biomarkers, both diagnostic, for risk stratification of APS, as well as therapeutic targets. In particular, LTA4H and NAMPT, which are known to play a role in pathophysiology of many inflammatory disorders, including SLE, and LTA4H has also been investigated as a potential therapeutic target (30–33). NAMPT, a DAMP, activates TLRs, which are known to play a critical role in pathophysiology of APS (4). TNFRSF21 (also known as death receptor 6 (DR6), a member of TNF receptor superfamily, is involved in immune regulation through NF-kB and has also been shown to be important in pathophysiology of many inflammatory disorders, including SLE (38).
Our findings also support a central role of immune activation in APS. Several immunologically relevant pathways were found to be activated encompassing both innate and adaptive immune systems. The most significant of these involve neutrophils (neutrophil activation, degranulation, and extracellular trap formation), phagocytosis activation, natural killer cell activation, platelet activation, and related pathways such as cytokine and interleukin signaling, JAK/STAT activation, and tyrosine kinase receptor signaling. Neutrophils have previously been reported to play an important role in pathophysiology of APS (4, 5, 10, 39). It remains unclear, though, whether neutrophils are the primary target of aPL or if neutrophil activation occurs as a secondary event. Anti-β2GP1, the pathologic antibodies implicated in APS, have been reported to activate multiple cellular components in the circulation, including platelets, endothelium, and neutrophils (4, 5, 11, 12, 40). Specifically, antibody-β2GP1 complex has been shown to interact and activate surface receptors such as glycoprotein 1bα, LRP8, TLRs. Our proteomics data also implicate activation of TLR, with evidence of activation of both TLR4-MyD88 and TLR4-TRIF pathways, as evidenced by the differentially abundant MAPK2K3, MAP3K7, BTK, IKBKG, PTPN11, BIRC2, SKP1, and UBE2 isoforms, particularly in MAPS (41). Our data also aligns with recent transcriptomics studies in APS, which includes bulk RNA sequencing or microarray analysis of whole blood, neutrophils, aortic valve tissue, and primary human endothelial cells treated with IgG fraction from APS patients (42–47). These studies identified and highlighted a role for interferon regulated genes and signaling (IFITs, IFNL3 etc.), cytokines (IL2, IL2R, IL6, IL15 etc.), chemokines (CCL13, CXCL10 etc.), other genes mediating both innate and adaptive immune responses (CYP26B1, LIFR, NLRPs, TRAF3, LILRA and B etc.), as well as ECM proteins (MMPs, SERPINs etc.), found to be significant in our proteomics analysis.
Proteomic abnormalities in TAPS and particularly MAPS provide evidence of both increasing intensity of thromboinflammation and cellular and tissue inflammation. This is evidenced by proteins belonging to the ECM organization, as well as intracellular vesicular, lysosome, and membrane trafficking, receptor mediated endocytosis, Rab GTPase activation, and various intracellular signaling pathways and metabolism. Notably, a growing body of literature supports a critical role played by ECM proteins in pathogenesis of systemic inflammatory diseases and chronic diseases that are known to occur through vascular and immune pathways (36, 37, 48, 49). We also found many plasma proteins in MAPS belonging to kidney-associated pathways, of particular interest in light of the kidney as a primary disease site in MAPS (3).
The proteomic alterations in OAPS appeared distinct, particularly when compared to TAPS and MAPS. This may reflect the modulation of immunoinflammatory milieu known to be associated with pregnancy (27, 28). Unlike other tissues, β2GP1 is known to have a high constitutive expression in extra-villous trophoblastic and syncytio-trophoblastic cells of the placenta, and may be essential for normal pregnancy (50–52). Could this differentially regulate the effect of anti-2GP1 antibodies and account for OAPS to have a distinct proteomic signature we noted? Moreover, regulated complement activation may be required for normal placentation, although dysregulated complement activation is a hallmark of APS as well as that of other pregnancy complications, such as pre-eclampsia (53). Given the role of β2GP1 as a complement regulator, it’s interaction with complement pathway may affect APS in pregnancy distinctively (54). These will require evaluation of larger cohorts of patients with OAPS, as well as further mechanistic studies.
Our study has some limitations. First, our sample size was relatively modest, but the consistency of our findings in the two cohorts provides meaningful insights that warrant further validation in larger cohorts of APS. Moreover, controls were not matched for age and sex, although the reproducibility of the observed patterns across the two demographically different cohorts supports the robustness of the most significant proteomics abnormalities in APS. While our data provide a comprehensive analysis of plasma proteome of APS and its subtypes, the exact mechanisms by which the identified inflammatory pathways evolve from aPL to either TAPS or MAPS remain unknown. Moreover, the most significant APS-associated proteins identified here need to be validated by specific immunoassays, which we did not perform. Notably, previously a high correlation between SomaScan and immunoassays has been reported (55, 56). Second, SomaScan 7K assay is known to contain an over-representation of secreted proteins. Mass spectrometry (MS)-based proteomics would be an ideal method for unbiased screening but carries low sensitivity. In fact, MS-based proteomics has previously been attempted on plasma, urine and extracellular vesicles in APS and a systematic review of 11 studies yielded only a combined 82 dysregulated proteins, belonging to cellular activation/degranulation and thrombosis pathways (57–60). Next, our samples were not drawn at the time of thromboembolic event. Although, the proteomic abnormalities we report may reflect the ‘baseline inflammation” associated with different APS subtypes, inducible proteins expected to be expressed only at the time of clinical events and relevant to thrombosis may be missed. Moreover, the trend in identified proteomic abnormalities is not known and it is not possible to exclude reverse causation, that is, some of the proteins may be a consequence of disease manifestation. The possibility of false positives also exists with simultaneous testing of thousands of proteins, but we used standard statistical approaches to control the false discovery rate, thereby minimizing type 1 errors. Lastly, our studies do not provide any evidence of the implicated upstream pathways or inflammatory cells directly involved in APS initiation. Despite these limitations, we provide the most comprehensive and up to date proteomic analysis of APS and its subtypes, and our findings provide important insights into the two-hit model of APS and identify potential new targets for risk stratification and therapeutic intervention in APS across the entire spectrum of clinical disease.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by The protocol was approved by the Institutional Review Boards of all participating institutions. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
AP: Data curation, Writing – review & editing, Methodology, Formal Analysis, Investigation, Conceptualization, Software, Funding acquisition. AB: Investigation, Writing – review & editing. LA: Writing – review & editing. JK: Writing – review & editing. MG: Writing – review & editing. IC: Writing – review & editing. DB: Writing – review & editing. RL-P: Writing – review & editing. HB: Writing – review & editing. NK: Writing – review & editing. MP: Writing – review & editing. RC: Writing – review & editing. VP: Writing – review & editing. PM: Writing – review & editing. HC: Writing – review & editing. RW: Writing – review & editing. MB: Writing – review & editing. GG: Writing – review & editing, Conceptualization. SG: Writing – review & editing. JH: Writing – review & editing. AL: Methodology, Funding acquisition, Writing – review & editing, Project administration, Supervision, Resources, Conceptualization. DE: Project administration, Writing – review & editing, Resources. AS: Investigation, Visualization, Data curation, Resources, Methodology, Validation, Conceptualization, Project administration, Supervision, Writing – original draft, Funding acquisition, Software, Formal Analysis, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Institute of Health (grant numbers K08HL150246) to AS. The work was additionally supported by The DeLuca Center for Innovation in Hematology Research at Yale Cancer Center, The Frederick A. Deluca Foundation, and an anonymous donor to the Classical Hematology program at Yale Cancer Center.
Acknowledgments
The authors thank the APS ACTION Network and Executive Committee for providing valuable input and guidance. The authors thank Ms. Sia Pilar Sharda Mendez for her kind assistance with Figure 5.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1676578/full#supplementary-material
References
1. Garcia D and Erkan D. Diagnosis and management of the antiphospholipid syndrome. N Engl J Med. (2018) 379:1290. doi: 10.1056/NEJMra1705454
2. Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, et al. Antiphospholipid syndrome. Nat Rev Dis Primers. (2018) 4:18005. doi: 10.1038/nrdp.2018.5
3. Barbhaiya M, Zuily S, Naden R, Hendry A, Manneville F, Amigo MC, et al. 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Ann Rheum Dis. (2023) 82:1258–70. doi: 10.1136/ard-2023-224609
4. Knight JS, Branch DW, and Ortel TL. Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. BMJ. (2023) 380:e069717. doi: 10.1136/bmj-2021-069717
5. Radic M and Pattanaik D. Cellular and molecular mechanisms of anti-phospholipid syndrome. Front Immunol. (2018) 9:969. doi: 10.3389/fimmu.2018.00969
6. Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, van Breda-Vriesman PJ, et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. (1990) 335:1544–7. doi: 10.1016/0140-6736(90)91374-J
7. Arad A, Proulle V, Furie RA, Furie BC, and Furie B. beta(2)-Glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood. (2011) 117:3453–9. doi: 10.1182/blood-2010-08-300715
8. de Groot PG and Urbanus RT. Screening: Guidelines for antiphospholipid antibody detection. Nat Rev Rheumatol. (2011) 8:125–6. doi: 10.1038/nrrheum.2011.188
9. Munzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, et al. NLRP3 inflammasome assembly in neutrophils is supported by PAD4 and promotes NETosis under sterile conditions. Front Immunol. (2021) 12:683803. doi: 10.3389/fimmu.2021.683803
10. Tan Y, Liu Q, Li Z, Yang S, and Cui L. Pyroptosis-triggered pathogenesis: New insights on antiphospholipid syndrome. Front Immunol. (2023) 14:1155222. doi: 10.3389/fimmu.2023.1155222
11. Bontadi A, Ruffatti A, Falcinelli E, Giannini S, Marturano A, Tonello M, et al. Platelet and endothelial activation in catastrophic and quiescent antiphospholipid syndrome. Thromb Haemost. (2013) 109:901–8. doi: 10.1160/TH12-03-0212
12. Borghi MO, Raschi E, Grossi C, Chighizola CB, and Meroni PL. Toll-like receptor 4 and beta2 glycoprotein I interaction on endothelial cells. Lupus. (2014) 23:1302–4. doi: 10.1177/0961203314536479
13. Brandt KJ, Fickentscher C, Boehlen F, Kruithof EK, and de Moerloose P. NF-kappaB is activated from endosomal compartments in antiphospholipid antibodies-treated human monocytes. J Thromb Haemost. (2014) 12:779–91. doi: 10.1111/jth.12536
14. Colasanti T, Alessandri C, Capozzi A, Sorice M, Delunardo F, Longo A, et al. Autoantibodies specific to a peptide of beta2-glycoprotein I cross-react with TLR4, inducing a proinflammatory phenotype in endothelial cells and monocytes. Blood. (2012) 120:3360–70. doi: 10.1182/blood-2011-09-378851
15. Babacic H, Christ W, Araujo JE, Mermelekas G, Sharma N, Tynell J, et al. Comprehensive proteomics and meta-analysis of COVID-19 host response. Nat Commun. (2023) 14:5921. doi: 10.1038/s41467-023-41159-z
16. Filbin MR, Mehta A, Schneider AM, Kays KR, Guess JR, Gentili M, et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep Med. (2021) 2:100287. doi: 10.1016/j.xcrm.2021.100287
17. Govaere O, Hasoon M, Alexander L, Cockell S, Tiniakos D, Ekstedt M, et al. A proteo-transcriptomic map of non-alcoholic fatty liver disease signatures. Nat Metab. (2023) 5:572–8. doi: 10.1038/s42255-023-00775-1
18. Walker KA, Chen J, Shi L, Yang Y, Fornage M, Zhou L, et al. Proteomics analysis of plasma from middle-aged adults identifies protein markers of dementia risk in later life. Sci Transl Med. (2023) 15:eadf5681. doi: 10.1126/scitranslmed.adf5681
19. Sevim E, Zisa D, Andrade D, Sciascia S, Pengo V, Tektonidou MG, et al. Characteristics of patients with antiphospholipid antibody positivity in the APS ACTION international clinical database and repository. Arthritis Care Res (Hoboken). (2022) 74:324–35. doi: 10.1002/acr.24468
20. Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PloS One. (2010) 5:e15004. doi: 10.1371/journal.pone.0015004
21. Rohloff JC, Gelinas AD, Jarvis TC, Ochsner UA, Schneider DJ, Gold L, et al. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther Nucleic Acids. (2014) 3:e201. doi: 10.1038/mtna.2014.49
22. Eldjarn GH, Ferkingstad E, Lund SH, Helgason H, Magnusson OT, Gunnarsdottir K, et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature. (2023) 622:348–58. doi: 10.1038/s41586-023-06563-x
23. Rooney MR, Chen J, Ballantyne CM, Hoogeveen RC, Boerwinkle E, Yu B, et al. Correlations within and between highly multiplexed proteomic assays of human plasma. Clin Chem. (2025). doi: 10.1093/clinchem/hvaf030
24. Kirsher DY, Chand S, Phong A, Nguyen B, Szoke BG, and Ahadi S. The current landscape of plasma proteomics: technical advances, biological insights, and biomarker discovery. bioRxiv. (2025) 8:279. doi: 10.1101/2025.02.14.638375
25. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. (2013) 14:128. doi: 10.1186/1471-2105-14-128
26. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. (2019) 10:1523. doi: 10.1038/s41467-019-09234-6
27. Forger F and Villiger PM. Immunological adaptations in pregnancy that modulate rheumatoid arthritis disease activity. Nat Rev Rheumatol. (2020) 16:113–22. doi: 10.1038/s41584-019-0351-2
28. Piccinni MP, Lombardelli L, Logiodice F, Kullolli O, Parronchi P, and Romagnani S. How pregnancy can affect autoimmune diseases progression? Clin Mol Allergy. (2016) 14:11. doi: 10.1186/s12948-016-0048-x
29. Pengo V, Ruffatti A, Legnani C, Testa S, Fierro T, Marongiu F, et al. Incidence of a first thromboembolic event in asymptomatic carriers of high-risk antiphospholipid antibody profile: a multicenter prospective study. Blood. (2011) 118:4714–8. doi: 10.1182/blood-2011-03-340232
30. Rohn TA, Numao S, Otto H, Loesche C, and Thoma G. Drug discovery strategies for novel leukotriene A4 hydrolase inhibitors. Expert Opin Drug Discov. (2021) 16:1483–95. doi: 10.1080/17460441.2021.1948998
31. Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. (2006) 203:837–42. doi: 10.1084/jem.20052371
32. Audrito V, Messana VG, and Deaglio S. NAMPT and NAPRT: two metabolic enzymes with key roles in inflammation. Front Oncol. (2020) 10:358. doi: 10.3389/fonc.2020.00358
33. Li M, Lai Y, Chen B, Guo C, Zhou M, Zhao S, et al. NAMPT is a metabolic checkpoint of IFNgamma-producing CD4(+) T cells in lupus nephritis. Mol Ther. (2023) 31:193–210. doi: 10.1016/j.ymthe.2022.09.013
34. Chaturvedi S, Brodsky RA, and McCrae KR. Complement in the pathophysiology of the antiphospholipid syndrome. Front Immunol. (2019) 10:449. doi: 10.3389/fimmu.2019.00449
35. Rodriguez-Pinto I, Moitinho M, Santacreu I, Shoenfeld Y, Erkan D, Espinosa G, et al. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun Rev. (2016) 15:1120–4. doi: 10.1016/j.autrev.2016.09.010
36. Bergmeier W and Hynes RO. Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb Perspect Biol. (2012) 4. doi: 10.1101/cshperspect.a005132
37. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. (2010) 10:712–23. doi: 10.1038/nri2852
38. Fujikura D, Ikesue M, Endo T, Chiba S, Higashi H, and Uede T. Death receptor 6 contributes to autoimmunity in lupus-prone mice. Nat Commun. (2017) 8:13957. doi: 10.1038/ncomms13957
39. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. (2015) 67:2990–3003. doi: 10.1002/art.39247
40. Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. (2017) 6. doi: 10.7554/eLife.24437
41. Fitzgerald KA and Kagan JC. Toll-like receptors and the control of immunity. Cell. (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
42. Verrou KM, Sfikakis PP, and Tektonidou MG. Whole blood transcriptome identifies interferon-regulated genes as key drivers in thrombotic primary antiphospholipid syndrome. J Autoimmun. (2023) 134:102978. doi: 10.1016/j.jaut.2022.102978
43. Knight JS, Meng H, Coit P, Yalavarthi S, Sule G, Gandhi AA, et al. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight. (2017) 2(18):e93897. doi: 10.1172/jci.insight.93897
44. Perez-Sanchez C, Barbarroja N, Messineo S, Ruiz-Limon P, Rodriguez-Ariza A, Jimenez-Gomez Y, et al. Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann Rheum Dis. (2015) 74:1441–9. doi: 10.1136/annrheumdis-2013-204600
45. Ugolini-Lopes MR, Torrezan GT, Gandara APR, Olivieri EHR, Nascimento IS, Okazaki E, et al. Enhanced type I interferon gene signature in primary antiphospholipid syndrome: Association with earlier disease onset and preeclampsia. Autoimmun Rev. (2019) 18:393–8. doi: 10.1016/j.autrev.2018.11.004
46. Plunde O, Svenungsson E, Ferrannini G, Franco-Cereceda A, and Back M. Antiphospholipid antibodies in patients with calcific aortic valve stenosis. Rheumatol (Oxford). (2023) 62:1187–96. doi: 10.1093/rheumatology/keac466
47. Patsouras M, Alexopoulou E, Foutadakis S, Tsiki E, Karagianni P, Agelopoulos M, et al. Antiphospholipid antibodies induce proinflammatory and procoagulant pathways in endothelial cells. J Transl Autoimmun. (2023) 6:100202. doi: 10.1016/j.jtauto.2023.100202
48. Sleeboom JJF, van Tienderen GS, Schenke-Layland K, van der Laan LJW, Khalil AA, and Verstegen MMA. The extracellular matrix as hallmark of cancer and metastasis: From biomechanics to therapeutic targets. Sci Transl Med. (2024) 16:eadg3840. doi: 10.1126/scitranslmed.adg3840
49. Zhou X, Zhang Y, and Wang N. Systematic identification of key extracellular proteins as the potential biomarkers in lupus nephritis. Front Immunol. (2022) 13:915784. doi: 10.3389/fimmu.2022.915784
50. Chamley LW, Allen JL, and Johnson PM. Synthesis of beta2 glycoprotein 1 by the human placenta. Placenta. (1997) 18:403–10. doi: 10.1016/S0143-4004(97)80040-9
51. La Rosa L, Meroni PL, Tincani A, Balestrieri G, Faden D, Lojacono A, et al. Beta 2 glycoprotein I and placental anticoagulant protein I in placentae from patients with antiphospholipid syndrome. J Rheumatol. (1994) 21:1684–93.
52. Miyakis S, Robertson SA, and Krilis SA. Beta-2 glycoprotein I and its role in antiphospholipid syndrome-lessons from knockout mice. Clin Immunol. (2004) 112:136–43. doi: 10.1016/j.clim.2004.02.014
53. Girardi G, Lingo JJ, Fleming SD, and Regal JF. Essential role of complement in pregnancy: from implantation to parturition and beyond. Front Immunol. (2020) 11:1681. doi: 10.3389/fimmu.2020.01681
54. Gropp K, Weber N, Reuter M, Micklisch S, Kopka I, Hallstrom T, et al. beta(2)-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. (2011) 118:2774–83. doi: 10.1182/blood-2011-02-339564
55. George MJ, Kleveland O, Garcia-Hernandez J, Palmen J, Lovering R, Wiseth R, et al. Novel insights into the effects of interleukin 6 antagonism in non-ST-segment-elevation myocardial infarction employing the SOMAscan proteomics platform. J Am Heart Assoc. (2020) 9:e015628. doi: 10.1161/JAHA.119.015628
56. Timsina J, Gomez-Fonseca D, Wang L, Do A, Western D, Alvarez I, et al. Comparative analysis of alzheimer's disease cerebrospinal fluid biomarkers measurement by multiplex SOMAscan platform and immunoassay-based approach. J Alzheimers Dis. (2022) 89:193–207. doi: 10.3233/JAD-220399
57. Araujo DM, Rodrigues CEM, Goncalves NGG, Rabelo-Junior CN, Lobo MDP, Moreira RA, et al. Proteins involved in the induction of procoagulant activity and autoimmune response in patients with primary antiphospholipid syndrome. Clin Appl Thromb Hemost. (2020) 26:1076029620905338. doi: 10.1177/1076029620905338
58. Anunciacion-Llunell A, Miro-Mur F, Esteve-Valverde E, Marques-Soares J, Pardos-Gea J, and Alijotas-Reig J. Proteomics and enriched biological processes in Antiphospholipid syndrome: A systematic review. Autoimmun Rev. (2021) 20:102982. doi: 10.1016/j.autrev.2021.102982
59. Tian W, Shi D, Zhang Y, Wang H, Tang H, Han Z, et al. Deep proteomic analysis of obstetric antiphospholipid syndrome by DIA-MS of extracellular vesicle enriched fractions. Commun Biol. (2024) 7:99. doi: 10.1038/s42003-024-05789-3
60. Zhou Z, You Y, Wang F, Sun Y, Teng J, Liu H, et al. Urine proteomics differentiate primary thrombotic antiphospholipid syndrome from obstetric antiphospholipid syndrome. Front Immunol. (2021) 12:702425. doi: 10.3389/fimmu.2021.702425
Keywords: antiphospholid syndrome, plasma proteomics, thromboinflammation, immune activation, coagulation, complement
Citation: Pine A, Butt A, Andreoli L, Knight JS, Gerosa M, Cecchi I, Branch DW, Lopez-Pedrera R, Belmont HM, Kello N, Petri M, Cervera R, Pengo V, Meroni PL, Cohen H, Willis R, Bertolccini ML, Goshua G, Gu S, Hwa J, Lee AI, Erkan D and Sharda AV (2025) A proteomic map of thromboinflammatory signatures in antiphospholipid syndrome: results from antiphospholipid syndrome alliance for clinical trials and international networking (APS ACTION) registry. Front. Immunol. 16:1676578. doi: 10.3389/fimmu.2025.1676578
Received: 30 July 2025; Accepted: 01 October 2025;
Published: 16 October 2025.
Edited by:
Davide Firinu, University of Cagliari, ItalyReviewed by:
Antonella Mameli, Department of General Multi-specialist Surgery at the University Hospital of Cagliari, ItalyCamila De Oliveira Vaz, Brigham and Women’s Hospital and Harvard Medical School, United States
Copyright © 2025 Pine, Butt, Andreoli, Knight, Gerosa, Cecchi, Branch, Lopez-Pedrera, Belmont, Kello, Petri, Cervera, Pengo, Meroni, Cohen, Willis, Bertolccini, Goshua, Gu, Hwa, Lee, Erkan and Sharda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anish V. Sharda, YW5pc2guc2hhcmRhQHlhbGUuZWR1