 Yingshi Bao1†
Yingshi Bao1† Yutong Su
Yutong Su Huaping Du
Huaping Du- 1Department of Neurology, Suzhou Ninth People’s Hospital, Soochow University, Suzhou, Jiangsu, China
- 2Department of Neurology, Qidong People’s Hospital, Qidong Liver Cancer Institute, Affiliated Qidong Hospital of Nantong University, Nantong, China
- 3The Second Clinical Medical College, Nanjing Medical University, Nanjing, Jiangsu, China
Chronic neuroinflammation is increasingly recognized not merely as a consequence of CNS pathology but as a driver of glioma initiation. Sustained immune activation, induced by trauma, infection, or neurodegeneration, reshapes the brain’s immune milieu in ways that favor malignant transformation. Persistent inflammation activates glial cells, triggers cytokine release, and disrupts the blood-brain barrier, permitting immune infiltration and dysfunction. These changes promote the accumulation and reprogramming of immunosuppressive populations, including regulatory T cells and myeloid-derived suppressor cells, while resident microglia and astrocytes adopt tumor-supportive phenotypes. We highlight signaling axes such as IL-6/STAT3, NF-κB, and TGF-β that connect immune dysregulation to epigenetic instability and the emergence of glioma-initiating cells. By tracing the progression from inflammation to tumorigenesis, we identify opportunities for early immune-based intervention, particularly in individuals with chronic neuroinflammatory conditions.
1 Introduction
For decades, the central nervous system (CNS) was viewed as immune-privileged, shielded by the blood–brain barrier (BBB), lacking conventional lymphatic drainage, and sparsely populated with professional antigen-presenting cells. This led to the belief that it was largely isolated from systemic immunity. This view has evolved. The CNS possesses a tightly regulated, responsive immune environment coordinated by resident glia, endothelial cells, and, under specific conditions, infiltrating peripheral immune cells (1).
Neuroinflammation, defined as immune activation within the CNS, may be triggered by infection, trauma, autoimmunity, or neurodegeneration. Acutely, it can be protective, facilitating repair and debris clearance. When prolonged or dysregulated, however, it becomes maladaptive, driving oxidative stress, synaptic dysfunction, glial scarring, and long-term microenvironmental damage (2). Recent research increasingly implicates chronic neuroinflammation not only as a consequence of CNS injury but as an active participant in oncogenic processes, particularly in glioma (3, 4).
Gliomas, and in particular glioblastomas, remain among the most aggressive primary brain tumors. Beyond canonical molecular hallmarks, including mutations in IDH1, amplification of EGFR, and loss of PTEN, the tumor immune microenvironment (TIME) critically shapes disease course and therapeutic resistance (5, 6). Chronic inflammation promotes genomic instability, epigenetic reprogramming, and immunosuppressive niches that enable the transformation of glial precursors into tumor-initiating cells (7). Epidemiological and preclinical evidence links chronic neuroinflammatory states with a possible increase in glioma risk; however, findings are heterogeneous and mechanisms remain under investigation.
In this review, we synthesize how chronic neuroinflammation contributes to gliomagenesis through three connected processes: (1) initiation of neuroinflammation and innate immune activation; (2) remodeling of the CNS immune landscape under chronic stress; and (3) immune-driven selection of resistant, tumor-initiating glial clones. We highlight intervention points along this continuum, focusing on early biomarkers and prevention-oriented strategies.
2 Neuroinflammatory triggers and immune initiation
Neuroinflammation begins when resident glia or infiltrating leukocytes detect damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) via pattern-recognition receptors (PRRs) expressed on microglia and astrocytes. Toll-like receptors (TLRs), especially TLR2 and TLR4, are prominent: TLR4 recognizes high-mobility group box 1 (HMGB1), a prototypical DAMP from necrotic neurons, which activates NF-κB and upregulates pro-inflammatory genes (8, 9).
In parallel, the nucleotide-binding domain-like receptor protein 3 (NLRP3) inflammasome senses cytosolic danger signals, including extracellular ATP, mitochondrial dysfunction, and misfolded proteins, leading to caspase-1 activation and maturation of IL-1β and IL-18 (10). PRR and inflammasome signaling initiates and sustains CNS immune responses and has been implicated in early gliomagenesis in animal models (11).
Microglia rapidly adopt a classically activated (M1-like) program characterized by iNOS, IL-1β, TNF-α, and IL-6, aiding pathogen clearance (12). However, prolonged activation sustains oxidative DNA damage in neighboring cells, fosters proliferation, and disrupts neuron–glia communication (13).
Astrocytes likewise respond to IL-1β and TNF-α by producing chemokines that increase BBB permeability and recruit monocytes and T cells, while secreting IL-6 and GM-CSF to reinforce glial activation (14). BBB breakdown allows continued influx of peripheral immune cells—CD4+/CD8+ T cells, NK cells, and myeloid-derived suppressor cells (MDSCs)—which can become exhausted or immunosuppressive (15, 16). IL-6–JAK/STAT3 signaling supports glial survival and expansion of stress-tolerant precursors, whereas TNF-α and IL-1β promote extracellular matrix remodeling, angiogenesis, and excitotoxicity (17).
Key triggers, receptors, cellular responses, and implications are summarized in Table 1. Together, these processes define the early phase of neuroinflammation: a complex, self-reinforcing network of glial activation, cytokine signaling, and immune cell recruitment. While initially aimed at preserving CNS integrity, persistent inflammation may generate a molecular and cellular environment primed for neoplastic transformation. Recognizing and intervening during this window of immune dysregulation could hold promise for glioma prevention or early therapeutic targeting.
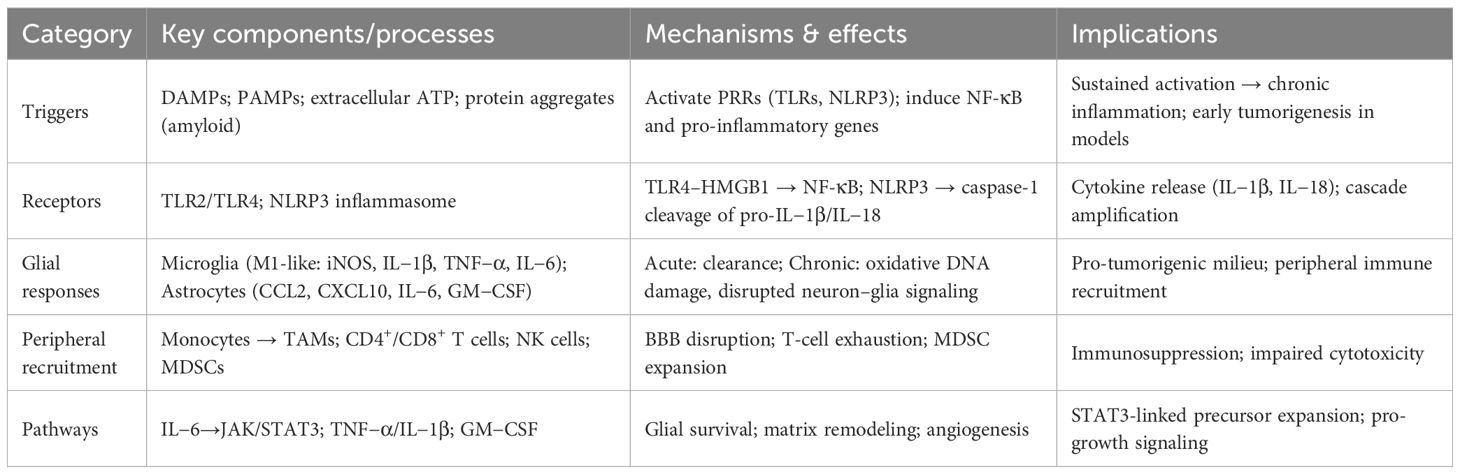
Table 1. Neuroinflammatory triggers and immune initiation in gliomagenesis.
3 Remodeling of the CNS immune microenvironment
Persistent neuroinflammation progressively remodels the CNS immune milieu into a permissive niche for tumor initiation, characterized by expansion of glioma-associated microglia/macrophages (GAMs), intensified astrocyte–immune crosstalk, and metabolic/checkpoint reprogramming. Below, we detail these processes.
3.1 Polarization of microglia and recruitment of macrophages
Microglia, the resident macrophages of the CNS, shift from a pro-inflammatory M1-like to an immunosuppressive M2-like state under chronic stimulation, secreting IL−10, TGF−β, growth factors (EGF, VEGF), and matrix metalloproteinases (MMPs) that promote tissue remodeling, angiogenesis, and suppression of cytotoxic immunity (18). Glioma-derived CSF1 and GM−CSF further regulate this transition (19, 20).
BBB disruption enables peripheral monocytes to infiltrate and differentiate into macrophages (21). Together with resident microglia, they form GAMs, which can constitute a substantial fraction of glioblastoma mass and exhibit transcriptional programs such as CD163, CD204, TREM2, ARG1 conferring immunotolerant, tumor−promoting functions (22).
3.2 Astrocyte–immune crosstalk
Astrocytes also play an important role in the process of immune remodeling, mainly through cytokine secretion and direct interaction with tumor cells and immune cells. In the chronic inflammatory environment, astrocytes are in a reactive state and upregulate STAT3 signaling, linked to immunosuppression via PD-L1 expression and CCL2-mediated Treg recruitment (23, 24). Through gap junctions, astrocytes can deliver glutathione and metabolic intermediates to adjacent glioma cells, enhancing oxidative-stress resistance (25). Context-dependent astrocyte states include neurotoxic A1 and tumor-supportive phenotypes secreting lipocalin-2, complement (C3), and MMPs, thereby promoting invasion and immune escape (15).
3.3 Neural regulation of immunity in the TME
Glioma associated immune cells, including microglia and macrophages, MDSCs and Tregs, are dynamically shaped by nociceptive, adrenergic, cholinergic and checkpoint mediated neuroregulatory pathways. Following tissue injury, peripheral nociceptive fibers release substance P and CGRP. Substance P acting through NK1R supports the survival of activated T cells, enhances proinflammatory cytokine production by macrophages and amplifies CCL5 driven neutrophil chemotaxis and migration. In contrast, CGRP drives macrophages toward an anti inflammatory M2 phenotype, inhibits NLRP3 inflammasome activation and IL 1β release in M1 macrophages and reduces cytokine secretion from macrophages and dendritic cells, thereby dampening antigen presentation and effector T cell activation. β-adrenergic signaling in tumor and myeloid cells induces immunomodulatory cytokines (IL-8, IL-6) and suppressive programs in macrophages while enhancing fatty-acid oxidation in MDSCs and impairing glycolytic reprogramming in CD8+ T cells (26–28). Cholinergic vagal signaling activates spleen resident memory CD4+ ChAT+ T cells, which release acetylcholine to modulate nicotinic receptors on macrophages and secrete trefoil factor 2 that restrains MDSC proliferation through CXCR4 signaling (29). In addition, nerve fibers in the prostate tumor microenvironment can express immune checkpoint molecules such as PD L1 that bind PD 1 on CD8+ T cells, promoting their depletion and weakening antitumor immunity (30). Figure 1 schematically summarizes neuro–immune–glioma crosstalk. Where observations are largely derived from established tumors, we explicitly note uncertainties about their timing relative to pre-tumor states.
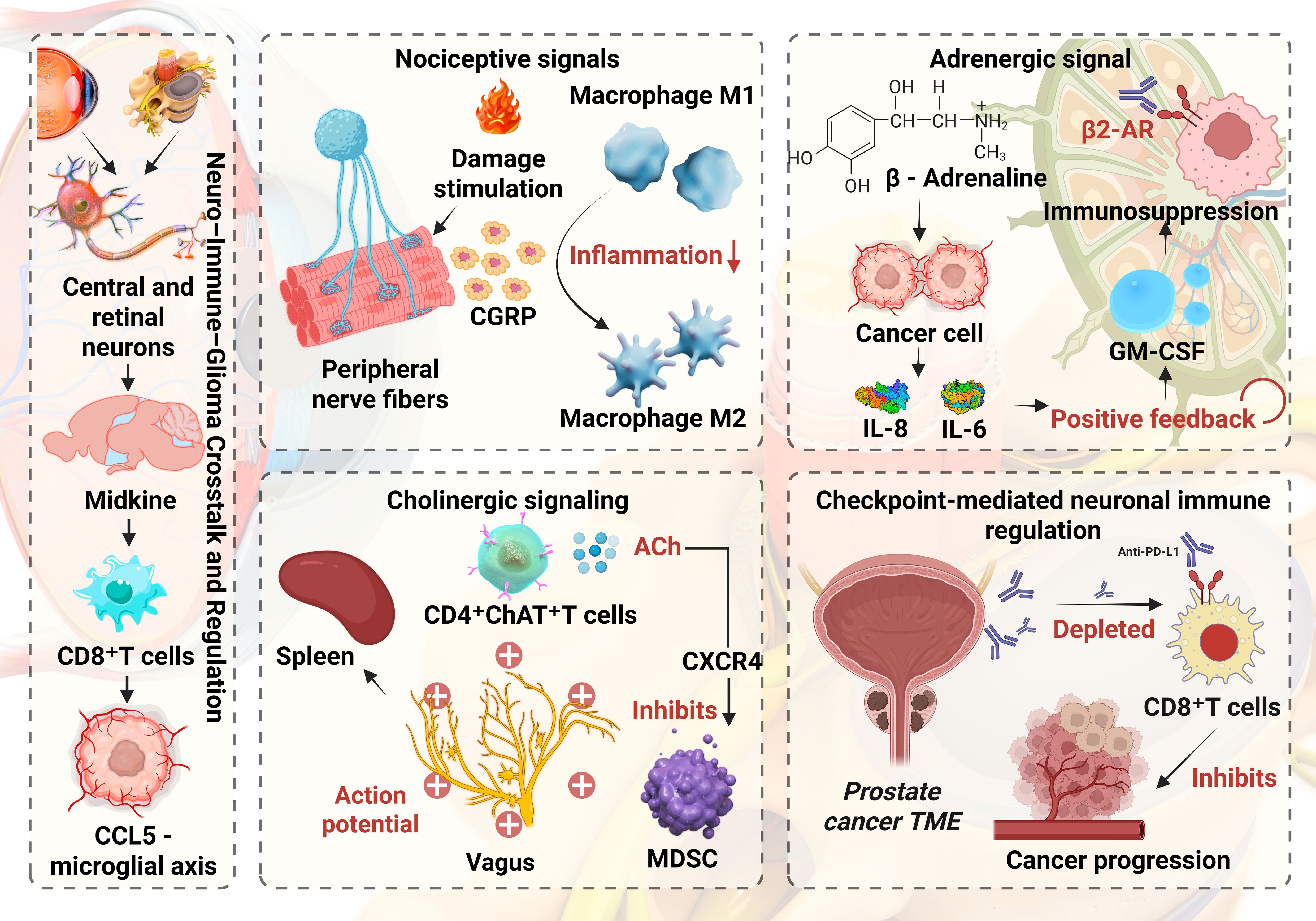
Figure 1. Neuro–Immune–Glioma crosstalk and regulation.
3.4 Immunosuppressive cell populations and metabolic reprogramming
Regulatory T cells (Tregs) accumulate in chronically inflamed CNS tissue and glioma, driven by chemokine axes such as CCL22–CCR4, CCL2–CCR2, and CXCL12–CXCR4 (31). Beyond increased numbers, Tregs exhibit functional specialization: effector Tregs expressing high CTLA-4 and TIGIT suppress dendritic-cell co-stimulation, while ICOS+ Tregs preferentially expand in IL-10–rich niches. Astrocyte and tumor-derived TGF-β stabilizes FOXP3 and enforces suppressive transcriptional programs. In pre-neoplastic settings, microglia-derived IL-10 and CSF1 may precondition perivascular niches to favor Treg retention, blunting early anti-tumor surveillance (32).
Myeloid-derived suppressor cells (MDSCs) encompass polymorphonuclear (PMN-) and monocytic (M-) subsets. PMN-MDSCs release reactive oxygen species and peroxynitrite, nitrating the T-cell receptor and diminishing antigen sensitivity. M-MDSCs deploy ARG1 and iNOS to deplete L-arginine and generate nitric oxide, inducing T-cell cycle arrest (33). Hypoxia and lactate stabilize HIF-1α and drive a ‘metabolic trap’—upregulating CD39/CD73 to convert ATP to adenosine, which signals via A2A receptors on T cells to inhibit cytotoxicity. This adenosinergic circuit emerges early during chronic inflammation when ATP release and ectonucleotidase expression are heightened (34).
Metabolic reprogramming compounds cellular suppression. Glioma and reactive glia increase glycolysis and glutaminolysis, acidifying the interstitium and impairing T-cell receptor signaling. IDO1-mediated tryptophan catabolism accumulates kynurenine, activating the aryl hydrocarbon receptor (AhR) in T cells and macrophages to reinforce tolerogenic states (35). Lipid uptake and oxidation in GAMs and MDSCs, potentiated by CPT1A, supports an M2-like phenotype and limits antigen presentation (36). NAD+/SIRT signaling and redox buffering (glutathione, thioredoxin) enable GICs and GAMs to withstand oxidative stress generated during chronic inflammation (37). Collectively, these cellular and metabolic programs not only sustain immunosuppression in established tumors but plausibly predate histological transformation, constituting a permissive niche for malignant initiation.
3.5 Recent advances in TAM reprogramming
Therapeutic re-education of tumor-associated macrophages (TAMs) aims to convert immunosuppressive M2-like states into inflammatory, antitumor phenotypes. CSF1R blockade reduces TAM survival and can shift transcriptional programs; however, compensatory recruitment and phenotypic plasticity limit durability. Combinatorial approaches with radiotherapy or oncolytic agents may enhance efficacy by providing danger signals that sustain M1-like activation (38).
Innate agonists, including TLR7/8 ligands, STING agonists, and CD40 agonists, promote antigen presentation and pro-inflammatory cytokine release. Localized delivery is critical to avoid systemic toxicity while overcoming the BBB. Brain-targeting nanoparticles that engage transferrin receptors or utilize RVG29/angiopep-2 peptides can ferry cargo across the BBB, co-delivering cytotoxics and immune adjuvants to both tumor cells and TAMs, thereby coordinating direct tumor kill with myeloid reprogramming (39, 40).
Phagocytosis checkpoints provide complementary targets. Antibodies against CD47 and SIRPα decouple inhibitory cues, permitting opsonization-dependent clearance by microglia/macrophages (41). Microglial receptors such as P2RY12 and TREM2 integrate purinergic and damage signals; modulating these pathways may augment antigen handling and crosstalk with dendritic cells for more effective T-cell priming (42, 43).
4 Immune-driven selection and tumorigenic transition
Chronic inflammation does not simply suppress immunity—it imposes selective pressures that favor glial clones equipped to evade or co-opt immune control. Over time, these pressures edit the antigenic landscape, reshape DNA repair and epigenetic programs, and culminate in the expansion of glioma-initiating cells (GICs).
4.1 Oncogenic signaling under immune pressure
IL-6/STAT3 and NF-κB function as central hubs that link inflammatory cues to survival, proliferation, and stemness. Sustained STAT3 signaling upregulates anti-apoptotic BCL2 family members and cyclins, while cooperating with HIF-1α under hypoxia to drive angiogenic programs (VEGF) (44). NF-κB activation downstream of TLRs and cytokine receptors induces matrix-remodeling enzymes (MMP2/9), chemokines, and immune checkpoints. These pathways collectively endow pre-malignant glial cells with resistance to stress and immune-mediated killing.
TGF-β can further promote the development of malignant evolution of glial cells by inducing epithelial mesenchymal transition (EMT)-like changes, conferring greater migratory capacity and resistance to immune-mediated killing (45). TGF-β can also downregulate the expression of MHC class I molecules and reduce the “visibility” of these cells to cytotoxic T lymphocytes (CTLs) (46).
4.2 Epigenetic alterations and DNA damage
Reactive oxygen and nitrogen species generated during chronic inflammation produce 8-oxoG lesions, single- and double-strand breaks, and replication stress. Activation of ATM/ATR pathways selects for clones with enhanced DNA damage responses or tolerance. APOBEC family cytidine deaminases, induced by inflammatory signaling, can accelerate mutational processes and genomic diversification (47). Inflammation reshapes chromatin landscapes. STAT3 and NF-κB recruit histone modifiers and DNA methyltransferases to promoter and enhancer regions, silencing tumor suppressors or antigen presentation machinery. Microglial cytokines and lactate influence histone acetylation in neighboring glia, promoting dedifferentiation traits associated with GIC phenotypes (48).
4.3 Immune editing and antigen presentation
Immune surveillance initially eliminates highly immunogenic clones. As selection proceeds, surviving populations reduce antigenicity or increase inhibitory ligand expression. PD-L1, galectin-9, and CD276 engage PD-1/TIM-3 receptors on T cells, reinforcing exhaustion. Simultaneously, defects in antigen processing, downregulation of β2-microglobulin, TAP1/2, and HLA molecules, decrease MHC-I presentation (49).
Adenosine-rich, hypoxic niches and persistent IL-10/TGF-β signaling favor an ‘immune silent’ state that resists dendritic-cell activation. Where cGAS–STING senses cytosolic DNA from damaged cells, chronic stimulation can paradoxically induce negative feedback and desensitization, blunting type I interferon responses critical for cross-priming (50).
4.4 Expansion and ecology of glioma-initiating cells
Under the pressure of inflammation and immune selectivity, GICs with stem-like characteristics gain survival advantages. GICs usually express nestin, Sox2 and CD44, showing significant resistance to oxidative stress, DNA damage and immune-mediated apoptosis (51). These cells are mostly located in chronic inflammatory areas, and the local microenvironment is rich in TGF-β, IL-6 and hypoxia signals, providing a supporting niche (52).
It is worth noting that these initial cell populations also have the mechanism of active inhibition of local immunity, such as the secretion of galectin-1 and prostaglandin E2, which further enhance the formation of the overall immunosuppressive microenvironment. Thus, immune-driven selection acts as a key filter in the inflamed CNS, promoting clonal expansion and evolution of glial cells and the emergence of malignant phenotypes. This transformation is gradual rather than abrupt, reflecting cumulative immune pressure, metabolic constraints, and molecular adaptations.
5 Discussion and future directions
Accumulating evidence indicates that chronic neuroinflammation does more than accompany glioma; it progressively reshapes the central nervous system immune milieu into a niche that permits malignant initiation and supports subsequent progression. Early pattern-recognition events and inflammasome activation trigger cytokine cascades and blood–brain barrier dysfunction, after which glial and myeloid populations adopt immunotolerant states. Microglia and infiltrating macrophages converge toward GAM phenotypes with pro-angiogenic and matrix-remodeling programs, astrocytes strengthen immune evasion through STAT3 activity and chemokine networks, and regulatory T cells together with MDSCs enforce suppression. Metabolic rewiring further entrenches dysfunction through lactate accumulation, adenosine signaling, and nutrient competition. In parallel, inflammatory hubs such as IL-6–STAT3 and NF-κB promote stem-like traits, while defects in antigen processing and upregulation of checkpoint ligands reduce immune visibility. These processes unfold in spatially organized niches and show temporal evolution from acute activation toward stable suppression, providing tractable windows for monitoring and intervention.
Clinical translation remains difficult in GBM because profound myeloid suppression, limited neoantigen load in subsets, and the physical constraints of the blood–brain barrier blunt responses to single-agent checkpoint blockade. A more effective strategy begins with microenvironment reconditioning, then mobilizes adaptive immunity. Upstream cytokine and growth-factor pathways such as IL-6-STAT3, TGF-β, and CSF1R represent rational control points; adenosine A2A and IDO1 constitute metabolic brakes that can be released; epigenetic modulators may restore antigen presentation and improve recognition. Myeloid reprogramming with TLR or STING agonists, CD40 engagement, or phagocytosis-checkpoint inhibition complements these approaches and should be deployed with attention to timing, for example peri-resection or immediately after radiotherapy when antigen release and trafficking are favorable. Regional delivery methods including convection-enhanced delivery, focused ultrasound opening, and intrathecal or intraventricular routes improve exposure while limiting systemic toxicity, and brain-targeted nanoparticles or depot biomaterials can sustain local immune modulation along resection margins or infiltrative tracts.
In recurrent disease, the phase 3 cohort of CheckMate 143 found no overall survival advantage for PD-1 blockade compared with bevacizumab and reported a higher objective response with bevacizumab, underscoring the difficulty of overcoming a suppressive ecosystem with single-agent checkpoint inhibition (53). In newly diagnosed disease, CheckMate 498 in MGMT-unmethylated glioblastoma showed no benefit for radiotherapy plus PD-1 blockade over the temozolomide-based standard, and CheckMate 548 in MGMT-methylated or indeterminate tumors did not improve survival when PD-1 blockade was added to chemoradiation (54, 55). These outcomes align with early cold immune microenvironments, lymphopenia from radiotherapy, steroid exposure, and limited trafficking across the blood–brain barrier that together blunt adaptive responses. Cellular therapies illustrate related constraints. An EGFRvIII-directed CAR-T study demonstrated tumor trafficking yet rapidly revealed antigen loss and adaptive resistance in situ (56). IL13Rα2-directed CAR-T achieved striking but transient regression in some patients, which highlights antigen heterogeneity and exhaustion (57). A large randomized trial of the EGFRvIII vaccine rindopepimut failed to improve survival in newly diagnosed disease, reinforcing the limitations of single-antigen strategies in a heterogeneous and suppressive context (58).
Much of the causal chain has been inferred from established tumors, which necessitates prospective cohorts of patients with chronic neuroinflammation, faithful models of pre-neoplastic states, and longitudinal tissue or cerebrospinal fluid sampling to separate cause from consequence. Inter-patient heterogeneity in autonomic tone, neuropeptide signaling, and metabolic programs is expected to shape therapeutic sensitivity and may warrant stage-aware and biomarker-guided protocols. Even with these caveats, a coherent model emerges in which sustained neuroinflammation seeds immunosuppressive ecosystems, selects for immune-evasive glial clones, and licenses gliomagenesis. Clarifying the earliest measurable deviations, aligning combinations to disease stage, and refining regional delivery are the most promising routes toward earlier diagnosis and preventive immunomodulation.
Author contributions
YB: Writing – review & editing, Conceptualization, Writing – original draft. YS: Visualization, Writing – original draft, Formal Analysis. ZC: Writing – original draft. TG: Writing – original draft. HD: Writing – original draft, Conceptualization, Writing – review & editing, Supervision. XJ: Writing – original draft, Resources, Visualization, Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer XH declared a shared parent affiliation with the author YS to the handling editor at the time of review.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ransohoff RM and Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. (2012) 12:623–35. doi: 10.1038/nri3265
2. Glass CK, Saijo K, Winner B, Marchetto MC, and Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
3. Hambardzumyan D, Gutmann DH, and Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. (2016) 19:20–7. doi: 10.1038/nn.4185
4. Chen Z and Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
5. Quail DF and Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
6. Hu X, Deng P, Ma M, Tang X, Qian J, Gong Y, et al. A machine learning model based on results of a comprehensive radiological evaluation can predict the prognosis of basal ganglia cerebral hemorrhage treated with neuroendoscopy. Front Neurol. (2024) 15:1406271. doi: 10.3389/fneur.2024.1406271
7. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. Csf-1r inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
8. Yang H, Wang H, and Andersson U. Targeting inflammation driven by hmgb1. Front Immunol. (2020) 11:484. doi: 10.3389/fimmu.2020.00484
9. Hu X, Qi D, Li S, Ye S, Chen Y, Cao W, et al. Development and validation of an interpretable machine learning model for prediction of the risk of clinically ineffective reperfusion in patients following thrombectomy for ischemic stroke. Ther Clin Risk Manage. (2025) 21:621–31. doi: 10.2147/tcrm.S520362
10. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. Nlrp3 is activated in alzheimer’s disease and contributes to pathology in app/ps1 mice. Nature. (2013) 493:674–8. doi: 10.1038/nature11729
11. Sim J, Park J, Moon JS, and Lim J. Dysregulation of inflammasome activation in glioma. Cell Commun Signal: CCS. (2023) 21:239. doi: 10.1186/s12964-023-01255-5
12. Prinz M and Priller J. The role of peripheral immune cells in the cns in steady state and disease. Nat Neurosci. (2017) 20:136–44. doi: 10.1038/nn.4475
13. Cherry JD, Olschowka JA, and O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflamm. (2014) 11:98. doi: 10.1186/1742-2094-11-98
14. Farina C, Aloisi F, and Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. (2007) 28:138–45. doi: 10.1016/j.it.2007.01.005
15. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
16. Engelhardt B and Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. (2012) 33:579–89. doi: 10.1016/j.it.2012.07.004
17. Yu H, Pardoll D, and Jove R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat Rev Cancer. (2009) 9:798–809. doi: 10.1038/nrc2734
18. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain Malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
19. Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, et al. Microglia are effector cells of cd47-sirpα Antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci U.S.A. (2019) 116:997–1006. doi: 10.1073/pnas.1721434116
20. Perelroizen R, Philosof B, Budick-Harmelin N, Chernobylsky T, Ron A, Katzir R, et al. Astrocyte immunometabolic regulation of the tumour microenvironment drives glioblastoma pathogenicity. Brain: J Neurol. (2022) 145:3288–307. doi: 10.1093/brain/awac222
21. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. (2017) 18:234. doi: 10.1186/s13059-017-1362-4
22. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. (2017) 77:2266–78. doi: 10.1158/0008-5472.Can-16-2310
23. Liddelow SA and Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
24. Linnerbauer M, Beyer T, Nirschl L, Farrenkopf D, Lößlein L, Vandrey O, et al. Pd-L1 positive astrocytes attenuate inflammatory functions of pd-1 positive microglia in models of autoimmune neuroinflammation. Nat Commun. (2023) 14:5555. doi: 10.1038/s41467-023-40982-8
25. Sin WC, Aftab Q, Bechberger JF, Leung JH, Chen H, and Naus CC. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene. (2016) 35:1504–16. doi: 10.1038/onc.2015.210
26. Nilsson MB, Armaiz-Pena G, Takahashi R, Lin YG, Trevino J, Li Y, et al. Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a src-dependent mechanism. J Biol Chem. (2007) 282:29919–26. doi: 10.1074/jbc.M611539200
27. Shahzad MM, Arevalo JM, Armaiz-Pena GN, Lu C, Stone RL, Moreno-Smith M, et al. Stress effects on fosb- and interleukin-8 (Il8)-driven ovarian cancer growth and metastasis. J Biol Chem. (2010) 285:35462–70. doi: 10.1074/jbc.M110.109579
28. Yang R, Lin Q, Gao HB, and Zhang P. Stress-related hormone norepinephrine induces interleukin-6 expression in ges-1 cells. Braz J Med Biol Res. (2014) 47:101–9. doi: 10.1590/1414-431x20133346
29. Khanmammadova N, Islam S, Sharma P, and Amit M. Neuro-immune interactions and immuno-oncology. Trends Cancer. (2023) 9:636–49. doi: 10.1016/j.trecan.2023.05.002
30. Mo RJ, Han ZD, Liang YK, Ye JH, Wu SL, Lin SX, et al. Expression of pd-L1 in tumor-associated nerves correlates with reduced cd8(+) tumor-associated lymphocytes and poor prognosis in prostate cancer. Int J Cancer. (2019) 144:3099–110. doi: 10.1002/ijc.32061
31. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished cd4 compartment explains cellular immune defects in patients with Malignant glioma. Cancer Res. (2006) 66:3294–302. doi: 10.1158/0008-5472.Can-05-3773
32. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, and Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. (2006) 8:261–79. doi: 10.1215/15228517-2006-008
33. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M, et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100a8/9 and arginase and suppress T cell function. Neuro Oncol. (2016) 18:1253–64. doi: 10.1093/neuonc/now034
34. Ott M, Tomaszowski KH, Marisetty A, Kong LY, Wei J, Duna M, et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.134386
35. Zhai L, Ladomersky E, Dostal CR, Lauing KL, Swoap K, Billingham LK, et al. Non-tumor cell ido1 predominantly contributes to enzyme activity and response to ctla-4/pd-L1 inhibition in mouse glioblastoma. Brain Behav Immun. (2017) 62:24–9. doi: 10.1016/j.bbi.2017.01.022
36. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.Cir-15-0036
37. Liang SP, Wang XZ, Piao MH, Chen X, Wang ZC, Li C, et al. Activated sirt1 contributes to dpt-induced glioma cell parthanatos by upregulation of nox2 and nat10. Acta Pharmacol Sin. (2023) 44:2125–38. doi: 10.1038/s41401-023-01109-3
38. Deng Y, Chen Q, Wan C, Sun Y, Huang F, Hu Y, et al. Microglia and macrophage metabolism: A regulator of cerebral gliomas. Cell Biosci. (2024) 14:49. doi: 10.1186/s13578-024-01231-7
39. Lin T, Zhao P, Jiang Y, Tang Y, Jin H, Pan Z, et al. Blood-brain-barrier-penetrating albumin nanoparticles for biomimetic drug delivery via albumin-binding protein pathways for antiglioma therapy. ACS Nano. (2016) 10:9999–10012. doi: 10.1021/acsnano.6b04268
40. Cui Y, Zhang M, Zeng F, Jin H, Xu Q, and Huang Y. Dual-targeting magnetic plga nanoparticles for codelivery of paclitaxel and curcumin for brain tumor therapy. ACS Appl Mater Interfaces. (2016) 8:32159–69. doi: 10.1021/acsami.6b10175
41. Afzal A, Afzal Z, Bizink S, Davis A, Makahleh S, Mohamed Y, et al. Phagocytosis checkpoints in glioblastoma: cd47 and beyond. Curr Issues Mol Biol. (2024) 46:7795–811. doi: 10.3390/cimb46080462
42. Zhong J, Xing X, Gao Y, Pei L, Lu C, Sun H, et al. Distinct roles of trem2 in central nervous system cancers and peripheral cancers. Cancer Cell. (2024) 42:968–84.e9. doi: 10.1016/j.ccell.2024.05.001
43. van Wageningen TA, Vlaar E, Kooij G, Jongenelen CAM, Geurts JJG, and van Dam AM. Regulation of microglial tmem119 and P2ry12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol Commun. (2019) 7:206. doi: 10.1186/s40478-019-0850-z
44. Brantley EC and Benveniste EN. Signal transducer and activator of transcription-3: A molecular hub for signaling pathways in gliomas. Mol Cancer Res. (2008) 6:675–84. doi: 10.1158/1541-7786.Mcr-07-2180
45. Heldin CH, Vanlandewijck M, and Moustakas A. Regulation of emt by tgfβ in cancer. FEBS Lett. (2012) 586:1959–70. doi: 10.1016/j.febslet.2012.02.037
46. Han J, Alvarez-Breckenridge CA, Wang QE, and Yu J. Tgf-β Signaling and its targeting for glioma treatment. Am J Cancer Res. (2015) 5:945–55.
47. Chen J, McKay RM, and Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. (2012) 149:36–47. doi: 10.1016/j.cell.2012.03.009
48. Esteller M. Cancer epigenetics: DNA methylation and chromatin alterations in human cancer. Adv Exp Med Biol. (2003) 532:39–49. doi: 10.1007/978-1-4615-0081-0_5
49. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
50. Low JT, Brown MC, Reitman ZJ, Bernstock JD, Markert JM, Friedman GK, et al. Understanding and therapeutically exploiting cgas/sting signaling in glioblastoma. J Clin Invest. (2024) 134. doi: 10.1172/jci163452
51. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
52. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell rna-seq highlights intratumoral heterogeneity in primary glioblastoma. Sci (New York NY). (2014) 344:1396–401. doi: 10.1126/science.1254257
53. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
54. Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated mgmt promoter: an international randomized phase iii trial. Neuro Oncol. (2023) 25:123–34. doi: 10.1093/neuonc/noac099
55. Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase iii trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated mgmt promoter. Neuro Oncol. (2022) 24:1935–49. doi: 10.1093/neuonc/noac116
56. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused egfrviii-directed car T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aaa0984
57. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
58. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, egfrviii-expressing glioblastoma (Act iv): A randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85. doi: 10.1016/s1470-2045(17)30517-x
Keywords: gliomagenesis, neuroinflammation, microglia, immunosuppression, STAT3 signaling, glioma-initiating cells
Citation: Bao Y, Chen Z, Su Y, Guo T, Du H and Jia X (2025) From neuroinflammation to gliomagenesis: immune drivers of malignant transformation in the CNS. Front. Immunol. 16:1682030. doi: 10.3389/fimmu.2025.1682030
Received: 08 August 2025; Accepted: 14 November 2025; Revised: 01 September 2025;
Published: 01 December 2025.
Edited by:
Robert Scheinman, University of Colorado, United StatesReviewed by:
Xiaolong Hu, Tongji University, ChinaCopyright © 2025 Bao, Chen, Su, Guo, Du and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huaping Du, ZHVodWFwaW5nMjI2QDEyNi5jb20=; Xianjun Jia, MTUyNjI5MDUwNDJAMTYzLmNvbQ==
†These authors have contributed equally to this work