 Mengze Hao1,2
Mengze Hao1,2 Xiaoli Zhao1,2
Xiaoli Zhao1,2 Xiaoyu Zhang1,2
Xiaoyu Zhang1,2 Weihua Zhai1,2Qiaoling Ma1,2
Weihua Zhai1,2Qiaoling Ma1,2 Donglin Yang1,2
Donglin Yang1,2 Aiming Pang1,2Sizhou Feng1,2
Aiming Pang1,2Sizhou Feng1,2 Yi He1,2*Erlie Jiang1,2Mingzhe Han1,2
Yi He1,2*Erlie Jiang1,2Mingzhe Han1,2- 1State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Haihe Laboratory of Cell Ecosystem, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China
- 2Tianjin Institutes of Health Science, Tianjin, China
Introduction: Mixed-phenotype acute leukemia (MPAL) is a rare subtype of acute leukemia with unfavorable outcome. There is no established optimal therapy regime.
Methods: We conducted a retrospective analysis in our transplant center to clarify the efficacy of allogeneic hematopoietic stem cell transplantation (allo-HSCT) in the treatment of MPAL.
Results: This study monitored 61 MPAL patients who underwent allo-HSCT at a single center in China. Haploidentical donor HSCT was 41, matched unrelated donor HSCT was 4, and matched sibling donor HSCT was 16. The median age at diagnosis was 32 years (range, 14-58). The two most common phenotypes were B-lymphoid/myeloid (n=33, 54.1%) and T-lymphoid/myeloid (n=22, 36.1%). In induction treatment, 50 (82.0%) patients received an ALL-like treatment protocol, and 15 of the 17 BCR::ABL1 positive patients received tyrosine kinase inhibitor (TKI) therapy. After induction treatment, 38 (62.3%) patients achieved complete remission (CR). Pre-HSCT 55/61 (90.2%) acquired complete remission (CR) and 46/61 (75.4%) turned minimal residual disease (MRD) -negative. The median follow up time was 28.2 months. The estimated 2-year overall survival (OS) rates after HSCT were 80.0% ± 6.0%. And the relapse-free survival (RFS) probabilities at 2-year were 68.0±7.0%. There was no significant difference in OS and RFS among different types of HSCT. Patients with MRD-positive pre-HSCT was associated with worse OS (P=0.022). Patients who achieved CR after induction therapy had a longer RFS (P=0.033).
Discussion: Allo-HSCT is effective in the treatment of MPAL especially in patients who achieved CR after induction therapy or who got MRD-negative pre-HSCT.
Introduction
Mixed-phenotype acute leukemia (MPAL) is a rare and heterogeneous disease, accounting for 2% to 5% of all acute leukemia (AL) (1). MPAL shows no clear evidence of differentiation along a single lineage, which is classified based on immunophenotypes into B-lymphoid/myeloid (B-M) and T-lymphoid/myeloid (T-M) subtypes (2). Furthermore, subtypes with specific genetic features, such as MPAL with BCR::ABL1 fusion or MPAL with KMT2A-rearranged, have been defined (2). BCR::ABL1 fusion is more common in adults, while KMT2A rearrangements are more prevalent in pediatric MPAL. These two subgroups account for approximately 19% to 28% of all MPAL cases (3, 4). The prognosis of MPAL is much worse than acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML), and the probability of recurrence rate was predicted to be higher in a meta-analysis (5).
The optimal treatment regimen for MPAL is still unclear. Previous reports have shown that induction treatment with an ALL regimen can improve remission rates (5). In the pre-transplant era, the prognosis for MPAL was poor, with a survival rate of approximately 20% (6). Consolidation treatment with allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the preferred approach (6, 7). Previous experiences suggested that a cluster of high-risk genetic features in MPAL was associated with worse outcomes such as BCR::ABL1 and KMT2A rearrangements and some complex karyotypes (3, 4, 8). The addition of a tyrosine kinase inhibitor (TKI) in the subset of BCR::ABL1-positive MPAL was well tolerated and recommended (9). A multicenter study by the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation (EBMT) recruited 519 de novo MPAL patients and reported a 3-year overall survival (OS) rate of 56.3% and a relapse-free survival (RFS) rate of 46.5% for those who underwent HSCT (6). Even with allo-HSCT, OS remains unsatisfactory, and the factors influencing prognosis warrant further investigation. This article aims to analyze the clinical, molecular, and cytogenetic characteristics of a group of 61 MPAL patients, with a particular focus on survival and prognostic factors post-allo-HSCT.
Materials and methods
Patients
Sixty-one consecutive patients, aged 14 years or older, with MPAL between August 2008 and December 2023, were recruited in this retrospective study, and all underwent allo-HSCT at the Stem Cell Transplantation Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences. All patients conformed to the definition of MPAL in the 2022 WHO classification of myeloid neoplasms and acute leukemia. All data were collected from clinical records. All cases were diagnosed using standard diagnostic methods, including morphological examination of peripheral blood (PB) and bone marrow (BM) smears, immunophenotyping by flow cytometry (FCM), conventional cytogenetics, and molecular studies. The FCM results at the time of diagnosis were retrospectively reviewed. The study was approved by the Medical Ethics Committee of the Institute of Hematology and Blood Diseases Hospital.
Treatment protocol for HSCT
All patients were conditioned with a myeloablative conditioning regimen (MAC) with or without total body irradiation (TBI), described as TFAC, TMC, and VBC regimens, and the transplant day was named as day 0. The TFAC conditioning regimen was comprised of TBI (10 Gy, days −9 to −7), fludarabine (30 mg/m2/day, days −6 to −4), cytarabine (2 g/m2/day, days −6 to −4) or idarubicin (12 mg/m2/day, days −6 to −4), and cyclophosphamide (CTX 40 mg/kg/day, days −3 to −2). The TMC conditioning regimen included TBI (10 Gy, days −8 to −6), melphalan (60 mg/m2/day, days −5 to −4), and CTX (40 mg/kg/day, days −3 to −2), and the VBC regimen included etoposide (20 mg/kg/day, days −8 to −7), busulfan (3.2 mg/kg/day, days −6 to −4), and CTX (40 mg/kg/day, days −3 to −2). Rabbit antithymocyte globulin (rATG 2.5 mg/kg/day, days −5 to −2) was added to the regimen in the haploidentical stem cell transplantation (HID-HSCT) and matched unrelated donor transplantation (MUD). All recipients received cyclosporine A or tacrolimus and short-term methotrexate as graft-versus-host disease (GVHD) prophylaxis. HID-HSCT recipients received mycophenolate mofetil consistent with our previous experience (10). The stem cell source was peripheral blood stem cells (PBSCs). Immunosuppressive agents can be reduced by 25%–30% at 2 to 3 months post-HSCT and discontinued within 6 months if GVHD has been fully resolved.
Definitions
Complex karyotypes were defined as the presence of three or more clonal structural chromosomal abnormalities, identified after culturing BM cells for 24 to 48 h in a tissue-culture medium using routine techniques. The date of granulocyte engraftment was the first day of three consecutive days with an absolute neutrophil value greater than 0.5 × 109/L. The time of platelet recovery was defined as the first of seven consecutive days with an absolute platelet count greater than 20 × 109/L in the absence of transfusion. BM aspirate was performed on day 0, day of neutrophil recovery, day 28, day 42, day 60, and day 90 to assess the disease status in the first 3 months after HSCT. Minimal residual disease (MRD) was monitored using FCM for all the patients with a sensitivity of 0.01%. Quantitative real-time polymerase chain reaction (qRT-PCR) was additionally performed only in those who were BCR::ABL1-positive (17 cases) or FLT3-ITD-positive (7 cases), with a sensitivity of 0.001% (11, 12). Less than 5% blasts in bone marrow were regarded as complete remission (CR), a blast count of more than 20% was defined as no remission (NR), and a blast count of 5%–19% was defined as partial remission (PR). The last follow-up was in November 2024. All patients had a median follow-up of 29.1 months (range 8.8–198.4 months). OS was defined as the time from the infusion to the last follow-up or death. RFS was defined as survival with no hematological relapse. Non-relapse mortality (NRM) was defined as death without previous relapse, and it was calculated by using a cumulative incidence function with death as a competing risk. AGVHD and chronic GVHD (cGVHD) were graded according to international criteria (13, 14).
Statistical analysis
The probabilities of OS and RFS were assessed using the Kaplan–Meier method, and differences between groups were compared using the log-rank test. The Cox proportional hazards regression model was applied in multivariate analysis to identify independent risk factors, including variables with a p-value <0.1, considered statistically significant. The covariates included in the Cox model were the response of induction therapy, MRD pre-HSCT, and conditioning regimen, and the response to induction therapy was treated as a time-dependent covariate. OS and RFS were analyzed using the Cox proportional hazards model. Considering competing risks, the cumulative incidence of relapse (CIR) was calculated by competing risk analysis. Moreover, the incidence of various infections (CMV, EBV, cystitis) as well as of GVHD (including aGVHD and cGVHD) was calculated by cumulative incidence. Appropriately, the chi-square test or Student’s t-test was used to compare the distribution of various parameters. Statistical analyses were performed using SPSS 27.0 and R software (version 3.4.3). P-values <0.05 were considered as a measure of statistical significance.
Results
Patient characteristics
Among the 61 patients, 33 (54.1%) were diagnosed with myeloid with B-cell marker (B-M) MPAL, 22 (36.1%) with T-lymphoid/myeloid (T-M) MPAL, and 6 (9.8%) with T-lymphoid/B-lymphoid/myeloid (T-B-M) or T-lymphoid/B-lymphoid (T-B) MPAL according to the WHO classification based on immune phenotype. Based on cytogenetics, 17 MPAL patients were BCR::ABL1-positive, and 6 patients had KMT2A rearrangement, with both phenotypes being B-M. There was a male prevalence (54.1% of patients), and the median age at diagnosis was 32 years (range 14–58). The median percentage of blast in BM was 79%, and the range was 20% to 98.5%. The range of blood cell analysis counts was very large (details in Table 1). All patients had hematopoietic cell transplantation-comorbidity index (HCT-CI) scores less than 1 and KPS scores not less than 80. The interval from diagnosis to HSCT ranges from 4 months to 15 months. Cytogenetic analysis was available in 52/61 patients. Karyotype was normal in 15 patients (24.6%) and complex in 12 (17.6%). Other cytogenetic abnormalities [such as +8, +21, +22, 20q−, t (9;22)] were found in 26 patients. Genetic data were available for 47/61 patients, with 7 patients having no gene mutations detected. More detailed information on gene mutations is shown in Figure 1. The most frequent gene mutations were RUNX1 (12 cases), FLT3-ITD (7 cases), ASXL1 (6 cases), PHF6 (5 cases), WT1 (5 cases), NOTCH1 (5 cases), and NRAS (5 cases). Detailed clinical characteristics are presented in Table 1.

Table 1. Characteristics of the 61 MPAL patients.

Figure 1. Gene mutations of 47 patients with mixed-phenotype acute leukemia (MPAL).
Chemotherapy and response
Fifty of the 61 patients (82%) received an ALL-like induction therapy based on the vindesine and prednisone (VP) regimen, 6 patients (9.8%) were treated using an AML-like induction based on cytarabine and daunorubicin/idarubicin (DA/IA), and the remaining 5 patients (8.2%) received both ALL-like and AML-like treatments. The CR rates after induction therapy of the three groups were 68.0% (34/50), 16.7% (1/6), and 60.0% (3/5), respectively (Supplementary File 1). Furthermore, we provided a detailed description of the evaluation of MRD after induction therapy by using FCM and qRT-PCR methods in Supplementary File 1. In total, 38 of 61 (62.3%) patients achieved a CR after induction, 6 (9.8%) PR, and 17 (27.9%) NR. Patients who received ALL-like therapy had higher CR rates after induction. However, there was no significant difference in statistics (P = 0.084%; OR = 0.269; 95% CI 0.069, 1.053). We recruited 17 BCR::ABL1-positive MPAL patients; 15 received TKI combined with induction chemotherapy: 8 with imatinib, 4 with dasatinib, 2 with orebatinib, and 1 with flumatinib. As a result, 12 cases had CR and 5 had NR after induction treatment. Two patients did not receive TKI during induction. One patient received both ALL-like and AML-like regimens for induction without TKI and had a CR. Consolidation chemotherapy was combined with TKI. Another patient received three cycles of AML-like induction without TKI but still achieved NR. In the fourth cycle, the patient received re-induction with TKI and achieved CR.
We conducted a univariate analysis on factors affecting response after induction chemotherapy, including a series of variables such as age (cutoff at 30 years), gender, induction treatment, gene mutation, karyotype (complex vs. others), and types of MPAL according to the WHO (immune phenotype). Patients with no gene mutation had a higher CR rate after induction therapy (7/7 vs. 22/40, P = 0.024). After induction, all 17 NR cases received the ALL-like re-induction therapy, with 13 achieving a CR and 8 cases (61.5%) being MRD-negative. Before allo-HSCT, 55/61 (90.2%) acquired a CR and 46/61 (75.4%) turned MRD-negative.
Allo-HSCT and survival
In the entire cohort, all 61 MPAL patients underwent allo-HSCT using PBSCs as the stem cell sources. Of these, 41 (67.2%) had HID-HSCT, 4 (6.6%) had MUD-HSCT, and 16 (26.2%) had matched sibling donor (MSD)-HSCT. All of them received myeloablative conditioning, with 49 patients (80.3%) receiving regimens that included TBI. The MRD of the infusion day was negative in 53 patients and positive in 8. The infusion number of mononuclear cells (MNCs) was 10.3 (4.29–18.78) × 108/kg, and the number of CD34-positive cells was 3.43 (1.93–11.64) × 106/kg. The median times to neutrophil and platelet engraftment were 13 (range 10–21) days and 14 (range 10–180) days, respectively.
Post-HSCT complications included the following: the cumulative incidence of CMV reactivation at 180 days was 42.7% ± 6.3% (Figure 2A), the cumulative incidence of EBV reactivation at 90 days was 13.1% ± 4.3% (Figure 2B), and the cumulative incidence of cystitis at 90 days was 29.5% ± 5.8% (Figure 2C). Grade II–IV aGVHD cumulative incidence at 180 days was 37.7% ± 6.2% (Figure 3A), and grade III–IV aGVHD cumulative incidence at 180 days was 18.0% ± 4.8% (Figure 3B). cGVHD cumulative incidence at 24 months was 26.7% ± 5.7% (Figure 3C), and extensive cGVHD cumulative incidence at 24 months was 16.4% ± 4.7% (Figure 3D).

Figure 2. (A) The cumulative incidence of CMV reactivation was 42.7% ± 6.3% at 180 days. (B) The cumulative incidence of EBV reactivation was 13.1% ± 4.3% at 90 days. (C) The cumulative incidence of cystitis was 29.5% ± 5.8% at 90 days.

Figure 3. (A) The cumulative incidence of grade II–IV acute graft-versus-host disease (aGVHD) at 180 days was 37.7% ± 6.2%. (B) The cumulative incidence of grade III–IV aGVHD at 180 days was 18.0% ± 4.8%. (C) The cumulative incidence of chronic graft-versus-host disease (cGVHD) at 24 months was 26.7% ± 5.7%. (D) The cumulative incidence of extensive cGVHD at 24 months was 16.4% ± 4.7%.
In the 61 MPAL patients, 9 cases were MRD-positive and 6 cases achieved NR. During the infusion day, 7 out of 9 became MRD-negative, and in 6 patients who achieved NR, 1 turned MRD-negative; however, the other 5 still achieved NR on day 0. The specific details are added in Supplementary File 2. Twenty-five (41.0%) patients received maintenance therapy after 2 to 3 months post-allo-HSCT with no aGVHD, no infection, and no stable blood count, including 11 with TKI, 11 with Venclexta, 1 with azacitidine, and 2 with chidamide. Of the 11 patients who were treated with TKIs, 3 patients received orebatinib, 7 received imatinib, and 1 received dasatinib. Only one patient was unable to tolerate imatinib due to nausea and vomiting, hence was switched to fumatinib for maintenance treatment. The remaining 24 patients were able to tolerate the maintenance therapy drugs, with a treatment duration ranging from 1.5 months to 30 months (Supplementary File 3). However, there were no statistically significant differences in the cumulative incidence of relapse or NRM between patients who received maintenance therapy and those who did not (Supplementary File 4A, B).
After HSCT, 17 (17/61, 27.9%) patients relapsed, and 12 patients accepted donor lymphocyte infusions (DLIs). Six patients became MRD-negative after DLI, while the other six still achieved NR. The specific details are added in Supplementary File 5. At the latest follow-up, 50 patients (82.0%) were alive, with 43 of these patients being in CR. Eleven patients (18.0%) have died, with the majority (10 patients, 90.9%) succumbing to relapse, and only one dying from infections.
The estimated 1-, 2-, and 5-year OS rates after HSCT were 89.0% ± 4.0%, 80.0% ± 6.0%, and 76.0% ± 7.0%, respectively. The RFS rates at 1, 2, and 5 years were 75.0% ± 6.0%, 68% ± 7.0%, and 68% ± 7.0%, respectively. Table 2 presents the specific prognostic data. The 3-year confidence interval (CI) for NRM was 2.3% ± 0.05% for the entire cohort (Supplementary File 4C). The 1- and 3-year cumulative recurrence rates were 23.7% ± 0.31% and 31.0% ± 0.43%, respectively (Supplementary File 4C). Furthermore, the survival curves for OS and RFS are depicted in Figures 4A, B.

Table 2. Univariate analysis of the OS and RFS of MPAL patients with allo-HSCT.
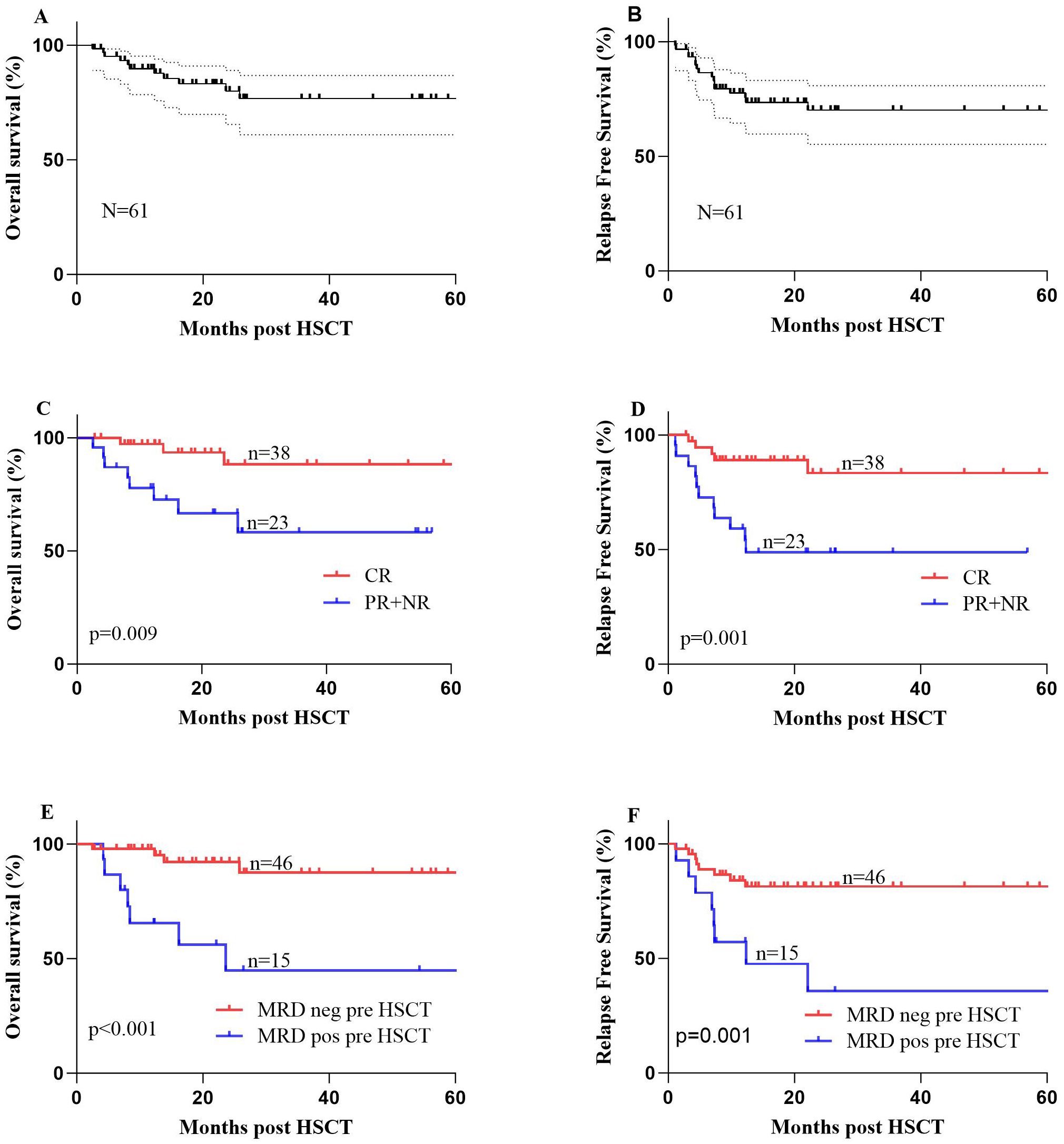
Figure 4. (A) The 1- and 2-year overall survival (OS) rates were 89.0% ± 4.0% and 80.0% ± 6.0%, respectively, for 61 patients with mixed-phenotype acute leukemia (MAPL) undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT). (B) The 1- and 2-year relapse-free survival (RFS) rates were 75.0% ± 6.0% and 68% ± 7.0%, respectively, for 61 patients with MAPL undergoing allo-HSCT. (C) OS of the two groups of patients with MPAL undergoing allo-HSCT: patients who achieved complete remission (CR) after induction therapy and patients who achieved partial remission (PR) or no remission (NR) after induction therapy. (D) RFS of the two groups of patients with MPAL undergoing allo-HSCT: patients who achieved CR after induction therapy and patients who achieved PR or NR after induction therapy. (E) OS of the two groups of patients with MPAL undergoing allo-HSCT: patients who were minimal residual disease (MRD)-negative before allo-HSCT and patients who were MRD-positive before allo-HSCT. (F) RFS of the two groups of patients with MPAL undergoing allo-HSCT: patients who were MRD-negative before allo-HSCT and patients who were MRD-positive before allo-HSCT.
Prognosticators for MPAL with allo-HSCT
Among the MPAL patients, we conducted univariate analyses for OS and RFS in the entire group, considering the following variables separately: age (with a cutoff at 30 years), gender, types of MPAL according to various classification systems, chromosome karyotype, status after induction therapy, MRD status pre-HSCT, conditioning regimen, donor type, gender match (F to M vs. others), ABO type match, allo-HSCT-related complications, and maintenance treatment. Consequently, patients who achieved CR after induction therapy had significantly longer OS (88.3% ± 6.6% vs. 66.6% ± 10.5%, P = 0.009) and better RFS (83.4% ± 7.3% vs. 46.6% ± 10.6%, P = 0.001) (Figures 4C, D). Additionally, the MRD status pre-HSCT and on the infusion day both played a significant role in both OS and RFS survival (negative vs. positive, P < 0.05) (Figures 4E, F). Recurrence affects the prognosis of OS, resulting in dismal survival rates (44.3% ± 12.4% vs. 96.7% ± 3.3%, P < 0.001).
In the multivariate analysis, a favorable variable for OS was achieving MRD negativity before allo-HSCT (P = 0.022, HR 0.22, 95% CI 0.06–0.80) (Table 3). According to RFS, CR after induction (P = 0.033, HR 0.30, 95% CI 0.10–0.91) was an independent protective factor (Table 4).

Table 3. Multivariate analysis of the OS of MPAL patients with allo-HSCT.
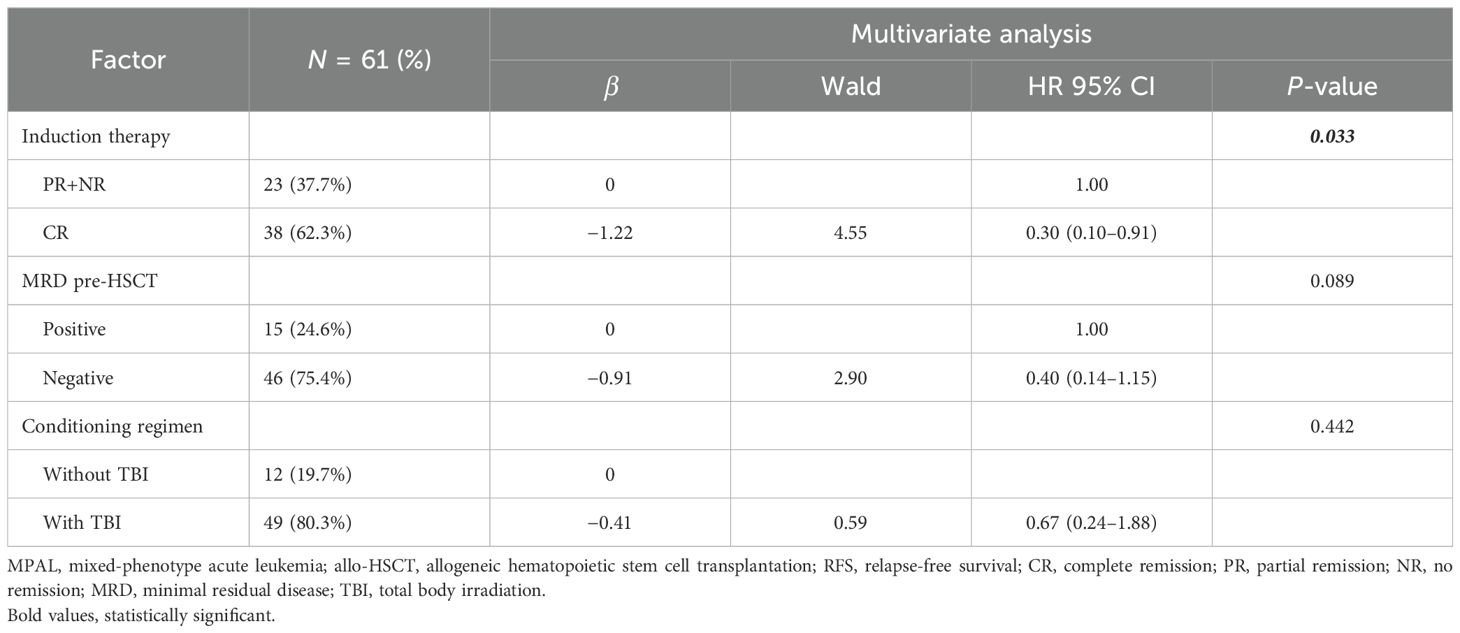
Table 4. Multivariate analysis of the RFS of MPAL patients with allo-HSCT.
Discussion
MPAL is a rare type of AL, characterized by the simultaneous display of more than one lineage, such as myeloid and B-lymphoid or T-lymphoid in MPAL (15–17). In addition to BCR::ABL1 and KMT2A-rearranged cases, ZNF384 and BCL11B rearrangements, as novel genetic findings, have been added as subtypes (15). We found that B-M and T-M MPAL are the most common types, consistent with earlier reports (5, 18). With the advancement of next-generation targeted sequencing and molecular biology technologies, our understanding of the gene mutation spectrum in MPAL patients has deepened. In MPAL, mutations are primarily found in genes related to epigenetic modification, signaling pathways, and transcription factors, and they vary depending on the patients’ ages. Pediatric MPAL primarily exhibits mutations in genes such as ZNF384, WT1, and CEBPA, while adults mainly have mutations in RUNX1, NOTCH1, and DNMT3A (19). In our data, no patient was younger than 14 years, and RUNX1, occurring in 12 cases, was the gene with the highest frequency. This mutation was considered a poor prognostic indicator in AL. Compared to mutations in ALL and AML, B-M MPAL had a similar expression profile to B-ALL, and T-M MPAL had a similar expression profile to ETP-ALL, both mainly featuring mutations in genes related to WT1, RAS, and the JAK-STAT pathway (19, 20). Neither PHF6 nor NOTCH1 mutations were found in B-M MPAL, but they were enriched in T-M MPAL. It has been reported that PHF6 and NOTCH1 mutations were often implicated in the development of T-ALL and less frequently in AML and other myeloid neoplasms (21). All of the above indicate the genetic differences among subgroups of MPAL. Extensive and in-depth exploration of the genomics of MPAL highlights its complexity, and this might lead to improvements in classification and nomenclature in the future.
There are still no confirmed therapeutic guidelines for MPAL. Until now, only a series of small retrospective studies have been conducted. A meta-analysis found that ALL regimens were more likely to achieve CRs after induction than AML regimens and that AML induction had poorer efficacy in multivariable analysis (5, 22). Another study with 49 MPAL patients showed similar survival rates despite different induction therapy types (23). Our data show that patients who received the ALL regimen achieved a higher CR rate; however, the P-values did not show statistical significance. In the pre-transplant era, the prognosis of MPAL patients remains poor, although remission could be achieved by chemotherapy. Research revealed that patients who received allo-HSCT lived longer, with a 2-year survival rate of 57.8%, as compared to 20.2% among patients who did not receive HSCT (23). In 2024, the authors renewed their data showing that HSCT patients had better progression-free survival (PFS) (P = 0.025) and OS (P = 0.011) compared to those not transplanted (24), especially those who achieved CR after induction therapy (24). In our cohort, all the patients underwent allo-HSCT, with 1- and 2-year OS rates of 89.0% ± 4.0% and 80.0% ± 6.0%, respectively. The survival rates were consistent with recent research findings (24), even better than prior literature (16). These better outcomes likely stem from our cohort’s younger median age of 32 years (no patient was older than 60) compared to the previous report’s median age of 49 years (range 18–62) (16). Moreover, the MRD-negative rate achieved by our patients before transplantation was higher (75.4% vs. 48.0%) (16).
In addition, we assessed the prognostic factors for MPAL patients after allo-HSCT. Several predictors with better OS were identified, such as achieving CR after induction therapy and being MRD-negative pre-HSCT or on the day of infusion. Moreover, MRD-negative pre-HSCT was an independent predictor for OS of MPAL patients. This is similar to a previous study, where patients who were MRD-negative pre-HSCT had superior survival post-HSCT of AML and ALL (16, 25, 26). However, there were no statistically significant differences in HSCT types—whether HID-HSCT, MUD-HSCT, or MSD-HSCT—making any of these a feasible strategy for MPAL patients.
However, relapse remains the major cause of treatment failure in AL patients who undergo allo-HSCT. Of the 61 patients, 17 had recurrence, 10 of whom died from relapse. These patients might benefit from post-transplantation maintenance therapy, especially high-risk AL patients, who are usually maintained for 1 or 2 years after allo-HSCT, particularly in the absence of GVHD (27, 28). In our study, 25 out of 61 patients received maintenance therapy after HSCT, including venetoclax, azacitidine, and TKIs (for BCR::ABL1-positive MPAL), with treatment durations ranging from 1.5 months to 1–2 years. Patients who received maintenance therapy seemed to have better survival (2-year survival rates: 91.7% ± 8.0% vs.71.8% ± 8.1%, P = 0.096) and a lower recurrence rate [5 in 25 (20.0%) vs. 12 in 36 (33.3%)].
As a single-center retrospective study, there are obvious limitations. In summary, MPAL is a heterogeneous lethal disease with no standardized guidelines for diagnosis and treatment. An ALL-like regimen should be considered for use in induction therapy. Allo-HSCT, whether HID-HSCT, MUD-HSCT, or MSD-HSCT, is a feasible strategy for MPAL patients, especially those who have achieved CR after induction therapy and those who are MRD-negative pre-HSCT. More prospective, multicenter, large-scale studies are still needed in the near future (29).
Data availability statement
The data analyzed in this study is subject to the following licenses/restrictions: Due to the nature of this research, participants of this study did not agree for their data to be shared publicly, so supporting data is not available. Requests to access these datasets should be directed to aGFvbWVuZ3plQGloY2Ftcy5hYy5jbg==.
Ethics statement
The studies involving humans were approved by Ethical committee of the Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
MHa: Writing – original draft. XLZ: Writing – review & editing. XYZ: Formal analysis, Data curation, Writing – review & editing. WZ: Writing – review & editing, Methodology. QM: Investigation, Writing – review & editing. DY: Writing – review & editing, Supervision. AP: Writing – review & editing, Data curation. SF: Conceptualization, Writing – review & editing. YH: Writing – review & editing. EJ: Writing – review & editing. MHa: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The work was funded by the Tianjin Natural Science Foundation, 23JCZXJC00220.
Acknowledgments
The authors are grateful to the patients for their participation.
Conflict of interest
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1691762/full#supplementary-material
Abbreviations
MPAL, mixed-phenotype acute leukemia; HSCT, hematopoietic stem cell transplantation; CR, complete remission; OS, overall survival; RFS, relapse-free survival; AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia; Mel, melphalan; CTX, cyclophosphamide; ATG, antithymocyte globulin; GVHD, graft-versus-host disease; MRD, minimal residual disease
References
1. Weinberg OK and Arber DA. Mixed-phenotype acute leukemia: historical overview and a new definition. Leukemia. (2010) 24:1844–51. doi: 10.1038/leu.2010.202
2. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. (2009) 114:937–51. doi: 10.1182/blood-2009-03-209262
3. Aggarwal N and Weinberg OK. Update on acute leukemias of ambiguous lineage. Clin Lab Med. (2021) 41:453–66. doi: 10.1016/j.cll.2021.03.016
4. Kurzer JH and Weinberg OK. Acute leukemias of ambiguous lineage: clarification on lineage specificity. Surg Pathol Clin. (2019) 12:687–97. doi: 10.1016/j.path.2019.03.008
5. Maruffi M, Sposto R, Oberley MJ, Kysh L, and Orgel E. Therapy for children and adults with mixed phenotype acute leukemia: a systematic review and meta-analysis. Leukemia. (2018) 32:1515–28. doi: 10.1038/s41375-018-0058-4
6. Munker R, Labopin M, Esteve J, Schmid C, Mohty M, and Nagler A. Mixed phenotype acute leukemia: outcomes with allogeneic stem cell transplantation. A retrospective study from the Acute Leukemia Working Party of the EBMT. Haematologica. (2017) 102:2134–40. doi: 10.3324/haematol.2017.174441
7. Munker R, Brazauskas R, Wang HL, De Lima M, Khoury HJ, Gale RP, et al. Allogeneic hematopoietic cell transplantation for patients with mixed phenotype acute leukemia. Biol Blood Marrow Transplant. (2016) 22:1024–9. doi: 10.1016/j.bbmt.2016.02.013
8. Kirtek T, Chen W, Laczko D, Bagg A, Koduru P, Foucar K, et al. Acute leukemias with complex karyotype show a similarly poor outcome independent of mixed, myeloid or lymphoblastic immunophenotype: A study from the Bone Marrow Pathology Group. Leuk Res. (2023) 130:107309. doi: 10.1016/j.leukres.2023.107309
9. Alexander TB and Orgel E. Mixed phenotype acute leukemia: current approaches to diagnosis and treatment. Curr Oncol Rep. (2021) 23:22. doi: 10.1007/s11912-020-01010-w
10. Zhang X, Zhao X, Chen S, Hao M, Zhang L, Gong M, et al. Addition of ruxolitinib to standard graft-versus-host disease prophylaxis for allogeneic stem cell transplantation in aplastic anemia patients. Bone Marrow Transplant. (2024) 59:997–1005. doi: 10.1038/s41409-024-02266-7
11. Sun Z, Chang Y, Huang X, Zhao X, Dong Y, Fang M, et al. Chinese consensus on minimal residual disease detection and interpretation of patients with acute lymphoblastic leukemia (2023). Zhonghua Xue Ye Xue Za Zhi. (2023) 44:267–75. doi: 10.3760/cma.j.issn.0253-2727.2023.04.002
12. Grob T, Sanders MA, Vonk CM, Kavelaars FG, Rijken M, Hanekamp DW, et al. Prognostic value of FLT3-internal tandem duplication residual disease in acute myeloid leukemia. J Clin Oncol. (2023) 41:756–65. doi: 10.1200/JCO.22.00715
13. Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 diagnosis and staging working group report. Biol Blood Marrow Transplant. (2015) 21:389–401.e1. doi: 10.1016/j.bbmt.2014.12.001
14. Sun Z, Zhu X, Li Y, Cheng Y, Chang Y, Huang X, et al. Chinese expert consensus on the diagnosis and treatment of acute graft-versus-host disease after hematopoietic stem cell transplantation (2024). Zhonghua Xue Ye Xue Za Zhi. (2024) 45:525–33. doi: 10.3760/cma.j.cn121090-20240608-00214
15. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
16. Lazzarotto D, Tanasi I, Vitale A, Piccini M, Dargenio M, Giglio F, et al. Multicenter retrospective analysis of clinical outcome of adult patients with mixed-phenotype acute leukemia treated with acute myeloid leukemia-like or acute lymphoblastic leukemia-like chemotherapy and impact of allogeneic stem cell transplantation: a Campus ALL study. Ann Hematol. (2023) 102:1099–109. doi: 10.1007/s00277-023-05162-0
17. Mansoor N, Javed O, Rafiq N, Aali A, and Meraj F. Mixed-phenotype acute leukemia, T/megakaryoblastic: does it really exist? J Hematop. (2023) 16:49–55. doi: 10.1007/s12308-023-00535-w
18. Patel SS and Weinberg OK. Diagnostic workup of acute leukemias of ambiguous lineage. Am J Hematol. (2020) 95:718–22. doi: 10.1002/ajh.25771
19. Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature. (2018) 562:373–9. doi: 10.1038/s41586-018-0436-0
20. Takahashi K, Wang F, Morita K, Yan Y, Hu P, Zhao P, et al. Integrative genomic analysis of adult mixed phenotype acute leukemia delineates lineage associated molecular subtypes. Nat Commun. (2018) 9:2670. doi: 10.1038/s41467-018-04924-z
21. Kurzer JH and Weinberg OK. PHF6 mutations in hematologic Malignancies. Front Oncol. (2021) 11:704471. doi: 10.3389/fonc.2021.704471
22. Wolach O and Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol. (2017) 24:139–45. doi: 10.1097/MOH.0000000000000322
23. Huang JJ, Appelbaum JS, Russell K, Shaw C, Eckel A, Chen X, et al. (2023). Treatment outcomes for adults with mixed phenotypic acute leukemia: A large single-Center retrospective study, in: The 65th ASH Annual Meeting Abstracts Blood, pp. 2838–40. CLINICAL AND EPIDEMIOLOGICAL, San Diego, California, United State.
24. Huang JJ, Appelbaum JS, Russell K, Shaw C, Eckel A, Chen X, et al. (2024). Hematopoietic stem cell transplant for adults with mixed phenotypic acute leukemia, in: 2024 ASCO HEMATOLOGIC MALIGNANCIES—LEUKEMIA, MYELODYSPLASTIC SYNDROMES, AND ALLOTRANSPLANT 6517 Rapid Oral Abstract Session Hematopoietic stem cell transplant for adults with mixed phenotypic acute leukemia. Chicago, United States.
25. Walter RB, Gooley TA, Wood BL, Milano F, Fang M, Sorror ML, et al. Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. J Clin Oncol. (2011) 29:1190–7. doi: 10.1200/JCO.2010.31.8121
26. Béné MC and Porwit A. Mixed phenotype/lineage leukemia: has anything changed for 2021 on diagnosis, classification, and treatment? Curr Oncol Rep. (2022) 24:1015–22. doi: 10.1007/s11912-022-01252-w
27. Al-Shaibani E, Novitzky-Basso I, Mattsson J, and Kim DDH. Post-transplant maintenance therapy in acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation harmonizing multiple therapeutic modalities including targeted therapy, immunotherapy and cellular therapy. Int J Hematol. (2023) 118:1–17. doi: 10.1007/s12185-023-03614-x
28. Xuan L and Liu Q. Maintenance therapy in acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol. (2021) 14:4. doi: 10.1186/s13045-020-01017-7
Keywords: mixed-phenotype acute leukemia (MPAL), allogeneic hematopoietic stem cell transplantation (allo-HSCT), prognosis, overall survival (OS), relapse-free survival (RFS), minimal residual disease (MRD)
Citation: Hao M, Zhao X, Zhang X, Zhai W, Ma Q, Yang D, Pang A, Feng S, He Y, Jiang E and Han M (2025) Allogeneic hematopoietic stem cell transplantation for mixed-phenotype acute leukemia: a single-center study. Front. Immunol. 16:1691762. doi: 10.3389/fimmu.2025.1691762
Received: 24 August 2025; Accepted: 29 October 2025;
Published: 18 November 2025.
Edited by:
Guido Moll, Charité University Medicine Berlin, GermanyReviewed by:
Yoshitaka Inoue, Kumamoto University Hospital, JapanVinícius de Molla, Federal University of São Paulo, Brazil
Copyright © 2025 Hao, Zhao, Zhang, Zhai, Ma, Yang, Pang, Feng, He, Jiang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi He, aHNjdF9odWF5dWFuQDE2My5jb20=