 Katja Sæterhaug Bye1
Katja Sæterhaug Bye1 Kristin Rian
Kristin Rian Terje Espevik
Terje Espevik Marit W. Anthonsen
Marit W. Anthonsen Maria Yurchenko
Maria Yurchenko- 1Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway
- 2Department of Infectious Diseases, Clinic of Medicine, St. Olavs Hospital HF, Trondheim University Hospital, Trondheim, Norway
Background: Human metapneumovirus (HMPV) is a major cause of acute respiratory disease in children, the elderly, and immunocompromised individuals. While pro-inflammatory cytokines and type I interferons (IFNs) are important for antiviral defense, excessive tumor necrosis factor (TNF) is associated with severe disease in HMPV and other respiratory infections. Hence, defining regulatory mechanisms by which HMPV induces TNF and IFN-β is important for therapeutic strategies in airway disease. The immunoregulatory receptors Toll-like receptor (TLR)4 and signaling lymphocytic activation molecule family 1 (SLAMF1) mediate TNF and IFN-β expression in response to LPS and Gram-negative bacteria, but their involvement in HMPV-stimulated cytokine expression is unclear.
Methods: We investigated the kinetics of TNF and IFNB1 expression in human monocyte-derived macrophages (MDMs) and THP-1 macrophage-like cells. The impact of SLAMF1 and TLR4 on TNF, IFNB1, and p38 MAPK was determined after their overexpression or knockout in THP-1 cells or silencing in MDMs.
Results: TLR4 knockout reduced TNF but not IFNB1 induced by HMPV, whereas SLAMF1 silencing reduced both cytokines. Overexpression of TLR4 or SLAMF1 enhanced p38 MAPK activation and TNF secretion, while silencing of TLR4 or SLAMF1 reduced p38 MAPK activation and TNF secretion. Pharmacological inhibition of p38 MAPK reduced both TNF and IFNB1, confirming its essential role in cytokine induction.
Conclusions: Together, our findings identify TLR4 and SLAMF1 as key regulators of early HMPV-induced inflammation via p38 MAPK. SLAMF1 additionally influences IFN-β responses and appears to affect viral replication dynamics. These insights suggest that targeting SLAMF1–TLR4 signaling may offer a therapeutic strategy to limit TNF-driven pathology in HMPV infection.
1 Introduction
Human metapneumovirus (HMPV) is an important human pathogen that can cause severe lower respiratory tract disease in infants, the elderly, and immunocompromised individuals, manifesting as bronchiolitis, pneumonia, or exacerbations of asthma or chronic obstructive pulmonary disease (1–3). HMPV is a negative−sense, single−stranded RNA virus of the Pneumoviridae family, with respiratory syncytial virus (RSV) being its closest relative (4). Reinfections with HMPV occur commonly throughout life (5), yet no vaccine or specific treatment for HMPV infection is currently available.
The innate immune system in the lungs detects viral infections through pattern recognition receptors (PRRs) expressed on cells such as airway epithelial cells and alveolar macrophages. These PRRs sense pathogen−associated molecular patterns (PAMPs) on viruses—such as surface glycoproteins, genomes, and replication intermediates—triggering an immediate immune response involving interferons (IFNs) and other inflammatory mediators that limit viral spread and recruit additional immune cells (6, 7). Depending on the infected cell type, different PRRs may be activated by HMPV to induce innate immune responses: the RIG−I−like receptors (RLRs); the endosomal Toll−like receptors (TLRs) TLR3, TLR7, and TLR8 (8, 9); and the LPS/Gram−negative bacteria sensing TLR4 (10, 11). Specific PRRs can generate distinct cytokine profiles depending on the downstream signaling complexes they engage. For instance, TLR4 can signal through either TIRAP/MyD88 or TRAM/TRIF complexes, leading to the production of pro−inflammatory cytokines or type I IFNs, respectively. TLR3 activation by its ligand preferentially promotes type I IFN production (12).
While respiratory epithelial cells are the primary targets of HMPV infection (13), HMPV can also infect dendritic cells and macrophages in the lungs (9, 14–16). Along with the main type I IFN-producing respiratory epithelial cells, alveolar macrophages initiate immune responses to HMPV by inducing type I IFNs and other pro−inflammatory cytokines, which generally contribute to viral clearance (17, 18). Macrophages are central regulators of immune balance and disease severity during infections with HMPV, RSV, influenza, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (14, 18–21).
Macrophages are well known as potent producers of TNF, particularly in inflammatory conditions and in response to PAMPs (22). TLR4 is crucial for TNF induction by bacteria and by LPS, a component of Gram−negative bacteria (12). Importantly, although TNF is a key pro−inflammatory cytokine critical for antiviral defenses, it also mediates hyperinflammation and acute lung injury in respiratory viral infections, as reported for influenza virus and SARS−CoV−2 (23–27). Similarly, we and others have shown that HMPV infection is accompanied by elevated TNF levels in nasopharyngeal aspirates and blood serum, which are associated with greater disease severity in hospitalized children (24, 28). However, the regulatory mechanisms underlying HMPV−induced TNF production remain incompletely understood.
A growing list of viruses is recognized to induce inflammatory responses via TLR4, including RSV, SARS−CoV−2, Ebola virus, influenza virus, and HMPV (10, 11, 29–34). TLR4 can sense viral surface glycoproteins and initiate downstream signaling that drives pro−inflammatory cytokine production (10, 31, 35, 36). For RSV and SARS−CoV−2, excessive TLR4 signaling has been implicated in lung pathology and heightened disease severity (33, 37).
We recently identified that the immune cell receptor SLAMF1 (signaling lymphocytic activation molecule family 1) enhances TLR4−mediated TNF and IFN-β induction by Escherichia coli in human macrophages (38). SLAMF1 is a co−stimulatory molecule expressed by immune cells, including macrophages, and regulates signal transduction networks essential for effective immune responses (39–44). Whether SLAMF1 regulates HMPV−induced TNF and IFN-β expression has not been explored. In this study, we set out to characterize the contribution of TLR4 and SLAMF1 to the induction of TNF and IFN-β in HMPV−infected human primary macrophages. Our results demonstrate that both TLR4 and SLAMF1 regulate an early HMPV−stimulated upregulation of the p38 MAPK–TNF axis, while TLR4 does not affect IFN-β induction in human macrophages. These findings reveal novel mechanisms controlling TNF and IFN-β output in human macrophages, which may be relevant for therapeutic strategies targeting pro−inflammatory signaling in HMPV−infected patients.
2 Materials and methods
2.1 Cell culture and inhibitors
The use of human buffy coats and serum from the blood bank at St. Olavs Hospital (Trondheim, Norway) was approved by the Regional Committee for Medical and Health Research Ethics (REC) in Central Norway (#2009/2245). Primary human monocytes were isolated from the buffy coat by adherence, as previously described (45). In brief, freshly prepared buffy coats were diluted by 100 mL of PBS and applied on top of Lymphoprep (Axis-Shield, Dundee, Scotland) according to the manufacturer’s instructions. Cells were counted using Z2 Coulter particle count and size analyzer (Beckman Coulter, Munich, Germany) on program B, resuspended in RPMI 1640 (Sigma-Aldrich, Merck, Darmstadt, Germany) supplemented with 5% of pooled human serum at a concentration of 6 × 106 per mL, and seeded to 24-well (0.5 mL per well) cell culture dishes. After a 60-min incubation for the surface adherence of monocytes, the dishes were washed three times with HBSS (Sigma-Aldrich, Merck) to remove non-adherent cells. Isolated cells were kept in RPMI 1640 medium supplemented with 10% human serum, 0.34 mM of L-glutamine, and 25 ng/mL of rhM-CSF (216-MC-025; R&D Systems, BioNordika, Oslo, Norway). THP-1 cells (ATCC) and generated THP-1 sublines were cultured in RMPI 1640 supplemented by 10% heat-inactivated FCS, 100 nM of penicillin/streptomycin (Thermo Fisher Scientific, Oslo, Norway), 5 μM of β-mercaptoethanol, 2 mM of L-glutamine, 10 mM of HEPES, 1 mM of sodium pyruvate, 4,500 mg/L of glucose, and 1,500 mg/L of sodium bicarbonate (Sigma-Aldrich, Merck). THP-1 cells (300,000 cells per/well, 24-well plates, in 0.5 mL of media per well) were differentiated with 60 ng/mL of PMA (Sigma-Aldrich, Merck) for 24 h, followed by 48 h in medium without PMA. The p38 MAPK inhibitor BIRB796 (doramapimod; Selleck Chemicals, #S1574, VWR International LLC, Norway) was dissolved in DMSO, aliquoted, and stored at −80°C to prevent repeated freeze–thaw cycles.
2.2 Generation of THP-1 sublines
For making TLR4 KO THP-1 and control THP-1 sublines, LentiCRISPRv2 plasmid (a gift from F. Zhang; 52961; Addgene; Sanjana et al., 2014) was ligated with 5′-AAACGCGTGAGACCAGAAAGCTGGC-3′ and 5′-CACCGAAGGTCCAAGTGCTCTAGAT-3′ for TLR4 and 5′-CACCGTTTGTAATCGTCGATACCC3′ and 5′-AAACGGGTATCGACGATTACAAAC-3′ for control non-targeting guiding RNA expression. Generation of TLR4 overexpressing control cells is described in (46). For SLAMF1 overexpression, SLAMF1Flag cDNA (38) was recloned to pLVX-EF1α-IRES-ZsGreen1 (Takara Bio, AH Diagnostics AS, Oslo, Norway). Packaging plasmids pMD2.G and psPAX2 were used for producing lentivirus (provided by D. Trono, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland; 12260 and 12259; Addgene). HEK293T cells (cultured in DMEM supplemented by 10% FCS, 100 IU/mL of penicillin, 100 μg/mL of streptomycin) were co-transfected with the packaging and lentiCRISPRv2 or the above-listed lentiviral pLVX constructs (empty vectors, or TLR4Flag pLVX, or SLAMF1Flag pLVX) using the GeneJuice transfection reagent (Merck), and the media was changed in 16 h. The lentivirus-containing supernatants were collected after another 48 h and used for transduction of THP-1 cells along with 8 µg/mL of protamine sulfate. Transduced THP-1 cells were then sorted based on ZsGreen co-expression 1 week after transduction using the BD FACS Aria III cell sorter (BD Biosciences, Oslo, Norway) with BD FACSDiva 8.0 software (BD Biosciences). All cell lines were regularly checked for mycoplasma contamination.
2.3 Virus propagation
Virus propagation was performed as described (16). Briefly, LLC-MK2 cells were inoculated with the clinical HMPV isolate NL/17/00 (A2) at a multiplicity of infection (MOI) of 0.01 in OptiMEM containing 2% FBS, 20 µg/mL of gentamicin, and 0.7 nM of glutamine. On days 7–8, the virus was harvested by freeze–thawing at −80°C, followed by purification on a 20% sucrose cushion and resuspension in OptiMEM (2% FBS). The virus titer was determined using a cell-based immunoassay. Purified virus particles were serially diluted (log10) on monolayers of LLC-MK2 cells in 96-well flat-bottom plates. After 4 days, cells were washed and stained with LIGHT DIAGNOSTICS™ HMPV direct fluorescence assay (Merck Millipore, Darmstadt, Germany), and foci-forming units were determined by manual counting.
2.4 In vitro HMPV infection
Cells were infected with HMPV A2 at MOI 1 in OptiMEM containing 2% FBS, 20 µg/mL of gentamicin, and 0.7 nM of glutamine. Cells were incubated with the virus for the indicated time.
2.5 siRNA treatment
Oligonucleotides used for silencing were AllStars negative control siRNA (SI03650318), FlexiTube siRNA Hs_SLAMF1_2 (SI00047250), and Hs_TLR4_2 (SI00151011; QIAGEN, Sollentuna, Sweden). On day 6 from isolation, cells were transfected with silencing oligonucleotides (20 nM final concentration) using Lipofectamine 3000 (L3000008, Invitrogen, Thermo Fisher Scientific, Oslo, Norway) as suggested by the manufacturer. Media was changed on day 7, transfection was repeated on day 8, media was changed on day 9, and HMPV infection was performed on day 10 to day 11.
2.6 qRT-PCR
Total RNA was isolated from the cells using QIAzol reagent (QIAGEN), and chloroform extraction was followed by purification on RNeasy Mini columns with DNAse digestion step (QIAGEN). cDNA was prepared with a Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), in accordance with the protocol of the manufacturer, using 400 ng of total RNA per sample. qPCR was performed with the PerfeCTa qPCR FastMix (Quanta Biosciences, VWR International, Oslo, Norway) in replicates and cycled in a StepOnePlus Real-Time PCR cycler (Thermo Fisher Scientific). The following TaqMan Gene Expression Assays (Applied Biosystems, Thermo Fisher Scientific, Oslo, Norway) were used: IFNβ (Hs01077958_s1), TNF (Hs00174128_m1), TBP (Hs00427620_m1), SLAMF1 (Hs00900288_m1), TLR4 (Hs00152939_m1), and CXCL10 (Hs01124251_g1). HMPV vRNA expression was analyzed by qRT-PCR using SybrGreen-based master mix Fast SYBR™ Green Master Mix (Thermo Fisher Scientific) and the following primers: HMPV-N (fwd) CATATAAGCATGCTATATTAAAAGAGTCTC, HMPV-N (rev) CCTATTTCTGCAGCATATTTGTAATCAG for HMPV-N and TBP (fwd) 5′-GAGCCAAGAGTGAAGAACAGTC-3′ and (rev) 5′-GCTCCCCACCATATTCTGAATCT-3′. Fold change in HMPV vRNA expression was calculated relative to the indicated infected sample in the figure legend. The level of TBP mRNA was used for normalization, and the results were presented as a relative expression compared with the control’s untreated sample. Relative expression was calculated using Pfaffl’s mathematical model (47). Graphs and statistical analyses were made with GraphPad Prism v9.1.2 (Dotmatics, Boston, MA, USA), with additional details provided in the figure legends and in Section 2.9.
2.7 Western blotting
Cell lysates for Western blotting (WB) analysis were prepared by simultaneous extraction of proteins and total RNA using QIAzol reagent (QIAGEN) as suggested by the manufacturer. The extracted total RNA was used for qRT-PCR, whereas protein samples were used for simultaneous analysis of protein expression/posttranslational modifications. Protein pellets after protein isolation were dissolved by heating the samples for 10 min at 95°C in a buffer containing 4 M of urea, 1% SDS (Sigma-Aldrich, Merck), and NuPAGE LDS Sample Buffer (4X) (Thermo Fisher Scientific), with a final 25 mM DTT in the samples. For SDS-PAGE, we used pre-cast gradient 4%–12% Bis-Tris protein gels NuPAGE Novex and 1X MOPS SDS running buffer (Thermo Fisher Scientific). Proteins from the gel were transferred to iBlot Transfer Stacks by using the iBlot Gel Transfer Device (Thermo Fisher Scientific). The blots were developed with the SuperSignal West Femto (Thermo Fisher Scientific) and visualized with the LI-COR ODYSSEY Fc Imaging System (LI-COR Biotechnology, Bad Homburg, Germany). For densitometry analysis of the WB bands, Odyssey Image Studio 5.2 software (LI-COR Biotechnology) was used, and the relative numbers of the bands’ intensity were normalized to the intensities of the respective loading-control protein (β-tubulin).
2.8 Antibodies and ELISA
The following primary antibodies were used for Western blotting: rabbit β-tubulin (ab6046) from Abcam; phospho-p38 MAPK (T180/Y182) (D3F9), phospho-STAT1 (Tyr701) (58D6), and anti-DYKDDDDK tag (D6W5B)/Flag tag from Cell Signaling Technology; mouse anti-HMPV Nucleoprotein from Abcam; and mouse STAT1 antibodies from BD Biosciences (#610185, Wokingham, UK). Secondary antibodies (HRP-linked) were from DAKO Denmark A/S (Glostrup, Denmark). The following ELISA assays were used: human CXCL10/IP-10 DuoSet ELISA (DY266) and human TNF DuoSet ELISA (DY210) from R&D Systems (Biotechne, Norway). Assays were performed as suggested by the manufacturer, and supernatants were stored at −80°C after collection and unfrozen on ice just before running the assays.
2.9 Statistical analysis
Data that were assumed to follow a log-normal distribution were log-transformed before statistical analysis. Quantification of gene expression by qRT-PCR was log-transformed and analyzed by repeated measures analysis of variance (RM-ANOVA) or a mixed model if there were missing data, followed by Holm–Šídák’s multiple comparisons post-test. ELISA and BioPlex data were analyzed using a Wilcoxon matched-pairs signed-rank test, a Mann–Whitney test, or a paired t-test. For the data from WB analysis, significance was evaluated by t-test with Welch’s correction. All graphs and analyses were generated with GraphPad Prism v10.1.0 (Dotmatics).
3 Results
3.1 HMPV induces TNF expression earlier than IFNB1 in THP−1 cells and human monocyte−derived macrophages
To determine the impact of TLR4 and SLAMF1 on HMPV-stimulated TNF and IFN-β responses in macrophages, we aimed to use THP-1 cells as a supporting model system to primary human monocyte-derived macrophages (MDMs), as THP-1 cells are more amenable to genetic manipulation (e.g., TLR4 and SLAMF1 knockout or overexpression studies). However, since transcriptional differences between THP-1 cells and MDMs have been reported (48, 49), we first compared HMPV-induced TNF and IFNB1 mRNA expression in THP-1 cells and MDMs. Human MDMs were differentiated from peripheral blood monocytes of healthy donors for 7 days, while THP−1 cells were differentiated into macrophage−like cells using phorbol 12-myristate 13-acetate (PMA). Both cell types were infected with HMPV (1–20 h) or treated with LPS, and lysates were collected at defined time points for parallel RNA and protein analyses.
The mRNA levels of IFNB1, TNF, viral nucleocapsid N (HMPV-N), SLAMF1, and TLR4 were quantified by RT-qPCR (Figures 1A, B, E, F). In parallel, phosphorylated and total STAT1 protein, as well as HMPV-N protein, was assessed by Western blotting (Figures 1C, D). STAT1 total levels may be induced by type I IFNs also in HMPV-infected cells (16, 50, 51), and we therefore analyzed both phosphorylated and total STAT1 levels. In both MDMs and THP-1 cells, HMPV-N mRNA accumulated progressively over time, with a slight delay in THP-1 cells compared with MDMs (Figures 1A, B, first panels). HMPV infection induced a gradual increase in IFNB1 expression in both cell types, with higher levels in MDMs than in THP-1 cells (Figures 1A, B, second panels). Consistently, phospho-STAT1 and total STAT1 protein levels increased toward the late stages of infection (20–24 h) in both cell types (Figures 1C, D), reflecting active type I IFN signaling (52).
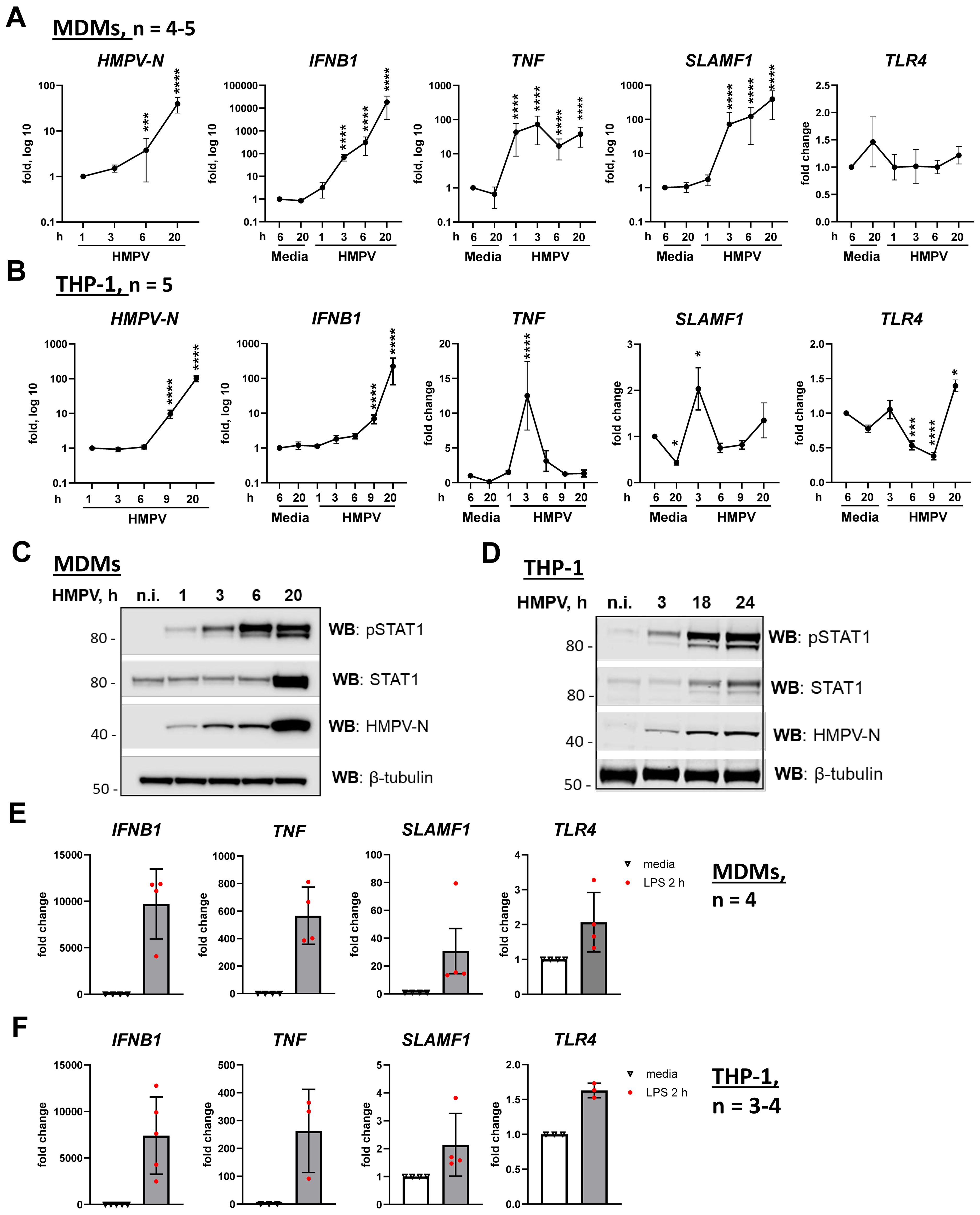
Figure 1. HMPV induces TNF mRNA expression at earlier time points of infection than IFNB1 in both MDMs and THP-1 cells. (A, B) HMPV-N vRNA expression and IFNB1, TNF, SLAMF1, and TLR4 mRNA expression were evaluated by qRT-PCR in MDMs or THP-1 cells (n = 5) infected with HMPV (MOI = 1). Results were normalized to non-infected samples (media 6 h) or, for HMPV-N vRNA, to the level detected 1 h after infection. Data are presented as mean ± SD, with statistical testing (comparison with media 6-h values) performed by two-way ANOVA on log-transformed data (***p < 0.001, ****p < 0.0001). (C, D) Western blot (WB) analysis was performed to determine the total and phosphorylated (Tyr701) STAT1 and HMPV-N protein expression by MDMs (C) or THP-1 cells (D) at different time points of infection. Representative images are presented for one out of four individual donors (C) or one out of four experiments for THP-1 cells (D). WB for β-tubulin was used as an endogenous loading control. (E, F) IFNB1, TNF, and SLAMF1 mRNA expression was evaluated by qRT-PCR in MDMs or THP-1 WT cells stimulated by LPS (100 ng/mL) for 2 h. (E, F) Data are presented as the mean of relative fold change ± SD.
Notably, TNF mRNA was strongly induced early during HMPV infection, peaking approximately 3 h in both MDMs and THP-1 cells (Figures 1A, B, third panels). While TNF levels subsequently declined in THP-1 cells between 6 and 20 h, MDMs maintained a higher expression during these later stages (Figures 1A, B). Strikingly, HMPV also induced SLAMF1 expression, with a marked upregulation in MDMs and a more moderate response in THP-1 cells (Figures 1A, B, fourth panels). In both cell types, SLAMF1 expression patterns paralleled those of TNF, with sustained induction in MDMs and a transient peak in THP-1 cells (Figures 1A, B, third and fourth panels).
To investigate whether modulation of TLR4 expression influences HMPV-induced responses, we examined TLR4 mRNA levels and observed that, while TLR4 expression in MDMs was not significantly affected by HMPV infection, TLR4 mRNA in THP-1 cells was significantly reduced at 6 and 9 h post-infection and subsequently upregulated at 20 h (Figures 1A, B, right panels). The transient reduction in TLR4 expression at 6 and 9 h in THP-1 cells could account, at least in part, for the more rapid decline in TNF mRNA induction observed in THP-1 cells compared with MDMs (Figures 1A, B, third panels). As expected and consistent with prior studies (53), TLR4 mRNA was modestly increased in response to LPS (Figures 1E, F, last panels).
In summary, TNF was induced earlier than IFNB1 following HMPV infection in both THP-1 cells and MDMs. However, when comparing THP-1 cells with MDMs, HMPV-mediated induction of IFNB1, TNF, and SLAMF1 mRNAs was higher in MDMs than in THP-1 cells, and the induction of TNF and SLAMF1 by HMPV declined more rapidly in THP-1 cells than in MDMs. Consistent with previous reports (48, 49), these findings underscore transcriptional differences between THP-1 cells and MDMs in their responses to inflammatory stimuli, demonstrating that during HMPV infection, particularly SLAMF1 but also TNF and IFNB1 are induced to higher levels in MDMs. Although these results highlight the limitations of using THP-1 cells as a standalone macrophage model, we consider THP-1 cells to represent a suitable supporting system to MDMs for investigating the roles of TLR4 and SLAMF1 in the regulation of IFNB1 and TNF induction, as both cell types exhibited comparable patterns of early TNF and progressive IFNB1 induction upon HMPV infection.
3.2 Knockout of TLR4 reduces HMPV−induced TNF expression without affecting IFNB1
TLR4 regulates the induction of both TNF and IFNB1 in response to LPS through different signaling complexes formed with TIRAP/MyD88 at the plasma membrane and TRAM/TRIF following endocytosis (12). To evaluate if TLR4 contributes to TNF and IFNB1 expression during HMPV infection, we next made use of a THP-1 TLR4 knockout subline (THP-1 TLR4 KO) alongside control THP-1 cells expressing non-targeting guide RNA. Both cell types were infected with HMPV for various time points, followed by analysis of HMPV-N (Figure 2A), TNF, and IFNB1 mRNA levels (Figure 2B) and measurement of secreted TNF and CXCL10 (Figure 2C). CXCL10 can be induced not only by type I interferons but also by other cytokines (54–57). To assess the contribution of type I IFN signaling to HMPV-induced CXCL10 expression, we examined the effect of anti-IFNAR neutralizing antibodies. Pretreatment with anti-IFNAR antibodies almost completely abrogated HMPV-induced CXCL10 expression (Supplementary Figure S1), indicating that CXCL10 induction is largely dependent on type I IFN signaling and thus reflects the levels of secreted type I IFNs. Our results showed that secreted TNF, but not CXCL10, was reduced in HMPV-infected THP-1 TLR4 KO cells (Figure 2C) and confirmed that TLR4 knockout did not impair HMPV-induced IFNB1 levels. HMPV-N mRNA expression was comparable between the control and TLR4 KO cells (Figure 2A), indicating that differences in cytokine expression were not due to altered viral replication.
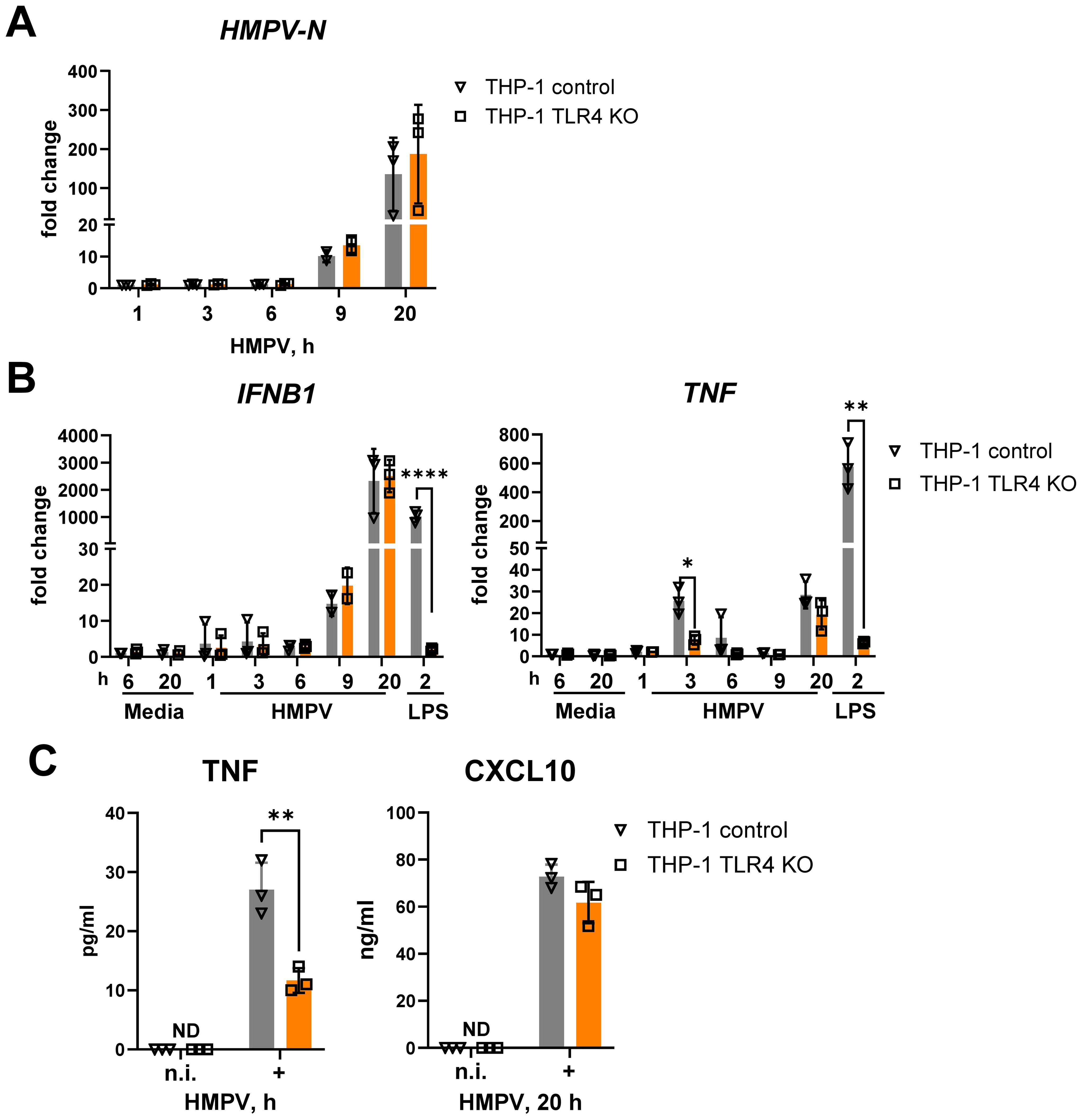
Figure 2. TLR4 knockout in THP-1 cells strongly reduces the expression of HMPV-mediated TNF mRNA and protein without affecting IFNB1 mRNA expression and IFN-β-dependent CXCL10 secretion. HMPV-N vRNA expression (A) and IFNB1 and TNF mRNA expression (B) were determined by qRT-PCR in THP-1 control cells and THP-1 TLR4 KO cells infected with HMPV (MOI = 1) for the indicated time points or stimulated by LPS for 2 h (n = 3). Results were normalized to non-infected (n.i.) samples. Data are presented as mean ± SD, with statistical testing performed by two-way ANOVA on log-transformed data (*p < 0.05, ****p < 0.0001). (C) TNF and CXCL10 secretion was determined by ELISA of supernatants from non-infected (n.i.) or HMPV-infected cells (20 h), and data are presented as mean ± SD (n = 3). Statistical significance was assessed using a paired t-test, and only significant results are indicated (**p < 0.01; ND, not detected).
TLR4 deficiency markedly reduced the early (3 h post-infection) HMPV-induced TNF mRNA expression, whereas HMPV-stimulated IFNB1 mRNA expression was unaffected (Figure 2B). As expected, TLR4 knockout abolished LPS-induced expression of both TNF and IFNB1 (Figure 2B). The selective effect of TLR4 knockout on HMPV-mediated TNF was further reflected at the protein level: secreted TNF was significantly reduced at 20 h post-infection in TLR4 KO cells, while secretion of the IFN-inducible chemokine CXCL10 remained unchanged (Figure 2C).
Together, these results show that in THP-1 cells, TLR4 is required for early HMPV-induced TNF expression but does not regulate HMPV-induced IFNB1.
3.3 TLR4 overexpression enhances HMPV−induced p38 MAPK activation and TNF and IFNB1 expression in THP−1 cells
We next examined how TLR4 overexpression affected HMPV-induced IFNB1 and TNF expression by comparing a previously generated (46) THP-1 subline overexpressing TLR4-Flag (THP-1 TLR4) with control THP-1 cells carrying an empty vector. TLR4 overexpression significantly increased HMPV-stimulated TNF and IFNB1 mRNA levels at later stages of infection (Figure 3A). Although HMPV-stimulated IFNB1 induction was not reduced in TLR4-deficient THP-1 cells (Figure 2B), overexpression of TLR4 enhanced IFNB1 expression (Figure 3A). The reason for this apparent discrepancy remains unclear; however, it is possible that other PRRs, such as DC-SIGN, may compensate for the absence of TLR4 in HMPV-mediated IFNB1 induction, as has been reported for other pathogen–PRR interactions (58, 59).
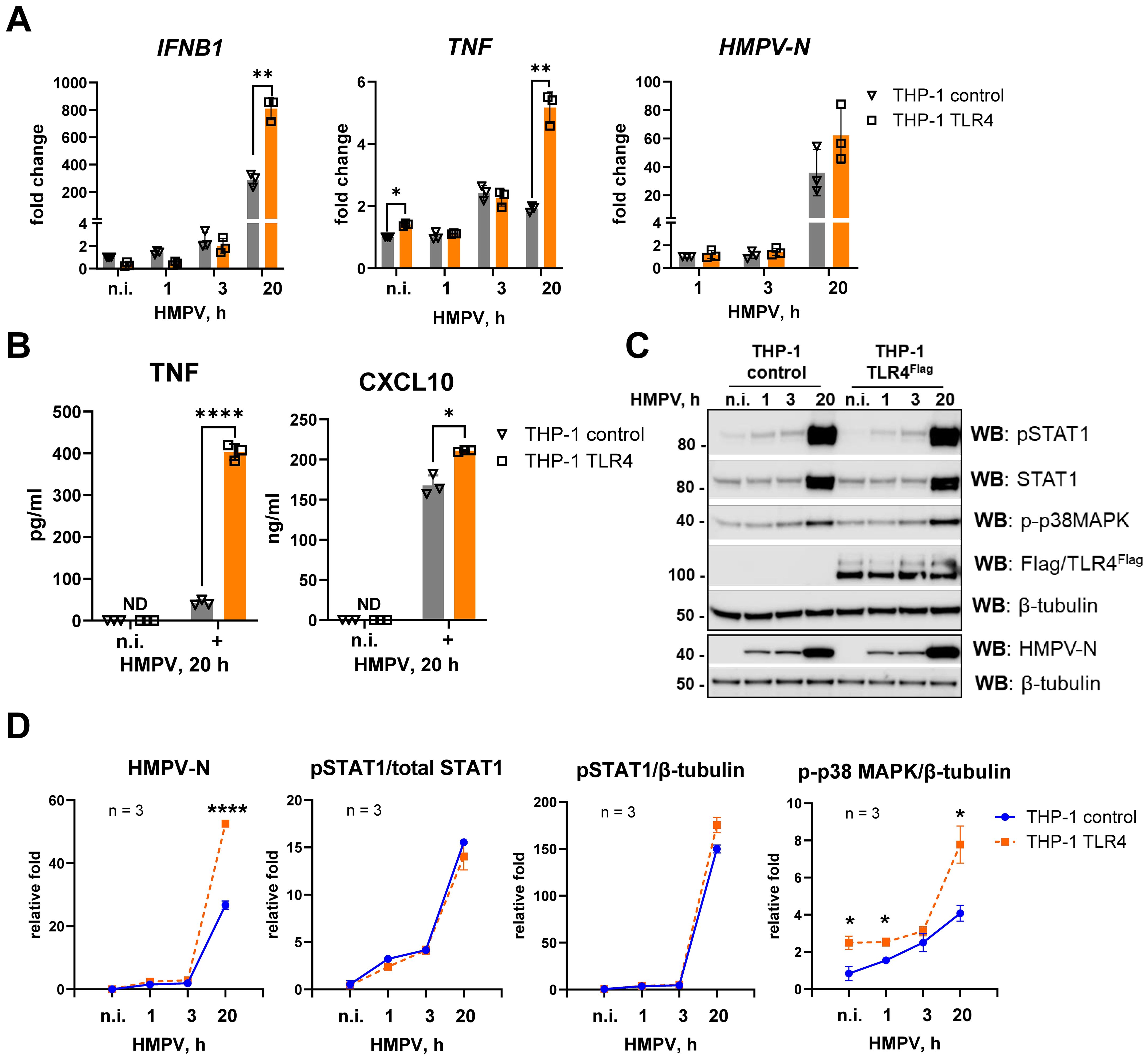
Figure 3. TLR4 overexpression in THP-1 cells enhances HMPV-stimulated TNF and IFNB1 induction and p38 MAPK activation. (A) HMPV-N vRNA expression and IFNB1 and TNF mRNA expression were determined by qRT-PCR in THP-1 control cells and THP-1 TLR4Flag cells infected by HMPV (MOI = 1) for the indicated time points or stimulated by LPS for 2 h (n = 3). The results were normalized to non-infected (n.i.) samples or, for HMPV-N, to the level detected 1 h after infection in control cells. Statistical testing was done by two-way ANOVA on log-transformed data (**p < 0.01). (B) TNF and CXCL10 cytokine secretion levels were determined by ELISA of supernatants from non-infected (n.i.) or HMPV-infected cells (20 h), and data are presented as mean ± SD (n = 3). Statistical significance was evaluated by paired t-test; significance level: *p < 0.05, ****p < 0.0001; ND, not detected. (C) WB analysis was performed to determine the expression of total and phosphorylated (Tyr701) STAT1, phospho-p38MAPK (Thr180/Tyr182), HMPV-N, and TLR4Flag in THP-1 sublines at different time points of HMPV infection (a representative image is shown, n = 4). WB for β-tubulin was used as an endogenous control. (D) Graphs show combined data for three experiments (analyzed in LiCor Odyssey software) of HMPV-N, pSTAT1, or p-p38 MAPK protein levels relative to β-tubulin or pSTAT1 relative to total STAT1. Statistical significance was assessed using a t-test with Welch’s correction, and only significant results are indicated (*p < 0.05, **p < 0.01).
HMPV-N mRNA and protein levels were also elevated in TLR4-overexpressing cells at 20 h post-infection (Figures 3A, C, D). In line with the mRNA data, secretion of TNF and CXCL10 was increased in TLR4-overexpressing THP-1 cells compared with controls (Figure 3B). In addition, phosphorylated STAT1, which we have previously shown to depend strongly on IFN receptor signaling in HMPV-infected MDMs (16), was modestly increased in TLR4-overexpressing cells (Figures 3C, D).
Overexpression of TLR4 in THP-1 cells was confirmed by immunoblotting (Figure 3C), and its functional activity was validated by enhanced induction of IFNB1 and TNF mRNA in response to LPS stimulation (Supplementary Figure S2).
The p38 MAPK pathway, known to be activated downstream of multiple PRRs and to regulate TLR4-mediated TNF expression in response to LPS (60, 61), had not been directly examined in the context of HMPV infection. Interestingly, we observed that HMPV triggered p38 MAPK phosphorylation, and this activation was significantly increased in TLR4-overexpressing THP-1 cells compared with control cells (Figures 3C, D).
Collectively, these findings indicate that TLR4 overexpression in THP-1 cells enhances HMPV-induced p38 MAPK activation and promotes increased TNF and IFNB1 expression in THP-1 cells.
3.4 SLAMF1 overexpression enhances HMPV−induced p38 MAPK activation and TNF secretion
In human macrophages, we have previously shown that SLAMF1 is critical for E. coli- and LPS-TLR4-mediated IFNB1 expression, while its effect on TNF expression was more modest (38). To investigate whether SLAMF1 influences HMPV-induced IFNB1 and TNF responses, we generated THP-1 cells overexpressing SLAMF1 (THP-1 SLAMF1) along with control cells with an empty vector (THP-1 control). SLAMF1 overexpression was confirmed by Western blotting (Supplementary Figure S3).
HMPV-stimulated IFNB1 mRNA levels and secretion of the IFN-inducible protein CXCL10 were largely unaffected by SLAMF1 overexpression in THP-1 cells. In contrast, TNF mRNA expression and TNF secretion were significantly increased in SLAMF1-overexpressing THP-1 cells (Figures 4A, B). Notably, TNF mRNA induction was enhanced as early as 3 h post-infection, with a corresponding increase in secreted TNF observed after 20 h of HMPV infection (Figures 4A, B). Additionally, HMPV-N mRNA levels were significantly higher in SLAMF1-overexpressing THP-1 cells at 20 h post-infection (Figure 4A).
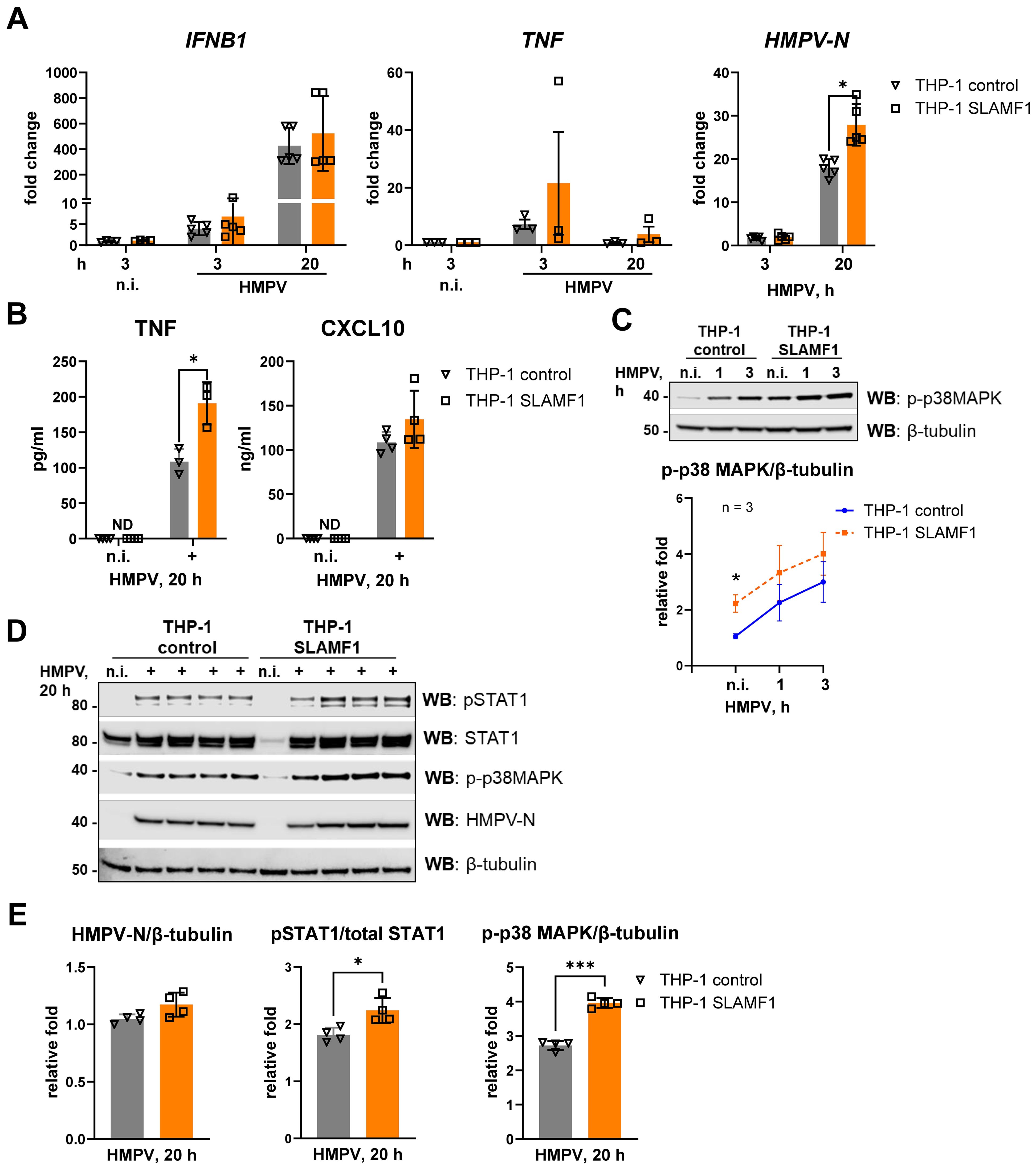
Figure 4. SLAMF1 overexpression in THP-1 cells increases HMPV-stimulated p38 MAPK activation and TNF secretion. (A) IFNB1, TNF, and HMPV-N vRNA mRNA expression was determined by qRT-PCR analysis of THP-1 control cells and THP-1 SLAMF1 cells infected with HMPV (MOI = 1) for the indicated time points (n = 3–5). Results were normalized to non-infected (n.i.) samples or, for HMPV-N, to the level detected 3 h after infection in control cells. Statistical testing was done by two-way ANOVA on log-transformed data (**p < 0.01). (B) TNF and CXCL10 cytokine secretion levels were determined by ELISA of supernatants from non-infected (n.i.) or HMPV-infected cells (20 h), and the data are presented as mean ± SD (n = 3–4). Statistical significance was evaluated paired t-test, with no significant results found; ND, not detected. (C, D) WB was performed to determine (C) the expression of phospho-p38MAPK (Thr180/Tyr182) at early time points of HMPV infection (representative image, n = 3) and (D, E) the expression of total and phosphorylated (Tyr701) STAT1, phospho-p38MAPK, and HMPV-N (results of four independent experiments are shown together on the blots, 20 h of infection) in THP-1 sublines. (C, D) β-Tubulin was used as an endogenous control. (E) Quantification of HMPV-N or p-p38 MAPK protein levels relative to β-tubulin or pSTAT1 relative to total STAT1 was performed using LiCor Odyssey software, combining data for the four experiments. Statistical significance was assessed using a paired t-test, and only significant results are indicated (*p < 0.05, **p < 0.01).
We have previously demonstrated that SLAMF1 is required for TLR4-driven activation of p38 MAPK (38). Consistent with this, SLAMF1 overexpression significantly enhanced HMPV-induced phosphorylation of p38 MAPK (Figures 4C–E), similar to the increased p38 MAPK activation observed in TLR4-overexpressing cells (Figure 3C). In agreement with the observation that SLAMF1 overexpression did not markedly increase HMPV-induced IFNB1 or CXCL10, phosphorylation of STAT1 by HMPV was modestly elevated in SLAMF1-overexpressing cells (Figures 4D, E). The reason why we do not observe a directly correlated output between IFNB1 levels and phosphorylated STAT1 may relate to different optimal kinetics of these responses and the experimental time points used for analysis.
Taken together, these findings show that SLAMF1 overexpression in THP-1 cells enhances HMPV-triggered p38 MAPK activation and TNF induction, while having only a limited effect on HMPV-induced IFNB1 expression in THP-1 cells.
3.5 TLR4 and SLAMF1 silencing in human MDMs reduces HMPV-induced TNF expression
While we used THP-1 to pinpoint effects of overexpression and knockout of SLAMF1 and TLR4 on TNF and IFNB1 induction, THP-1 showed lower HMPV-stimulated induction of SLAMF1, TNF, and IFNB1 and had been shown to be phenotypically different from MDMs (48, 49). Hence, for translational significance, it is of utmost importance to validate findings from THP-1 cells in human primary MDMs. Hence, we next examined the effects of TLR4 and SLAMF1 silencing on HMPV-induced TNF and IFNB1 expression in human primary MDMs derived from healthy donors (n = 6). As we find that commercially available antibodies for TLR4 and SLAMF1 have low specificity in immunoblotting, efficient silencing was confirmed by significantly reducing TLR4 and SLAMF1 mRNA levels at all tested infection time points, as well as reducing LPS-induced TNF and IFNB1 in siTLR4-treated MDMs (Supplementary Figure S4). The LPS-mediated cytokine response is completely dependent on TLR4 expression and could serve as an additional readout for TLR4 expression (62). The efficiency of our SLAMF1 siRNA in MDMs has previously been validated by confocal microscopy (38).
Silencing of SLAMF1 or TLR4 had minimal impact on HMPV-induced IFNB1 mRNA expression, except for a reduction observed in SLAMF1-silenced MDMs at 20 h post-infection (Figure 5A). In contrast, HMPV-induced TNF mRNA expression in MDMs was significantly reduced at 3 h post-infection following silencing of either SLAMF1 or TLR4 (Figure 5B). To evaluate the secreted type I IFN levels in SLAMF1- and TLR4-silenced MDMs after 20 h of HMPV infection (Figure 5A), we assessed CXCL10 expression, which critically depends on type I IFN signaling in HMPV-infected MDMs (Supplementary Figure 1). Consistent with the reduced IFNB1 mRNA levels in SLAMF1-silenced MDMs, CXCL10 induction was significantly decreased upon SLAMF1 depletion (Figure 5C), indicating that secreted type I IFNs are indeed reduced in HMPV-infected SLAMF1-depleted MDMs. Similarly, in line with the reduced TNF mRNA induction, TNF secretion was markedly diminished in TLR4- or SLAMF1-depleted MDMs across all examined time points (Figure 5D).
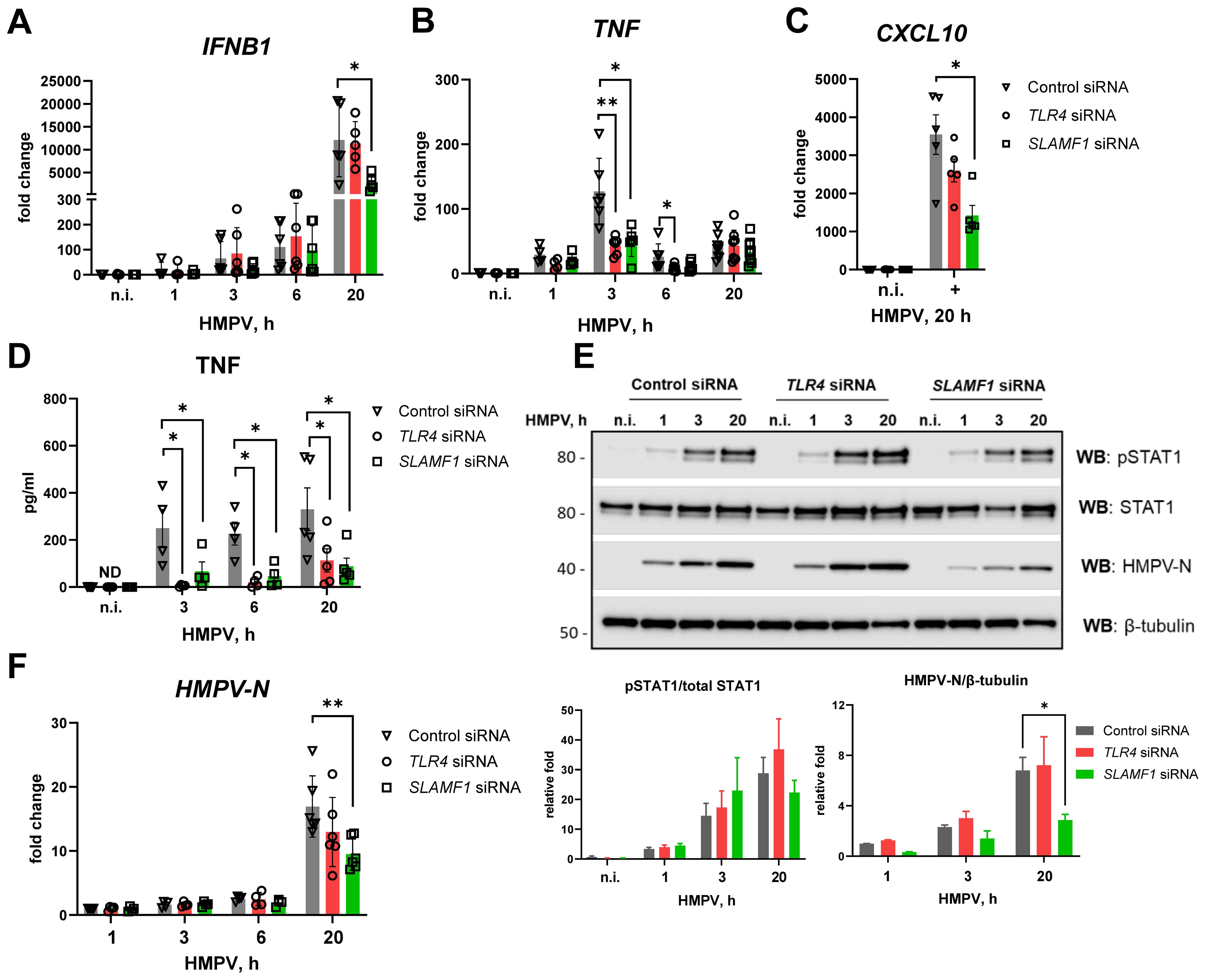
Figure 5. TLR4 or SLAMF1 silencing in human MDMs reduces HMPV-mediated TNF expression, while only SLAMF1 silencing decreases IFNB1 induction. Primary human MDMs were transfected with control, TLR4, and SLAMF1 siRNA oligos and infected with HMPV for the indicated time points (n = 5–8). (A, B) Expression of IFNB1 (A), TNF (B), and CXCL10 (C) mRNA was evaluated by qRT-PCR, and the results were normalized to non-infected (n.i.) samples. (D) TNF cytokine secretion level was determined by ELISA of supernatants from non-infected (n.i.) or HMPV-infected cells (20 h). (E) WB analysis (representative image, n = 4) was performed to determine total and phosphorylated (Tyr701) STAT1 levels and HMPV-N protein expression. β-Tubulin was used as an endogenous control for loading. Graphs show combined data for quantification of HMPV-N protein level relative to β-tubulin or pSTAT1 relative to total STAT1 (LiCor Odyssey software). (F) HMPV-N vRNA expression was determined by qRT-PCR, and the results were normalized to the level detected after 1 h of infection of control cells. Data on graphs are presented as mean of relative fold change ± SEM (A–D, F) or ± SD (E). Statistical testing was done by two-way ANOVA on log-transformed data (A–C, F) or by a multiple Wilcoxon test (D) or by a paired t-test (E), and only significant results are indicated (*p < 0.05, **p < 0.01; ND, not detected).
Knockdown of TLR4 or SLAMF1 did not substantially alter HMPV-induced STAT1 phosphorylation at early time points, although SLAMF1 depletion significantly reduced STAT1 phosphorylation at 20 h post-infection (Figure 5E), consistent with the reduced HMPV-N mRNA and protein levels in these MDMs (Figures 5E, F). Comparing these results to THP-1 cells, we note that SLAMF1 affected late IFNB1 induction in MDMs but did not affect IFNB1 induction in THP-1 cells (Figure 4), suggesting that SLAMF1 may be expressed at higher levels in HMPV-infected MDMs (Figure 1) and hence impact IFNB1 expression in human MDMs.
Collectively, these results indicate that both TLR4 and SLAMF1 are required for the early induction of TNF mRNA by HMPV in human MDMs, while only SLAMF1 contributes to HMPV-mediated IFNB1 expression at later stages of infection.
3.6 HMPV−induced p38 MAPK activation is regulated by TLR4 and SLAMF1 and drives TNF and IFNB1 induction in human macrophages
We observed that HMPV infection induces p38 MAPK phosphorylation in THP-1 cells, and this effect is enhanced by TLR4 and SLAMF1 (Figures 3, 4). To further assess the contribution of these receptors to HMPV-mediated p38 MAPK activation in human primary MDMs, we examined p38 MAPK phosphorylation in MDMs transfected with TLR4 or SLAMF1 siRNAs and subsequently infected with HMPV at different time points. HMPV infection triggered robust p38 MAPK phosphorylation in MDMs as early as 1 h post-infection, with activation slightly reduced but maintained throughout 20 h (Figure 6A). Silencing either TLR4 or SLAMF1 reduced HMPV-induced p38 MAPK phosphorylation from the earliest analyzed time point (Figure 6A), indicating that both receptors contribute to early p38 MAPK activation in MDMs, possibly through the TLR4–TIRAP–MyD88 signaling axis, which is positively regulated by SLAMF1 in human macrophages (38).
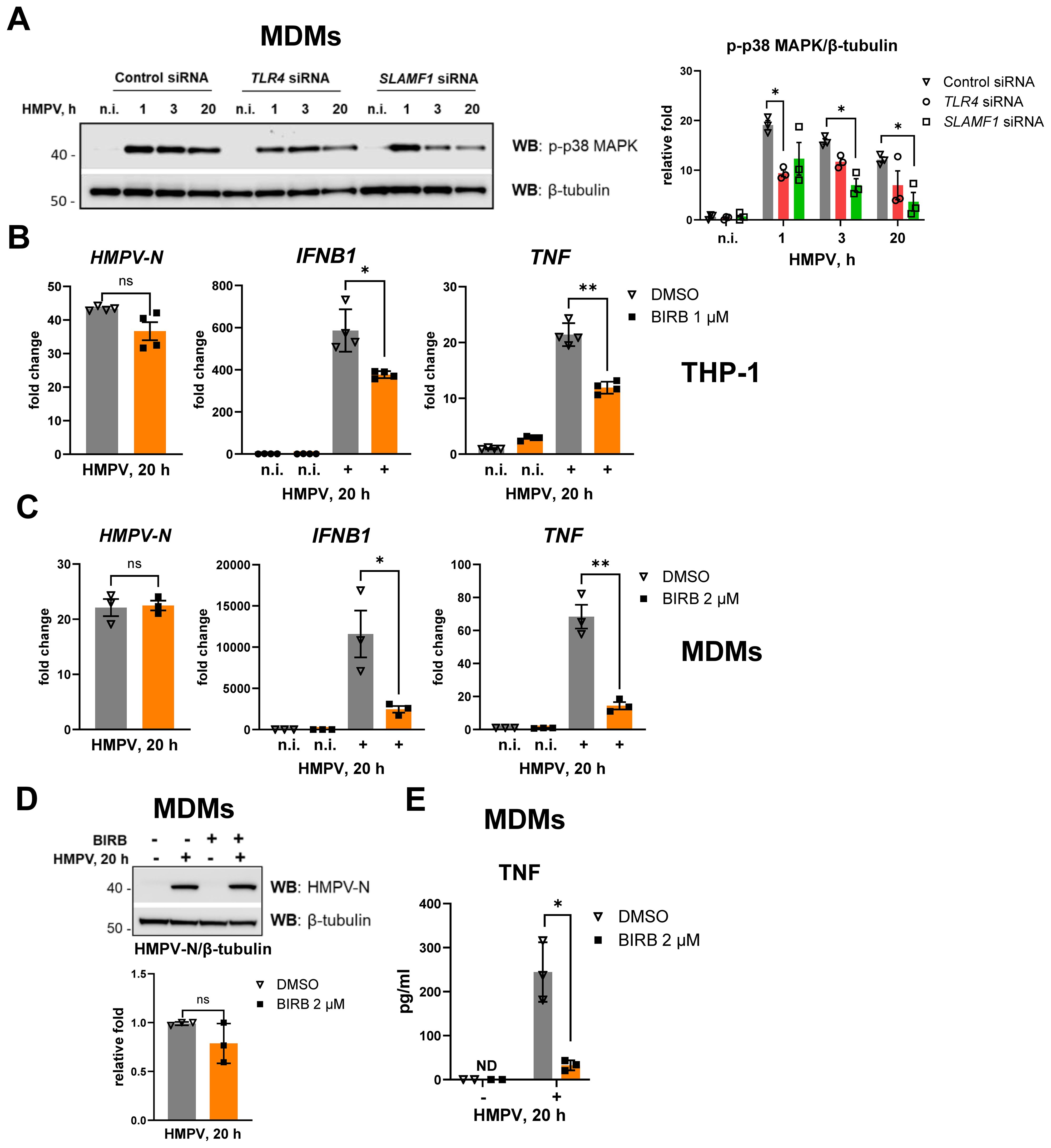
Figure 6. Silencing of SLAMF1 or TLR4 inhibits HMPV-mediated phosphorylation of p38 MAPK, and p38 MAPK inhibition decreases HMPV-induced IFNB1 and TNF expression. (A) WB analysis of phospho-p38 MAPK (Thr180/Tyr182) in lysates of primary human MDMs transfected with siRNA oligos (control, TLR4, and SLAMF1 siRNA) and infected with HMPV (representative image, n = 3). The graph shows combined data for all the experiments with phospho-p38 MAPK levels normalized to β-tubulin (loading control). (B, C) HMPV-N vRNA expression and IFNB1 and TNF mRNA expression were determined by qRT-PCR in THP-1 cells or primary human MDMs pretreated with DMSO (control) or the BIRB796 p38 MAPK inhibitor for 30 min before HMPV infection (MOI = 1) for 20 h (n = 3). Results were normalized to non-infected (n.i.) samples and for HMPV-N to the level detected 1 h after infection in DMSO-treated cells. Data are presented as mean relative fold change ± SD (THP-1 cells, B) or ± SEM (MDMs, C). Statistical testing was done by unpaired t-test (ns, not significant; *p < 0.05, **p < 0.01). (D) WB analysis was performed to determine HMPV-N protein levels in MDMs pretreated with DMSO or BIRB796 and infected with HMPV for 20 h (n = 3). The graph represents combined data for all experiments, normalized to β-tubulin (loading control). Statistical significance was evaluated using a paired t-test; significance level (ns, not significant). (E) TNF secretion levels were examined by ELISA in the supernatants from non-infected (n.i.) or HMPV-infected cells (20 h), with data presented as mean ± SD (n = 3); ND, not detected. Statistical significance was evaluated using a paired t-test, and only significant results are indicated (*p < 0.05).
To evaluate the functional role of p38 MAPK activation in HMPV-induced cytokine expression, THP-1 cells and MDMs were pretreated with the p38 MAPK inhibitor BIRB796 prior to HMPV infection for 20 h. Inhibition of p38 MAPK significantly reduced the induction of TNF and IFNB1 mRNAs in HMPV-infected THP-1 cells (Figure 6B) and macrophages (Figure 6C), while HMPV-N RNA (Figures 6B, C) and protein levels (Figure 6D) remained unchanged. Consistent with the reduced TNF mRNA, TNF secretion was strongly impaired by p38 MAPK inhibition in MDMs (Figure 6E). The cytotoxicity of BIRB796 at the concentrations used in Figures 6B–E (1–2 μM) was assessed by LDH release assay, and no cytotoxic effects were detected (Supplementary Figure S5A). To evaluate the specificity of BIRB796, given that it may partially inhibit JNKs (63), we examined the effects of the additional p38 MAPK inhibitors SB202190 and SB203580 on HMPV-induced IFNB1 and TNF expression (Supplementary Figure S5B). SB202190 and SB203580 specifically inhibit p38 MAPK activity without affecting JNK activity (64–67). Both inhibitors reduced HMPV-induced IFNB1 and TNF expression to a similar extent as BIRB796 (Supplementary Figure S5B). Finally, we confirmed that BIRB796 abrogated the phosphorylation of the downstream p38 MAPK target MK2/MAPKAPK2 while having no effect on the phosphorylation of JNKs or the JNK target protein ATF2 (Supplementary Figure S5C). Collectively, these findings indicate that the inhibitory effect of BIRB796 on HMPV-induced IFNB1 and TNF expression results from specific inhibition of p38 MAPK signaling.
Collectively, these findings demonstrate that in THP-1 cells and human MDMs, TLR4 and SLAMF1 contribute to HMPV-mediated p38 MAPK activation, leading to TNF expression, and that p38 MAPK contributes to IFNB1 induction in both THP-1 cells and MDMs.
4 Discussion
In this study, we identify TLR4 and SLAMF1 as immune receptors that regulate the expression of the pro-inflammatory cytokine TNF during HMPV infection in human primary macrophages. Both TLR4 and SLAMF1 contribute to activation of the p38 MAPK pathway, which we show is an important signaling step for HMPV-mediated TNF induction.
Understanding the mechanisms controlling TNF expression during viral infection is critical, as this cytokine has been linked to severe disease caused by major respiratory viruses such as SARS-CoV-2, influenza virus, and the pneumoviruses RSV and HMPV (68–70). Moreover, TNF is a principal mediator of RSV-induced disease, and elevated nasal TNF levels correlate with severity in infants with RSV bronchiolitis (70). For HMPV, we and others (24, 28) have reported that higher TNF levels in nasopharyngeal aspirates and blood are associated with pneumonia and bronchiolitis severity, supporting a critical role for TNF in HMPV-induced lung pathology. While TNF contributes to viral clearance early in infection, as shown for influenza virus and RSV (71, 72), excessive TNF, together with IFN-γ, can drive cell death, tissue damage, and mortality, as demonstrated in SARS-CoV-2 infection (68). The involvement of a TLR4–SLAMF1–p38 MAPK axis in early TNF induction by HMPV has not been reported and could aid therapeutic options to regulate TNF levels.
In this study, we employed THP-1 cells as a supporting system to MDMs, as THP-1 cells are more amenable to genetic manipulation. Notably, HMPV infection induced higher levels of IFNB1, TNF, and SLAMF1 expression in MDMs than in THP-1 cells. Moreover, TNF induction declined more rapidly in THP-1 cells. These observations are consistent with previous reports demonstrating transcriptional differences between MDMs and THP-1 cells (73) and underscore the importance of validating findings obtained in THP-1 cells using primary macrophages.
Our results further show that HMPV triggers TNF and IFNB1 expression with distinct kinetics in both MDMs and THP-1 cells: TNF peaks early, whereas IFNB1 gradually accumulates over time. This suggests that different receptors mediate these responses. Indeed, we found that TLR4 overexpression in THP-1 cells, or conversely, TLR4 depletion in THP-1 cells or MDMs, respectively, enhanced or reduced TNF induction by HMPV, while the effects on IFNB1 expression varied depending on whether TLR4 was overexpressed or silenced. TLR4 overexpression in THP-1 cells enhanced HMPV-induced IFNB1 expression, whereas TLR4 knockdown in THP-1 cells or MDMs did not significantly affect IFNB1 levels. The basis for this discrepancy remains unclear, but it is possible that alternative PRRs, such as DC-SIGN, may compensate for the absence of TLR4 in HMPV-mediated IFNB1 induction, as reported for other pathogen–PRR interactions (58, 59). Nevertheless, in both THP-1 cells and MDMs, TLR4 expression levels exerted a more pronounced effect on TNF induction than on IFNB1 expression. This aligns with previous work showing that cytoplasmic RIG-I-like receptors are major drivers of IFN-β in macrophages and dendritic cells infected with HMPV and other RNA viruses (74–76). We therefore propose that TLR4 (together with SLAMF1) predominantly regulates early TNF induction, whereas RIG-I-like receptors contribute to progressive IFN-β production as viral RNA and replication intermediates accumulate (Figure 7).
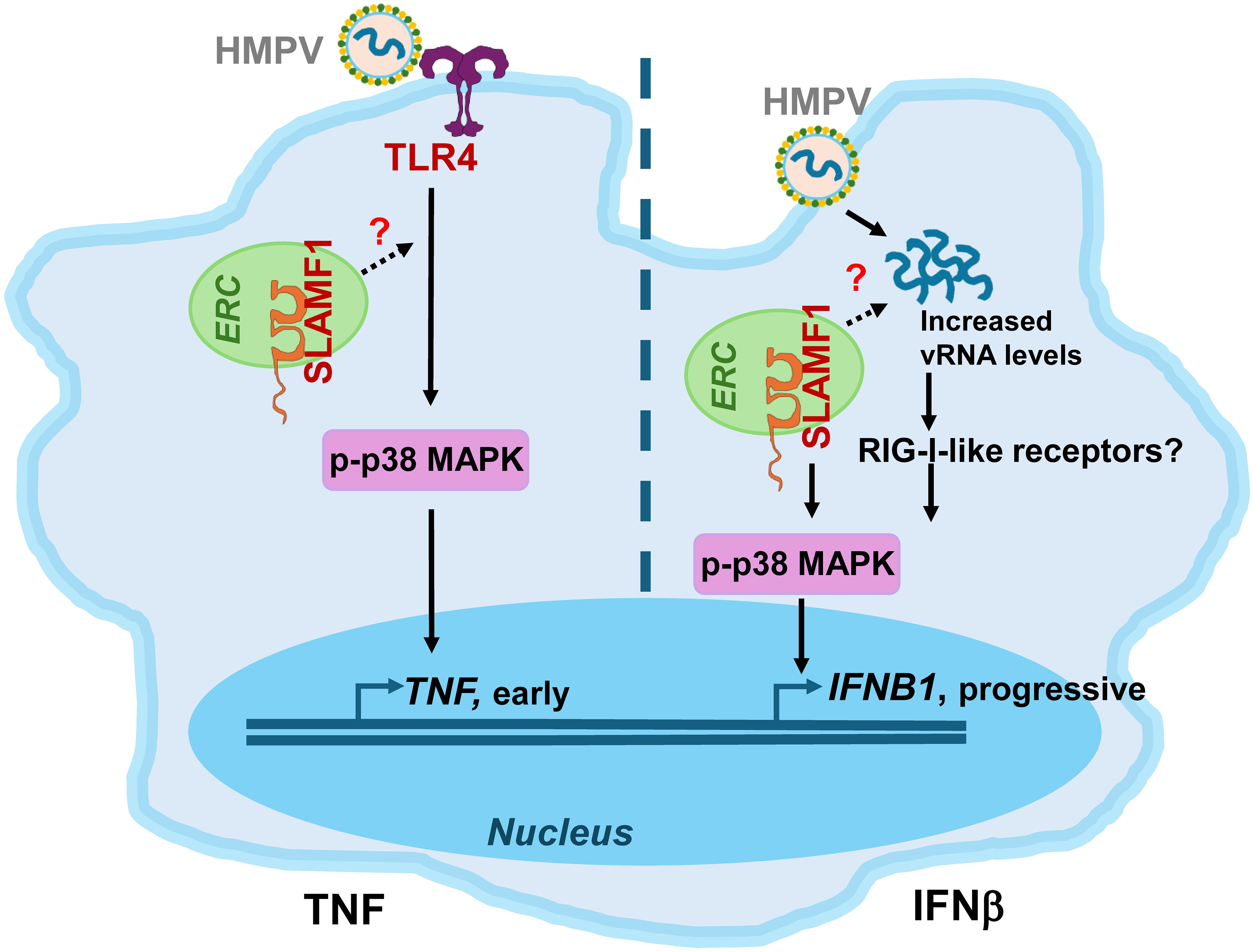
Figure 7. Proposed model of TNF and IFNB1 induction by HMPV. SLAMF1 is localized in the endocytic recycling compartment (ERC) in unstimulated human MDMs (38). Left panel, TNF induction: SLAMF1 and TLR4 stimulate early p38 MAPK phosphorylation, driving early TNF induction in both THP-1 cells and MDMs. SLAMF1 could facilitate TLR4-mediated signaling to p38 MAPK-TNF induction by ERC-mediated trafficking mechanisms, similar to that previously shown for LPS-driven p38 MAPK-TNF induction in primary human macrophages (38). SLAMF1 enhances p38 MAPK-TNF in both THP-1 cells and MDMs. Right panel, IFNB1 induction: HMPV infection and increased HMPV vRNA levels enhance p38 MAPK-regulated IFNB1 induction in both THP-1 cells and MDMs, potentially via RIG-I-like receptors. SLAMF1 could contribute to the increased vRNA levels and IFNB1 induction and appears to play a more prominent role in human MDMs than in THP-1 cells. HMPV, human metapneumovirus; TLR4, Toll-like receptor 4; vRNA, viral RNA; RIG-I-like receptors, retinoic acid-inducible gene-I-like receptors; MAPK, mitogen-activated protein kinase.
Our findings also reinforce the emerging role of TLR4 in recognizing viral components. Several viral glycoproteins, including those from SARS-CoV-2, RSV, Ebola virus, and dengue virus, activate TLR4-dependent signaling (29, 31, 77, 78). For HMPV, the fusion protein was shown to be a potent TLR4 agonist that elicits inflammatory responses in monocytes, while the HMPV surface glycoprotein G modulated cytokine induction in dendritic cells via TLR4 signaling (10, 79). Although the clinical relevance of TLR4-mediated TNF production remains incompletely defined, TLR4 knockout mice show reduced manifestations of HMPV disease (11), and TLR4 polymorphisms that are associated with reduced function correlate with enhanced RSV disease susceptibility (80). Furthermore, the failure of a formalin-inactivated RSV vaccine candidate was linked to insufficient TLR4 activation, whereas the addition of TLR4 agonists improved its protection in animal models (78, 81).
In this study, we found that in HMPV-infected human macrophages, silencing of both TLR4 and SLAMF1 reduced TNF expression. We have previously shown that SLAMF1 enhances TLR4-mediated TNF expression and secretion by E. coli and LPS in human macrophages (38). Accordingly, the reduced levels of HMPV-induced TNF expression and secretion in SLAMF1-silenced MDMs could be explained by the regulatory role of SLAMF1 in TLR4-mediated signaling. Mechanistically, we speculate that SLAMF1, which we have previously shown to be localized in the endocytic recycling compartment (ERC) in human macrophages (38), enhances TLR4 signaling in response to HMPV, hence stimulating early p38 MAPK activation and subsequent TNF induction (schematically depicted in Figure 7). This is based on our current observation that SLAMF1 overexpression in THP-1 cells amplified HMPV-induced p38 activation and TNF output, that p38 and TNF secretion in MDMs was reduced by SLAMF1 silencing, and our previous findings showing that SLAMF1 overexpression increases TLR4-mediated p38 MAPK phosphorylation (38). Moreover, p38 MAPK activity was required for both TNF and IFNB1 induction by HMPV (Figures 6B, C). However, although SLAMF1 overexpression in THP-1 cells enhanced p38 MAPK-dependent TNF induction (Figure 4), it did not significantly increase IFNB1 expression in HMPV-infected THP-1 cells (Figure 4). We propose that the SLAMF1–p38 axis exerts a stronger influence on TNF than on IFNB1, as p38 MAPK is a key component of a positive feedback loop to TNF induction involving TNF signaling (82). In contrast, IFNB1 induction primarily depends on interferon regulatory factors, with p38 MAPK acting in a modulatory capacity, as demonstrated for the RNA virus—Sendai virus (82–84).
When comparing THP-1 cells and MDMs, we found that the SLAMF1–p38 MAPK axis enhanced TNF induction in both models. Conversely, HMPV-induced p38 MAPK phosphorylation, IFNB1, and CXCL10 expression were significantly reduced in siSLAMF1-treated MDMs (Figures 5, 6), whereas SLAMF1 overexpression in THP-1 cells did not significantly enhance IFNB1 or CXCL10 secretion (Figure 4). These findings may reflect inherent differences in SLAMF1 function between MDMs and THP-1 cells.
We further observed that STAT1 phosphorylation in HMPV-infected macrophages peaks at later time points (20–24 h), whereas p38 MAPK phosphorylation occurs as early as 1 h post-infection and remains sustained, both in MDMs and THP-1 cells. To our knowledge, p38 MAPK involvement in HMPV-mediated cytokine induction has not been previously described. Here, silencing TLR4 or SLAMF1 in MDMs reduced HMPV-induced p38 MAPK activation at early time points, indicating that HMPV triggers signaling downstream of both receptors early during infection in MDMs. Inhibition of p38 MAPK markedly reduced both TNF and IFNB1 induction, both in MDMs and in THP-1 cells (Figure 6), suggesting that p38 MAPK activity is required not only for early TLR4-driven TNF induction but may also contribute to IFN-β production downstream of RIG-I signaling at later stages, consistent with the findings for the Sendai virus (85).
Notably, at later time points of HMPV infection (20 h), SLAMF1 silencing in human MDMs also reduced IFNB1 expression, whereas TLR4 silencing did not. Also, upon SLAMF1 depletion, we observed reduced HMPV-N mRNA and protein levels at late time points of infection, which could suggest that SLAMF1 affects viral uptake or replication that could lead to altered IFNB1 expression. Indeed, SLAMF1 has been proposed as a measles virus receptor (86, 87) and has been implicated in virus uptake mechanisms such as macropinocytosis in immune cells (88). Although we did not address HMPV entry in this study, the decrease in HMPV levels after SLAMF1 knockdown warrants further investigation.
Overall, our results identify distinct regulatory mechanisms for HMPV−mediated TNF and IFN-β in human macrophages, with TLR4 and SLAMF1 driving early p38 MAPK activation and TNF induction, while SLAMF1 also influences late IFN-β responses in MDMs. As suggested for SARS−CoV−2 and RSV, therapeutic modulation of TNF−mediated inflammation could be a promising strategy to limit tissue damage and immunopathology in HMPV infection. Thus, future studies aimed at exploring pharmacologic modulators of TLR4–SLAMF1–p38 MAPK signaling may provide new opportunities to mitigate excessive TNF production and improve clinical outcomes in patients with severe HMPV infection.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
KB: Writing – review & editing, Data curation, Investigation, Formal analysis, Writing – original draft. KR: Methodology, Writing – original draft, Investigation. LR: Investigation, Methodology, Writing – original draft. TE: Writing – review & editing, Resources, Funding acquisition. MY: Data curation, Resources, Writing – original draft, Supervision, Methodology, Visualization, Investigation, Conceptualization, Writing – review & editing, Funding acquisition. MA: Writing – review & editing, Project administration, Visualization, Funding acquisition, Resources, Supervision, Conceptualization, Methodology, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by the Research Council of Norway through its Centers of Excellence funding scheme grant 223255/F50 (to TE), the Liaison Committee for Education, Research and Innovation in Central Norway innovation researcher grant 90794301 (to MY), Horizon Europe 2020 research and innovation grant No. 813343 (to MA).
Acknowledgments
We thank Dr. Jørgen Stenvik (NTNU, Norway) for providing the BIRB796 p38 MAPK inhibitor, Dr. Simon Loevenich (NTNU, Norway) for providing the cell material, and ViroNovative and B. van den Hoogen (Erasmus MC, Rotterdam) for the LLC-MK2 cells and the HMPV isolate NL/17/00 (A2).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1697494/full#supplementary-material
References
1. Boivin G, Abed Y, Pelletier G, Ruel L, Moisan D, Cote S, et al. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J Infect Dis. (2002) 186:1330–4. doi: 10.1086/344319
2. van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, et al. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med. (2001) 7:719–24. doi: 10.1038/89098
3. van den Hoogen BG, Osterhaus DM, and Fouchier RA. Clinical impact and diagnosis of human metapneumovirus infection. Pediatr Infect Dis J. (2004) 23:S25–32. doi: 10.1097/01.inf.0000108190.09824.e8
4. Ribo-Molina P, van Nieuwkoop S, Mykytyn AZ, van Run P, Lamers MM, Haagmans BL, et al. Human metapneumovirus infection of organoid-derived human bronchial epithelium represents cell tropism and cytopathology as observed in in vivo models. mSphere. (2024) 9:e0074323. doi: 10.1128/msphere.00743-23
5. Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, et al. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N Engl J Med. (2004) 350:443–50. doi: 10.1056/NEJMoa025472
6. Ivashkiv LB and Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
7. Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, et al. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. (2002) 76:5532–9. doi: 10.1128/JVI.76.11.5532-5539.2002
8. Soto JA, Galvez NMS, Benavente FM, Pizarro-Ortega MS, Lay MK, Riedel C, et al. Human metapneumovirus: mechanisms and molecular targets used by the virus to avoid the immune system. Front Immunol. (2018) 9:2466. doi: 10.3389/fimmu.2018.02466
9. Kolli D, Velayutham TS, and Casola A. Host-viral interactions: role of pattern recognition receptors (PRRs) in human pneumovirus infections. Pathogens. (2013) 2:232–63. doi: 10.3390/pathogens2020232
10. Kolli D, Bao X, Liu T, Hong C, Wang T, Garofalo RP, et al. Human metapneumovirus glycoprotein G inhibits TLR4-dependent signaling in monocyte-derived dendritic cells. J Immunol. (2011) 187:47–54. doi: 10.4049/jimmunol.1002589
11. Velayutham TS, Kolli D, Ivanciuc T, Garofalo RP, and Casola A. Critical role of TLR4 in human metapneumovirus mediated innate immune responses and disease pathogenesis. PLoS One. (2013) 8:e78849. doi: 10.1371/journal.pone.0078849
12. Fitzgerald KA and Kagan JC. Toll-like receptors and the control of immunity. Cell. (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
13. Zhao Y, Chahar HS, Komaravelli N, Dossumbekova A, and Casola A. Human metapneumovirus infection of airway epithelial cells is associated with changes in core metabolic pathways. Virology. (2019) 531:183–91. doi: 10.1016/j.virol.2019.03.011
14. Guerrero-Plata A, Casola A, Suarez G, Yu X, Spetch L, Peeples ME, et al. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol. (2006) 34:320–9. doi: 10.1165/rcmb.2005-0287OC
15. Kuiken T, van den Hoogen BG, van Riel DA, Laman JD, van Amerongen G, Sprong L, et al. Experimental human metapneumovirus infection of cynomolgus macaques (Macaca fascicularis) results in virus replication in ciliated epithelial cells and pneumocytes with associated lesions throughout the respiratory tract. Am J Pathol. (2004) 164:1893–900. doi: 10.1016/S0002-9440(10)63750-9
16. Loevenich S, Spahn AS, Rian K, Boyartchuk V, and Anthonsen MW. Human metapneumovirus induces IRF1 via TANK-binding kinase 1 and type I IFN. Front Immunol. (2021) 12:563336. doi: 10.3389/fimmu.2021.563336
17. Martinez-Espinoza I, Bungwon AD, and Guerrero-Plata A. Human metapneumovirus-induced host microRNA expression impairs the interferon response in macrophages and epithelial cells. Viruses. (2023) 15:2272. doi: 10.3390/v15112272
18. Guilliams M, Lambrecht BN, and Hammad H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol. (2013) 6:464–73. doi: 10.1038/mi.2013.14
19. Kolli D, Gupta MR, Sbrana E, Velayutham TS, Chao H, Casola A, et al. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am J Respir Cell Mol Biol. (2014) 51:502–15. doi: 10.1165/rcmb.2013-0414OC
20. Marvin SA, Russier M, Huerta CT, Russell CJ, and Schultz-Cherry S. Influenza virus overcomes cellular blocks to productively replicate, impacting macrophage function. J Virol. (2017) 91:e01417-16. doi: 10.1128/JVI.01417-16
21. Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature. (2022) 606:585–93. doi: 10.1038/s41586-022-04802-1
22. Falvo JV, Tsytsykova AV, and Goldfeld AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun. (2010) 11:27–60. doi: 10.1159/000289196
23. Lucas R, Hadizamani Y, Enkhbaatar P, Csanyi G, Caldwell RW, Hundsberger H, et al. Dichotomous role of tumor necrosis factor in pulmonary barrier function and alveolar fluid clearance. Front Physiol. (2021) 12:793251. doi: 10.3389/fphys.2021.793251
24. Malmo J, Moe N, Krokstad S, Ryan L, Loevenich S, Johnsen IB, et al. Cytokine profiles in human metapneumovirus infected children: identification of genes involved in the antiviral response and pathogenesis. PLoS One. (2016) 11:e0155484. doi: 10.1371/journal.pone.0155484
25. Pandey P and Karupiah G. Targeting tumour necrosis factor to ameliorate viral pneumonia. FEBS J. (2022) 289:883–900. doi: 10.1111/febs.15782
26. Patel BV, Wilson MR, O'Dea KP, and Takata M. TNF-induced death signaling triggers alveolar epithelial dysfunction in acute lung injury. J Immunol. (2013) 190:4274–82. doi: 10.4049/jimmunol.1202437
27. Shi X, Zhou W, Huang H, Zhu H, Zhou P, Zhu H, et al. Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice. Crit Care. (2013) 17:R301. doi: 10.1186/cc13171
28. Xiang WQ, Li L, Wang BH, Ali AF, and Li W. Profiles and predictive value of cytokines in children with human metapneumovirus pneumonia. Virol J. (2022) 19:214. doi: 10.1186/s12985-022-01949-1
29. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. (2000) 1:398–401. doi: 10.1038/80833
30. Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. (2010) 3:291–300. doi: 10.1038/mi.2010.6
31. Halajian EA, LeBlanc EV, Gee K, and Colpitts CC. Activation of TLR4 by viral glycoproteins: A double-edged sword? Front Microbiol. (2022) 13:1007081. doi: 10.3389/fmicb.2022.1007081
32. Shinya K, Ito M, Makino A, Tanaka M, Miyake K, Eisfeld AJ, et al. The TLR4-TRIF pathway protects against H5N1 influenza virus infection. J Virol. (2012) 86:19–24. doi: 10.1128/JVI.06168-11
33. Sahanic S, Hilbe R, Dunser C, Tymoszuk P, Loffler-Ragg J, Rieder D, et al. SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages: A possible correlation with strong pro-inflammatory responses in severe COVID-19. Heliyon. (2023) 9:e21893. doi: 10.1016/j.heliyon.2023.e21893
34. Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. (2021) 31:818–20. doi: 10.1038/s41422-021-00495-9
35. Fontes-Dantas FL, Fernandes GG, Gutman EG, De Lima EV, Antonio LS, Hammerle MB, et al. SARS-CoV-2 Spike protein induces TLR4-mediated long-term cognitive dysfunction recapitulating post-COVID-19 syndrome in mice. Cell Rep. (2023) 42:112189. doi: 10.1016/j.celrep.2023.112189
36. Rallabhandi P, Phillips RL, Boukhvalova MS, Pletneva LM, Shirey KA, Gioannini TL, et al. Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2. mBio. (2012) 3:e00218-12. doi: 10.1128/mBio.00218-12
37. Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, et al. Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Dis. (2004) 189:2057–63. doi: 10.1086/420830
38. Yurchenko M, Skjesol A, Ryan L, Richard GM, Kandasamy RK, Wang N, et al. SLAMF1 is required for TLR4-mediated TRAM-TRIF-dependent signaling in human macrophages. J Cell Biol. (2018) 217:1411–29. doi: 10.1083/jcb.201707027
39. Calderon J, Maganto-Garcia E, Punzon C, Carrion J, Terhorst C, and Fresno M. The receptor Slamf1 on the surface of myeloid lineage cells controls susceptibility to infection by Trypanosoma cruzi. PLoS Pathog. (2012) 8:e1002799. doi: 10.1371/journal.ppat.1002799
40. Dragovich MA and Mor A. The SLAM family receptors: Potential therapeutic targets for inflammatory and autoimmune diseases. Autoimmun Rev. (2018) 17:674–82. doi: 10.1016/j.autrev.2018.01.018
41. Kohn EM, Dos Santos Dias L, Dobson HE, He X, Wang H, Klein BS, et al. SLAMF1 is dispensable for vaccine-induced T cell development but required for resistance to fungal infection. J Immunol. (2022) 208:1417–23. doi: 10.4049/jimmunol.2100819
42. Sintes J, Romero X, Marin P, Terhorst C, and Engel P. Differential expression of CD150 (SLAM) family receptors by human hematopoietic stem and progenitor cells. Exp Hematol. (2008) 36:1199–204. doi: 10.1016/j.exphem.2008.03.015
43. Wang N, Satoskar A, Faubion W, Howie D, Okamoto S, Feske S, et al. The cell surface receptor SLAM controls T cell and macrophage functions. J Exp Med. (2004) 199:1255–64. doi: 10.1084/jem.20031835
44. Zhong MC, Lu Y, Qian J, Zhu Y, Dong L, Zahn A, et al. SLAM family receptors control pro-survival effectors in germinal center B cells to promote humoral immunity. J Exp Med. (2021) 218:e20200756. doi: 10.1084/jem.20200756
45. Husebye H, Aune MH, Stenvik J, Samstad E, Skjeldal F, Halaas O, et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity. (2010) 33:583–96. doi: 10.1016/j.immuni.2010.09.010
46. Nilsen KE, Zhang B, Skjesol A, Ryan L, Vagle H, Boe MH, et al. Peptide derived from SLAMF1 prevents TLR4-mediated inflammation in vitro and in vivo. Life Sci Alliance. (2023) 6:e202302164. doi: 10.26508/lsa.202302164
47. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. (2001) 29:e45. doi: 10.1093/nar/29.9.e45
48. Bosshart H and Heinzelmann M. THP-1 cells as a model for human monocytes. Ann Transl Med. (2016) 4:438. doi: 10.21037/atm.2016.08.53
49. Pinto SM, Kim H, Subbannayya Y, Giambelluca MS, Bosl K, Ryan L, et al. Comparative proteomic analysis reveals varying impact on immune responses in phorbol 12-myristate-13-acetate-mediated THP-1 monocyte-to-macrophage differentiation. Front Immunol. (2021) 12:679458. doi: 10.3389/fimmu.2021.679458
50. Lehtonen A, Matikainen S, and Julkunen I. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J Immunol. (1997) 159:794–803. doi: 10.4049/jimmunol.159.2.794
51. Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. (2013) 32:2751–63. doi: 10.1038/emboj.2013.203
52. Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, Saalmuller A, et al. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc Natl Acad Sci U S A. (2008) 105:8944–9. doi: 10.1073/pnas.0801794105
53. Wan J, Shan Y, Fan Y, Fan C, Chen S, Sun J, et al. NF-kappaB inhibition attenuates LPS-induced TLR4 activation in monocyte cells. Mol Med Rep. (2016) 14:4505–10. doi: 10.3892/mmr.2016.5825
54. Brownell J, Bruckner J, Wagoner J, Thomas E, Loo YM, Gale M Jr., et al. Direct, interferon-independent activation of the CXCL10 promoter by NF-kappaB and interferon regulatory factor 3 during hepatitis C virus infection. J Virol. (2014) 88:1582–90. doi: 10.1128/JVI.02007-13
55. Koetzler R, Zaheer RS, Wiehler S, Holden NS, Giembycz MA, and Proud D. Nitric oxide inhibits human rhinovirus-induced transcriptional activation of CXCL10 in airway epithelial cells. J Allergy Clin Immunol. (2009) 123:201–8.e9. doi: 10.1016/j.jaci.2008.09.041
56. Yu M and Levine SJ. Toll-like receptor, RIG-I-like receptors and the NLRP3 inflammasome: key modulators of innate immune responses to double-stranded RNA viruses. Cytokine Growth Factor Rev. (2011) 22:63–72. doi: 10.1016/j.cytogfr.2011.02.001
57. Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, and Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. (2005) 289:L85–95. doi: 10.1152/ajplung.00397.2004
58. Nish S and Medzhitov R. Host defense pathways: role of redundancy and compensation in infectious disease phenotypes. Immunity. (2011) 34:629–36. doi: 10.1016/j.immuni.2011.05.009
59. Gillespie L, Gerstenberg K, Ana-Sosa-Batiz F, Parsons MS, Farrukee R, Krabbe M, et al. DC-SIGN and L-SIGN are attachment factors that promote infection of target cells by human metapneumovirus in the presence or absence of cellular glycosaminoglycans. J Virol. (2016) 90:7848–63. doi: 10.1128/JVI.00537-16
60. West AP, Koblansky AA, and Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. (2006) 22:409–37. doi: 10.1146/annurev.cellbio.21.122303.115827
61. Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH, et al. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediators Inflamm. (2014) 2014:352371. doi: 10.1155/2014/352371
62. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
63. Kuma Y, Sabio G, Bain J, Shpiro N, Marquez R, and Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J Biol Chem. (2005) 280:19472–9. doi: 10.1074/jbc.M414221200
64. Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. (1995) 364:229–33. doi: 10.1016/0014-5793(95)00357-f
65. Engelman JA, Lisanti MP, and Scherer PE. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J Biol Chem. (1998) 273:32111–20. doi: 10.1074/jbc.273.48.32111
66. Eyers PA, Craxton M, Morrice N, Cohen P, and Goedert M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem Biol. (1998) 5:321–8. doi: 10.1016/S1074-5521(98)90170-3
67. Nemoto S, Xiang J, Huang S, and Lin A. Induction of apoptosis by SB202190 through inhibition of p38beta mitogen-activated protein kinase. J Biol Chem. (1998) 273:16415–20. doi: 10.1074/jbc.273.26.16415
68. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-coV-2 infection and cytokine shock syndromes. Cell. (2021) 184:149–68.e17. doi: 10.1016/j.cell.2020.11.025
69. Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, and Straus SE. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest. (1998) 101:643–9. doi: 10.1172/JCI1355
70. Santos LD, Antunes KH, Muraro SP, de Souza GF, da Silva AG, Felipe JS, et al. TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during respiratory syncytial virus infection. Eur Respir J. (2021) 57:2003764. doi: 10.1183/13993003.03764-2020
71. Rutigliano JA and Graham BS. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J Immunol. (2004) 173:3408–17. doi: 10.4049/jimmunol.173.5.3408
72. Seo SH and Webster RG. Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J Virol. (2002) 76:1071–6. doi: 10.1128/JVI.76.3.1071-1076.2002
73. Kohro T, Tanaka T, Murakami T, Wada Y, Aburatani H, Hamakubo T, et al. A comparison of differences in the gene expression profiles of phorbol 12-myristate 13-acetate differentiated THP-1 cells and human monocyte-derived macrophage. J Atheroscler Thromb. (2004) 11:88–97. doi: 10.5551/jat.11.88
74. Goutagny N, Jiang Z, Tian J, Parroche P, Schickli J, Monks BG, et al. Cell type-specific recognition of human metapneumoviruses (HMPVs) by retinoic acid-inducible gene I (RIG-I) and TLR7 and viral interference of RIG-I ligand recognition by HMPV-B1 phosphoprotein. J Immunol. (2010) 184:1168–79. doi: 10.4049/jimmunol.0902750
75. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. (2006) 441:101–5. doi: 10.1038/nature04734
76. Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A. (2010) 107:1512–7. doi: 10.1073/pnas.0912986107
77. Shirato K and Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. (2021) 7:e06187. doi: 10.1016/j.heliyon.2021.e06187
78. Marr N and Turvey SE. Role of human TLR4 in respiratory syncytial virus-induced NF-kappaB activation, viral entry and replication. Innate Immun. (2012) 18:856–65. doi: 10.1177/1753425912444479
79. Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. (2011) 17:195–9. doi: 10.1038/nm.2262
80. Richard K, Piepenbrink KH, Shirey KA, Gopalakrishnan A, Nallar S, Prantner DJ, et al. A mouse model of human TLR4 D299G/T399I SNPs reveals mechanisms of altered LPS and pathogen responses. J Exp Med. (2021) 218:e20200675. doi: 10.1084/jem.20200675
81. Delgado MF, Coviello S, Monsalvo AC, Melendi GA, Hernandez JZ, Batalle JP, et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. (2009) 15:34–41. doi: 10.1038/nm.1894
82. Brinkman BM, Telliez JB, Schievella AR, Lin LL, and Goldfeld AE. Engagement of tumor necrosis factor (TNF) receptor 1 leads to ATF-2- and p38 mitogen-activated protein kinase-dependent TNF-alpha gene expression. J Biol Chem. (1999) 274:30882–6. doi: 10.1074/jbc.274.43.30882
83. Wang J, Basagoudanavar SH, Wang X, Hopewell E, Albrecht R, Garcia-Sastre A, et al. NF-kappa B RelA subunit is crucial for early IFN-beta expression and resistance to RNA virus replication. J Immunol. (2010) 185:1720–9. doi: 10.4049/jimmunol.1000114
84. Popli S, Chakravarty S, Fan S, Glanz A, Aras S, Nagy LE, et al. IRF3 inhibits nuclear translocation of NF-kappaB to prevent viral inflammation. Proc Natl Acad Sci U S A. (2022) 119:e2121385119. doi: 10.1073/pnas.2121385119
85. Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, et al. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. J Biol Chem. (2009) 284:10774–82. doi: 10.1074/jbc.M807272200
86. Tatsuo H, Ono N, Tanaka K, and Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. (2000) 406:893–7. doi: 10.1038/35022579
87. Tatsuo H, Ono N, and Yanagi Y. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J Virol. (2001) 75:5842–50. doi: 10.1128/JVI.75.13.5842-5850.2001
Keywords: virus, airway disease, hyperinflammation, TNF, toll-like receptor 4, HMPV, SLAMF1
Citation: Sæterhaug Bye K, Rian K, Ryan L, Espevik T, Anthonsen MW and Yurchenko M (2025) The immune receptors TLR4 and SLAMF1 regulate TNF release by human metapneumovirus in human macrophages. Front. Immunol. 16:1697494. doi: 10.3389/fimmu.2025.1697494
Received: 02 September 2025; Accepted: 28 October 2025;
Published: 19 November 2025.
Edited by:
Andrew Foey, University of Plymouth, United KingdomReviewed by:
Mohammad Arish, University of Virginia, United StatesRoshan Ghimire, Oklahoma State University, United States
Copyright © 2025 Sæterhaug Bye, Rian, Ryan, Espevik, Anthonsen and Yurchenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marit W. Anthonsen, bWFyaXQudy5hbnRob25zZW5AbnRudS5ubw==; bWFyaWlhLnl1cmNoZW5rb0BudG51Lm5v
†These authors have contributed equally to this work and share last authorship