 Xiaopeng Yun
Xiaopeng Yun Li Zhang
Li Zhang- Department of Public Security, Shanxi Police College, Taiyuan, China
Studying the thermal decomposition of energetic materials at high temperatures can provide detailed reaction and mechanistic information, which is critical for understanding the reactivity of energetic materials, designing mixed explosives, and achieving improved safety. In this work, the effects of temperature and pressure on graphene (Gr)-based HMX crystals were investigated using ReaxFF molecular dynamics simulations. The thermal decomposition processes of perfect HMX crystals, HMX crystals with (001), (010), or (100) crystal planes, and HMX/Gr mixed systems were studied at high temperatures and pressures. In the mixed systems, different configurations of HMX molecules adsorbed on the Gr surface were confirmed by theoretical calculation methods. With the pressure ranging from atmospheric pressure to 31 GPa, 3, 5, and 3 configurations of HMX adsorbed on the Gr surface were identified for the (001)/Gr, (010)/Gr, and (100)/Gr systems, respectively. The time-dependent curves for the evolution of fragments, intermediates, and pyrolysis products were analyzed. The rate constant for the thermal decomposition of HMX was found to be significantly affected by the addition of Gr. In particular, the thermal decomposition reaction was strongly inhibited in the (010)/Gr system. This result indicates that Gr promotes an anisotropic thermal effect, resulting from the steric hindrance of the NO2 functional groups and the interaction between Gr and HMX molecules. Gr also affected the initial reaction pathway of homolytic N–NO2 bond cleavage, with C=O, C–OH, and C–OC bonds on the Gr surface participating in the formation of nitro radicals and HONO.
Introduction
Energy materials (EMs), such as explosives, pyrotechnics, and propellants, play an important role in many fields including aeronautics and the defense industry (Fried et al., 2001). Because of their high density, energy, and tension, EMs are widely used in applications ranging from engineering blasting, automobile air bags, and oil exploitation to weapons and rocket propellants (Singh, 2005). As pure EMs are highly sensitive, they are typically mixed with binders, additives, and desensitizers to improve the thermal decomposition mechanism, tensile mechanical properties, and sensitivity. Considering the excellent properties of graphene (Gr) (Allen et al., 2010), such as a large specific surface area, high thermal conductivity, and high electrical conductivity, graphene-based materials (GBMs), such as Gr, graphene oxide (GO), and reduced graphene oxide (rGO), and EM/GBM mixtures have received considerable attention (Chen et al., 2006). Recently, GBMs have been proposed as additives and desensitizers to replace graphite and other compounds in 1,3,5-triamino-2,4,6-trinitrobenzene (TATB)-based (Lin et al., 2017) and hexanitrohexaazaisowurtzitane (HNIW or CL-20)-based (Liu et al., 2014; Yu et al., 2014) plastic-bonded explosives (PBXs).
Various studies (Liu et al., 2014; Yu et al., 2014) have focused on elucidating the origin of the reduced mechanical sensitivity and thermal stability of GBM/CL-20 mixtures. Owing to their high specific surface areas, GBMs were found to provide a large buffer space when EM/GBM mixtures encounter mechanical shock and impact. The thermal decomposition process has also been studied at low temperatures. Owing to the good thermal conductivity of Gr, heat flow can be quickly transferred from Gr to the PBX surface, thus promoting the decomposition of PBX and reducing its thermal stability. At high temperatures, the oxygen-containing functional groups on the surface of GO decompose, releasing OH radicals and transferring some of the heat flow to the PBX, which also promotes PBX decomposition. In contrast, because most of the oxygen-containing groups on the surface of rGO are reduced, PBX thermal stability is not affected by the addition of the rGO powder.
For 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX) and GO composites, the activation energy is significantly increased by the addition of GO to HMX crystals. Furthermore, the thermal stability of HMX explosives is increased (Li et al., 2013) by a flame-retardant effect resulting from the metal salts used in the GO preparation process (Shi and Li, 2011). The desensitizing effect of GO sheets is better than that of fullerene and carbon nanotubes (CNTs). Niu et al. (2017) prepared HMX/rGO/graphite mixtures via an in situ chemical reduction coating method. The obtained results revealed that GO sheets and graphite could be used as co-desensitizers in HMX explosives. These previous studies indicate that thermal reactions at high temperatures and pressures depend strongly on the preparation method and conditions. In particular, the addition of GO to HMX and CL-20 has an opposite effect on the thermal stability. However, the thermal decomposition mechanisms of Gr and GO in EM/GBM mixtures under extreme conditions are not well understood because of the lack of experimental data.
Theoretical methods, such as quantum mechanical and molecular dynamics simulations, have been applied to investigate the thermal decomposition mechanisms of the EM/GBM mixture and clarify the effects of GBM addition on the sensitivity of EMs. To investigate the mechanisms of functionalized graphene sheets (FGSs) dispersed in liquid nitromethane (NM), Liu et al (2012) performed ab initio molecular dynamics simulations. The results showed that the thermal decomposition of NM molecules is greatly accelerated in the presence of FGSs, resulting in the formation of water, nitrogen, and carbon dioxide. Various reaction pathways, such as proton or oxygen exchange, were identified, and the FGSs were found to act as a catalyst in the thermal decomposition process. Zhang et al. (2019) determined the thermal stability of GO, 4,4′-Azo-1,2,4-triazole (ATRZ), and GO-ATRZ using ReaxFF-lg force field molecular dynamics simulations. Because of the strong space effect of GO, the activation energy of GO-ATRZ was 16.1 kJ/mol higher than that of pure ATRZ, indicating that GO can improve the thermal stability of ATRZ. Thus, GO can be combined with ATRZ as a desensitizer. For 1,1-diamino-2,2-dinitroethene (FOX-7)-GO composites (Su et al., 2019), which is an ideal prototype system, the interaction interface was studied using a dispersion-corrected density functional approach. The results revealed that both interfacial charge transfer from FOX-7 to GO and hydrogen bonding contribute to relatively strong interactions between FOX-7 and GO.
The thermal processes of EM/GBM mixtures at high temperatures and pressures, which are more complex than those of pure EMs, are accompanied by the release of a large amount of energy. Furthermore, experimental verification of the intermediates and initial reaction pathways in mixed systems under extreme conditions has been challenging. The complex decomposition processes of EM/GBM mixtures involve various unimolecular and bimolecular reactions. As Gr is the simplest type of GBM, elucidating the role of Gr in mixtures with EMs can provide a basis for understanding decreased sensitivity and complex detonation reactions. ReaxFF-lg molecular dynamics simulation (RMDS), which was first proposed by Liu et al (2011), has been used to study the reactions of HMX (Wu et al., 2020), 2,4,6-triamino-5-nitropyrimidine-1,3-dioxide (ICM-102) (Yang et al., 2020), TATB (Hamilton et al., 2019), CL-20 (Wang et al., 2019), and CL-20/trinitrotoluene (TNT) (Ren et al., 2019). Furthermore, these results have been compared with quantum mechanics calculations and experimental findings.
In the present work, RMDS was used to study the molecular configurations of HMX molecules adsorbed on Gr surfaces as well as the thermal decomposition of HMX/Gr systems at high temperatures. This work focused on the thermal behavior of perfect HMX crystals, HMX crystals with different crystal planes, and HMX/Gr mixtures. An in-depth understanding of the effect of Gr on complex explosive chemical mechanisms and various factors that influence the degradation pathways is essential for the preparation, storage, transportation, and use of mixed explosives.
Model Building and Computational Approach
A perfect HMX crystal structure was obtained from experimental X-ray data (Choi and Boutin, 1970). Shock sensitivity of HMX is different along different directions (Menikoff et al., 2005). Furthermore, compression of β-HMX using a diamond anvil cell has revealed that the lattice parameters of a single crystal are anisotropic (Hooks et al., 2006). In addition, COMPASS-based molecular dynamics simulations have shown that the interactions between HMX and a binder are different, depending on the crystallographic plane of HMX (Xiao et al., 2005).
To investigate the effects of the HMX crystal plane and Gr on thermal decomposition of HMX/Gr mixtures, we built a series of models. Along the x, y, and z directions, a 4 × 2 × 3 perfect HMX supercell and an 8 × 8 × 1 graphite supercell were employed by expanding the unit cells of HMX and graphite. Then, (001), (010), and (100) structures were obtained by cleaving the perfect HMX supercell along the (001), (010), and (100) crystal planes, respectively. Thus, the total number of HMX molecules for each crystal plane was 48 (1344 atoms). The initial lattice parameters of perfect HMX (P-HMX), the (001), (010), and (100) crystal planes of HMX (P-001, P-010, and P-100, respectively), and Gr mixtures with the (001), (010), and (100) crystal planes of HMX (G-001, G-010, and G-100, respectively) are listed in Table 1.
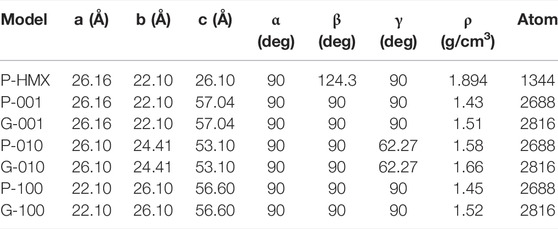
TABLE 1. Initial supercells in the calculated systems.
Because the HMX cell structure was taken from neutron diffraction data, the space group of β-HMX unit cell structure is P21/C. The periodic boundary conditions of HMX supercell structure for a perfect crystal were the same as those of traditional molecular dynamics. The HMX structure and HMX/Gr mixed structure along different crystal planes, which were built on HMX supercell structure and graphite supercell structure, belong to the P1 space group.
In this article, based on the principle of size matching, the spatial structures of HMX and graphene with different crystal planes were obtained. It can be seen from Table1 that the values of axis a and axis b of all structures are about 20 Å. Taking the HMX/Gr modeling process along the (001) plane as an example, after cutting the perfect HMX supercell structure, the (001) lattice parameter of the two-dimensional structure was 26.16 × 22.10 Å. The lattice parameters of the graphene unit cell were 2.46 Å × 2.46 Å × 6.8 Å, and then the lattice parameter becomes 19.68 × 19.68 × 6.8 Å after expanding eight times, eight times, and one time along the a-axis, b-axis, and c-axis, respectively. Currently, the graphite supercell structure matches the size of the HMX supercell structure with different crystal planes. Therefore, the graphene structure composed of 128 carbon atoms is selected as the simulation object in this article.
Figure 1 outlines the processes used to establish the P-001 and G-001 models. For example, the P-001 model, which contains 96 HMX molecules, was built by combining two (001) plane structures. The P-010 and P-100 models were established using the same methods. The HMX/Gr models were built using HMX crystal plane structures containing 96 HMX molecules and one Gr molecule. The Gr molecule, which was obtained from the 8 × 8 × 1 graphite supercell, was placed between two HMX crystal plane structures. In the HMX/Gr models, the mass fractions of Gr and HMX were 5.1 and 94.9%, respectively.
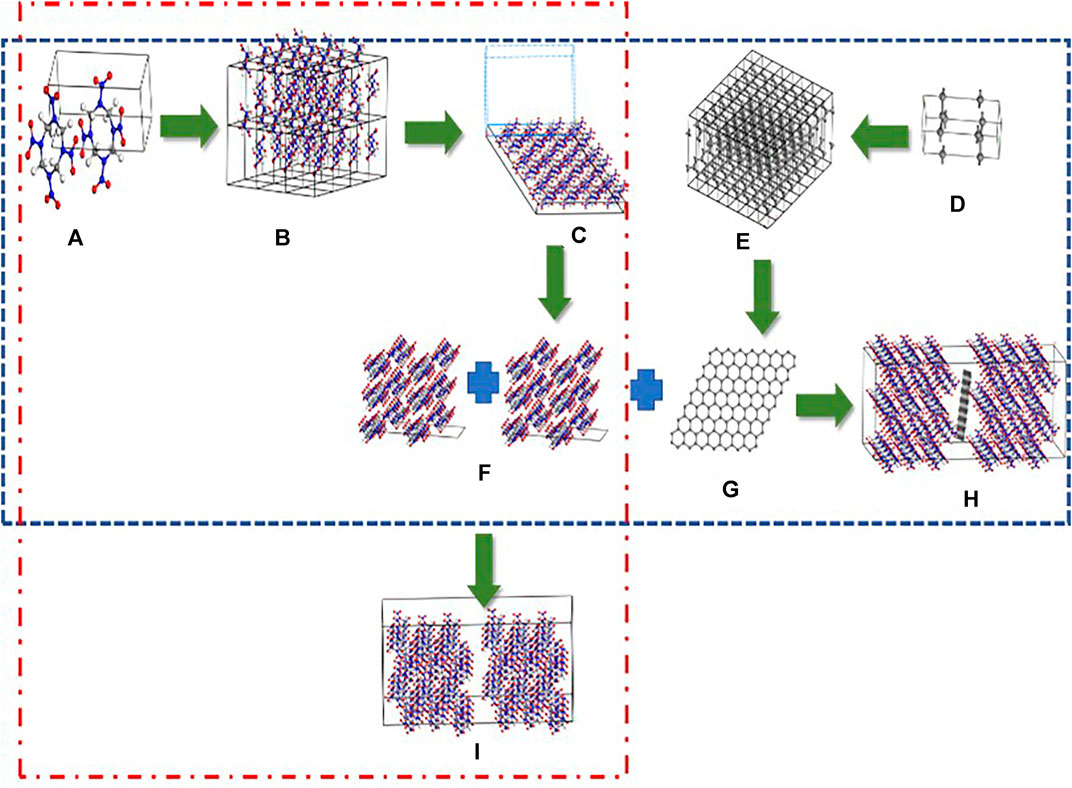
FIGURE 1. Process for establishing the G-001 (h) and P-001 1) supercells. (A) and (D) unit cells of β-HMX and graphite, (B) and (E) 4 × 2 × 3 β-HMX supercell and 8 × 8 × 1 graphite supercell, (C) cleavage of the perfect β-HMX crystal along the (001) crystal plane, (F) and (G) the structures of the (001) crystal plane and graphene, (H) the final structure of G-001, and (I) final structure of P-001. The red dotted line, which consists of (A), (B), (C), (F), and (H), represents the establishing process of the P-001 model. The blue dotted line shows the establishing process of the G-001 model.
In this work, the energy minimization of HMX and HMX/Gr models without relaxing the lattice parameters was performed to release internal structural stress from the seven supercells. The method of the structural optimization process was the limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm. The maximum number of the minimization step and convergence criterion were 1000 and 4.184 (kJ/mol)/Å, respectively.
Subsequently, a two-step RMDS consisting of a 25 ps isothermal–isobaric (NPT) molecular dynamics simulation (0.25 fs time step) and a 25 ps isothermal–isochoric (NVT) molecular dynamics simulation (0.25 fs time step) was used to relax the seven structures at room temperature with the pressure ranging from atmospheric pressure to 31 GPa with the Nose–Hoover chain thermostat (50 fs damping constant) and the NHCP anisotropic barostat (500 fs damping constant), which represents an anisotropic Parrinello–Rahman–Hoover (RPH) NPT ensemble algorithm coupled with the Martyna–Tuckerman–Tobias–Klein (MTTK) algorithm to control the pressure fluctuation in the RMDS.
Finally, based on the NVT ensemble, the total 100 ps RMDS was calculated to investigate thermal decomposition at 3000 K, with a time step of 0.1 fs. By analyzing the behaviors of the HMX and Gr molecules during the evolution of the RMDS, the effect of Gr on the mixed explosives could be elucidated.
Results and Discussion
Validation of the ReaxFF-lg Force Field
For molecular dynamics simulations, it is necessary to confirm the applicability of the force field to the calculated system. Many studies have examined the applicability of the ReaxFF force field to HMX and other EMs. It has been found that the equation of the state of HMX (Liu et al., 2011) and the reaction process under different loading conditions (Hamilton et al., 2019; Ren et al., 2019; Wang et al., 2019; Wu et al., 2020; Yang et al., 2020) are well described by this force field. However, the applicability of the ReaxFF force field to mixed systems composed of HMX and Gr has not been reported.
To verify the suitability of the ReaxFF-lg force field for HMX/Gr mixtures, RMDS (25 ps NPT + 25 ps NVT) was performed for the seven HMX systems at 300 K and atmospheric pressure. The lattice parameters and density determined for the P-HMX unit cell are summarized in Table 2 and compared with results obtained experimentally or with other theoretical methods. The density obtained using RMDS was 1.808 g/cm3, which is in good agreement with the results of previous experiments (Choi and Boutin, 1970; Menikoff et al., 2005; Xiao et al., 2005; Hooks et al., 2006; Peng et al., 2014; He et al., 2015) and DFT-D2 calculations (Peng et al., 2014).
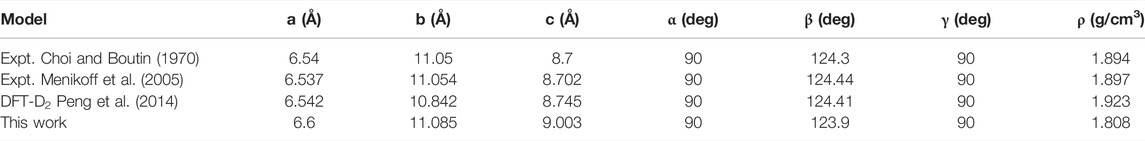
TABLE 2. Lattice parameters of the P-HMX unit cell obtained using various experimental and theoretical methods.
To verify the applicability of the ReaxFF-lg force field to the reactions between graphene molecule and HMX molecules (supporting information of Supplementary Figures S1–S5), two compared system models composed of single-molecule HMX (Supplementary Figure S1A) and single-molecule HMX and graphene (Supplementary Figures S1B–K) were established, and then the initial reaction pathway of N–NO2 bond breaking of the HMX molecule to form a nitro radical in the two models was calculated by the method of structure optimization and potential energy surface scanning methods. The results (Supplementary Figures S2–S5) show that graphene does not affect the initial reaction pathway of HMX.
The lattice parameters predicated by RMDS for the different HMX crystal planes and HMX/Gr mixtures are summarized in Table 3. The density, which is typically the main factor affecting thermal decomposition processes, was approximately 1.8 g/cm3 for all the investigated systems. As shown in Table 3, the a-axis, b-axis, and α, β, and γ values changed slightly, whereas the c-axis value changed abruptly, indicating that the c-axis is easily compressed.
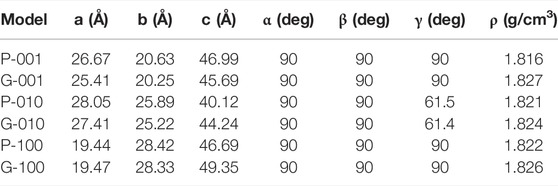
TABLE 3. Lattice parameters of different HMX crystal planes and HMX/Gr mixtures.
Notably, the interaction between HMX and Gr in the HMX/Gr mixtures can be expressed by the binding energy, which is positively correlated with compatibility and stability. In other words, a larger binding energy is expected for mixed explosives with greater compatibility and stability. The binding energy can be calculated as:
where Ebind is the binding energy between HMX and Gr, Einteraction is the interaction energy between HMX and Gr, Etotal is the potential energy of the HMX/Gr system, and EHMX and EGr are the potential energies of HMX and Gr, which can be obtained by removing Gr and HMX from the HMX/Gr system. To compare the effect of graphene on the binding energies of HMX/Gr systems, the binding energy of each carbon atom in graphene was obtained by dividing 126 carbon atoms of Gr. Based on the RMDS results, G-010 and G-100 were predicted to have the largest and smallest binding energies in Table 4, respectively. The binding energies of three-mixture models were also calculated by classic molecular dynamic simulations, which were based on the COMPASS force field (supporting information of Supplementary Table S1). The results show the same order with the ReaxFF force field results. He et al. (2015) investigated the interactions between HMX with different crystal plane directions and hydroxyl-terminated polybutadiene (HTPB), which is a typical binder in PBXs. They proposed that the formation energy between HMX crystals and HTPB decreased in the order (010) ˃ (001) ˃ (100). These results indicated that strong interactions, such as hydrogen bonds, can be formed between Gr or HTPB and HMX crystals, especially with the (010) crystal plane.
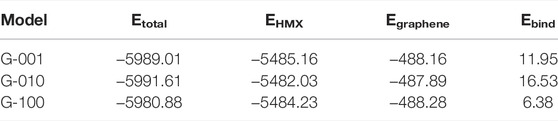
TABLE 4. Normalized binding energies (kJ/mol) of HMX/Gr per carbon atom in Gr.
Evolution of HMX Molecules
The decomposition of solid HMX crystals at high temperatures produce many small fragments and products, as demonstrated using the RMDS (Liu et al., 2011) and experiments (Niu et al., 2017). Figure 2 shows the decay of the number of HMX molecules during thermal decomposition of the HMX/Gr mixtures at 3000 K. For all the investigated systems, the same trend was observed during the initial stage (0–0.3 ps). At longer simulation times (0.31–1.8 ps), the HMX molecules gradually disappeared via several initial reactions pathways with different rate constants.
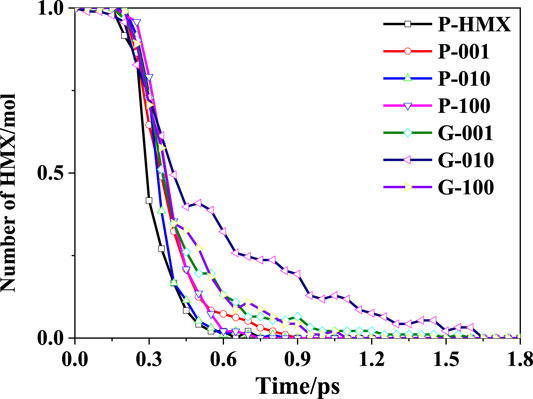
FIGURE 2. Decay of HMX molecules versus simulation time at 3000 K.
Based on the first-order phase transition nucleation theory and an interatomic interaction model via Monte Carlo simulation (Los et al., 2015), the melting temperature of Gr is 4510 K, which is higher than that of graphite. In particular, an investigation of chemical bond breaking and formation using ab initio molecular dynamics calculations (AIMD) suggested that Gr cannot melt when the system is heated up to 4500 K, leading to a quasi-2D liquid state (Ganz et al., 2017). Thus, in the current study, Gr was not reactive during the thermal decomposition of the HMX/Gr systems at 3000 K. Nevertheless, it was confirmed that Gr and the HMX crystal face affect the decomposition process of HMX molecules in the various calculated systems.
For the first stage, which has been proposed to have a critical effect on the thermal decomposition process, the rate constant can be calculated using the number evolution of HMX molecules using a first-order rate equation (Wang et al., 2019):
where N(t) and N0 are the numbers of HMX molecules at simulation times t and t0, respectively. Figure 3 shows the rate constant, k1, for the decay of HMX molecules. These results suggest that both the crystal plane and Gr affect HMX decomposition, with smaller rate constants observed for P-001, P-100, G-001, G-010, and G-100 than for P-HMX. Furthermore, the rate constants for the HMX/Gr mixtures are smaller than those for HMX alone with the same crystal plane. The (010) crystal plane accelerates the thermal decomposition of HMX molecules in P-010, whereas the rate constant abruptly decreases when Gr adds to the same HMX crystal plane. The differences in the rate constants for the (001), (010), and (100) crystal planes and the corresponding HMX/Gr mixtures are 0.68, 5.17, and 0.69 ps−1, respectively. Thus, Gr was found to have a considerable desensitization effect on HMX/Gr mixtures, especially with the (010) crystal plane.
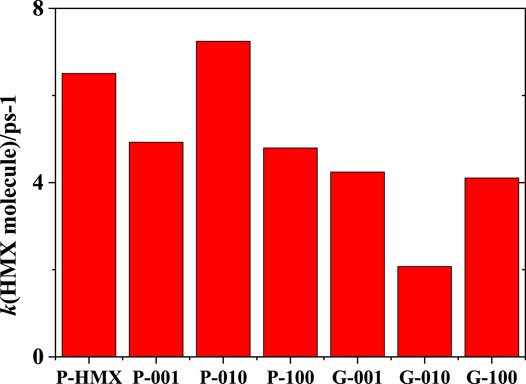
FIGURE 3. Rate constants for the decay of HMX molecules at 3000 K.
Both experimental and theoretical studies have shown that HMX exhibits anisotropic responses under shock loading. Menikoff et al. (2005) measured the wave profiles of HMX, which is an elastic–plastic material, and calculated the nonlinear and transient wave behavior using a rate-dependent elastic–plastic model. The reported effective yield strength values for the (011) and (010) crystal planes are 0.18 and 0.31 GPa, respectively. Ge et al. (2014) performed AIMD simulations of HMX in conjunction with the multiscale shock technique under different lattice vectors. A comparison of shock wave propagation along the a, b, and c lattice vectors revealed the smallest sliding rate for lattice vector b, suggesting that the anisotropy of HMX under shock loading can mainly be attributed to lattice vector b. Interestingly, our current results suggest that Gr also has anisotropic reaction responses under thermal loading depending on the orientation of the HMX crystal plane.
Key Intermediate Fragments
The decomposition pathways, intermediates, and the products of single molecules and crystal structures under different loadings have been analyzed using DFT calculations (Lewis et al., 2000a; Lewis et al., 2000b; Lyman et al., 2002; Zhang et al., 2003; Cobos, 2005; Sharia and Kuklja, 2010), AIMD simulations (Manaa et al., 2002; Ge et al., 2012; Ge et al., 2014), and RMDS (Zhou and Huang, 2011). For HMX, the initial pathways are homolytic cleavage of the N–NO2 bond to produce a NO2 radical, migration of a H atom from methylene to form HONO, breaking of four C–N bonds to form CH2N2O2, isomerization of NO2 with subsequent C–N bond breaking to release N2O2 fragments, and C–H bond dissociation. As theoretical simulations (Los et al., 2015; Ganz et al., 2017) have indicated that Gr is not reactive at temperatures lower than 4510 K, for the HMX/Gr systems in this study, all the intermediate fragments are considered to originate from HMX molecules.
The evolution of the fragment number with simulation time is commonly used to study the mechanisms of EMs under different conditions. We calculated the fragments in the seven models using a bond order of 0.3 at 3000 K. NO2, HONO, N2O2, HNO3, and NO were found to be the main intermediates during thermal decomposition of the HMX and HMX/Gr systems, which is consistent with other calculations (Manaa et al., 2002; Zhou and Huang, 2011; Ge et al., 2012) and experimental results (Shaw and Walker, 1977; Behrens and Bulusu, 1991). The time evolutions of NO2 radical fragments at 3000 K are presented in Figure 4 for the HMX crystal planes and HMX/Gr systems. The maximum number of NO2 fragments per HMX molecule observed for P-HMX, P-001, P-010, P-100, G-001, G-010, and G-100 are 1.67, 1.48, 1.59, 1.54, 1.25, 0.93, and 1.12 mol, respectively.
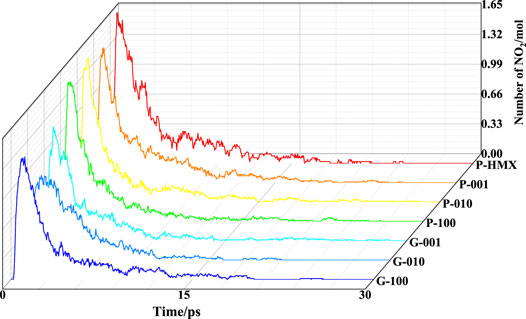
FIGURE 4. Evolution of NO2 radical fragments at 3000 K.
Taking the NO2 fragments observed for P-HMX as the benchmark, the maximum numbers of NO2 fragments in the other six calculated systems are lower. The differences in these values between P-HMX and P-001, P-010, P-100, G-001, G-010, and G-100 are 0.19, 0.06, 0.13, 0.42, 0.74, and 0.55 mol, respectively. The addition of Gr caused obvious differences in the NO2 radical evolution curves with time. Figure 5 shows the G-001, G-010, and G-100 models viewed in the XZ plane [i.e., along the (010) direction]. Among the HMX/Gr systems, the steric hindrance is largest in G-010 because of the NO2 functional groups. Owing to steric hindrance by the NO2 functional groups and interactions between Gr and HMX, the presence of Gr can inhibit the initial decomposition reaction pathways of HMX, with a directional and selective effect along the (010) crystal plane.
FIGURE 5. Models of G-001 (A), G-010 (B), and G-100 (C) in the XZ plane.
To further evaluate the effect of the crystal plane and Gr on HMX/Gr thermal decomposition, the evolutions of HONO, N2O2, HNO3, and NO are obtained, as shown in Figure 6. In all the calculated systems, more gradual changes were observed for the HNO3, NO, and N2O2 fragments than for HONO.
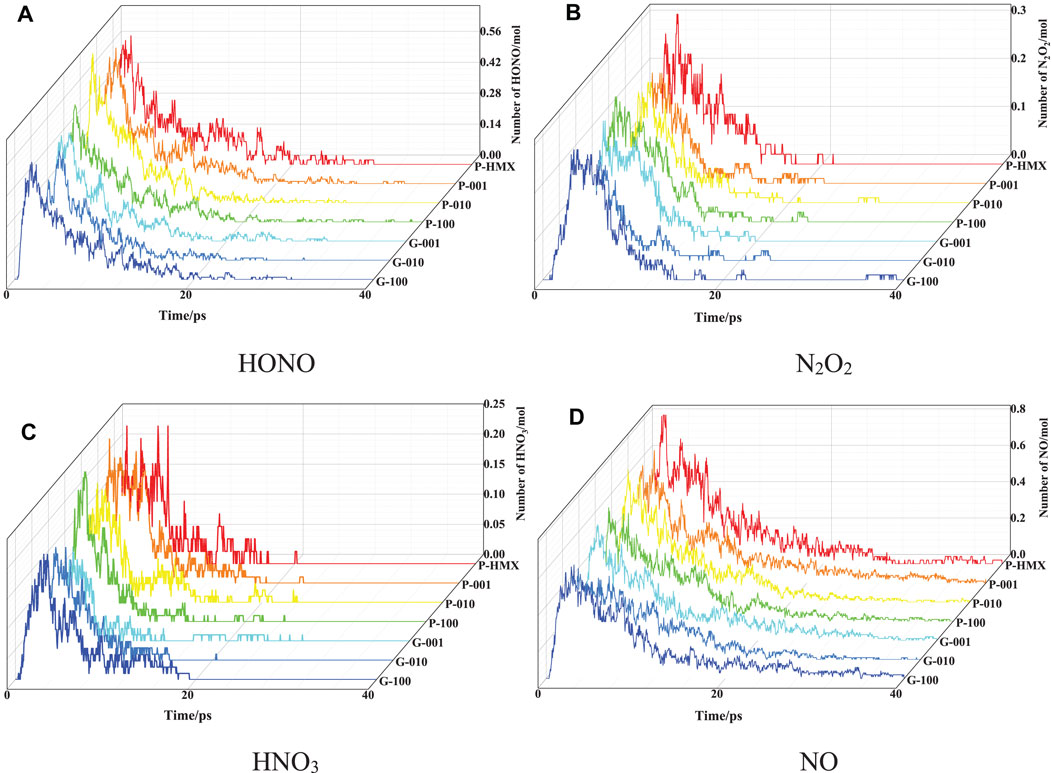
FIGURE 6. Evolution of intermediates at 3000 K. (A) HONO, (B) N2O2, (C) HNO3, and (D) NO.
The rate constant for each intermediate was calculated using Equation (2) in the simulation time range corresponding to the decay from the maximum population to zero at 3000 K as shown in Table 5. For both HONO and N2O2, the rate constants are higher for the HMX/Gr systems than for P-HMX. However, the NO rate constant was similar for all seven calculated systems. Furthermore, the NO2 rate constants show complex behavior. The NO2 rate constants for P-010 and P-100 are significantly higher than that for P-HMX.
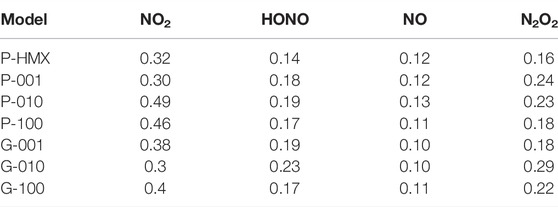
TABLE 5. Rate constants (ps−1) of intermediates at 3000 K.
The HMX crystal planes and Gr did not affect the initial decomposition pathways of HMX molecules in any of the systems. However, the trends of intermediates and the rate constants in the HMX/Gr systems change abruptly relative to those of P-HMX and HMX with different crystal planes.
Fragment Formation on Gr Surfaces
The adsorption of HMX molecules on Gr in the HMX/Gr systems was investigated at room temperature in the pressure range from atmospheric pressure to 31 GPa. For G-001, G-010, and G-100, we found 3, 5, and 3 different configurations for the binding of HMX molecules on Gr. At atmospheric pressure, two configurations were observed for the adsorption of HMX molecules on Gr, as shown in Figure 7. All adsorbed HMX molecules were located at the Gr edge. In G-001 and G-100, two oxygen atoms in the nitro functional groups of three HMX molecules were adsorbed on the Gr surface, which can be described as three C4H8O6N8O(ads)2 molecules (Figures 7A,C). In contrast, in G-010, one oxygen atom in the nitro functional group of one HMX molecule was adsorbed on the Gr surface, described as one C4H8O7N8O(ads) molecule (Figure 7B). This change in configuration shows that the interactions between HMX and Gr differ depending on the crystal plane.
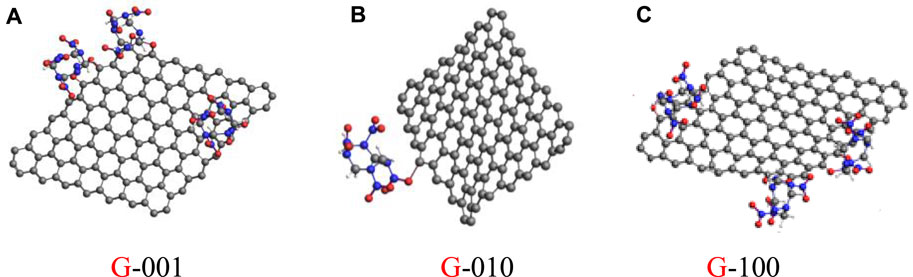
FIGURE 7. HMX configurations on the HMX/Gr systems at atmospheric pressure. (A) G-001, (B) G-010, and (C) G-100.
To study the effect of pressure on the amount of HMX molecules adsorbed on the Gr surface, the structures of the HMX/Gr supercells were calculated at 300 K and different pressures using the structures at atmospheric pressure and room temperature as the starting point. The number distribution of HMX molecules adsorbed on the Gr surface was analyzed. The results showed that the nitro functional groups on the A-axis and E-axis of a single HMX molecule could be adsorbed on the surface of Gr. In the pressure range of 1–31 GPa, along the (001) plane, HMX molecules could be adsorbed on the Gr surface with two different configurations. In the first configuration, one oxygen atom in the nitro functional group was adsorbed on the Gr surface, described as C4H8O7N8O(ads) (Figure 8A). In the second configuration, one N atom and two O atoms in the nitro functional group were adsorbed on the Gr surface, described as C4H8O6N7N(ads)O(ads)2 (Figure 8B). In the same pressure range, along the (100) plane, HMX molecules could be adsorbed on the Gr surface via an N atom in the CN ring, described as C4H8O8N7N(ads) (Figure 8C). In the second configuration, two HMX molecules were adsorbed on the Gr surface via one oxygen atom in the nitro functional group, described as C4H8O7N8O(ads), with one molecule located above the Gr surface and the other below, as shown in Figure 8D.
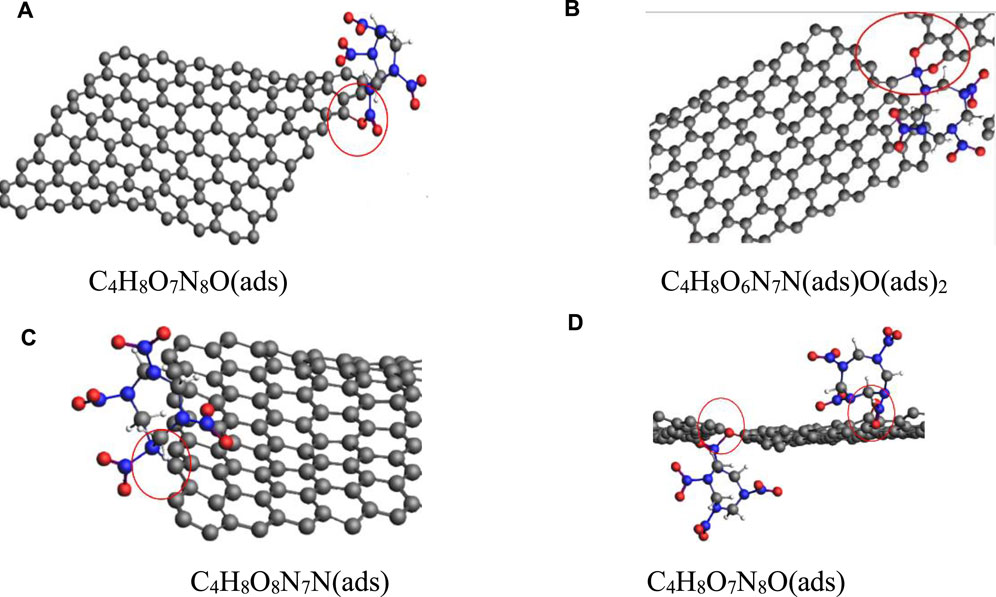
FIGURE 8. HMX configurations on G-001 (A,B) and G-100 (C,D) in the pressure range of 1–31 GPa.
In the same pressure range, along the (010) plane, four configurations were obtained for the adsorption of a single HMX molecule on the Gr surface, as shown in Figure 9. In the first configuration, two oxygen atoms in a nitro functional group were adsorbed on the Gr surface, described as C4H8O6N8O(ads)2 (Figure 9A). In the second configuration, a nitrogen atom in the CN ring was adsorbed on the Gr surface, described as C4H8O8N7N(ads) (Figure 9B). In the third configuration, an oxygen atom in a nitro functional group and an N atom in the CN ring were adsorbed on the Gr surface, described as C4H8O7N7O(ads)N(ads) (Figure 9C). In the fourth configuration, four oxygen atoms from two nitro groups were adsorbed on the Gr surface, described as C4H8O4N8O(ads)4 (Figure 9D). Compared with G-001 and G-100, HMX adsorption on G-010 at high pressures is more complex. For G-010, two nitro functional groups in a HMX molecule can bind to Gr, which is an important factor affecting the stability of HMX under extreme conditions.
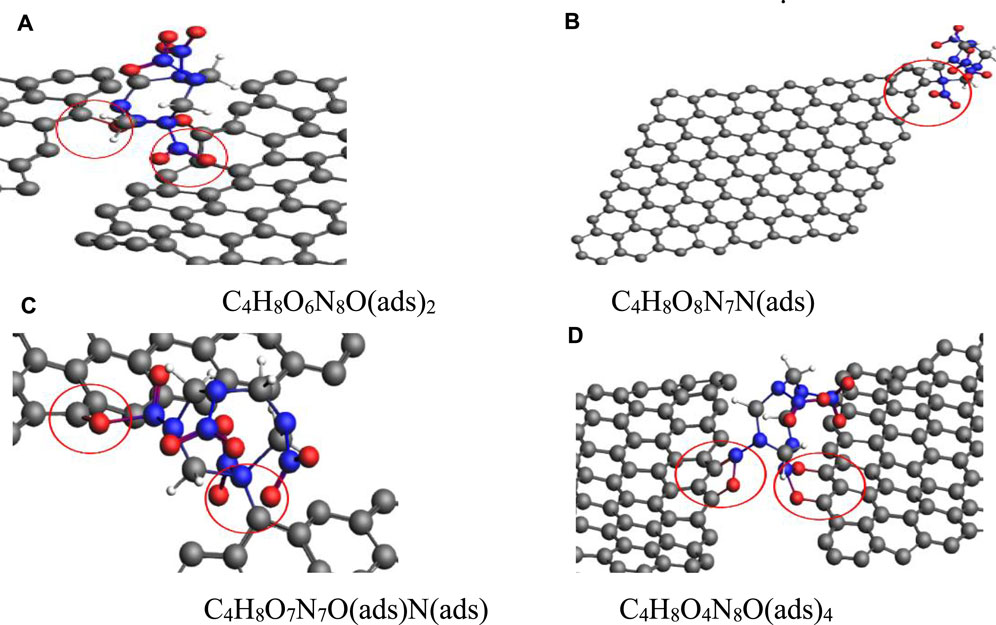
FIGURE 9. HMX configurations on G-010 in the pressure range of 1–31 GPa. (A) C4H8O6N8O(ads)2, (B) C4H8O8N7N(ads), (C) C4H8O7N7O(ads)N(ads), and (D) C4H8O4N8O(ads)4.
Figure 10 shows the number of HMX molecules adsorbed on the Gr surface in each HMX/Gr system in the pressure range from atmospheric pressure to 31 GPa. At all pressures except 19, 21, and 23 GPa, fewer HMX molecules were adsorbed on the Gr surface along the (010) plane than along the (001) and (100) planes.
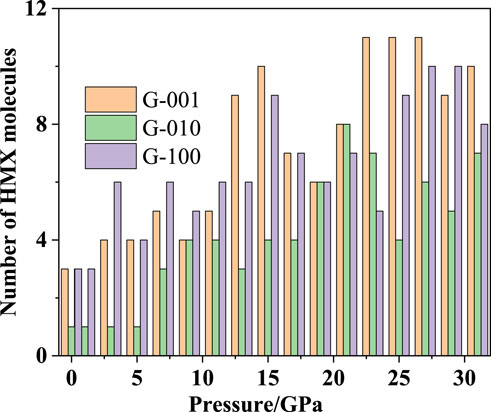
FIGURE 10. Number of HMX molecules adsorbed on Gr in HMX/Gr systems with the pressure ranging from atmospheric pressure to 31 GPa.
In the condensed phase at 3000 K, the chemical processes occurring in the HMX/Gr systems were more complex than those in the pure HMX supercells, and the initial reaction pathways of the HMX molecules were different. Moreover, the fragments produced in the HMX/Gr systems clearly depended on the crystal plane. Table 6 shows the main fragments formed on the Gr surface at 3500 K and 100 ps. The fragments are described using the notation A × B(ads)C, where A represents the number of fragments on the Gr surface, B(ads) represents the types of atoms bonded to the Gr surface, and C represents the number of bonded B atoms. For G-001, G-010, and G-100, 25, 34, and 31 decomposition product fragments, respectively, were generated on the Gr surface.

TABLE 6. Types and amounts of fragments formed on the Gr surface.
For G-001, the most common fragment was O(ads), whereas for G-010 and G-100 O(ads), the major fragments were OC(ads) and HO (ads). This change indicates that at high temperatures, the fragments generated by HMX react with the C atoms on the Gr surface to form different kinds of products, which reduces the reaction rate constant of HMX molecules. Thus, by prolonging the decomposition time of HMX molecules, the addition of Gr can improve the sensitivity of HMX explosives.
To further analyze the debris properties adsorbed on the surface of graphene, the total amount of fragments in different mixture systems was counted and classified by the thermal decomposition products without carbon atoms absorbed on the Gr surface [represented as C0, likes HN(ads), HO(ads), HOO(ads), CNO(ads), CH2O3O(ads), and NN(ads)], the one carbon atom bonded to the Gr surface [represented as C1, likes C13H3O9C(ads) and C10H2O8C(ads)] and containing two or more carbon atoms absorbed on the Gr surface [represents as C2, C3O3C(ads)2O(ads), C6H2O5NC(ads)2]. The final products such as N2, CO2, and CO can be absorbed on the Gr surface, while the H2O fragments cannot be absorbed on the Gr surface. The largest relative molecular weight fragments absorbed on the Gr surface of G-001, G-010, and G-100 are C21H7O15N3C(ads)3, C26H3O20N3C(ads)8O(ads)2, and C12H3O8NC(ads)5N(ads), respectively.
The results show that not only the number of fragments adsorbed on the surface of graphene in the G-010 system is the largest but also the carbon cluster, which is C26H3O20N3C(ads)8O(ads)2 with the largest relative molecular weight, is adsorbed on the surface of graphene in the G-010 system. Figure 11 is the percentage of different fragments absorbed on Gr in G-001, G-010 and G-100. The percentage of C0 fragments in G-100 is larger than the same percentage of C0 fragments in G-001 and G-010. In addition, in G-001, G-010, and G-100 systems, the products without carbon atoms account for 31.58, 52.81, and 53.57%, respectively.
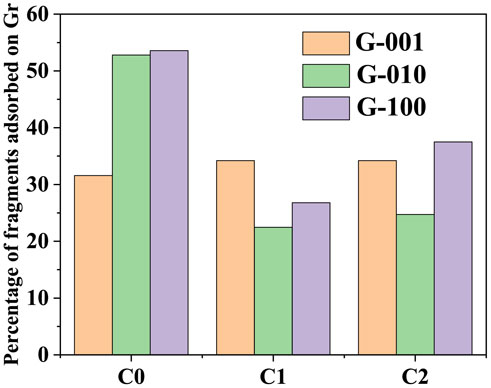
FIGURE 11. Percentage of fragments absorbed on Gr.
Figure 12 shows the time evolution of the O(ads) population in the HMX/Gr systems. Similar trends were observed for all the HMX/Gr systems. The amount of O(ads) increases quickly from 0 to 10.2 ps, decreased slowly between 10.3 and 71.5 ps, and then reaches an equilibrium value at 71.6–100 ps. The maximum O(ads) amount decreases in the following order: G-010 > G-100 > G-001.
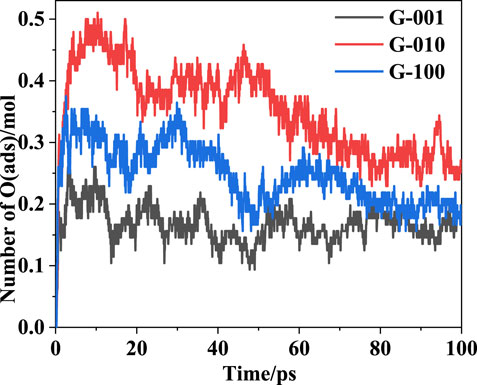
FIGURE 12. Evolution of O(ads) in HMX/Gr systems at 3000 K.
The time evolutions of HO(ads) and OC(ads) populations in the HMX/Gr systems are displayed in Figure 13. For G-001, G-010, and G-100 at 3000 K, the number of HO(ads) fragments fluctuated at 4, 8, and 6, respectively, whereas the number of OC(ads) fragments fluctuated at 3.5, 8.3, and 7.0, respectively. These results indicate that the HO(ads) and OC(ads) amounts reached equilibrium during the simulation time with the same order as O(ads), but no obvious peak values were observed for HO(ads) and OC(ads).
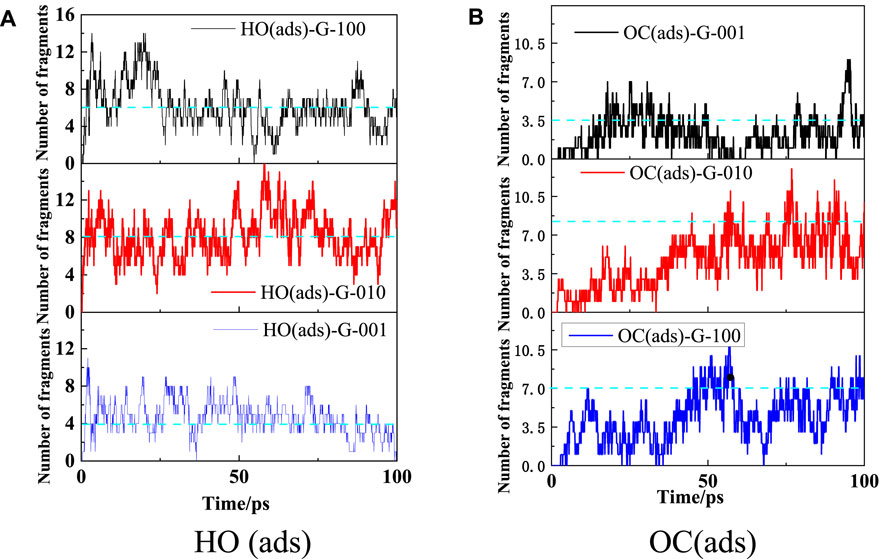
FIGURE 13. Evolution of HO(ads) (A) and OC(ads) (B) in HMX/Gr systems at 3000 K.
The fragment evolution results indicate that the initial decomposition pathways in the HMX/Gr systems at 3000 K involve the reaction of C atoms in Gr with the intermediates, resulting in the formation of C=O, C–OH, and C–CO bonds between C atoms and nitro radicals, hydroxyl radicals, and CO. A comparison of the product distributions for the HMX/Gr systems shows that the type and quantity of Gr products are maximized along the (010) crystal plane. Thus, the decomposition reactions in the HMX/Gr systems at high temperature are directional and selective, which provides theoretical guidance for the formulation of Gr-based explosives with improved safety.
Final Products
The final products in the thermal decomposition process were analyzed using a bond-order cutoff of 0.3. The evolution of various products (N2, H2O, CO2, and CO) during the simulation time is presented in Figure 14. These results suggest that these small products appear quickly at the beginning of the simulation (0–20 ps) and then reach a plateau at 20.1–100 ps. This behavior implies that Gr and the HMX crystal planes do not affect the kinetic parameters of the final products.
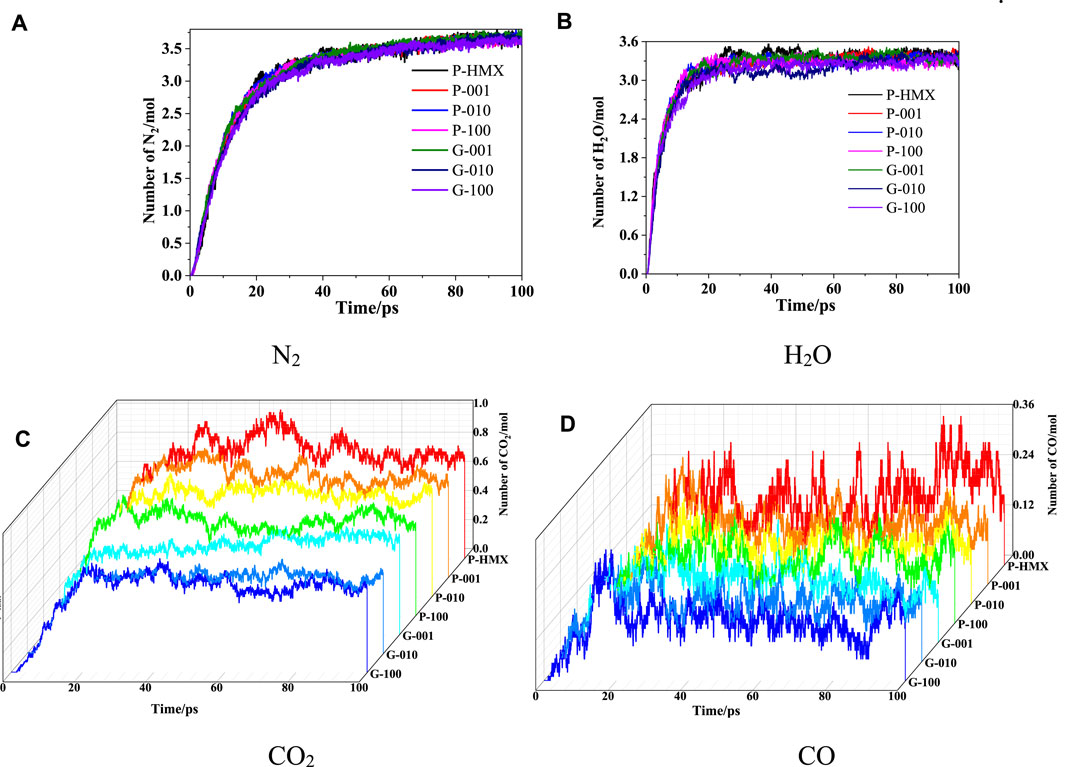
FIGURE 14. Evolution of the final products in HMX/Gr systems at 3000 K. (A) N2, (B) H2O, (C) CO2, and (D) CO.
Conclusion
In this work, RMDS was used to study the initial thermal decomposition reactions in HMX/Gr mixtures with different crystal planes. The analyses of the lattice parameters, binding energies, rate constants, and the evolution of intermediates and products revealed that the thermal stability of HMX is strongly dependent on the crystal plane and Gr.
In particular, the thermal stability of HMX was found to be more sensitive to the (010) crystal plane than the (001) and (100) planes, with the addition of Gr significantly lowering the reaction rate constants for HMX molecules. Monitoring the evolution of various intermediates, including NO2 radicals, HONO, NO, N2O2, and HNO3, at 3000 K showed that the addition of Gr to HMX along the (010) crystal plane increased the maximum amounts of these intermediates. Furthermore, these results indicated that the homolytic cleavages of N–NO2 and HONO elimination are the primary pathways for HMX decomposition in HMX/Gr systems in the early stages at high temperatures.
Notably, the addition of Gr along the (010) crystal plane was found to suppress NO2 radical formation. At pressures ranging from atmospheric pressure to 31 GPa at 300 K, HMX molecules were mostly adsorbed on the Gr surface via oxygen and nitrogen atoms in the nitro functional groups or nitrogen atoms in the HMX ring. Based on the intermediate population analysis for the HMX/Gr systems, the addition of Gr did not change the mechanism of HMX thermal decomposition, but the amount of product fragments decreased in the order G-010 > G-100 > G-001. The carbon atoms in Gr can easily form C=O, C–OH, and C–CO bonds to produce O(ads), HO(ads), and OC(ads) fragments, with the greatest effects observed for G-010.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
XY participated in the calculation of the mixed system and the production of some charts, and LZ was responsible for the overall conception, calculated the simulations process, and wrote the manuscript.
Funding
The authors thank the Shanxi Province Teaching Reform Project of Colleges and Universities under Grant No.2020405.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the Shanxi Province Teaching Reform Project of Colleges and Universities under Grant No. 2020405.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmech.2022.851198/full#supplementary-material
References
Allen, M. J., Tung, V. C., and Kaner, R. B. (2010). Honeycomb Carbon: A Review of Graphene. Chem. Rev. 110 (1), 132–145. doi:10.1021/cr900070d
Behrens, R., and Bulusu, S. (1991). Thermal Decomposition of Energetic Materials. 2. Deuterium Isotope Effects and Isotopic Scrambling in Condensed-Phase Decomposition of Octahydro-1,3,5,7-Tetranitro-1,3,5,7-Tetrazocine. J. Phys. Chem. 95 (15), 5838–5845. doi:10.1021/j100168a025
Chen, L-Y., Zhao, S-X., Yang, P-J., Heng, S-Y., Li, W., and Huang, X-W. (2006). The Coating and Desensitization of CL-20. Chin. J. Energetic Mater. 14 (3), 171–173.
Choi, C. S., and Boutin, H. P. (1970). A Study of the Crystal Structure of β-Cyclotetramethylene Tetranitramine by Neutron Diffraction. Acta Crystallogr. Sect B 26 (9), 1235–1240. doi:10.1107/s0567740870003941
Cobos, C. J. (2005). DFT Study of the Thermochemistry of Gas-Phase 1,3,5,7-Tetranitro-1,3,5,7-Tetraazacyclooctane (β-HMX). J. Mol. Struct. THEOCHEM 714 (2-3), 147–152. doi:10.1016/j.theochem.2004.09.042
Fried, L. E., Manaa, M. R., Pagoria, P. F., and Simpson, R. L. (2001). Design and Synthesis of Energetic Materials. Annu. Rev. Mater. Res. 31, 291–321. doi:10.1146/annurev.matsci.31.1.291
Ganz, E., Ganz, A. B., Yang, L.-M., and Dornfeld, M. (2017). The Initial Stages of Melting of Graphene between 4000 K and 6000 K. Phys. Chem. Chem. Phys. 19 (5), 3756–3762. doi:10.1039/c6cp06940a
Ge, N.-N., Wei, Y.-K., Ji, G.-F., Chen, X.-R., Zhao, F., and Wei, D.-Q. (2012). Initial Decomposition of the Condensed-Phase β-HMX under Shock Waves: Molecular Dynamics Simulations. J. Phys. Chem. B 116 (46), 13696–13704. doi:10.1021/jp309120t
Ge, N.-N., Wei, Y.-K., Song, Z.-F., Chen, X.-R., Ji, G.-F., Zhao, F., et al. (2014). Anisotropic Responses and Initial Decomposition of Condensed-Phase β-HMX under Shock Loadings via Molecular Dynamics Simulations in Conjunction with Multiscale Shock Technique. J. Phys. Chem. B 118 (29), 8691–8699. doi:10.1021/jp502432g
Hamilton, B. W., Kroonblawd, M. P., Islam, M. M., and Strachan, A. (2019). Sensitivity of the Shock Initiation Threshold of 1,3,5-Triamino-2,4,6-Trinitrobenzene (TATB) to Nuclear Quantum Effects. J. Phys. Chem. C 123 (36), 21969–21981. doi:10.1021/acs.jpcc.9b05409
He, Z.-H., Chen, J., Ji, G.-F., Liu, L.-M., Zhu, W.-J., and Wu, Q. (2015). Dynamic Responses and Initial Decomposition under Shock Loading: A DFTB Calculation Combined with MSST Method for β-HMX with Molecular Vacancy. J. Phys. Chem. B 119 (33), 10673–10681. doi:10.1021/acs.jpcb.5b05081
Hooks, D. E., Hayes, D. B., Hare, D. E., Reisman, D. B., Vandersall, K. S., Forbes, J. W., et al. (2006). Isentropic Compression of Cyclotetramethylene Tetranitramine (HMX) Single Crystals to 50GPa. J. Appl. Phys. 99 (12), 124901. doi:10.1063/1.2203411
Lewis, J. P., Glaesemann, K. R., VanOpdorp, K., and Voth, G. A. (2000). Ab Initio Calculations of Reactive Pathways for α-Octahydro-1,3,5,7-Tetranitro-1,3,5,7-Tetrazocine (α-HMX). J. Phys. Chem. A. 104 (48), 11384–11389. doi:10.1021/jp002173g
Lewis, J. P., Sewell, T. D., Evans, R. B., and Voth, G. A. (2000). Electronic Structure Calculation of the Structures and Energies of the Three Pure Polymorphic Forms of Crystalline HMX. J. Phys. Chem. B 104 (5), 1009–1013. doi:10.1021/jp9926037
Li, R., Wang, J., Shen, J. P., Hua, C., and Yang, G. C. (2013). Preparation and Characterization of Insensitive HMX/Graphene Oxide Composites. Propellants Explos. Pyrotech. 38 (6), 798–804. doi:10.1002/prep.201200199
Lin, C., He, G., Liu, J., Pan, L., and Liu, S. (2017). Enhanced Non-Linear Viscoelastic Properties of Polymer Bonded Explosives Based on Graphene and a Neutral Polymeric Bonding Agent. Cent. Eur. J. Energ. Mater. 14 (4), 788–805. doi:10.22211/cejem/78753
Liu, J., Ren, H., Jiao, Q.-J., and Yu, L. (2014). The Influence of GO/RGO on the Thermal Decomposition of HNIW. Integr. Ferroelectr. 152 (1), 127–136. doi:10.1080/10584587.2014.902252
Liu, L.-M., Car, R., Selloni, A., Dabbs, D. M., Aksay, I. A., and Yetter, R. A. (2012). Enhanced Thermal Decomposition of Nitromethane on Functionalized Graphene Sheets: Ab Initio Molecular Dynamics Simulations. J. Am. Chem. Soc. 134 (46), 19011–19016. doi:10.1021/ja3058277
Liu, L., Liu, Y., Zybin, S. V., Sun, H., and Goddard, W. A. (2011). ReaxFF-lg: Correction of the ReaxFF Reactive Force Field for London Dispersion, with Applications to the Equations of State for Energetic Materials. J. Phys. Chem. A. 115 (40), 11016–11022. doi:10.1021/jp201599t
Los, J. H., Zakharchenko, K. V., Katsnelson, M. I., and Fasolino, A. (2015). Melting Temperature of Graphene. Phys. Rev. B 91 (4), 045415. doi:10.1103/physrevb.91.045415
Lyman, J. L., Liau, Y.-C., and Brand, H. V. (2002). Thermochemical Functions for Gas-phase, 1,3,5,7-Tetranitro-1,3,5,7-Tetraazacyclooctane (HMX), its Condensed Phases, and its Larger Reaction Products. Combustion and Flame 130 (3), 185–203. doi:10.1016/s0010-2180(02)00364-4
Manaa, M. R., Fried, L. E., Melius, C. F., Elstner, M., and Frauenheim, T. (2002). Decomposition of HMX at Extreme Conditions: A Molecular Dynamics Simulation. J. Phys. Chem. A. 106 (39), 9024–9029. doi:10.1021/jp025668+
Menikoff, R., Dick, J. J., and Hooks, D. E. (2005). Analysis of Wave Profiles for Single-Crystal Cyclotetramethylene Tetranitramine. J. Appl. Phys. 97 (2), 023529. doi:10.1063/1.1828602
Niu, C., Jin, B., Peng, R., Shang, Y., and Liu, Q. (2017). Preparation and Characterization of Insensitive HMX/rGO/G Composites via In Situ Reduction of Graphene Oxide. RSC Adv. 7 (51), 32275–32281. doi:10.1039/c7ra03863a
Peng, Q., Rahul,, , Wang, G., Liu, G. R., and De, S. (2014). Structures, Mechanical Properties, Equations of State, and Electronic Properties of β-HMX under Hydrostatic Pressures: A DFT-D2 Study. Phys. Chem. Chem. Phys. 16 (37), 19972–19983. doi:10.1039/c4cp02193b
Ren, C., Li, X., and Guo, L. (2019). Chemical Insight on Decreased Sensitivity of CL-20/TNT Cocrystal Revealed by ReaxFF MD Simulations. J. Chem. Inf. Model. 59 (5), 2079–2092. doi:10.1021/acs.jcim.8b00952
Sharia, O., and Kuklja, M. M. (2010). Ab Initio Kinetics of Gas Phase Decomposition Reactions. J. Phys. Chem. A. 114 (48), 12656–12661. doi:10.1021/jp108065c
Shaw, R., and Walker, F. E. (1977). Estimated Kinetics and Thermochemistry of Some Initial Unimolecular Reactions in the thermal Decomposition of 1,3,5,7-Tetranitro-1,3,5,7-Tetraazacyclooctane in the Gas Phase. J. Phys. Chem. 81 (25), 2572–2576. doi:10.1021/j100540a040
Shi, Y., and Li, L.-J. (2011). Chemically Modified Graphene: Flame Retardant or Fuel for Combustion? J. Mater. Chem. 21 (10), 3277–3279. doi:10.1039/c0jm02953j
Singh, H. (2005). Current Trend of R&D in the Field of High Energy Materials (HEMs) - An Overview. Explosion 15 (3), 120–133.
Su, Y., Sun, Y., and Zhao, J. (2019). Interaction Mechanisms of Insensitive Explosive FOX-7 and Graphene Oxides from Ab Initio Calculations. Nanomaterials 9 (9), 1290. doi:10.3390/nano9091290
Wang, F., Chen, L., Geng, D., Lu, J., and Wu, J. (2019). Molecular Dynamics Simulations of an Initial Chemical Reaction Mechanism of Shocked CL-20 Crystals Containing Nanovoids. J. Phys. Chem. C 123 (39), 23845–23852. doi:10.1021/acs.jpcc.9b06137
Wu, J., Yang, L., Li, Y., Sultan, M., Geng, D., and Chen, L. (2020). Microscopic Mechanisms of Femtosecond Laser Ablation of HMX from Reactive Molecular Dynamics Simulations. J. Phys. Chem. C 124 (21), 11681–11693. doi:10.1021/acs.jpcc.0c01567
Xiao, J., Fang, G., Ji, G., and Xiao, H. (2005). Simulation Investigations in the Binding Energy and Me-Chanical Properties of HMX-Based Polymer-Bonded Explosives. Chin. Sci Bull 50 (1), 21–26. doi:10.1360/982004-147
Yang, K., Chen, L., Liu, D., Lu, J., Xiao, Y., Geng, D., et al. (2020). Anisotropic Initial Reaction Mechanism and Sensitivity Characterization of the Layered Crystal Structure Explosive ICM-102 under Shock Loading. J. Phys. Chem. C 124 (19), 10367–10375. doi:10.1021/acs.jpcc.0c01840
Yu, L., Ren, H., Guo, X.-Y., Jiang, X.-B., and Jiao, Q.-J. (2014). A Novel ε-HNIW-Based Insensitive High Explosive Incorporated with Reduced Graphene Oxide. J. Therm. Anal. Calorim. 117 (3), 1187–1199. doi:10.1007/s10973-014-3928-7
Zhang, C., Fu, X., Li, J., Fan, X., and Zhang, G. (2019). Desensitizing Effect of Graphene Oxide on Thermolysis Mechanisms of 4,4′-Azo-1,2,4-Triazole Studied by Reactive Molecular Dynamics Simulations. J. Phys. Chem. A. 123 (7), 1285–1294. doi:10.1021/acs.jpca.8b10087
Zhang, S., Nguyen, H. N., and Truong, T. N. (2003). Theoretical Study of Mechanisms, Thermodynamics, and Kinetics of the Decomposition of Gas-Phase α-HMX (Octahydro-1,3,5,7-Tetranitro-1,3,5,7-Tetrazocine). J. Phys. Chem. A. 107 (16), 2981–2989. doi:10.1021/jp030032j
Keywords: graphene, thermal decomposition, crystal plane, anisotropy, absorption
Citation: Yun X and Zhang L (2022) Temperature and Pressure Effects on HMX/Graphene via ReaxFF Molecular Dynamics Simulations. Front. Mech. Eng 8:851198. doi: 10.3389/fmech.2022.851198
Received: 05 February 2022; Accepted: 11 April 2022;
Published: 09 June 2022.
Edited by:
Seunghwa Ryu, Korea Advanced Institute of Science and Technology, South KoreaReviewed by:
Hyungjun Kim, Korea Advanced Institute of Science and Technology, South KoreaZhao Qin, Syracuse University, United States
Copyright © 2022 Yun and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhang, emhhbmdsaTM5Mjk1NjQ0M0AxNjMuY29t