
 Norbert Suttorp
Norbert Suttorp
- Department of Internal Medicine/Infectious Diseases and Pulmonary Medicine, Charité – Universitätsmedizin Berlin, Berlin, Germany
Listeria monocytogenes is an intracellular, Gram-positive bacterium that can cause life-threatening illness especially in immunocompromised individuals and newborns. The pathogen propagates within the cytosol of various host cells after escaping from the phagosomal compartment depending on the cytolysin listeriolysin O. While L. monocytogenes can manipulate the endocytic and many host-cell signaling cascades to its advantage, host cells are however capable of detecting Listeria infection at different cellular compartments by expressing innate immune receptors that trigger antibacterial defense pathways. These receptors include the Toll-like receptors, NOD-like receptors (NLRs), and cytosolic DNA sensors. Some NLRs as well as the DNA sensor AIM2 form multiprotein complexes called inflammasomes. Inflammasomes regulate caspase-1-dependent production of the key inflammatory cytokines IL-1β and IL-18 as well as pyroptotic cell death in L. monocytogenes-infected cells. This review describes the current knowledge about innate immune sensing and inflammasome activation in Listeria infection.
Listeria Monocytogenes – Infection and Intracellular Life-Style
Listeria monocytogenes is a flagellated Gram-positive bacterium that can cause life-threatening illness characterized by gastroenteritis, meningitis, encephalitis, materno-fetal, and perinatal infections. Infection with L. monocytogenes occurs through ingestion of contaminated food, such as unpasteurized dairy products and undercooked meats (Allerberger and Wagner, 2010). Listeria cross the intestinal barrier by invading intestinal epithelial cells, reaching the liver as well as spleen via the lymphoid system and the blood, where they are internalized by splenic and hepatic macrophages. During severe infections, the bacteria disseminate via the blood and cross the blood–brain barrier resulting in infections of the meninges and the brain. Furthermore it can cross the fetoplacental barrier in pregnant women which leads to infection of the fetus. L. monocytogenes is able to invade different non-phagocytic cells and is resistant to intracellular killing by macrophages after phagocytosis (Hamon et al., 2006; Barbuddhe and Chakraborty, 2009). In the intestinal tract, L. monocytogenes invades epithelial cells via interaction of its virulence protein internalin A (InlA) with epithelial cadherin (E-cadherin), leading to bacterial internalization within a membrane-bound vacuole (Mengaud et al., 1996; Schubert et al., 2002). In contrast, internalin B (InlB) binds to c-Met, a receptor tyrosine kinase and the natural receptor for hepatocyte growth factor (HGF) and thus promotes invasion of multiple mammalian cell types (Shen et al., 2000; Veiga and Cossart, 2005). InlB has been implicated in murine liver colonization after intravenous infection (Dramsi et al., 1995; Shen et al., 2000). In the vacuole, a decreased pH activates the pore-forming toxin listeriolysin O (LLO) that destroys the phagosomal membrane and the bacterium subsequently escapes into the cytosol (Bielecki et al., 1990). This allows the bacterium to replicate in the cytosol to high numbers. Cytosolic Listeria express ActA to induce host-cell actin polymerization and direct spread from cell to cell, thereby escaping immune detection and defense at the extracellular compartment (Tilney and Portnoy, 1989; Domann et al., 1992; Kocks et al., 1992). In addition to the cytosolic replication, a vacuolar growth of some bacteria might play a role for persistent infection in mice (Birmingham et al., 2008). Overall, L. monocytogenes manipulates the endocytic and many host-cell signaling cascades in order to replicate. On the other hand, host cells possess surveillance systems at different cellular compartments capable of detecting Listeria infection and activating defense pathways which in most cases might control infection.
Sensing of L. Monocytogenes by Pattern Recognition Receptors of the Innate Immune System
Upon infection with L. monocytogenes, innate immune responses are rapidly triggered and are essential for host survival (Pfeffer et al., 1993; Krull et al., 1997; Unanue, 1997). However, the activation of innate immunity by pattern recognition receptors (PRRs) in response to infection with L. monocytogenes is still not completely understood. In general, the membrane-bound Toll-like receptors (TLRs), as well as the cytosolic nuclear oligomerization domain (NOD)-like receptors (NLRs) and DNA sensors are critical for innate defense by recognizing conserved structures of microorganisms (Corr and O’Neill, 2009; Opitz et al., 2009; Takeuchi and Akira, 2010). Some of these PRRs activate signaling pathways leading to activation of transcription factors such as NF-κB and/or IFN regulatory factor 3 (IRF3) which direct upregulation of proinflammatory genes such as TNFα, IL-8 and pro-IL-1β, or type I IFNs, respectively. Other PRRs form protein complexes called inflammasomes that regulate production of IL-1β and IL-18 at a post-translational level and trigger the caspase-1-dependent inflammatory cell death (pyroptosis; Bergsbaken et al., 2009; Schroder and Tschopp, 2010).
Lipoproteins of L. monocytogenes are recognized by TLR2 at the cell surface (Machata et al., 2008). Some studies showed that mice deficient in TLR2 were more susceptible to L. monocytogenes infection with increased bacterial loads and reduced activation of macrophages, compared to wild-type mice (Torres et al., 2004). Other studies, however, did not reveal differences in susceptibility of TLR2-knockout and wild-type mice to wild-type Listeria (Edelson and Unanue, 2002; Gekara et al., 2009). The adapter molecule MyD88, that signals downstream of most TLRs and of the IL-1 as well as IL-18 receptors, has been shown to be essential for innate immunity to L. monocytogenes (Seki et al., 2002). Mice deficient in MyD88 displayed a higher susceptibility to L. monocytogenes infection than mice lacking either IFN-γ or both IL-12 and IL-18 (Edelson and Unanue, 2002; Seki et al., 2002). Furthermore, L. monocytogenes flagellin activates TLR5. However, since some L. monocytogenes strains do not express flagellin at 37°C, and bacteria mutants deficient in flagellin show an unaltered virulence, the role of TLR5 in recognition of Listeria remains unclear (Hayashi et al., 2001; Way et al., 2004).
In the cytosol, peptidoglycan fragments of L. monocytogenes are sensed by NOD1 and NOD2 leading to expression of proinflammatory genes and antimicrobial peptides (Kobayashi et al., 2005; Opitz et al., 2006; Park et al., 2007; Mosa et al., 2009). This NOD1/2-stimulated gene expression is dependent on the receptor interacting protein-2 (Rip-2) as well as NF-κB and p38 mitogen-activated protein kinase (Chin et al., 2002; Kobayashi et al., 2002). Accordingly, mice deficient in NOD1 or Rip-2 show increased susceptibility toward Listeria infection (Chin et al., 2002; Mosa et al., 2009).
Another surveillance mechanism that detects intracellular L. monocytogenes is mediated by a yet-to-be-identified cytosolic PRR that triggers a type I IFN response (O’Riordan et al., 2002; Stockinger et al., 2002; McCaffrey et al., 2004). This PRR possibly detects Listeria DNA, although known DNA sensors such as AIM2 or DAI/ZBP1 are most likely not involved (Auerbuch et al., 2004; Carrero et al., 2004; O’Connell et al., 2004; Stetson and Medzhitov, 2006; Leber et al., 2008; Lippmann et al., 2008; Rathinam et al., 2010). The Listeria-mediated type I IFN production occurs through a pathway dependent on the adapter molecule STING, the serine threonine kinase TBK1 and the transcription factor IRF3 (Stockinger et al., 2004; O’Connell et al., 2005; Ishikawa et al., 2009). Of note, this pathway appears to be detrimental for L. monocytogenes infections in vivo since mice deficient in IRF3 or the type I IFN receptor are more resistant than wild-type mice toward the bacterial infection (Auerbuch et al., 2004; Carrero et al., 2004; O’Connell et al., 2004).
In addition to the above mentioned PRRs that mainly control immune responses to Listeria infection via transcriptional upregulation of inflammatory genes, some NLRs as well as other cytosolic receptors regulate production of the key proinflammatory cytokines IL-1β and IL-18 at a post-translational level and stimulate pyroptosis in L. monocytogenes-infected cells (see below).
Role of Inflammasome Pathways in L. Monocytogenes Infection
The NLR protein family consists of over 20 members in mammals. They are all composed of a C-terminal leucine-rich repeat domain, a central nucleotide-binding domain, and of an N-terminal effector domain (Franchi et al., 2009). This effector domain is either a caspase-recruitment domain (CARD) or pyrin domain (PYD; Figure 1). Some NLR members that contain a PYD (this NLR subgroup is called NLRP), and the NLR member NLRC4 which contains a CARD are able to form inflammasomes (Schroder and Tschopp, 2010). These protein complexes also include the CARD-bearing caspase-1 and, in most cases, the CARD- and PYD-containing adapter molecule ASC. Homophilic interactions between respective domains in the NLRs, ASC, and caspase-1 are necessary for inflammasome activation. The NLRP protein NLRP3 plays a central role in caspase-1 activation in response to a variety of exogenous and endogenous stimuli, such as bacterial pore-forming toxins, ATP, uric acid crystals, cholesterol crystals, and alum (Martinon et al., 2002, 2006; Kanneganti et al., 2006; Mariathasan et al., 2006; Duewell et al., 2010). While it appears unlikely that these NLRP3 activators directly bind to NLRP3, the exact signal(s) that stimulates NLRP3 itself is still a matter of debate. Some studies suggest that lysosomal damage and the accompanying release of lysosomal cathepsins lead to the activation of the NLRP3 pathway (Halle et al., 2008; Hornung et al., 2008). Another model proposes that the generation of reactive oxygen species by mitochondria is an event upstream of NLRP3 activation (Dostert et al., 2008; Zhou et al., 2010). Future studies are required to clarify the underlying mechanism of NLRP3 activation (see also Stutz et al., 2009; Schroder and Tschopp, 2010). The cytosolic NLRC4 detects bacterial flagellin and the presence of type III secretion systems (Franchi et al., 2006; Miao et al., 2006, 2010). Upon activation, NLRs oligomerize and recruit procaspase-1 via the CARD domain, directly or indirectly via the adaptor protein ASC (Mariathasan et al., 2004). The assembled inflammasome then mediates caspase-1 activation. Activated caspase-1 post-translationally processes pro-IL-1β as well as pro-IL-18 to their mature forms and stimulates pyroptosis (Martinon et al., 2002; Bergsbaken et al., 2009). Recently, the HIN-200 family member AIM2 has been identified as a cytosolic double-stranded DNA (dsDNA) sensor that induces caspase-1-dependent IL-1β maturation and thus is the first non-NLR family member forming an inflammasome (Burckstummer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Roberts et al., 2009). In contrast to the NLRs, oligomerization of the AIM2 complex presumably is mediated by clustering upon direct binding to the ligand dsDNA, to which AIM2 binds via its C-terminal HIN-domain (Burckstummer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009). The AIM inflammasome is composed of AIM2, ASC, and caspase-1.
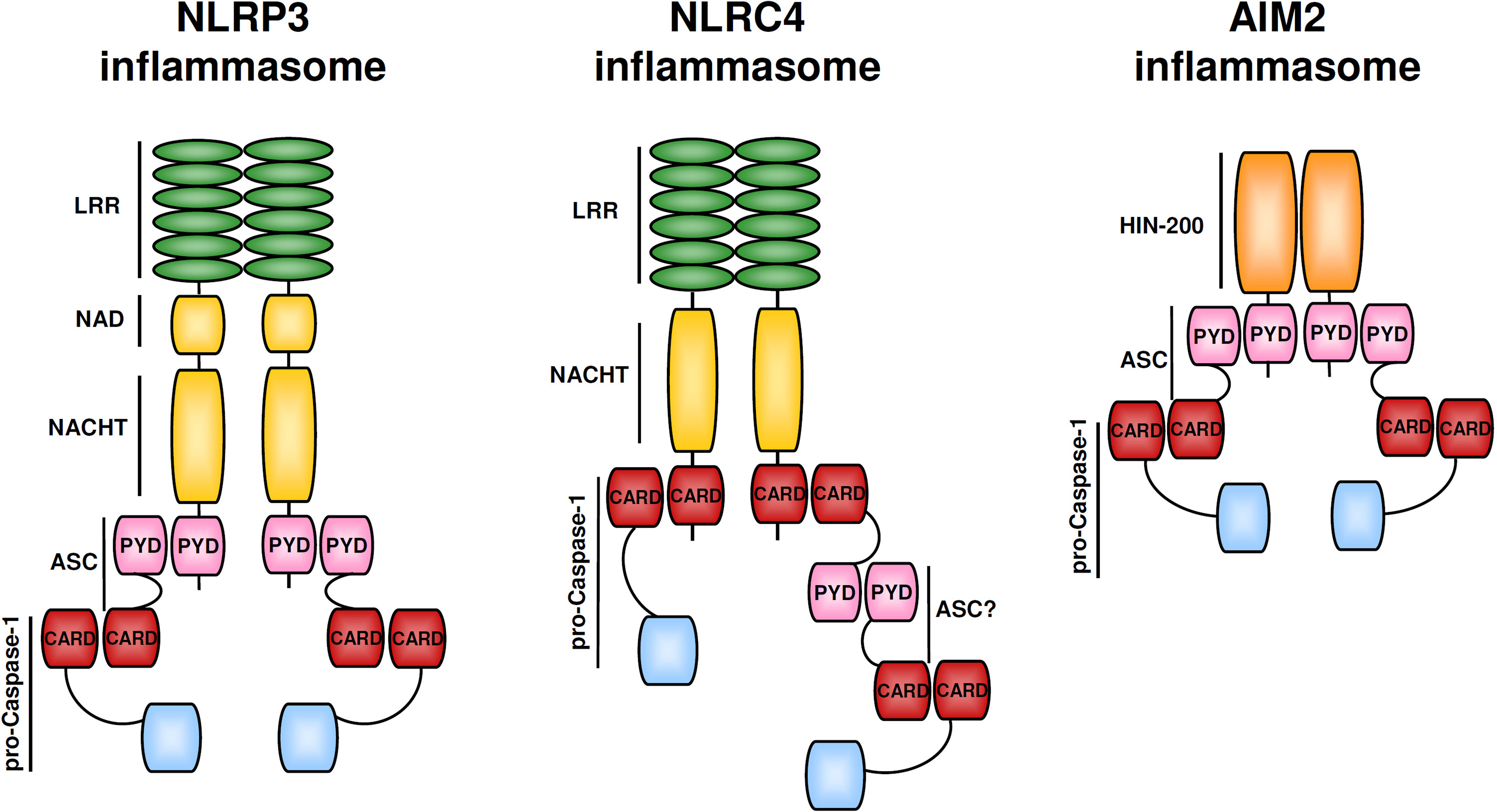
Figure 1. NLRP3, NLRC4, and AIM2 inflammasomes. Inflammasomes are assembled by self-oligomerizing proteins. The NLRP3 inflammasome consists of NLRP3, ASC, and caspase-1. NLRC4 can directly interact with procaspase-1 but maximal NLRC4 inflammasome activation might require ASC. The AIM2 inflammasome is composed of AIM2, ASC, and caspase-1. The PYD domain of AIM2 interacts with the PYD of ASC via homotypic PYD–PYD interactions, so that the ASC CARD domain can recruit procaspase to the complex. Domains: CARD, caspase-recruitment domain; HIN-200 domain; LRR, leucine-rich repeat; NACHT, nucleotide-binding, and oligomerization domain; PYD, pyrin domain.
In addition to the above-mentioned stimuli, L. monocytogenes infection also leads to a strong activation of caspase-1, production of IL-1β as well as IL-18, and to caspase-1-dependent cell death (Tsuji et al., 2004; Ozoren et al., 2006; Cervantes et al., 2008). Listeria eradication in the early phase of infection was impaired in caspase-1-deficient mice. These mutant mice showed a prominent decrease in production of IL-18, and as a consequence, of IFNγ (Tsuji et al., 2004). The first inflammasome identified to be activated by L. monocytogenes was the NLRP3 inflammasome (Mariathasan et al., 2006). In mouse macrophages infected with L. monocytogenes, NLRP3 as well as the adapter ASC were essential for caspase-1 activation and secretion of IL-1β and IL-18 (Mariathasan et al., 2006; Ozoren et al., 2006; Figure 2). Furthermore, mouse macrophages infected with L. monocytogenes deficient for the toxin listeriolysin O (LLO) did not secrete IL-1β and IL-18 (Mariathasan et al., 2006; Ozoren et al., 2006; Hara et al., 2008). Similarly, NLRP3, ASC, and LLO were required for IL-1β production in human peripheral blood mononuclear cells (PBMCs; Meixenberger et al., 2010). The LLO-mediated phagosomal rupture and release of cathepsin B into the cytosol might be involved in NLRP3 activation in human and murine cells (Meixenberger et al., 2010). However, several papers showed that L. monocytogenes activates caspase-1 through additional inflammasomes besides the NLRP3 complex.
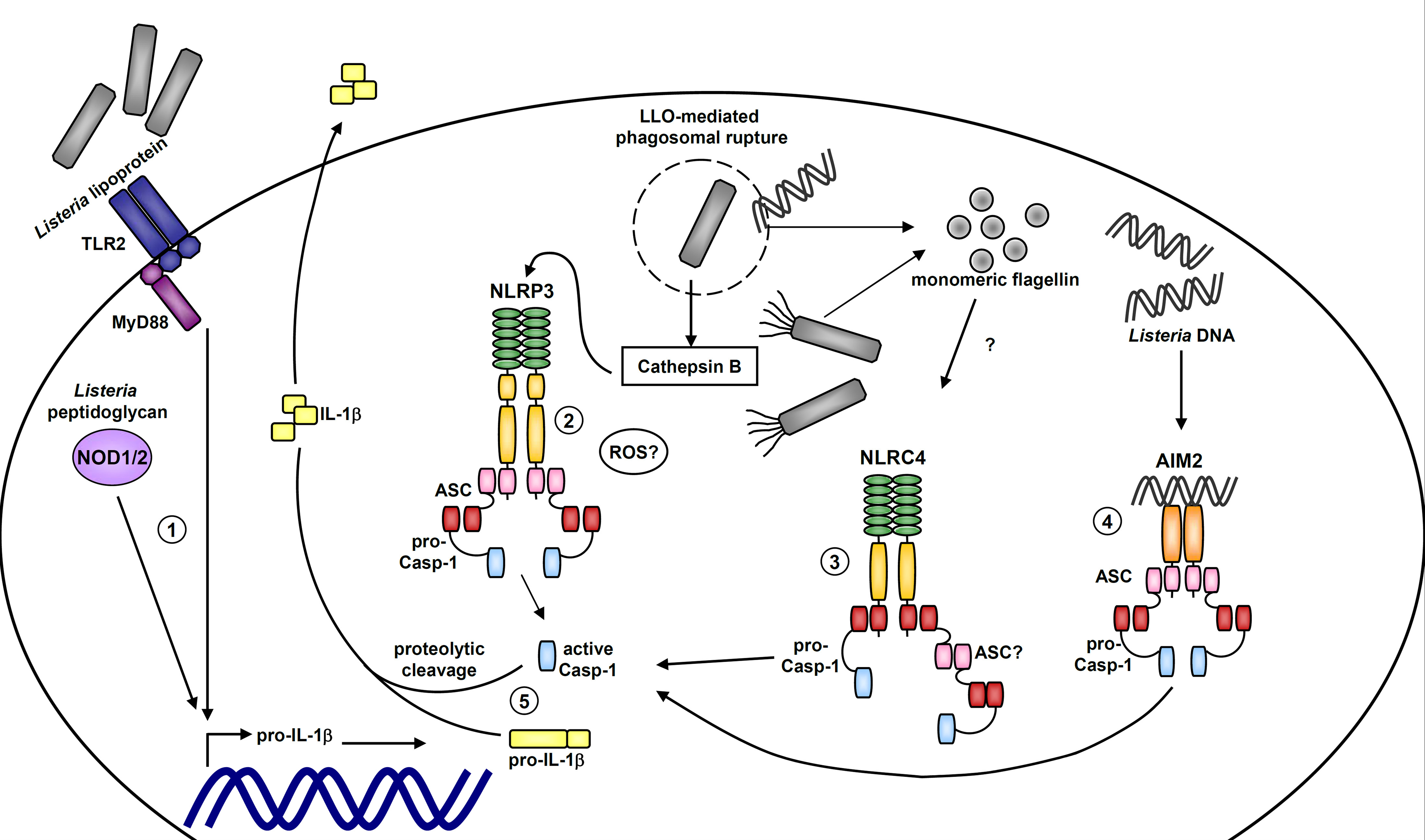
Figure 2. Model of inflammasome activation by Listeria monocytogenes. Activation of the extracellular TLR2 pathway via listerial lipoprotein and intracellular detection of bacterial peptidoglycans by NOD1/2 lead to upregulation of pro-IL-1β transcription (1). After bacterial internalization within a membrane-bound vacuole, listeriolysin O (LLO) leads to phagosomal rupture followed by escape of Listeria into the cytosol as well as cathepsin B release. Lysosomal damage and cathepsin B release (and perhaps other signals such as ROS) probably activate the NLRP3 inflammasome (2). Flagellin expression by L. monocytogenes or monomeric flagellin in the cytosol is detected by the NLRC4 inflammasome (3). Listerial DNA in the cytosol is sensed by the AIM2 inflammasome (4). Upon inflammasome activation, caspase-1 is autoactivated and mediates processing and secretion of the proinflammatory cytokines IL-1β and IL-18 (5; IL-18 is not depicted).
NLRC4 has been shown to be required for caspase-1 activation in infections with different bacteria expressing flagellin (Franchi et al., 2006; Miao et al., 2006), and some studies showed a partial impairment of caspase-1 activation and IL-1β production in NLRC4-deficient cells infected with L. monocytogenes (Warren et al., 2008; Wu et al., 2010). Other papers, however, found no evidence for a critical role of NLRC4 in Listeria-mediated caspase-1 activation (Kim et al., 2010; Meixenberger et al., 2010). It is known that the expression of flagellin is strictly inhibited at 37°C in some L. monocytogenes strains (Grundling et al., 2004; Way et al., 2004) and these differences in flagellin expression among bacterial strains are most likely responsible for the different results regarding NLRC4 involvement in L. monocytogenes infection.
In addition, several recent studies argue for a critical role of AIM2 in the recognition of L. monocytogenes in mouse macrophages via sensing Listeria DNA in the cytosol (Kim et al., 2010; Rathinam et al., 2010; Tsuchiya et al., 2010; Warren et al., 2010; Wu et al., 2010). Knockdown of AIM2 in wild-type macrophages resulted in a reduced L. monocytogenes-stimulated caspase-1 activation, IL-1β secretion, and cell death (Kim et al., 2010). Caspase-1 activation was completely inhibited in NLRP3-deficient macrophages treated with AIM2 siRNA. Accordingly, macrophages lacking AIM2 showed a lower but not abrogated caspase-1 activation and IL-1β production after L. monocytogenes infection (Rathinam et al., 2010). It was indicated that lysis of some Listeria in the host-cell cytosol – maybe as a result of vacuolar acidification and lysosomal fusion before vacuolar escape or yet undefined antimicrobial mechanisms – leads to release of bacterial DNA and activation of the AIM2 inflammasome (Sauer et al., 2010; Warren et al., 2010). Listeria DNA colocalized with AIM2 and ASC specks in the host cytosol (Warren et al., 2010; Wu et al., 2010). The AIM2 inflammasome together with other inflammasomes trigger IL-1β production as well as a weak pyroptotic cell death. L. monocytogenes mutants showing a higher amount of autolysis or Listeria treated with bactericidal antibiotics triggered a stronger AIM2-dependent cell death and IL-1β production (Sauer et al., 2010). Together, these data show that AIM2 is activated by Listeria DNA which triggers caspase-1 activation, cell death, and secretion of IL-1 family cytokines. Considering that AIM2 is an IFN-stimulated gene, the findings of AIM2 involvement in Listeria-mediated inflammasome activation fit well to the previous observation that type I IFN signaling was required for strong L. monocytogenes-stimulated IL-1β and IL-18 secretion (Henry et al., 2007).
Thus, multiple inflammasomes are involved in sensing L. monocytogenes infection. Warren et al. (2010) recently showed that NLRP3 most likely is temporally activated first, probably detecting vacuolar rupture. NLRC4 and AIM2 get activated at a later time point as flagellin monomers and bacterial DNA are released into the cytosol. Some studies, however, differ to some extent in conclusions regarding functional importance of the three different inflammasomes in L. monocytogenes infection (Franchi et al., 2007; Warren et al., 2008, 2010; Kim et al., 2010; Meixenberger et al., 2010; Rathinam et al., 2010; Sauer et al., 2010; Wu et al., 2010). This is likely attributed to the use of different Listeria strains that might vary in the expression and extent of release of the agonists of NLRP3, NLRC4, and AIM2. Moreover, some studies used LPS-primed macrophages in their experiments to induce strong pro-IL-1β and NLRP3 expression, whereas others performed infections in unprimed cells.
Inflammasomes can be seen as major sentinels of the innate immune defense against L. monocytogenes. They contribute to pathogen sensing and control post-translational processing of the inflammatory cytokines IL-1β and IL-18. IL-1β and IL-18 activate via the IL-1 and IL-18 receptors a MyD88-dependent signaling and subsequent NF-κB- and MAPK-regulated gene expression. Indeed, signals stimulated by LLO leading to IL-1β/IL-18 production, activation of the IL-1/IL-18 receptors and of MyD88-dependent signaling have been shown to compensate for lack of TLR2 in Listeria infection in mice (Gekara et al., 2009). IL-18 is crucial for IFN-γ induction, which is essential for the innate intracellular defense against L. monocytogenes as well as for the T cell-mediated acquired immunity (Tsuji et al., 2004). The significance of IL-1 in Listeria infections has been demonstrated by several groups. It has been shown that the blockade of IL-1 receptor exacerbates the disease and mice lacking caspase-1 or the IL-1β receptor are significantly more susceptible to infections with L. monocytogenes (Havell et al., 1992; Labow et al., 1997; Tsuji et al., 2004). Moreover, IL-1 receptor antagonist (IL-1Ra)-deficient mice and IL-1Ra-overproducing mice are less or more susceptible, respectively, to Listeriosis (Hirsch et al., 1996).
Conclusion
The interaction of L. monocytogenes with host cells is complex and involves entry of the bacterium to different cellular compartments as well as a multilayered host-cell surveillance system capable of detecting infection. NLR and AIM2 inflammasomes are key players of this innate immune surveillance system. They are directly and indirectly activated by various PAMPs and virulence factors expressed by L. monocytogenes and mediate production of key inflammatory cytokines that alert the immune system, as well as host-cell death which helps to constrain bacterial infection.
Conflict of Interest Statement:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Trinad Chakraborty as well as members of the lab for helpful discussions. Work in the laboratory is supported by the Deutsche Forschungsgemeinschaft (projects SFB-TR84/B1 to Norbert Suttorp and OP 86/5-1, OP 86/7-1 and SFB-TR84/A1 to Bastian Opitz).
References
Allerberger, F., and Wagner, M. (2010). Listeriosis: a resurgent foodborne infection. Clin. Microbiol. Infect. 16, 16–23.
Auerbuch, V., Brockstedt, D. G., Meyer-Morse, N., O’Riordan, M., and Portnoy, D. A. (2004). Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 200, 527–533.
Barbuddhe, S. B., and Chakraborty, T. (2009). Listeria as an enteroinvasive gastrointestinal pathogen. Curr. Top. Microbiol. Immunol. 337, 173–195.
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109.
Bielecki, J., Youngman, P., Connelly, P., and Portnoy, D. A. (1990). Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature 345, 175–176.
Birmingham, C. L., Canadien, V., Kaniuk, N. A., Steinberg, B. E., Higgins, D. E., and Brumell, J. H. (2008). Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature 451, 350–354.
Burckstummer, T., Baumann, C., Bluml, S., Dixit, E., Durnberger, G., Jahn, H., Planyavsky, M., Bilban, M., Colinge, J., Bennett, K. L., and Superti-Furga, G. (2009). An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272.
Carrero, J. A., Calderon, B., and Unanue, E. R. (2004). Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 200, 535–540.
Cervantes, J., Nagata, T., Uchijima, M., Shibata, K., and Koide, Y. (2008). Intracytosolic Listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell. Microbiol. 10, 41–52.
Chin, A. I., Dempsey, P. W., Bruhn, K., Miller, J. F., Xu, Y., and Cheng, G. (2002). Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature 416, 190–194.
Corr, S. C., and O’Neill, L. A. (2009). Listeria monocytogenes infection in the face of innate immunity. Cell. Microbiol. 11, 703–709.
Domann, E., Wehland, J., Rohde, M., Pistor, S., Hartl, M., Goebel, W., Leimeister-Wachter, M., Wuenscher, M., and Chakraborty, T. (1992). A novel bacterial virulence gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J. 11, 1981–1990.
Dostert, C., Petrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677.
Dramsi, S., Biswas, I., Maguin, E., Braun, L., Mastroeni, P., and Cossart, P. (1995). Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol. Microbiol. 16, 251–261.
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., Abela, G. S., Franchi, L., Nunez, G., Schnurr, M., Espevik, T., Lien, E., Fitzgerald, K. A., Rock, K. L., Moore, K. J., Wright, S. D., Hornung, V., and Latz, E. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361.
Edelson, B. T., and Unanue, E. R. (2002). MyD88-dependent but toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J. Immunol. 169, 3869–3875.
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513.
Franchi, L., Amer, A., Body-Malapel, M., Kanneganti, T. D., Ozoren, N., Jagirdar, R., Inohara, N., Vandenabeele, P., Bertin, J., Coyle, A., Grant, E. P., and Nunez, G. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582.
Franchi, L., Kanneganti, T. D., Dubyak, G. R., and Nunez, G. (2007). Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 282, 18810–18818.
Franchi, L., Warner, N., Viani, K., and Nunez, G. (2009). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128.
Gekara, N. O., Dietrich, N., Lyszkiewicz, M., Lienenklaus, S., and Weiss, S. (2009). Signals triggered by a bacterial pore-forming toxin contribute to toll-like receptor redundancy in Gram-positive bacterial recognition. J. Infect. Dis. 199, 124–133.
Grundling, A., Burrack, L. S., Bouwer, H. G., and Higgins, D. E. (2004). Listeria monocytogenes regulates flagellar motility gene expression through MogR, a transcriptional repressor required for virulence. Proc. Natl. Acad. Sci. U.S.A. 101, 12318–12323.
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., Fitzgerald, K. A., Latz, E., Moore, K. J., and Golenbock, D. T. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865.
Hamon, M., Bierne, H., and Cossart, P. (2006). Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 4, 423–434.
Hara, H., Tsuchiya, K., Nomura, T., Kawamura, I., Shoma, S., and Mitsuyama, M. (2008). Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J. Immunol. 180, 7859–7868.
Havell, E. A., Moldawer, L. L., Helfgott, D., Kilian, P. L., and Sehgal, P. B. (1992). Type I IL-1 receptor blockade exacerbates murine listeriosis. J. Immunol. 148, 1486–1492.
Hayashi, F., Smith, K. D., Ozinsky, A., Hawn, T. R., Yi, E. C., Goodlett, D. R., Eng, J. K., Akira, S., Underhill, D. M., and Aderem, A. (2001). The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103.
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J., and Monack, D. M. (2007). Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994.
Hirsch, E., Irikura, V. M., Paul, S. M., and Hirsh, D. (1996). Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc. Natl. Acad. Sci. U.S.A. 93, 11008–11013.
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., Latz, E., and Fitzgerald, K. A. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518.
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., Fitzgerald, K. A., and Latz, E. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856.
Ishikawa, H., Ma, Z., and Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792.
Kanneganti, T. D., Ozoren, N., Body-Malapel, M., Amer, A., Park, J. H., Franchi, L., Whitfield, J., Barchet, W., Colonna, M., Vandenabeele, P., Bertin, J., Coyle, A., Grant, E. P., Akira, S., and Nunez, G. (2006). Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236.
Kim, S., Bauernfeind, F., Ablasser, A., Hartmann, G., Fitzgerald, K. A., Latz, E., and Hornung, V. (2010). Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40, 1545–1551.
Kobayashi, K., Inohara, N., Hernandez, L. D., Galan, J. E., Nunez, G., Janeway, C. A., Medzhitov, R., and Flavell, R. A. (2002). RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416, 194–199.
Kobayashi, K. S., Chamaillard, M., Ogura, Y., Henegariu, O., Inohara, N., Nunez, G., and Flavell, R. A. (2005). Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734.
Kocks, C., Gouin, E., Tabouret, M., Berche, P., Ohayon, H., and Cossart, P. (1992). L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68, 521–531.
Krull, M., Nost, R., Hippenstiel, S., Domann, E., Chakraborty, T., and Suttorp, N. (1997). Listeria monocytogenes potently induces up-regulation of endothelial adhesion molecules and neutrophil adhesion to cultured human endothelial cells. J. Immunol. 159, 1970–1976.
Labow, M., Shuster, D., Zetterstrom, M., Nunes, P., Terry, R., Cullinan, E. B., Bartfai, T., Solorzano, C., Moldawer, L. L., Chizzonite, R., and McIntyre, K. W. (1997). Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J. Immunol. 159, 2452–2461.
Leber, J. H., Crimmins, G. T., Raghavan, S., Meyer-Morse, N. P., Cox, J. S., and Portnoy, D. A. (2008). Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog. 4, e6. doi: 10.1371/journal.ppat.0040006
Lippmann, J., Rothenburg, S., Deigendesch, N., Eitel, J., Meixenberger, K., van Laak, V., Slevogt, H., N’guessan, P. D., Hippenstiel, S., Chakraborty, T., Flieger, A., Suttorp, N., and Opitz, B. (2008). IFNbeta responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell. Microbiol. 10, 2579–2588.
Machata, S., Tchatalbachev, S., Mohamed, W., Jansch, L., Hain, T., and Chakraborty, T. (2008). Lipoproteins of Listeria monocytogenes are critical for virulence and TLR2-mediated immune activation. J. Immunol. 181, 2028–2035.
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., Roose-Girma, M., Erickson, S., and Dixit, V. M. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218.
Mariathasan, S., Weiss, D. S., Newton, K., McBride, J., O’Rourke, K., Roose-Girma, M., Lee, W. P., Weinrauch, Y., Monack, D. M., and Dixit, V. M. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232.
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241.
McCaffrey, R. L., Fawcett, P., O’Riordan, M., Lee, K. D., Havell, E. A., Brown, P. O., and Portnoy, D. A. (2004). A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc. Natl. Acad. Sci. U.S.A. 101, 11386–11391.
Meixenberger, K., Pache, F., Eitel, J., Schmeck, B., Hippenstiel, S., Slevogt, H., N’Guessan, P., Witzenrath, M., Netea, M. G., Chakraborty, T., Suttorp, N., and Opitz, B. (2010). Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J. Immunol. 184, 922–930.
Mengaud, J., Ohayon, H., Gounon, P., Mege, R.-M., and Cossart, P. (1996). E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84, 923–932.
Miao, E. A., Alpuche-Aranda, C. M., Dors, M., Clark, A. E., Bader, M. W., Miller, S. I., and Aderem, A. (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575.
Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau, R., Lorang, C. G., Warren, S. E., Leaf, I. A., and Aderem, A. (2010). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107, 3076–3080.
Mosa, A., Trumstedt, C., Eriksson, E., Soehnlein, O., Heuts, F., Janik, K., Klos, A., Dittrich-Breiholz, O., Kracht, M., Hidmark, A., Wigzell, H., and Rottenberg, M. E. (2009). Nonhematopoietic cells control the outcome of infection with Listeria monocytogenes in a nucleotide oligomerization domain 1-dependent manner. Infect. Immun. 77, 2908–2918.
O’Connell, R. M., Saha, S. K., Vaidya, S. A., Bruhn, K. W., Miranda, G. A., Zarnegar, B., Perry, A. K., Nguyen, B. O., Lane, T. F., Taniguchi, T., Miller, J. F., and Cheng, G. (2004). Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 200, 437–445.
O’Connell, R. M., Vaidya, S. A., Perry, A. K., Saha, S. K., Dempsey, P. W., and Cheng, G. (2005). Immune activation of type I IFNs by Listeria monocytogenes occurs independently of TLR4, TLR2, and receptor interacting protein 2 but involves TNFR-associated NF kappa B kinase-binding kinase 1. J. Immunol. 174, 1602–1607.
O’Riordan, M., Yi, C. H., Gonzales, R., Lee, K. D., and Portnoy, D. A. (2002). Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 13861–13866.
Opitz, B., Eitel, J., Meixenberger, K., and Suttorp, N. (2009). Role of Toll-like receptors, NOD-like receptors and RIG-I-like receptors in endothelial cells and systemic infections. Thromb. Haemost. 102, 1103–1109.
Opitz, B., Puschel, A., Beermann, W., Hocke, A. C., Forster, S., Schmeck, B., van Laak, V., Chakraborty, T., Suttorp, N., and Hippenstiel, S. (2006). Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J. Immunol. 176, 484–490.
Ozoren, N., Masumoto, J., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Erturk, I., Jagirdar, R., Zhu, L., Inohara, N., Bertin, J., Coyle, A., Grant, E. P., and Nunez, G. (2006). Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J. Immunol. 176, 4337–4342.
Park, J. H., Kim, Y. G., McDonald, C., Kanneganti, T. D., Hasegawa, M., Body-Malapel, M., Inohara, N., and Nunez, G. (2007). RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol. 178, 2380–2386.
Pfeffer, K., Matsuyama, T., Kundig, T. M., Wakeham, A., Kishihara, K., Shahinian, A., Wiegmann, K., Ohashi, P. S., Kronke, M., and Mak, T. W. (1993). Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73, 457–467.
Rathinam, V. A., Jiang, Z., Waggoner, S. N., Sharma, S., Cole, L. E., Waggoner, L., Vanaja, S. K., Monks, B. G., Ganesan, S., Latz, E., Hornung, V., Vogel, S. N., Szomolanyi-Tsuda, E., and Fitzgerald, K. A. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402.
Roberts, T. L., Idris, A., Dunn, J. A., Kelly, G. M., Burnton, C. M., Hodgson, S., Hardy, L. L., Garceau, V., Sweet, M. J., Ross, I. L., Hume, D. A., and Stacey, K. J. (2009). HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060.
Sauer, J. D., Witte, C. E., Zemansky, J., Hanson, B., Lauer, P., and Portnoy, D. A. (2010). Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7, 412–419.
Schubert, W. D., Urbanke, C., Ziehm, T., Beier, V., Machner, M. P., Domann, E., Wehland, J., Chakraborty, T., and Heinz, D. W. (2002). Structure of internalin, a major invasion protein of Listeria monocytogenes, in complex with its human receptor E-cadherin. Cell 111, 825–836.
Seki, E., Tsutsui, H., Tsuji, N. M., Hayashi, N., Adachi, K., Nakano, H., Futatsugi-Yumikura, S., Takeuchi, O., Hoshino, K., Akira, S., Fujimoto, J., and Nakanishi, K. (2002). Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 169, 3863–3868.
Shen, Y., Naujokas, M., Park, M., and Ireton, K. (2000). InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103, 501–510.
Stetson, D. B., and Medzhitov, R. (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103.
Stockinger, S., Materna, T., Stoiber, D., Bayr, L., Steinborn, R., Kolbe, T., Unger, H., Chakraborty, T., Levy, D. E., Muller, M., and Decker, T. (2002). Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J. Immunol. 169, 6522–6529.
Stockinger, S., Reutterer, B., Schaljo, B., Schellack, C., Brunner, S., Materna, T., Yamamoto, M., Akira, S., Taniguchi, T., Murray, P. J., Muller, M., and Decker, T. (2004). IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J. Immunol. 173, 7416–7425.
Stutz, A., Golenbock, D. T., and Latz, E. (2009). Inflammasomes: too big to miss. J. Clin. Invest. 119, 3502–3511.
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820.
Tilney, L. G., and Portnoy, D. A. (1989). Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109, 1597–1608.
Torres, D., Barrier, M., Bihl, F., Quesniaux, V. J., Maillet, I., Akira, S., Ryffel, B., and Erard, F. (2004). Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect. Immun. 72, 2131–2139.
Tsuchiya, K., Hara, H., Kawamura, I., Nomura, T., Yamamoto, T., Daim, S., Dewamitta, S. R., Shen, Y., Fang, R., and Mitsuyama, M. (2010). Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J. Immunol. 185, 1186–1195.
Tsuji, N. M., Tsutsui, H., Seki, E., Kuida, K., Okamura, H., Nakanishi, K., and Flavell, R. A. (2004). Roles of caspase-1 in Listeria infection in mice. Int. Immunol. 16, 335–343.
Unanue, E. R. (1997). Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunol. Rev. 158, 11–25.
Veiga, E., and Cossart, P. (2005). Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat. Cell Biol. 7, 894–900.
Warren, S. E., Armstrong, A., Hamilton, M. K., Mao, D. P., Leaf, I. A., Miao, E. A., and Aderem, A. (2010). Cutting edge: cytosolic bacterial DNA activates the inflammasome via Aim2. J. Immunol. 185, 818–821.
Warren, S. E., Mao, D. P., Rodriguez, A. E., Miao, E. A., and Aderem, A. (2008). Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 180, 7558–7564.
Way, S. S., Thompson, L. J., Lopes, J. E., Hajjar, A. M., Kollmann, T. R., Freitag, N. E., and Wilson, C. B. (2004). Characterization of flagellin expression and its role in Listeria monocytogenes infection and immunity. Cell. Microbiol. 6, 235–242.
Wu, J., Fernandes-Alnemri, T., and Alnemri, E. S. (2010). Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30, 693–702.
Keywords: Listeria, innate immunity, NOD-like receptor, inflammasome, AIM2
Citation: Eitel J, Suttorp N and Opitz B (2011) Innate immune recognition and inflammasome activation in Listeria monocytogenes infection. Front. Microbio. 1:149. doi: 10.3389/fmicb.2010.00149
Received: 01 November 2010;
Paper pending published: 16 November 2010;
Accepted: 29 December 2010;
Published online: 14 January 2011.
Edited by:
Amal Amer, The Ohio State University, USAReviewed by:
Stephanie M. Seveau, The Ohio State University, USAAlfredo G. Torres, University of Texas Medical Branch, USA
Copyright: © 2011 Eitel, Suttorp and Opitz. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Bastian Opitz, Department of Internal Medicine/Infectious Diseases and Pulmonary Medicine, Charité – Universitätsmedizin Berlin, Augustenburger Platz 1, 13353 Berlin, Germany. e-mail:YmFzdGlhbi5vcGl0ekBjaGFyaXRlLmRl