
- Department of Biological Sciences, Purdue University, West Lafayette, IN, USA
Programmed cell death is considered the ultimate solution for the host to eliminate infected cells, leading to the abolishment of the niche for microbial replication and the ablation of infection. Thus, it is not surprising that successful pathogens have evolved diverse strategies to reprogram the cell death pathways for their proliferation. Using effector proteins translocated by the Dot/Icm type IV secretion system, the facultative intracellular pathogen Legionella pneumophila manipulates multiple host cellular processes to create a niche within host cells to support its replication. Investigation in the past decade has established that in mammalian cells this bacterium actively modulates two host cell death pathways, namely the canonical apoptotic pathway controlled by the mitochondrion and the pyroptotic pathway controlled by the Nod-like receptor Naip5 and the Ipaf inflammasome. In this review, I will discuss the recent progress in understanding the mechanisms the bacterium employs to interfere with these host cell death pathways and how such modulation contribute to the intracellular life cycle of the pathogen.
Introduction
For intracellular pathogens whose proliferation requires nutritional supplies from the host cell cytosol, the death of the cell before the completion of a productive infection is disastrous. Furthermore, cells actively executing the apoptotic processes often secrete chemical signals or display specific molecules on their surface so that phagocytes can recognize and engulf them, leading to the termination of infection. Even if such engulfment did not occur, pathogens released prior to mature infection often are not primed for the second round infection and can be recognized and destroyed more easily by the immune system. Therefore, it is not unexpected to learn that hijacking host cell death pathways constitutes an important pathogenic strategy for almost all well adapted intracellular pathogens.
As detailed in the several excellent articles of this review series, Legionella pneumophila is a facultative intracellular pathogen that uses similar strategies to replicate in phylogenetically distant eukaryotic cells, ranging from amebae to human alveolar macrophages. Within these evolutionarily distant host cells, the L. pneumophila-containing vacuole (LCV) undertakes a unique maturation pathway characterized by the evasion of endocytic fusion and the interception of membrane trafficking vesicles originating from the endoplasmic reticulum (ER; Isberg et al., 2009; Hubber and Roy, 2010). As the bacterium begins to multiply, active acquisition of membrane materials from the ER-derived vesicles compensates the expansion of the LCV. The result of such remodeling is the formation of a compartment morphologically and cell biologically resembling the ER (Isberg et al., 2009). Whereas the interaction with the host membrane trafficking pathways is probably the best understood process during intracellular L. pneumophila growth, it is becoming clear that modulation of several other pathways, including lipid metabolism, autophagy, ubiquitination, and host cell death, is also critical for successful infection (Hubber and Roy, 2010). To accomplish this feat, L. pneumophila delivers more than 200 bacterial proteins via the Dot/Icm type IV secretion system into host cells where they engage in distinct host pathways to facilitate the biogenesis of the LCV permissive for bacterial replication (Ensminger and Isberg, 2009). Here, I will discuss recent progress in the interplays between L. pneumophila and the host cell death pathways and how such interplays contribute to successful bacterial infection in mammalian cells.
Host Cell Death Pathways and Their Regulation
In mammalian cells, programmed cell death is divided into at least four categories: apoptosis, pyroptosis, necrosis, and necroptosis (Fink and Cookson, 2005; Vandenabeele et al., 2010). Apoptosis is the best characterized programmed cell death mode; it plays critical roles in development, maintaining tissue homeostasis, shaping the immune repertoire, and restricting the progress of infections (Danial and Korsmeyer, 2004). This cell death mode can be initiated by two distinct but partially overlapping pathways: the extrinsic, receptor-mediated pathway and the intrinsic mitochondrial pathway (Salvesen and Riedl, 2008). In both cases, biochemical cascades triggered by extracellular ligands or intracellular damage led to the activation of caspases, which are a family of cysteine-dependent aspartate-specific proteases. These enzymes mediate most of the apoptotic program and some of them can be blocked by inhibitor of apoptotic proteins (IAPs; Scott et al., 2005). The mitochondrion is the central controlling site for the intrinsic apoptotic pathway because it harbors cytochrome c and second mitochondrion-derived activator of caspase (SMAC or DIABLO).
The release of cytochrome c into the cytosol leads to the assembly of a supramolecular complex known as the apoptosome, which initiates the caspase activation cascade (Riedl and Salvesen, 2007). In parallel, SMAC neutralizes the caspase-inhibitory activity of XIAP, thereby indirectly contributing to the maximal activation of the caspase cascade (Riedl and Salvesen, 2007). The release of these two apoptosis-initiating molecules is caused by perturbation of the integrity of the outer mitochondrial membrane (OMM), which is delicately regulated by members of the Bcl-2 protein family. Based on their roles in controlling apoptosis, Bcl-2 family proteins can be divided into two subsets: pro- and anti-apoptotic molecules. Members of this protein family can form homo- as well as heterodimers (Chipuk et al., 2010). Indeed, the formation of heterodimers between pro- and anti-apoptotic members, a process that alters the cellular ratios between these two subsets of proteins, determines at least in part the susceptibility of cells to a death signal (Cory and Adams, 2002). Members of the Bcl-2 family share two common features. First, they possess up to four conserved Bcl-2 homology (BH) domains, designated BH1, BH2, BH3, and BH4 (Chipuk et al., 2010); However, a number of pro-apoptotic Bcl-2 family proteins, such as Bid, Bim, Bad, and BNIP3 contain only the BH3 domain and are classified as the “BH3-only” subfamily (Chipuk et al., 2010). Second, most members of this protein family contain a carboxy-terminal hydrophobic domain, which in many cases is critical for their biological activities by membrane insertion and membrane remodeling (Lomonosova and Chinnadurai, 2008). Upon sensing cell stress caused by various insults such as DNA damage, cytokine deprivation, or infection, these BH3-only proteins trigger the insertion of the two pro-apoptotic, pore-forming proteins BAX and/or BAK, into the OMM (Chinnadurai et al., 2008), causing the release of the cytochrome c and SMAC. Members of the pro-survival proteins, including Bcl-2, BCL-XL, and MCL1, inhibit apoptosis by directly sequestering BAK, BAX, and BH3-only proteins to prevent permeabilization of OMM (Chipuk et al., 2010).
Regulation of Host Cell Death Pathways by L. Pneumophila
Apoptosis plays an important role in the defense against pathogens on the level of both the reaction of an individual host cell to an invading microorganism and the reacting immune system (Creagh et al., 2003). Accordingly, successful pathogens have evolved different but often equally effective mechanisms to manipulate host cell death pathways to benefit their proliferation. Such manipulation can be achieved by targeting the activity of one or more host proteins critical in each step of the apoptotic pathways. For example, many viruses code for proteins that specifically inhibit apoptosis of infected cells by directly interacting with pro-apoptotic members of the BH3-only proteins (Roulston et al., 1999; Everett and McFadden, 2002). Similarly, some obligate intracellular bacterial pathogens such as Chlamydia trachomatis and Rickettsia rickettsii actively inhibit apoptosis (Clifton et al., 1998; Fan et al., 1998). Of particular interest is that C. trachomatis prevents infected cells from undergoing apoptosis by specifically degrading members of the pro-death BH3-only proteins (Dong et al., 2005), probably by the Chlamydia protease-like factor (CPAF; Pirbhai et al., 2006).
Accumulating evidence indicates that L. pneumophila is able to manipulate host cell death pathways by targeting regulatory molecules with diverse mechanisms at different points of the signaling cascade. Earlier studies suggest that L. pneumophila actively induces apoptosis of infected cells via the activation of the executioner caspase, caspase-3 (Gao and Au Kwaik, 1999a,b; Molmeret et al., 2004). In permissive cell lines such as U937 or human peripheral blood monocytes, infection by L. pneumophila caused high level apoptosis within the first 3 h of bacterial uptake, and, in some cases, apoptotic cells reached 100% (Gao and Abu Kwaik, 1999b). However, a later study found that despite vigorous bacterial replication over a time span of 13 h, the proportion of apoptotic cells did not significantly increase in permissive macrophages, suggesting that in permissive host cells, L. pneumophila actively inhibits infected cells from undergoing apoptosis (Derre and Isberg, 2004). This notion was validated by the discovery of SdhA and SidF, two Dot/Icm substrates involved in the inhibition of host cell death. Mouse bone marrow macrophages infected with a sdhA mutant became apoptotic, displaying increased nuclear degradation, mitochondrial disruption, membrane permeability, and caspase activation, indicating a role for SdhA in inhibiting host cell death (Laguna et al., 2006). Interestingly, the requirement for SdhA is cell-type specific because the growth defect was less severe in the ameba Dictyostelium discoideum or the more permissive U937 cell-derived macrophages (Laguna et al., 2006). SdhA appears to be multifunctional because it also plays a key role in the suppression of type I interferons (IFN) response in macrophages infected by L. pneumophila (Monroe et al., 2009). Although less severe, cells infected by mutants lacking SidF also exhibited higher levels of apoptosis, which led to marginal but detectable defects in intracellular growth (Banga et al., 2007). SidF interacts and inhibits the cell death-inducing activity of BNIP3 and Bcl-rambo, two non-canonical pro-apoptotic members of Bcl-2 protein family (Banga et al., 2007; Figure 1). Both mitochondrial proteins, BNIP3 and Bcl-rambo appears to induce apoptosis by different mechanisms. BNIP3 appear to play a role in cellular response to stress (Chinnadurai et al., 2008). On the other hand, Bcl-rambo induces cell apoptosis specifically blocked by the caspase inhibitors, IAPs (Kataoka et al., 2001). Thus, it is attempting to speculate that SidF plays a role in making infected cells less sensitive to stress caused by L. pneumophila infection. Nevertheless, the activities of SdhA and SidF indicate that L. pneumophila inhibits host apoptosis by directly targeting host proteins involved in controlling the cell death pathways at the mitochondrion (Figure 1).
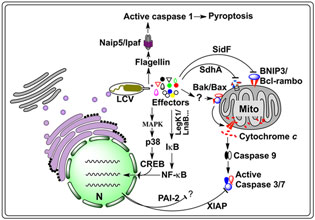
Figure 1. Host cell death pathways targeted by L. pneumophila. Internalized L. pneumophila translocates a large number of effectors into host cytosol via the Dot/Icm type IV secretion system. A yet unidentified set of effectors trigger an imbalance between the pro-death and pro-survival members of the Bcl-2 protein family, leading to the insertion of Bax/Bak into the mitochondrial out membrane, thus the release of cytochrome c and subsequent activation of the caspases 3 and 7. Another set of effectors, including LegK1 and LnaB, activate NF-κB, most likely by initiating the kinase cascade that ultimately causes phosphorylation and subsequent degradation of IκB, the inhibitor of NF-κB, leading to nucleus translocation of NF-κB and the induction of anti-apoptotic genes such as Xiap and Pai-2, whose product inhibits cell death by targeting caspases 3 and 7. Activation of the MAP kinase pathway by another set of unknown effectors can lead to similar effects. A third set of proteins such as SidF and SdhA, which inhibit host cell death by targeting pro-apoptotic proteins BNIP3 and Bcl-rambo or by unknown mechanisms. In non-permissive mouse macrophages, flagellin reached the cytosol probably via the Dot/Icm transporter is sensed by the NLR receptor Naip5, which together with Ipaf and the inflammasome activates caspase-1, leading to pyroptotic cell death. CREB, cAMP response element-binding protein; LCV, Legionella containing vacuole; MAPK, mitogen-activated protein kinases; Mito, mitochondrion; N, nucleus; Naip5, NLR family, apoptosis inhibitory protein 5; PAI-2, plasminogen activator inhibitor-2; XIAP, X-linked inhibitor of apoptosis protein.
Another layer of cell death inhibition mechanisms utilized by L. pneumophila is revealed by experiments designed to examine host gene expression profiles in response to low dose bacterial challenge (Abu-Zant et al., 2007; Losick et al., 2010). Several groups of genes known to be directly or indirectly involved in regulating host cell death were significantly induced, including stress response genes such as heat shock protein genes s and pro-survival members of the Bcl-2 protein family (Losick and Isberg, 2006; Abu-Zant et al., 2007). Interestingly, the most striking induction was observed in a collection of anti-apoptotic genes positively regulated at a transcriptional level by the regulator nuclear factor-κB (NF-κB; Losick and Isberg, 2006; Abu-Zant et al., 2007; Figure 1). Consistently, in human and permissive mouse macrophages, infection by wild type L. pneumophila led to nuclear translocation of NF-κB (Losick and Isberg, 2006; Abu-Zant et al., 2007). Thus, L. pneumophila is able to inhibit mammalian cell death by increasing the levels of anti-apoptotic proteins at transcriptional level. Two lines of evidence indicate that the induction of these anti-apoptotic genes is important for productive L. pneumophila infection. First, bacterial challenge of macrophages lacking one such anti-apoptotic gene, the plasminogen activator inhibitor-2 (PAI-2), led to significantly higher levels of apoptosis and reduction in bacterial replication (Losick and Isberg, 2006). Second, inhibition of nuclear translocation of NF-κB by genetic or pharmaceutical agents caused extensive cell death upon low dose bacterial challenge (Losick and Isberg, 2006). These anti-apoptotic proteins arrest host cell death by various mechanisms. Proteins like Bcl-2 can interact and inhibit the activity of several pro-death BH3-only proteins (Chipuk et al., 2010). On the other hand, IAPs such as XIAP can directly neutralize the activity of caspase-3 and -7 (Scott et al., 2005).
Interestingly, the observed NF-κB activation is involved in a signaling pathway independent of the Toll-like receptor (TLR) adaptor MyD88 and the cytoplasmic sensor Nod1, but is absolutely dependent upon the Dot/Icm secretion system (Losick and Isberg, 2006). These observations suggest the existence of Dot/Icm substrates capable of activating NF-κB. Consistent with this notion, LegK1 and LnaB, two Dot/Icm substrates with such activity have been identified in screenings using NF-κB responsive reporters and constructs that direct the expression of individual bacterial genes (Ge et al., 2009; Losick et al., 2010; Figure 1). Whereas the biochemical mechanisms of LnaB is unknown, LegK1, appears to directly target IκBα and other IκB family of inhibitors including p100 in the non-canonical NF-κB pathway by phosphorylation (Ge et al., 2009). Similar to most characterized Dot/Icm substrates, deletion of legK1 or lnaB resulted in little or only partial reduction in NF-κB activation. Consistently, such mutants did not exhibit defects in intracellular bacterial growth (Ge et al., 2009; Losick et al., 2010). Several reasons can account for the lack of growth defect phenotypes of these mutants. First, proteins such as SdhA and SidF that target host cell death proteins can provide the protection in these mutants. Second, other yet unidentified effectors may contribute to the activation of NF-κB during the infection by these mutants. This is very likely because functional redundancy has been observed in effectors targeting other cellular pathways important for L. pneumophila intracellular growth (Isberg et al., 2009). Third, as documented in a recent report, NF-κB activated by a non-canonical mechanism may also contribute to the induction of anti-apoptotic genes. In the study, Fontana et al. (2011) found that in conjunction with classic pathogen associated molecular pattern (PAMP) molecules, inhibition of host protein synthesis by several Dot/Icm substrates led to prolonged activation of NF-κB, thus strong induction of a set of inflammatory cytokines such as interleukin (IL)-23 and granulocyte–macrophage colony stimulating factor. Such activation was achieved by the failure to resynthesize IκB, the short-lived inhibitor of the NF-κB transcription factor in the presence of these protein synthesis inhibitors (Fontana et al., 2011). Finally, some substrates of the Dot/Icm transporter also potently activate the MAP kinase pathway, which could also lead to induction of the anti-apoptotic genes such as pai-2 (Shin et al., 2008). Given the high level of conservation in the MAPK pathway in eukaryotes, it is not unexpected that the activation of this pathway also occurs in the ameba D. discoideum (Li et al., 2009). Clearly, inhibition of host cell apoptosis by L. pneumophila is achieved by collective activities of various bacterial proteins; these proteins either directly or indirectly, reprogram the various cell death pathways to ensure maximal bacterial replication.
In contrast to the many lines of evidence directly supporting active inhibition of host cell death by L. pneumophila, evidence pointing to the induction of apoptosis by this bacterium mostly is indirect. The apoptotic phenotypes associated with host cells infected with mutants lacking one or more cell death inhibiting Dot/Icm substrates such as SdhA and SidF suggested the existence of effectors capable of inducing cell death (Laguna et al., 2006; Banga et al., 2007). Apoptosis induced by L. pneumophila is extremely apparent in specialized phagocytes such as dendritic cells. In these cells, infection by L. pneumophila induced a caspase-3-dependent apoptotic pathway that aborted intracellular bacterial replication in the early phase of infection (Nogueira et al., 2009; Figure 1). Interestingly, dendritic cells from mice deficient in Bak and Bax (Bak−/− Bax−/−) or mice overexpressing the pro-survival protein Bcl-2 are able to support intracellular bacterial infection without undergoing apoptosis (Nogueira et al., 2009), indicating that infection by L. pneumophila activates the mitochondrial pathway of apoptosis by inducing an imbalance between the pro-apoptotic and pro-survival members of Bcl-2 protein family. The activation of the mitochondrial apoptotic pathway also occurs in macrophages but at a much slower pace (Abu-Zant et al., 2005; Nogueira et al., 2009). The drastic differences between macrophages and dendritic cells in response to cell death stimuli such as those caused by L. pneumophila infection may be due to higher level or more active of the putative receptors or sensor proteins responsible for engaging the cell death signals from the bacterium in the latter cell type. It has been proposed that the sensitivity to pathogens exhibited by dendritic cells serves a protective role to the host by preventing infectious agents from using these cells as vehicles to reach deep tissues of the organism (Nogueira et al., 2009).
The induction of host cell death by bacterial toxins has been well documented. For example, Shiga toxins trigger apoptosis in many cell types, probably by inducing stress in the ER through inhibition of protein synthesis (Lee et al., 2008). Although a number of Dot/Icm substrates toxic to both yeast and mammalian cells have been identified, none of them has been shown to specifically induce host cell death. For example, at least five L. pneumophila proteins capable of inhibiting host protein synthesis have been reported. When overexpressed, these proteins are highly toxic to host cells (Belyi et al., 2006, 2008; Shen et al., 2009; Fontana et al., 2011). However, whether these proteins play roles in the induction of host cell death under infection conditions is unknown. It is worth noting that a mutant lacking all five genes failed to induce prolonged NF-κB activation, suggesting that these toxic proteins contribute to protect host cell death during infection (Fontana et al., 2011). Such outcomes clearly are opposite to the phenotypes observed by overexpressing these proteins in host cells. Nevertheless, since the Dot/Icm transporter but not bacterial replication is required for the activation of the apoptotic pathway (Abu-Zant et al., 2005; Nogueira et al., 2009), substrates of this transporter must be involved in the activation process. Either the activities of one or more Dot/Icm substrates or the “stress” caused by the collective impact of multiple effectors, or a combination of both can be the mechanism used by the bacterium for such activation. Interestingly, macrophages lacking Bak and Bax or overexpressing Bcl-2 still undergo extensive apoptosis upon being challenged by the sdhA mutant (Nogueira et al., 2009), suggesting that L. pneumophila is able to activate a cell death pathway independent of several critical components of the mitochondrial apoptotic pathway.
Besides the mitochondrial apoptotic pathway, L. pneumophila also induces pyroptosis, a form of inflammatory cell death in macrophages from mice harboring a functional Naip5 allele (Fortier et al., 2005). Naip5 is a member of the Nod-like receptor (NLR) family, and an important component of the cytosolic protein complexes called inflammasomes, which induce autoactivation of caspase-1 (Figure 1). The NLRs are considered functionally equivalent to the TLRs localized on the surface or within endosomes of immune cell (Davis et al., 2011), which recognize components of the pathogen called PAMPs such as bacterial flagellin, peptidoglycan, and nucleic acid variants (West et al., 2006). Caspase-1 induced pyroptosis, which is accompanied by the release of mature IL-1β and IL-18 and other cytokines, and is inherently proinflammatory (Fink and Cookson, 2006). The observation that flagellin deficient L. pneumophila mutants gained the ability to grow productively in macrophages expressing functional Naip5 without activating caspase-1-dependent cell death indicates that flagellin activates Naip5 in the cytoplasm (Molofsky et al., 2006; Ren et al., 2006). A subsequent study using a retroviral transduction to express flagellin directly in macrophages clearly showed that flagellin is necessary and sufficient for the pyroptosis induction via a pathway controlled by the Ipaf inflammasome (Lightfield et al., 2008). Consistently, macrophages from Naip5-deficient mice completely failed to activate caspase-1 and were able to support robust growth of wild type L. pneumophila, further indicating that flagellin-mediated pyroptosis induction absolutely requires a functional Naip5 (Lightfield et al., 2008). Thus, the induction of pyroptosis in immune cells from restrictive mice by L. pneumophila flagellin is “accidental,” but has provided an excellent model to dissect the host immune surveillance mechanisms (Vance, 2010).
Potential Benefits of L. Pneumophila-Induced Apoptosis
Clearly, in permissive mammalian cells, infection by L. pneumophila activates the classic mitochondrial apoptotic pathway and possibly other yet unrecognized cell death processes, leading to activation of several caspases, including caspases 3 and 7. Concurrently, the bacterium employs a combination of mechanisms to inhibit infected cells from fully executing the apoptotic cascade to allow productive bacterial replication. Whereas the benefit of inhibiting apoptosis by L. pneumophila is obvious, the benefit for activation this pathway is less clear. Activated caspases may participate in cellular processes in not directly related to cell death but important for the biogenesis of the LCV. For example, active caspase-3 appears to cleave Rabaptin-5, an Rab5 effector, and cell treated with caspase-3 inhibitor (DEVD-fmk) or the pan inhibitor of caspases (Z-VAD-fmk) abolished intracellular bacterial growth (Molmeret et al., 2004). However, in contrast to this observation, macrophages from permissive mice (A/J) lacking caspase-3 (Casp3−/−) still support robust intracellular bacterial growth (Zamboni et al., 2006; Nogueira et al., 2009). These discrepancies may result from the non-specificity of the caspase inhibitors or the different host cells used in these studies. Recently, Srikanth et al. (2010) showed that cleavage of bacterial effectors by caspase-3 is important for pathogenicity of Salmonella enterica serovar Typhimurium. It is possible that active caspases also participate in cellular processes that are not directly related to cell death but are important in the biogenesis of the LCV.
Concluding Remarks and Prospectives
A number of important questions remain outstanding in our understanding of the modulation of host cell death pathways by L. pneumophila. First, it is believed that amebae but not metazoan play a role in the evolution of L. pneumophila pathogenicity. However, NF-κB signaling and many components of apoptosis have now been documented to be targeted by proteins of this bacterium are not present in amebae cells. It is possible that L. pneumophila co-evolved with some as yet unrecognized lower metazoan. In support of this notion, two recent studies found that L. pneumophila can infect Caenorhabditis elegans (Brassinga et al., 2010; Komura et al., 2010), an organism with well-defined cell death pathways (Conradt, 2009). Alternatively, L. pneumophila can acquire genes specific for metazoan pathways from other more adapted mammalian pathogens which also are parasites of amebae (Moliner et al., 2010). Second, the nature of the bacterial signals that triggers the mitochondrial cell death pathway remains elusive. Particularly, the sdhA mutant appears to elicit a cell death pathway independent of the canonical mitochondrial pathway controlled by the Bcl-2 protein family (Nogueira et al., 2009). The identification of these proteins will undoubtedly facilitate the search of the host “receptors” that directly sense the bacterial signals. Research in this field aiming at characterizing both pro-survival and pro-death L. pneumophila proteins and their host targets will surely generate more exciting discoveries in years to come.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Studies in our laboratory are supported by grants R01AI069344, K02AI085403, and R21AI092043 and R56AI090142 from the National Institutes of Health (NIH).
References
Abu-Zant, A., Jones, S., Asare, R., Suttles, J., Price, C., Graham, J., and Kwaik, Y. A. (2007). Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell. Microbiol. 9, 246–264.
Abu-Zant, A., Santic, M., Molmeret, M., Jones, S., Helbig, J., and Abu Kwaik, Y. (2005). Incomplete activation of macrophage apoptosis during intracellular replication of Legionella pneumophila. Infect. Immun. 73, 5339–5349.
Banga, S., Gao, P., Shen, X., Fiscus, V., Zong, W. X., Chen, L., and Luo, Z. Q. (2007). Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl. Acad. Sci. U.S.A. 104, 5121–5126.
Belyi, Y., Niggeweg, R., Opitz, B., Vogelsgesang, M., Hippenstiel, S., Wilm, M., and Aktories, K. (2006). Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. U.S.A. 103, 16953–16958.
Belyi, Y., Tabakova, I., Stahl, M., and Aktories, K. (2008). Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol. 190, 3026–3035.
Brassinga, A. K., Kinchen, J. M., Cupp, M. E., Day, S. R., Hoffman, P. S., and Sifri, C. D. (2010). Caenorhabditis is a metazoan host for Legionella. Cell. Microbiol. 12, 343–361.
Chinnadurai, G., Vijayalingam, S., and Gibson, S. B. (2008). BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene 27(Suppl. 1), S114–S127.
Chipuk, J. E., Moldoveanu, T., Llambi, F., Parsons, M. J., and Green, D. R. (2010). The BCL-2 family reunion. Mol. Cell 37, 299–310.
Clifton, D. R., Goss, R. A., Sahni, S. K., van Antwerp, D., Baggs, R. B., Marder, V. J., Silverman, D. J., and Sporn, L. A. (1998). NF-kappa B-dependent inhibition of apoptosis is essential for host cellsurvival during Rickettsia rickettsii infection. Proc. Natl. Acad. Sci. U.S.A. 95, 4646–4651.
Conradt, B. (2009). Genetic control of programmed cell death during animal development. Annu. Rev. Genet. 43, 493–523.
Cory, S., and Adams, J. M. (2002). The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656.
Creagh, E. M., Conroy, H., and Martin, S. J. (2003). Caspase-activation pathways in apoptosis and immunity. Immunol. Rev. 193, 10–21.
Davis, B. K., Wen, H., and Ting, J. P. (2011). The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735.
Derre, I., and Isberg, R. R. (2004). Macrophages from mice with the restrictive Lgn1 allele exhibit multifactorial resistance to Legionella pneumophila. Infect. Immun. 72, 6221–6229.
Dong, F., Pirbhai, M., Xiao, Y., Zhong, Y., Wu, Y., and Zhong, G. (2005). Degradation of the proapoptotic proteins Bik, Puma, and Bim with Bcl-2 domain 3 homology in Chlamydia trachomatis-infected cells. Infect. Immun. 73, 1861–1864.
Ensminger, A. W., and Isberg, R. R. (2009). Legionella pneumophila Dot/Icm translocated substrates: a sum of parts. Curr. Opin. Microbiol. 12, 67–73.
Everett, H., and McFadden, G. (2002). Poxviruses and apoptosis: a time to die. Curr. Opin. Microbiol. 5, 395–402.
Fan, T., Lu, H., Hu, H., Shi, L., McClarty, G. A., Nance, D. M., Greenberg, A. H., and Zhong, G. (1998). Inhibition of apoptosis in Chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 187, 487–496.
Fink, S. L., and Cookson, B. T. (2005). Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916.
Fink, S. L., and Cookson, B. T. (2006). Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 8, 1812–1825.
Fontana, M. F., Banga, S., Barry, K. C., Shen, X., Tan, Y., Luo, Z.-Q., and Vance, E. V. (2011). Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathogen 7, e1001289. doi: 10.1371/journal.ppat.1001289
Fortier, A., Diez, E., and Gros, P. (2005). Naip5/Birc1e and susceptibility to Legionella pneumophila. Trends Microbiol. 13, 328–335.
Gao, L. Y., and Abu Kwaik, Y. (1999a). Activation of caspase 3 during Legionella pneumophila-induced apoptosis. Infect. Immun. 67, 4886–4894.
Gao, L. Y., and Abu Kwaik, Y. (1999b). Apoptosis in macrophages and alveolar epithelial cells during early stages of infection by Legionella pneumophila and its role in cytopathogenicity. Infect. Immun. 67, 862–870.
Ge, J., Xu, H., Li, T., Zhou, Y., Zhang, Z., Li, S., Liu, L., and Shao, F. (2009). A Legionella type IV effector activates the NF-κB pathway by phosphorylating the IkappaB family of inhibitors. Proc. Natl. Acad. Sci. U.S.A. 106, 13725–13730.
Hubber, A., and Roy, C. R. (2010). Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26, 261–283.
Isberg, R. R., O’Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24.
Kataoka, T., Holler, N., Micheau, O., Martinon, F., Tinel, A., Hofmann, K., and Tschopp, J. (2001). Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J. Biol. Chem. 276, 19548–19554.
Komura, T., Yasui, C., Miyamoto, H., and Nishikawa, Y. (2010). Caenorhabditis elegans as an alternative model host for Legionella pneumophila, and protective effects of Bifidobacterium infantis. Appl. Environ. Microbiol. 76, 4105–4108.
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N., and Isberg, R. R. (2006). A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18745–18750.
Lee, S. Y., Lee, M. S., Cherla, R. P., and Tesh, V. L. (2008). Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 10, 770–780.
Li, Z., Dugan, A. S., Bloomfield, G., Skelton, J., Ivens, A., Losick, V., and Isberg, R. R. (2009). The amoebal MAP kinase response to Legionella pneumophila is regulated by DupA. Cell Host Microbe 6, 253–267.
Lightfield, K. L., Persson, J., Brubaker, S. W., Witte, C. E., von Moltke, J., Dunipace, E. A., Henry, T., Sun, Y. H., Cado, D., Dietrich, W. F., Monack, D. M., Tsolis, R. M., and Vance, R. E. (2008). Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178.
Lomonosova, E., and Chinnadurai, G. (2008). BH3-only proteins in apoptosis and beyond: an overview. Oncogene 27(Suppl. 1), S2–S19.
Losick, V. P., Haenssler, E., Moy, M. Y., and Isberg, R. R. (2010). LnaB: a Legionella pneumophila activator of NF-kappaB. Cell. Microbiol. 12, 1083–1097.
Losick, V. P., and Isberg, R. R. (2006). NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189.
Moliner, C., Fournier, P. E., and Raoult, D. (2010). Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol. Rev. 34, 281–294.
Molmeret, M., Zink, S. D., Han, L., Abu-Zant, A., Asari, R., Bitar, D. M., and Abu Kwaik, Y. (2004). Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella-containing phagosome. Cell. Microbiol. 6, 33–48.
Molofsky, A. B., Byrne, B. G., Whitfield, N. N., Madigan, C. A., Fuse, E. T., Tateda, K., and Swanson, M. S. (2006). Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104.
Monroe, K. M., McWhirter, S. M., and Vance, R. E. (2009). Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5, e1000665. doi: 10.1371/journal.ppat.1000665
Nogueira, C. V., Lindsten, T., Jamieson, A. M., Case, C. L., Shin, S., Thompson, C. B., and Roy, C. R. (2009). Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS Pathog. 5, e1000478. doi: 10.1371/journal.ppat.1000478
Pirbhai, M., Dong, F., Zhong, Y., Pan, K. Z., and Zhong, G. (2006). The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 281, 31495–31501.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Riedl, S. J., and Salvesen, G. S. (2007). The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 8, 405–413.
Roulston, A., Marcellus, R. C., and Branton, P. E. (1999). Viruses and apoptosis. Annu. Rev. Microbiol. 53, 577–628.
Scott, F. L., Denault, J. B., Riedl, S. J., Shin, H., Renatus, M., and Salvesen, G. S. (2005). XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 24, 645–655.
Shen, X., Banga, S., Liu, Y., Xu, L., Gao, P., Shamovsky, I., Nudler, E., and Luo, Z. Q. (2009). Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell. Microbiol. 11, 911–926.
Shin, S., Case, C. L., Archer, K. A., Nogueira, C. V., Kobayashi, K. S., Flavell, R. A., Roy, C. R., and Zamboni, D. S. (2008). Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4, e1000220. doi: 10.1371/journal.ppat.1000220
Srikanth, C. V., Wall, D. M., Maldonado-Contreras, A., Shi, H. N., Zhou, D., Demma, Z., Mumy, K. L., and McCormick, B. A. (2010). Salmonella pathogenesis and processing of secreted effectors by caspase-3. Science 330, 390–393.
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714.
West, A. P., Koblansky, A. A., and Ghosh, S. (2006). Recognition and signaling by toll-like receptors. Annu. Rev. Cell. Dev. Biol. 22, 409–437.
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and `Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Keywords: apoptosis, caspase, Bcl-2 protein, infection, effector, type IV secretion
Citation: Luo Z.-Q (2011) Striking a balance: modulation of host cell death pathways by Legionella pneumophila. Front. Microbio. 2:36. doi: 10.3389/fmicb.2011.00036
Received: 15 January 2011;
Paper pending published: 01 February 2011;
Accepted: 10 February 2011;
Published online: 23 February 2011.
Edited by:
Carmen Buchrieser, Pasteur Institute, FranceReviewed by:
Dario S. Zamboni, Universidade de São Paulo, Brazil;Elizabeth L. Hartland, The University of Melbourne, Australia
Copyright: © 2011 Luo. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Zhao-Qing Luo, Department of Biological Sciences, Purdue University, 915 West State Street, West Lafayette, IN 47907, USA. e-mail:bHVvekBwdXJkdWUuZWR1