

 Kevin Visconti2,3 and Gary K. Schoolnik2,3
Kevin Visconti2,3 and Gary K. Schoolnik2,3
- 1 Department of Microbiology, School of Medicine, University of Colorado Denver, Aurora, CO, USA
- 2 Division of Infectious Diseases and Geographic Medicine, Department of Medicine, Stanford Medical School, Stanford, CA, USA
- 3 Department of Microbiology and Immunology, Stanford Medical School, Stanford, CA, USA
The bacteriostatic and bactericidal effects and the transcriptional response of Mycobacterium tuberculosis to representative oxidative and nitrosative stresses were investigated by growth and survival studies and whole genome expression analysis. The M. tuberculosis reaction to a range of hydrogen peroxide (H2O2) concentrations fell into three distinct categories: (1) low level exposure resulted in induction of a few highly sensitive H2O2-responsive genes, (2) intermediate exposure resulted in massive transcriptional changes without an effect on growth or survival, and (3) high exposure resulted in a muted transcriptional response and eventual death. M. tuberculosis appears highly resistant to DNA damage-dependent, mode-one killing caused by low millimolar levels of H2O2 and only succumbs to overwhelming levels of oxidative stress observed in mode-two killing. Nitric oxide (NO) exposure initiated much the same transcriptional response as H2O2. However, unlike H2O2 exposure, NO exposure induced dormancy-related genes and caused dose-dependent bacteriostatic activity without killing. Included in the large shared response to H2O2 and NO was the induction of genes encoding iron–sulfur cluster repair functions including iron acquisition. Stress regulons controlled by IdeR, Sigma H, Sigma E, and FurA comprised a large portion of the response to both stresses. Expression of several oxidative stress defense genes was constitutive, or increased moderately from an already elevated constitutive level, suggesting that bacilli are continually primed for oxidative stress defense.
Introduction
During the course of infection, Mycobacterium tuberculosis (Mtb) must cope with a variety of host-mediated stresses, in particular, the antibacterial properties of macrophages. Macrophages produce antimicrobial reactive oxygen and nitrogen species (ROS and RNS) via NADPH oxidase (NOX2/gp91phox) and inducible nitric oxide synthase (iNOS; Ehrt and Schnappinger, 2009).
In vitro experiments demonstrate no effect on Mtb intracellular growth in Phox-deficient macrophages, suggesting wild-type Mtb is resistant to the inhibitory effects of the macrophage oxidative burst (Chan et al., 1992). Mice deficient in Phox are partially inhibited in their ability to control Mtb growth in an aerosolized infection model before the onset of specific immunity (Cooper et al., 2000), suggesting a role for ROS in the control of Mtb early in the infectious process. By contrast, iNOS deficient mice, which lack inducible NO production, are highly susceptible to Mtb infection (Yang et al., 2009). In addition, treatment of Mtb-infected mice with the iNOS inhibitors given during acute or chronic infection causes mice to rapidly succumb to the disease (Flynn et al., 1998; Nathan, 2002). Furthermore, inhibition of the iNOS modulating cytokine, TNFα, leads to reactivation of Mtb from persistent murine infection (Mohan et al., 2001) and reactivation of latent human disease (Gardam et al., 2003). Although the role of NO in human tuberculosis remains unsettled evidence supporting its importance has come from a variety of areas (Nathan and Shiloh, 2000) including the demonstration that human granulomas contain iNOS, endothelial-NOS, and nitrotyrosine, a compound whose accumulation indicates production of NO (Nathan, 2002). Additionally, the ability of human alveolar macrophages to kill Mtb is dependent on the activity of iNOS, and the human macrophages taken from healthy subjects latently infected with Mtb produce NO, controlling the growth of the bacteria (Yang et al., 2009). The presence of NO within human granulomas could contribute to host resistance since in vitro experiments demonstrate direct RNS-mediated bacteriostatic (Firmani and Riley, 2002; Ouellet et al., 2002; Voskuil et al., 2003) and bactericidal activity (Nathan, 2002). Mice deficient in both phox and iNOS are much more susceptible to Mtb infection than either mutant alone, which would indicate that RNS and ROS protect the host in a partially redundant fashion (Shiloh and Nathan, 2000). However, there was little overlap seen between response to either NO or H2O2 when cultures exposed to either stress was analyzed by 2-dimensional gel electrophoresis and compared to unexposed cultures (Garbe et al., 1996).
Mtb utilizes a range of mechanisms to defend against ROS and RNS including direct scavenging of the reactive species and the repair and protection of proteins and DNA (Ehrt and Schnappinger, 2009). The resistance of Mtb to ROS is partly due to the thick Mtb cell wall containing lipoarabinomannan (LAM) and cyclopropanated mycolic acids, as well as phenolic glycolipid I (PGL-1), which act as potent scavengers of oxygen radicals (Flynn and Chan, 2001). In addition, Mtb produces the ROS scavenging enzymes catalase (KatG; Manca et al., 1999; Ng et al., 2004), superoxide dismutases (SodA and C; Jackett et al., 1978; Piddington et al., 2001; Tullius et al., 2001), and the peroxidase and peroxynitrite reductase complex of AhpC, AhpD, SucB(DlaT), and Lpd (Bryk et al., 2002). A dlaT mutant is also hyper-susceptible to RNS (Shi and Ehrt, 2006), and cells with a deletion mutant in ahpC are more susceptible to ROS (Springer et al., 2001; Master et al., 2002). The thioredoxin/thioredoxin reductase systems from Mtb also contributes to the reduction of peroxides (Zhang et al., 1999). The low-molecular-weight antioxidant mycothiol takes the place of glutathione found in many bacteria and is important in maintaining a reduced environment and resistance to oxidative stress (Buchmeier et al., 2006). The sulfur assimilation pathway plays a role in oxidative and nitrosative stress as a cysH mutant deficient in cysteine and methionine synthesis is more sensitive to these stresses (Senaratne et al., 2006). Mtb DNA is directly protected from ROS by the DNA binding protein, Lsr2 (Colangeli et al., 2009). Additional RNS resistance mechanisms include the catalytic detoxification of NO achieved by the truncated hemoglobin (trHbN) in an oxygen-dependant reaction (Ouellet et al., 2002; Pathania et al., 2002). The presence of trHbN protects aerobic respiration from inhibition by NO in Mycobacterium smegmatis (Pathania et al., 2002). Deletion of genes that encode methionine sulfoxide reductase (msrA; St John et al., 2001; Lee et al., 2009), the Mtb proteasome (prcBA; Darwin et al., 2003; Gandotra et al., 2007), nucleotide excision repair (uvrB; Darwin et al., 2003), and F-420 biosynthesis (fbiC; Darwin et al., 2003) are also hyper-susceptible to RNS. Alpha crystalline (acr), and bacterioferritin (bfrB), as well as the DosR regulon are upregulated upon addition of NO (Garbe et al., 1999; Ohno et al., 2003), but the role of these proteins in NO protection is little understood.
Defense against oxidative stress in Escherichia coli is largely controlled by the OxyR and SoxR regulators (Imlay, 2008). Genes regulated by OxyR include the catalase gene, katG, for detoxification of H2O2, and the alkyl hydroperoxide reductase genes, ahpCF, for detoxification of alkyl peroxides. Mtb possesses many of the same genes that encode ROS defense mechanisms in other bacteria, including katG (catalase), sodA, (superoxide dismutase), ahpCDE (alkyl peroxide reductase), thioredoxin, and thioredoxin reductase genes. However, the regulation of these genes by Mtb differs markedly from their regulation in enteric bacteria. It was originally proposed that Mtb is capable of only a limited transcriptional response to oxidative and nitrosative stress compared to enteric bacteria, due to an incomplete repertoire of regulatory factors (Deretic et al., 1995; Sherman et al., 1995; Zahrt and Deretic, 2002). Previous work done by Garbe et al. (1996) would indicate that there is very little transcriptional response to H2O2 in Mtb as demonstrated on 2-dimensional gel electrophoresis, and genes involved in detoxifying oxidative species are not upregulated early in a mouse model when the oxidative burst is occurring (Shi et al., 2008). In particular, the gene encoding OxyR has sustained multiple mutations, and thus this primary regulator of the oxidative stress response in other bacteria is a pseudogene in Mtb and other closely related species in the Mtb complex (Deretic et al., 1995; Sherman et al., 1995). By contrast, a functional copy of oxyR is present in several other mycobacteria including M. leprae, M. avium, and M. marinum (Pagan-Ramos et al., 1998). SoxR regulates expression of sodA and other ROS responsive genes in E. coli, but no apparent homolog of soxR exists in the Mtb genome (Cole et al., 1998). Alternative Mtb transcriptional regulators have been implicated in ROS defense: FurA (Milano et al., 2001; Pym et al., 2001; Zahrt et al., 2001), IdeR (Dussurget et al., 1996; Rodriguez et al., 2002), CarD (Stallings et al., 2009), and Sigma H (Fernandes et al., 1999; Raman et al., 2001), as well as genes involved in iron acquisition (Jang et al., 2009).
Although NO and H2O2 both play a role in controlling Mtb infection, not much is understood on how the bacteria transcriptionally respond to different concentrations of these two stimuli. An increased understanding of the interaction between Mtb and chemicals used by the host to control it could shed light on which genes are important for bacterial persistence mechanisms. In order to determine how Mtb is differentially affected by ROS and RNS, we designed experiments to examine the concentration-dependent effects of a broad range of doses of ROS and RNS on Mtb growth and survival. We also determined the transcriptional response of Mtb to H2O2 and NO at these same doses.
Materials and Methods
Culture Conditions
Mtb clinical isolate isolated from human sputum, 1254 (American Type Culture Collection 51910), 7H9 medium (supplemented with bovine serum albumin, NaCl, glucose, and glycerol), 250 ml vented tissue culture flasks positioned upright, 90 rpm shaking, and a starting culture density of OD 0.15 were used, unless otherwise indicated. RNA samples isolated from OD 0.15 cultures of the Mtb strain being assayed on the same day were used for the reference sample in each experiment. Cells were collected with a 4-min centrifugation step and frozen on dry ice.
H2O2 and NO Exposure
Mtb cultures were exposed to a single doses of H2O2 or the NO donors DETA/NO (diethylenetriamine/nitric oxide adduct) or spermidine-NONOate (Sigma Co.). Doses of 0.0, 0.05, 0.5, 1.0, 5, 10, 50, and 200 mM H2O2; 0.005, 0.05, 0.5, 1.0, and 5.0 mM DETA/NO; and 5 mM spermidine-NONOate were used. Stock solutions of H2O2 were prepared in H2O and NO donors were suspended in 0.01 N NaOH. Optical density 600 nm and colony forming units (CFU) were monitored after addition of stress. Cells were sampled 40 min or 4 h following H2O2 and DETA/NO addition and total RNA was isolated and compared by microarray to RNA from untreated cells but prepared at the same time and from the same parent culture. Three independent experiments were conducted for each H2O2 and DETA/NO concentration.
RNA Isolation
RNA isolation and microarray analysis were performed as previously described (Wilson et al., 2001). Briefly, cell pellets were suspended in 1 ml Trizol reagent (Gibco-BRL) and transferred to 2 ml screw cap tubes containing 0.5 ml 0.1 mm diameter zirconia/silica beads (BioSpec Products). Three 30 s pulses in a bead beater disrupted cells. Tubes were placed in ice between bead beating steps. Cell debris was separated by a 55-s centrifugation. The supernatant was transferred to 2 ml Heavy Phase Lock Gel I tubes (Eppendorf), containing 300 μl of chloroform, and inverted rapidly for 15 s and incubated 2 min with periodic inversion. Samples were centrifuged 5 min and the aqueous phase was added to 270 μl of isopropanol, followed by addition of 270 μl of (0.8 M Na citrate, 1.2 M NaCl). Samples were incubated 10 min to overnight at 4°C and centrifuged for 15 min at 4°C. The RNA pellets were washed with 1 ml 75% ethanol, centrifuged 5 min and air-dried for 20 min. Following suspension of the RNA pellets in 90 μl of water, 10 μl of DNase I 10× buffer and 6 units of DNase I (Ambion) were added and the samples were incubated for 30 min. Final purification of RNA was by RNeasy column (Qiagen).
cDNA Labeling and Microarray Hybridization
Both a PCR gene product microarray and a 70-mer oligonucleotide-based microarray (tuberculosis oligonucleotide set [Qiagen]) were used. Labeled cDNA was prepared as follows: 1.5 μg of total RNA and 4.4 μg of random oligonucleotide hexamers were incubated 2 min at 98°C, cooled on ice, combined with Stratascript RTase buffer, 0.5 mM dATP, dGTP, and dCTP, 0.02 mM dTTP, 1.5 nmol Cy3 or Cy5-dUTP (Amersham), and 1.8 μl Stratascript RTase (Stratagene) in a total volume of 25 μl and incubated 10 min at 25°C followed by 90 min at 42°C. Labeled cDNA was diluted in 400 μl TE and purified by microcon-10 (Amicon) filtration. Ten microliter hybridization solution (labeled cDNA, 5 μg tRNA, 3.8× SSC, 0.27% SDS) was sealed under a coverslip with rubber cement and hybridized overnight at 65°C for the DNA microarray. Oligonucleotide microarrays were first prehybridized for 1 h in 5X SSC, 1% BSA, and 0.1% SDS and washed with H2O and isopropanol. Following the prehybridization, 10 μl hybridization solution (labeled cDNA, 5 μg tRNA, 2× SSC, 25% formamide, 0.1% SDS) was hybridized overnight at 54°C. Slides were washed once for 2 min in 0.05% SDS, 1× SSC, washed twice for 2 min in 0.06× SSC, centrifuged at 600 rpm for 2 min, and were immediately scanned.
Microarray Data Analysis
Microarrays were scanned using a GenePix 4000A (Axon Instruments). The intensities of the two dyes at each spot were quantified using ScanAlyze (Michael Eisen1). All gene specific spots on the microarray other than those whose induction ratio was in the top or bottom 5% were used to normalize the intensities of Cy3 and Cy5 from each spot. After Cy3 and Cy5 channel normalization we eliminated large percentage fluctuations in low background spot values by adjusting low signal intensity spots to a minimum noise value. The noise value for each channel was determined by calculating the average intensity value for the 20% lowest intensity spots, and then every value below this average noise value was raised to the noise value. Microarray-determined ratios were calculated from three biological replicates and one to two microarrays for each biological replicate. Significance analysis of microarrys (SAM)2 was used to determine statistically significant regulated genes under H2O2 and NO stress (Tusher et al., 2001). Genes included in Table A1 in Appendix conformed to a highly stringent selection criteria. They exhibited at least a fourfold induction or repression ratio in at least one condition and had a SAM corresponding false discovery q value of zero.
Results
Effect of H2O2 and NO on Growth and Viability
Mtb clinical isolate 1254, growing in mid-logarithmic phase (optical density, OD, of 0.15), was exposed to a range of initial H2O2 concentrations from 50 μM to 200 mM (Figure 1). Culture OD was monitored for 72 h as a measure of growth, and CFU were determined at 4 and 24 h as a measure of cell viability. Exposure to 0.05, 0.5, and 5.0 mM H2O2 had no effect on culture growth or viability. A 10-mM exposure caused only a slight growth defect, while exposure to 50 and 200 mM resulted in a significant decrease in culture OD and complete sterilization of the cultures by 24 h. A small fraction of bacilli were still viable (∼0.1%) 4 h post 50 mM H2O2 exposure, however, by 24 h no colonies were recovered on petri plates. OD600 measurements did not reach its minimum value until 72 h.
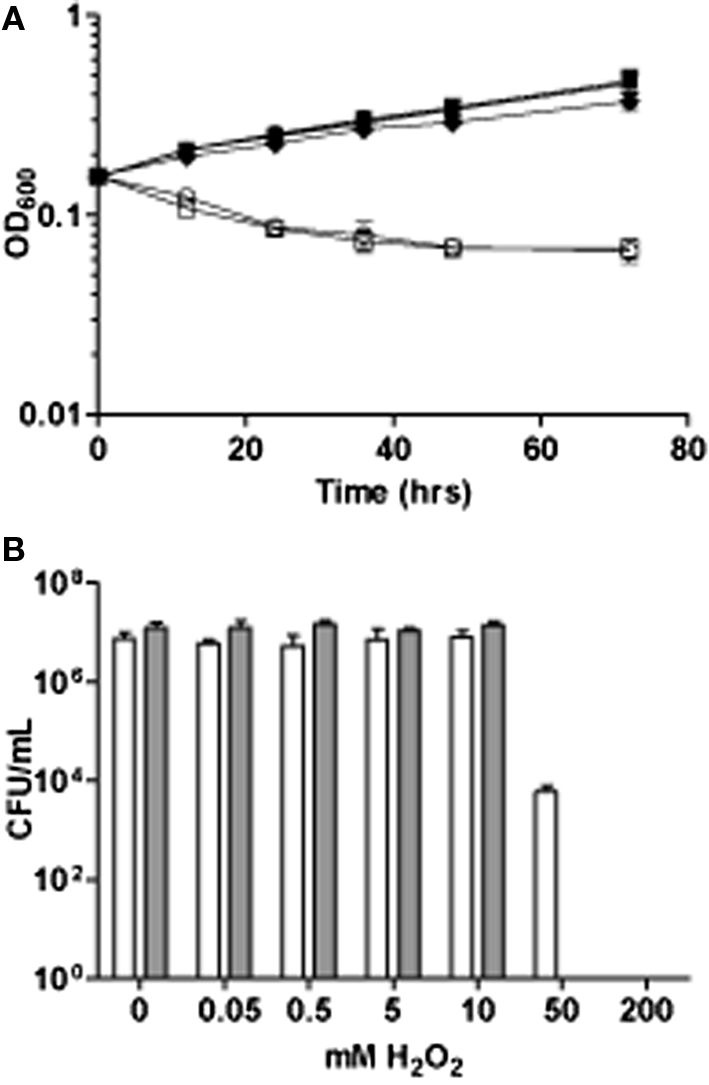
Figure 1. The effect of H2O2 stress on growth and survival. (A) Growth of Mtb for 72 h post exposure to H2O2 Filled circles, H2O control, filled squares, 0.05; filled triangles 0.5, filled downward triangles, 5.0: filled diamonds, 10.0; open circles 50; and open squares 200 mM H2O2(B) Survival of Mtb monitored by colony forming units 4 h, white bars, and 24 h, gray bars, hours post exposure to a range of H2O2 doses between 0.05 and 200 mM.
Mtb strain 1254 was also exposed to a range of concentrations between 5 μM and 5 mM of the slow release NO donor (DETA/NO: diethylenetriamine/nitric oxide adduct; Sigma Co.). DETA/NO is reported to release NO with a half-life of 20 h at pH 7.0. We determined the half-life of DETA/NO to be 5.5 h under our standard growth conditions (Voskuil et al., 2003). Exposure to 5 and 50 μM DETA/NO had no effect on culture growth, whereas 0.5, 1.0, and 5.0 mM DETA/NO had a dose-dependent, long-term effect on culture growth (Figure 2A). Previously, we demonstrated that exposure to intermediate levels of NO (0.5 and 1.0 mM DETA/NO) results in a 12- to 24-h temporary cessation of growth (Voskuil et al., 2003). Growth resumed after depletion of the NO donor (∼18 h for 0.5 mM DETA/NO); however, the resumptive growth rate was less than maximal for the medium based on the growth rate of cultures exposed to non-inhibitory concentrations of NO.
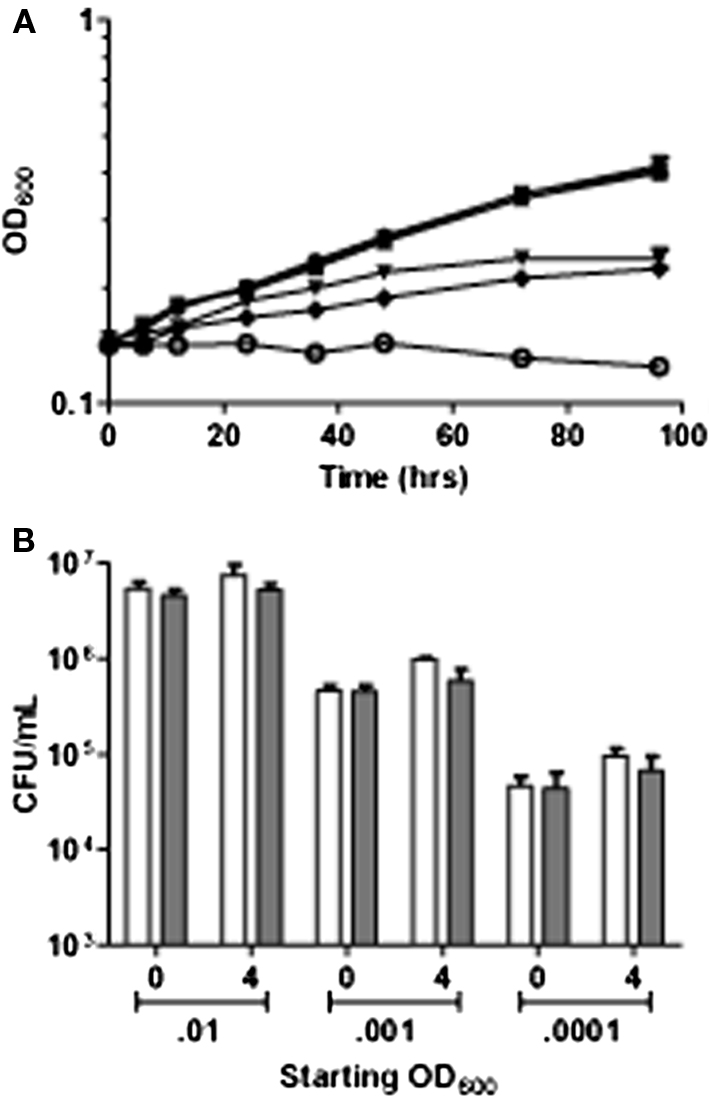
Figure 2. The effect of NO stress on growth and survival. (A) Growth of Mtb for 96 h post exposure to slow release NO donor DETA/NO. Filled circles, 0.01 N NaOH control; filled squares, 0.005: filled triangles 0.05; filled downward triangles, 0.5; filled diamonds, 1.0; and open circles 5 mM DETA/NO (B) Survival of Mtb over a range of low starting cell densities (0.01, 0.001, and 0.0001 OD) monitored by colony forming units. White bars indicate 0.01 N NaOH controls and gray bars indicate exposure to 5 mM of the rapid release NO donor spermidine-NONOate Time points were taken before addition of NO donor (0) and after 4 h of exposure to NO donor (4).
Exposure to all NO concentrations had little effect on cell viability, ascertained by viable plate counts, assayed 4 and 24 h post NO addition. A slight decrease in CFU was seen in the 1- and 5-mM DETA/NO exposed cultures after 24 h, data published previously (Voskuil et al., 2003). However, this could be attributed to lower growth rates in cultures exposed to the two highest concentrations of DETA/NO rather than cell death (Voskuil et al., 2003). To further investigate the bacteriostatic activity of NO, we exposed low cell numbers to high levels of the rapid NO donor Spermidine-NONOate. Viability was not affected even after exposure of bacterial cultures at OD values of 0.01, 0.001, and 0.0001 to 5 mM Spermidine-NONOate resulting in delivery of 10 mM NO (Figure 2B). Therefore, exposure to NO under standard laboratory conditions mediates a dose-dependent bacteriostatic effect with little apparent killing even at levels much higher than required for bacteriostasis.
Expression Patterns Induced by H2O2
Changes in mRNA expression patterns after exposure to H2O2 were monitored by oligonucleotide DNA microarray analysis. Mtb cultures were grown and exposed to H2O2 as described in Section “Materials and Methods.” The global expression patterns of genes that respond to H2O2 are depicted in Table A1. At 0.05 and 0.5 mM H2O2 few gene expression changes were observed. The furA–katG–rv1907c three gene operon and genes involved in iron acquisition were the most sensitive to low concentrations of H2O2 (Table A1). At intermediate concentrations of H2O2 (5 and 10 mM) a dramatic effect on gene expression was observed with hundreds of genes induced and repressed. Interestingly, at lethal concentrations of H2O2 (50 and 200 mM) a significantly muted transcriptional response was observed (Table A1). Many genes induced by intermediate concentrations of H2O2 were either not induced or were repressed during exposure to lethal concentrations of H2O2. Conversely, some genes were induced to a greater degree at the lethal concentrations.
Expression Patterns Induced by NO
NO-induced gene expression changes were monitored by two microarray formats: a PCR product (amplicon) DNA microarray; and, an oligonucleotide microarray. Both formats produced similar qualitative regulation ratios. We examined exposure to 0.5 mM DETA/NO by both platforms and similar results were obtained; however, the oligonucleotide platform produced on average an increased level of regulation (Table A1). The most striking difference between the H2O2 and NO expression profiles was the dramatic induction of genes controlled by the dormancy survival regulator, DosR (Figure 3A; Table S1 in Supplementary Material; Voskuil et al., 2003). The expression of many of these genes using RNA from the same experiments as those used in this paper has been previously confirmed under the same conditions via qRT-PCR (Voskuil et al., 2003). 0.05 mM of DETA/NO resulted in strong induction of the DosR regulon, but did not slow growth. Exposure to 0.05 mM DETA/NO resulted in the differential regulation of only a few genes which are not part of the DosR regulon. NO serves a signaling function which leads to the induction of the DosR regulon, even before an effect on growth (i.e., bacteriostasis) is evident. At concentrations of DETA/NO above 0.05 mM, in addition to the DosR regulon induction and bacteriostatic response seen at lower doses, a more typical oxidative stress gene expression pattern emerged. Expression data demonstrated that a large number of genes induced by NO concentration >0.05 mM were also induced by H2O2 (Table A1).
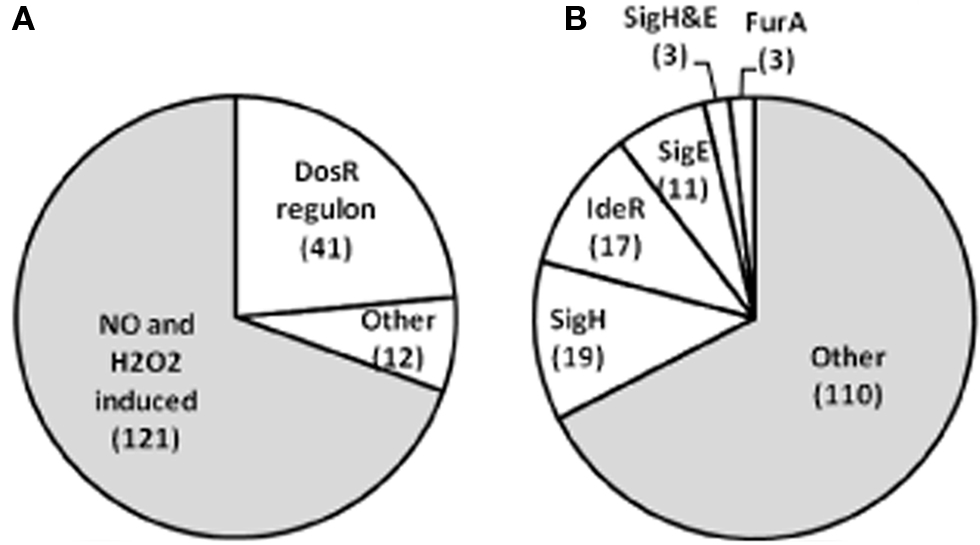
Figure 3. Distribution of genes regulated in response to NO and H2O2. (A) 174 were induced at least 2-fold after 40 min exposure to 0.005, 0.05, 0.5, 1.0, or 5.0 mM DETA/NO and fulfilled the criteria for inclusion in Table A1. Forty-one of the 174 genes are members of the previously published DosR regulon (Voskuil et al., 2003) and are therefore not included in Table A1. A majority of genes were regulated by both NO and H2O2 and 53 genes were regulated solely by NO including the 41 the DosR regulon genes (B) 163 genes from Table A1 were induced by H2O2, of these 42 were not induced by NO. Four transcriptional regulators: SigH (Imlay, 2003), IdeR (Rodriguez et al., 2002), SigH (Park and Imlay, 2003), and FurA (this study and Pym et al., 2001; Zahrt et al., 2001) control genes that encompass 33% of the H2O2 response.
The ROS and RNS Expression Profile
In the absence of a functional OxyR, three other transcriptional regulators (SigH, IdeR, and DosR) appear to have a large role in shaping the response to ROS and RNS in Mtb. Expression of sigH is induced up to 22-fold by 50 mM H2O2 and sixfold by 5 mM DETA/NO. ideR expression is moderately induced by both stresses as measured by the oligonucleotide microarray (3.2-fold by 10 mM H2O2 and 3.4-fold by 0.5 mM DETA/NO). However, these ideR induction ratios were below the cutoff required for inclusion in Table A1. None of the DosR regulon genes was regulated in response to H2O2 (Table S1). The DosR-regulated genes were not included in Table A1 as their induction by NO has been reported (Voskuil et al., 2003). Genes controlled by SigH, IdeR, SigE, and FurA account for a large number of the genes induced by H2O2 and NO (Figure 3B). However, 14 additional regulators are also induced by H2O2 at least fourfold and 7 regulators by NO; including the global transcription factor Sigma B (Table A1).
Differential Expression of Genes Which Confer Resistance to ROS and/or RNS
The Mtb genome encodes several proteins known to be involved with or implicated in resistance to ROS and/or RNS (Imlay, 2008; Ehrt and Schnappinger, 2009). Expression of some of these genes was found to be highly induced in response to H2O2 and NO. These include the genes encoding catalase (katG), thioredoxin reductase (trxB2), and the thioredoxin genes (thiX, trxB1, and trxC). Maximum induction of katG, trxB2, thiX, trxB1, and trxC by exposure to 5–50 mM H2O2, as measured by the oligonucleotide microarray, was 12, 30, 16, 58, and 13-fold, respectively. Maximum induction of the same genes by exposure to 0.5–5 mM DETA/NO, as measured by the amplicon microarray, was 3.2, 2.8, 3.2, 9.3, and 2.1-fold, respectively. Other genes predicted or proven to be involved with ROS and/or RNS resistance were not highly differentially expressed and thus not included in Table A1. ahpC did not fit the strict criteria for inclusion in Table A1, although it had a maximum induction level of 3.2 by NO, and is important for resistance against oxidative stress in Mtb (Springer et al., 2001; Master et al., 2002).
Even though many genes implicated in ROS and RNS stress resistance were not highly induced by H2O2 or NO, their basal expression levels (as measured by the relative intensity of the microarray signal) were high even in the absence of exogenous ROS or RNS. In particular, the average micorarray signal intensities from both the amplicon and oligonucleotide microarrays were in the top 4, 12, 8, 11, 16, and 10% of all genes for sodA, sodC, katG, ahpD, ahpE, and ahpC, respectively.
A Common Oxidative and Nitrosative Stress Response
The breadth of the shared transcriptional response to H2O2 and NO suggests that a common set of mechanisms plays a role during adaptation to ROS and RNS (Table A1). The most pronounced of these are genes controlled by the global transcription regulators IdeR and Sigma H (Figure 3B). In addition, a large number of the genes which are negatively regulated by both H2O2 and NO encode general housekeeping functions. The most repressed genes include 12 ribosomal protein genes and nuoD and nuoE, encoding NADH dehydrogenase subunits (Table A1), indicating a general slowing of growth.
NO and H2O2 both strongly induce genes from rv1460 to rv1466; the annotated functions of these genes suggest that RNS and ROS stimulate a concerted effort to replace or repair iron–sulfur clusters. Exposure to even low levels of H2O2 or NO-induced genes involved in iron acquisition (mbt cluster), while the bfrB gene, encoding the iron-storage protein bacterioferritin, was repressed. After exposure to higher concentrations of H2O2 or NO, the induction of the mbt
cluster was consistently less than that observed during exposure to intermediate levels of these stresses.
The H2O2-Specific Response
A notable difference between the response of Mtb to H2O2 and NO was the lack of a DNA damage response observed after initial NO exposure. By contrast, H2O2 induced several genes involved in DNA repair: recA, radA, and alkA. alkA encodes a methylated-DNA–protein–cysteine methyltransferase, which repairs alkylated and methylated guanine and thymine residues by a suicide reaction where the alkyl or methyl group is transferred to a cysteine in the enzyme (Samson, 1992). rv3201c and rv3202c were also induced by H2O2; these genes encode subunits of an ATP-dependent helicase homologous to UvrD helicases, which are also involved in DNA repair. Several transposase and resolvase genes from insertion sequences and phages, were also induced by H2O2 (rv0605, rv0606, rv0829, rv0850, rv2885c, rv2978c, rv2979c, and rv3827c); some of these were previously shown to respond to DNA damage (Boshoff et al., 2003; Rand et al., 2003). Boshoff et al. (2003) also demonstrated induction of DNA damage responsive genes (including recA, radA, alkA, rv3202c) by DNA damage from by UV irradiation and mitomycin.
Although most of the above mentioned genes were not induced by a 40-min exposure to NO, most were weakly induced after 4 h of NO exposure, suggesting a moderate level of DNA damage after extended NO exposure. Interestingly, these genes were not induced (or were induced significantly less) at lethal levels of H2O2. For example, recA was induced 4.4- and 4.1-fold by 5 and 10 mM H2O2 but only 0.8 and 0.5-fold at 50 and 200 mM H2O2; and radA was induced 9.3 and 9.5-fold at 5 and 10 mM H2O2, but only 1.0- and 1.1-fold at 50 and 200 mM H2O2.
Another striking difference between the H2O2 and NO expression profiles is the apparent exclusive induction of several PE and PPE genes by H2O2. Four such genes reside between rv1806-9 (PE20, PPE31, PPE32, PPE33); the first two in this group are induced over 40-fold. A fifth gene in the PE/PPE families, PE33 (rv3650), was found to be induced 9-fold. By contrast to several of the DNA repair genes, these genes were not induced after 40 min or 4 h of NO exposure and even appeared to be slightly repressed. We previously demonstrated that regulation of genes in the PE and PPE families is highly variable under a variety of conditions and largely independent of other family members (Voskuil et al., 2004).
Genes encoding a variety of other functions were highly regulated by H2O2, but not by NO, including genes involved in propionate metabolism [rv1130 and rv1131 (gltA1)] and the leuD and leuC genes involved in leucine biosynthesis, which were induced 20–40-fold. Interestingly, the resuscitation-promoting factor gene rpfA was highly repressed at 5 and 10 mM H2O2, but not at higher concentrations of H2O2 or after short-term NO exposure. However, after 4 h of 0.5 mM DETA/NO exposure, rpfA was also repressed.
The NO-Specific Response
Most genes that respond to high, but not to low levels of NO (i.e., NO-responsive genes other than the DosR regulon, which uniquely responds to low levels of NO) were also regulated in response to H2O2 (Table A1). One hundred seventy-four genes were induced in response to DETA/NO (Figure 3A). Forty-one of these genes are members of the DosR regulon (not included in Table A1). Of the remaining 133 NO-induced genes, 121 were also induced by H2O2. Accordingly, only 12 genes (cyp138, rv0330c, rv0967, sigE, rv1684, rv2033c, rv2036, rv2038c, rv2125, rv2620c, rv2621c, and lpqA) were selectively induced by NO but not H2O2. Thus, other than the DosR regulon, 121 of the 133 genes which were differentially regulated by higher concentrations of NO were also regulated by H2O2. Taken together, these results reflect the oxidative stress capacity of NO at high levels.
Discussion
A large number of genes have been identified as important in resistance to oxidative and nitrosative stress (Ehrt and Schnappinger, 2009). Interestingly, many of these genes are not induced by H2O2 or NO. The presence in the Mtb genome of a disrupted copy of oxyR predicts that genes normally repressed by OxyR in other species will exhibit higher basal expression in Mtb. In turn, higher basal expression levels would correlate with reduced induction ratios in response to oxidative stress. The lack of genes that respond to oxidative stress has led to the conclusion that Mtb is not capable of a robust response to this stress (Sherman et al., 1995; Garbe et al., 1996; Zahrt and Deretic, 2002; Shi et al., 2008). We find this to be the case for several genes that are commonly associated with oxidative stress resistance. These exhibit a relatively high basal level of expression even in the absence of exogenous ROS. Expression of katG and ahpC was maintained at a high basal level, although it was also further induced by addition of stress. In the absence of ROS generated by activated macrophages, a basal level of expression by ROS detoxification genes might be warranted to cope with oxidative stress emanating from metabolism in the presence of molecular oxygen. Most aerobic organisms, including some mycobacteria species (Pagan-Ramos et al., 2006; Imlay, 2008), employ OxyR to keep transcription at a low basal level until exogenous stress is encountered. The success of Mtb as a pathogen suggests that the loss of OxyR and the resulting constitutive expression of a portion of the oxidative stress response is not debilitating, but instead is likely to have been a propitious event that conferred a selective advantage. However, even in the absence of OxyR-mediated regulation, Mtb is capable of a large concerted transcriptional response to H2O2 as demonstrated in Table A1.
Several studies have indicated significant Mtb resistance to oxidative stress in vitro and in vivo. Our data confirm the high tolerance of Mtb for H2O2. H2O2 exposure resulted in no appreciable effect on Mtb growth or viability until exposures reached levels greater than 10 mM; suggesting Mtb is resistant to all but extreme levels. Low levels of H2O2 (0.5 mM) initiated the regulation of only the furA–katG and the mbt clusters, which are repressed by the regulators FurA and IdeR, respectively. Both of these regulators appear to be highly sensitive to the presence of H2O2, a consequence of their oxidative-sensitive metal-centers. At 5 and 10 mM H2O2, growth and viability were still largely unaffected. However, the large number and amplitude of the genes induced at these concentrations indicates at least a transient stress from H2O2 exposure. The response includes genes controlled by four transcriptional regulators previously implicated in ROS resistance [FurA (Zahrt et al., 2001), IdeR (Dussurget et al., 1996; Rodriguez et al., 2002), Sigma H (Fernandes et al., 1999; Raman et al., 2001), and Sigma E (Wu et al., 1997)] as well as the induction of 14 additional transcriptional regulators by H2O2. Thus, the response of Mtb to stress-inducing levels of H2O2 is broad and involves many regulatory networks. At very high doses (50 and 200 mM), H2O2 is lethal and the transcriptional response is significantly reduced and altered in comparison to effects mediated by non-lethal levels of H2O2. At first glance, the reduced response to lethal concentrations of H2O2 would seem to be most plausibly attributed to a collapse of the transcriptional machinery in dying bacilli. However, some genes are induced, or are more highly induced, in response to lethal levels of H2O2 compared to their expression during exposure to intermediate (non-lethal) concentrations of H2O2. Additionally, results from the study of high millimolar concentrations of H2O2 may not be physiologically relevant, unless the high concentration mimics the accumulation of oxidative damage after prolonged exposure to lower levels, which is likely observed under lethal conditions. Taken together, our results demonstrate that Mtb exhibits three, concentration-dependent H2O2 transcriptional responses: (1) a low dose response marked by induction of the most sensitive systems (H2O2 scavenging and iron acquisition); (2) a massive response to high, but sub-lethal levels of H2O2; and (3) a muted response to lethal levels of H2O2. The amount of peroxynitrite (formed by the reaction between equimolar amounts of NO and superoxide within the phagosome) produced per hour per macrophage is approximately 6 fmol (Ischiropoulos et al., 1992), which is approximately the same as the 5-fmol experienced by each cell of Mtb when 0.5 mM H2O2 was added, although it is still difficult to determine what specific level of ROS and RNS Mtb experiences in vivo over time. It is informative that many of the genes listed in Table A1 upregulated under mid-range concentrations of NO (0.5–5 mM) or H2O2 (5–10 mM) were previously found to be induced by Mtb during infection of activated macrophages (Schnappinger et al., 2003), indicating that the levels of ROS and RNS used in these experiments are most likely similar to what is experienced by the bacteria within the macrophage.
The second and third responses of Mtb to H2O2 are reminiscent of the two modes of E. coli killing by H2O2 first described by Imlay and Linn (1986). They demonstrated that E. coli killed by 1–2 mM H2O2 is due to DNA damage and was termed mode-one killing. Mode-two killing occurs at levels above 20 mM where multiple cellular components are targeted and killing is dependent on exposure time, and occurred when other cellular damage accumulated to a lethal level. Our data are partially in agreement with the results from E. coli indicating that high levels of H2O2 interfere with a H2O2-mediated mechanism that damages DNA. One of the main differences in the H2O2 susceptibility of E. coli and Mtb, however, appears to be the resistance of Mtb to mode-one killing. Induction of Mtb DNA damage repair mechanisms indicates that DNA damage occurs at 5 and 10 mM H2O2. However the pathogen is not killed at these concentrations and thus the damage does not result in DNA-dependent mode-one killing which typifies the effects of H2O2 on E. coli. We hypothesize that one mechanism by which Mtb avoids mode-one killing is by maintaining a very slow growth rate. An Mtb doubling time of 12–24 h (compared to 20 min for E. coli) allows significantly more time for DNA repair to occur before a DNA lesion has a chance to disrupt DNA replication and exert its lethal effects.
Previous work has indicated that the response to H2O2 shares very little in common with the response to NO in Mtb (Garbe et al., 1996). This study, however, used 2-dimensional gel electrophoresis to analyze response to these stresses, which is much less sensitive than microarray technology. It is interesting that much of the transcriptional response of Mtb to H2O2 and NO was remarkably similar at higher concentrations of and longer exposure times to DETA/NO (Table A1), but their effects on growth and survival are markedly different. Mtb either quickly adapts to H2O2 stress and continues to grow, or it dies. NO slowed or stopped Mtb growth but had little effect on survival even at high NO to bacilli ratios. RNS are known to kill under acidic conditions (Darwin and Nathan, 2005; Rhee et al., 2005). Our data indicate that exposure to NO is not lethal under standard laboratory conditions of aerobic growth and medium composition. However, sub-optimal conditions, such as those found in a phagosome, may well be more relevant to disease than growth in optimal laboratory medium. A recent study also indicates NO is lethal under anaerobic conditions and our unpublished data support this finding (Singh et al., 2008). It is well established that NO has both short-term reversible and long-term irreversible effects on respiratory components (Brown, 2001). NO will reversibly inhibit cytochrome c oxidase by competing with oxygen for binding, but after several hours of exposure NO irreversibly inactivates cytochrome c oxidase and other respiratory proteins including the NADH reductase. Immediate cessation of growth following NO exposure is most likely a result of reversible inhibition of cytochrome c oxidase and thus, of aerobic respiration (Voskuil et al., 2003), while the longer term effects on growth result from less reversible damage to the bacteria or medium. Another possible mechanism could involve the DosR regulon, which is induced by NO, but not H2O2 (Voskuil et al., 2003). DosR regulon proteins may modulate the growth rate, directly or indirectly.
Similar to what was found in the present study, iron acquisition genes were induced in M. bovis BCG cultures upon addition of H2O2 (Jang et al., 2009). Induction by H2O2 and NO of several genes and clusters of genes, point to an effect by ROS and RNS on iron–sulfur metabolism and iron–sulfur cluster synthesis or repair. Iron–sulfur cluster prosthetic groups (Fe2S2, Fe3S3, and Fe4S4) undergo oxidation–reduction reactions. Therefore, they are often part of electron transfer proteins in addition to fulfilling Fe, O2, 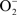 , and NO sensing roles (Beinert et al., 1997; Beinert and Kiley, 1999). Due to their sensitivity to oxidation, iron–sulfur cluster containing proteins are highly susceptible to oxidative and nitrosative stress (Beinert and Kiley, 1999). Restoration of an oxidatively damaged iron–sulfur center entails the synthesis of separate units and their insertion into the corresponding protein. H2O2 and NO strongly induce the SUF six gene operon (rv1460–rv1466) that encodes the alternative mycobacterial iron–sulfur cluster machinery (Huet et al., 2005). Pathways for the accumulation of the iron and sulfur precursors of iron–sulfur clusters are also upregulated. Two genes involved in cysteine biosynthesis cysK2 and cysM were also induced. Cysteine provides the sulfur for iron–sulfur center construction. Two other genes highly induced by H2O2, but not NO, are also involved in cysteine metabolism. These are cysD and cysN, which encode sulfate adenylyltransferase subunits involved in the activation of sulfate for cysteine biosynthesis (Pinto et al., 2004). Iron acquisition is indicated by the induction of the mbt gene cluster (rv2377c-84 and rv2386c), which encodes the biosynthesis of the iron chelating siderophore mycobactin. Repression of bfrB, encoding the iron-storage protein bacterioferritin, is also consistent with the need to acquire and use, not store, available iron. IdeR negatively regulates the mbt cluster and positively regulates bfrB when iron levels are high (Gold et al., 2001; Rodriguez et al., 2002). The oxidation of the iron-center of IdeR by H2O2 or NO appears to mimic low-iron conditions by affecting a “chemical” knockout of this protein, resulting in the expression of IdeR-repressed genes, and repression of IdeR-induced genes.
, and NO sensing roles (Beinert et al., 1997; Beinert and Kiley, 1999). Due to their sensitivity to oxidation, iron–sulfur cluster containing proteins are highly susceptible to oxidative and nitrosative stress (Beinert and Kiley, 1999). Restoration of an oxidatively damaged iron–sulfur center entails the synthesis of separate units and their insertion into the corresponding protein. H2O2 and NO strongly induce the SUF six gene operon (rv1460–rv1466) that encodes the alternative mycobacterial iron–sulfur cluster machinery (Huet et al., 2005). Pathways for the accumulation of the iron and sulfur precursors of iron–sulfur clusters are also upregulated. Two genes involved in cysteine biosynthesis cysK2 and cysM were also induced. Cysteine provides the sulfur for iron–sulfur center construction. Two other genes highly induced by H2O2, but not NO, are also involved in cysteine metabolism. These are cysD and cysN, which encode sulfate adenylyltransferase subunits involved in the activation of sulfate for cysteine biosynthesis (Pinto et al., 2004). Iron acquisition is indicated by the induction of the mbt gene cluster (rv2377c-84 and rv2386c), which encodes the biosynthesis of the iron chelating siderophore mycobactin. Repression of bfrB, encoding the iron-storage protein bacterioferritin, is also consistent with the need to acquire and use, not store, available iron. IdeR negatively regulates the mbt cluster and positively regulates bfrB when iron levels are high (Gold et al., 2001; Rodriguez et al., 2002). The oxidation of the iron-center of IdeR by H2O2 or NO appears to mimic low-iron conditions by affecting a “chemical” knockout of this protein, resulting in the expression of IdeR-repressed genes, and repression of IdeR-induced genes.
Despite the need to acquire iron to repair iron–sulfur centers, the powerful expression of iron scavenging genes during oxidative stress was unexpected as free ferrous [Fe(II)] iron reacts with H2O2 to form extremely reactive and damaging hydroxyl radicals owing to the Fenton reaction (Imlay, 2003). Indeed, derepression of iron acquisition genes in E. coli results in iron overload and an increase in oxidative stress (Imlay, 2003). Cysteine also promotes DNA damage by favoring the conversion of intracellular ferric [Fe(III)] iron to ferrous iron thus making it available to react with H2O2 to produce hydroxyl radicals (Park and Imlay, 2003). Consequently, both the uptake of iron and the synthesis of cysteine should promote DNA damage via Fenton chemistry. However, the need for iron and cysteine to repair damaged iron- and cysteine-containing proteins appears to super-cede the negative effects of hydroxyl radical formation at intermediate levels of H2O2 and NO stress.
Conclusion
Exposure to H2O2 or NO initiated an overlapping robust transcriptional response marked by the induction of a number of genes including those involved in iron–sulfur cluster synthesis and other cellular repair mechanisms. NO exposure was bacteriostatic, while H2O2 exposure was not bacteriostatic at concentrations below 50 mM while at or above this concentration H2O2 was bactericidal. It appears Mtb is resistant to DNA damage-mediated killing by H2O2 as no effect on growth or viability was observed after exposure to H2O2 levels that highly induced DNA repair mechanisms. We are currently investigating the mechanisms utilized by Mtb to resist DNA damage-mediated killing by H2O2. The high basal resistance of Mtb to oxidative and nitrosative stress seems to be a combination of the intrinsic resistance of the Mtb cell wall, the constitutive expression of genes that encode ROS and RNS scavenging functions, induction of genes that encode for repair of oxidized proteins, and induction of DNA repair mechanisms.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Kimberly Chong, Dirk Schnappinger, and Sangeeta Balakrishnan for DNA microarray assistance. We also thank T. B. K. Reddy for assistance with microarray database submission. The Walter V. and Idun Y. Berry Foundation, the American Lung Association, and NIH (RO1 AI061505) awarded to Martin I. Voskuil, NIH (RO1 AI44826) and the Defense Advanced Research Projects Agency (BAA-00-33) awarded to Gary K. Schoolnik, and NIH training grant (T32 AI052066-07) awarded to Iona L. Bartek supported this work.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/cellular_and_infection_microbiology/10.3389/fmicb.2011.00105/abstract
Complete microarray expression data can be obtained from the TB database (TBDB: http://www.tbdb.org; Reddy et al., 2009) or the NCBI Gene Expression Omnibus series number GSE16146.
Footnotes
References
Beinert, H., Holm, R. H., and Munck, E. (1997). Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 277, 653–659.
Beinert, H., and Kiley, P. J. (1999). Fe-S proteins in sensing and regulatory functions. Curr. Opin. Chem. Biol. 3, 152–157.
Boshoff, H. I., Reed, M. B., Barry, C. E. III, and Mizrahi, V. (2003). DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 113, 183–193.
Brown, G. C. (2001). Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. Acta 1504, 46–57.
Bryk, R., Lima, C. D., Erdjument-Bromage, H., Tempst, P., and Nathan, C. (2002). Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 295, 1073–1077.
Buchmeier, N. A., Newton, G. L., and Fahey, R. C. (2006). A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J. Bacteriol. 188, 6245–6252.
Chan, J., Xing, Y., Magliozzo, R. S., and Bloom, B. R. (1992). Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 175, 1111–1122.
Colangeli, R., Haq, A., Arcus, V. L., Summers, E., Magliozzo, R. S., McBride, A., Mitra, A. K., Radjainia, M., Khajo, A., Jacobs, W. R. Jr., Salgame, P., and Alland, D. (2009). The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc. Natl. Acad. Sci. U.S.A. 106, 4414–4418.
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S., Barry III, C. E., Tekaia, F., Badcock, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R., Devlin, K., Feltwell, T., Gentles, S., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Krogh, A., McLean, J., Moule, S., Murphy, L., Oliver, K., Osborne, J., Quail, M. A., Rajandream, M.-A., Rogers, J., Rutter, S., Seeger, K., Skelton, J., Squares, R., Squares, S., Sulston, J. E., Taylor, K., Whitehead, S., and Barrell, B. G. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544.
Cooper, A. M., Segal, B. H., Frank, A. A., Holland, S. M., and Orme, I. M. (2000). Transient loss of resistance to pulmonary tuberculosis in p47 (phox-/-) mice. Infect. Immun. 68, 1231–1234.
Darwin, K. H., Ehrt, S., Gutierrez-Ramos, J. C., Weich, N., and Nathan, C. F. (2003). The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302, 1963–1966.
Darwin, K. H., and Nathan, C. F. (2005). Role for nucleotide excision repair in virulence of Mycobacterium tuberculosis. Infect. Immun. 73, 4581–4587.
Deretic, V., Philipp, W., Dhandayuthapani, S., Mudd, M. H., Curcic, R., Garbe, T., Heym, B., Via, L. E., and Cole, S. T. (1995). Mycobacterium tuberculosis is a natural mutant with an inactivated oxidative-stress regulatory gene: implications for sensitivity to isoniazid. Mol. Microbiol. 17, 889–900.
Dussurget, O., Rodriguez, M., and Smith, I. (1996). An ideR mutant of Mycobacterium smegmatis has derepressed siderophore production and an altered oxidative-stress response. Mol. Microbiol. 22, 535–544.
Ehrt, S., and Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 11, 1170–1178.
Fernandes, N. D., Wu, Q. L., Kong, D., Puyang, X., Garg, S., and Husson, R. N. (1999). A mycobacterial extracytoplasmic sigma factor involved in survival following heat shock and oxidative stress. J. Bacteriol. 181, 4266–4274.
Firmani, M. A., and Riley, L. W. (2002). Reactive nitrogen intermediates have a bacteriostatic effect on Mycobacterium tuberculosis in vitro. J. Clin. Microbiol. 40, 3162–3166.
Flynn, J. L., Scanga, C. A., Tanaka, K. E., and Chan, J. (1998). Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 160, 1796–1803.
Gandotra, S., Jang, S., Murray, P. J., Salgame, P., and Ehrt, S. (2007). Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobacterium tuberculosis. Infect. Immun. 75, 5127–5134.
Garbe, T. R., Hibler, N. S., and Deretic, V. (1996). Response of Mycobacterium tuberculosis to reactive oxygen and nitrogen intermediates. Mol. Med. 2, 134–142.
Garbe, T. R., Hibler, N. S., and Deretic, V. (1999). Response to reactive nitrogen intermediates in Mycobacterium tuberculosis: induction of the 16-kilodalton alpha-crystallin homolog by exposure to nitric oxide donors. Infect. Immun. 67, 460–465.
Gardam, M. A., Keystone, E. C., Menzies, R., Manners, S., Skamene, E., Long, R., and Vinh, D. C. (2003). Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect. Dis. 3, 148–155.
Gold, B., Rodriguez, G. M., Marras, S. A., Pentecost, M., and Smith, I. (2001). The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Mol. Microbiol. 42, 851–865.
Huet, G., Daffe, M., and Saves, I. (2005). Identification of the Mycobacterium tuberculosis SUF machinery as the exclusive mycobacterial system of [Fe-S] cluster assembly: evidence for its implication in the pathogen’s survival. J. Bacteriol. 187, 6137–6146.
Imlay, J. A. (2008). Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 77, 755–776.
Imlay, J. A., and Linn, S. (1986). Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J. Bacteriol. 166, 519–527.
Ischiropoulos, H., Zhu, L., and Beckman, J. S. (1992). Peroxynitrite formation from macrophage-derived nitric oxide. Arch. Biochem. Biophys. 298, 446–451.
Jackett, P. S., Aber, V. R., and Lowrie, D. B. (1978). Virulence and resistance to superoxide, low pH and hydrogen peroxide among strains of Mycobacterizum tuberculosis. J. Gen. Microbiol. 104, 37–45.
Jang, H. J., Nde, C., Toghrol, F., and Bentley, W. E. (2009). Microarray analysis of Mycobacterium bovis BCG revealed induction of iron acquisition related genes in response to hydrogen peroxide. Environ. Sci. Technol. 43, 9465–9472.
Lee, W. L., Gold, B., Darby, C., Brot, N., Jiang, X., de Carvalho, L. P., Wellner, D., St John, G., Jacobs, W. R. Jr., and Nathan, C. (2009). Mycobacterium tuberculosis expresses methionine sulphoxide reductases A and B that protect from killing by nitrite and hypochlorite. Mol. Microbiol. 71, 583–593.
Manca, C., Paul, S., Barry, C. E. III, Freedman, V. H., and Kaplan, G. (1999). Mycobacterium tuberculosis catalase and peroxidase activities and resistance to oxidative killing in human monocytes in vitro. Infect. Immun. 67, 74–79.
Master, S. S., Springer, B., Sander, P., Boettger, E. C., Deretic, V., and Timmins, G. S. (2002). Oxidative stress response genes in Mycobacterium tuberculosis: role of ahpC in resistance to peroxynitrite and stage-specific survival in macrophages. Microbiology 148, 3139–3144.
Milano, A., Forti, F., Sala, C., Riccardi, G., and Ghisotti, D. (2001). Transcriptional regulation of furA and katG upon oxidative stress in Mycobacterium smegmatis. J. Bacteriol. 183, 6801–6806.
Mohan, V. P., Scanga, C. A., Yu, K., Scott, H. M., Tanaka, K. E., Tsang, E., Tsai, M. M., Flynn, J. L., and Chan, J. (2001). Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect. Immun. 69, 1847–1855.
Nathan, C. (2002). Inducible nitric oxide synthase in the tuberculous human lung. Am. J. Respir. Crit. Care Med. 166, 130–131.
Nathan, C., and Shiloh, M. U. (2000). Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848.
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D., and McKinney, J. D. (2004). Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302.
Ohno, H., Zhu, G., Mohan, V. P., Chu, D., Kohno, S., Jacobs, W. R. Jr., and Chan, J. (2003). The effects of reactive nitrogen intermediates on gene expression in Mycobacterium tuberculosis. Cell. Microbiol. 5, 637–648.
Ouellet, H., Ouellet, Y., Richard, C., Labarre, M., Wittenberg, B., Wittenberg, J., and Guertin, M. (2002). Truncated hemoglobin HbN protects Mycobacterium bovis from nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 99, 5902–5907.
Pagan-Ramos, E., Master, S. S., Pritchett, C. L., Reimschuessel, R., Trucksis, M., Timmins, G. S., and Deretic, V. (2006). Molecular and physiological effects of mycobacterial oxyR inactivation. J. Bacteriol. 188, 2674–2680.
Pagan-Ramos, E., Song, J., McFalone, M., Mudd, M. H., and Deretic, V. (1998). Oxidative stress response and characterization of the oxyR-ahpC and furA-katG loci in Mycobacterium marinum. J. Bacteriol. 180, 4856–4864.
Park, S., and Imlay, J. A. (2003). High levels of intracellular cysteine promote oxidative DNA damage by driving the fenton reaction. J. Bacteriol. 185, 1942–1950.
Pathania, R., Navani, N. K., Gardner, A. M., Gardner, P. R., and Dikshit, K. L. (2002). Nitric oxide scavenging and detoxification by the Mycobacterium tuberculosis haemoglobin, HbN in Escherichia coli. Mol. Microbiol. 45, 1303–1314.
Piddington, D. L., Fang, F. C., Laessig, T., Cooper, A. M., Orme, I. M., and Buchmeier, N. A. (2001). Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect. Immun. 69, 4980–4987.
Pinto, R., Tang, Q. X., Britton, W. J., Leyh, T. S., and Triccas, J. A. (2004). The Mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a tri-functional sulfate-activating complex. Microbiology 150, 1681–1686.
Pym, A. S., Domenech, P., Honore, N., Song, J., Deretic, V., and Cole, S. T. (2001). Regulation of catalase-peroxidase (KatG) expression, isoniazid sensitivity and virulence by furA of Mycobacterium tuberculosis. Mol. Microbiol. 40, 879–889.
Raman, S., Song, T., Puyang, X., Bardarov, S., Jacobs, W. R. Jr., and Husson, R. N. (2001). The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J. Bacteriol. 183, 6119–6125.
Rand, L., Hinds, J., Springer, B., Sander, P., Buxton, R. S., and Davis, E. O. (2003). The majority of inducible DNA repair genes in Mycobacterium tuberculosis are induced independently of RecA. Mol. Microbiol. 50, 1031–1042.
Reddy, T. B., Riley, R., Wymore, F., Montgomery, P., DeCaprio, D., Engels, R., Gellesch, M., Hubble, J., Jen, D., Jin, H., Koehrsen, M., Larson, L., Mao, M., Nitzberg, M., Sisk, P., Stolte, C., Weiner, B., White, J., Zachariah, Z. K., Sherlock, G., Galagan, J. E., Ball, C. A., and Schoolnik, G. K. (2009). TB database: an integrated platform for tuberculosis research. Nucleic Acids Res. 37, D499–D508.
Rhee, K. Y., Erdjument-Bromage, H., Tempst, P., and Nathan, C. F. (2005). S-nitroso proteome of Mycobacterium tuberculosis: enzymes of intermediary metabolism and antioxidant defense. Proc. Natl. Acad. Sci. U.S.A. 102, 467–472.
Rodriguez, G. M., Voskuil, M. I., Gold, B., Schoolnik, G. K., and Smith, I. (2002). ideR, an essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 70, 3371–3381.
Samson, L. (1992). The suicidal DNA repair methyltransferases of microbes. Mol. Microbiol. 6, 825–831.
Schnappinger, D., Ehrt, S., Voskuil, M. I., Liu, Y., Mangan, J. A., Monahan, I. M., Dolganov, G., Efron, B., Butcher, P. D., Nathan, C., and Schoolnik, G. K. (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704.
Senaratne, R. H., De Silva, A. D., Williams, S. J., Mougous, J. D., Reader, J. R., Zhang, T., Chan, S., Sidders, B., Lee, D. H., Chan, J., Bertozzi, C. R., and Riley, L. W. (2006). 5’-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol. Microbiol. 59, 1744–1753.
Sherman, D. R., Sabo, P. J., Hickey, M. J., Arain, T. M., Mahairas, G. G., Yuan, Y., Barry, C. E. III, and Stover, C. K. (1995). Disparate responses to oxidative stress in saprophytic and pathogenic mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 92, 6625–6629.
Shi, L., Sohaskey, C. D., North, R. J., and Gennaro, M. L. (2008). Transcriptional characterization of the antioxidant response of Mycobacterium tuberculosis in vivo and during adaptation to hypoxia in vitro. Tuberculosis (Edinb.) 88, 1–6.
Shi, S., and Ehrt, S. (2006). Dihydrolipoamide acyltransferase is critical for Mycobacterium tuberculosis pathogenesis. Infect. Immun. 74, 56–63.
Shiloh, M. U., and Nathan, C. F. (2000). Reactive nitrogen intermediates and the pathogenesis of Salmonella and mycobacteria. Curr. Opin. Microbiol. 3, 35–42.
Singh, R., Manjunatha, U., Boshoff, H. I., Ha, Y. H., Niyomrattanakit, P., Ledwidge, R., Dowd, C. S., Lee, I. Y., Kim, P., Zhang, L., Kang, S., Keller, T. H., Jiricek, J., and Barry, C. E. III. (2008). PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395.
Springer, B., Master, S., Sander, P., Zahrt, T., McFalone, M., Song, J., Papavinasasundaram, K. G., Colston, M. J., Boettger, E., and Deretic, V. (2001). Silencing of oxidative stress response in Mycobacterium tuberculosis: expression patterns of ahpC in virulent and avirulent strains and effect of ahpC inactivation. Infect. Immun. 69, 5967–5973.
St John, G., Brot, N., Ruan, J., Erdjument-Bromage, H., Tempst, P., Weissbach, H., and Nathan, C. (2001). Peptide methionine sulfoxide reductase from Escherichia coli and Mycobacterium tuberculosis protects bacteria against oxidative damage from reactive nitrogen intermediates. Proc. Natl. Acad. Sci. U.S.A. 98, 9901–9906.
Stallings, C. L., Stephanou, N. C., Chu, L., Hochschild, A., Nickels, B. E., and Glickman, M. S. (2009). CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138, 146–159.
Tullius, M. V., Harth, G., and Horwitz, M. A. (2001). High extracellular levels of Mycobacterium tuberculosis glutamine synthetase and superoxide dismutase in actively growing cultures are due to high expression and extracellular stability rather than to a protein-specific export mechanism. Infect. Immun. 69, 6348–6363.
Tusher, V. G., Tibshirani, R., and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121.
Voskuil, M. I., Schnappinger, D., Rutherford, R., Liu, Y., and Schoolnik, G. K. (2004). Regulation of the Mycobacterium tuberculosis PE/PPE genes. Tuberculosis (Edinb.) 84, 256–262.
Voskuil, M. I., Schnappinger, D., Visconti, K. C., Harrell, M. I., Dolganov, G. M., Sherman, D. R., and Schoolnik, G. K. (2003). Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713.
Wilson, M., Voskuil, M., Schnappinger, D., and Schoolnik, G. K. (2001). Functional genomics of Mycobacterium tuberculosis using DNA microarrays. Methods Mol. Med. 54, 335–357.
Wu, Q. L., Kong, D., Lam, K., and Husson, R. N. (1997). A mycobacterial extracytoplasmic function sigma factor involved in survival following stress. J. Bacteriol. 179, 2922–2929.
Yang, C. S., Yuk, J. M., and Jo, E. K. (2009). The role of nitric oxide in mycobacterial infections. Immune Netw. 9, 46–52.
Zahrt, T. C., and Deretic, V. (2002). Reactive nitrogen and oxygen intermediates and bacterial defenses: unusual adaptations in Mycobacterium tuberculosis. Antioxid. Redox Signal. 4, 141–159.
Zahrt, T. C., Song, J., Siple, J., and Deretic, V. (2001). Mycobacterial FurA is a negative regulator of catalase-peroxidase gene katG. Mol. Microbiol. 39, 1174–1185.
Keywords: Mycobacterium tuberculosis, reactive oxygen species, reactive nitrogen species, microarray, nitric oxide, hydrogen peroxide
Citation: Voskuil MI, Bartek IL, Visconti K and Schoolnik GK (2011) The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbio. 2:105. doi: 10.3389/fmicb.2011.00105
Received: 31 January 2011;
Accepted: 27 April 2011;
Published online: 13 May 2011.
Edited by:
Adel M. Talaat, University of Wisconsin Madison, USAReviewed by:
Thomas C. Zahrt, Medical College of Wisconsin, USASarah Ward, University of Wisconsin Madison, USA
Gyanu Lamichhane, Johns Hopkins University, USA
Copyright: © 2011 Voskuil, Bartek, Visconti and Schoolnik. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Martin I. Voskuil, Department of Microbiology, School of Medicine, University of Colorado Denver, P18-9115, 12800 East 19th Avenue, P.O. Box 6511, Aurora, CO 80045, USA. e-mail:bWFydGluLnZvc2t1aWxAdWNkZW52ZXIuZWR1