 Joanne Engel1,2*
Joanne Engel1,2* Yonatan Eran1
Yonatan Eran1
- 1 Department of Medicine, University of California at San Francisco, San Francisco, CA, USA
- 2 Department of Microbiology and Immunology, University of California at San Francisco, San Francisco, CA, USA
The lumenal surfaces of human body are lined by a monolayer of epithelia that together with mucus secreting cells and specialized immune cells form the mucosal barrier. This barrier is one of the most fundamental components of the innate immune system, protecting organisms from the vast environmental microbiota. The mucosal epithelium is comprised of polarized epithelial cells with distinct apical and basolateral surfaces that are defined by unique set of protein and lipid composition and are separated by tight junctions. The apical surface serves as a barrier to the outside world and is specialized for the exchange of materials with the lumen. The basolateral surface is adapted for interaction with other cells and for exchange with the bloodstream. A wide network of proteins and lipids regulates the formation and maintenance of the epithelium polarity. Many human pathogens have evolved virulence mechanisms that target this network and interfere with epithelial polarity to enhance binding to the apical surface, enter into cells, and/or cross the mucosal barrier. This review highlights recent advances in our understanding of how Pseudomonas aeruginosa, an important opportunistic human pathogen that preferentially infects damaged epithelial tissues, exploits the epithelial cell polarization machinery to enhance infection.
The Mucosal Barrier
The mucosal surfaces of our body are a primary component of our innate immune system and serve as a barrier against endogenous microflora as well as against external pathogens. This barrier is made of polarized epithelial cells, specialized immune cells, and secreted mucus. Many pathogens have evolved strategies to circumvent this barrier, including entering into cells or traveling through them by transcytosis, crossing through intercellular junctions, or directly disrupting the barrier by killing cells in the epithelium (Kazmierczak et al., 2001a).
The mucosal barrier epithelium is comprised of one or more layers of epithelial cells that have specialized and distinct apical and basolateral surfaces, separated by tight junctions (TJs), that form selective permeability barriers between biological compartments (Wang and Margolis, 2007; Martin-Belmonte and Mostov, 2008). The apical surface faces the lumen of the cavity, while the basolateral surface faces adjoining cells and the underlying basement membrane. The apical and basolateral membrane domains are distinguished by unique assemblies of proteins and lipids, creating specific membrane domains with distinct roles in formation and maintenance of barrier function, as well as the myriad of physiological barrier functions, such as nutrient exchange.
The apical surface contains transporters and enzymes that are specialized to interact with the external environment. The outer leaflet of the apical surface is highly enriched in glycosphingolipids and cholesterol. The basolateral plasma membrane of the epithelial cell contains many transporters and receptors that are involved in uptake of nutrients and hormones from the circulation. The basolateral surface can be divided into lateral domains, which contact other cells, and basal domains, which contact the basement membrane and blood vessels. The lateral surface contains specialized cell–cell contact domains, including TJs and adherens junctions (AJs).
The TJ is located at the apical-most region of the lateral surface and defines the boundary between the apical and basolateral surfaces (Ebnet, 2008). The TJ serves two functions. First, it acts as a “gate” or “barrier” to prevent paracellular diffusion between the cells. This function enables the epithelial monolayer to restrict permeability to solutes or larger particles, including pathogens. Second, the TJ acts as a fence to prevent diffusion or intermixing of plasma membrane components between the apical and basolateral domains. The TJ contains three classes of integral membrane proteins: occludins, claudins, and JAMs, each of which forms homophilic interactions that are responsible for gate function of the TJ. The TJ is attached to the cytoskeleton by a set of adaptor proteins including zonula occludens protein 1 (ZO-1).
The AJ lies underneath the TJ. The AJ consists mainly of classical cadherin family members and nectins, which are integral membrane proteins whose large extracellular domains interact in a homophilic or heterophilic manner to connect adjacent cells. Cadherins are linked to the cytoskeleton through β-catenin, α-catenin, and p120-catenin. In addition to providing the structural “linking” of neighboring cells, cadherins function as organizing nodes for multiprotein complexes that regulate cell–cell contacts, an essential function for morphogenesis and remodeling of tissues and organs (Meng and Takeichi, 2009).
A wide network of proteins and lipids regulates the formation and maintenance of epithelial cell polarity. The first step in the formation of apical–basolateral polarity is the formation of cell–cell junctions. E-cadherins from adjacent cells interact to create homophilic intercellular adhesions. Activation of small Rho GTPase family members, leads to cytoskeleton rearrangement and recruitment of structural and regulatory proteins, resulting in the formation of mature TJs and AJs (Iden and Collard, 2008). Junction maturation is coupled to the development of apical–basolateral asymmetry in the cell, where the newly formed AJ serves as a site for basolateral protein sorting (Yeaman et al., 2004). Maintenance of cell polarity and junction integrity involves continuous sensing of external cues such as extracellular matrix content and cell–cell contacts. These cues are translated into cellular signals that are received by a regulatory core of three protein complexes: Par3/Par6/aPKC, the Crumbs complex (Crumbs-3, PALS1, and PATJ), and the Scribble complex (Scribble, LGL1/2, and DLG1). The mutually exclusive localization of these three complexes helps to stabilize apical–basolateral polarity (Bryant and Mostov, 2008; Iden and Collard, 2008).
In addition to the asymmetric distribution of key polarity proteins described above, phosphoinositides have emerged as important determinants of membrane identity. These lipids bind to specific protein domains, particularly to those involved in the regulation of the cytoskeleton. In mammalian cells, phosphatidylinositol 4,5-bisphosphate (PIP2) is found primarily on the apical surface whereas phosphatidylinositol 3,4,5-trisphosphate (PIP3) localizes to the basolateral surface (Martin-Belmonte and Mostov, 2008).
The apical–basolateral polarity regulation system is increasingly recognized as an important target for pathogens. Our understanding of the interactions between pathogens with the mucosal barrier has been greatly aided by the use of epithelial cell lines, such as dog kidney (MDCK) cells, Calu-3 (a cell line derived from a human adenocarcinoma), and 16HBE (derived from human bronchial epithelial cells) cells that grow as a single confluent monolayer and recapitulate the development of polarized epithelium when grown on porous filter supports (transwells; Mostov et al., 2005). When grown at high densities under these conditions, the cells can obtain nutrients from the basolateral medium and will form polarized epithelium with distinct apical and basolateral surfaces and functional TJs and AJs within 24 h. With continued culture, cell polarity develops further. One advantage of this system is that pathogen–epithelial interactions can be studied without confounding effects contributed by immune cells. Furthermore, by using confluent monolayers, it is possible to compare microbe interactions between the apical and basolateral surfaces without having to take into account the effect of increased access to the basolateral surface that occurs in subconfluent cells or in the setting of epithelial injury (Kazmierczak et al., 2001a). Finally, some of the epithelial cells, including MDCK cells and primary mouse alveolar type II cells, can be grown as three-dimensional (3D) cysts when cultured on extracellular matrix in which the basolateral surface faces outward (Bryant and Mostov, 2008). These models may more closely mimic organs; in addition, they facilitate the examination of interactions of pathogens with the basolateral surface in the absence of the porous filter support (Barrila et al., 2010; Bucior et al., 2010). These reductionist systems provide a platform to analyze host–pathogen interactions, which can then be further validated in animal studies.
Pathogens Subvert Cell Polarity: An Emerging Theme
Important human mucosal pathogens, including viruses and bacteria, have been shown to directly target components of the polarity regulation network. Here we discuss a few recently described examples (Figure 1). Neisseria meningitidis, a devastating and common cause of bacterial meningitis, recruits Par3/Par6/aPKC to subvert junctional proteins at the endothelial cell surface. Ectopic intercellular junctional domains are formed at the site of binding of microcolonies by subverting an endothelial specific G-protein coupled receptor, the β2-adrenergic receptor, and an associated scaffolding protein, β-arrestin (Coureuil et al., 2010). This event leads to depletion of junctional proteins at the cell–cell interface and opening of intercellular junctions at the brain–endothelial interface, potentially explaining the tropism of this pathogen for the human central nervous system (Coureuil et al., 2009). Notably, these events are not observed in epithelial cells, as they appear to require the endothelial specific proteins VE-cadherin and β2-adrenergic receptor. Enteropathogenic Escherichia coli, a leading cause of diarrhea in children in the third world, induces PIP2 and PIP3 clustering at the bacterial-induced actin-rich pedestal in MDCK cells (Sason et al., 2009; Smith et al., 2010). CagA, the only known Helicobacter pylori effector translocated through its type IV secretion system, recruits the TJ proteins ZO-1 and JAM-A to the site of bacterial attachment in polarized MDCK cells (Amieva et al., 2003). CagA also directly interacts with and inhibits the serine/threonine kinase Par-1, causing it to dissociate from the membrane and leading to junction and polarity defects (Saadat et al., 2007). These events may contribute to the H. pylori-induced disorganization of gastric epithelial architecture and subsequent mucosal damage, inflammation, and cancer (Saadat et al., 2007). The disruption of epithelial barrier polarity by CagA has also been reported to create a nutrient-rich niche for H. pylori replication at the apical surface, allowing growth of microcolonies directly over the intercellular junctions (Tan et al., 2009). Listeria monocytogenes, a Gram-positive, food-borne pathogen uses E-cadherin as a binding receptor and as an internalization platform. Binding of the bacterial surface protein internalin A to E-cadherin initiates a process of actin rearrangement and AJ protein recruitment near the bacterial binding site that stimulates endocytosis and internalization of the bacteria (Bonazzi et al., 2009). Several viruses, including adenovirus, α-herpes viruses, reoviruses, and Hepatitis C target junctional complexes and polarity regulators as well (Bergelson, 2009).
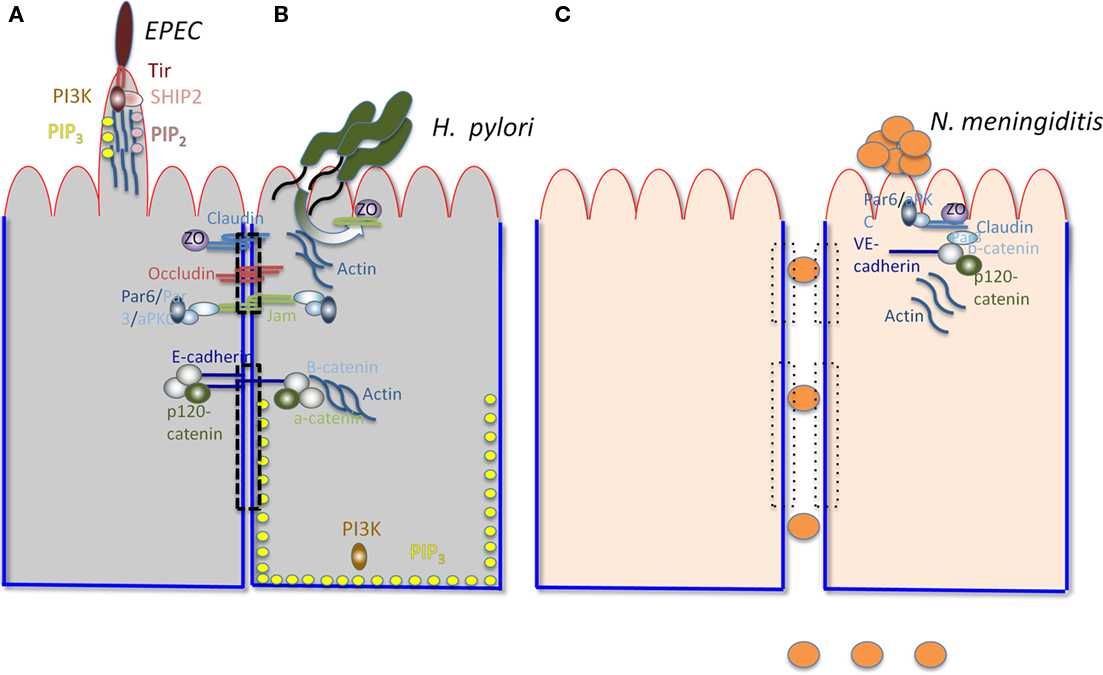
Figure 1. Interactions of various pathogens with polarized cells. The apical surface is outlined in red and the basolateral surface in blue. The TJ (upper) and AJ (lower) are indicated by a dashed rectangle. The major components of TJs (claudin, occludin, ZO, and JAMs) and AJs (E-cadherin, b-catenin, and a-catenin) are shown. The Par3/Par6/aPKC complex is shown to associate with JAMs. Actin is associated with AJs. PI3K and PIP3 are associated with the BL surface. (A) Illustrates relevant characteristics of EPEC-induced pedestals. PI3K, PIP3, SHIP2, and PIP2 are recruited by Tir (the translocated intimin receptor) to the actin-containing pedestal, along with actin, Arp2/3, Nck, and N-wasp (not shown). (B) Illustrates H. pylori recruiting junctional components, including JAMs, ZO-1, and Par (not shown) to form a replicative niche at the AP surface. (C) Represents endothelial cells, shows N. meningitidis disrupting intracellular junctions and breaching the blood–brain barrier by recruiting components of the TJ and the AJ (including Par-6, aPKC, Par-3, Claudin, ZO-5, VE-cadherin, b-catenin, and p-120 catenin) to the site of binding of the bacterial microcolony at the AP surface. The loss of the TJ and AJ is illustrated by the rectangles with dotted lines.
Pseudomonas aeruginosa, an Opportunistic Pathogen, Exploits Epithelial Damage and Loss of Polarity
For opportunistic pathogens, of which Pseudomonas aeruginosa is an important example, the mucosal barrier represents a formidable challenge to bacterial-mediated damage or entry. However, in the setting of injured or incompletely polarized epithelium, P. aeruginosa can initiate colonization and unleash its arsenal of potent virulence factors, which include the type III secretion system (T3SS) and its secreted effectors (Engel, 2003; Engel and Balachandran, 2009). Indeed, this Gram-negative pathogen is a leading cause of nosocomial infections in hospitalized patients (Mandell et al., 2010). Its predilection for injured tissue explains its propensity to cause ventilator-associated pneumonia, skin infections in burn patients or at the site of surgical incisions, and catheter-related infections, amongst others. P. aeruginosa is also a cause of chronic lung infections and ultimately death in patients with cystic fibrosis (CF; Mandell et al., 2010). Studies of the interaction of P. aeruginosa with polarized epithelium in culture and in vivo show that the degree of polarity significantly affects the final outcome of infection. Bacterial-induced host cell cytotoxicity or internalization is enhanced when bacteria are added to cells whose polarity has been altered by various manipulations (Fleiszig et al., 1997, 1998; Kazmierczak et al., 2001b, 2004). Finally, P. aeruginosa preferentially adheres to, enters, and injures wounded epithelium (Yamaguchi and Yamada, 1991; Zahm et al., 1991; Tsang et al., 1994; de Bentzmann et al., 1996a,b,c; Geiser et al., 2001). In this review, we describe selected recent advances in our understanding of P. aeruginosa interactions with polarized epithelium at each step of the infection process.
First Minutes of Infection: Binding to the Epithelium
The primary adhesins for P. aeruginosa binding to epithelial cells include type IV pili (TFP), polarly localized pili that can extend and retract, and flagella, that power swimming motility. For both adhesins, the molecular details of attachment to polarized epithelium remain to be clearly defined (Engel, 2007). In recent exciting studies, the role of retractile TFP in adherence of P. aeruginosa (strain PAK) to human lung epithelial primary cells grown as pseudostratified epithelium on a porous filter support was further examined (Heiniger et al., 2010). The authors compared infection of the apical surface of the intact pseudostratified epithelium to infection of mechanically injured cells, in which the cells comprising the basolateral layer of the pseudostratified epithelium are exposed. Binding to the intact pseudostratified epithelium required TFP but was not dependent upon pilin retraction. In contrast, pilin retraction (powered by the ATPase PilT) as well as the surface pilin associated protein PilY1, was required to penetrate into the basolateral portions of the monolayer. In the future, it will be of interest to determine whether PilY1 can directly bind to eukaryotic cells, whether antibodies directed against PilY1 can block this binding, and whether PilY1 binds specifically to one or more host cell proteins.
Bucior et al. (2010) examined the binding and subsequent downstream events during exposure of P. aeruginosa to the apical or basolateral surfaces of epithelial cells grown at various states of polarization. Confluent monolayers of MDCK and Calu-3 cells were plated on transwells and grown for different lengths of time to recapitulate various stages of polarization. These cells were infected with two different P. aeruginosa strains (PA01 and PAK) and the role of glycans or heparan sulfate proteoglycans (HSPGs) was examined. N-glycans were of special interest because previous work had revealed that MDCK cells with defects in cell surface glycosylation were resistant to P. aeruginosa infection, indicating that bacteria might require N-glycans for initial adhesion and subsequent host cell injury (Apodaca et al., 1995). HSPGs were examined because they are enriched at the basolateral surface. Indeed, the authors found that N-glycan chains at the apical surface were necessary and sufficient for P. aeruginosa binding, invasion, and cytotoxicity to MDCK and Calu-3 cells grown at various states of polarization. Enhanced expression and/or expression of more complex mannose-containing glycans increased binding, invasion, and cytotoxicity toward well-polarized epithelial cell monolayers, whereas pharmacologic inhibition of N-glycosylation or infection of concanavalin A-resistant MDCK cells resulted in decreased binding, invasion, and cytotoxicity. At the basolateral surface, the sulfation of heparan sulfate (HS) chains of HSPGs was found to be critical for P. aeruginosa binding, cytotoxicity, and invasion. In incompletely polarized epithelium, HSPG abundance was increased at the apical surface, explaining at least in part the increased susceptibility of injured epithelium to P. aeruginosa colonization and damage. Using MDCK cells grown as 3D cysts as a model for epithelial organs, P. aeruginosa was shown to specifically co-localized with HS-rich areas at the basolateral membrane. P. aeruginosa was also shown to bind HS chains and N-glycans coated onto plastic surfaces, with the highest binding affinity toward HS chains, suggesting that N-glycans and HS may be major regulators of P. aeruginosa binding to apical and basolateral membranes, respectively. Together, these findings demonstrate that P. aeruginosa recognizes distinct receptors on the apical and basolateral surface of polarized epithelium.
In the future, it will be of interest to examine whether TFP and flagella play different roles in adhesion at the apical versus basolateral surface, whether they display different binding specificities toward N-glycans and HSPGs, and whether these specificities are observed in vivo as well. It would also be informative to test whether N-glycosylation or HSPGs are upregulated in lungs of CF patients. Identifying the specific N-glycosylated apical receptor for P. aeruginosa will be an important step for our understanding of P. aeruginosa pathogenesis. Even in the absence of a defined apical receptor, this work suggests that therapies that target both N-glycosylation and HSPG synthesis, that compete with binding to N-glycans or HSPGs (such as simple sugars or heparin), or that restore polarized segregation of these molecules may be useful adjuncts to more standard antibiotic therapy.
The Early Hours: Subversion of Epithelial Polarity to Enhance Infection
Pseudomonas aeruginosa binding activates a central host signaling molecule, phosphatidylinositol 3-kinase (PI3K), which is required for the synthesis of PIP3 and for activation of a downstream effector, the serine/threonine kinase Akt. Activation of the PI3K/PIP3/Akt pathway was shown to be necessary and sufficient for P. aeruginosa entry from the apical surface of polarized epithelial cells (Kierbel et al., 2005a). Interestingly, PIP3 has recently emerged as a key polarity regulator that serves as a scaffold at the basolateral cytoplasm (Di Paolo and De Camilli, 2006; Gassama-Diagne et al., 2006). In follow-up studies, Kierbel and colleagues (Gassama-Diagne et al., 2006; Kierbel et al., 2007) have shown that P. aeruginosa subverts the PI3K/PIP3/Akt pathway to transform a patch of the apical surface into one with basolateral characteristics and to gain entry from the apical surface (Figure 2). In polarized monolayers, P. aeruginosa binds near cell–cell junctions, where it activates and recruits PI3K to the apical surface. This event leads to a remarkable remodeling of the apical membrane in which protrusions enriched for PIP3 and actin form at the apical surface at the site of bacterial binding. These protrusions are deficient in apical membrane markers and contain basolateral constituents. Notably, no disruption of the TJs could be detected, suggesting that the bacterial-induced rerouting of basolateral markers involved transcytosis rather than diffusion. Consistent with this notion, a dominant negative mutant of dynamin also blocked delocalization of basolateral proteins to the bacterial-induced apical protrusion. The end result is that this bacterium transforms apical into basolateral membrane, creating a local microenvironment that facilitates its colonization and entry into to the mucosal barrier.
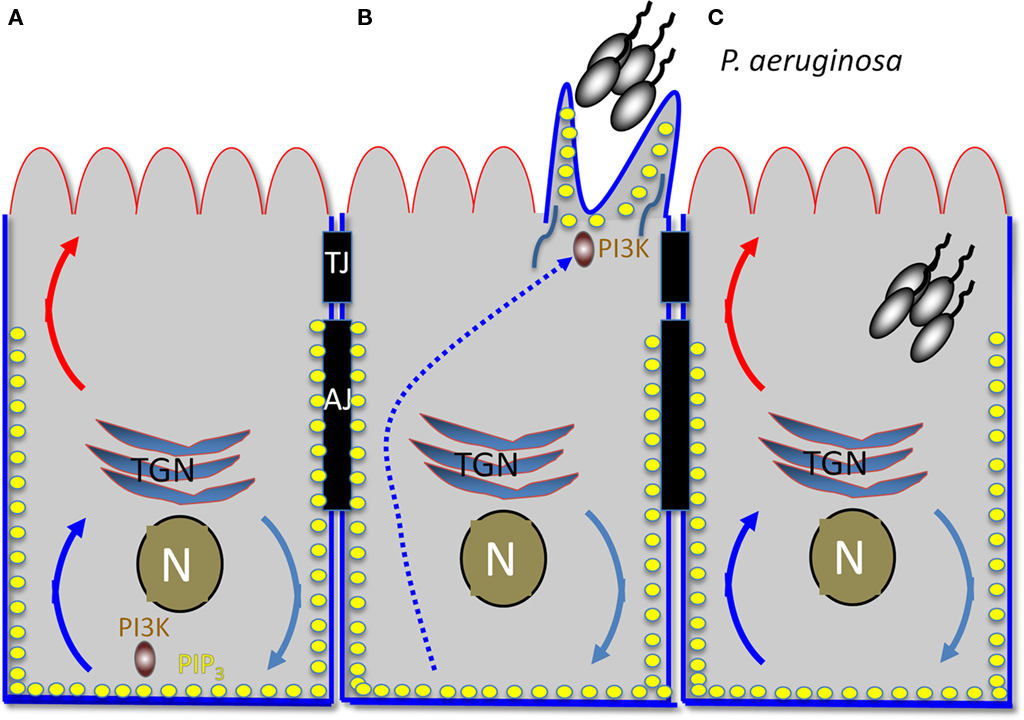
Figure 2. Early hours of P. aeruginosa infection: subversion of host cell polarity to transform apical into a basolateral membrane. (A) Diagram of polarized epithelial cell. The apical surface is outlined in red and the basolateral surface in blue. TJ, tight junction; AJ, adherens junction; TGN, trans-Golgi network; N, nucleus. Turquoise, blue, and red curved arrows represent vesicular trafficking. PIP3 is represented by the yellow dots. (B) Protrusion formation. An aggregate of P. aeruginosa recruits PI3K to the apical surface, generating local production of PIP3. BL recycling of proteins is redirected to the AP surface, creating a basolateral environment at the apical surface. (C) An aggregate of P. aeruginosa is internalized into the host cell, possibly at least in part through the transient protrusion.
Since apical–basolateral polarity is highly regulated in epithelial cells, it will be interesting to identify the initial events that lead to protrusion formation. Preliminary results suggest that polarity regulators are recruited to the bacterial binding site prior to PI3K (unpublished results). Analysis of the dynamics of protrusion formation, determining whether AJs or TJs are formed at the site of aggregate binding, and elucidating the molecular mechanisms by which they are redirected to the apical surface, will be key to understanding PA-induced apical protrusions.
For both P. aeruginosa and N. meningitidis, protrusion formation involves aggregates of bacteria as opposed to individual bacteria. It is possible that a threshold number of bacteria are required to activate this response or that these structures represent a specific host response to a physically large cluster. While bacterial aggregates are observed in stationary phase cultures in the absence of host cells (Allesen-Holm et al., 2006), it will be interesting to determine whether they are preformed or whether they form in response to host cells, as suggested by earlier work that described pack-swarming motility (Dacheux et al., 2001). Finally, comparison of the interactions of the polarized epithelium with single bacteria versus aggregates, the requirement for the major adhesins TFP and flagella, and the role of the T3SS in the formation of protrusions will be of interest to examine.
What function does protrusion formation serve and who benefits from it? One attractive possibility is that the bacteria use these protrusions for entry. Indeed, treatments that inhibit entry, such as inhibitors of PI3K or of the actin cytoskeleton, also inhibit protrusion formation (Kierbel et al., 2005b). Alternatively or in addition, protrusion formation may represent a response that could be beneficial to the host. For example, recruitment of basolateral constituents to the apical surface could trigger the innate immune response in a spatially restricted manner. The relationship of the PI3K/Akt pathway to other signaling pathways activated upon P. aeruginosa binding and internalization, including translocation of NF-kB to the nucleus and cytokine production, remains to be explored.
Late Infection: Disruption of Cell–Cell Contacts in the Epithelium or Simply Cell Death?
An important question in the study P. aeruginosa pathogenesis is the fate of the adherent bacteria, whether they be aggregates or single bacterial cells. For chronic infection, such as in lungs of CF patients, P. aeruginosa is thought to grow as biofilms in the thick layer of mucus overlying airway epithelial cells rather than adhering and invading directly into to the airway epithelium (Moreau-Marquis et al., 2008). In contrast, during acute infection, bacterial internalization, host cell destruction, and penetration to the basolateral surface are observed (Kazmierczak et al., 2001a). The general trend emerging from these studies is that loss of cell polarity enhances adhesion and favors subsequent downstream events, including cell death, disruption of cell junctions, inhibition of wound repair, and/or bacterial internalization (Kazmierczak et al., 2001a; Garrity-Ryan et al., 2004; Bucior et al., 2010), but differences in cell types, infection conditions and in the spectrum of T3SS toxins influences the outcome. For example, ExoU-secreting strains are associated with rapid host cell death by necrosis; ExoS-secreting P. aeruginosa strains lead to slower host cell death with features of apoptosis; and strains engineered to produce only ExoT lead to apoptotic cell death by ADP ribosylation of Crk (Alaoui-El-Azher et al., 2006; Shafikhani et al., 2008; Hauser, 2009).
Out of the four known P. aeruginosa T3SS effectors proteins, ExoS and ExoT are logical candidates for interfering with cellular polarity and disrupting cell junction integrity (Engel and Balachandran, 2009; Hauser, 2009). These two highly homologous bifunctional enzymes encode an N-terminal GTPase-activating protein (GAP) domain with activity toward Rho family GTPases and a C-terminal ADP ribosyltransferase domain. Each domain interacts with central components of the polarity regulation network. The GAP domains of both ExoT and ExoS inactivate the RhoA family GTPases, Ccd42, Rac1, and RhoA. The ExoS ADPRT domain targets a variety of effectors involved in regulating the actin cytoskeleton, including Ras, Ral, Rabs, and ERM proteins, while the ADPRT domain of ExoT specifically targets CrkI and CrkII, scaffolding proteins associated with focal adhesions (Sun et al., 2004).
Recent studies from the Prince lab have examined in more detail the effect of T3SS effectors on epithelial barrier function (Soong et al., 2008). Human airway cells grown on transwell filters were infected with various informative isogenic mutants of PAK, a representative non-ExoU-producing strain. ExoS was found to be necessary and sufficient to alter the integrity of TJs in the absence of host cell death, as measured by accessibility to biotin, permeability to small and large molecular weight dextrans, bacterial transmigration, and the absence of LDH release or trypan blue staining under the conditions of the experiments. In PAK strains engineered to express only ExoS, the ADPRT domain was necessary and sufficient to alter epithelial cell permeability. PAK strains expressing ExoS disrupted ZO-1, occludin, and ezrin localization, decreased membrane-associated occludin, and blocked ezrin phosphorylation as would be predicted from ADP ribosylation of ezrin. The authors conclude that the ADPRT domain of ExoS is sufficient to disrupt epithelial barrier integrity. Of note, the exact mechanism by which ExoS disrupts TJs remains to be determined. It could represent a general cellular response to ExoS-mediated cell death or to the disruption of the actin cytoskeleton, or it could be a direct consequence of ExoS on junctional components.
Summary
An emerging theme in microbial pathogenesis is the recognition that pathogens exploit or disrupt components of the mucosal barrier in order to facilitate colonization, to create a specialized niche for replication where they remain shielded from the host immune response, and/or to disseminate to distant tissues or to new hosts. Host cell polarity determinants are logical targets for pathogens, as control of epithelial cell polarity requires constant sensing of external cues. In addition, the signaling nodes emanating from polarity determinants may interface with the host innate immune response, although this connection has just begun to be explored. This review highlights recent advances and suggests future directions in furthering our understanding of how P. aeruginosa exploit the host polarity machinery to establish infection. These studies may lead us to discover new bacterial virulence mechanisms and therapeutic targets against this highly antibiotic resistance pathogen. Finally, it will extend our understanding of the initiation and maintenance of epithelial cell polarity, an area important in development, cancer biology, and pathogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (PO1 AI53194 and RO1 AI065902). During part of this time, Yonatan Eran was supported by an EMBO long-term fellowship.
References
Alaoui-El-Azher, M., Jia, J., Lian, W., and Jin, S. (2006). ExoS of Pseudomonas aeruginosa induces apoptosis through a Fas receptor/caspase 8-independent pathway in HeLa cells. Cell. Microbiol. 8, 326–338.
Allesen-Holm, M., Barken, K. B., Yang, L., Klausen, M., Webb, J. S., Kjelleberg, S., Molin, S., Givskov, M., and Tolker-Nielsen, T. (2006). A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 59, 1114–1128.
Amieva, M. R., Vogelmann, R., Covacci, A., Tompkins, L. S., Nelson, W. J., and Falkow, S. (2003). Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300, 1430–1434.
Apodaca, G., Bomsel, M., Lindstedt, R., Engel, J., Frank, D., Mostov, K., and Wiener-Kronish, J. (1995). Characterization of Pseudomonas aeruginosa-induced MDCK cell injury: glycosylation defective host cells are resistant to bacterial killing. Infect. Immun. 63, 1541–1551.
Barrila, J., Radtke, A. L., Crabbe, A., Sarker, S. F., Herbst-Kralovetz, M. M., Ott, C. M., and Nickerson, C. A. (2010). Organotypic 3D cell culture models: using the rotating wall vessel to study host–pathogen interactions. Nat. Rev. Microbiol. 8, 791–801.
Bergelson, J. M. (2009). Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host Microbe 5, 517–521.
Bonazzi, M., Lecuit, M., and Cossart, P. (2009). Listeria monocytogenes internalin and E-cadherin: from bench to bedside. Cold Spring Harb. Perspect. Biol. 1, a003087.
Bryant, D. M., and Mostov, K. E. (2008). From cells to organs: building polarized tissue. Nat. Rev. Mol. Cell Biol. 9, 887–901.
Bucior, I., Mostov, K., and Engel, J. N. (2010). Pseudomonas aeruginosa-mediated damage requires distinct receptors at the apical and basolateral surfaces of the polarized epithelium. Infect. Immun. 78, 939–953.
Coureuil, M., Lecuyer, H., Scott, M. G., Boularan, C., Enslen, H., Soyer, M., Mikaty, G., Bourdoulous, S., Nassif, X., and Marullo, S. (2010). Meningococcus hijacks a beta2-adrenoceptor/beta-arrestin pathway to cross brain microvasculature endothelium. Cell 143, 1149–1160.
Coureuil, M., Mikaty, G., Miller, F., Lecuyer, H., Bernard, C., Bourdoulous, S., Dumenil, G., Mege, R. M., Weksler, B. B., Romero, I. A., Couraud, P. O., and Nassif, X. (2009). Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science 325, 83–87.
Dacheux, D., Goure, J., Chabert, J., Usson, Y., and Attree, I. (2001). Pore-forming activity of type III system-secreted proteins leads to oncosis of Pseudomonas aeruginosa-infected macrophages. Mol. Microbiol. 40, 76–85.
de Bentzmann, S., Plotkowski, C., and Puchelle, E. (1996a). Receptors in the Pseudomonas aeruginosa adherence to injured and repairing airway epithelium. Am. J. Respir. Crit. Care Med. 154, S155–S162.
de Bentzmann, S., Roger, P., Dupuit, F., Bajolet-Laudinat, O., Fuchey, C., Plotkowski, M. C., and Puchelle, E. (1996b). Asialo GM1 is a receptor for Pseudomonas aeruginosa adherence to regenerating respiratory epithelial cells. Infect. Immun. 64, 1582–1588.
de Bentzmann, S., Roger, P., and Puchelle, E. (1996c). Pseudomonas aeruginosa adherence to remodelling respiratory epithelium. Eur. Respir. J. 10, 2145–2150.
Di Paolo, G., and De Camilli, P. (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657.
Ebnet, K. (2008). Organization of multiprotein complexes at cell–cell junctions. Histochem. Cell Biol. 130, 1–20.
Engel, J. (2007). “Pseudomonas aeruginosa internalization by non-phagocytic cells,” in Pseudomonas: A Model System in Biology, eds. J.-L. Ramos and A. Filloux (New York, NY: Springer) 343–368.
Engel, J., and Balachandran, P. (2009). Role of Pseudomonas aeruginosa type III effectors in disease. Curr. Opin. Microbiol. 12, 61–66.
Engel, J. N. (2003). “Molecular pathogenesis of acute Pseudomonas aeruginosa infections,” in Severe Infections Caused by Pseudomonas aeruginosa, eds A. Hauser and J. Rello (New York: Kluwer Academic/Plenum Press), 201–230.
Fleiszig, S. M., Vallas, V., Jun, C. H., Mok, L., Balkovetz, D. F., Roth, M. G., and Mostov, K. E. (1998). Susceptibility of epithelial cells to Pseudomonas aeruginosa invasion and cytotoxicity is upregulated by hepatocyte growth factor. Infect. Immun. 66, 3443–3446.
Fleiszig, S. M., Wiener-Kronish, J. P., Miyazaki, H., Vallas, V., Mostov, K. E., Kanada, D., Sawa, T., Yen, T. S., and Frank, D. W. (1997). Pseudomonas aeruginosa-mediated cytotoxicity and invasion correlate with distinct genotypes at the loci encoding exoenzyme S. Infect. Immun. 65, 579–586.
Garrity-Ryan, L., Shafikhani, S., Balachandran, P., Nguyen, L., Oza, J., Jakobsen, T., Sargent, J., Fang, X., Cordwell, S., Matthay, M. A., and Engel, J. N. (2004). The ADP ribosyltransferase domain of Pseudomonas aeruginosa ExoT contributes to its biological activities. Infect. Immun. 72, 546–558.
Gassama-Diagne, A., Yu, W., Ter Beest, M., Martin-Belmonte, F., Kierbel, A., Engel, J., and Mostov, K. (2006). Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat. Cell Biol. 8, 963.
Geiser, T. K., Kazmierczak, B. I., Garrity-Ryan, L. K., Matthay, M. A., and Engel, J. N. (2001). Pseudomonas aeruginosa ExoT inhibits in vitro lung epithelial wound repair. Cell. Microbiol. 3, 223–236.
Hauser, A. R. (2009). The type III secretion system of Pseudomonas aeruginosa : infection by injection. Nat. Rev. Microbiol. 7, 654–665.
Heiniger, R. W., Winther-Larsen, H. C., Pickles, R. J., Koomey, M., and Wolfgang, M. C. (2010). Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell. Microbiol. 12, 1158–1173.
Iden, S., and Collard, J. G. (2008). Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell Biol. 9, 846–859.
Kazmierczak, B., Mostov, K., and Engel, J. (2001a). Interaction of bacterial pathogens with polarized epithelium. Annu. Rev. Microbiol. 55, 407–435.
Kazmierczak, B. I., Jou, T.-S., Mostov, K., and Engel, J. (2001b). Rho-GTPase activity modulates Pseudomonas aeruginosa internalization by epithelial cells. Cell. Microbiol. 3, 85–98.
Kazmierczak, B. I., Mostov, K., and Engel, J. N. (2004). Epithelial cell polarity alters Rho-GTPase responses to Pseudomonas aeruginosa. Mol. Biol. Cell 15, 411–419.
Kierbel, A., Gassam, A., Mostov, K., and Engel, J. (2005a). The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol. Biol. Cell 16, 2577.
Kierbel, A., Gassama-Diagne, A., Mostov, K., and Engel, J. N. (2005b). The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol. Biol. Cell 16, 2577–2585.
Kierbel, A., Gassama-Diagne, A., Rocha, C., Radoshevich, L., Olson, J., Mostov, K., and Engel, J. (2007). Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J. Cell Biol. 177, 21–27.
Mandell, G. L., Bennett, J. E., and Dolin, R. (2010). Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 7th Edn. Philadelphia, PA: Churchill Livingstone/Elsevier.
Martin-Belmonte, F., and Mostov, K. (2008). Regulation of cell polarity during epithelial morphogenesis. Curr. Opin. Cell Biol. 20, 227–234.
Meng, W., and Takeichi, M. (2009). Adherens junction: molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 1, a002899.
Moreau-Marquis, S., Stanton, B. A., and O’Toole, G. A. (2008). Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm. Pharmacol. Ther. 21, 595–599.
Mostov, K., Brakeman, P., Datta, A., Gassama, A., Katz, L., Kim, M., Leroy, P., Levin, M., Liu, K., Martin, F., O’Brien, L. E., Verges, M., Su, T., Tang, K., Tanimizu, N., Yamaji, T., and Yu, W. (2005). Formation of multicellular epithelial structures. Novartis Found. Symp. 269, 193–200; discussion 200–205, 223–230.
Saadat, I., Higashi, H., Obuse, C., Umeda, M., Murata-Kamiya, N., Saito, Y., Lu, H., Ohnishi, N., Azuma, T., Suzuki, A., Ohno, S., and Hatakeyama, M. (2007). Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 447, 330–333.
Sason, H., Milgrom, M., Weiss, A. M., Melamed-Book, N., Balla, T., Grinstein, S., Backert, S., Rosenshine, I., and Aroeti, B. (2009). Enteropathogenic Escherichia coli subverts phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate upon epithelial cell infection. Mol. Biol. Cell 20, 544–555.
Shafikhani, S. H., Morales, C., and Engel, J. (2008). The Pseudomonas aeruginosa type III secreted toxin ExoT is necessary and sufficient to induce apoptosis in epithelial cells. Cell. Microbiol. 10, 994–1007.
Smith, K., Humphreys, D., Hume, P. J., and Koronakis, V. (2010). Enteropathogenic Escherichia coli recruits the cellular inositol phosphatase SHIP2 to regulate actin-pedestal formation. Cell Host Microbe 7, 13–24.
Soong, G., Parker, D., Magargee, M., and Prince, A. S. (2008). The type III toxins of Pseudomonas aeruginosa disrupt epithelial barrier function. J. Bacteriol. 190, 2814–2821.
Sun, J., Maresso, A. W., Kim, J. J., and Barbieri, J. T. (2004). How bacterial ADP-ribosylating toxins recognize substrates. Nat. Struct. Mol. Biol. 11, 868–876.
Tan, S., Tompkins, L. S., and Amieva, M. R. (2009). Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog. 5, e1000407. doi: 10.1371/journal.ppat.1000407
Tsang, K. W. T., Rutman, A., Tanaka, E., Lundt, V., Dewar, A., Cole, P. J., and Wilson, R. (1994). Interaction of Pseudomonas aeruginosa with human respiratory mucosa in vitro. Eur. Respir. J. 7, 1746–1753.
Wang, Q., and Margolis, B. (2007). Apical junctional complexes and cell polarity. Kidney Int. 72, 1448–1458.
Yamaguchi, T., and Yamada, H. (1991). Role of mechanical injury on airway surface in the pathogenesis of Pseudomonas aeruginosa. Am. Rev. Respir. Dis. 144, 1147–1152.
Yeaman, C., Grindstaff, K. K., and Nelson, W. J. (2004). Mechanism of recruiting Sec6/8 (exocyst) complex to the apical junctional complex during polarization of epithelial cells. J. Cell. Sci. 117, 559–570.
Keywords: Pseudomonas aeruginosa, microbial pathogenesis, host–pathogen interactions, cell polarity, epithelial barrier, tight junctions, adherens junctions
Citation: Engel J and Eran Y (2011) Subversion of mucosal barrier polarity by Pseudomonas aeruginosa. Front. Microbio. 2:114. doi: 10.3389/fmicb.2011.00114
Received: 18 February 2011; Paper pending published: 18 March 2011;
Accepted: 09 May 2011; Published online: 26 May 2011.
Edited by:
Dara Frank, Medical College of Wisconsin, USAReviewed by:
Steve Blanke, University of Illinois at Urbana-Champaign, USALee-Ann H. Allen, University of Iowa, USA
Copyright: © 2011 Engel and Eran. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Joanne Engel, University of California San Francisco, Box 0654, 513 Parnassus Avenue, San Francisco, CA 94143-0654, USA. e-mail:amVuZ2VsQG1lZGljaW5lLnVjc2YuZWR1