 Emily M. Mallick1
Emily M. Mallick1 Michael J. Brady2 Steven A. Luperchio3 Vijay K. Vanguri4 Loranne Magoun2 Hui Liu2 Barbara J. Sheppard3
Michael J. Brady2 Steven A. Luperchio3 Vijay K. Vanguri4 Loranne Magoun2 Hui Liu2 Barbara J. Sheppard3 Jean Mukherjee5 Art Donohue-Rolfe5 Saul Tzipori5
Jean Mukherjee5 Art Donohue-Rolfe5 Saul Tzipori5 John M. Leong1,2* and David B. Schauer3,6†
John M. Leong1,2* and David B. Schauer3,6†
- 1 Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA, USA
- 2 Department of Molecular Biology and Microbiology, Tufts University School of Medicine, Boston, MA, USA
- 3 Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
- 4 Department of Pathology, University of Massachusetts Medical School, Worcester, MA, USA
- 5 Division of Infectious Diseases, Tufts University School of Veterinary Medicine, North Grafton, MA, USA
- 6 Division of Comparative Medicine, Massachusetts Institute of Technology, Cambridge, MA, USA
Upon binding to intestinal epithelial cells, enterohemorrhagic Escherichia coli (EHEC), enteropathogenic E. coli (EPEC), and Citrobacter rodentium trigger formation of actin pedestals beneath bound bacteria. Pedestal formation has been associated with enhanced colonization, and requires intimin, an adhesin that binds to the bacterial effector translocated intimin receptor (Tir), which is translocated to the host cell membrane and promotes bacterial adherence and pedestal formation. Intimin has been suggested to also promote cell adhesion by binding one or more host receptors, and allelic differences in intimin have been associated with differences in tissue and host specificity. We assessed the function of EHEC, EPEC, or C. rodentium intimin, or a set of intimin derivatives with varying Tir-binding abilities in animal models of infection. We found that EPEC and EHEC intimin were functionally indistinguishable during infection of gnotobiotic piglets by EHEC, and that EPEC, EHEC, and C. rodentium intimin were functionally indistinguishable during infection of C57BL/6 mice by C. rodentium. A derivative of EHEC intimin that bound Tir but did not promote robust pedestal formation on cultured cells was unable to promote C. rodentium colonization of conventional mice, indicating that the ability to trigger actin assembly, not simply to bind Tir, is required for intimin-mediated intestinal colonization. Interestingly, streptomycin pre-treatment of mice eliminated the requirement for Tir but not intimin during colonization, and intimin derivatives that were defective in Tir-binding still promoted colonization of these mice. These results indicate that EPEC, EHEC, and C. rodentium intimin are functionally interchangeable during infection of gnotobiotic piglets or conventional C57BL/6 mice, and that whereas the ability to trigger Tir-mediated pedestal formation is essential for colonization of conventional mice, intimin provides a Tir-independent activity during colonization of streptomycin pre-treated mice.
Introduction
The family of attaching and effacing (AE) pathogens consists of enterohemorrhagic Escherichia coli (EHEC), enteropathogenic E. coli (EPEC), and Citrobacter rodentium. EHEC colonizes the large intestine and can result in diarrhea, hemorrhagic colitis, and life-threatening hemolytic uremic syndrome (Kaper et al., 2004; Pennington, 2010). The highly related EPEC colonizes the small intestine and is a causative agent of infantile diarrhea in the developing world (Chen and Frankel, 2005; Spears et al., 2006). C. rodentium, is a related murine pathogen that typically colonizes the large intestine and causes transmissible murine colonic hyperplasia, characterized by colonic epithelial cell proliferation and high rates of mortality in suckling animals (reviewed in Luperchio and Schauer, 2001; Mundy et al., 2005).
The three pathogens are so-named AE pathogens because they each colonize the intestinal epithelium by inducing in host cells “AE lesions,” which consist of effacement of brush border microvilli, intimate adherence of bacteria, and polymerization of actin into a pedestal-like extension of the epithelial cell beneath the bound bacterium (Moon et al., 1983; for review, see Kaper et al., 2004). Bacteria entirely incapable of generating AE lesions are severely defective for colonization and disease (Donnenberg et al., 1993a,b; Schauer and Falkow, 1993b; Tzipori et al., 1995; Deng et al., 2003; Ritchie et al., 2003), while bacteria still capable of intimate attachment but defective selectively for pedestal formation are moderately attenuated (Ritchie et al., 2008; Crepin et al., 2010).
The ability to generate the AE phenotype by these organisms requires the locus of enterocyte effacement (LEE), a pathogenicity island that encodes a type III secretion system (TTSS) and several translocated effectors (Kenny and Finlay, 1995; McDaniel et al., 1995). Translocated intimin receptor (Tir), a type III translocated effector critical for intimate bacterial attachment and actin pedestal formation, becomes localized in the host plasma membrane with the N- and C-terminal cytoplasmic domains (for review, see Lommel et al., 2004; Frankel and Phillips, 2008; Campellone, 2010). The central, extracellular domain is recognized by the outer membrane adhesin intimin, encoded by the eae gene (Jerse et al., 1990; Donnenberg and Kaper, 1991). The intimin N-terminus promotes outer membrane localization and multimerization, whereas the intimin C-terminus encodes its adhesive activities (Frankel et al., 1995; Liu et al., 1999; Batchelor et al., 2000; Luo et al., 2000; Yi et al., 2010).
It has been postulated that in addition to binding Tir, intimin possesses host receptor adhesive activities that also contribute to colonization. For instance, the intimin related Yersinia pseudotuberculosis invasin protein binds to β1-chain integrins (Isberg et al., 1987), and EPEC intimin was shown to be capable of recognizing β1-chain integrins, albeit with apparently much lower affinity (Frankel et al., 1996a). Nucleolin is recognized by EHEC intimin (Sinclair and O’Brien, 2002) and localized beneath cell-associated EPEC during infection of cultured monolayers (Dean and Kenny, 2011). Finally, intimin but not Tir, contributes to the disruption of epithelial barrier function (Dean and Kenny, 2004), suggesting the existence of Tir-independent functions of intimin.
Although EPEC and EHEC intimin have been demonstrated to be interchangeable for pedestal formation on cultured cells, intimin exhibits considerable allelic variation in the C-terminal domain responsible for adhesive activity (Frankel et al., 1994), and the intimin alleles from the canonical EHEC, EPEC, and C. rodentium strains are distinct and have been associated with differences in function. For example, although tissue tropism during infection of human intestinal explants is multifactorial, infection of intestinal tissue ex vivo suggests that intimin of EHEC O157:H7 (also known as intimin γ) promotes colonization of different epithelial types than intimin of canonical EPEC (intimin α) or C. rodentium (intimin β) (Phillips and Frankel, 2000; Fitzhenry et al., 2002; Girard et al., 2005; Mundy et al., 2007). In addition, whereas wild type EHEC colonizes the large bowel of gnotobiotic piglets, an EHEC strain harboring a plasmid expressing EPEC intimin acquired the additional ability to colonize the small intestine (Tzipori et al., 1995). Allelic differences may also contribute to differences in species host range, because whereas C. rodentium expressing EPEC intimin is able to efficiently colonize Swiss NIH and C3H/HeJ mice (Schauer and Falkow, 1993b; Frankel et al., 1996b; Hartland et al., 2000), C. rodentium expressing a derivative EPEC intimin harboring the adhesive domain of EHEC intimin provided only poor colonization function in these animals (Hartland et al., 2000; Mundy et al., 2007).
To gain insight into the critical activities of intimin, we assessed the in vivo functionality of EHEC, EPEC, or C. rodentium intimin, or a set of EHEC intimin derivatives with varying Tir-binding abilities. Colonization was not detectably altered by allele-specific differences in intimin in two animal infection models. Notably, whereas the ability to trigger Tir-mediated pedestal formation was found to be essential for colonization of conventional mice, intimin provided a Tir-independent function during colonization of antibiotic pre-treated mice.
Materials and Methods
Media, Bacterial Strains, and Growth Conditions
Bacteria were stored in Luria–Bertani (LB) broth (American Bioanalytical, Natick, MA, USA) with 50% glycerol at either −80° or −135°C. Bacteria were grown at 37°C in LB broth, in Antibiotic Medium 3 (Difco, Laboratories, Detroit, MI, USA), on LB agar (American Bioanalytical, Natick, MA, USA), on MacConkey lactose agar, or on eosin–methylene blue agar (Difco Laboratories, Lawrence, KS, USA). Where indicated, kanamycin, chloramphenicol, gentamicin, ampicillin, and zeocin were added at final concentrations of 20, 10, 100, 100 or 750, and 75 μg/ml respectively. The bacterial strains and plasmids used in this study are listed in Tables 1 and 2.

Table 1. Bacterial strains used in this study.

Table 2. Description of plasmids used in study.
Construction of pCVD438 (pIntEPEC) Derivatives
Genomic DNA from EHEC O157:H7 strain EDL933 and plasmid pIntEPEC were purified using standard methodologies. All primers used to construct these pCVD438 derivatives are listed in Table 3. To construct pHL69 (pIntEHEC), a two-step amplification (fusion PCR) was performed. First, the Pfu Turbo PCR system (Stratagene, La Jolla, CA, USA) was used to generate three fragments: the region 5′ of the eaeEPEC open reading frame (ORF) in pIntEPEC (from the HindIII site to the eae start codon), the region 3′ of the eaeEPEC ORF in pIntEPEC (from the eae stop codon to the SalI site), and the coding region of the eaeEHEC gene from EDL933. To generate the first two fragments, pIntEPEC was restriction enzyme digested with BstEII to delete an internal 1,847-bp fragment (nucleotides 585–2432 of eaeEPEC) and used as the PCR template. The coding region of EDL933 eaeEHEC was amplified from genomic DNA. Each primer at a junction point in the fusion PCR was tailed with a 9- to 15-bp overhanging sequence from the neighboring fragment. The amplification products were isolated using the QIAquick PCR purification kit (QIAgen Inc., Valencia, CA, USA), mixed, and subjected to a second round of amplification with the pACYC184 primers listed in Table 3. The fusion PCR product was confirmed by DNA sequencing and ligated into the corresponding HindIII/SalI site of pIntEPEC, effectively replacing the eaeEPEC ORF of pIntEPEC with that of EHEC EDL933. The ligation mix was introduced into laboratory strains of E. coli by high-voltage electroporation and chloramphenicol-resistant clones were isolated.

Table 3. DNA sequences of oligonucleotides used in this study.
A similar methodology was used to generate pInv, pInv–Int395, pInv–Int181, and pInv–Int100 (Table 2). In the case of pInv, the coding region of the Y. pseudotuberculosis inv gene was amplified from pRI203. For the latter three plasmids, the hybrid invasin–intimin alleles had been previously generated and cloned into pT7-4 (Liu et al., 1999). These pT7-4 derivatives, pHL35, pHL49, and pHL55, respectively, were used as template DNA to amplify the coding region for pInv–Int395, pInv–Int181, and pInv–Int100, respectively.
Generation of C. rodentium Strains
Plasmid DNA was isolated using the QIAprep Spin Miniprep Kit (QIAgen, Valencia, CA, USA) and quantified by UV spectrophotometry. Plasmids were introduced into the C. rodentiumΔeae strain by chemical transformation using calcium chloride. Successful transformants were selected with chloramphenicol and were confirmed to be carrying the proper plasmid.
Generation of an EHEC Strain Encoding EPEC Intimin
A three-way PCR was performed using templates encoding EHEC cesT with 3′ tail encoding EPEC eae (∼750 bp), EPEC eae (∼2.8 kb) with 3′ EHEC tail, and EHEC 3′UTR (∼960 bp). The PCR product was cloned into pGEM7xf(+), linearized by BamHI and XbaI digestion, and transformed into KM48 harboring pTP223. Transformants were screened by sucrose resistance and chloramphenicol sensitivity. Candidates were confirmed by PCR and sequencing.
Generation of C. rodentiumΔtir
The C. rodentium tir deletion mutant was made using a slightly modified version of the one-step PCR-based gene activation protocol (Datsenko and Wanner, 2000). Briefly, a tertiary PCR product containing the zeocin cassette and its promoter flanked by 576 bp homology upstream of the start of tir and 608 base pairs downstream of the stop of tir was generated using three template PCR products, A + B, C + D, and E + F. Product A + B was generated using primers F-A-Tir-KO and R-B-Tir-KO (Table 3) with genomic C. rodentium (DBS100) DNA (isolated using a kit by Promega, Madison, WI, USA) as a template. Primers F-C-Tir-KO-zeo and R-D-Tir-KO-zeo were used to amplify a MluI/EcoRI cut pDONORzeo fragment to generate the 504-bp PCR product C + D. Primers F-E-Tir-KO and R-F-Tir-KO were used to amplify genomic C. rodentium DNA to make PCR product E + F. This 1.65 kb PCR product was electroporated into C. rodentium (DBS100) containing the lambda-red plasmid, pKD46, and recombinants were selected for by plating on LB plates supplemented with 750 μg/ml ampicillin. Replacement of tir with the zeocin cassette and its promoter was confirmed by PCR using the following primers: F-Tir-Ext, R-Tir-Ext, F-Tir-Int-323–342, R-Tir-Int-819–838, F-Tir-Int-590–610, R-Tir-Int-1049–1069, F-Zeo-Int, and R-Zeo-Int (see Table 3).
Purification of EHEC Tir and Generation of an Anti-EHEC Tir Antibody
pDV205 (ampR) is a derivative of pET21 that contains EHEC tir with a histidine tag. For expression and purification of EHEC Tir, BL21 DE3+ pDV205 was cultured in 2x YT media at 37°C to an OD600 of 0.6–0.7 then induced with 1 mM IPTG for 3 h at 37°C. The culture was spun at 4,420 × g, 20 min at 4°C, and the supernatant was discarded. The pellet was resuspended in 2.5 ml lysis buffer per gram wet weight. Four milligrams of lysozyme was added and the sample was sonicated (Branson Sonifier 450, Branson Ultrasonics Corporation, Danbury, CT, USA) Duty cycle 70, output 3, 10 s bursts × 6 cycles). The lysate was then centrifuged at 16,100 × g, 20 min at 4°C, and the supernatant was run on a QIAgen Ni-NTA Agarose column (QIAgen, Valencia, CA, USA) and the resulting EHEC Tir was eluted and quantified using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
For antibody production, all inoculations were completed subcutaneously with 30 ng of purified EHEC Tir in 50 μl with 50 μl of adjuvant for a total inoculation volume of 100 μl. Female BALB/c mice (Jackson Laboratories, Bar Harbor, ME, USA) were inoculated with EHEC Tir supplemented with Imject Freund’s Complete Adjuvant (Thermo Scientific, Rockford, IL, USA). At days 14 and 28, mice were boosted with EHEC Tir supplemented with Imject Freund’s Incomplete Adjuvant (Thermo Scientific, Rockford, IL, USA). Mice were sacrificed on day 38 and blood was harvested via cardiac bleed, allowed to clot at room temperature for 30 min, and then was spun twice at 8,600 × g, 15 min at 4°C. Serum was then aliquoted and stored at −80°C.
Invasion Assays
Entry into cultured cells was measured in a manner similar to that described previously (Finlay and Falkow, 1988). Briefly, HEp-2 cells (ATCC CCL-23) were seeded in 24-well plates and grown overnight at 37°C in 5% CO2 and Dulbecco’s Modified Eagles medium supplemented with 10% heat inactivated calf serum and 2 mM L-glutamine (DMEM). Bacteria were grown overnight in LB, diluted in fresh cell culture medium, and inoculated at an approximate multiplicity of infection (MOI) of 100. The cells were incubated for 3 h at 37°C in 5% CO2 and then washed with phosphate-buffered saline (PBS). Fresh cell culture medium supplemented with gentamicin was added to each well and the cells were incubated at 37°C in 5% CO2 for 1 h. The cells were washed with PBS, lysed with 1% Triton X-100 in PBS, and diluted to 1 ml with LB broth. Percent invasion for each well was determined by plating dilution series.
Infection of Monolayers Pre-Infected with EPEC eae
The “prime and challenge” assay is a modification of a previously described bacterial adherence assay (Rosenshine et al., 1996; Liu et al., 1999) and was used to quantify intimin–Tir interactions in vitro. Briefly, nearly confluent monolayers of HEp-2 cells were washed twice in PBS, and RHFM [RPMI-1640 supplemented with 20 mM HEPES, pH 7.0, 2% fetal bovine serum (FBS), and 0.5% D-mannose] was added to the monolayers. The monolayers were then mock-infected or infected with the EPEC eae mutant strain JPN15.96 at MOI of ∼200 and incubated at 37°C in 5% CO2 for 3 h. JPN15.96 is capable of high efficiency translocation of Tir into mammalian cells, but unable to form pedestals due to the absence of intimin. The monolayers were washed twice with PBS and then incubated for 1 h at 37°C in 5% CO2 with 100 μg/ml gentamicin in DMEM/HEPES to kill remaining bacteria. The monolayers were washed three times in PBS and then infected with the appropriate “challenge” bacterium (i.e., C. rodentium, C. rodentiumΔeae, or C. rodentiumΔeae expressing EPEC intimin, EHEC intimin, invasin, or an invasin–intimin hybrid) diluted in fresh DMEM/HEPES at an MOI of 100 for 3 h at 37° in 5% CO2. At the conclusion of the assay, monolayers were washed six times with PBS, lysed with 1% Triton X-100 in PBS, and diluted to 1 ml with LB broth. Percent adherence for each well was determined by plating dilution series.
The prime and challenge assay was also adapted to evaluate actin pedestal formation mediated by intimin and its derivatives by seeding HEp-2 cells onto coverslips. At the conclusion of the assay described above, coverslips were washed six times with PBS, fixed with 2.5% paraformaldehyde, and permeabilized with 0.1% Triton X-100 in PBS. Coverslips were then double fluorescently labeled for F-actin and bacteria and examined on a Nikon Eclipse E600 microscope. F-actin was labeled with Texas red-conjugated phalloidin (Molecular Probes, Eugene, OR, USA) and visualized with a 580-nm dichroic filter. C. rodentium were labeled with an anti-C. rodentium polyclonal rabbit antibody raised against C. rodentium strain DBS100 and visualized with a Cascade blue-conjugated goat anti-rabbit IgG antibody (Molecular Probes, Eugene, OR, USA) using a 400-nm dichroic filter. Coverslips labeled only with anti-C. rodentium primary antibody and the Cascade blue-conjugated secondary antibody showed no crossover when viewed with the 580-nm filter. Image acquisition was performed with the Spot program (Diagnostics Instruments, software version 3.0.4) and imported into Adobe Photoshop 5.0.
Pedestal, Bacterial Binding, and Tir Focusing Assays
The FAS assay of (Nicholls et al., 2000), as modified for C. rodentium (Newman et al., 1999) was the basis for this assay. A single colony from each strain was grown in 1 ml media (+/− antibiotic) for 8 h. Cultures were diluted 1:500 into 5 ml DMEM supplemented with 0.1 M HEPES (pH 7.0; +/− antibiotic) and incubated at 37°C without agitation with 5% CO2 for 12–15 h. Cell monolayers were prepared by splitting 95–100% confluent mouse embryonic fibroblasts (MEFs) into 24-well culture plates containing sterile glass coverslips followed by overnight growth at 37°C with 5% CO2. Prior to seeing onto culture plates, MEFs were maintained in MEF cell culture media [DMEM (hi glucose) + 10% FBS with penicillin, streptomycin, and glutamine] at 37°C, 5% CO2. For infections, cell monolayers were washed twice with sterile PBS followed by addition of FAS media containing 25 μl of overnight cultured C. rodentium to each well. Plates were spun at 700 g for 10 min then incubated at 37°C with 5% CO2 for 3 h. After 1.5 h, plates were spun again at 700 g for an additional 10 min to insure proper bacterial binding to cells. After 3 h, cells were washed twice with sterile PBS and 0.5 ml pre-warmed FAS media was added to each well and cells were then incubated for an additional 3 h. Cells were then washed five times with sterile PBS, fixed with 4% PFA for 30 min, washed, permeabilized with 0.1% Triton X-100, and stained with anti-EHEC Tir antibody (1:500) as a primary antibody for 30 min. The samples were washed and then stained for 30 min with anti-Mouse 488 secondary antibody (Invitrogen, Carlsbad, CA, USA; 1:200), phalloidin (Molecular Probes, OR, USA; 1:100), and DAPI (1:500). After washing cells an additional three times with PBS, the coverslips were mounted on slides using ProLong® Gold anti-fade reagent (Invitrogen, Eugene, OR, USA). For binding assays, the number of bacteria per cell was counted for 25 random cells. Counting was done in triplicate and the mean, median, and maximum number of bacteria bound to cells was determined. The stratification of the number of cells with the given amount of bound bacteria in each interval (0, 1–5, 6–10, 11–15, 16–20, and >20) was also determined.
Immunoblotting
Preparation of bacterial cell lysates was performed as described previously (Campellone et al., 2002; Brady et al., 2007). Samples were resolved by 10% SDS PAGE and transferred to PVDF membrane. Membranes were blocked in PBS supplemented with 5% milk for 30 min prior to treatment with sheep anti-EHEC Intimin (1:500), goat anti-EPEC Intimin (1:2000), rabbit anti-OmpA, or rabbit anti-O157 antiserum (1:750, Difco Laboratories, Lawrence, KS, USA) for 2 h, washed and treated with secondary antibody as previously described (Campellone et al., 2004a; Campellone and Leong, 2005; Brady et al., 2007).
Immunofluorescence Microscopy of EHEC Strains
To ensure an EHEC strain expressing EPEC intimin can generate actin pedestals and that intimin plasmids used to overexpress EHEC and EPEC intimin are functional to complement a deletion of EHEC intimin by FAS, HeLa cell monolayers were infected with indicated strains, fixed, and permeabilized as described previously (Campellone et al., 2002). For each strain, qualitative scoring of F-actin pedestals was performed as indicated. Approximately equal numbers of actin pedestals were observed for EHECΔeae expressing EHEC intimin (pHL6) or EPEC intimin (pHL4).
Mouse Infection Studies
Mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and housed in the UMMS animal facility. All animal procedures were done in compliance with the University of Massachusetts Medical School IACUC. Female, eight-week-old C57BL/6J mice were gavaged with PBS or ∼2 × 109 of overnight culture of C. rodentium strain specified in 100 μl PBS. Inoculum concentrations were confirmed by serial dilution plating. C. rodentium fecal shedding was determined by serial dilution plating of fecal slurries (10% w/v in PBS) on Maconkey agar or LB agar with selection for zeocin, kanamycin, chloramphenicol, or kanamycin and chloramphenicol. For streptomycin pre-treatment experiments, mice were given 5 mg/ml streptomycin for 48 h prior to infection.
Mouse Tissue Collection and Histology
At necropsy the large intestine (colon through cecum) was fixed in 10% buffered formalin, dehydrated, and embedded in paraffin. Five-micron sections stained with hematoxylin and eosin were evaluated and scored blindly by a board-certified pathologist (Vijay Vanguri). Assessment of mucosal hyperplasia was targeted to areas of comparable muscularis propria thickness in order to reduce error from differences in planes of section.
Transmission Electron Microscopy
Mouse intestinal tissue samples were taken at various time points post-infection and fixed in 2.5% glutaraldehyde in 0.05 M Sodium Phosphate buffer, pH 7.2. Samples were processed and analyzed at the University of Massachusetts Medical School Electron Microscopy core facility according to standard procedures. Briefly, fixed samples were moved into fresh 2.5% glutaraldehyde in 0.05 M Sodium Phosphate buffer and left overnight at 4°C. The samples were then rinsed twice in the same fixation buffer and post-fixed with 1% osmium tetroxide for 1 h at room temperature. Samples were then washed twice with DH2O for 20 min at 4°C and then dehydrated through a graded ethanol series of 20% increments, before two changes in 100% ethanol. Samples were then infiltrated first with two changes of 100% Propylene Oxide and then with a 1:1 mix of propylene oxide: SPI-Pon 812 resin. The following day three changes of fresh 100% SPI-Pon 812 resin were done before the samples were polymerized at 68°C in plastic capsules. The samples were then reoriented and thin sections were placed on copper support grids and stained with lead citrate and uranyl acetate. Sections were examined using the FEI Tecani 12 BT with 80 kV accelerating voltage, and images were captured using a Gatan TEM CCD camera.
Piglet Infections
Gnotobiotic piglets were derived and infected orally with TUV93-0, which is deficient for the expression of Stx, thus eliminating the potentially confounding toxigenic effects on colonization (Tzipori et al., 1995), as described previously (Campellone et al., 2007; Ritchie et al., 2008; Brady et al., 2011). To quantify bacteria in the small intestine, the small intestine was divided into five parts and each section was viewed individually for the presence of bacteria and AE lesions. If bacteria or AE lesions were found in any of these sections, they were given the score of “+.” The average score was then determined for the small intestines of piglets in each group. A score of “+” indicates the presence of bacteria and AE lesions in at least one of the five sections of the small intestine in each of the piglets, a score of “+/−” indicates the presence of bacteria and AE lesions in at least one of the five sections of the small intestine in at least half of the piglets, and a score of “−” indicates that fewer than half of the piglets had bacteria and AE lesions in at least one of the five sections of the small intestine and also includes piglets that did not become colonized. To enumerate bacteria in the spiral colon and cecum, each section was scored for the presence of bacteria and AE lesions. A score of “+” indicates that every piglet in the group had bacteria and AE lesions present, a score of “+/−” indicates that at least half of the piglets in the group had bacteria and AE lesions present, and a score of “−” indicates that fewer than half of the piglets had bacteria and AE lesions present.
Statistical Analysis
Data were analyzed using GraphPad Prism. Comparison of multiple groups was performed using one-way analysis of variance (ANOVA) with Bonferroni multiple comparison post-tests. Statistical significance of differences between two groups was evaluated using two-tailed unpaired t-tests. In all tests p-values below 0.05 (*), 0.01 (**), and 0.001 (***) were considered statistically significant, unless indicated otherwise. In all graphs, error bars represent SEM.
Results
Precise Chromosomal Replacement of EHEC eae with EPEC eae Does Not Alter Tissue Tropism in Piglets
Plasmid complementation of an EHEC eae mutant with EPEC eae results in a strain with altered tissue tropism that colonizes the small bowel of piglets (Tzipori et al., 1995). To determine if EPEC and EHEC intimin might be functionally interchangeable when expressed from the endogenous chromosomal locus, we precisely replaced the EHEC eae allele in EHEC TUV93-0 with the eae allele from EPEC strain JPN15/pMAR7 (Figure 1A). Immunoblotting confirmed that the resulting strain, LM-1, produced EPEC intimin (Figure 1B). As predicted from previous work demonstrating that EHEC and EPEC intimin are functionally interchangeable for in vitro pedestal formation (DeVinney et al., 1999), LM-1 generated actin pedestals on cultured cells (Figure 1C).
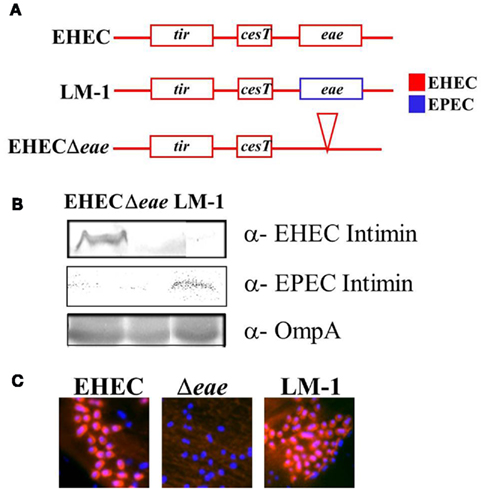
Figure 1. An EHEC strain containing EPEC intimin on the chromosome is functional for pedestal formation. (A) The endogenous EHEC eae gene was precisely replaced by EPEC eae to create LM-1. (B) Expression of EHEC and EPEC intimin in wild type EHEC, EHECΔeae, and LM-1 was assessed by western blot. Blotting with anti-OmpA was included as a loading control. (C) HeLa cells were infected with the indicated strain and the monolayers were stained with DAPI (blue) to visualize bound bacteria and phalloidin (red) to visualize actin pedestals.
To test the function of EPEC intimin during infection by EHEC, gnotobiotic piglets were infected orally with LM-1 and after 72 h, infection was assessed by histological analysis of the small and large intestines. Upon infection with wild type EHEC, piglets suffered diarrheal illness and bacteria were associated with extensive segments of the cecal and colonic epithelium (Table 4). Wild type EHEC were only occasionally associated with the epithelium of the small intestine. In contrast, infection of gnotobiotic piglets with EHECΔeae did not result in detectable bacterial association with epithelium of any intestinal segment, and correspondingly, the animals did not suffer from diarrhea. Notably, EHEC strain LM-1, which expresses EPEC intimin, colonized the epithelium of the cecum and spiral colon at levels indistinguishable from that of wild type EHEC, and like the wild type strain, induced diarrheal illness (Table 4). In contrast to the previous finding that EHEC expressing EPEC intimin from a plasmid could efficiently colonize the small intestine of gnotobiotic piglets (Tzipori et al., 1995), bacteria were vanishingly sparse in the small intestine (Table 4). These results demonstrate that EPEC intimin, when expressed in EHEC from the endogenous chromosomal locus, can provide intimin function during intestinal infection of piglets, but does not influence tissue tropism in this model.

Table 4. An EHEC strain carrying a precise chromosomal replacement of eae with EPEC eae displays piglet intestinal tropism indistinguishable from wild type.
EHEC Intimin Can Promote Murine Colonization and Disease by C. rodentium
We further assessed ability of EHEC intimin to complement a C. rodentiumΔeae mutant for colonization and intestinal disease in mice. Previous work demonstrated that C. rodentiumΔeae harboring pCVD438, which encodes EPEC intimin, was fully virulent in Swiss NIH and C3H/HeJ mice (Hartland et al., 2000; Mundy et al., 2007). In contrast, C. rodentiumΔeae expressing an intimin hybrid carrying the N-terminal 554 residues of EPEC intimin and the C-terminal 380 residues of EHEC intimin colonized mice 100-fold less efficiently and did not cause the colonic hyperplasia that is characteristic of C. rodentium infection (Hartland et al., 2000; Mundy et al., 2007).
To compare the colonization function of EPEC and EHEC intimin in mice, we precisely replaced the EPEC eae coding sequence of pCVD438 (which for simplicity we herein refer to as “pIntEPEC”) with the EHEC eae coding sequence to create pIntEHEC (i.e., pHL69; Table 2). C57BL/6 mice were infected with approximately 5 × 109 CFU of C. rodentium, C. rodentiumΔeae, C. rodentiumΔeae/pIntEPEC, or C. rodentiumΔeae/pIntEHEC. Mice infected with C. rodentium reached peak colonization levels seven days post-infection with approximately 109 bacteria per gram of feces before being cleared from mice by approximately 15–20 days after inoculation, presumably due to the development of an adaptive immune response (Ghaem-Maghami et al., 2001; Vallance et al., 2002; Simmons et al., 2003; Figure 2A and data not shown). In contrast, the number of C. rodentiumΔeae in the stool peaked at three days post-infection, reaching less than 106 bacteria per gram of feces, and quickly diminished, never rising to more than 104 bacteria per gram of feces thereafter (Figure 2A). Similar to what was previously reported (Frankel et al., 1996b; Hartland et al., 2000; Mundy et al., 2007), EPEC intimin, when expressed in C. rodentiumΔeae promoted high level colonization with comparable kinetics to wild type C. rodentium (Figure 2A). Additionally, intestinal sections from rodents infected with C. rodentiumΔeae expressing EPEC intimin showed colonic mucosal hyperplasia, goblet cell depletion, acute inflammation, erosions, and degenerative epithelial changes on the surface and in the crypts (Figure 2B). Interestingly, we also found that EHEC intimin, when expressed in C. rodentiumΔeae, promoted colonization, colonic hyperplasia, and intestinal damage indistinguishable from wild type C. rodentium (Figure 2) and generated AE lesions morphologically identical to wild type C. rodentium (Figure 3). Thus, EHEC intimin, in spite of being significantly divergent in sequence from C. rodentium intimin, could provide complete intimin function in this model.
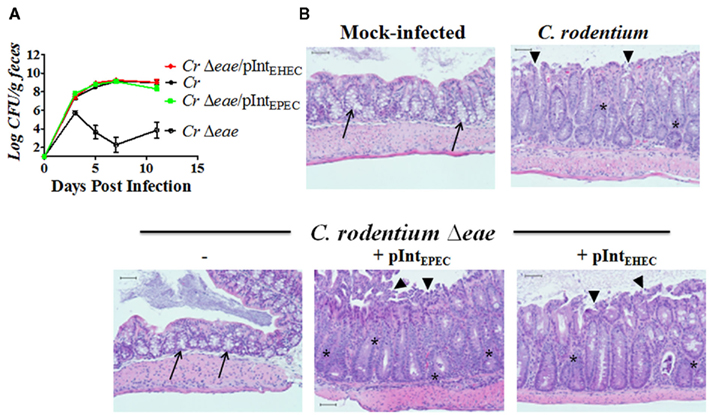
Figure 2. Enterohemorrhagic E. coli intimin is able to restore colonization and disease in a C. rodentium eae mutant. (A) Colonization of eight-week-old C57BL/6 mice by C. rodentium (“Cr”), C. rodentiumΔeae, C. rodentiumΔeae/pIntEPEC, or C. rodentiumΔeae/pIntEHEC was determined by viable stool counts. Shown are the averages CFU (±SEM) of five mice. Data shown are representative of one of three independent experiments. (B) Hematoxylin and eosin stained large intestinal sections from mock-infected mice or mice infected with indicated strain are shown under 100× magnification. Arrow heads point to ragged areas of the surface epithelium at sites of surface erosion. Arrows point to areas of many goblet cells. Asterisks indicate areas of active inflammation. Scale bars measure 50 μm.
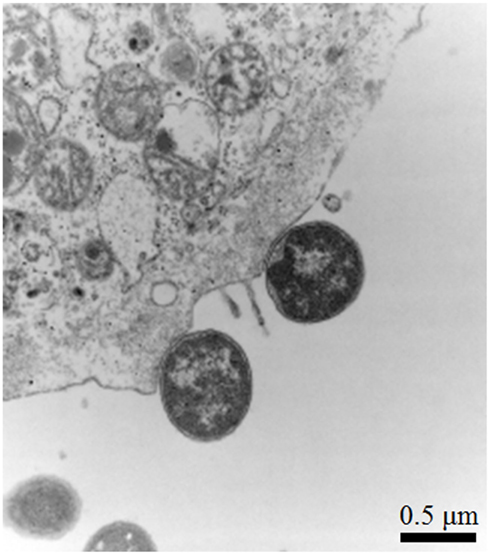
Figure 3. Enterohemorrhagic E. coli intimin can complement a C. rodentium eae mutant for AE lesion formation in vivo. Transmission electron micrograph showing AE pedestal formation on colonic epithelial cells from the distal colon of mice infected with C. rodentiumΔeae/pIntEHEC. Original magnification, 25,000×. Bar, 0.5 μm.
EHEC Intimin and Tir-Binding Invasin–Intimin Fusions Can Mediate Host Cell Adherence by C. rodentium
We next characterized the ability of C. rodentiumΔeae expressing EHEC intimin to attach to host cells that express high levels of Tir on their surface, which more sensitively assesses Tir–intimin interactions compared to conventional infection assays (Liu et al., 2002). Pre-infection (i.e., “priming”) of cells with an EPECΔeae mutant permits efficient delivery of Tir to the eukaryotic cell (Rosenshine et al., 1996; Liu et al., 1999). After gentamycin treatment and washing to kill and remove bound bacteria, infection (i.e., “challenge”) of these monolayers with intimin-expressing bacteria permits sensitive assessment of bacterial binding mediated by Tir–intimin interaction (Rosenshine et al., 1996; Liu et al., 1999). Utilizing this assay, C. rodentiumΔeae/pIntEHEC bound to pre-infected monolayers indistinguishably from that of wild type C. rodentium, and approximately 20-fold more efficiently than C. rodentiumΔeae (Figure 4A).
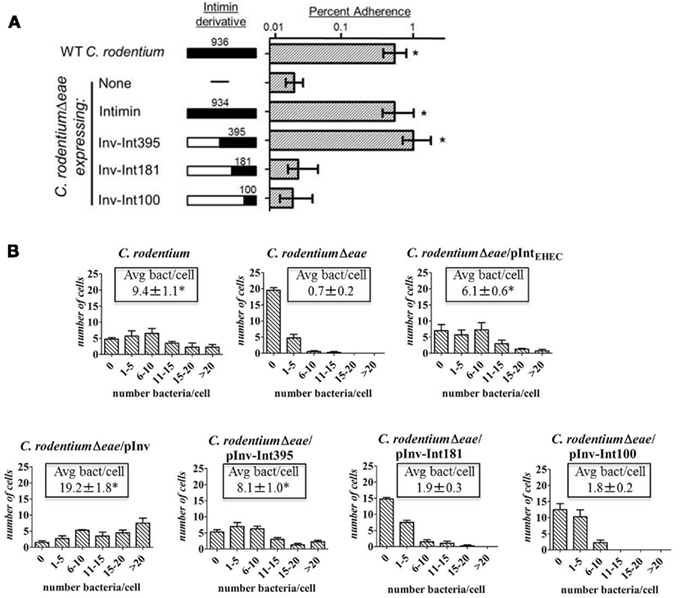
Figure 4. A hybrid containing the C-terminal 395 amino acids of EHEC intimin can complement a C. rodentium eae mutant for host cell attachment. (A) HEp-2 cells pre-infected with an EPECΔeae strain were infected with wild type C. rodentium, C. rodentiumΔeae, or C. rodentiumΔeae strains expressing EHEC intimin, invasin, or the indicated invasin–EHEC intimin hybrids (Inv–Int395, Inv–Int181, and Inv–Int100), and stably bound bacteria were determined (Materials and Methods). Data are shown as the mean ± SEM and represent two independent assays with at least three replicates per assay. Asterisk indicates binding significantly (p < 0.05) higher than C. rodentiumΔeae of C. rodentiumΔeae expressing pInv–Int181 or pInv–Int100, determined by two-tailed unpaired t-test. (B) Mouse embryonic fibroblasts were infected with the indicated C. rodentium strains and the number of bound bacteria per cell was counted for four sets of 25 randomly selected cells. Shown are the number of cells with the given number (0, 1–5, 6–10, 11–15, 16–20, and >20) of bound bacteria. The experiment was performed in triplicate. Shown in box is the mean number of bacteria per cell ± SEM. Asterisk indicates a significant difference (p < 0.05) compared to C. rodentiumΔeae, determined using a one-way ANOVA test.
We next characterized the minimal region of EHEC intimin required for Tir-mediated cell adherence when expressed in C. rodentium. The intimin related protein Y. pseudotuberculosis invasin, which binds to β1-chain integrins (Isberg and Leong, 1990; Frankel et al., 1996a), was previously used to deliver various portions of EHEC intimin to the surface of the bacteria in the form of invasin–intimin hybrid proteins (Liu et al., 1999). C. rodentiumΔeae expressing invasin was capable of high efficiency entry into cultured epithelial cells, strongly suggesting that invasin can be expressed in functional form on the surface of C. rodentium (data not shown). Thus, we determined the minimal region of EHEC intimin required for cell adherence by C. rodentium by expression of invasin–intimin fusions in this bacterium. C. rodentiumΔeae producing Inv–Int395, a hybrid that contains the C-terminal 395 amino acids of intimin, bound to pre-infected monolayers at wild type levels, implying that this hybrid promotes Tir-binding when expressed in C. rodentiumΔeae, as it does when expressed in E. coli K12 (Figure 4A; Liu et al., 1999). In contrast, Inv–Int181, which was previously shown to retain some Tir-binding activity when expressed in E. coli K12 (Liu et al., 1999), and Inv–Int100, bound poorly to monolayers pre-infected with the EPECΔeae mutant (Figure 4A). These data suggest that the minimal functional domain of EHEC intimin that can confer high level Tir-binding on C. rodentium is located in the C-terminal 395 amino acids.
To determine if the functional Tir-binding domain defined by the more elaborate prime and challenge assay described above also functioned to promote cell attachment by C. rodentium during simple infection, we infected MEFs with C. rodentiumΔeae expressing invasin or EHEC intimin derivatives and determined the number of bacteria bound to each of 25 randomly selected cells. Cells infected with wild type C. rodentium harbored an average of more than nine bacteria per cell, and fewer than five of the 25 cells were entirely free of bacteria (Figure 4B). Intimin was required for binding, because approximately 20 of the 25 cells infected with C. rodentiumΔeae were bacteria-free, and virtually no cells harbored more than five bacteria (Figure 4B). EHEC intimin, when expressed in C. rodentiumΔeae, was able to restore binding capabilities of the mutant strain, with an average of approximately six bacteria per cell (Figure 4B).
We similarly tested the ability of the invasin–intimin hybrids to bind to cells. Invasin, when expressed on the surface of C. rodentiumΔeae, dramatically enhanced the ability of the bacteria to associate with cells (p < 0.05), with an average of approximately 19 bacteria per cell, consistent with its ability to promote high-level host cell interaction (Figure 4B; Isberg and Leong, 1990). C. rodentiumΔeae expressing Inv–Int395, which mediated attachment of C. rodentium in the prime and challenge assay (Figure 4A), bound to mammalian cells statistically indistinguishably from wild type C. rodentium, with an average of approximately eight bacteria bound to each cell (Figure 4B). On average, fewer than five of the 25 cells were bacteria-free. In contrast, C. rodentium expressing Inv–Int181 and Inv–Int100 bound to cells indistinguishably from C. rodentiumΔeae, with an average of less than two bacteria per cell and the majority of cells harboring fewer than five bacteria (Figure 4B). These data indicate that EHEC intimin can complement a C. rodentiumΔeae mutant for cell attachment, and that the binding function of this protein is encompassed by the C-terminal 395 amino acids.
EHEC Intimin, but Not a Tir-Binding Invasin–Intimin Hybrid, Promotes Tir Clustering and Pedestal Formation by C. rodentium
Given the ability of EHEC intimin and the hybrid Inv–Int395 to promote binding of C. rodentium to mammalian cells, we next tested their ability to promote Tir clustering and pedestal formation upon infection of mammalian cells. As expected, cells infected with C. rodentiumΔeae demonstrated virtually no bound bacteria and after staining monolayers with anti-EHEC Tir, only one quarter of the rare bound bacteria were associated with somewhat diffuse foci of Tir (Figure 5, column 2; and data not shown). In contrast, upon infection with wild type C. rodentium, 95% of bound bacteria were associated with intensely stained Tir foci, and 98% of bacteria generated actin pedestals (Figure 5, “Cr”). These defects in Tir clustering and pedestal formation were fully complemented by a plasmid expressing EHEC intimin, as well as by a plasmid expressing EPEC intimin, which was included as a positive control (Figure 5, “pIntEHEC” and “pIntEPEC”, respectively). Thus, EHEC intimin appears to provide full function for cell binding, Tir clustering, and pedestal formation when expressed in C. rodentium.
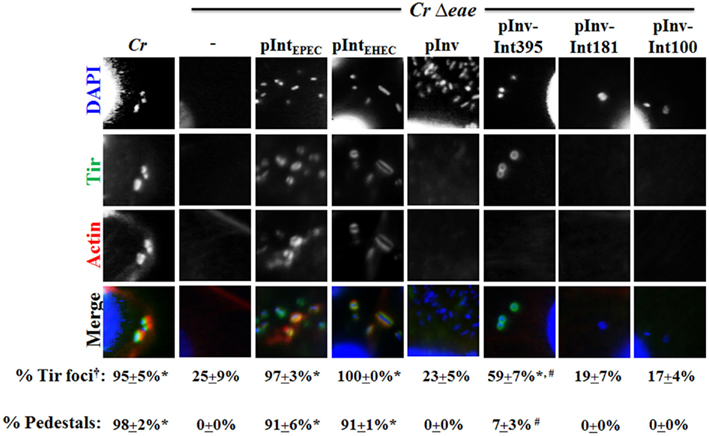
Figure 5. Enterohemorrhagic E. coli intimin, but not a Tir-binding invasin–intimin hybrid, promotes Tir clustering and pedestal formation by C. rodentium. Monolayers were infected with the indicated C. rodentium (“Cr”) strains and stained with DAPI to visualize bacteria (blue), anti-EHEC Tir antibody (green), and fluorescent phalloidin (red) to detect F-actin. Twenty-five bound bacteria were scored for their association with Tir foci or pedestals. †Note that when quantifying the percentage of cells infected with C. rodentiumΔeae that had Tir foci and/or actin pedestals associated with bound bacteria, many more cells needed to be counted given the inability of this strain to adhere efficiently to cells. Data represent quadruplicate samples. Asterisk indicates that percent positive bacteria is significantly (p < 0.05) greater than that for C. rodentiumΔeae. “#” Indicates that percent positive bacteria is significantly (p < 0.05) lower than that for C. rodentium/pIntEHEC.
To define the region of EHEC intimin critical for Tir clustering and pedestal formation, in parallel we infected monolayers with C. rodentiumΔeae expressing invasin–intimin hybrids. C. rodentiumΔeae expressing invasin or either of the two fusion proteins, Inv–Int181 or Inv–Int100, that were unable to promote Tir-mediated cellular attachment by C. rodentium, resulted in detectable Tir foci no more frequently than was found for C. rodentiumΔeae (Figure 5, “pInv”, “pInv–Int181”, or “pInv–Int100”). In contrast, 59% of C. rodentiumΔeae expressing Inv–Int395, which promotes binding to C. rodentium to mammalian cells (Figure 4), were associated with Tir foci (Figure 5, “pInv–Int395”), a percentage that was both significantly higher than that of C. rodentiumΔeae and significantly lower than that of wild type C. rodentium or C. rodentiumΔeae expressing wild type EHEC intimin. The partial complementation of Tir clustering by pInv–Int395 was also reflected in the low (i.e., 7%) frequency of pedestal formation (Figure 5, “pInv–Int395”). Thus, although the expression of Inv–Int395 in C. rodentium resulted in high-level Tir-mediated cell binding, it did not result in efficient Tir clustering or pedestal formation in cultured mammalian cells.
A Tir-Binding Invasin–Intimin Fusion Protein Does Not Promote Murine Colonization by C. rodentium and Results in Minimal Colonic Hyperplasia
After characterizing the invasin–intimin hybrids in vitro, we next determined their ability to complement a C. rodentiumΔeae mutant for colonization and disease in vivo. As shown above (Figure 2A), after oral gavage of C57BL/6 mice with approximately 5 × 109 C. rodentiumΔeae/pIntEHEC, C. rodentiumΔeae expressing EHEC intimin demonstrated wild type levels of colonization, colonic hyperplasia, goblet cell depletion, and abundant inflammation (Figures 6A,B). In contrast, in most instances C. rodentiumΔeae/pInv displayed colonization kinetics indistinguishable from C. rodentiumΔeae (Figure 6A) and did not trigger colitis (Figure 6B), showing that bacterial binding to β1-chain integrins, even by a high affinity ligand such as invasin (Tran Van Nhieu and Isberg, 1993) is not sufficient to promote C. rodentium colonization. (Occasionally, C. rodentiumΔeae/pInv colonized mice somewhat better than C. rodentiumΔeae, but always at a level several orders of magnitude lower that wild type C. rodentium or C. rodentium expressing EHEC intimin and usually just above the limit of detection, i.e., 100 CFU/g feces; data not shown).
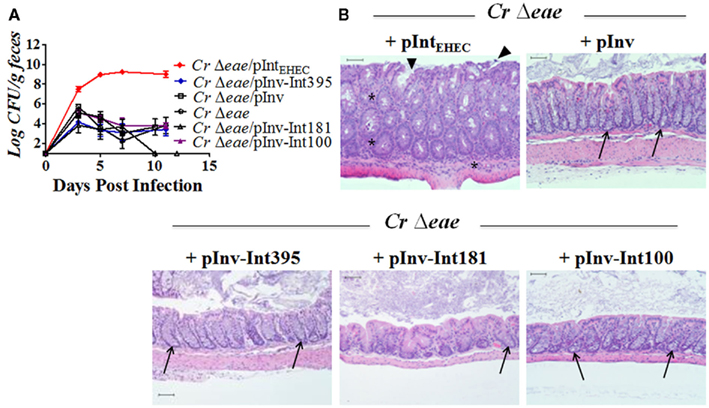
Figure 6. A Tir-binding invasin–intimin fusion protein does not promote murine colonization by C. rodentium. (A) Eight-week-old C57BL/6 mice were inoculated with indicated C. rodentium strains by oral gavage and intestinal colonization was determined by viable stool counts. Shown are the averages CFU (±SEM) of groups of five mice. Each strain was tested in at least three independent experiments. The data displayed includes data from two different experiments, one in which C. rodentiumΔeae/pInv and C. rodentiumΔeae were analyzed and another in which the remaining strains were tested. The combined results are overlaid on the graph for ease of comparison. (B) Hematoxylin and eosin stained large intestinal sections from mice infected with designated strain were analyzed upon 100× magnification. Scale bars measure 50 μm. Arrow heads indicate foci of epithelial surface disruption. Arrows indicate areas of goblet cells and asterisks indicate foci of active inflammation.
As expected, C. rodentiumΔeae expressing Inv–Int181 or Inv–Int100, neither of which mediated mammalian cell attachment, Tir clustering, or pedestal formation in vitro, colonized mice no better than C. rodentiumΔeae and resulted in no detectable intestinal histopathology (Figures 6A,B). Notably, C. rodentiumΔeae expressing Inv–Int395, which promoted Tir-mediated attachment to cultured monolayers but did not cluster Tir efficiently or trigger pedestal formation on cultured cells, was as defective as C. rodentiumΔeae in colonization and triggered virtually no intestinal damage (Figures 6A,B). The inability of Inv–Int395 to demonstrate wild type intimin function during mammalian infection demonstrates that the ability of intimin to bind Tir, in the absence of efficiently clustering Tir and triggering pedestal formation, is insufficient to promote colonization and disease.
Intimin But Not Tir is Required for C. rodentium Colonization of Mice Pre-Treated with Streptomycin
Streptomycin pre-treatment of mice permits EHEC, which is not normally an efficient mouse pathogen, to both colonize mice and cause toxigenic disease (Wadolkowski et al., 1990a,b; Melton-Celsa et al., 1996; Mohawk and O’Brien, 2011). Pilot experiments indicated that brief streptomycin pre-treatment of out-bred Swiss-Webster mice facilitated C. rodentium infection (David B. Schauer, Joseph Newman, and Steven A. Luperchio, unpublished observations), so we pre-treated C57BL/6 mice with streptomycin for 48 h prior to infection with C. rodentium strains that display different capacities to generate actin pedestals. Wild type C. rodentium colonized these mice with high efficiency, reaching ∼108–109/g feces by day three after inoculation and persisting at that level for ∼15 days (Figure 7A, “Cr”). Intestinal colonization diminished after day 15 post-infection, but unlike infection of untreated mice, easily detectable CFU were present until the experiment was terminated at 31 days post-infection. Although C. rodentiumΔeae was present in the stool at high levels at three days post-infection, this strain was not capable of stable colonization (Figure 7A, “CrΔeae”). The colonization defect was fully complemented by plasmid-borne EHEC or EPEC intimin for at least the first 15 days post-infection (Figure 7A, “CrΔeae/pIntEHEC” or “CrΔeae/pIntEPEC,” respectively). At later time points, complementation was not complete, as bacteria expressing either EPEC or EHEC intimin were cleared somewhat more rapidly than wild type C. rodentium. These data indicate even when the flora is disrupted by antibiotic pre-treatment, intimin is required for maximal murine colonization. In addition, the late time point differences notwithstanding, EHEC or EPEC intimin were able to provide wild type colonization function for the first two weeks of infection.
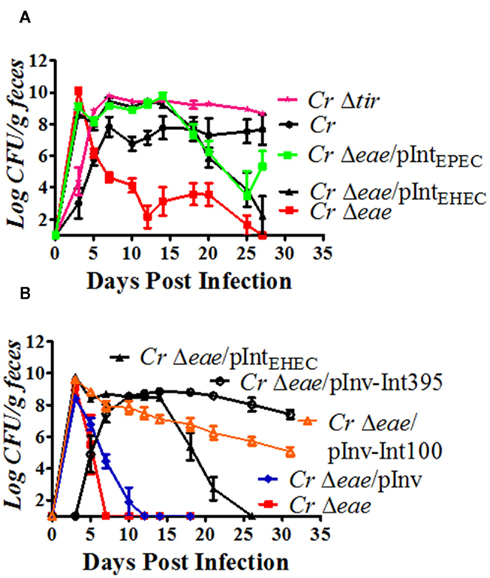
Figure 7. Intimin but not Tir is required for colonization of streptomycin pre-treated mice by C. rodentium. (A,B) Eight-week-old C57BL/6 mice were pre-treated with streptomycin in their drinking water for 48 h prior to being inoculated with approximately 5 × 109 CFU of indicated C. rodentium strains by oral gavage (see Materials and Methods), with the exception of C. rodentiumΔtir in (A), in which approximately 106 CFU were inoculated. Colonization was determined by viable stool counts, and shown are the averages CFU (±SEM) of three to five mice per group. [Note that the slightly lower level of colonization by C. rodentiumΔtir in (A) at three days post-infection was likely due to the lower inoculum used]. Each strain was tested in at least two independent experiments.
To determine whether the requirement for intimin was a reflection of its ability to interact with Tir, we inoculated streptomycin pre-treated mice with a C. rodentiumΔtir mutant. Notably, whereas Tir is essential for colonization by C. rodentium in conventional mice (Deng et al., 2003), we found that Tir was dispensable in streptomycin pre-treated mice. In fact, fecal counts of C. rodentiumΔtir reached approximately 1010 CFU/gram of feces by six days post-infection and remained highly elevated thereafter (Figure 7A, “CrΔtir”). Clearly, intimin provides a Tir-independent colonization function in this infection model.
To test whether the potential integrin binding activity of intimin might be the Tir-independent activity that provides this function, we infected streptomycin pre-treated mice with C. rodentiumΔeae harboring pInv, which expresses the β1-chain integrin ligand invasin. C. rodentiumΔeae/pInv, like C. rodentiumΔeae, was present at high levels transiently (at post-infection day three) in the stool of infected mice but was typically unable to sustain infection, suggesting that integrin binding of intimin did not contribute to colonization function (Figure 7B, “CrΔeae/pInv”). (As was the case for infection of untreated C57BL/6 mice, occasionally the expression of invasin promoted a level colonization several orders of magnitude lower than C. rodentium that expressed endogenous or EHEC intimin; data not shown).
Finally, to determine what region of intimin was required to promote infection of antibiotic pre-treated mice, we infected mice with a C. rodentiumΔeae/pInv–Int395 or C. rodentiumΔeae/pInv–Int100. We found that expression of either Inv–Int395, which binds Tir, or Inv–Int100, which does not, was sufficient to promote stable high level colonization (Figure 7B, “CrΔeae/pInv–Int395” and “CrΔeae/pInv–Int100”). In some instances, the degree of colonization by C. rodentiumΔeae/pInv–Int100 was somewhat less than C. rodentiumΔeae/pInv–Int395 after ten days post-infection, but remained >∼106/g feces throughout the 25- to 31-days of infection (Figure 7B and data not shown). Thus, it appears that the C-terminal 100 residues of intimin contain the activity that promotes colonization of antibiotic pre-treated mice. The finding that this portion of intimin is incapable of binding to Tir is consistent with the hypothesis that a Tir-independent function of intimin provides acritical colonization activity in animals with altered flora.
Discussion
Intimin, encoded by eae, is an adhesin essential for colonization by AE pathogens (Donnenberg et al., 1993a; Schauer and Falkow, 1993b; Tzipori et al., 1995; Dean-Nystrom et al., 1998; Ritchie et al., 2003). Allelic variation of intimin has been associated with differences in colonization of intestinal samples ex vivo (Fitzhenry et al., 2002; Mundy et al., 2007), and inoculation of gnotobiotic piglets with an EHEC strain harboring a plasmid encoding EPEC intimin resulted in colonization of the small intestine, a property not possessed by the same strain harboring a plasmid encoding EHEC intimin (Tzipori et al., 1995). To avoid potential effects related solely to high copy number expression of intimin, in this study we precisely replaced the endogenous (chromosomal) eae coding sequence of EHEC with EPEC eae. In fact, we found that the recombinant strain was incapable of efficiently colonizing the small intestine and infected the piglet indistinguishably from its isogenic wild type EHEC parent. A previous study utilizing a newborn (not gnotobiotic) piglet model also failed to note differences in tissue tropism due to intimin allele, although that study was not designed to carefully assess tissue tropism (Donnenberg et al., 1993a). Preliminary findings suggest that neither overexpression of EHEC nor EPEC intimin is associated with clear change in EHEC tissue tropism in gnotobiotic piglets, suggesting that simple overexpression does not account for the previously described tropism differences (Michael J. Brady and Saul Tzipori, unpublished observations).
To assess potential functional differences specific to intimin to particular alleles in a second animal model, we infected mice with C. rodentium strains expressing endogenous intimin, or intimin of canonical EPEC or EHEC strains. We found that EHEC intimin, like EPEC intimin, promoted efficient intestinal colonization and wild type disease. Notably, previous studies indicated that C. rodentium expressing a chimeric intimin composed of the 554 N-terminal residues of EPEC intimin fused to the 380 C-terminal residues of EHEC intimin colonized the murine intestine approximately a 100-fold less efficiently than wild type C. rodentium and was entirely defective for inducing disease (Hartland et al., 2000; Mundy et al., 2007). The N-terminal 554 residues of EPEC and EHEC intimin are 97% identical, and although we cannot rule out the possibility that an important (perhaps Tir-binding) activity of EHEC intimin is diminished by the exchange of its N-terminus for that of EPEC intimin, an alternative hypothesis is that differences in the murine host strain contribute to the apparent discrepant results; previous studies demonstrating allele-specific differences utilized Swiss NIH or C3H/Hej mice (Hartland et al., 2000; Mundy et al., 2007), whereas we utilized C57BL/6 mice in the current study. Additionally, anecdotal evidence suggests that the degree of murine infection by C. rodentium is dependent on the commercial source of mice (M. McBee, personal communication), a factor that might influence the composition of the intestinal microbiome. Regardless, the demonstration here that EHEC intimin can confer apparently full biological function in this infection model provides a facile experimental system in which to assess either EHEC intimin function or therapeutic strategies targeting this important virulence factor.
Intimin has the capacity to recognize mammalian proteins, such as β1-chain integrins or nucleolin, activities that have been postulated to facilitate colonization by promoting initial bacterial attachment to mucosal surfaces (Frankel et al., 1996a; Sinclair and O’Brien, 2002). Expression of invasin, a high affinity β1-chain integrin ligand, conferred on C. rodentium the ability to enter mammalian cells in vitro, indicating that the invasin integrin binding domain was presented in a functional form on the bacterial surface. Invasin did not, however, provide consistent intimin function in promoting murine colonization by C. rodentium, indicating that the β1-chain integrin binding activity is not the sole activity of intimin required for biological function.
The above finding is predicted from the demonstration that an essential receptor for intimin is the type III effector Tir, which becomes localized in the plasma membrane after translocation (Kenny et al., 1997). Tir-binding by intimin is required for high level attachment to cultured mammalian cells (Liu et al., 1999), and Tir-deficient mutants of AE pathogens, like intimin mutants, are incapable of intestinal colonization (Marches et al., 2000; Deng et al., 2003; Ritchie et al., 2003). We found that C. rodentium expressing EHEC intimin was capable of Tir-mediated binding to mammalian cells. A C. rodentium strain expressing the intimin–invasin chimera Inv–Int395, which contains three intimin immunoglobulin-like domains and the C type lectin-like domain (Frankel et al., 1995; Luo et al., 2000), also efficiently bound to mammalian cells. C. rodentium expressing Inv–Int181, which contains a region that is capable of binding Tir when expressed as a recombinant protein, did not bind primed cells or unmanipulated cells. Previous analysis of recombinant intimin derivatives revealed that sequences N-terminal to the minimal 181-residue Tir-recognition domain may influence binding (Liu et al., 1999). Interestingly however, a laboratory E. coli K12 strain expressing Inv–Int181, in contrast to C. rodentium expressing this chimera, bound to mammalian cells that had been pre-infected with EPEC (Liu et al., 1999). LPS O antigen can sterically hinder the recognition of invasin in the outer membrane (Voorhis et al., 1991), and the strain-specific difference in the activity of Inv–Int181 may reflect differences in O antigen length and/or density.
Intimin-mediated clustering of Tir in the plasma membrane promotes the formation of an actin assembly complex beneath bound bacteria, leading to actin assembly (Campellone et al., 2004a; Touze et al., 2004). Indeed, C. rodentium expressing EHEC intimin not only bound to monolayers that were primed with EPECΔeae, but also triggered robust actin assembly. In contrast, whereas C. rodentium expressing Inv–Int395 was capable of Tir-mediated binding to mammalian monolayers, this interaction did not trigger efficient pedestal formation. Given the extensive sequence differences between invasin and intimin, any number of factors may account for the partial function of Inv–Int395 in pedestal formation. However, multimeric interactions greatly facilitate the efficiency of actin assembly (Blasutig et al., 2008; Padrick et al., 2008; Sallee et al., 2008), and it is possible that Tir-binding by C. rodentium producing Inv–Int395 did not result in the high-density membrane clustering of Tir that triggers efficient actin assembly. Consistent with this hypothesis, C. rodentium expressing Inv–Int395 generated foci of Tir in infected cells at a significantly lower efficiency did C. rodentium expressing full-length C. rodentium, EPEC, or EHEC intimin. Indeed, whereas latex beads coated at high concentrations with C-terminal fragments of purified EHEC intimin stimulated robust actin assembly on monolayers pre-infected with EPECΔeae, E. coli K12 strains expressing invasin–intimin hybrids containing the equivalent regions of intimin did not (Liu et al., 1999).
The finding that C. rodentium expressing either EHEC intimin or Inv–Int395 both bound to Tir but only the former triggered actin pedestal formation in vitro, provided an opportunity to determine if pedestal formation correlated with intestinal colonization. Indeed, whereas C. rodentium expressing EHEC intimin was capable of colonizing mice to levels indistinguishable from levels attained by wild type C. rodentium, C. rodentium expressing Inv–Int395 colonized mice at levels a million-fold lower, barely above background, and were cleared from the mice by day five post-infection. C. rodentiumΔeae expressing Inv–Int395 also was not associated with any manifestations of disease. A correlation between the ability to generate robust pedestal in vitro and efficient colonization of the mammalian host has previously been described. First, an EHEC strain lacking a second translocated effector, EspFU (also known as TccP), remains capable of Tir translocation and intimin-mediated binding but is incapable of stimulating robust actin assembly (Campellone et al., 2004b; Garmendia et al., 2004), and is defective (albeit mildly) in late-stage colonization in infant rabbits (Ritchie et al., 2008). Second, a C. rodentium encoding a mutant Tir that binds intimin but is deficient in downstream actin signaling is out-competed late in infection by a wild type strain during co-infection experiments (Crepin et al., 2010). Thus, mutants of any of the three bacterial factors directly involved in pedestal formation, intimin, Tir, and EspFU, that specifically diminish pedestal formation in vitro also diminish colonization in vivo. A genetically engineered C. rodentium strain that generates pedestals using an additional mechanism did not display a competitive advantage over a wild type strain (Girard et al., 2009), suggesting that even though inefficient pedestal formation is associated with diminished colonization, pedestal formation enhanced over wild type levels does not lead to enhanced colonization. The means by which a threshold pedestal formation activity may be required for maximal intestinal colonization is currently unclear, but the observation that the EHECΔespFU mutant formed smaller than wild type aggregates on the intestinal epithelium of piglets and showed a late colonization defect in infant rabbits (Ritchie et al., 2008) suggests that pedestals may stabilize bacterial interaction with the mucosal surface or otherwise promote expansion of the infectious niche at that site.
Whereas Tir-mediated actin pedestal formation appears to contribute to colonization of gnotobiotic piglets and conventional mice, infection of streptomycin pre-treated revealed a Tir-independent function of intimin. Intimin function in this model was largely independent of intimin allele – C. rodentium expressing EPEC or EHEC intimin colonized these mice at high levels for at least two weeks, although they both showed somewhat diminished colonization relative to wild type C. rodentium beyond that time point. Most notably, whereas C. rodentiumΔeae was incapable of stably colonizing antibiotic-treated mice, C. rodentiumΔtir colonized these mice at least as well as wild type C. rodentium. The ability of intimin to bind host cells or components of host cells such as β1-chain integrins or nucleolin has been previously mapped to C-terminal portions of intimin (Frankel et al., 1994, 1996a; Sinclair and O’Brien, 2002, 2004). We found that the colonization activity of intimin in this infection model was retained by intimin derivative sharboring C-terminal regions of intimin, even if such derivatives (e.g., Inv–Int100, containing only the C-terminal 100 residues of intimin) were incapable of binding Tir. Intimin also contributes to the disruption of epithelial barrier function in vitro in a Tir-independent manner (Dean and Kenny, 2004). Notably, the Tir-independent intimin activities in cell attachment or barrier disruption have been characterized entirely by in vitro assays, and the contribution of these activities to colonization and disease during mammalian infection has not been documented. A comparison of these Tir-independent intimin in vitro activities to intimin function in promoting colonization of streptomycin-treated mice might provide insight into novel activities of intimin relevant to colonization of a mammalian host.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Joseph Newman for helpful discussion and technical assistance. We thank the UMASS DERC core facility for fixing, sectioning, and staining all tissue sections and also Lara Strittmatter and Greg Hendricks of the UMass Electron Microscopy Core Facility. The anti-EHEC intimin antibody was a generous gift from Alison O’Brien and the anti-OmpA antibody was a kind gift from Carol Kumamoto. This work was supported by PHS grant CA63112 from the NCI to David B. Schauer and R01-AI46454 to John M. Leong. Steven A. Luperchio was supported by NIEHS Training Grant ES07020.
References
Barthold, S. W., Coleman, G. L., Bhatt, P. N., Osbaldiston, G. W., and Jonas, A. M. (1976). The etiology of transmissible murine colonic hyperplasia. Lab. Anim. Sci. 26, 889–894.
Batchelor, M., Prasannan, S., Daniell, S., Reece, S., Connerton, I., Bloomberg, G., Dougan, G., Frankel, G., and Matthews, S. (2000). Structural basis for recognition of the translocated intimin receptor (Tir) by intimin from enteropathogenic Escherichia coli. EMBO J. 19, 2452–2464.
Blasutig, I. M., New, L. A., Thanabalasuriar, A., Dayarathna, T. K., Goudreault, M., Quaggin, S. E., Li, S. S., Gruenheid, S., Jones, N., and Pawson, T. (2008). Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol. Cell Biol. 28, 2035–2046.
Brady, M. J., Campellone, K. G., Ghildiyal, M., and Leong, J. M. (2007). Enterohaemorrhagic and enteropathogenic Escherichia coli Tir proteins trigger a common Nck-independent actin assembly pathway. Cell. Microbiol. 9, 2242–2253.
Brady, M. J., Radhakrishnan, P., Liu, H., Magoun, L., Murphy, K. C., Mukherjee, J., Donohue-Rolfe, A., Tzipori, S., and Leong, J. M. (2011). Enhanced actin pedestal formation by enterohemorrhagic Escherichia coli O157:H7 adapted to the mammalian host. Front. Microbiol. 2:226. doi: 10.3389/fmicb.2011.00226
Campellone, K. G. (2010). Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia coli: Tir, EspFu and actin pedestal assembly. FEBS J. 277, 2390–2402.
Campellone, K. G., Giese, A., Tipper, D. J., and Leong, J. M. (2002). A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol. Microbiol. 43, 1227–1241.
Campellone, K. G., and Leong, J. M. (2005). Nck-independent actin assembly is mediated by two phosphorylated tyrosines within enteropathogenic Escherichia coli Tir. Mol. Microbiol. 56, 416–432.
Campellone, K. G., Rankin, S., Pawson, T., Kirschner, M. W., Tipper, D. J., and Leong, J. M. (2004a). Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J. Cell Biol. 164, 407–416.
Campellone, K. G., Robbins, D., and Leong, J. M. (2004b). EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. Cell 7, 217–228.
Campellone, K. G., Roe, A. J., Lobner-Olesen, A., Murphy, K. C., Magoun, L., Brady, M. J., Donohue-Rolfe, A., Tzipori, S., Gally, D. L., Leong, J. M., and Marinus, M. G. (2007). Increased adherence and actin pedestal formation by dam-deficient enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 63, 1468–1481.
Chen, H. D., and Frankel, G. (2005). Enteropathogenic Escherichia coli: unravelling pathogenesis. FEMS Microbiol. Rev. 29, 83–98.
Crepin, V. F., Girard, F., Schuller, S., Phillips, A. D., Mousnier, A., and Frankel, G. (2010). Dissecting the role of the Tir:Nck and Tir:IRTKS/IRSp53 signalling pathways in vivo. Mol. Microbiol. 75, 308–323.
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645.
Dean, P., and Kenny, B. (2004). Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol. Microbiol. 54, 665–675.
Dean, P., and Kenny, B. (2011). Cell-surface nucleolin is sequestered into EPEC microcolonies and may play a role during infection. Microbiology 157, 1761–1767.
Dean-Nystrom, E. A., Bosworth, B. T., Moon, H. W., and O’Brien, A. D. (1998). Escherichia coli O157:H7 requires intimin for enteropathogenicity in calves. Infect. Immun. 66, 4560–4563.
Deng, W., Vallance, B. A., Li, Y., Puente, J. L., and Finlay, B. B. (2003). Citrobacter rodentium translocated intimin receptor (Tir) is an essential virulence factor needed for actin condensation, intestinal colonization and colonic hyperplasia in mice. Mol. Microbiol. 48, 95–115.
DeVinney, R., Stein, M., Reinscheid, D., Abe, A., Ruschkowski, S., and Finlay, B. B. (1999). Enterohemorrhagic Escherichia coli O157:H7 produces Tir, which is translocated to the host cell membrane but is not tyrosine phosphorylated. Infect. Immun. 67, 2389–2398.
Donnenberg, M. S., and Kaper, J. B. (1991). Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59, 4310–4317.
Donnenberg, M. S., Tacket, C. O., James, S. P., Losonsky, G., Nataro, J. P., Wasserman, S. S., Kaper, J. B., and Levine, M. M. (1993a). Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J. Clin. Invest. 92, 1412–1417.
Donnenberg, M. S., Tzipori, S., McKee, M. L., O’Brien, A. D., Alroy, J., and Kaper, J. B. (1993b). The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J. Clin. Invest. 92, 1418–1424.
Finlay, B. B., and Falkow, S. (1988). Comparison of the invasion strategies used by Salmonella cholerae-suis, Shigella flexneri and Yersinia enterocolitica to enter cultured animal cells: endosome acidification is not required for bacterial invasion or intracellular replication. Biochimie 70, 1089–1099.
Fitzhenry, R. J., Pickard, D. J., Hartland, E. L., Reece, S., Dougan, G., Phillips, A. D., and Frankel, G. (2002). Intimin type influences the site of human intestinal mucosal colonisation by enterohaemorrhagic Escherichia coli O157:H7. Gut 50, 180–185.
Frankel, G., Candy, D. C., Everest, P., and Dougan, G. (1994). Characterization of the C-terminal domains of intimin-like proteins of enteropathogenic and enterohemorrhagic Escherichia coli, Citrobacter freundii, and Hafnia alvei. Infect. Immun. 62, 1835–1842.
Frankel, G., Candy, D. C., Fabiani, E., Adu-Bobie, J., Gil, S., Novakova, M., Phillips, A. D., and Dougan, G. (1995). Molecular characterization of a carboxy-terminal eukaryotic-cell-binding domain of intimin from enteropathogenic Escherichia coli. Infect. Immun. 63, 4323–4328.
Frankel, G., Lider, O., Hershkoviz, R., Mould, A. P., Kachalsky, S. G., Candy, D., Cahalon, L., Humphries, M. J., and Dougan, G. (1996a). The cell-binding domain of intimin from enteropathogenic Escherichia coli binds to beta1 integrins. J. Biol. Chem. 271, 20359–20364.
Frankel, G., Phillips, A. D., Novakova, M., Field, H., Candy, D. C., Schauer, D. B., Douce, G., and Dougan, G. (1996b). Intimin from enteropathogenic Escherichia coli restores murine virulence to a Citrobacter rodentium eaeA mutant: induction of an immunoglobulin A response to intimin and EspB. Infect. Immun. 64, 5315–5325.
Frankel, G., and Phillips, A. D. (2008). Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell. Microbiol. 10, 549–556.
Garmendia, J., Phillips, A. D., Carlier, M. F., Chong, Y., Schuller, S., Marches, O., Dahan, S., Oswald, E., Shaw, R. K., Knutton, S., and Frankel, G. (2004). TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 6, 1167–1183.
Ghaem-Maghami, M., Simmons, C. P., Daniell, S., Pizza, M., Lewis, D., Frankel, G., and Dougan, G. (2001). Intimin-specific immune responses prevent bacterial colonization by the attaching-effacing pathogen Citrobacter rodentium. Infect. Immun. 69, 5597–5605.
Girard, F., Batisson, I., Frankel, G. M., Harel, J., and Fairbrother, J. M. (2005). Interaction of enteropathogenic and Shiga toxin-producing Escherichia coli and porcine intestinal mucosa: role of intimin and Tir in adherence. Infect. Immun. 73, 6005–6016.
Girard, F., Crepin, V. F., and Frankel, G. (2009). Modelling of infection by enteropathogenic Escherichia coli strains in lineages 2 and 4 ex vivo and in vivo by using Citrobacter rodentium expressing TccP. Infect. Immun. 77, 1304–1314.
Hartland, E. L., Huter, V., Higgins, L. M., Goncalves, N. S., Dougan, G., Phillips, A. D., MacDonald, T. T., and Frankel, G. (2000). Expression of intimin gamma from enterohemorrhagic Escherichia coli in Citrobacter rodentium. Infect. Immun. 68, 4637–4646.
Isberg, R. R., and Leong, J. M. (1990). Multiple β1 chain integrins are receptors for invasin, a protein that promoted bacterial penetration into mammalian cells. Cell 60, 861–871.
Isberg, R. R., Voorhis, D. L., and Falkow, S. (1987). Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell 50, 769–778.
Jerse, A. E., Yu, J., Tall, B. D., and Kaper, J. B. (1990). A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. U.S.A. 87, 7839–7843.
Kaper, J. B., Nataro, J. P., and Mobley, H. L. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140.
Kenny, B., DeVinney, R., Stein, M., Reinscheid, D. J., Frey, E. A., and Finlay, B. B. (1997). Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91, 511–520.
Kenny, B., and Finlay, B. B. (1995). Protein secretion by enteropathogenic Escherichia coli is essential for transducing signals to epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 92, 7991–7995.
Liu, H., Magoun, L., Luperchio, S., Schauer, D. B., and Leong, J. M. (1999). The Tir-binding region of enterohaemorrhagic Escherichia coli intimin is sufficient to trigger actin condensation after bacterial-induced host cell signalling. Mol. Microbiol. 34, 67–81.
Liu, H., Radhakrishnan, P., Magoun, L., Prabu, M., Campellone, K. G., Savage, P., He, F., Schiffer, C. A., and Leong, J. M. (2002). Point mutants of EHEC intimin that diminish Tir recognition and actin pedestal formation highlight a putative Tir binding pocket. Mol. Microbiol. 45, 1557–1573.
Lommel, S., Benesch, S., Rohde, M., Wehland, J., and Rottner, K. (2004). Enterohaemorrhagic and enteropathogenic Escherichia coli use different mechanisms for actin pedestal formation that converge on N-WASP. Cell. Microbiol. 6, 243–254.
Luo, Y., Frey, E. A., Pfuetzner, R. A., Creagh, A. L., Knoechel, D. G., Haynes, C. A., Finlay, B. B., and Strynadka, N. C. (2000). Crystal structure of enteropathogenic Escherichia coli intimin-receptor complex. Nature 405, 1073–1077.
Luperchio, S. A., and Schauer, D. B. (2001). Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 3, 333–340.
Marches, O., Nougayrede, J. P., Boullier, S., Mainil, J., Charlier, G., Raymond, I., Pohl, P., Boury, M., De Rycke, J., Milon, A., and Oswald, E. (2000). Role of Tir and intimin in the virulence of rabbit enteropathogenic Escherichia coli serotype O103:H2. Infect. Immun. 68, 2171–2182.
McDaniel, T. K., Jarvis, K. G., Donnenberg, M. S., and Kaper, J. B. (1995). A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 92, 1664–1668.
Melton-Celsa, A. R., Darnell, S. C., and O’Brien, A. D. (1996). Activation of Shiga-like toxins by mouse and human intestinal mucus correlates with virulence of enterohemorrhagic Escherichia coli O91:H21 isolates in orally infected, streptomycin-treated mice. Infect. Immun. 64, 1569–1576.
Mohawk, K. L., and O’Brien, A. D. (2011). Mouse models of Escherichia coli O157:H7 infection and Shiga toxin injection. J. Biomed. Biotechnol. 2011, 258185.
Moon, H. W., Whipp, S. C., Argenzio, R. A., Levine, M. M., and Giannella, R. A. (1983). Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 41, 1340–1351.
Mundy, R., MacDonald, T. T., Dougan, G., Frankel, G., and Wiles, S. (2005). Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706.
Mundy, R., Schuller, S., Girard, F., Fairbrother, J. M., Phillips, A. D., and Frankel, G. (2007). Functional studies of intimin in vivo and ex vivo: implications for host specificity and tissue tropism. Microbiology 153, 959–967.
Murphy, K. C., and Campellone, K. G. (2003). Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4, 11. doi: 10.1186/1471-2199-4-11
Newman, J. V., Zabel, B. A., Jha, S. S., and Schauer, D. B. (1999). Citrobacter rodentium espB is necessary for signal transduction and for infection of laboratory mice. Infect. Immun. 67, 6019–6025.
Nicholls, L., Grant, T. H., and Robins-Browne, R. M. (2000). Identification of a novel genetic locus that is required for in vitro adhesion of a clinical isolate of enterohaemorrhagic Escherichia coli to epithelial cells. Mol. Microbiol. 35, 275–288.
Padrick, S. B., Cheng, H. C., Ismail, A. M., Panchal, S. C., Doolittle, L. K., Kim, S., Skehan, B. M., Umetani, J., Brautigam, C. A., Leong, J. M., and Rosen, M. K. (2008). Hierarchical regulation of WASP/WAVE proteins. Mol. Cell 32, 426–438.
Phillips, A. D., and Frankel, G. (2000). Intimin-mediated tissue specificity in enteropathogenic Escherichia coli interaction with human intestinal organ cultures. J. Infect. Dis. 181, 1496–1500.
Riley, L. W., Remis, R. S., Helgerson, S. D., McGee, H. B., Wells, J. G., Davis, B. R., Hebert, R. J., Olcott, E. S., Johnson, L. M., Hargrett, N. T., Blake, P. A., and Cohen, M. L. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308, 681–685.
Ritchie, J. M., Brady, M. J., Riley, K. N., Ho, T. D., Campellone, K. G., Herman, I. M., Donohue-Rolfe, A., Tzipori, S., Waldor, M. K., and Leong, J. M. (2008). EspFU, a type III-translocated effector of actin assembly, fosters epithelial association and late-stage intestinal colonization by E. coli O157:H7. Cell. Microbiol. 10, 836–847.
Ritchie, J. M., Thorpe, C. M., Rogers, A. B., and Waldor, M. K. (2003). Critical roles for stx2, eae, and Tir in enterohemorrhagic Escherichia coli-induced diarrhea and intestinal inflammation in infant rabbits. Infect. Immun. 71, 7129–7139.
Rosenshine, I., Ruschkowski, S., Stein, M., Reinscheid, D. J., Mills, S. D., and Finlay, B. B. (1996). A pathogenic bacterium triggers epithelial signals to form a functional bacterial receptor that mediates actin pseudopod formation. EMBO J. 15, 2613–2624.
Sallee, N. A., Rivera, G. M., Dueber, J. E., Vasilescu, D., Mullins, R. D., Mayer, B. J., and Lim, W. A. (2008). The pathogen protein EspF(U) hijacks actin polymerization using mimicry and multivalency. Nature 454, 1005–1008.
Schauer, D. B., and Falkow, S. (1993a). Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect. Immun. 61, 2486–2492.
Schauer, D. B., and Falkow, S. (1993b). The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect. Immun. 61, 4654–4661.
Simmons, C. P., Clare, S., Ghaem-Maghami, M., Uren, T. K., Rankin, J., Huett, A., Goldin, R., Lewis, D. J., MacDonald, T. T., Strugnell, R. A., Frankel, G., and Dougan, G. (2003). Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 71, 5077–5086.
Sinclair, J. F., and O’Brien, A. D. (2002). Cell surface-localized nucleolin is a eukaryotic receptor for the adhesin intimin-gamma of enterohemorrhagic Escherichia coli O157:H7. J. Biol. Chem. 277, 2876–2885.
Sinclair, J. F., and O’Brien, A. D. (2004). Intimin types alpha, beta, and gamma bind to nucleolin with equivalent affinity but lower avidity than to the translocated intimin receptor. J. Biol. Chem. 279, 33751–33758.
Spears, K. J., Roe, A. J., and Gally, D. L. (2006). A comparison of enteropathogenic and enterohaemorrhagic Escherichia coli pathogenesis. FEMS Microbiol. Lett. 255, 187–202.
Tabor, S., and Richardson, C. C. (1985). A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U.S.A. 82, 1074–1078.
Touze, T., Hayward, R. D., Eswaran, J., Leong, J. M., and Koronakis, V. (2004). Self-association of EPEC intimin mediated by the beta-barrel-containing anchor domain: a role in clustering of the Tir receptor. Mol. Microbiol. 51, 73–87.
Tran Van Nhieu, G., and Isberg, R. R. (1993). Bacterial internalization mediated by β1 chain integrins is determined by ligand affinity and receptor density. EMBO J. 12, 1887–1895.
Tzipori, S., Gunzer, F., Donnenberg, M. S., de Montigny, L., Kaper, J. B., and Donohue-Rolfe, A. (1995). The role of the eaeA gene in diarrhea and neurological complications in a gnotobiotic piglet model of enterohemorrhagic Escherichia coli infection. Infect. Immun. 63, 3621–3627.
Vallance, B. A., Deng, W., Knodler, L. A., and Finlay, B. B. (2002). Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. 70, 2070–2081.
Vingadassalom, D., Kazlauskas, A., Skehan, B., Cheng, H. C., Magoun, L., Robbins, D., Rosen, M. K., Saksela, K., and Leong, J. M. (2009). Insulin receptor tyrosine kinase substrate links the E. coli O157:H7 actin assembly effectors Tir and EspF(U) during pedestal formation. Proc. Natl. Acad. Sci. U.S.A. 106, 6754–6759.
Voorhis, D. L., Dillon, S., Formal, S. B., and Isberg, R. R. (1991). An O antigen can interfere with the function of the Yersinia pseudotuberculosis invasin protein. Mol. Microbiol. 5, 317–325.
Wadolkowski, E. A., Burris, J. A., and O’Brien, A. D. (1990a). Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 58, 2438–2445.
Wadolkowski, E. A., Sung, L. M., Burris, J. A., Samuel, J. E., and O’Brien, A. D. (1990b). Acute renal tubular necrosis and death of mice orally infected with Escherichia coli strains that produce Shiga-like toxin type II. Infect. Immun. 58, 3959–3965.
Yanisch-Perron, C., Vieira, J., and Messing, J. (1985). Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33, 103–119.
Keywords: Citrobacter rodentium, intimin, enterohemorrhagic Escherichia coli, invasin, enteropathogenic Escherichia coli
Citation: Mallick EM, Brady MJ, Luperchio SA, Vanguri VK, Magoun L, Liu H, Sheppard BJ, Mukherjee J, Donohue-Rolfe A, Tzipori S, Leong JM and Schauer DB (2012) Allele- and Tir-independent functions of intimin in diverse animal infection models. Front. Microbio. 3:11. doi: 10.3389/fmicb.2012.00011
Received: 05 April 2011; Accepted: 07 January 2012;
Published online: 31 January 2012.
Edited by:
Elizabeth L. Hartland, The University of Melbourne, AustraliaReviewed by:
Olivier Marchès, Queen Mary University of London, UKAlan Phillips, Institute of Child Health, UK
Copyright: © 2012 Mallick, Brady, Luperchio, Vanguri, Magoun, Liu, Sheppard, Mukherjee, Donohue-Rolfe, Tzipori, Leong and Schauer. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: John M. Leong, Department of Molecular Biology and Microbiology, Tufts University School of Medicine, 136 Harrison Avenue, Boston, MA 02111, USA. e-mail:am9obi5sZW9uZ0B0dWZ0cy5lZHU=
†Deceased