Dendritic cells during Epstein Barr virus infection
 Christian Münz
Christian Münz- Viral Immunobiology, Institute of Experimental Immunology, University of Zurich, Zurich, Switzerland
Epstein Barr virus (EBV) causes persistent infection in more than 90% of the human adult population and is associated with 2% of all tumors in humans. This γ-herpes virus infects primarily human B and epithelial cells, but it has been reported to be sensed by dendritic cells (DCs) during primary infection. These activated DCs are thought to contribute to innate restriction of EBV infection and initiate EBV-specific adaptive immune responses via cross-priming. The respective evidence and their potential importance for EBV-specific vaccine development will be discussed in this review.
Infection and Tumorigenesis by Epstein Barr Virus
Epstein Barr virus (EBV) was discovered 50 years ago in a cell line (EB1) from an African child with Burkitt’s lymphoma (Epstein et al., 1964). Despite this association with lymphomas and carcinomas, including Hodgkin’s lymphoma and nasopharyngeal carcinoma (Kutok and Wang, 2006; Cesarman, 2014), EBV is carried without symptoms by the vast majority of persistently infected individuals, which account for more than 90% of the adult human population (Rickinson et al., 2014). EBV-associated malignancies arise with increased frequency in immunosuppressed patients, for example after transplantation (post-transplant lymhoproliferative disease or PTLD), immunosuppressive co-infections such as HIV, or primary genetic immunodeficiencies (like X-linked lymphoproliferative disease or XLP). These findings indicate that asymptomatic chronic infection with EBV results in part from continuous virus-specific immune control. Mainly cellular immunity by natural killer (NK) and T cells seems to mediate this immune control (Rickinson et al., 2014), and some EBV-associated malignancies can even be cured by adoptive transfer of EBV-specific T-cell lines (Gottschalk et al., 2005). Some evidence has been provided that dendritic cells (DCs) sense EBV infection and are involved in the priming of these protective innate and adaptive immune responses. This evidence and its relevance for EBV-specific vaccine development will be discussed in this review.
Selective Host Cell Tropism of EBV
Dendritic cells are probably not initiating EBV-specific immune control after getting directly infected by the virus. Although it has been reported that EBV can enter monocyte precursors of DCs, no EBV antigen expression could be found in these studies and only CMV-promoter-driven green fluorescent protein (GFP) expression of recombinant EBV was detected after infection (Li et al., 2002; Guerreiro-Cacais et al., 2004). Indeed, the main host cell of EBV is the human B cell. In healthy EBV carriers, memory B cells seem to constitute the site of long-term persistence (Babcock et al., 1998). Latency 0 in these memory B cells is associated with no viral protein expression but transcription of EBV encoded small RNAs (EBERs) and micro RNAs (miRNAs). EBV uses its envelope glycoprotein gp 350 to attach to complement receptors 1 and 2 (CD35 and CD21) on the surface of B cells, uses gp42 binding to MHC class II molecules and finally the trimeric complex of gH, gL, and gB for fusion with the membrane (Connolly et al., 2011). The B-cell compartment is reached by EBV after transmission via saliva in the tonsils. Naïve B-cell infection at these sites is associated with the expression of eight latent EBV proteins and the non-translated RNAs (Babcock et al., 2000). This latency III or growth program drives infected B cells into proliferation and is present in PTLD and HIV-associated diffuse large B cell lymphomas (DLBCL). The six EBV nuclear antigen (EBNA1, 2, 3A, 3B, 3C, and LP) and two latent membrane proteins (LMP1 and LMP2) are sufficiently immunogenic, so that tumors expressing all of these only emerge under severe immunosuppression. One outcome of this EBV-driven activation of naïve B cells is thought to be their differentiation into germinal center B cells. In these centroblasts and centrocytes, only three EBV proteins retain their expression (EBNA1, LMP1, and LMP2; Babcock et al., 2000). This latency II pattern, which is also found in Hodgkin’s lymphoma, was proposed to rescue EBV-infected B cells from deletion by mimicking B-cell receptor engagement and T-cell help via CD40 signaling by LMP2 and LMP1, respectively. Therefore, EBV rescues its infected B cells from the germinal center reaction in order to gain access into the long-lived memory B-cell pool. From there, the virus reactivates into lytic replication and infectious particle production after B-cell receptor stimulation. Indeed, lytic EBV replication has primarily been found in plasma cells (Laichalk and Thorley-Lawson, 2005) and B-cell receptor cross-linking can initiate replication in some Burkitt’s lymphoma cell lines (Takada, 1984). If this reactivation occurs in mucosal secondary lymphoid tissues, the virus can be secreted into saliva and transmitted to new individuals.
Efficient transmission, however, might require an additional amplification step in epithelial cells, which have been found to be preferentially infected by free virus from the basolateral side (Tugizov et al., 2003). Integrin binding by BMRF2 and gH/gL for gB-mediated fusion might mediate this epithelial cell infection and B-cell-produced EBV seems to be particularly good at it (Borza and Hutt-Fletcher, 2002). This basolateral infection during shedding into saliva might give rise to the EBV-associated carcinomas at mucosal sites, including nasopharyngeal carcinoma. The biased tropism of EBV toward B and epithelial cells suggests that DCs are most likely not directly infected by EBV, but process viral particles and dying EBV-infected B and epithelial cells for both immune detection of infection and initiation of innate and adaptive immune responses.
Innate Immune Recognition of EBV
Epstein Barr virus is a double-stranded DNA virus of the lymphocryptovirus subgroup of γ-herpesviridae. As such, the viral particle carries double-stranded linear DNA without much methylation, which can be detected by the toll-like receptor (TLR) 9 (Casanova et al., 2011). Indeed, EBV DNA triggers TLR9-mediated recognition of the virus in plasmacytoid DCs, B cells, and monocytes (Fiola et al., 2010; Severa et al., 2013; Younesi et al., 2014). Once EBV enters B cells, it circularizes its DNA to episomes, which then get heavily methylated (Woellmer et al., 2012). Therefore, viral DNA of dying EBV-infected B cells is probably invisible to TLR9. In contrast to mice, human conventional DCs (cDCs) do not express TLR9 (Iwasaki and Medzhitov, 2004). Instead TLR2 and 3 have been implicated in EBV recognition by macrophages and conventional DCs (Gaudreault et al., 2007; Ariza et al., 2009; Iwakiri et al., 2009). While the TLR2 ligand of EBV remains enigmatic, EBERs have been proposed as TLR3 ligands (Iwakiri et al., 2009). It appears that EBERs are released from infected B cells in complex with the EBER-binding protein La. Apart from TLR3-binding, EBERs can also stimulate the intracellular pathogen associated molecular pattern (PAMP) receptor retinoic acid-inducible gene 1 (RIG-I; Samanta et al., 2006). Both TLR-3 and RIG-I recognize double-stranded RNA (dsRNA) and EBERs seem to form hairpin structures that allow their recognition by these two intra- and extracellular receptors for dsRNA. Therefore, EBV seems to stimulate both pDCs and cDCs by viral DNA in viral particles and viral RNA released from infected cells, respectively (Figure 1).
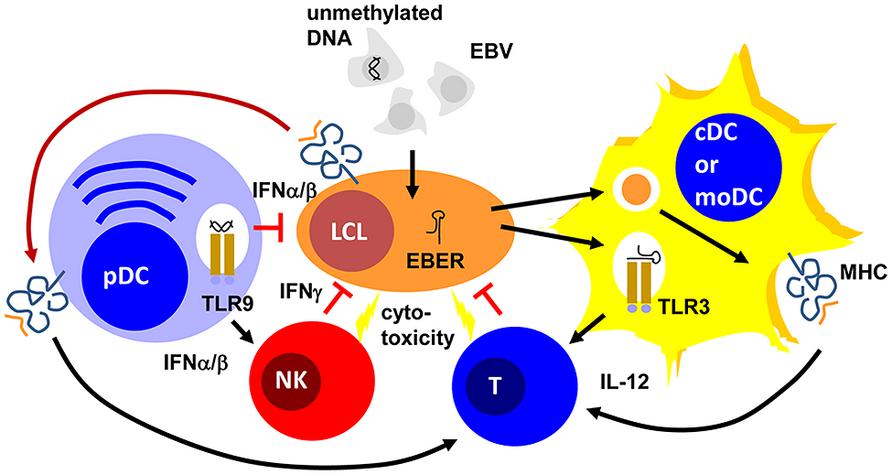
FIGURE 1. Plasmacytoid, conventional and monocyte-derived DCs might contribute to EBV specific immune control. Unmethylated DNA of EBV particles and EBERs of EBV-infected B cells (LCLs) mature plasmacytoid (pDCs) and conventional or monocyte-derived DCs (cDCs or moDCs) via TLR9 or TLR3 stimulation, respectively. These mature pDC and cDC or moDC populations activate natural killer (NK) and T cells via type I interferon (IFNα/β) or interleukin 12 (IL-12) secretion, respectively. For T-cell stimulation by MHC presentation they acquire EBV antigens either via phagocytosis of dying LCLs (for cDCs and moDCs) or trogocytosis of EBV epitope presenting MHC complexes (pDCs). The activated NK and primed T cells then delay primary EBV infection via IFNγ and kill infected cells. PDCs can also delay primary EBV infection via IFNα/β production.
Innate Immune Control of EBV
These DC populations seem to play significant roles during primary EBV infection. Along these lines pDCs are potent sources of type I interferons (IFNα and β; Reizis et al., 2011). In particular, human pDCs produce high levels of IFNα2 and α14 (Meixlsperger et al., 2013). IFNα and β have been found to restrict B-cell transformation by EBV during the first 24 h of infection (Lotz et al., 1985). While this study suggested that the protective type I IFN effect directly targeted infected B cells, a PBMC transfer model into SCID mice suggested that the IFNα/β-dependent effect was mediated via NK cell activation and EBV-specific memory T cells (Lim et al., 2006). In this study, PBMC reconstituted SCID mice were challenged with EBV infection with and without prior deletion or enrichment of pDCs in the transferred PBMCs. They observed pDC- and TLR9-dependent IFNα production in response to primary EBV infection. Furthermore, EBV-induced lymphoma formation was observed after pDC depletion and this was mediated by decreased NK and EBV-specific memory T-cell activation in the transferred PBMCs of healthy EBV carriers. Therefore, type I IFN, probably produced primarily by pDCs during primary EBV infection, seems to have a protective function against EBV-induced B-cell transformation, early by directly targeting B cells and later by activating protective lymphocyte populations.
One of these protective lymphocyte populations are NK cells. Their activity is stimulated by DCs during viral infections in mice (Lucas et al., 2007). In particular, surface presentation of IL-15 is important for this NK cell activation by DCs. Similarly, human DCs are able to activated NK cells (Ferlazzo et al., 2002). IL-12, IL-15, and IFNα are primarily involved in NK cell activation by human monocyte-derived DCs (moDCs; Ferlazzo et al., 2004; Strowig et al., 2008). This NK cell activation occurs most potently after TLR3-mediated maturation of moDCs and preferentially stimulates CD56bright killer immunoglobulin-like receptor (KIR)-negative NK cells (Brilot et al., 2007; Strowig et al., 2008). In tonsils, the primary site of EBV infection, this NK cell subset produces large amounts of type II IFN (IFN; Strowig et al., 2008; Lünemann et al., 2013). IFNγ can restrict primary B-cell transformation by EBV during the first 3–4 days (Lotz et al., 1985; Strowig et al., 2008; Lünemann et al., 2013). It seems to delay LMP1 expression during the first 3–5 days after primary EBV infection of B cells (Strowig et al., 2008). Accordingly, DC stimulation of NK cells restricts B-cell transformation by EBV in vitro, especially when the NK cells are derived from tonsils and are part of the CD56brightKIR- NK cell subset (Strowig et al., 2008; Lünemann et al., 2013). Apart from this cytokine-mediated delay of B-cell transformation, NK cells might also directly kill infected B cells undergoing lytic EBV replication (Pappworth et al., 2007; Chijioke et al., 2013). This restricts lytically EBV replicating B cells in vitro and in vivo in a mouse model of human immune component reconstitution after CD34+ hematopoietic progenitor cell (HPC) transfer (Pappworth et al., 2007; Chijioke et al., 2013). In this mouse model, NK cell activation can be also achieved by TLR3 agonist injection (Strowig et al., 2010) and this adjuvant elicits potent DC maturation (Meixlsperger et al., 2013). Thus, DCs mediate innate immune control during EBV infection by IFNα/β production of pDCs and activate NK cells that delay B-cell transformation via IFNγ and eliminate lytic EBV replication by killing of virus-producing cells (Figure 1).
DCs in the Priming of Adaptive EBV-Specific Immune Control
Apart from innate lymphocyte activation during EBV infection, DCs are most likely also involved in the priming of EBV-specific, protective T-cell responses (Rickinson et al., 2014). Indeed, in vitro EBV infection of B cells is very inefficient in priming EBV-specific T cells from PBMCs of EBV-negative donors (Bickham et al., 2003). However, addition of autologous moDCs allows priming of EBV-specific T cells in these cultures. For this purpose, DCs presumably cross-present EBV antigens from dying EBV-infected B cells in these cultures. Indeed, such dying EBV-transformed B cells can be presented on MHC class I and II molecules of moDCs for CD8+ and CD4+ T-cell stimulation, respectively (Münz et al., 2000; Subklewe et al., 2001). However, some observations call this prominent role of DCs in the priming of EBV-specific T-cell responses into question. For example, EBV-transformed lymphoblastoid B cell lines (LCLs) were able to prime EBV-specific CD4+ T cells at low frequencies, but these could be expanded after CD25 targeted selection (Savoldo et al., 2002). Furthermore, it was found that CD8+ T cells primarily recognize early, but not late lytic EBV antigens, apart from some prominent latent EBV antigens (Hislop et al., 2007). Indeed, only subdominant CD8+ T-cell responses were documented against late lytic EBV antigens (Abbott et al., 2013), while CD4+ T-cell responses against late lytic antigens can be observed (Adhikary et al., 2006). Since EBV encoded inhibitors of MHC class I antigen presentation get expressed during early viral gene expression and, therefore, would primarily prevent late lytic antigen presentation on MHC class I, this hierarchy in lytic EBV antigen recognition by CD8+ T cells was taken as an indication that EBV infected cells prime this CD8+ T-cell hierarchy. An alternative explanation, however, could be that DCs prime these different EBV specificities similarly by cross-presentation, and the preference for early lytic EBV antigen recognition then is established by amplification of the respective T-cell responses via restimulation by EBV-infected B cells. A similar amplification was recently observed for the EBNA1 antigen targeted to the endocytic receptor DEC-205 on DCs and B cells (Leung et al., 2013b). Among the human DC subsets, priming of EBV-specific T-cell responses has been ascribed or demonstrated primarily for phagocytic DC subsets. These would include CD1c+ or CD141+ cDCs, and moDCs. However, a recent study also reported that pDCs might trogocytose MHC class I peptide complexes, presenting EBV epitopes (Bonaccorsi et al., 2014). This cross-dressing with LCL-derived MHC class I complexes is also sufficient to stimulate EBV-specific CD8+ T cells. Therefore, different DC populations could contribute to EBV-specific T-cell priming to establish protective EBV-specific immune control in healthy carriers of this human tumor virus.
Conclusion and Outlook
These EBV-specific T cells are clearly the protective entity during the adaptive immune responses against EBV (Rickinson et al., 2014). How they are primed requires further investigation, because vaccination against EBV should probably engage the respective DC populations both by adjuvant choice as well as antigen targeting to the relevant DC subsets. Indeed with the advent of mice with reconstituted human immune system compartments, which recapitulate primary EBV infection and EBV-associated lymphomagenesis (Leung et al., 2013a), it becomes feasible to define DC populations that are involved in the priming of protective immune responses in vivo. In this preclinical model, CD4+ and CD8+ T cells mediate immune control over EBV infection and B-cell lymphoma development (Strowig et al., 2009) and protective EBV-specific CD4+ T cells can be primed with vaccine candidates (Gurer et al., 2008; Meixlsperger et al., 2013). Therefore, it should be feasible to define important DC populations that initiate EBV-specific immune control by for example antibody depletion (Meixlsperger et al., 2013), in order to then refine vaccination approaches that protect from EBV infection challenge. With such smart vaccine formulations that are directed against the most relevant DC populations EBV negative adolescents with a high risk to suffer symptomatic EBV infection could be vaccinated and their predisposition to develop Hodgkin’s lymphoma or multiple sclerosis attenuated (Hjalgrim et al., 2003; Thacker et al., 2006).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in the author’s laboratory is supported by Cancer Research Switzerland (KFS-3234-08-2013), the Association for International Cancer Research (11-0516), KFSPMS, and KFSPHLD of the University of Zurich, the Baugarten Foundation, the Sobek Foundation, Fondation Acteria, the Wellcome Trust, the Leukaemia and Lymphoma Research, the Medical Research Council and the Swiss National Science Foundation (310030_143979 and CRSII3_136241).
References
Abbott, R. J., Quinn, L. L., Leese, A. M., Scholes, H. M., Pachnio, A., and Rickinson, A. B. (2013). CD8+ T cell responses to lytic EBV infection: late antigen specificities as subdominant components of the total response. J. Immunol. 191, 5398–5409. doi: 10.4049/jimmunol.1301629
Adhikary, D., Behrends, U., Moosmann, A., Witter, K., Bornkamm, G. W., and Mautner, J. (2006). Control of Epstein-Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J. Exp. Med. 203, 995–1006. doi: 10.1084/jem.20051287
Ariza, M. E., Glaser, R., Kaumaya, P. T., Jones, C., and Williams, M. V. (2009). The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182, 851–859. doi: 10.4049/jimmunol.182.2.851
Babcock, G. J., Decker, L. L., Volk, M., and Thorley-Lawson, D. A. (1998). EBV persistence in memory B cells in vivo. Immunity 9, 395–404. doi: 10.1016/S1074-7613(00)80622-6
Babcock, J. G., Hochberg, D., and Thorley-Lawson, A. D. (2000). The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13, 497–506. doi: 10.1016/S1074-7613(00)00049-2
Bickham, K., Goodman, K., Paludan, C., Nikiforow, S., Tsang, M. L., Steinman, R. M.,et al. (2003). Dendritic cells initiate immune control of Epstein-Barr virus transformation of B lymphocytes in vitro. J. Exp. Med. 198, 1653–1663. doi: 10.1084/jem.20030646
Bonaccorsi, I., Morandi, B., Antsiferova, O., Costa, G., Oliveri, D., Conte, R.,et al. (2014). Membrane transfer from tumor cells overcomes deficient phagocytic ability of plasmacytoid dendritic cells for the acquisition and presentation of tumor antigens. J. Immunol. 192, 824–832. doi: 10.4049/jimmunol.1301039
Borza, C. M., and Hutt-Fletcher, L. M. (2002). Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8, 594–599. doi: 10.1038/nm0602-594
Brilot, F., Strowig, T., Roberts, S. M., Arrey, F., and Münz, C. (2007). NK cell survival mediated through the regulatory synapse with human dendritic cells requires IL-15Ralpha. J. Clin. Invest. 117, 3316–3329. doi: 10.1172/JCI31751
Casanova, J. L., Abel, L., and Quintana-Murci, L. (2011). Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu. Rev. Immunol. 29, 447–491. doi: 10.1146/annurev-immunol-030409-101335
Cesarman, E. (2014). Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 9, 349–372. doi: 10.1146/annurev-pathol-012513-104656
Chijioke, O., Muller, A., Feederle, R., Barros, M. H., Krieg, C., Emmel, V.,et al. (2013). Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. 5, 1489–1498. doi: 10.1016/j.celrep.2013.11.041
Connolly, S. A., Jackson, J. O., Jardetzky, T. S., and Longnecker, R. (2011). Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9, 369–381. doi: 10.1038/nrmicro2548
Epstein, M. A., Achong, B. G., and Barr, Y. M. (1964). Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1, 702–703. doi: 10.1016/S0140-6736(64)91524-7
Ferlazzo, G., Thomas, D., Pack, M., Paludan, C., Schmid, D., Strowig, T.,et al. (2004). Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc. Natl. Acad. Sci. U.S.A. 101, 16606–16611. doi: 10.1073/pnas.0407522101
Ferlazzo, G., Tsang, M. L., Moretta, L., Melioli, G., Steinman, R. M., and Münz, C. (2002). Human dendritic cells activate resting NK cells and are recognized via the NKp30 receptor by activated NK cells. J. Exp. Med. 195, 343–351. doi: 10.1084/jem.20011149
Fiola, S., Gosselin, D., Takada, K., and Gosselin, J. (2010). TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 185, 3620–3631. doi: 10.4049/jimmunol.0903736
Gaudreault, E., Fiola, S., Olivier, M., and Gosselin, J. (2007). Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 81, 8016–8024. doi: 10.1128/JVI.00403-07
Gottschalk, S., Rooney, C. M., and Heslop, H. E. (2005). Post-transplant lymphoproliferative disorders. Annu. Rev. Med. 56, 29–44. doi: 10.1146/annurev.med.56.082103.104727
Guerreiro-Cacais, A. O., Li, L., Donati, D., Bejarano, M. T., Morgan, A., Masucci, M. G.,et al. (2004). Capacity of Epstein-Barr virus to infect monocytes and inhibit their development into dendritic cells is affected by the cell type supporting virus replication. J. Gen. Virol. 85, 2767–2778. doi: 10.1099/vir.0.80140-0
Gurer, C., Strowig, T., Brilot, F., Pack, M., Trumpfheller, C., Arrey, F.,et al. (2008). Targeting the nuclear antigen 1 of Epstein Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood 112, 1231–1239. doi: 10.1182/blood-2008-03-148072
Hislop, A. D., Taylor, G. S., Sauce, D., and Rickinson, A. B. (2007). Cellular responses to viral infection in humans: lessons from epstein-barr virus. Annu. Rev. Immunol. 25, 587–617. doi: 10.1146/annurev.immunol.25.022106.141553
Hjalgrim, H., Askling, J., Rostgaard, K., Hamilton-Dutoit, S., Frisch, M., Zhang, J. S.,et al. (2003). Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N. Engl. J. Med. 349, 1324–1332. doi: 10.1056/NEJMoa023141
Iwakiri, D., Zhou, L., Samanta, M., Matsumoto, M., Ebihara, T., Seya, T.,et al. (2009). Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 206, 2091–2099. doi: 10.1084/jem.20081761
Iwasaki, A., and Medzhitov, R. (2004). Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995. doi: 10.1038/ni1112
Kutok, J. L., and Wang, F. (2006). Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 1, 375–404. doi: 10.1146/annurev.pathol.1.110304.100209
Laichalk, L. L., and Thorley-Lawson, D. A. (2005). Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79, 1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005
Leung, C., Chijioke, O., Gujer, C., Chatterjee, B., Antsiferova, O., Landtwing, V.,et al. (2013a). Infectious diseases in humanized mice. Eur. J. Immunol. 43, 2246–2254. doi: 10.1002/eji.201343815
Leung, C. S., Maurer, M. A., Meixlsperger, S., Lippmann, A., Cheong, C., Zuo, J.,et al. (2013b). Robust T-cell stimulation by Epstein-Barr virus-transformed B cells after antigen targeting to DEC-205. Blood 121, 1584–1594. doi: 10.1182/blood-2012-08-450775
Li, L., Liu, D., Hutt-Fletcher, L., Morgan, A., Masucci, M. G., and Levitsky, V. (2002). Epstein-Barr virus inhibits the development of dendritic cells by promoting apoptosis of their monocyte precursors in the presence of granulocyte macrophage-colony-stimulating factor and interleukin-4. Blood 99, 3725–3734. doi: 10.1182/blood.V99.10.3725
Lim, W. H., Kireta, S., Russ, G. R., and Coates, P. T. (2006). Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection and delay EBV-related mortality in humanized NOD-SCID mice. Blood 109, 1043–1050. doi: 10.1182/blood-2005-12-024802
Lotz, M., Tsoukas, C. D., Fong, S., Carson, D. A., and Vaughan, J. H. (1985). Regulation of Epstein-Barr virus infection by recombinant interferons. Selected sensitivity to interferon-gamma. Eur. J. Immunol. 15, 520–525. doi: 10.1002/eji.1830150518
Lucas, M., Schachterle, W., Oberle, K., Aichele, P., and Diefenbach, A. (2007). Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26, 503–517. doi: 10.1016/j.immuni.2007.03.006
Lünemann, A., Vanoaica, L. D., Azzi, T., Nadal, D., and Münz, C. (2013). A distinct subpopulation of human natural killer cells restricts B cell transformation by the Epstein-Barr virus. J. Immunol. 191, 4989–4995. doi: 10.4049/jimmunol.1301046
Meixlsperger, S., Leung, C. S., Ramer, P. C., Pack, M., Vanoaica, L. D., Breton, G.,et al. (2013). CD141+ dendritic cells produce prominent amounts of IFN-alpha after dsRNA recognition and can be targeted via DEC-205 in humanized mice. Blood 121, 5034–5044. doi: 10.1182/blood-2012-12-473413
Münz, C., Bickham, K. L., Subklewe, M., Tsang, M. L., Chahroudi, A., Kurilla, M. G.,et al. (2000). Human CD4+ T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 191, 1649–1660. doi: 10.1084/jem.191.10.1649
Pappworth, I. Y., Wang, E. C., and Rowe, M. (2007). The switch from latent to productive infection in Epstein-Barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 81, 474–482. doi: 10.1128/JVI.01777-06
Reizis, B., Bunin, A., Ghosh, H. S., Lewis, K. L., and Sisirak, V. (2011). Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol. 29, 163–183. doi: 10.1146/annurev-immunol-031210-101345
Rickinson, A. B., Long, H. M., Palendira, U., Münz C., and Hislop, A. (2014). Cellular immune controls over Epstein-Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol. 35, 159–169. doi: 10.1016/j.it.2014.01.003
Samanta, M., Iwakiri, D., Kanda, T., Imaizumi, T., and Takada, K. (2006). EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 25, 4207–4214. doi: 10.1038/sj.emboj.7601314
Savoldo, B., Cubbage, M. L., Durett, A. G., Goss, J., Huls, M. H., Liu, Z.,et al. (2002). Generation of EBV-specific CD4+ cytotoxic T cells from virus naive individuals. J. Immunol. 168, 909–918. doi: 10.4049/jimmunol.168.2.909
Severa, M., Giacomini, E., Gafa, V., Anastasiadou, E., Rizzo, F., Corazzari, M.,et al. (2013). EBV stimulates TLR- and autophagy-dependent pathways and impairs maturation in plasmacytoid dendritic cells: implications for viral immune escape. Eur. J. Immunol. 43, 147–158. doi: 10.1002/eji.201242552
Strowig, T., Brilot, F., Arrey, F., Bougras, G., Thomas, D., Muller, W. A.,et al. (2008). Tonsilar natural killer cells restrict B cell transformation by the Epstein Barr virus via IFN-γ. PLoS Pathog. 4:e40027. doi: 10.1371/journal.ppat.0040027
Strowig, T., Chijioke, O., Carrega, P., Arrey, F., Meixlsperger, S., Ramer, P. C.,et al. (2010). Human NK cells of mice with reconstituted human immune system components require preactivation to acquire functional competence. Blood 116, 4158–4167. doi: 10.1182/blood-2010-02-270678
Strowig, T., Gurer, C., Ploss, A., Liu, Y. F., Arrey, F., Sashihara, J.,et al. (2009). Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J. Exp. Med. 206, 1423–1434. doi: 10.1084/jem.20081720
Subklewe, M., Paludan, C., Tsang, M. L., Mahnke, K., Steinman, R. M., and Münz, C. (2001). Dendritic cells cross-present latency gene products from Epstein-Barr virus-transformed B cells and expand tumor-reactive CD8+ killer T cells. J. Exp. Med. 193, 405–411. doi: 10.1084/jem.193.3.405
Takada, K. (1984). Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33, 27–32. doi: 10.1002/ijc.2910330106
Thacker, E. L., Mirzaei, F., and Ascherio, A. (2006). Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann. Neurol. 59, 499–503. doi: 10.1002/ana.20820
Tugizov, S. M., Berline, J. W., and Palefsky, J. M. (2003). Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 9, 307–314. doi: 10.1038/nm830
Woellmer, A., Arteaga-Salas, J. M., and Hammerschmidt, W. (2012). BZLF1 Governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. 8:e1002902. doi: 10.1371/journal.ppat.1002902
Younesi, V., Shirazi, F. G., Memarian, A., Amanzadeh, A., Jeddi-Tehrani, M., and Shokri, F. (2014). Assessment of the effect of TLR7/8, TLR9 agonists and CD40 ligand on the transformation efficiency of Epstein-Barr virus in human B lymphocytes by limiting dilution assay. Cytotechnology 66, 95–105. doi: 10.1007/s10616-013-9542-x
Keywords: plasmacytoid dendritic cells, conventional dendritic cells, monocyte-derived dendritic cells, natural killer cells, T cells
Citation: MüNz C (2014) Dendritic cells during Epstein Barr virus infection. Front. Microbiol. 5:308. doi: 10.3389/fmicb.2014.00308
Received: 08 April 2014; Paper pending published: 22 May 2014;
Accepted: 04 June 2014; Published online: 20 June 2014.
Edited by:
Laura Hertel, Children’s Hospital Oakland Research Institute, USAReviewed by:
Stephen Gottschalk, Baylor College of Medicine, USAAndrew Hislop, University of Birmingham, UK
Copyright © 2014 Münz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Münz, Viral Immunobiology, Institute of Experimental Immunology, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland e-mail:Y2hyaXN0aWFuLm11ZW56QHV6aC5jaA==